

Abstract
Synaptic ribbons are thought to provide vesicles for continuous release in some retinal nonspiking neurons, yet recent studies indicate that genetic removal of the ribbon has little effect on release kinetics. To investigate vesicle replenishment at synaptic ribbons, we used total internal reflection fluorescence microscopy to image synaptic vesicles and ribbons in retinal bipolar cells of goldfish (Carassius auratus) of both sexes. Analysis of vesicles released by trains of 30 ms depolarizations revealed that most releasable vesicles reside within 300 nm of the ribbon center. A single 30 ms step to 0 mV was sufficient to deplete the membrane-proximal vesicle pool, while triggering rapid stepwise movements of distal vesicles along the ribbon and toward the plasma membrane. Replenishment only becomes rate-limiting for recovery from paired-pulse depression for interstimulus intervals shorter than 250 ms. For longer interstimulus intervals, vesicle movement down the ribbon is fast enough to replenish released vesicles, but newly arrived vesicles are not release-ready. Notably, the rates of vesicle resupply and maturation of newcomers are among the fastest measured optically at any synapse. Lastly, our data show that the delay in vesicle departure increases and vesicle speed decreases with multiple stimuli. Our results support a role for ribbons in the supply of vesicles for release, provide direct measurements of vesicle movement down the ribbon, and suggest that multiple factors contribute to paired-pulse depression.
SIGNIFICANCE STATEMENT Synaptic ribbons are macromolecular scaffolds that tether synaptic vesicles close to release sites in nonspiking neurons of the retina and cochlea. Because these neurons release neurotransmitter continuously, synaptic ribbons are assumed to act as platforms for supplying vesicles rapidly in the face of prolonged stimulation. Yet, ribbon synapses suffer from profound paired-pulse depression, which takes seconds to subside. We investigated the mechanistic origin of this phenomenon by directly imaging triggered vesicle movement and release at ribbon sites in retinal bipolar cells, and find that, although ribbon synapses deliver and prime vesicles faster than most conventional synapses, both vesicle absence and vesicle priming contribute to the long recovery from paired-pulse depression.
Keywords: bipolar cells, retina, ribbon synapses, synaptic depression, TIRF microscopy, vesicle release
Introduction
In nonspiking cells of the retina, cochlea, and lateral line, synaptic ribbons harbor a dense array of glutamatergic synaptic vesicles near the plasma membrane directly opposite postsynaptic receptors. Because synaptic ribbons reside in neurons that release for prolonged periods of time, it has long been suggested that ribbon transport provides vesicles for uninterrupted continuous release (for review, see Matthews and Fuchs, 2010; Moser et al., 2020). However, more recently, this idea has been challenged.
Ribeye is the most abundant protein in synaptic ribbons, and frame-shifting mutations to both ribeye genes in zebrafish leads to morphologic changes and mislocalization of ribbons in lateral line hair cells, but minimal effects on exocytosis, even in response to very long stimuli (Lv et al., 2016). Similarly, genetic disruption of ribeye in mouse leads to loss of all synaptic ribbons without change in the kinetics of release in bipolar cells (Maxeiner et al., 2016), or in the kinetics or amount of release in inner hair cells (Becker et al., 2018; Jean et al., 2018). Moreover, despite the abundance of vesicles near ribbon sites, retinal ribbon-type synapses exhibit profound depression in response to pairs of stimuli, suggesting that ribbons may not be especially adept at resupplying vesicles (Mennerick and Matthews, 1996; Sakaba et al., 1997; von Gersdorff and Matthews, 1997; Gomis et al., 1999; Burrone and Lagnado, 2000; DeVries, 2000; Singer and Diamond, 2003; Rabl et al., 2006; Choi et al., 2008; Innocenti and Heidelberger, 2008).
If, indeed, synaptic ribbons mark the location of exocytosis (Llobet et al., 2003; Midorikawa et al., 2007; Zenisek, 2008; Snellman et al., 2011; Vaithianathan and Matthews, 2014) and are responsible for the turnover of synaptic vesicles (LoGiudice et al., 2008; Matthews and Fuchs, 2010; Vaithianathan and Matthews, 2014), the time course of recovery from paired-pulse depression could reflect slow replenishment of release sites, slow maturation of newly arrived ribbon-attached vesicles, or both. In a previous study, Vaithianathan et al. (2016) demonstrated vectorial transport biased toward the membrane in response to depolarizing stimuli. We sought to take advantage of the superior signal-to-noise properties of total internal reflection fluorescence (TIRF) microscopy to better define the properties of this transport process. In addition, we investigated which mechanism, depletion or maturation, underlies the long recovery from synaptic depression in goldfish retinal bipolar cells.
By directly imaging released and docked vesicles at synaptic ribbon sites with TIRF microscopy, we found that vesicles on ribbons move toward the membrane in a rapid unidirectional manner and that vesicle replenishment only becomes rate-limiting for recovery from paired-pulse depression if interstimulus intervals are shorter than 250 ms. For longer interstimulus intervals, vesicle movement down the ribbon is fast enough to replenish released vesicles, but newcomer vesicles are not competent for release, indicating that both vesicle absence and biochemical steps downstream of vesicle arrival contribute to depression.
These observations are in stark contrast with those on conventional synapses of the hippocampus and auditory system, which report much slower resupply and vesicle priming rates (Midorikawa and Sakaba, 2015, 2017). Interestingly, a recently published report measuring vesicle trafficking and release in cerebellar mossy fiber terminals found rapid resupply of vesicles on the order of 190 ms following a single stimulus, suggesting that vesicle replenishment can be fast in the absence of a ribbon as well, albeit with much fewer vesicles (Miki et al., 2020). Together, our results point to the ribbon's role in the continuous supply of large numbers of vesicles for release.
Materials and Methods
All experiments were approved by the Yale Animal Care and Use Committee and were performed according to the ARVO Statement for the Use of Animals in Ophthalmic and Visual Research.
Cell preparation
Goldfish bipolar neurons were prepared as previously described (Zenisek et al., 2002; Joselevitch and Zenisek, 2009). Briefly, adult goldfish (Carassius auratus) were decapitated and eyes enucleated and hemisected. Retinas were isolated, cut into four to six pieces, and incubated at room temperature for 20 min in a low-Ca2+ solution designed to stop exocytosis, containing the following (in mM): 120 NaCl, 0.5 CaCl2, 2.5 KCl, 1.0 MgCl2, 10 glucose, 10 HEPES, 0.75 EGTA (260 mOsm, pH adjusted to 7.4 with NaOH), plus 1100 units/ml hyaluronidase (type V, Sigma Millipore). Next, the pieces of retina were washed in low-Ca2+ solution and placed for 30-35 min in a digestion medium consisting of low-Ca2+ solution with 35 units/ml papain (lyophilized powder; Sigma Millipore) and 0.5 mg/ml cysteine. Finally, the pieces of retina were rinsed and placed in low-Ca2+ solution in an oxygenated environment at 14°C until dissociated. Retinas were triturated mechanically with a fire-polished glass Pasteur pipette and plated onto a highly refractive coverslip (n488 = 1.78; Plan Optik AG) for recording and imaging. All recordings were performed within 90 min of dissociation.
FM1-43 loading, imaging, and data acquisition
For imaging studies, a selected cell terminal was puffed for 5 s with a solution containing 3–5 μM FM1-43 (Invitrogen) and the following (in mM): 2.5 CaCl2, 25 KCl, 97.5 NaCl, 1.0 MgCl2, 1 Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid, Sigma Millipore), and 10 HEPES (250 mOsm, pH adjusted to 7.4 with NaOH). This solution is expected to depolarize the terminals mildly to stimulate exocytosis and endocytosis, labeling a small fraction (1%–2%) of the total number of vesicles in each terminal and thus allowing us to resolve individual vesicles (Rouze and Schwartz, 1998; Zenisek et al., 2000). To estimate the likelihood of resolving individual events with this method, we calculated the frequency of “failures” in our experiments by determining the number of events per ribbon per stimulus across experiments. Visible events occurred at a rate of 0.14/ribbon/stimulus, which would predict that two labeled vesicles would be detectable at single ribbons 1.95% of the time.
The neuron was then washed by local superfusion for 30-60 min with the low-Ca2+ solution designed to stop exocytosis. In some experiments, 1 mM ADVASEP-7 (Sigma Millipore) was added to this washing solution in an effort to reduce background staining (Kay et al., 1999). After the wash, the superfusion was switched to the recording solution, containing the following (in mM): 120 NaCl, 2.5 CaCl2, 2.5 KCl, 1.0 MgCl2, 10 glucose, 10 HEPES, and 2 glutathione (260 mOsm, pH adjusted to 7.4 with NaOH).
For patch-clamp recordings, 8–12 MΩ thick-walled borosilicate electrodes (BF150-86-10HP, Sutter Instrument) were pulled with a P-97 Brown/Flaming Puller (Sutter Instrument) and filled with a solution containing the following (in mM): 100 Cs-methanesulfonate, 10 TEACl, 4 MgCl2, 10 HEPES, 0.5 EGTA, 10 ATP-Mg, 1 GTP-Li, and 1 glutathione (230 mOsm, pH adjusted to 7.2 with CsOH). For synaptic ribbon visualization, the pipette solution also contained 5 μM of a ribeye-binding peptide (rhodamine + EQTVPVDLSVARDR, molecular weight 1997.75), synthesized by the W.M. Keck Facility at Yale University.
Cells were viewed through an inverted microscope (IX70, Olympus) modified for through-the-objective TIRFM (Axelrod, 2001). A 488 nm wavelength beam from a solid-state laser (Coherent) or Argon-gas laser (Coherent) was used to image FM1-43 fluorescence, while a 561 nm laser (CV Melles Griot) was used to visualize the fluorescence of the ribeye-binding peptide. Both beams were expanded and focused off-axis onto the back focal plane of a 1.65 NA objective (Apo ×100 O HR, Olympus). Two shutters (Uniblitz) placed in the optical paths controlled the illumination. After leaving the objective, light entered immersion oil of high refractive index (n488 = 1.78, Cargille Labs) and then the glass coverslip of similar refractive index. The beam was totally internally reflected at the interface between the glass and the solution or cell, generating an evanescent field with a decay constant of ∼50 nm. Fluorescence was recorded using an on-chip amplified CCD camera (Cascade 512B, Photometrics). Image sequences were captured at either 30 Hz or 33 Hz with MetaMorph Software (Molecular Devices, RRID:SCR_002368).
Imaging data analysis
Results were analyzed with MetaMorph and MATLAB (The MathWorks, RRID:SCR_001622). To find the location of docked vesicles and ribbons, images were averaged and fit to a 2D-Gaussian function on an inclined plane. Each individual frame was also fit to a 2D-Gaussian function, and the SE in the location of the center in the x and y axes was used to estimate by propagation of errors the inaccuracy in determining the center of the object. For high-resolution localization, vesicles and/or ribbons with an error >20 nm in location were discarded. For other types of analyses, all vesicles within 300 nm of the nearest ribbon were used.
Movies with fusing, captured, or moving vesicles were visually selected, and square regions centered on the vesicle were excised for analysis using MATLAB subroutines. To estimate the timing of vesicle departure and arrival, the fluorescence of vesicles was measured in a 0.6-µm-diameter ROI and subtracted from the background fluorescence within a 1.2-µm-diameter concentric annulus. Fluorescence data points were fit with a Boltzmann function of the following form:
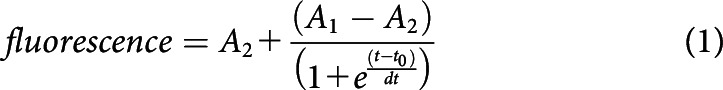 |
where A2 (maximal amplitude), A1 (minimal amplitude), t0 (function midpoint or half-maximal time), and dt are free parameters. The t value corresponding to 90% of A2 was defined as the arrival time of newly immobilized vesicles and calculated as follows:
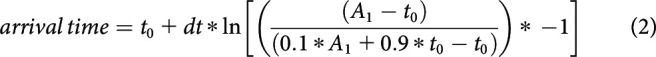 |
Similarly, the moment vesicles started moving after a voltage stimulus was called estimated departure time and defined as the t value corresponding to 10% of A2. It was calculated as follows:
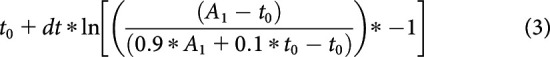 |
Other forms of analysis for estimating vesicle dynamics are described in the text.
