

Abstract
Despite an increasing burden of osteoarthritis in developed societies, target discovery has been slow and there are currently no approved disease-modifying osteoarthritis drugs. This lack of progress is due in part to a series of misconceptions over the years: that osteoarthritis is an inevitable consequence of ageing, that damaged articular cartilage cannot heal itself, and that osteoarthritis is driven by synovial inflammation similar to that seen in rheumatoid arthritis. Molecular interrogation of disease through ex-vivo tissue analysis, in-vitro studies, and preclinical models have radically reshaped the knowledge landscape. Inflammation in osteoarthritis appears to be distinct from that seen in rheumatoid arthritis. Recent randomised controlled trials, using treatments repurposed from rheumatoid arthritis, have largely been unsuccessful. Genome-wide studies point to defects in repair pathways, which accords well with recent promise using growth factor therapies or Wnt pathway antagonism. Nerve growth factor has emerged as a robust target in osteoarthritis pain in phase 2–3 trials. These studies, both positive and negative, align well with those in preclinical surgical models of osteoarthritis, indicating that pathogenic mechanisms identified in mice can lead researchers to valid human targets. Several novel candidate pathways are emerging from preclinical studies that offer hope of future translational impact. Enhancing trust between industry, basic, and clinical scientists will optimise our collective chance of success.
Introduction
The global impact of osteoarthritis, the most common form of joint disease in developed societies, is predicted to rise steadily as obesity and longevity increase.1 Osteoarthritis is a substantial societal burden, associated with increased mortality and frequently complicated by multimorbidity and polypharmacy.2, 3, 4 The recent acceptance of osteoarthritis as a serious disease has helped to drive the therapeutic agenda forward, to garner support from academia and industry, and to influence health-care prioritisation.5 The market for symptomatic and disease-modifying treatments is huge, and yet relatively little progress has been made thus far in bringing new treatments to patients.
Osteoarthritis research can be broadly divided into clinical and basic categories. Clinical research includes pathology, epidemiology, and interventional studies in humans, whereas basic research encompasses the study of molecular pathogenesis through in-vitro systems, preclinical models, and large-scale omics (ie, genomics, transcriptomics, proteomics, and metabolomics) studies. Osteoarthritis is a mechanically driven disease. This notion is compellingly described in the epidemiological literature6 and confirmed in basic science studies, which have shown the highly mechanosensitive nature of joint tissues,7, 8, 9, 10, 11, 12 the activation of inflammatory signalling by mechanical injury,12, 13 the dependence on mechanics in preclinical osteoarthritis,14, 15 and the involvement of mechanosensing mechanisms in in-vivo pathogenesis.16, 17 Several other important causal factors—such as obesity, age, and genetics—affect the ability of joint tissues to withstand mechanical stress over a lifetime and affect the ability to repair damaged tissues. These factors might also increase the risk of osteoarthritis in ways that are independent of mechanics. For example, osteoarthritis in non-weightbearing joints is increased in obese individuals,18 possibly due to low-grade systemic inflammation,19, 20 which might be linked to the gut microbiome.21
Various impediments are recognised in osteoarthritis drug development. Osteoarthritis is an insidious and heterogeneous disease. These qualities inevitably mean that clinical trials are often prohibitively expensive, and raise the possibility that one target might not work for all. Molecular pathogenesis also has its challenges. Molecular tools have needed to be refined to work in paucicellular, matrix-rich tissues, such as articular cartilage. Low access to human tissue at early stages of disease has necessitated a reliance on preclinical models, which has also required substantial refinement, largely involving moving away from disease models involving chemical induction methods (eg, monosodium iodoacetate, papain, and collagenase injection) in favour of those induced by surgical destabilisation of the joint.22 In the past 15 years, target discovery in osteoarthritis has increased substantially, particularly through large, agnostic omic studies using end-stage human disease tissue and through molecular validation facilitated by preclinical mouse models and clinical trials. There has also been considerable research into methodological tools for improving clinical outcome measures and osteoarthritis trial design.23 In this Review, recent successes and failures in osteoarthritis clinical trials are considered in parallel with preclinical advances. Together, these different types of research are helping to unravel the complexities of osteoarthritis pathogenesis and to provide future targeting strategies with a higher chance of translational success.
Targeting inflammation in osteoarthritis
Support for the involvement of inflammation in osteoarthritis comes from clinical observation (joint line tenderness, synovial thickening, and episodic joint effusion) and radiographic evidence of synovial hypertrophy and bone marrow oedema (by MRI and ultrasound) that are associated with clinical outcome.24, 25, 26 Additionally, various inflammatory molecules—including cytokines, chemokines, and metalloproteinases—have been measured in osteoarthritis cartilage and synovium.27, 28
Clinicians distinguish between inflammatory arthritis and osteoarthritis through a relative paucity of leucocytes in osteoarthritis synovial fluid, which are predominantly monocytes (in osteoarthritis) rather than neutrophils (in rheumatoid arthritis). Patients with osteoarthritis typically complain of less than 30 min early morning stiffness and show a modest systemic inflammatory response.29 These features are used clinically to aid the diagnosis of osteoarthritis. Whether low-grade inflammation contributes to osteoarthritis pathogenesis, both in terms of pain and structural disease, has been subject to heated debate over the years. Several randomised controlled trials that address different aspects of inflammation have been recently conducted. All the tested drugs derive from experience in rheumatoid arthritis where there is proven efficacy for such therapies.
Corticosteroids
Intra-articular corticosteroids are widely used in clinical practice in osteoarthritis, although few historical studies have applied stringent placebo-controlled, randomised, and double-blind assessments. In hand osteoarthritis, a randomised controlled trial30 of intra-articular triamcinalone hexacetomide (a long-acting steroid preparation) plus lidocaine (a local anaesthetic) showed clinical improvement up to 12 weeks following injection of the drug compared with lidocaine alone. This result met the primary outcome of the study, albeit in only two of eight co-primary endpoints.30 For both groups, the injected joint was splinted for 48 h immediately after treatment. A phase 2b trial31 of an extended-release intra-articular steroid showed greater efficacy than placebo in pain outcomes for knee osteoarthritis at several time points, even though the primary endpoint (pain at 12 weeks) was not met. Combined phase 2–3 studies of this preparation have showed an acceptable safety profile and a reduction in use of rescue pain medication (paracetamol or acetaminophen).32 In 2017, this preparation received approval by the US Food and Drug Administration (FDA) for clinical use under the trade name Zilretta.
Oral prednisolone has also been tested in hand osteoarthritis. The first study by Wenham and colleagues,33 in which 70 patients were randomly assigned to receive 5 mg of prednisolone or placebo daily, found no statistically significant improvement in pain at 4 or 12 weeks. However, a 2019 study,34 in which patients were randomly assigned to receive 10 mg of prednisolone or placebo daily for 6 weeks, showed significant improvement in patient-reported pain and function at the primary endpoint of 6 weeks. Symptoms returned rapidly after withdrawal of the drug. The study found a reduction in synovial thickness, but no improvement in synovitis by MRI or power doppler assessment, making the primary target tissue of the drug unclear.34
Few studies have attempted to examine the long-term effects of corticosteroids on joints. In a randomised controlled trial by McAlindon and colleagues,35 140 patients with knee osteoarthritis were randomly assigned to receive intra-articular injections of triamcinolone or saline once every 3 months for 2 years. Clinical outcomes were assessed every 3 months, and cartilage damage was measured by MRI at 2 years. No clinical benefit was seen for any of the outcome measures compared with placebo, although it is possible that the periodicity of follow-up caused transient responses to be missed (ie, if responses returned to baseline by 3 months). Importantly, this study showed a small but statistically significant increase in cartilage volume loss, raising concerns about the effect of repeated and long-term corticosteroid use on joint health.35 Similar findings were also shown using data derived from the Osteoarthritis Initiative.36 A cautious approach to intra-articular steroid is indicated by a “conditional type 1B recommendation” for this treatment in the 2019 guidelines from the Osteoarthritis Research Society International for non-surgical treatment of hip and knee osteoarthritis.37
Disease-modifying anti-rheumatic drugs
Both hydroxychloroquine and methotrexate are used in patients with rheumatoid arthritis and, less commonly, on an individual-patient basis in osteoarthritis. Two randomised controlled trials using oral hydroxychloroquine in hand osteoarthritis have been published, neither of which met the primary study endpoint of reduction in pain.38, 39 Additionally, no clinical response was seen in a predefined substudy in which patients were stratified by the presence or absence of power doppler signal, which is indicative of a more inflammatory phenotype.38 The PROMOTE study40 has reported by abstract41 a small difference in pain in those with knee osteoarthritis taking methotrexate, although the effect size was not deemed clinically meaningful. A small randomised controlled trial42 of 64 patients with hand osteoarthritis taking 10 mg of methotrexate failed to show a beneficial effect on pain, the primary outcome, although some changes to the evolution of joint remodelling were suggested in the reported abstract. A meta-analysis43 has concluded no efficacy of conventional synthetic disease-modifying anti-rheumatic drugs across all joint osteoarthritis.
Anticytokine therapies
An absence of efficacy was also evident in four randomised controlled trials in hand osteoarthritis that targeted either tumour necrosis factor or interleukin (IL)-144, 45, 46, 47 and two trials in knee osteoarthritis targeting IL-1,48, 49 one of which used an intra-articular approach. Despite promise from various small open-label studies, none of the randomised controlled trials met their primary study endpoints, suggesting that classical cytokine-driven inflammation is at the root of neither pain nor structural damage in osteoarthritis. These results are in accordance with preclinical data in which gene deletion of IL-1β,50 the IL-1-converting enzyme, IL-1R (Vincent, unpublished data), tumour necrosis factor,51 or inflammasome pathway components (which lead to processing of IL-1-family cytokines)52 does not confer protection from osteoarthritis after surgical joint destabilisation. Despite a strong rationale based on in-vitro studies, evidence to support a direct pathogenic role for IL-1 in osteoarthritis pathogenesis appears, in retrospect, to have been weak.53
Other putative inflammatory targets from preclinical models
These studies force us to conclude that classical inflammation, of the type that is pathogenic in rheumatoid arthritis, does not drive osteoarthritis. One exception to this notion might be IL-6. Although the osteoarthritis phenotype has been inconsistently reported in IL-6 knockout mice,54, 55 therapeutic studies suggest that neutralisation of IL-6 modifies disease in murine osteoarthritis.56 A clinical trial using tocilizumab, an IL-6-receptor-neutralising antibody, in hand osteoarthritis completed in 2019 but has not yet been reported (registered with ClinicalTrials.gov, NCT02477059).
Targeting the proteases that degrade the articular cartilage extracellular matrix has long been regarded as an attractive approach to disease modification in osteoarthritis. A disintegrin and metalloproteinase with thrombospondin motif (Adamts)-5 was identified as the principal aggrecan-degrading enzyme in mice,57 and in humans ADAMTS-5 also mediates proteolytic activity in osteoarthritis chondrocytes58 (possibly also involving ADAMTS-4).59 Aggrecanase inhibition is being re-explored, after companies had abandoned earlier studies at the preclinical phase because of adverse cardiovascular events, using an anti-Adamts-5 monoclonal antibody.60 A good safety profile and evidence of target engagement with a small molecule inhibitor61 is now being followed by phase 2 studies in knee osteoarthritis, with structural disease as the primary outcome (registered with ClinicalTrials.gov, NCT03595618).
Activation of other components of the innate immune system might have more important pathogenic roles in disease, and some of these components have been examined in preclinical osteoarthritis (table 1 ). Several chemokine family members have been explored after joint destabilisation, with some having disease-modifying effects in murine osteoarthritis (table 1). These proteins are expressed by chondrocytes and have chondroprotective and disease-causing roles, not always correlating with cell infiltration of the joint. They therefore probably act in both canonical and non-canonical ways.68, 69, 70 C-C motif chemokine 2 (Ccl2) and its receptor, C-C chemokine receptor type 2 (Ccr2), are the best validated of these targets. Constitutive deletion of Ccl2 or Ccr2 delays and suppresses pain severity in preclinical osteoarthritis but has little effect on cartilage damage when induced in animals that are 10 weeks old.63, 64 However, a reduction in structural disease has been seen when older (aged 20 weeks) Ccr2 knockout mice are subjected to joint destabilisation,65 and when pharmacological Ccr2 inhibition is delivered.67 In another study,66 structure modification was observed when a Ccr2 antagonist was given either between 1–4 or 4–8 weeks after joint destabilisation, but not when given between 8–12 or 1–8 weeks after. A reduction in pain behaviour was observed over short (3 week) and long (12 week) periods of treatment at all stages of disease. Blocking transforming growth factor α (Tgfα) signalling, a strong inducer of Ccl2 in the rodent osteoarthritis joint, also reduced structural disease after joint destabilisation in the rat.67 TGFα is of particular interest because it has been identified as a candidate gene for determining cartilage thickness and osteoarthritis risk in humans.77, 78 These results point towards a role for Ccl2 and Ccr2 in murine osteoarthritis pain and a possible role in structural progression.
Table 1.
Putative innate immune targets showing disease modification in preclinical studies
| Target tested | Study details | Cartilage modifying? | Symptom modifying? | |
|---|---|---|---|---|
| Complement | ||||
| Wang et al62 | C5 and Cd59a | Knockout data confirmed by pharmacological approach | Yes | Not examined |
| Chemokines | ||||
| Miotla Zarebska et al,63 Miller et al,64 Raghu et al,65 Longobardi,66 and Appleton et al67 | Ccl2 or Ccr2 | Constitutive gene deletion inconsistent across different studies but appearing to show structure modification at later time points; pharmacological studies point towards a key treatment window | Inconsistent | Yes |
| Takebe et al,68 and Raghu et al65 | Ccr5 or Ccl5 | Inconsistent cartilage degradation scores; neither study showed a difference in synovitis scores after gene deletion | Inconsistent | Not examined |
| Sambamurthy et al69 | Ccr7 | Modest structural role, knockout mice have reduced pain behaviour | Yes | Yes |
| Sherwood et al70 | Cxcr2 | Structural increase at 8 weeks in knockout mice (ie, protective) | Yes | Not examined |
| Qin et al71 | Cxcr4 | Inhibition in bone abrogates surgically induced osteoarthritis | Yes | Not examined |
| Mechanoflammation | ||||
| Choi et al72 | IkB-zeta subunit of Nf-κB | Over-expression worsens disease; conditional detection leads to decreased disease (both on Col2 promoter) | Yes | Not examined |
| Kobayashi et al73 | RelA (p65) Nf-κB transcription factor | Dual action of RelA in disease: heterozygotes protected; homozygotes showed increased disease through prevention of anti-apoptotic mechanisms induced by Pik3r1 (a Gwas hit for cartilage thickness) | Yes | Not examined |
| Culley et al74 | Ikka | Conditional knockout (aggrecan Cre) disease protection associated with increased apoptosis | Yes | Not examined |
| Ismail et al75 | Jnk2 | Chondroprotection observed at 4 weeks, 8 weeks, and 12 weeks after surgery | Yes | Not examined |
| Mast-cell activation | ||||
| Wang et al76 | c-Kit and Mcl1 | Deletion produces functional deletion of c-Kit-dependent and c-Kit-independent mast cells; chondroprotection also observed with Apc366, a tryptase inhibitor | Yes | Not examined |
| Wang et al76 | Igh7 and Fcer1 | Both genes target IgE-mediated activation of mast cells, indicating that IgE-induced mast-cell activation drives osteoarthritis pathology | Yes | Not examined |
Two mRNA studies of human synovium have been done in individuals stratified by having painful or non-painful osteoarthritis.27, 79 One of these studies27 identified CCL2 as being significantly up-regulated in painful disease. CCR2 antagonism in osteoarthritis pain has been explored clinically (registered with ClinicalTrials.gov, NCT00689273), although the results of the study do not appear to have been reported. A clinical study examining TGFα blockade is currently recruiting (registered with ClinicalTrials.gov, NCT04456686). All current osteoarthritis disease-modifying drug trials are summarised in table 2 .
Table 2.