To evaluate the distance traveled by a vesicle each time it makes a step toward the membrane, we calculated the vertical displacement for each vesicle (Dz) from the ratio of fluorescence between the start and end of the movement (A2/A1), according to a method described previously (Zenisek et al., 2000) as follows:
 |
where d is the decay constant of the evanescent field (50 nm in our experimental conditions).
Vesicle speed was determined by fitting the fluorescence profile of individual vesicles (or the mean fluorescence profile of all vesicles) to a single exponential followed by an abrupt stop, using a least-squares method for error minimization.
Experimental design and statistical analysis
Animals of both sexes were used in this study. Statistical analyses were performed using Origin Pro 8 software (OriginLab, RRID:SCR_014212). Data were checked for normality with Shapiro–Wilk tests. Pairwise comparison of non-normal data was performed with two-tailed Mann–Whitney tests; accordingly, medians are reported in addition to means.
Results
To investigate the properties of vesicle transport in bipolar cell synaptic terminals, synaptic vesicles in freshly dissociated bipolar cells from the goldfish retina were labeled with FM1-43, which loads into vesicles by endocytosis (Betz et al., 1992), and imaged using TIRF microscopy. Since vesicles are packed into the synaptic terminal at a higher density than can be resolved using light microscopy, labeling was restricted to a small fraction of the total vesicle pool to visualize individual synaptic vesicles (Zenisek et al., 2000). This was achieved by exposing cells briefly to FM1-43 in the presence of 25 mM K+ (see Materials and Methods). After loading with the dye, bipolar cells were voltage-clamped at the cell soma. In many experiments, a ribeye-binding peptide conjugated with rhodamine was added to the patch pipette (Zenisek et al., 2004; Joselevitch and Zenisek, 2009), allowing us to visualize ribbon sites in the same cells.
Immobilized synaptic vesicles gather at ribbon sites
Synaptic vesicles were visible as diffraction-limited spots after labeling. As described previously (Zenisek et al., 2000; Holt et al., 2004; Zenisek, 2008), most vesicles fluctuate in intensity with time and were visible for <200 ms, whereas a subset of vesicles retained a steady fluorescence intensity, indicating that they were immobile. When bipolar cells were periodically subjected to 30 ms step depolarizations from −60 to 0 mV, fusion of immobilized vesicles could be seen as a localized increase in spot fluorescence followed by the formation of a cloud of dye, as reported previously (Zenisek et al., 2000). An example of such an event is illustrated in Figure 1A, B, as well as in Movies 1 and 2.
Figure 1.
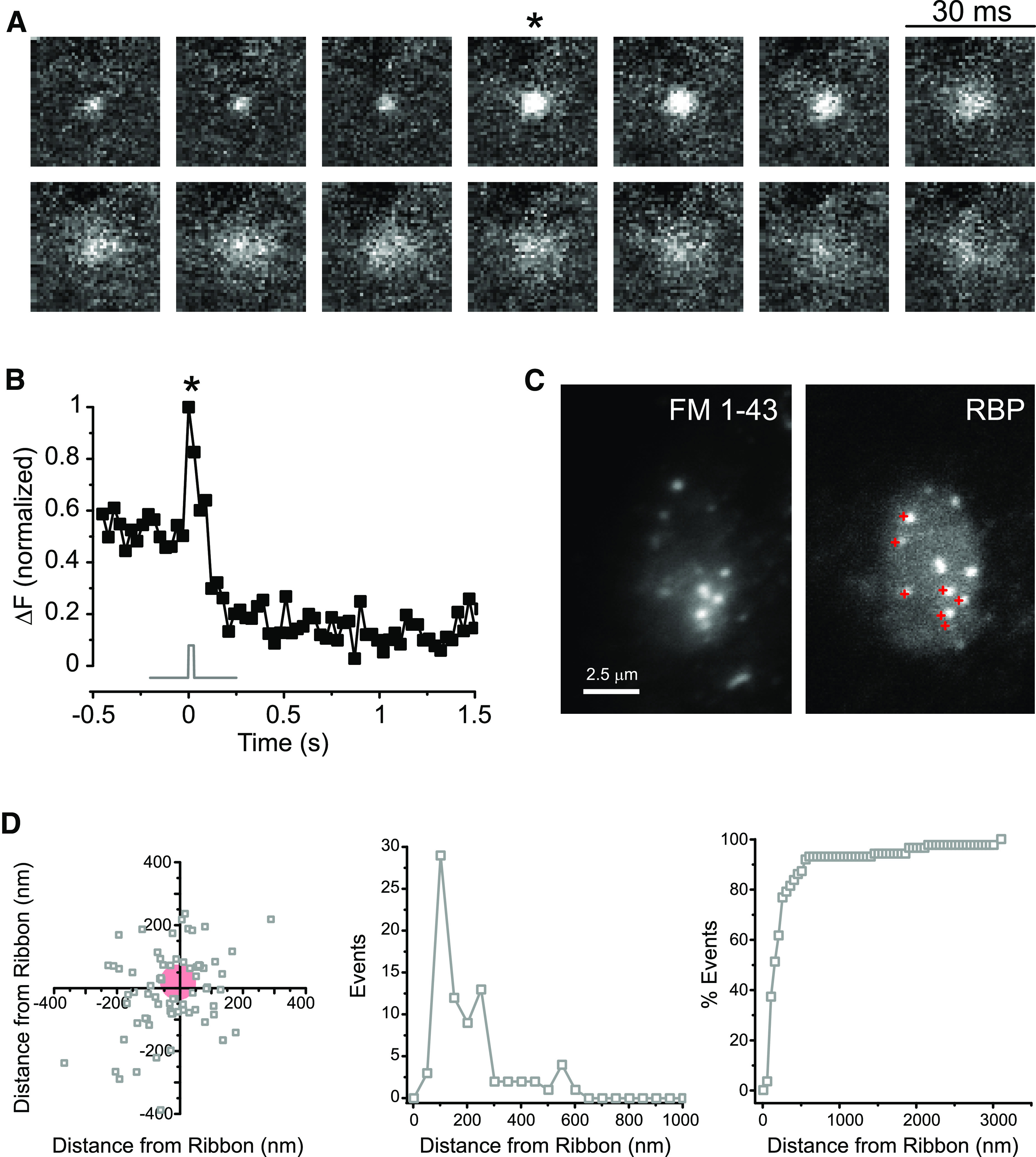
Released vesicles concentrate at ribbon sites. A, Consecutive video images from a docked vesicle that underwent fusion. Released vesicles appear as a cloud of dye and may be visible at the cell membrane for a long period of time. *The frame of a single 30 ms depolarization from −60 to 0 mV. B, The normalized fluorescence of the vesicle in A is plotted in relation to the timing of the voltage stimulus (gray trace). The release of vesicular contents is seen as an increase in fluorescence and correlates well to the change in membrane potential. *The timing of depolarization in A. C, Release events in a bipolar cell terminal seen with FM1-43 (left) correlate well with ribbons labeled in the same terminal by the ribeye-binding peptide (RBP, right). Red crosses in the RBP image represent released vesicles. The FM image is an average of 85 frames, and the RBP image is an average of 10 frames. D, High-resolution scatter plot (left), simple histogram (middle), and cumulative histogram (right) of 87 events observed in 25 records from 9 bipolar cells, showing that most vesicles docked within 300 nm of labeled ribbons. These active release zones correspond roughly to the base of synaptic ribbons. Red circle represents region devoid of vesicles near the center of the ribbon.
Squares represent the coordinates of the approach of two synaptic vesicles and fusion of one of them in response to a 30 ms voltage step from –60 to 0 mV. Movie is slowed 4.7×; frames were low pass filtered to remove high spatial frequency pixel noise.
Squares represent the coordinates of the fusion of three resident vesicles and subsequent approach and release of newcomers in response to a 30 ms voltage step from –60 to 0 mV. Movie is slowed 1.57×; frames were low pass filtered to remove high spatial frequency pixel noise.
Previous studies using optical techniques indicated that docked vesicles preferentially localize near ribbons (Zenisek, 2008; Vaithianathan et al., 2016) and that brief stimuli give rise to fusion events mostly near active zones (Zenisek et al., 2000) or labeled ribbon sites (Midorikawa et al., 2007), whereas longer depolarizations elicit extrasynaptic and nonvesicular fusion events (Zenisek et al., 2000; Coggins et al., 2007; Midorikawa et al., 2007). Like these previous reports, we found that vesicle docking and exocytosis colocalized well with ribbon sites. Figure 1C (left) shows a map of the location of fusing vesicles in one terminal (see Movie 3), marked as red crosses and superimposed on a TIRF image of the synaptic ribbon locations taken just after stimulating exocytosis (Fig. 1C, right). As can be observed in this example, fusion events were confined to the vicinity of synaptic ribbons.
Bipolar cell terminal loaded with FM1-43 and submitted to four consecutive 30 ms depolarizing steps from –60 to 0 mV at 480 ms interstimulus intervals. White circle represents timing of the stimulus. The first frame of the movie is the same terminal imaged with the 561 nm laser, showing the location of the labeled ribbons (same as in Fig. 1C, RBP). Movie is slowed 2×; frames were low pass filtered to remove high spatial frequency pixel noise.
To investigate the association between vesicles and ribbon in more detail, we mapped the distance of each vesicle relative to the nearest ribbon at high spatial resolution. For this, we localized the center of each fluorescent spot to subpixel accuracy, a strategy used to visualize small movements of molecular motors (Yildiz et al., 2003) and used by super-resolution microscopy techniques (Toomre and Bewersdorf, 2010). Overall, 79% of vesicles that could be localized with an error ≤20 nm (see Materials and Methods) were found within 300 nm of the center of the nearest ribbon, which agrees well with both the estimated location of release-ready vesicles in relation to Ca2+ channels and the spatial spread of Ca2+ microdomains at these ribbon synapses under similar experimental conditions (Burrone et al., 2002; Beaumont et al., 2005). The distribution of these vesicles relative to the ribbon center is shown as a scatter plot in Figure 1D (left), and replotted as a histogram of distances in Figure 1D (middle) and as a cumulative histogram in Figure 1D (right).
Since the base of a synaptic ribbon is ∼400 nm long (von Gersdorff et al., 1996), we propose that each of these active release zones corresponds to the base of a synaptic ribbon. It is interesting to note that we found a void of ∼50 nm radius in the center of the scatter plot (Fig. 1D, left). This roughly corresponds to the width of the ribbon along its minor axis (von Gersdorff et al., 1996) and suggests that vesicles do not dock directly underneath synaptic ribbons, probably because this is prevented by the protein complexes involved in anchoring ribbons at the membrane (Dick et al., 2003; tom Dieck et al., 2005). Indeed, the region directly beneath the ribbon appears devoid of vesicles in electron micrographs of bipolar cells (von Gersdorff et al., 1996). Evidently, bipolar cell synaptic release is mostly confined to ribbon sites when triggered by brief stimuli, as previously proposed (von Gersdorff et al., 1996; Zenisek et al., 2000; Midorikawa et al., 2007; Coggins and Zenisek, 2009; Datta et al., 2017). In the following sections, we refer to active zones as these sites of concentrated synaptic release and pool data from both ribeye-labeled cells, where direct correlation to ribbon sites is possible, and unlabeled cells, where the signal-to-noise ratio allows for better imaging of vesicles. The remaining analysis in this paper was restricted to this ribbon-associated pool of vesicles.