Registered active drug trials in osteoarthritis
| Study type and number of participants | Drug | Target or drug type | Route | Trial status | Primary outcome | Secondary outcomes | |
|---|---|---|---|---|---|---|---|
| NCT04456686 | Phase 2 randomised controlled trial; 125 participants | LY3016859 | Transforming growth factor α and epiregulin | Intravenous | Recruiting | Pain (numeric rating scale) | Function |
| NCT04447898 | Phase 1 randomised controlled trial; 24 participants | PPV-06 vaccination | Interleukin-6 | Subcutaneous | Not yet recruiting | Safety | Not given |
| NCT04385303 | Phase 3 randomised controlled trial; 726 participants | Lorecivivint (SM04690) | Wnt pathway | Intra-articular | Recruiting | Pain (numeric rating scale) | Function |
| NCT03928184 | Phase 3 randomised controlled trial; 725 participants | Lorecivivint (SM04690) | Wnt pathway | Intra-articular | Recruiting | Pain (numeric rating scale) | Function; structure |
| NCT04318041 | Phase 3 randomised controlled trial; 128 participants | Diacerin | Unknown or anti-inflammatory | Oral | Not yet recruiting | Structure (MRI) | Function |
| NCT04303026 | Phase 3 randomised controlled trial; 70 participants | Zoledronic acid | Osteoclast activity | Intravenous | Recruiting | Pain (Visual analogue scale) | Function; structure |
| NCT04231318 | Phase 3 randomised controlled trial; 231 participants | Cingal | Triamcinalone plus hyaluronate | Intra-articular | Not yet recruiting | Pain (WOMAC) | Not given |
| NCT04224584 | Phase 2 crossover controlled trial; 40 participants | Duloxetine | CNS reuptake inhibitor | Oral | Recruiting | Pressure pain threshold | Not given |
| NCT04117893 | Phase 4 randomised open-label trial; 150 participants | Duloxetine plus hyaluronic acid plus triamcinolone | CNS reuptake inhibitor plus corticosteroid plus hyaluronan | Oral and intra-articular | Not yet recruiting | Pain (average pain scores) | Function |
| NCT04261049 | Open-label trial; 35 participants | Zilretta | Corticosteroid (slow release) | Intra-articular | Not yet recruiting | Muscle strength; function and gait | Not given |
| NCT04123561 | Phase 3 randomised controlled trial; 500 participants | TLC599 | Corticosteroid (slow release) | Intra-articular | Recruiting | Pain (WOMAC) | Function |
| NCT04120402 | Phase 2 randomised controlled trial; 238 participants | EP-104IAR | Corticosteroid (slow release) | Intra-articular | Not yet recruiting | Pain (WOMAC) | Function |
| NCT04097379 | Phase 2 randomised controlled trial; 40 participants | LRX712 | Not disclosed; pro-regenerative | Intra-articular | Not yet recruiting | Structure (sodium cartilage content by MRI) | Pharmaco-kinetics |
| NCT03913442 | Phase 4 randomised controlled trial; 120 participants | Colchicine | Anti-inflammatory; precise mechanism disputed | Oral | Recruiting | Pain (visual analogue scale) | Function |
| NCT03815448 | Randomised controlled trial; 200 participants | Methotrexate | Immunosuppressant (folate antagonist) | Oral | Recruiting | Synovitis (MRI); pain (visual analogue scale) | Function |
| NCT01927484 | Randomised controlled trial; 120 participants | Methotrexate | Immunosuppressant (folate antagonist) | Oral | Not yet recruiting* | Pain (visual analogue scale) | Function |
| NCT02905799 | Phase 3 randomised controlled trial; 164 participants | Resveratrol | Anti-ageing or anti-inflammatory (multiple proposed mechanisms of action) | Oral | Recruiting | Pain (numeric rating scale) | Function |
| NCT04119687 | Phase 1 open-label trial; 24 participants | FX201 | Interleukin-1 receptor antagonist gene therapy | Intra-articular | Recruiting | Safety | Biodistribution |
| NCT02790723 | Phase 1 open label; 9 participants | Sc-rAAV2.5IL-1Ra | Interleukin-1 receptor antagonist gene therapy | Intra-articular | Recruiting | Safety | Not given |
| NCT02471118 | Phase 2 crossover randomised controlled trial; 100 participants | Adalimumab | Anti-tumour necrosis factor | Subcutaneous | Recruiting | OARSI/OMERACT response | Pain; function |
| NCT03595618 | Phase 2 randomised controlled trial; 928 participants | GLPG1972 | ADAMTS-5 inhibitor | Oral | Not yet recruiting | Structure (cartilage thickness by MRI) | Other structure; function and pain |
Data taken from ClinicalTrials.gov on July 24, 2020.
Trial terminated due to COVID-19.
Other types of innate immune activation might be important in osteoarthritis pathogenesis but have as yet only been explored as targets in preclinical models (table 1). Components of the common terminal pathway of complement activation are strongly up-regulated in the synovial fluid of individuals with osteoarthritis,80 with evidence of the formation of membrane attach complex within human osteoarthritis cartilage.62 Deletion of C5 (an upstream activator of the common pathway) in mice led to reduced disease severity after joint destabilisation, whereas deletion of an inhibitor of terminal activation (Cd59a) led to increased disease severity.62 The same research group also identified mast-cell activity as a pathogenic mediator in murine osteoarthritis.76 Mast-cell activation has previously been described in the osteoarthritis joint,81 and is associated with structural disease.25
Inflammasome activation is purported to have a role in osteoarthritis, especially when disease is complicated by a crystal arthropathy. However, studies in mice in which components of the inflammasome pathway (activated by crystals) were genetically deleted failed to show a role for inflammasome in surgically induced osteoarthritis.82, 83 Several groups have examined the role of alarmins in osteoarthritis, through deletion of Toll-like receptors, S100 proteins, or advanced glycosylation end productspecific receptors. Collectively, these studies do not support a role for these molecules in surgically induced murine osteoarthritis.82, 84 Many preclinical studies in this area of research remain unpublished, and this reporting bias has been unhelpful for research over the years.85
My own work, and work arising from the Centre of Osteoarthritis Pathogenesis at the Kennedy Institute of Rheumatology (Oxford, UK) has highlighted an important role for what has been termed mechanoflammation,86 showing that mechanical injury directly drives inflammatory signalling and inflammatory genes in joint tissues, including the articular cartilage and synovium.87, 88 Joint immobilisation after destabilisation surgery attenuates the induction of pathogenic proteases and prevents osteoarthritis development.14 Mechanoflammation drives TGFβ-activated kinase (TAK1) and downstream activation of the inflammatory mitogen-activated protein kinases (JNK and p38) and nuclear factor κB (NF-κB).12 NF-κB signalling pathway has long been considered an important inducer of inflammatory gene regulation in osteoarthritis. It is a complex pathway with canonical and non-canonical pathways that mediate anti-apoptotic and pro-inflammatory functions. This characterisation has been confirmed in vivo in a dose-dependent manner, in which heterozygous deletion of RelA (p65), a transcription factor activated upon canonical NF-κB activation, resulted in chondroprotection, whereas homozygous deletion led to accelerated disease through the suppression of apoptosis.73 Accelerated disease resulting from homozygous deletion of p65 was mediated through decreased expression of the anti-apoptoic gene Pik3r1, itself a candidate gene arising from a genome-wide association study for cartilage thickness.89 Deletion of Ikkα (which inhibits κB-kinase-α, an upstream NF-κB pathway activator) leads to disease protection and anti-apoptotic effects in vivo.74 Although NF-κB might be important in transcriptional regulation of proteases in osteoarthritis, JNK activation controls the bioavailability of aggrecanase activity in vitro58 and in vivo,75 by a mechanism that appears to involve re-uptake of aggrecanases by the cell surface scavenger receptor, low-density lipoprotein receptor-related protein.90, 91 Targeting protease activity through metal cation symporter Zip8, a zinc transporter, has also been shown in murine osteoarthritis.92 Zip8 is regulated by the hypoxia transcription factor Hif2α,93 which has also been shown to be disease-modifying in preclinical osteoarthritis models.94
Promoting anabolism and repair in osteoarthritis
The inability of articular cartilage to repair is famously attributed to William Hunter who stated in 1743 that “…ulcerated Cartilage is universally allowed to be a very troublesome disease…and when destroyed, it is never recovered”.95 The essence of this statement has been reiterated in textbooks for decades, but recent years have seen a paradigm shift. Improved MRI imaging indicates that asymptomatic focal defects in the joint surface are much more common than previously suggested, and prospective studies conclude that around 30% of focal cartilage defects spontaneously regress over time.96 Regression of osteoarthritis, as measured by Kellgren and Lawrence x-ray score during a 14-year period, has been documented in the Chingford Women's cohort,97 and preclinical studies show evidence of intrinsic repair of focal cartilage defects in a mouse strain (genetic) and age-dependent manner.98, 99
Load-altering procedures
The best clinical evidence of intrinsic cartilage repair in individuals with osteoarthritis comes from open-label studies of joint distraction. The largest study to date involved 20 patients. Applying a distraction frame for 6 weeks across the osteoarthritis knee joint resulted in an impressive clinical response (reduced pain and improved function assessed by the Western Ontario and McMaster Universities Osteoarthritis Index [WOMAC]) and regrowth of tissue that resembled articular cartilage by MRI at 1 year and 2 years.100, 101 Extended follow-up of this cohort showed that trial participants were less likely than a disease-matched osteoarthritis population to undergo joint replacement surgery.102 Similar, albeit smaller, studies have been done by other groups.103 The procedure results in a reduction of compressive load through the joint and complete prevention of surface shear stress (ie, no joint flexion). These concepts fit well with observations in mice, in which immobilising the knee joint in a fully extended position prevents protease regulation and protects the mouse from osteoarthritis after joint destabilisation. Maintaining some compressive force is likely to be more effective than complete joint immobilisation because it promotes the release of matrix-bound chondroprotective growth factors, such as fibroblast growth factor (FGF) 2.14, 16 When the synovial fluid levels of candidate molecules were examined over the course of joint distraction, out of ten analytes examined, only two, FGF2 and TGFβ (both pro-regenerative growth factors), predicted a good clinical response.104
High tibial osteotomy, whereby a wedge of bone is removed from the top of the tibia (usually) to correct valgus–varus joint malalignment, is also associated with clinical improvement.105 Moreover, when studies have examined the cartilage macroscopically through arthrotomy, histologically, or by MRI, evidence of cartilage regeneration is observed in the now off-loaded part of the joint.106, 107, 108
Intra-articular FGF18
Sperifermin is a truncated form of FGF18. The FGFs form a large family of pleiotropic growth factors implicated in a range of physiological and pathological processes, including embryonic development, tissue repair, and cancer.109 Whereas FGF2 is promiscuous, binding to all four FGF receptors (FGFRs), FGF18 is thought to be more selective for FGFR3, which is the chondroprotective FGFR in murine osteoarthritis studies.110, 111, 112 Of note, polymorphic variants in FGFR3 have been identified in two genome-wide association studies: a population study77 associating a polymorphic variant with articular cartilage thickness, and another study113 that identified it as an at-risk allele in osteoarthritis. The latter study also identified FGF18 as a candidate gene associated with osteoarthritis risk.113
In 2014, a proof-of-concept study114 was reported in which 192 individuals with osteoarthritis were randomly assigned to receive three doses of intra-articular sprifermin (recombinant truncated form of FGF18) or placebo, with follow-up at 6 months and 12 months. The study failed to meet its primary endpoint (a difference in articular cartilage thickness in the central medial femoro-tibial compartment), but it did show delayed loss of cartilage overall and thickening in the lateral compartment.114 In 2019, the FORWARD trial,115 in which 549 participants received intra-articular sprifermin every 6 months or 12 months, or placebo, reported a significant increase in total femoro-tibial cartilage volume compared with placebo at 2-year follow-up, albeit without significant clinical improvement. In a recent post-hoc analysis116 of the original trial data (thus far reported in abstract form), sprifermin treatment showed a statistically significant clinical and structural improvement over among a subgroup of 161 patients who were defined as being at high risk of progression. Although these studies do not specifically show reversal of cartilage damage (ie, true repair), they do show that damage can be arrested and therefore indicate a structure-modifying osteoarthritis drug. Whether these drugs turn out to be true disease-modifying osteoarthritis drugs is not yet clear. The apparent discordance between structure and symptoms in osteoarthritis is discussed later in this Review.
Intra-articular Wnt inhibitor
Wnts are a complex family of cellular signalling molecules that direct a broad range of cellular responses, particularly regarding bone development. Wnts are activated upon mechanical stress of articular cartilage117, 118 and are thought to drive the dedifferentiated chondrocyte phenotype, bone remodelling, and induction of catabolic enzymes seen in osteoarthritis.119, 120, 121 Canonical Wnt signalling involves stabilisation of the signalling molecule beta-catenin within the cell. Interfering with beta-catenin has shown conflicting outcomes in experimental osteoarthritis, indicating that this molecule is not readily amenable to therapeutic translation.122 Interfering with natural inhibitors of Wnt signalling in mice, such as Dkk1 and Dot1l, reveals the disease-modifying potential of this pathway.123, 124, 125, 126 SM04690 is a synthetic Wnt inhibitor with an undisclosed (unknown) primary mechanism of action that has shown success in murine models of osteoarthritis.127, 128 A phase 1 study of a single intra-articular dose of SM04690 in 61 participants with moderate osteoarthritis showed acceptable safety, with exploratory clinical endpoints that showed a positive trend towards improvement in pain and joint space narrowing.129 A phase 2 study of 455 individuals with unilateral knee osteoarthritis, although not reaching its primary endpoint (improvement of WOMAC pain at week 13), showed improvement in pain and an increase in joint space indicative of disease modification, which was especially evident in people with unilateral disease.130 In May 2019, the phase 3 studies were launched (registered on ClinicalTrials.gov, NCT04385303).
Other putative anabolic targets from preclinical studies
Recent studies in murine osteoarthritis identify the transcriptional coactivator Yap and WW domain-containing transcription regulator protein 1 (Taz) pathway as a strong chondroprotective mechanism. Yap and Taz are both transcription factors that are activated by Hippo signalling, a highly conserved pathway thought to be involved in cellular mechanotransduction.131 Genetic and pharmacological enhancement of this pathway protects joints from osteoarthritis after joint destabilisation,132 which might in part be due to it controlling the generation of chondroprogenitor cells arising from the synovium.133 The Yap–Taz pathway also reciprocally controls Tak1132 (strongly induced by cartilage injury), and this might be an important mechanism by which inflammation suppresses repair (figure 1 ).
Figure 1.
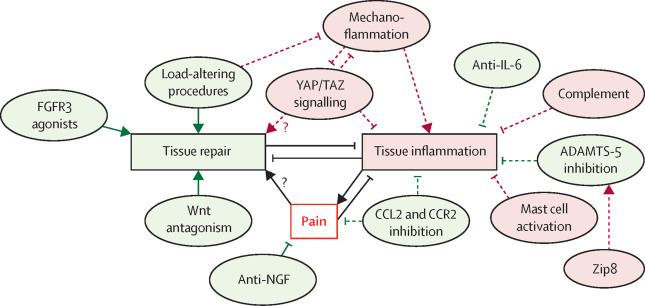
Emerging therapeutic targets in osteoarthritis
Pathological targets largely cluster into those promoting repair, those neutralising pain, and those suppressing tissue inflammation (leading in turn to degradation). A reciprocal relationship exists between inflammatory and repair pathways in the osteoarthritis joint, both of which affect pain. Green circles indicate targets that show efficacy in mouse experiments and for which therapeutic strategies are being tested in clinical trials, and red circles indicate putative pathways identified in mice that have not yet been tested in clinical studies. Solid lines represent those with proven efficacy in human studies, and dashed lines indicate where clinical study outcomes are not yet known. Arrows indicate promotion, and flat line-ends represent suppression. Question marks indicate where connection is speculative. Load-altering procedures include surgical joint distraction and wedge osteotomy to correct joint malalignment, which probably suppress mechanoflammation. Peripheral pain arises from joint pathology and might suppress tissue inflammation and enable tissue repair by preventing mechanical overload of the joint. Zip8 is a zinc transporter that controls protease regulation in chondrocytes. FGFR=fibroblast growth factor receptor. IL=interleukin. YAP=transcriptional coactivator Yap. TAZ=WW domain-containing transcription regulator protein 1. ADAMTS=a disintegrin and metalloproteinase with thrombospondin motif. CCL=C-C motif chemokine. CCR=C-C chemokine receptor. Zip8=metal cation symporter Zip8. NGF=nerve growth factor.