Vesicles move down the ribbon on stimulation
Bipolar cells exhibit at least two kinetic components of exocytosis in response to step depolarizations to 0 mV (Mennerick and Matthews, 1996; Sakaba et al., 1997; Neves and Lagnado, 1999; Zenisek et al., 2000; Singer and Diamond, 2003). The fastest component of exocytosis is rate-limited by the activation of the Ca2+ current and depleted with a time constant of few milliseconds (Mennerick and Matthews, 1996; Sakaba et al., 1997; Singer and Diamond, 2003). The rapidity of this component of release coupled with its relatively low sensitivity to Ca2+ (>10 μm for fast release) (Heidelberger et al., 1994; Beaumont et al., 2005) strongly suggests that it must arise from vesicles closest to the Ca2+ channels at or near the base of the ribbon. A second, slower component is depleted with a time constant of >250 ms (Mennerick and Matthews, 1996; Sakaba et al., 1997; Burrone and Lagnado, 2000) and was suggested to arise from vesicles both at active zones and at outlier locations (Mennerick and Matthews, 1996; Sakaba et al., 1997; Zenisek et al., 2000; Llobet et al., 2003; Singer and Diamond, 2003; Midorikawa et al., 2007; Zenisek, 2008; Mehta et al., 2014; Datta et al., 2017). This notion is further supported by the facts that (1) increasing the concentration of Ca2+ buffers affects the ultrafast pool less than the slower pool (Mennerick and Matthews, 1996; Sakaba et al., 1997; Pan et al., 2001; Burrone et al., 2002; Singer and Diamond, 2003; Beaumont et al., 2005) because the latter responds to global Ca2+ changes in the terminal, whereas the former responds to the size of Ca2+ microdomains close to sites of Ca2+ entry (Beaumont et al., 2005), and that (2) Ca2+ channels are tightly localized to ribbon sites (Zenisek et al., 2003).
We therefore looked more directly at the possibility that vesicles move down the ribbon in response to short stimuli. Bipolar cells were depolarized from −60 to 0 mV for 30 ms to elicit exocytosis of the vesicles docked at the base of the ribbons and subsequent replenishment. Vesicle movement toward the coverslip, as indicated by an increase in vesicle fluorescence without lateral spread of dye fluorescence, often followed these brief depolarizations (Fig. 2A). The limits to allowable geometries imposed by the plasma-membrane laying on the glass substrate suggest that ribbons were biased toward perpendicular to the coverslip in our experiments, at least within the depth of our evanescent field. Since (1) we looked specifically at regions around ribbons (Fig. 1C,D) and (2) a large number of vesicles were visible and immobile at a distance from the membrane before the stimulus or before release, whereas cytoplasmic vesicles exhibit high mobility (Zenisek et al., 2000; Holt et al., 2004; LoGiudice et al., 2008; Zenisek, 2008), it seems most likely these bound vesicles are ribbon-resident vesicles. It also makes it unlikely that, in most cases, vesicles moved laterally from the cytosol directly into the TIRF field. We cannot exclude, however, that vesicles from the cytosol attached to the ribbon at sites beyond our evanescent field, such as the apical half of the ribbon, as suggested by Vaithianathan and Matthews (2014), and then made their way down the ribbon and toward the membrane.
Figure 2.
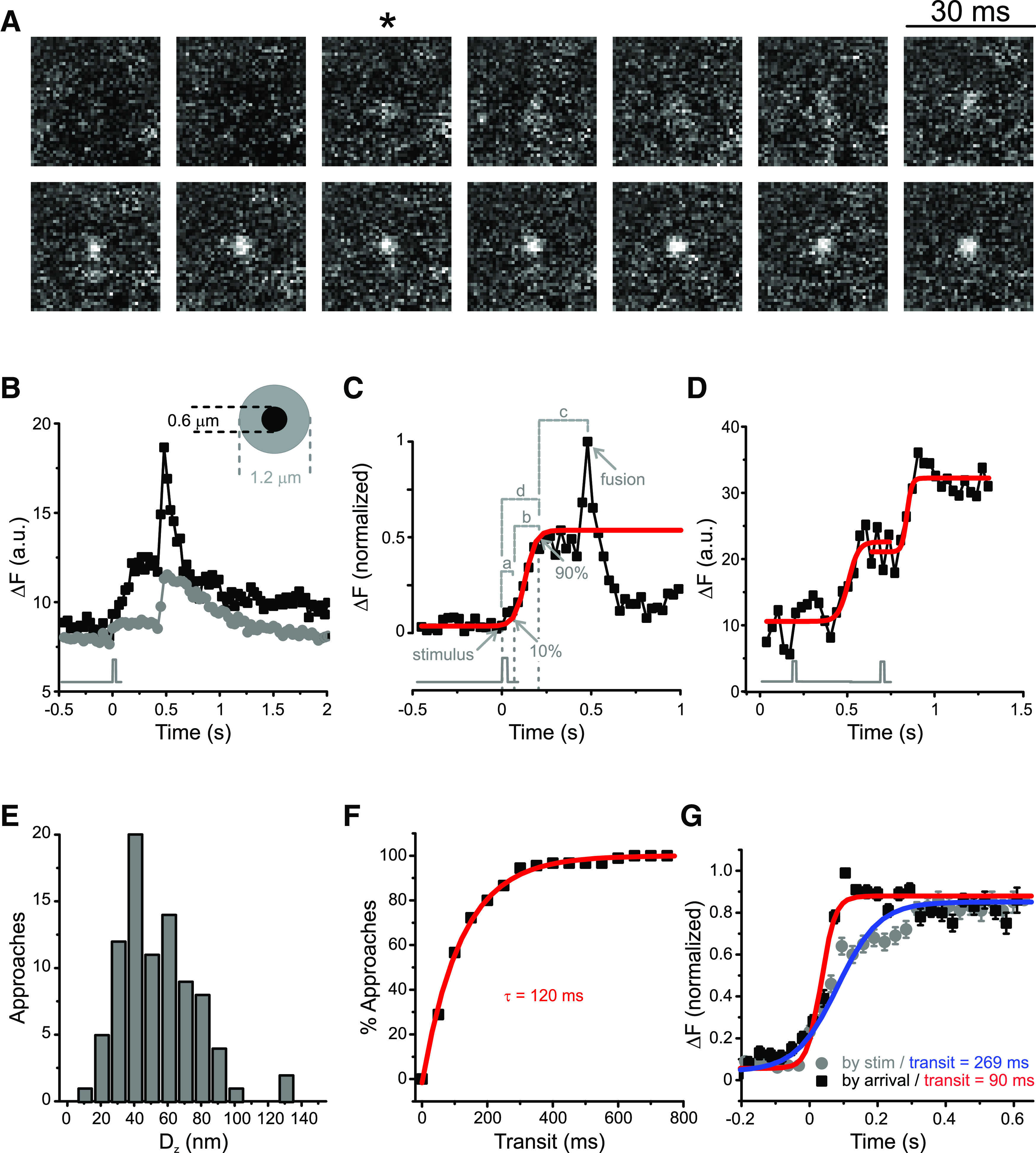
Vesicles move down the ribbon on stimulation. A, Voltage stimuli trigger capture and movement of vesicles move down the ribbon. Consecutive video images from a newly immobilized vesicle. *The frame of a single 30 ms depolarization from −60 to 0 mV. B, Quantifying triggered movement along the synaptic ribbon. Gray trace represents the timing of a single 30 ms depolarization from −60 to 0 mV. The fluorescence of a newly immobilized vesicle (black symbols) increases as the vesicle approaches the membrane, and this increase is strictly correlated with stimulus timing. The fluorescence is confined to a 0.6-µm-diameter circle (inset); the fluorescence of the surrounding area (gray symbols) does not increase in a similar fashion. C, The fluorescence profile of individual approaches (black squares) was fitted with a Boltzmann function (red trace), and the following parameters were measured for each vesicle: (a) delay for departure, defined as the difference between the estimated departure time (10% of the maximal amplitude of the Boltzmann function) and the stimulus time (gray trace under graph); (b) transit time, defined as the difference between the arrival time (90% of the maximal amplitude of the Boltzmann function) and the estimated departure time; (c) sitting time, defined as difference between the fusion time and the arrival time; and (d) delay for arrival, defined as the difference between the arrival time and the stimulus time. D, Example of a vesicle that makes two consecutive steps toward the membrane. Each step could be well fit by a Boltzmann function (red traces) with 127 and 61 ms transit times, respectively. Black trace represents stimulus timing. E, Triggered vertical displacement along the ribbon. Distance traveled by each of 94 approaches within a single step toward the membrane, calculated from the ratio of fluorescence between the start and end of the movement (see Materials and Methods). F, The cumulative distribution of transit times for 90 vesicle approaches can be described by an exponential function with time constant τ = 120 ms (red line, R2 = 0.99). G, Mean fluorescence profile of 92 approaches aligned to the arrival time (black squares) or to the stimulus time (gray circles). Boltzmann fits to these profiles (red and blue traces) yielded distinct transit times: 90 ms when vesicles were aligned to the arrival time and 269 ms when vesicles were aligned to the stimulus time. This means that vesicles can reach the membrane within 90 ms but start moving with a variable delay after the cell is depolarized. Error bars indicate SEM.
Figure 2B plots the fluorescence profile of the vesicle in Figure 2A (black symbols) and of a contiguous annulus (gray symbols), used to estimate background fluorescence (for details of this analysis, see Materials and Methods). A minor rise of peripheral fluorescence on stimulation, such as the one in Figure 2B (gray symbols), was seen in some vesicles and has two major sources: (1) fluorescence from the vesicle itself that contaminates the annulus because of scattered light and/or imperfect centering of the ROI on the vesicle, and (2) dye from vesicles fusing outside of the ROIs can diffuse into both the annulus and the circle encompassing the vesicle. Errors arising from dye diffusion are minimized by using the annulus to correct for changes in local background. The timing of vesicle arrivals at active zones was determined as previously described (Zenisek et al., 2000) by fitting the fluorescence of approaching vesicles to a sigmoidal function (Fig. 2C, red trace). The estimated departure time was defined as when that fit reached 10% of its maximum and the arrival time as when it reached 90% of the asymptote. The time vesicles spent moving toward the membrane was calculated as the difference between the estimated departure time and arrival time and was called transit time (Fig. 2C, b). Other measured parameters were as follows: delay for departure (Fig. 2C, a), defined as the difference between the estimated departure time and the stimulus time; sitting time (Fig. 2C, c), or the difference between the fusion time and the arrival time; and delay for arrival (Fig. 2C, d), defined as the difference between the arrival time and the stimulus time.
The movement of approaching vesicles at active zones was directionally biased toward the membrane, since 97 increases in vesicle fluorescence were observed in the frames following a depolarization and no stepwise decreases in fluorescence were observed (40 movies, 13 cells). While in our experiments most vesicles made single approaches toward the membrane, in some cases vesicles occasionally appeared to move from a more distal to a more proximal position twice in the same location in response to two depolarizations, suggesting a vesicle making two steps toward the membrane (n = 17 vesicles, 11 movies, 7 cells). An example is shown in Figure 2D. Each of these double approaches could be fit by the same sigmoidal function, yielding similar individual transit times (115.5 ± 46.2 ms after the stimulus for the first approach and 151.2 ± 46.3 ms for the second, mean ± SEM), consistent with stepwise movements toward the cell membrane. One likely explanation for these two distinct movement patterns (i.e., smooth translocation vs stepwise motion) is that a labeled vesicle would move directly toward the membrane if the release site beneath was empty, but it would stop at an intermediate position if there was an unlabeled vesicle occupying the bottom of the ribbon. This is a plausible scenario because most ribbons will have only one labeled vesicle with the FM loading technique used here (Zenisek et al., 2002).
Figure 2E depicts a histogram of the measured distances for 90 approaches (39 movies, 12 cells). It is noteworthy that the histogram peaks ∼40 nm (median = 45.7 nm, mean ± SD = 53.2 ± 36.5), a vertical displacement similar to the distance measured between vesicle centers on the ribbon (von Gersdorff et al., 1996). Since vertical displacement was calculated as the ratio between the final and initial fluorescence values (see Materials and Methods), it was subject to the signal-to-noise ratio in our recordings, which was variable. To estimate the ambiguity in our estimation of distance, we measured the mean fluorescence and SEM of a sample of 30 vesicles before their approaches (i.e., at baseline) and after arrival (plateau) and calculated the uncertainty by propagation of errors. On average, we found that the SEM was 11% of the fluorescence value at baseline and 5% at the plateau, which would equate to a rough estimate of 14 nm variability.
The cumulative distribution of transit times for the same dataset (Fig. 2F) indicates that most vesicles spent ∼120 ms in transit once they started moving. Figure 2G shows the average fluorescence of 92 such approaches aligned to the voltage stimulus (gray circles) or to the arrival time (black squares). When aligned to the arrival time, the resulting mean intensity profile could be fit by a sigmoidal function with an apparent transit time of 90 ms. When the same data were aligned to the triggering stimulus, however, the resulting curve is broader, with an apparently longer transit time (269 ms), indicating that, although approaches tend to be fast, not all vesicles start moving at the same time after a stimulus. This variable delay for departure could reflect the translocation of vesicles originally residing at different distances from the membrane, some of which are beyond our evanescent field and are only detected mid-transit. In this case, one would expect vesicles with larger delays for departure to have originated from further away and thus take longer to reach the membrane and should have larger delays for arrival. One might also expect events with larger delays to be detected after release of the bottom row of vesicles when depletion necessitates recruitment from more distal regions of the ribbon (i.e., after the first stimulus of a train). We next set out to investigate this possibility.
Some vesicles start moving later
We applied trains of 30 ms depolarizations from −60 mV to 0 mV with different interstimulus intervals (240, 250, and 480 ms) to deplete the bottom row of vesicles and elicit distinct degrees of replenishment, as reported previously (Mennerick and Matthews, 1996; Sakaba et al., 1997; von Gersdorff and Matthews, 1997; Gomis et al., 1999), and analyzed the distribution of delays for departure (Fig. 3A), transit times (Fig. 3B), and delays for arrival (Fig. 3C) for vesicle approaches triggered within the first 240 ms following a depolarization. Since at departure the net change in fluorescence is quite small (i.e., 10% of the Boltzmann function used to fit these profiles; Fig. 2C), the determination of its timing is prone to ambiguities induced by the signal-to-noise ratio of our recordings; occasionally, the departure artifactually preceded the depolarization. To compensate for such uncertainties, we have used the estimated departure times obtained from the Boltzmann fits to assign vesicles to their respective triggering stimuli, and included in this analysis vesicles that apparently started moving up to two frames before each stimulus frame.
Figure 3.
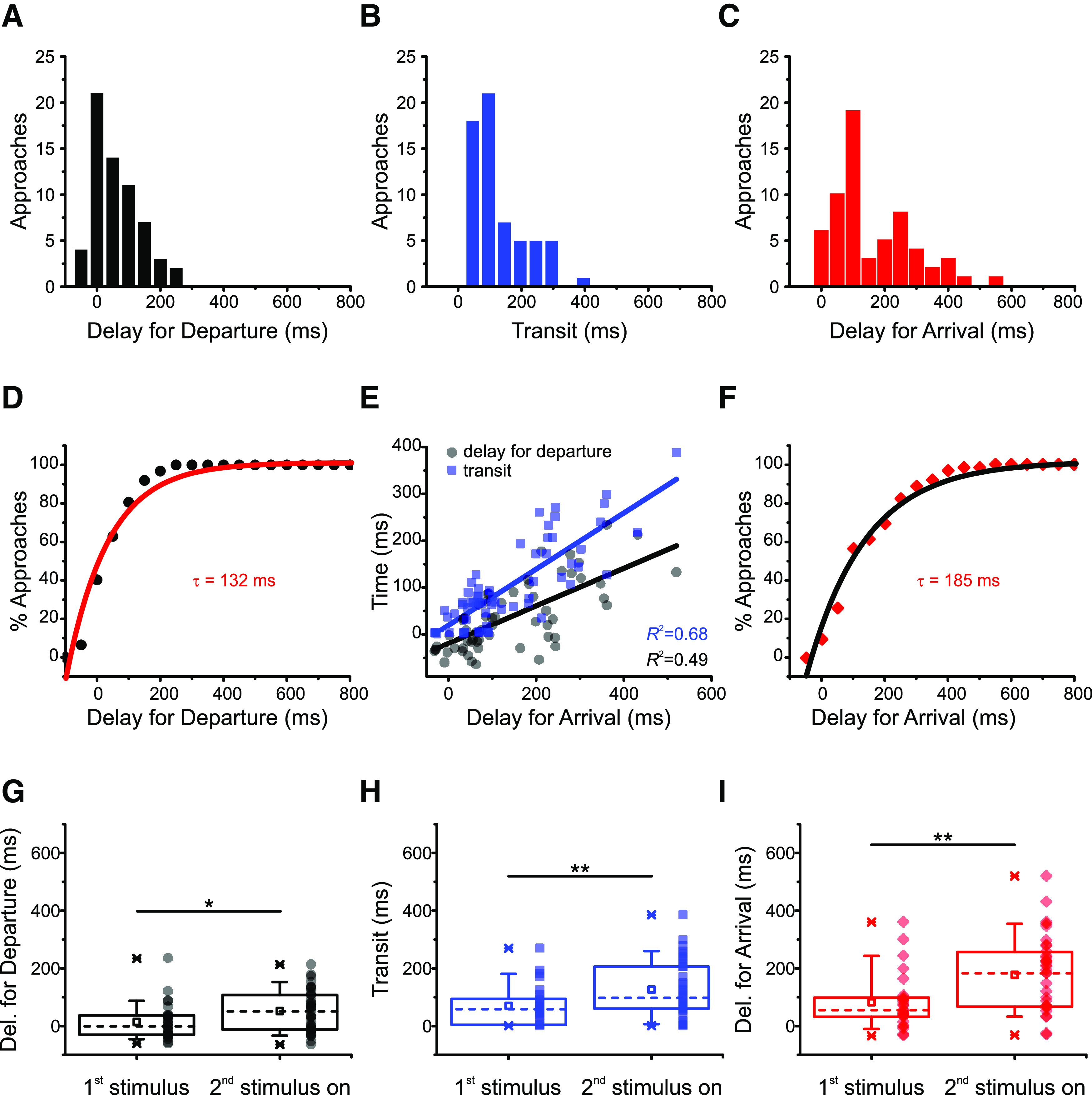
Arrival depends on how long vesicles travel and when vesicles depart. A, Distribution of delays for departure in 62 vesicle approaches (11 cells, 27 movies). The variability in delays for departure is larger than that of transit times (coefficient of variation = 1.99 for delays for departure 0.89 for transit times). B, Distribution of transits for the same vesicles in A; 74% of observed vesicles arrived at the membrane within 150 ms. C, Distribution of delays for arrival in the same vesicles of A and B. D, The cumulative distribution of delays for departure for the vesicle approaches in A can be described by an exponential function with time constant τ = 132 ms (red line, R2 = 0.96). E, Both transit times (black squares) and delays for departure (gray circles) contribute to delays for arrival. Straight lines indicate linear regressions to the data (R2 = 0.66 and R2 = 0.44, respectively). F, The cumulative distribution of delays for arrival for the vesicle approaches in C can be described by an exponential function with time constant τ = 185 ms (black line, R2 = 0.98). G, Delays for departure, (H) transit times, and (I) delays for arrival are influenced by stimulus history. Median values (indicated by the dashed lines) are smaller for the first stimulus compared with the remainder applied in all trains. Squares represent means. Boxes enclose the interquartile range (between the 25th and 75th percentiles). Bars represent the 10th and 90th percentiles. Stars represent the minimum and maximum. Significance: *p = 0.031 for delays for departure; **p = 0.009 for transit times; **p = 0.003 for delays for arrival; two-tailed Mann–Whitney test.
Figure 3A shows that, although 80% of observed vesicles started moving within 100 ms of the trigger (τ = 132 ms, R2 = 0.96; Fig. 3D), the distribution of delays for departure exhibited variability, which can also be seen in the distribution of transit times (Fig. 3B). The coefficient of variation of the former distribution (defined as the mean divided by the SD) was larger: cv = 1.99 for delays for departure and cv = 0.89 for transit times. As a result, the timing of delays for arrival was also variable (Fig. 3C; cv = 0.91). Both transit times and delays for departure contribute to delays for arrival because the latter correlate with both variables, the correlation with transit times being higher (Fig. 3E; adjusted R2 values depicted in the figure). Indeed, 82% of vesicles made their way to the membrane within 250 ms (τ = 185 ms, R2 = 0.98; Fig. 3F), which reflects the combination of time constants for departure (Fig. 3D) and transit (Fig. 2F).
We next investigated whether the stimulus history influences delays for departure (Fig. 3G), transit times (Fig. 3H), and delays for arrival (Fig. 3I), by comparing vesicle approaches in response to the first stimulus separately in relation to the remainder stimuli of each train. As shown in Figure 3G, delays for departure were significantly shorter for the first stimulus (median = −1 ms for the first stimulus and 51 ms for the remainder, two-tailed Mann–Whitney U = 321, n1<>n2, p = 0.031). The negative median reflects noise in the fluorescence data; as mentioned previously, some vesicles appear to start moving before the triggering stimulus. Similarly, Figure 3H shows that transit times were also shorter for vesicles departing after the first stimulus (median = 58 ms for the first stimulus and 98 ms for the remainder, two-tailed Mann–Whitney U = 289, n1<>n2, p = 0.009). This result suggests that vesicles wait longer to move and need longer to reach the membrane after a second stimulus than following a first stimulus. Finally, delays for arrival were likewise smaller for vesicle approaches triggered by the first stimulus of each train (Fig. 3I; median = 56 ms for the first stimulus and 184 ms for the remainder, two-tailed Mann–Whitney U = 267, n1<>n2, p = 0.003).
Although longer delays for departure and delays for arrival in response to later stimuli could be explained by vesicles moving from outside of our evanescent field after depletion of the first row of vesicles by the first depolarization of a train, longer transit times cannot because transit times are calculated once vesicles reach the evanescent field and become visible. Longer transit times after repetitive stimulation must therefore reflect a slowing down of movement after the first stimulus of a train. We therefore set out to estimate how fast synaptic vesicles move down the ribbon under our experimental conditions.
Estimate of vesicle speed on the ribbon
We used three different methods to estimate vesicle speed, illustrated in Figure 4. The first method consisted of fitting the mean fluorescence aligned by arrival time of the 92 approaches in Figure 2G (Fig. 4A, black squares) to an exponential function followed by an abrupt stop (Fig. 4A, gray circles). Using the time constant of the fit and the measured length constant of our evanescent field, we were able to estimate a speed of each vesicle in transit. This method yielded an estimate of 870 nm/s. Alternatively, we used the same model in Figure 4A to individually estimate the speed of the approaches in Figure 2E-G (gray, Fig. 4B; the subset of 62 events analyzed in Fig. 3 is shown in purple). With this method, the calculated speeds were 608 ± 28 nm/s for n = 90 and 623 ± 35 nm/s for n = 62 (mean ± SEM; medians depicted in the figure).