Targeting nerve growth factor to treat osteoarthritis pain
Nerve growth factor (NGF) has long been known to sensitise pain fibres and, in doing so, enhance the firing rate of nociceptors in response to mechanical and thermal stimuli. NGF is also known to be a neurotrophic factor, directing the growth of new nerves.134 The use of anti-NGF neutralising antibodies to inhibit osteoarthritis pain has been heralded as a huge breakthrough for osteoarthritis patients who have struggled for years with inadequate pain relief. Several biological drugs targeting NGF, all delivered systemically (intravenously or subcutaneously), have been tested in phase 2 studies, with a meta-analysis showing efficacy across the different studies.135 Two companies have now published phase 2–3 studies using NGF neutralising antibodies,136, 137, 138 with fasinumab and tanezumab showing efficacy over placebo. Concerns over patients developing rapidly progressive osteoarthritis in index and non-index joints (ranging from 2 to 10% according to dose and study) forced the FDA to introduce mitigation strategies, which included reducing highest doses and prohibiting the use of concomitant non-steroidal anti-inflammatory drugs. The community now awaits a decision from the FDA on whether this class of drug, which was designated as fast track in 2017, will be approved for patient use.
Other strategies to target NGF signalling have also been tested. In 2019, two randomised controlled trials targeting high affinity nerve growth factor receptor (TrkA), the receptor through which NGF signals, were published.139, 140 In one study,139 215 participants were randomly assigned to receive twice daily oral dosing with ASP7962, placebo or naproxen for 4 weeks. The study failed to meet its primary endpoint (WOMAC pain subscore).139 A second study140 randomly assigned 104 participants to intraarticular GZ389988A or placebo. This study did show improved pain outcomes of the drug compared with placebo, although the effect size was small and of questionable clinical value.140 Neither study was accompanied by evidence of target tissue drug bioavailability.
Anti-NGF clinical trials align well with evidence of NGF-mediated pain-like behaviour in rodent osteoarthritis. Pain-like behaviour can be measured by evoked or non-evoked methods. Like humans, rodents will spontaneously off-load the damaged joint when experiencing pain, and this behaviour can be measured by assessing the amount of weight transmitted through each hind limb. Using this technique, mice have been shown to display two phases of pain behaviour after joint destabilisation: an initial post-operative phase that resolves after 1 week, and a later phase that starts only once there is significant joint damage (10 weeks after destabilisation of the medial meniscus or 8 weeks after partial meniscectomy).141, 142, 143 Late osteoarthritis pain in rodents is Ngf-dependent144, 145 and tumour necrosis factor-independent.145 The driver of NGF-dependent late osteoarthritis pain is unclear, but Ngf mRNA upregulation occurs in the articular cartilage rather than bone or meniscus, and there is very little inflammatory gene regulation in the joint during this time.146 Although this observation might be surprising in view of broadly held views that osteoarthritis pain originates from inflammatory processes in the synovium or subchondral bone, emerging molecular data from human tissue also support the notion that cartilage is the principal source of NGF in the osteoarthritis joint. Using agnostic approaches, NGF was not regulated in the synovium of individuals with painful compared with non-painful osteoarthritis,27, 79 and it was not found in bone marrow lesions from samples taken at the time of arthroplasty.147 NGF was found to be regulated in damaged articular cartilage in early microarray studies of osteoarthritis cartilage,148 and it defines one of seven subsets of chondrocytes identified by single-cell sequencing of human osteoarthritis cartilage.149 NGF is regulated by direct cartilage injury (mechanoflammation) in a TAK1-dependent manner, and it is tempting to speculate that damage to chondrocytes near the osteochondral junction is an important trigger for the NGF-driven neoinnervation of the articular cartilage that is seen late in both murine and human disease.150, 151 Neoinnervation of this region also requires a permissive subchondral bone to support axonal extension. This neoinnervation has recently been shown to be dependent upon Netrin-1, secreted by osteoclasts during the course of murine osteoarthritis.152 An overall model for the development of pain in osteoarthritis has been proposed.153
Conclusions
There are many reasons to be optimistic about new therapeutic developments in osteoarthritis. Although it is true that much of what has been learned in the past few years from clinical studies is what not to use in disease, these negative studies have been highly informative in reminding the medical community that osteoarthritis is distinct from inflammatory arthritidies, such as rheumatoid arthritis. Research has shown that inflammation in osteoarthritis is nuanced and that classical immunomodulatory pathways are not good targets, but that there are several other inflammatory pathways awaiting clinical exploration, including those driven by direct mechanical injury of the cartilage (so-called mechanoflammation), complement, and mast cells.
The nature and role of inflammation in osteoarthritis pathogenesis thus remains unclear. Clarification is crucially important, not only so that we can develop appropriate targeted therapies for patients, but also to decide whether patients require stratification before treatment. There has been a popular move to try to phenotype patients, with a view to personalising their treatment to improve the efficacy of a given drug. However, these phenotypes currently lack cohesion; some are defined by clinical features (eg, inflammatory osteoarthritis), and others by co-morbidity (eg, metabolic osteoarthritis), precipitating factor (eg, post-traumatic osteoarthritis), or anatomical site (eg, hand osteoarthritis, hip osteoarthritis). There is little or no evidence that stratification by any of these features changes the response to treatment. Further carefully considered phenotypes that take into consideration molecular pathways are probably required. Large-scale molecular endotyping of patient samples is currently in its infancy, but will probably help.
Clinical successes point towards a focus on regenerative or anabolic pathways rather than inflammatory ones (figure 1). This suggestion fits well with preclinical studies, although the reciprocal relationship between repair and inflammation in the chondrocyte suggests that targeting one will probably affect the other.132 Recent large genome-wide association studies in osteoarthritis also support the concept that osteoarthritis is a failure of repair. Several at-risk loci have been attributed to genes in the TGFβ and FGF pathways, and there is a notable absence of loci that predict the regulation of classical inflammatory genes.113, 154 Newer targets identified by genome studies, including the retinoic acid pathway, also look promising.155
NGF-targeting for pain relief is the target closest to being ready to use in osteoarthritis. Clinical success in late osteoarthritis indicates that analgesia occurs largely as a result of nociceptor desensitisation. It remains to be seen whether interfering with this pathway at earlier stages of the disease could affect the neoinnervation of the cartilage due to the neurotrophic functions of NGF, and to what extent this could prevent painful disease from developing. This type of strategy would need to be considered in the context of current safety concerns around the development of rapidly progressive osteoarthritis, which remains a real concern. Other molecules that appear to have a role in the neoinnervation of the osteochondral junction in osteoarthritis models include Netrin-1,152 a molecule secreted by osteoclasts that guides axonal growth through the subchondral bone. Blocking bone remodelling with a bisphosphonate early in murine osteoarthritis development appears to block pain without affecting structural disease, according well with clinical studies in osteoarthritis in which bisphosphonates are not disease-modifying when given in established disease.156, 157
One major outstanding issue remains the apparent discordance between structural and symptomatic disease, which raises questions about whether validated drugs need to be able to, or indeed could ever, target both. Whether different joint pathologies give rise to different types of symptoms at different stages of disease is currently unknown, as is the relative contribution of factors that drive central sensitisation of pain. Of the few examples available at this stage, cartilage structure-modifying drugs (eg, sprifermin) mainly arrest disease progression rather than regenerating the cartilage, so perhaps symptoms could not be expected to reverse. Where structural damage appears to reverse (eg, after joint distraction), symptoms also appear to improve (albeit with no placebo control). Targeting pain alone is unlikely to improve structure in the short term and might worsen damage through mechanical overuse. In preclinical models, there tends to be better accordance between structural damage and pain-like behaviour,153 with some clear examples emerging that might identify true disease-modifying osteoarthritis drugs of the future, such as those involving the YAP–TAZ pathway.
Finally, it is reassuring to conclude that, where there is overlap, research in surgical preclinical osteoarthritis models aligns well with findings in clinical trials (figure 2 ). This concordance provides valuable validation of the models and will help develop mutual trust between the different osteoarthritis research disciplines. It is increasingly difficult to claim that mouse osteoarthritis is fundamentally different to human osteoarthritis, or that post-traumatic osteoarthritis does not inform age-related disease in humans. Part of this reassurance has emerged through improved awareness of bias mitigation in clinical and preclinical studies.158 It is also partly due to the acceptance that osteoarthritis has disease-specific molecular targets. Regardless, this is an important time for osteoarthritis research, with tangible translational benefits within reach.