Figure 4.
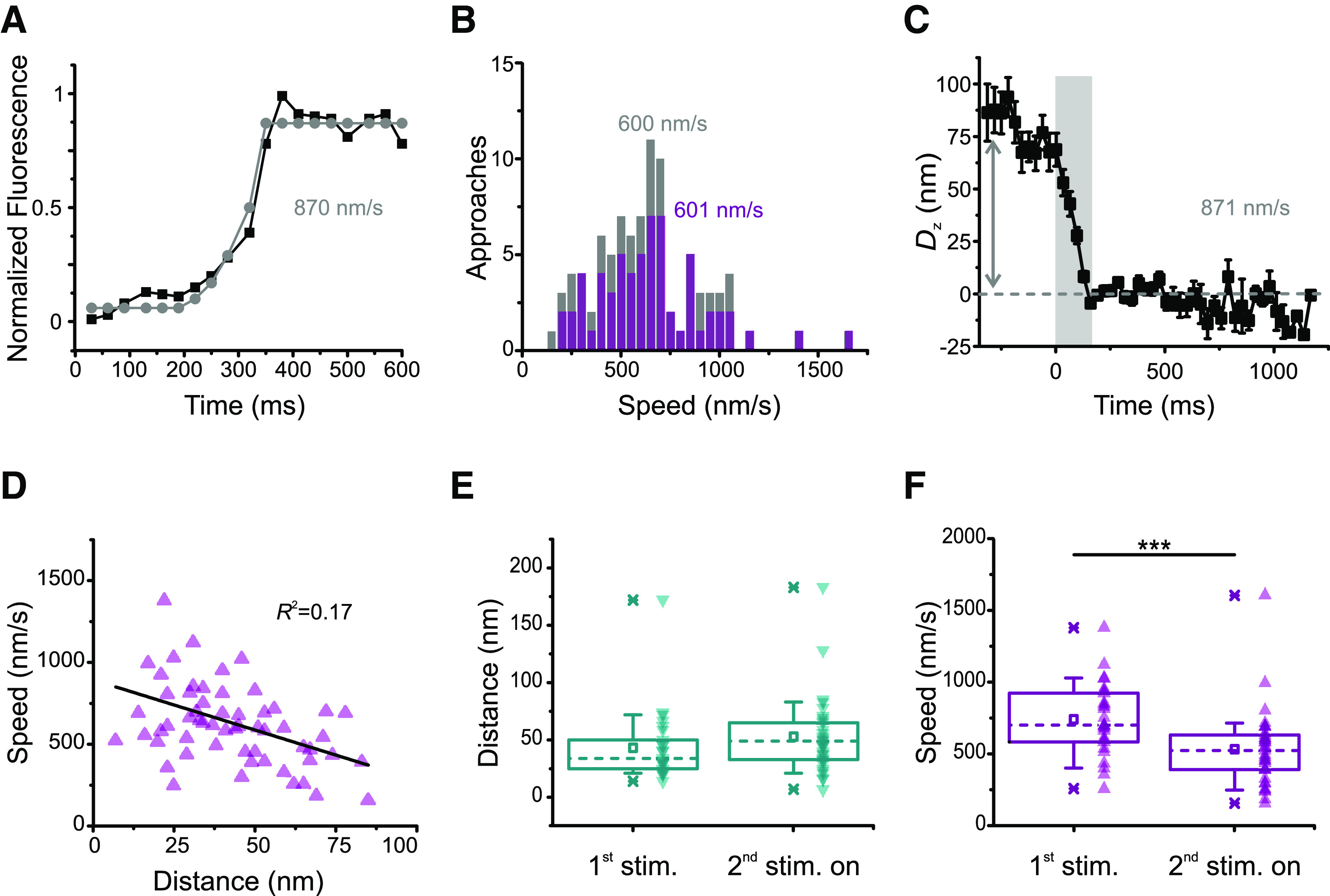
Methods for estimating vesicle speed. A, The mean fluorescence profile of 92 approaches aligned to the arrival time (black squares, same as the black squares in Fig. 2G) was modeled with an exponential rise followed by an abrupt stop (gray circles), yielding a speed estimate of 870 nm/s. B, Distribution of the fluorescence of individual approaches fit with the model in A for n = 90 (gray, whole dataset, same as in Fig. 2E–G) or n = 62 (purple, same dataset as in Fig. 3). Average speeds were 608 ± 28 and 623 ± 35 nm/s, respectively (mean ± SEM). Medians depicted in the figure. C, Vertical displacement for the fluorescence of 92 approaches aligned to arrival time (black squares in Fig. 2G). Error bars indicate SEM. The mean total displacement was 78.46 nm (arrow); dividing this number by the mean transit for the dataset in Figure 2G (90 ms) yields a speed estimate of 871 nm/s. D, Vesicle speed does not correlate well with vertical displacement. Same dataset as in Figure 3 (n = 62 approaches, 27 movies, 11 cells). R2 depicted in the figure. E, There is no significant difference in distance traveled for first stimulus and subsequent stimuli. Although median (dashed lines) and mean (squares) speeds are larger from the second stimulus on, the difference is not statistically significant (U = 344.5, p = 0.069, two-tailed Mann–Whitney test). Boxes enclose the interquartile range (between the 25th and 75th percentiles). Bars represent the 10th and 90th percentiles. Stars represent minimum and maximum. F, Stimulus history influences speed. Both medians (dashed lines) and means (squares) are smaller from the second stimulus on. Boxes enclose the interquartile range (between the 25th and 75th percentiles). Bars represent the 10th and 90th percentiles. Stars represent minimum and maximum. ***Significance (U = 718, p = 0.00036, two-tailed Mann–Whitney test).
Last, we also calculated speed from the vertical displacement (Dz) of the same 92 approaches aligned to the arrival time (Fig. 2G, black squares), based on the relative fluorescence of the vesicle before and after its approach, and according to the formula described in Materials and Methods. Figure 4C depicts a plot of the mean vertical distance traveled by these vesicles toward the membrane, showing that they traveled the depth of our evanescent field within <250 ms (gray area). The mean vertical displacement in this graph is 78 nm (arrow); dividing this number by the mean transit of the vesicles aligned to arrival time (90 ms; Fig. 2G, red line) yields a speed estimate of 871 nm/s. All three methods produced values close to the 800 nm/s estimate obtained previously during a continuous stimulus (Zenisek et al., 2000), which let more Ca2+ into the cell than our brief depolarizations. This suggests that the extra Ca2+ during a prolonged stimulus does not make vesicles move faster.
Figure 4D shows that vesicle speed does not correlate well with vertical displacement. This reflects that, while the distances traveled were not influenced by stimulus history (Fig. 4E; median = 34 nm for the first stimulus and 49 nm for the remainder, two-tailed Mann–Whitney U = 344.5, n1<>n2, p = 0.069), speeds were significantly higher for the first stimulus (Fig. 4F; median = 701 nm/s for the first stimulus and 524 nm/s for all subsequent stimuli, two-tailed Mann–Whitney U = 718, n1<>n2, p = 0.00036). To summarize, the results shown so far indicate that, from the second stimulus on, vesicles travel the same distance, depart later, take longer to reach the membrane, and travel slower.
Triggered vesicle replenishment is incomplete for short interstimulus intervals
To investigate further how quickly vesicles are replenished, we compared the numbers of stimulus-elicited fusion events and newly arrived vesicles at active zones for trains of 30 ms steps from −60 mV to 0 mV at different interstimulus intervals (60, 120, 250, and 480 ms). The results obtained with the 250 ms interstimulus interval protocol are depicted in Figure 5A, B. Figure 5A shows a histogram illustrating when fusion events (black columns) and new vesicle arrivals (white columns) occurred in response to a series of four 30 ms depolarizations to 0 mV at 4 Hz. Fusion events were time-locked to the depolarizations, with no events occurring during the interval between stimuli. As previously described, the synapse exhibited depression (Mennerick and Matthews, 1996; Sakaba et al., 1997; von Gersdorff and Matthews, 1997; Gomis et al., 1999); the number of released vesicles decreased drastically from the first stimulus to the remainder in the train. Figure 5B plots the data in Figure 5A as a cumulative histogram. Of note, the number of recruited vesicles following a particular depolarization (white circles) mimicked the number of fusion events observed during the same stimulus (black squares), indicating little vesicle depletion during the stimulus protocol.
Figure 5.

Timing of vesicle exocytosis and translocation in response to train of stimuli. A, Histogram showing timing of fusion events (black columns) and capture events (white columns) in response to series of four 30 ms depolarizations from −60 to 0 mV at 4 Hz. B, Bottom, Timing of depolarizations. Data from 4 cells, 19 movies. Cumulative histogram of the data in A. Black squares represent fusion events. White circles represent captured vesicles. Gray trace represents the timing of each voltage step. During the trains, the total number of newly recruited vesicles follows closely the number of released vesicles. C, Cumulative histogram of fusion events (black squares) and capture events (white circles) for the protocol with 480 ms interstimulus interval (n = 5 cells, 6 movies). Also, here the number of immobilized vesicles follows closely the number of released vesicles. D, Cumulative histogram of fusion events (black squares) and capture events (white squares) for the protocol with 120 ms interstimulus interval (n = 5 cells, 5 movies). For this protocol, the number of immobilized vesicles falls short of the number of released vesicles at all times during the voltage train but catches up by the end of the movie. E, Cumulative histogram of fusion events (black squares) and capture events (white squares) for the protocol with 60 ms interstimulus interval (n = 4 cells, 5 movies). Here, vesicle depletion is most pronounced. F, Fractional vesicle occupancy and fusion events for all depolarizations after the first one in a train, normalized to the number of fusion events in the first depolarization. The occupancy at each depolarization (available) was estimated by taking the number of newcomer vesicles and subtracting the number of vesicles lost by exocytosis. Replenishment maintains an available pool of vesicles for longer intervals but fails to fully replenish vesicles following shorter intervals (white bars).
Figure 5C-E shows the results obtained with three other interstimulus intervals: 480 ms (Fig. 5C), 120 ms (Fig. 5D), and 60 ms (Fig. 5E). While there are clear signs of vesicle depletion at interstimulus intervals <250 ms (Fig. 5D,E), for trains of lower frequency the number of newly immobilized vesicles followed closely the number of released vesicles (Fig. 5A-C). Figure 5F shows the number of available vesicles and the number of released vesicles relative to Step 1 for all subsequent steps at different train frequencies. To determine the number of available vesicles, the number of newly added newcomer vesicles were counted, and the number lost via exocytosis were subtracted from that number for each step in a train after the initial step. The results show that ribbons facilitate nearly complete vesicle resupply for intervals >250 ms between pulses but fail to keep up when intervals are shorter. Therefore, a dearth of vesicles contributes to depression for these short interstimulus intervals. At longer intervals, however, vesicles are present, but still unable to undergo exocytosis. The time needed for fusion after recruitment, or sitting time, could play a role in this phenomenon.
Newly arrived vesicles are not fusion-competent
We next investigated how long it takes for a vesicle to fuse once it has arrived. To do so, we measured all vesicle arrival times using the sigmoidal fit approach highlighted above and measured the delay between each arrival time and subsequent stimuli. This delay was defined as the sitting time. For each sitting time, we determined which fraction of vesicles fused in response to a depolarization. Figure 6A shows the results for the 47 vesicles with highest signal-to-noise ratio in our dataset, binned for different delays (n = 119 events, 47 vesicles, 36 movies, 13 cells).
Figure 6.

Newly arrived vesicles are not fusion-competent. A, Probability of vesicle fusion for different intervals between the time of vesicle arrival and fusion (sitting time). Vesicle fusion rate increased with sitting time (n = 119 events, 47 vesicles; 5 vesicles had sitting time >1000 ms and are not included in the graph). B, Same dataset as in A, plotted as a cumulative probability of fusion for all vesicles with each sitting time or briefer. Within the first 90 ms after arrival at the membrane, the probability of fusion is zero. The graph could be fitted with a single exponential function with time constant of 77 ms (inset, y0 = 0.28 A1 = −0.82; R2 = 0.85).
To look at fusion competence, we calculated the probability of release, that is, how likely a newcomer vesicle was to fuse once it arrived at the membrane, regardless of stimulus. Figure 6B shows a cumulative histogram of release probability as a function of sitting time for the same data as in Figure 6A. The fusion probability showed time dependence, with no vesicles fusing in response to stimuli within 90 ms of their arrival (as in Fig. 6A). The cumulative probability graph in Figure 6B could be fitted with a single exponential function with a time constant of 77 ms (inset: adjusted R2 = 0.85). These data indicate that indeed newly arrived vesicles are not yet ready to be released and progressively become more release-ready with time.
Together, the distribution of delays for arrival (Fig. 3C), combined with the low probability of fusion of newly arrived vesicles (Fig. 6A,B), suggests that most vesicles may need 100-350 ms to approach the membrane, depending on departure point, and a subset become release-ready in another 200-500 ms once they have arrived. This would yield recovery times in the order of 300-850 ms, close to the fastest time constant for recovery from paired-pulse depression reported for the same cell type elsewhere (Gomis et al., 1999).
Discussion
Unidirectional flow of vesicles on the ribbon
Our results directly show that vesicles move down the membrane-proximal portion of the synaptic ribbon in response to depolarization in a unidirectional manner, and that delivery of vesicles to the membrane is not sufficient to make these vesicles release-ready. Based on its morphology, the synaptic ribbon has long been hypothesized to be a vesicle-transporting organelle (Bunt, 1971; Gray and Pease, 1971). Our study is consistent with this hypothesis, since the vesicles we observed moved in all cases toward the membrane with no retrograde motion, indicating an extreme directional bias in their movement direction.
An elegant previous study using confocal imaging of sparsely labeled vesicles in the analogous bipolar cell in zebrafish was the first to show direct evidence for vesicles moving on a ribbon (Vaithianathan et al., 2016). The authors showed a milder directional bias in vesicle movement on the ribbon, with most vesicles moving toward the membrane, but a significant portion moving away (Vaithianathan et al., 2016). While species differences are a formal possibility, we view this as unlikely. We instead consider two, not mutually exclusive, other explanations for this difference: (1) vesicles at distal sites, invisible to TIRF imaging, behave differently than the ones nearest the membrane; and (2) the signal-to-noise ratio with confocal imaging prevented precise enough determination of vesicle location in some vesicles, causing these vesicles to appear to go in a retrograde direction.
Our approach makes use of the high signal-to-noise ratio afforded by TIRF microscopy and the labeling of vesicles with many fluorophores to resolve nm-scale movements of individual vesicles. In a previous publication, we estimated a frame-to-frame jitter of 3.8 nm in the z direction for immobilized vesicles, which may reflect either noise or genuine movement and can be thought of as an upper limit for localization in the z direction (Zenisek, 2008). The work of Vaithianathan et al. (2019) tracked individual vesicles, likely containing a single fluorescent molecule, at an average resolution of 27 nm, with many docked vesicles showing jitter in localization of >50 nm. This suggests that the ∼30 to 40 nm movements we observe here may be obscured in their experiments by the noise for some vesicles.
However, it should be noted that our superior resolution comes at a cost of not being able to observe distal locations on the ribbon. Our TIRF microscope generates an evanescent field with a length constant of ∼50 nm, allowing us to visualize vesicles at distances up to ∼100 nm and perhaps less, depending on the signal-to-noise ratio of the labeling. The ribbon in bipolar cells projects distally into the cell up to 150 nm, making much of the back half invisible to our imaging, indicating that distal vesicles moving in a retrograde direction would not be detected in our studies.
Vesicle movement in response to stimulus trains
In this study, we investigated the properties of vesicle transport and docking in bipolar cell ribbon-type presynaptic terminals. We find that docked vesicles are concentrated on the ribbon and that ribbon-associated vesicles move rapidly along the ribbon in response to repetitive stimuli. We also found that the vesicle delay for departure, transit time, and vesicle speed slowed with repeated stimuli, while distances traveled were not significantly different. Since Ca2+ levels lower dramatically with distance from open Ca2+ channels (Roberts, 1993; Naraghi and Neher, 1997), Ca2+ concentration is expected to drop at more distal locations on the ribbon. Hence, one explanation for this finding would be that the trigger for initiation of movement is Ca2+-dependent and that lower Ca2+ concentrations at distal sites lead to a slower initiation of movement. Alternatively, distal vesicles may have to wait for vesicles beneath them to move before they can begin movement.
Interestingly, the number of departures mirrored the number of fusion events in the preceding stimulus. Since the bipolar cell L-type Ca2+ channels are slowly inactivating (Heidelberger and Matthews, 1992; Tachibana et al., 1993), and Ca2+ levels return to basal levels over many seconds (Kobayashi et al., 1995; Zenisek and Matthews, 2000; Zenisek et al., 2003), Ca2+ levels rise with recurring depolarizations (Kobayashi et al., 1995). If Ca2+ were the trigger for movement, one would not expect the initiation of movement and translocation itself to be slowed down with repeated stimulation. Hence, exocytosis and presumably vacancies at the base of the ribbon rather than Ca2+ itself appear to be the determinant for the number of vesicles recruited (Graydon et al., 2014). While our results favor a vacancy model, experiments will be necessary to distinguish between these two models, since our experiments and analyses were not designed to investigate the Ca2+ dependence of replenishment.
Numerous studies have shown that Ca2+ speeds up functional replenishment as measured electrophysiologically (von Ruden and Neher, 1993; Mennerick and Matthews, 1996; Gomis et al., 1999; Sakaba and Neher, 2001a,b; Singer and Diamond, 2006; Babai et al., 2010; Graydon et al., 2011). In that context, our results are surprising in that vesicle resupply slows as Ca2+ is presumably building within the terminal. This difference may reflect Ca2+-dependent factors that govern release that are independent of vesicle translocation. Alternatively, or in addition, Ca2+-dependent enhancement of vesicle replenishment may only partially compensate for other processes that slow vesicle replenishment with repeated stimuli. To disambiguate these possibilities, one would need to manipulate Ca2+ and/or Ca2+ buffers in future TIRF experiments.
Vesicle depletion and depression at bipolar cell synapses
We show here that the number of newcomer vesicles correlated with the number of vesicles released during each stimulus for interstimulus intervals ≥250 ms, indicating that vesicle replenishment is not rate-limiting for those interstimulus intervals. For shorter interstimulus intervals, vesicle depletion played a prominent role in synaptic depression.
Although the repopulation of ribbon active zones was rapid, newly immobilized vesicles were not immediately available for release. Similar results were recently reported in an elegant optical study of cerebellar mossy fibers (Miki et al., 2020), the fastest type of conventional synapse recorded (Delvendahl and Hallermann, 2016). After paired-pulse stimulation, newcomer vesicles in these synapses need ≥400 ms after arrival at the membrane to become fusion-competent (Miki et al., 2020), a number within the 200-500 ms range reported here. However, the results are not directly comparable to ours here, since (1) the authors only probed single pairs of depolarizations spaced 100 ms apart and most of the release recorded was asynchronous to the second depolarization, and (2) most fusion events in that preparation are not demarcated by a diffusing cloud of dye, making it challenging to distinguish asynchronous events from vesicle departure events. It remains to be seen how much time is required for cerebellar mossy fiber synapses to become primed for synchronous release. Interestingly, the results from the cerebellar mossy fibers stand in stark contrast to those of conventional synapses of the hippocampus and calyx of Held, which fail to show priming of newcomer vesicles within seconds after arrival (Midorikawa and Sakaba, 2015, 2017). The fact that the mean priming time constant in cerebellar mossy fibers is still 2 times larger than the lower limit in our measurements suggests that ribbons may accelerate priming, in addition to helping vesicle translocation, but more experiments will be required to sort this out.
Our results directly show that vesicles move down the synaptic ribbon in response to depolarization, as previously shown by Vaithianathan et al. (2016), and that delivery of vesicles to the membrane is not sufficient to make these vesicles release-ready. Together, the time vesicles need to arrive at the membrane, and the time they need to become release-ready might explain the recovery times from paired-pulse synaptic depression.
Paired-pulse depression and vesicle availability
Many synapses exhibit short-term synaptic depression in response to two closely spaced stimuli. Considerable evidence indicates that depression arises from a depletion of the pool of vesicles ready for immediate release, and its recovery reflects a refractory period in which new vesicles replenish this pool (Zucker and Regehr, 2002). It is not known, however, to what extent depression reflects the physical absence of a vesicle and whether the physical replenishment of these vesicles reflects recovery from depression. Here, we investigated the relationship between vesicle release, replenishment, and synaptic depression in goldfish retinal mixed-input bipolar cells by directly imaging single vesicles using TIRF microscopy. The synaptic terminal of a goldfish mixed-input bipolar cell contains ∼30-70 ribbons (von Gersdorff et al., 1996; Holt et al., 2004; Zenisek et al., 2004), each carrying ∼100 synaptic vesicles (von Gersdorff et al., 1996). In addition, there are hundreds of thousands of vesicles scattered randomly throughout the cytosol (von Gersdorff et al., 1996).
Based on the work of many investigators, at least three components of exocytosis have been identified: a small rapid component, which is exhausted with a time constant of ∼1.5 ms (Mennerick and Matthews, 1996; Sakaba et al., 1997; von Gersdorff et al., 1998; Neves and Lagnado, 1999), a slower component that is exhausted with a time constant of ∼300 ms (von Gersdorff and Matthews, 1994; Sakaba et al., 1997; von Gersdorff et al., 1998; Gomis et al., 1999), and a sustained component (Lagnado et al., 1996; Rouze and Schwartz, 1998). Similar results have been observed in recordings from mouse and rat bipolar cells (Singer and Diamond, 2006; Zhou et al., 2006). Since the combined size of the fast and the slow component of exocytosis is strikingly similar to the total number of vesicles on the ribbon and the size of the fast component is similar to the number of vesicles at the base of the ribbon, these two components or pools are thought to represent the fusion of all vesicles tethered to the ribbons before the stimulus (von Gersdorff et al., 1996), the fast component corresponding to fusion of vesicles at the base of the ribbon. Our results here agree well with this idea: we show that single 30 ms depolarizations cause exocytosis of membrane proximal vesicles that are rapidly replenished within 150-350 ms after the termination of the pulse.
Retinal bipolar cells exhibit paired-pulse depression with a time constant of recovery that depends on stimulus strength (Mennerick and Matthews, 1996; von Gersdorff and Matthews, 1997; Gomis et al., 1999; Burrone and Lagnado, 2000; Singer and Diamond, 2006). One study using membrane capacitance reported that recovery from paired-pulse depression in response to a 20 ms stimulus followed by two time constants of 0.64 and 31 s, with ∼70% of the vesicles exhibiting the longer time constant (Gomis et al., 1999). Our results show that vesicles removed by exocytosis are replenished in ∼100–350 ms and need another 200–500 ms to mature, which would explain the fastest time constant found by these authors.
The lower release probability of newly immobilized vesicles
The data presented here indicate that, although vesicle replenishment is fast, newly immobilized vesicles are not immediately competent for fusion. This results in a decreased release probability of these captured vesicles compared with vesicles that had been docked for some time. There are some hypotheses that try to explain this phenomenon. In some systems, a positional priming step is required to localize vesicles to Ca2+ channels (Hwang et al., 2013). Consequently, the Ca2+ concentration would be diminished at the new release sites, leading to an apparent reduction in release probability until channels can find their way to the vesicles. Our results could be explained by such a mechanism. An alternative explanation for this change in release probability is that synaptic vesicles are intrinsically heterogeneous, and that release probability is modulated actively according to the stimulation history of a particular synapse (Burrone and Lagnado, 2000; Wolfel et al., 2007). This possibility is related to the fact that fusion requires vesicles to undergo a maturation or priming process that includes more steps than immobilization itself, bringing vesicles closer to being releasable, yet preventing them from being released spontaneously (Sorensen, 2004).