Figure 2.
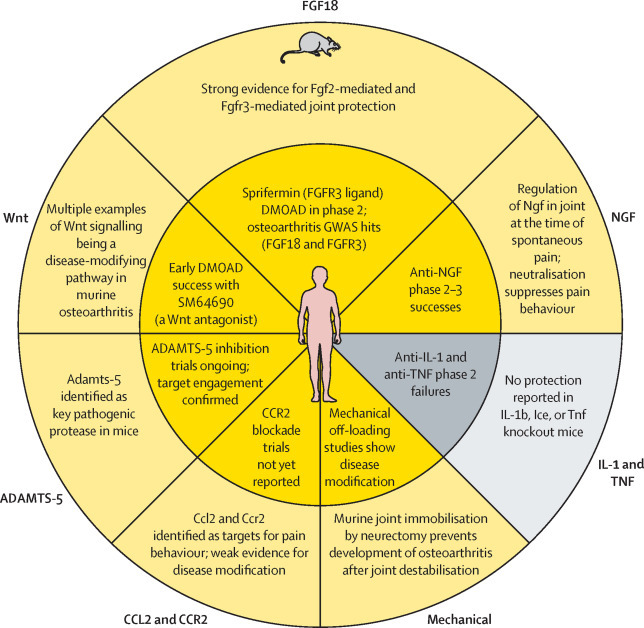
Concordance between tested targets in mouse and human osteoarthritis studies
Several pathways have been or are being tested in human osteoarthritis, having also been tested in murine surgical models. Yellow indicates therapies that show treatment success or target engagement in each study. Grey shows therapies that have failed to modify symptomatic or structural disease. Note that there is 100% concordance between mouse (outer ring) and human (inner ring) studies. FGF=fibroblast growth factor. FGFR=fibroblast growth factor receptor. DMOAD=disease-modifying osteoarthritis drug. GWAS=genome-wide association study. NGF=nerve growth factor. IL=interleukin. TNF=tumour necrosis factor. ICE=capsase-1/interleukin-1 converting enzyme. CCL=C-C motif chemokine. CCR=C-C chemokine receptor. ADAMTS=A disintegrin and metalloproteinase with thrombospondin motif.
Search strategy and selection criteria
This is a narrative Review based on clinical trials done in hand and knee or hip osteoarthritis by searching PubMed with the terms “Phase” with “Trial” and “Osteoarthritis” in the title from Jan 1, 2012, to July 31, 2019. Further information was sought through Clinicaltrials.gov, by searching for “osteoarthritis” studies in which the intervention was “drug”. Preclinical studies were interrogated through Skeletalvis.ncl.ac.uk. This Review is not intended to be a comprehensive review of all clinical trials in osteoarthritis or all pathways identified through murine studies. Rather, its intention is to focus on those targets for which there is overlap between murine and human studies. Additionally, the Review highlights a few emerging pathways that have strong preclinical evidence for a role in pathogenesis and which could be amenable to clinical targeting. Inevitably, an exercise of this sort reflects the author's personal views on pathogenesis, based on 20 years of working with preclinical surgical models, human tissue, and patients with osteoarthritis.
Acknowledgments
Acknowledgments
TLV's lab is supported by the Centre for Osteoarthritis Pathogenesis, Versus Arthritis (grant numbers 20205 and 21621). I would like to thank Dr Elizabeth Thompson for help with figures.
Declaration of interests
TLV has consulted for GlaxoSmithKline, Mundipharma, and Union Chimique Belge in the past 3 years on an ad-hoc basis. She directs the STEpUP osteoarthritis consortium, which has financial support from Samumed, Fidia, and Galapagos.
References
- 1.Oo WM, Yu SP-C, Daniel MS, Hunter DJ. Disease-modifying drugs in osteoarthritis: current understanding and future therapeutics. Expert Opin Emerg Drugs. 2018;23:331–347. doi: 10.1080/14728214.2018.1547706. [DOI] [PubMed] [Google Scholar]
- 2.Losina E, Paltiel AD, Weinstein AM, et al. Lifetime medical costs of knee osteoarthritis management in the United States: impact of extending indications for total knee arthroplasty. Arthritis Care Res (Hoboken) 2015;67:203–215. doi: 10.1002/acr.22412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kluzek S, Sanchez-Santos MT, Leyland KM, et al. Painful knee but not hand osteoarthritis is an independent predictor of mortality over 23 years follow-up of a population-based cohort of middle-aged women. Ann Rheum Dis. 2016;75:1749–1756. doi: 10.1136/annrheumdis-2015-208056. [DOI] [PubMed] [Google Scholar]
- 4.Neogi T. The epidemiology and impact of pain in osteoarthritis. Osteoarthritis Cartilage. 2013;21:1145–1153. doi: 10.1016/j.joca.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hawker GA. Osteoarthritis is a serious disease. Clin Exp Rheumatol. 2019;37(suppl 120):3–6. [PubMed] [Google Scholar]
- 6.Brandt KD, Dieppe P, Radin EL. Commentary: is it useful to subset “primary” osteoarthritis? A critique based on evidence regarding the etiopathogenesis of osteoarthritis. Semin Arthritis Rheum. 2009;39:81–95. doi: 10.1016/j.semarthrit.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Quinn TM, Schmid P, Hunziker EB, Grodzinsky AJ. Proteoglycan deposition around chondrocytes in agarose culture: construction of a physical and biological interface for mechanotransduction in cartilage. Biorheology. 2002;39:27–37. [PubMed] [Google Scholar]
- 8.Kim YJ, Sah RL, Grodzinsky AJ, Plaas AH, Sandy JD. Mechanical regulation of cartilage biosynthetic behavior: physical stimuli. Arch Biochem Biophys. 1994;311:1–12. doi: 10.1006/abbi.1994.1201. [DOI] [PubMed] [Google Scholar]
- 9.Lee RB, Wilkins RJ, Razaq S, Urban JP. The effect of mechanical stress on cartilage energy metabolism. Biorheology. 2002;39:133–143. [PubMed] [Google Scholar]
- 10.Li X, Han L, Nookaew I, et al. Stimulation of Piezo1 by mechanical signals promotes bone anabolism. eLife. 2019;8:8. doi: 10.7554/eLife.49631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vincent T, Hermansson M, Bolton M, Wait R, Saklatvala J. Basic FGF mediates an immediate response of articular cartilage to mechanical injury. Proc Natl Acad Sci USA. 2002;99:8259–8264. doi: 10.1073/pnas.122033199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ismail HM, Didangelos A, Vincent TL, Saklatvala J. Rapid activation of transforming growth factor β-activated kinase 1 in chondrocytes by phosphorylation and K63—linked polyubiquitination upon injury to animal articular cartilage. Arthritis Rheumatol. 2017;69:565–575. doi: 10.1002/art.39965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fanning PJ, Emkey G, Smith RJ, Grodzinsky AJ, Szasz N, Trippel SB. Mechanical regulation of mitogen-activated protein kinase signaling in articular cartilage. J Biol Chem. 2003;278:50940–50948. doi: 10.1074/jbc.M305107200. [DOI] [PubMed] [Google Scholar]
- 14.Burleigh A, Chanalaris A, Gardiner MD, et al. Joint immobilization prevents murine osteoarthritis and reveals the highly mechanosensitive nature of protease expression in vivo. Arthritis Rheum. 2012;64:2278–2288. doi: 10.1002/art.34420. [DOI] [PubMed] [Google Scholar]
- 15.Kim JH, Lee G, Won Y, et al. Matrix cross-linking-mediated mechanotransduction promotes posttraumatic osteoarthritis. Proc Natl Acad Sci USA. 2015;112:9424–9429. doi: 10.1073/pnas.1505700112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chia S-L, Sawaji Y, Burleigh A, et al. Fibroblast growth factor 2 is an intrinsic chondroprotective agent that suppresses ADAMTS-5 and delays cartilage degradation in murine osteoarthritis. Arthritis Rheum. 2009;60:2019–2027. doi: 10.1002/art.24654. [DOI] [PubMed] [Google Scholar]
- 17.O'Conor CJ, Griffin TM, Liedtke W, Guilak F. Increased susceptibility of Trpv4-deficient mice to obesity and obesity-induced osteoarthritis with very high-fat diet. Ann Rheum Dis. 2013;72:300–304. doi: 10.1136/annrheumdis-2012-202272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Visser AW, Ioan-Facsinay A, de Mutsert R, et al. Adiposity and hand osteoarthritis: the Netherlands Epidemiology of Obesity study. Arthritis Res Ther. 2014;16:R19. doi: 10.1186/ar4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kroon FPB, Veenbrink AI, de Mutsert R, et al. The role of leptin and adiponectin as mediators in the relationship between adiposity and hand and knee osteoarthritis. Osteoarthritis Cartilage. 2019;27:1761–1767. doi: 10.1016/j.joca.2019.08.003. [DOI] [PubMed] [Google Scholar]
- 20.Huang ZY, Stabler T, Pei FX, Kraus VB. Both systemic and local lipopolysaccharide (LPS) burden are associated with knee OA severity and inflammation. Osteoarthritis Cartilage. 2016;24:1769–1775. doi: 10.1016/j.joca.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boer CG, Radjabzadeh D, Medina-Gomez C, et al. Intestinal microbiome composition and its relation to joint pain and inflammation. Nat Commun. 2019;10 doi: 10.1038/s41467-019-12873-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vincent TL, Williams RO, Maciewicz R, Silman A, Garside P. Mapping pathogenesis of arthritis through small animal models. Rheumatology (Oxford) 2012;51:1931–1941. doi: 10.1093/rheumatology/kes035. [DOI] [PubMed] [Google Scholar]
- 23.Messier SP, Callahan LF, Golightly YM, Keefe FJ. OARSI clinical trials recommendations: design and conduct of clinical trials of lifestyle diet and exercise interventions for osteoarthritis. Osteoarthritis Cartilage. 2015;23:787–797. doi: 10.1016/j.joca.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Damman W, Liu R, Bloem JL, Rosendaal FR, Reijnierse M, Kloppenburg M. Bone marrow lesions and synovitis on MRI associate with radiographic progression after 2 years in hand osteoarthritis. Ann Rheum Dis. 2017;76:214–217. doi: 10.1136/annrheumdis-2015-209036. [DOI] [PubMed] [Google Scholar]
- 25.deLange-Brokaar BJ, Ioan-Facsinay A, Yusuf E, et al. Pain in knee osteoarthritis patients associates with distinct pattern of synovitis. Arthritis Rheumatol. 2014;67:733–740. doi: 10.1002/art.38965. [DOI] [PubMed] [Google Scholar]
- 26.Neogi T, Guermazi A, Roemer F, et al. Association of joint inflammation with pain sensitization in knee osteoarthritis: the multicenter osteoarthritis study. Arthritis Rheumatol. 2016;68:654–661. doi: 10.1002/art.39488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wyatt LA, Nwosu LN, Wilson D, et al. Molecular expression patterns in the synovium and their association with advanced symptomatic knee osteoarthritis. Osteoarthritis Cartilage. 2019;27:667–675. doi: 10.1016/j.joca.2018.12.012. [DOI] [PubMed] [Google Scholar]
- 28.Aigner T, Zien A, Gehrsitz A, Gebhard PM, McKenna L. Anabolic and catabolic gene expression pattern analysis in normal versus osteoarthritic cartilage using complementary DNA-array technology. Arthritis Rheum. 2001;44:2777–2789. doi: 10.1002/1529-0131(200112)44:12<2777::aid-art465>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 29.Vincent TL, Watt FE. Osteoarthritis. Medicine. 2014;42:213–219. [Google Scholar]
- 30.Spolidoro Paschoal NO, Natour J, Machado FS, de Oliveira HA, Furtado RN. Effectiveness of triamcinolone hexacetonide intraarticular injection in interphalangeal joints: a 12-week randomized controlled trial in patients with hand osteoarthritis. J Rheumatol. 2015;42:1869–1877. doi: 10.3899/jrheum.140736. [DOI] [PubMed] [Google Scholar]
- 31.Conaghan PG, Cohen SB, Berenbaum F, Lufkin J, Johnson JR, Bodick N. Brief report: a phase iib trial of a novel extended-release microsphere formulation of triamcinolone acetonide for intraarticular injection in knee osteoarthritis. Arthritis Rheumatol. 2018;70:204–211. doi: 10.1002/art.40364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kivitz AJ, Conaghan PG, Cinar A, Lufkin J, Kelley SD. Rescue analgesic medication use by patients treated with triamcinolone acetonide extended-release for knee osteoarthritis pain: pooled analysis of three phase 2/3 randomized clinical trials. Pain Ther. 2019;8:271–280. doi: 10.1007/s40122-019-0125-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wenham CYJ, Hensor EMA, Grainger AJ, et al. A randomized, double-blind, placebo-controlled trial of low-dose oral prednisolone for treating painful hand osteoarthritis. Rheumatology (Oxford) 2012;51:2286–2294. doi: 10.1093/rheumatology/kes219. [DOI] [PubMed] [Google Scholar]
- 34.Kroon FPB, Kortekaas MC, Boonen A, et al. Results of a 6-week treatment with 10 mg prednisolone in patients with hand osteoarthritis (HOPE): a double-blind, randomised, placebo-controlled trial. Lancet. 2019;394:1993–2001. doi: 10.1016/S0140-6736(19)32489-4. [DOI] [PubMed] [Google Scholar]
- 35.McAlindon TE, LaValley MP, Harvey WF, et al. Effect of intra-articular triamcinolone vs saline on knee cartilage volume and pain in patients with knee osteoarthritis: a randomized clinical trial. JAMA. 2017;317:1967–1975. doi: 10.1001/jama.2017.5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeng C, Lane NE, Hunter DJ, et al. Intra-articular corticosteroids and the risk of knee osteoarthritis progression: results from the Osteoarthritis Initiative. Osteoarthritis Cartilage. 2019;27:855–862. doi: 10.1016/j.joca.2019.01.007. [DOI] [PubMed] [Google Scholar]
- 37.Bannuru RR, Osani MC, Vaysbrot EE, et al. OARSI guidelines for the non-surgical management of knee, hip, and polyarticular osteoarthritis. Osteoarthritis Cartilage. 2019;27:1578–1589. doi: 10.1016/j.joca.2019.06.011. [DOI] [PubMed] [Google Scholar]
- 38.Kingsbury SR, Tharmanathan P, Keding A, et al. Hydroxychloroquine effectiveness in reducing symptoms of hand osteoarthritis: a randomized trial. Ann Intern Med. 2018;168:385–395. doi: 10.7326/M17-1430. [DOI] [PubMed] [Google Scholar]
- 39.Lee W, Ruijgrok L, Boxma-de Klerk B, et al. Efficacy of hydroxychloroquine in hand osteoarthritis: a randomized, double blind, placebo-controlled trial. Arthritis Care Res (Hoboken) 2018;70:1320–1325. doi: 10.1002/acr.23471. [DOI] [PubMed] [Google Scholar]
- 40.Kingsbury SR, Tharmanathan P, Arden NK, et al. Pain reduction with oral methotrexate in knee osteoarthritis, a pragmatic phase iii trial of treatment effectiveness (PROMOTE): study protocol for a randomized controlled trial. Trials. 2015;16:77. doi: 10.1186/s13063-015-0602-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kingsbury SR, Tharmanathan P, Keding A, et al. Significant pain reduction with oral methotrexate in knee osteoarthritis; results from a randomised controlled phase III trial of treatment effectiveness. Arthritis Rheumatol. 2018;70(suppl 10):S84–S85. [Google Scholar]
- 42.Ferrero S, Wittoek R, Allado E, et al. Methotrexate in patients with hand osteoarthritis refractory to analgesics: a randomised, double-blind, placebo controlled trial. Ann Rheum Dis. 2019;78(suppl 2):163. [Google Scholar]
- 43.Persson MSM, Sarmanova A, Doherty M, Zhang W. Conventional and biologic disease-modifying anti-rheumatic drugs for osteoarthritis: a meta-analysis of randomized controlled trials. Rheumatology (Oxford) 2018;57:1830–1837. doi: 10.1093/rheumatology/key131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kloppenburg M, Ramonda R, Bobacz K, et al. Etanercept in patients with inflammatory hand osteoarthritis (EHOA): a multicentre, randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2018;77:1757–1764. doi: 10.1136/annrheumdis-2018-213202. [DOI] [PubMed] [Google Scholar]