Comparison with conventional synapses
To help understand the role of the synaptic ribbon in synaptic transmission, it is useful to compare the vesicle dynamics in ribbon-type and non–ribbon-type synapses. In response to trains of 30 ms depolarizations, new vesicles moved toward the membrane to repopulate the base of the ribbon within 100-350 ms, which was followed by a period of 200-500 ms before vesicles became fusion-competent. Previous studies using prolonged stimuli also showed similar delays between vesicle arrival and exocytosis in salamander rods (90 ms) (Chen et al., 2013) and bipolar cells (200 ms) (Zenisek et al., 2000) and also show fast replenishment.
Striking differences emerge when comparing TIRF measurements at conventional synapses with those we measure here. In hippocampal mossy fiber boutons (Midorikawa and Sakaba, 2017) and in the isolated calyx of Held (Midorikawa and Sakaba, 2015), the physical replacement of vesicles was a comparatively slow process, taking 4-5 s, 40- to 50-fold slower than ribbon synapses. Despite the slow replenishment of vesicles to the membrane, recovery from depression is relatively fast via the priming of previously docked vesicles. This priming of predocked vesicles occurs over a similar time scale as the maturation of newcomers in our study or in photoreceptors; but in the case of conventional synapses, Midorikawa and Sakaba (2015, 2017) found that newcomer vesicles did not become release-ready over the time of their imaging, indicating that the transition from vesicle arrival to a preprimed state was too slow to measure in conventional synapses.
An exception among conventional synapses seems to be cerebellar mossy fibers, which are optimized to signal at extremely high (>1 kHz) frequencies (Delvendahl and Hallermann, 2016). Similar to the bipolar cell ribbon synapses studied here, they can quickly deliver vesicles to the membrane after a single depolarization, and newly arrived vesicles mature much faster than in other conventional synapses (Miki et al., 2020). These synapses apparently have developed alternative mechanisms to enable quick vesicle replenishment and maturation, similar to our observations in a ribbon synapse. The fastest time constant reported for vesicle delivery at cerebellar mossy fibers (188 ms) (Miki et al., 2020) is similar to that of delays for arrival of our pooled data (Fig. 3F), but more than 3 times slower that the median delay for arrival in response to the first stimulus of a train (56 ms; Fig. 3I), and the time constant for priming (440 ms) is almost 5 times slower than that of retinal bipolar cells (77 ms; Fig. 6B), although, as discussed above, differences in experimental protocols make a direct comparison difficult.
These results suggest that ribbon synapses are adept at rapid vesicle resupply and priming. The role of ribbons in catalyzing vesicle maturation is also congruent with evidence that ribbon-tethered vesicles are insensitive to ATP-γ-S (Heidelberger et al., 2002) and with experiments where acute photodamage to the ribbon eliminates release downstream of its ability to bind new vesicles (Snellman et al., 2011). However, given the new results from cerebellar mossy fiber synapses (Miki et al., 2020) and the unchanged kinetics of release from ribbon synapses in ribeye KO animals (Maxeiner et al., 2016; Becker et al., 2018; Jean et al., 2018), it may be that these properties may not require the ribbon itself.
While our results support ribbon synapses as specialists in rapid and efficient delivery of release-ready vesicles, it remains uncertain as to why genetic removal of ribeye leads to profound effects on ribbon morphology, but mild effects on kinetics of continuous exocytosis (Lv et al., 2016; Maxeiner et al., 2016; Becker et al., 2018; Jean et al., 2018). We suggest three hypotheses to explain the results. (1) That the structure itself is relatively unimportant for vesicle delivery and preparation and that the specialized proteins in ribbon synapses, some of which could also be present in fast conventional synapses, such as those from cerebellar mossy fibers, can fulfill the role in the absence of a ribbon. Indeed, while ribeye is necessary for formation of normal ribbons, it seems dispensable for retaining normal release kinetics in hair cells and mouse bipolar cells. (2) Genetic and/or homeostatic compensation in KO animals can overcome the loss of the ribbon to return release kinetics to normal, perhaps invoking other mechanisms in addition to vesicle trafficking. (3) The ribbon's role in goldfish mixed-input bipolar cells is different from that in other cell types. Future experiments will be needed to address these possibilities.
Footnotes
This work was supported by National Institutes of Health Grants EY021195 and EY003821, Yale University Vision Core EY026878, and Fundacao de Amparo a Pesquisa do Estado de Sao Paulo 2010/16469-0 to C.J.
The authors declare no competing financial interests.
References
- Axelrod D. (2001) Total internal reflection fluorescence microscopy in cell biology. Traffic 2:764–774. 10.1034/j.1600-0854.2001.21104.x [DOI] [PubMed] [Google Scholar]
- Babai N, Bartoletti TM, Thoreson WB (2010) Calcium regulates vesicle replenishment at the cone ribbon synapse. J Neurosci 30:15866–15877. 10.1523/JNEUROSCI.2891-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont V, Llobet A, Lagnado L (2005) Expansion of calcium microdomains regulates fast exocytosis at a ribbon synapse. Proc Natl Acad Sci USA 102:10700–10705. 10.1073/pnas.0501961102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker L, Schnee ME, Niwa M, Sun W, Maxeiner S, Talaei S, Kachar B, Rutherford MA, Ricci AJ (2018) The presynaptic ribbon maintains vesicle populations at the hair cell afferent fiber synapse. Elife 7:e30241 10.7554/eLife.30241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ, Mao F, Bewick GS (1992) Activity-dependent fluorescent staining and destaining of living vertebrate motor nerve terminals. J Neurosci 12:363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunt AH. (1971) Enzymatic digestion of synaptic ribbons in amphibian retinal photoreceptors. Brain Res 25:571–577. 10.1016/0006-8993(71)90461-6 [DOI] [PubMed] [Google Scholar]
- Burrone J, Lagnado L (2000) Synaptic depression and the kinetics of exocytosis in retinal bipolar cells. J Neurosci 20:568–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J, Neves G, Gomis A, Cooke A, Lagnado L (2002) Endogenous calcium buffers regulate fast exocytosis in the synaptic terminal of retinal bipolar cells. Neuron 33:101–112. 10.1016/s0896-6273(01)00565-7 [DOI] [PubMed] [Google Scholar]
- Chen MH, Van Hook MJ, Zenisek D, Thoreson WB (2013) Properties of ribbon and non-ribbon release from rod photoreceptors revealed by visualizing individual synaptic vesicles. J Neurosci 33:2071–2086. 10.1523/JNEUROSCI.3426-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SY, Jackman S, Thoreson WB, Kramer RH (2008) Light regulation of Ca2+ in the cone photoreceptor synaptic terminal. Vis Neurosci 25:693–700. 10.1017/s0952523808080814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggins M, Zenisek D (2009) Evidence that exocytosis is driven by calcium entry through multiple calcium channels in goldfish retinal bipolar cells. J Neurophysiol 101:2601–2619. 10.1152/jn.90881.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggins MR, Grabner CP, Almers W, Zenisek D (2007) Stimulated exocytosis of endosomes in goldfish retinal bipolar neurons. J Physiol 584:853–865. 10.1113/jphysiol.2007.140848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta P, Gilliam J, Thoreson WB, Janz R, Heidelberger R (2017) Two pools of vesicles associated with synaptic ribbons are molecularly prepared for release. Biophys J 113:2281–2298. 10.1016/j.bpj.2017.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvendahl I, Hallermann S (2016) The cerebellar mossy fiber synapse as a model for high-frequency transmission in the mammalian CNS. Trends Neurosci 39:722–737. 10.1016/j.tins.2016.09.006 [DOI] [PubMed] [Google Scholar]
- DeVries SH. (2000) Bipolar cells use kainate and AMPA receptors to filter visual information into separate channels. Neuron 28:847–856. 10.1016/s0896-6273(00)00158-6 [DOI] [PubMed] [Google Scholar]
- Dick O, tom Dieck S, Altrock WD, Ammermüller J, Weiler R, Garner CC, Gundelfinger ED, Brandstätter JH (2003) The presynaptic active zone protein bassoon is essential for photoreceptor ribbon synapse formation in the retina. Neuron 37:775–786. 10.1016/s0896-6273(03)00086-2 [DOI] [PubMed] [Google Scholar]
- Gomis A, Burrone J, Lagnado L (1999) Two actions of calcium regulate the supply of releasable vesicles at the ribbon synapse of retinal bipolar cells. J Neurosci 19:6309–6317. 10.1523/JNEUROSCI.19-15-06309.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EG, Pease HL (1971) On understanding the organisation of the retinal receptor synapses. Brain Res 35:1–15. 10.1016/0006-8993(71)90591-9 [DOI] [PubMed] [Google Scholar]
- Graydon CW, Cho S, Li GL, Kachar B, Von Gersdorff H (2011) Sharp Ca2+ nanodomains beneath the ribbon promote highly synchronous multivesicular release at hair cell synapses. J Neurosci 31:16637–16650. 10.1523/JNEUROSCI.1866-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graydon CW, Zhang J, Oesch NW, Sousa AA, Leapman RD, Diamond JS (2014) Passive diffusion as a mechanism underlying ribbon synapse vesicle release and resupply. J Neurosci 34:8948–8962. 10.1523/JNEUROSCI.1022-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger R, Matthews G (1992) Calcium influx and calcium current in single synaptic terminals of goldfish retinal bipolar neurons. J Physiol 447:235–256. 10.1113/jphysiol.1992.sp019000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger R, Heinemann C, Neher E, Matthews G (1994) Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature 371:513–515. 10.1038/371513a0 [DOI] [PubMed] [Google Scholar]
- Heidelberger R, Sterling P, Matthews G (2002) Roles of ATP in depletion and replenishment of the releasable pool of synaptic vesicles. J Neurophysiol 88:98–106. 10.1152/jn.2002.88.1.98 [DOI] [PubMed] [Google Scholar]
- Holt M, Cooke A, Neef A, Lagnado L (2004) High mobility of vesicles supports continuous exocytosis at a ribbon synapse. Curr Biol 14:173–183. 10.1016/j.cub.2003.12.053 [DOI] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JR, Joung JK (2013) Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol 31:227–229. 10.1038/nbt.2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenti B, Heidelberger R (2008) Mechanisms contributing to tonic release at the cone photoreceptor ribbon synapse. J Neurophysiol 99:25–36. 10.1152/jn.00737.2007 [DOI] [PubMed] [Google Scholar]
- Jean P, Lopez de la Morena D, Michanski S, Tobon LM, Chakrabarti R, Picher MM, Neef J, Jung S, Gultas M, Maxeiner S, Neef A, Wichmann C, Strenzke N, Grabner C, Moser T (2018) The synaptic ribbon is critical for sound encoding at high rates and with temporal precision. Elife 7:e29275 10.7554/eLife.29275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joselevitch C, Zenisek D (2009) Imaging exocytosis in retinal bipolar cells with TIRF microscopy. J Vis Exp 28:1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay AR, Alfonso A, Alford S, Cline HT, Holgado AM, Sakmann B, Snitsarev VA, Stricker TP, Takahashi M, Wu LG (1999) Imaging synaptic activity in intact brain and slices with FM1-43 in C. elegans, lamprey, and rat. Neuron 24:809–817. 10.1016/S0896-6273(00)81029-6 [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Sakaba T, Tachibana M (1995) Potentiation of Ca2+ transients in the presynaptic terminals of goldfish retinal bipolar cells. J Physiol 482:7–13. 10.1113/jphysiol.1995.sp020495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagnado L, Gomis A, Job C (1996) Continuous vesicle cycling in the synaptic terminal of retinal bipolar cells. Neuron 17:957–967. 10.1016/s0896-6273(00)80226-3 [DOI] [PubMed] [Google Scholar]
- Llobet A, Cooke A, Lagnado L (2003) Exocytosis at the ribbon synapse of retinal bipolar cells studied in patches of presynaptic membrane. J Neurosci 23:2706–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoGiudice L, Sterling P, Matthews G (2008) Mobility and turnover of vesicles at the synaptic ribbon. J Neurosci 28:3150–3158. 10.1523/JNEUROSCI.5753-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv C, Stewart WJ, Akanyeti O, Frederick C, Zhu J, Santos-Sacchi J, Sheets L, Liao JC, Zenisek D (2016) Synaptic ribbons require ribeye for electron density, proper synaptic localization, and recruitment of calcium channels. Cell Rep 15:2784–2795. 10.1016/j.celrep.2016.05.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews G, Fuchs P (2010) The diverse roles of ribbon synapses in sensory neurotransmission. Nat Rev Neurosci 11:812–822. 10.1038/nrn2924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxeiner S, Luo F, Tan A, Schmitz F, Sudhof TC (2016) How to make a synaptic ribbon: RIBEYE deletion abolishes ribbons in retinal synapses and disrupts neurotransmitter release. EMBO J 35:1098–1114. 10.15252/embj.201592701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta B, Ke JB, Zhang L, Baden AD, Markowitz AL, Nayak S, Briggman KL, Zenisek D, Singer JH (2014) Global Ca2+ signaling drives ribbon-independent synaptic transmission at rod bipolar cell synapses. J Neurosci 34:6233–6244. 10.1523/JNEUROSCI.5324-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Matthews G (1996) Ultrafast exocytosis elicited by calcium current in synaptic terminals of retinal bipolar neurons. Neuron 17:1241–1249. 10.1016/s0896-6273(00)80254-8 [DOI] [PubMed] [Google Scholar]
- Miki T, Midorikawa M, Sakaba T (2020) Direct imaging of rapid tethering of synaptic vesicles accompanying exocytosis at a fast central synapse. Proc Natl Acad Sci USA 117:14493–14502. 10.1073/pnas.2000265117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midorikawa M, Sakaba T (2015) Imaging exocytosis of single synaptic vesicles at a fast CNS presynaptic terminal. Neuron 88:492–498. 10.1016/j.neuron.2015.09.047 [DOI] [PubMed] [Google Scholar]
- Midorikawa M, Sakaba T (2017) Kinetics of releasable synaptic vesicles and their plastic changes at hippocampal mossy fiber synapses. Neuron 96:1033–1040. 10.1016/j.neuron.2017.10.016 [DOI] [PubMed] [Google Scholar]
- Midorikawa M, Tsukamoto Y, Berglund K, Ishii M, Tachibana M (2007) Different roles of ribbon-associated and ribbon-free active zones in retinal bipolar cells. Nat Neurosci 10:1268–1276. 10.1038/nn1963 [DOI] [PubMed] [Google Scholar]
- Moser T, Grabner CP, Schmitz F (2020) Sensory processing at ribbon synapses in the retina and the cochlea. Physiol Rev 100:103–144. 10.1152/physrev.00026.2018 [DOI] [PubMed] [Google Scholar]
- Naraghi M, Neher E (1997) Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci 17:6961–6973. 10.1523/JNEUROSCI.17-18-06961.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves G, Lagnado L (1999) The kinetics of exocytosis and endocytosis in the synaptic terminal of goldfish retinal bipolar cells. J Physiol 515:181–202. 10.1111/j.1469-7793.1999.181ad.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan ZH, Hu HJ, Perring P, Andrade R (2001) T-type Ca2+ channels mediate neurotransmitter release in retinal bipolar cells. Neuron 32:89–98. 10.1016/S0896-6273(01)00454-8 [DOI] [PubMed] [Google Scholar]
- Rabl K, Cadetti L, Thoreson WB (2006) Paired-pulse depression at photoreceptor synapses. J Neurosci 26:2555–2563. 10.1523/JNEUROSCI.3667-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts WM. (1993) Spatial calcium buffering in saccular hair cells. Nature 363:74–76. 10.1038/363074a0 [DOI] [PubMed] [Google Scholar]
- Rouze NC, Schwartz EA (1998) Continuous and transient vesicle cycling at a ribbon synapse. J Neurosci 18:8614–8624. 10.1523/JNEUROSCI.18-21-08614.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Neher E (2001a) Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron 32:1119–1131. 10.1016/S0896-6273(01)00543-8 [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E (2001b) Quantitative relationship between transmitter release and calcium current at the calyx of Held synapse. J Neurosci 21:462–476. 10.1523/JNEUROSCI.21-02-00462.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Tachibana M, Matsui K, Minami N (1997) Two components of transmitter release in retinal bipolar cells: exocytosis and mobilization of synaptic vesicles. Neurosci Res 27:357–370. 10.1016/s0168-0102(97)01168-1 [DOI] [PubMed] [Google Scholar]
- Singer JH, Diamond JS (2003) Sustained Ca2+ entry elicits transient postsynaptic currents at a retinal ribbon synapse. J Neurosci 23:10923–10933. 10.1523/JNEUROSCI.23-34-10923.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer JH, Diamond JS (2006) Vesicle depletion and synaptic depression at a mammalian ribbon synapse. J Neurophysiol 95:3191–3198. 10.1152/jn.01309.2005 [DOI] [PubMed] [Google Scholar]
- Snellman J, Mehta B, Babai N, Bartoletti TM, Akmentin W, Francis A, Matthews G, Thoreson W, Zenisek D (2011) Acute destruction of the synaptic ribbon reveals a role for the ribbon in vesicle priming. Nat Neurosci 14:1135–1141. 10.1038/nn.2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen JB. (2004) Formation, stabilisation and fusion of the readily releasable pool of secretory vesicles. Pflugers Arch 448:347–362. [DOI] [PubMed] [Google Scholar]
- Tachibana M, Okada T, Arimura T, Kobayashi K, Piccolino M (1993) Dihydropyridine-sensitive calcium current mediates neurotransmitter release from bipolar cells of the goldfish retina. J Neurosci 13:2898–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- tom Dieck S, Altrock WD, Kessels MM, Qualmann B, Regus H, Brauner D, Fejtová A, Bracko O, Gundelfinger ED, Brandstätter JH (2005) Molecular dissection of the photoreceptor ribbon synapse: physical interaction of Bassoon and RIBEYE is essential for the assembly of the ribbon complex. J Cell Biol 168:825–836. 10.1083/jcb.200408157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toomre D, Bewersdorf J (2010) A new wave of cellular imaging. Annu Rev Cell Dev Biol 26:285–314. 10.1146/annurev-cellbio-100109-104048 [DOI] [PubMed] [Google Scholar]
- Vaithianathan T, Matthews G (2014) Visualizing synaptic vesicle turnover and pool refilling driven by calcium nanodomains at presynaptic active zones of ribbon synapses. Proc Natl Acad Sci USA 111:8655–8660. 10.1073/pnas.1323962111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaithianathan T, Henry D, Akmentin W, Matthews G (2016) Nanoscale dynamics of synaptic vesicle trafficking and fusion at the presynaptic active zone. eLife 5:e13245 10.7554/eLife.13245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaithianathan T, Wollmuth LP, Henry D, Zenisek D, Matthews G (2019) Tracking newly released synaptic vesicle proteins at ribbon active zones. iScience 17:10–23. 10.1016/j.isci.2019.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G (1994) Dynamics of synaptic vesicle fusion and membrane retrieval in synaptic terminals. Nature 367:735–739. 10.1038/367735a0 [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G (1997) Depletion and replenishment of vesicle pools at a ribbon-type synaptic terminal. J Neurosci 17:1919–1927. 10.1523/JNEUROSCI.17-06-01919.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Vardi E, Matthews G, Sterling P (1996) Evidence that vesicles on the synaptic ribbon of retinal bipolar neurons can be rapidly released. Neuron 16:1221–1227. 10.1016/s0896-6273(00)80148-8 [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Sakaba T, Berglund K, Tachibana M (1998) Submillisecond kinetics of glutamate release from a sensory synapse. Neuron 21:1177–1188. 10.1016/S0896-6273(00)80634-0 [DOI] [PubMed] [Google Scholar]
- von Ruden L, Neher E (1993) A Ca-dependent early step in the release of catecholamines from adrenal chromaffin cells. Science 262:1061–1065. 10.1126/science.8235626 [DOI] [PubMed] [Google Scholar]
- Wolfel M, Lou X, Schneggenburger R (2007) A mechanism intrinsic to the vesicle fusion machinery determines fast and slow transmitter release at a large CNS synapse. J Neurosci 27:3198–3210. 10.1523/JNEUROSCI.4471-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR (2003) Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science 300:2061–2065. 10.1126/science.1084398 [DOI] [PubMed] [Google Scholar]
- Zenisek D. (2008) Vesicle association and exocytosis at ribbon and extraribbon sites in retinal bipolar cell presynaptic terminals. Proc Natl Acad Sci USA 105:4922–4927. 10.1073/pnas.0709067105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenisek D, Matthews G (2000) The role of mitochondria in presynaptic calcium handling at a ribbon synapse. Neuron 25:229–237. 10.1016/S0896-6273(00)80885-5 [DOI] [PubMed] [Google Scholar]
- Zenisek D, Steyer JA, Almers W (2000) Transport, capture and exocytosis of single synaptic vesicles at active zones. Nature 406:849–854. 10.1038/35022500 [DOI] [PubMed] [Google Scholar]
- Zenisek D, Steyer JA, Feldman ME, Almers W (2002) A membrane marker leaves synaptic vesicles in milliseconds after exocytosis in retinal bipolar cells. Neuron 35:1085–1097. 10.1016/s0896-6273(02)00896-6 [DOI] [PubMed] [Google Scholar]
- Zenisek D, Davila V, Wan L, Almers W (2003) Imaging calcium entry sites and ribbon structures in two presynaptic cells. J Neurosci 23:2538–2548. 10.1523/JNEUROSCI.23-07-02538.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenisek D, Horst NK, Merrifield C, Sterling P, Matthews G (2004) Visualizing synaptic ribbons in the living cell. J Neurosci 24:9752–9759. 10.1523/JNEUROSCI.2886-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou ZY, Wan QF, Thakur P, Heidelberger R (2006) Capacitance measurements in the mouse rod bipolar cell identify a pool of releasable synaptic vesicles. J Neurophysiol 96:2539–2548. 10.1152/jn.00688.2006 [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64:355–405. 10.1146/annurev.physiol.64.092501.114547 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Squares represent the coordinates of the approach of two synaptic vesicles and fusion of one of them in response to a 30 ms voltage step from –60 to 0 mV. Movie is slowed 4.7×; frames were low pass filtered to remove high spatial frequency pixel noise.
Squares represent the coordinates of the fusion of three resident vesicles and subsequent approach and release of newcomers in response to a 30 ms voltage step from –60 to 0 mV. Movie is slowed 1.57×; frames were low pass filtered to remove high spatial frequency pixel noise.
Bipolar cell terminal loaded with FM1-43 and submitted to four consecutive 30 ms depolarizing steps from –60 to 0 mV at 480 ms interstimulus intervals. White circle represents timing of the stimulus. The first frame of the movie is the same terminal imaged with the 561 nm laser, showing the location of the labeled ribbons (same as in Fig. 1C, RBP). Movie is slowed 2×; frames were low pass filtered to remove high spatial frequency pixel noise.