- 45.Kloppenburg M, Peterfy C, Haugen IK, et al. Phase IIa, placebo-controlled, randomised study of lutikizumab, an anti-interleukin-1α and anti-interleukin-1β dual variable domain immunoglobulin, in patients with erosive hand osteoarthritis. Ann Rheum Dis. 2019;78:413–420. doi: 10.1136/annrheumdis-2018-213336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chevalier X, Ravaud P, Maheu E, et al. Adalimumab in patients with hand osteoarthritis refractory to analgesics and NSAIDs: a randomised, multicentre, double-blind, placebo-controlled trial. Ann Rheum Dis. 2015;74:1697–1705. doi: 10.1136/annrheumdis-2014-205348. [DOI] [PubMed] [Google Scholar]
- 47.Verbruggen G, Wittoek R, Vander Cruyssen B, Elewaut D. Tumour necrosis factor blockade for the treatment of erosive osteoarthritis of the interphalangeal finger joints: a double blind, randomised trial on structure modification. Ann Rheum Dis. 2012;71:891–898. doi: 10.1136/ard.2011.149849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fleischmann RM, Bliddal H, Blanco FJ, et al. A phase ii trial of lutikizumab, an anti-interleukin-1α/β dual variable domain immunoglobulin, in knee osteoarthritis patients with synovitis. Arthritis Rheumatol. 2019;71:1056–1069. doi: 10.1002/art.40840. [DOI] [PubMed] [Google Scholar]
- 49.Chevalier X, Goupille P, Beaulieu AD, et al. Intraarticular injection of anakinra in osteoarthritis of the knee: a multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2009;61:344–352. doi: 10.1002/art.24096. [DOI] [PubMed] [Google Scholar]
- 50.Clements KM, Price JS, Chambers MG, Visco DM, Poole AR, Mason RM. Gene deletion of either interleukin-1beta, interleukin-1beta-converting enzyme, inducible nitric oxide synthase, or stromelysin 1 accelerates the development of knee osteoarthritis in mice after surgical transection of the medial collateral ligament and partial medial meniscectomy. Arthritis Rheum. 2003;48:3452–3463. doi: 10.1002/art.11355. [DOI] [PubMed] [Google Scholar]
- 51.Fukai A, Kamekura S, Chikazu D, et al. Lack of a chondroprotective effect of cyclooxygenase 2 inhibition in a surgically induced model of osteoarthritis in mice. Arthritis Rheum. 2012;64:198–203. doi: 10.1002/art.33324. [DOI] [PubMed] [Google Scholar]
- 52.Bougault C, Gosset M, Houard X, et al. Stress-induced cartilage degradation does not depend on the NLRP3 inflammasome in human osteoarthritis and mouse models. Arthritis Rheum. 2012;64:3972–3981. doi: 10.1002/art.34678. [DOI] [PubMed] [Google Scholar]
- 53.Vincent TL. IL-1 in osteoarthritis: time for a critical review of the literature. F1000 Res. 2019;8:1–8. doi: 10.12688/f1000research.18831.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ryu J-H, Yang S, Shin Y, Rhee J, Chun C-H, Chun J-S. Interleukin-6 plays an essential role in hypoxia-inducible factor 2α-induced experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum. 2011;63:2732–2743. doi: 10.1002/art.30451. [DOI] [PubMed] [Google Scholar]
- 55.de Hooge ASK, van de Loo FAJ, Bennink MB, Arntz OJ, de Hooge P, van den Berg WB. Male IL-6 gene knock out mice developed more advanced osteoarthritis upon aging. Osteoarthritis Cartilage. 2005;13:66–73. doi: 10.1016/j.joca.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 56.Latourte A, Cherifi C, Maillet J, et al. Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann Rheum Dis. 2017;76:748–755. doi: 10.1136/annrheumdis-2016-209757. [DOI] [PubMed] [Google Scholar]
- 57.Glasson SS, Askew R, Sheppard B, et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature. 2005;434:644–648. doi: 10.1038/nature03369. [DOI] [PubMed] [Google Scholar]
- 58.Ismail HM, Yamamoto K, Vincent TL, Nagase H, Troeberg L, Saklatvala J. Interleukin-1 acts via the JNK-2 signaling pathway to induce aggrecan degradation by human chondrocytes. Arthritis Rheumatol. 2015;67:1826–1836. doi: 10.1002/art.39099. [DOI] [PubMed] [Google Scholar]
- 59.Song R-H, Tortorella MD, Malfait A-M, et al. Aggrecan degradation in human articular cartilage explants is mediated by both ADAMTS-4 and ADAMTS-5. Arthritis Rheum. 2007;56:575–585. doi: 10.1002/art.22334. [DOI] [PubMed] [Google Scholar]
- 60.Larkin J, Lohr T, Elefante L, et al. The highs and lows of translational drug development: antibody-mediated inhibition of ADAMTS-5 for osteoarthritis disease modification. Osteoarthritis Cartilage. 2014;22:S483–S484. doi: 10.1016/j.joca.2015.02.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deckx HM, Hatch S, Robberechts M, et al. A safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) study with increasing oral doses of GLPG1972 administered daily for 29 days shows a strong biomarker effect in patients with knee and/or hip OA. Ann Rheum Dis. 2018;77:795. [Google Scholar]
- 62.Wang Q, Rozelle AL, Lepus CM, et al. Identification of a central role for complement in osteoarthritis. Nat Med. 2011;17:1674–1679. doi: 10.1038/nm.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miotla Zarebska J, Chanalaris A, Driscoll C, et al. CCL2 and CCR2 regulate pain-related behaviour and early gene expression in post-traumatic murine osteoarthritis but contribute little to chondropathy. Osteoarthritis Cartilage. 2017;25:406–412. doi: 10.1016/j.joca.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miller RE, Tran PB, Das R, et al. CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc Natl Acad Sci USA. 2012;109:20602–20607. doi: 10.1073/pnas.1209294110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Raghu H, Lepus CM, Wang Q, et al. CCL2/CCR2, but not CCL5/CCR5, mediates monocyte recruitment, inflammation and cartilage destruction in osteoarthritis. Ann Rheum Dis. 2017;76:914–922. doi: 10.1136/annrheumdis-2016-210426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Longobardi L, Temple JD, Tagliafierro L, et al. Role of the C-C chemokine receptor-2 in a murine model of injury-induced osteoarthritis. Osteoarthritis Cartilage. 2017;25:914–925. doi: 10.1016/j.joca.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Appleton CT, Usmani SE, Pest MA, Pitelka V, Mort JS, Beier F. Reduction in disease progression by inhibition of transforming growth factor α-CCL2 signaling in experimental posttraumatic osteoarthritis. Arthritis Rheumatol. 2015;67:2691–2701. doi: 10.1002/art.39255. [DOI] [PubMed] [Google Scholar]
- 68.Takebe K, Rai MF, Schmidt EJ, Sandell LJ. The chemokine receptor CCR5 plays a role in post-traumatic cartilage loss in mice, but does not affect synovium and bone. Osteoarthritis Cartilage. 2015;23:454–461. doi: 10.1016/j.joca.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sambamurthy N, Nguyen V, Smalley R, et al. Chemokine receptor-7 (CCR7) deficiency leads to delayed development of joint damage and functional deficits in a murine model of osteoarthritis. J Orthop Res. 2018;36:864–875. doi: 10.1002/jor.23671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sherwood J, Bertrand J, Nalesso G, et al. A homeostatic function of CXCR2 signalling in articular cartilage. Ann Rheum Dis. 2015;74:2207–2215. doi: 10.1136/annrheumdis-2014-205546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qin HJ, Xu T, Wu HT, et al. SDF-1/CXCR4 axis coordinates crosstalk between subchondral bone and articular cartilage in osteoarthritis pathogenesis. Bone. 2019;125:140–150. doi: 10.1016/j.bone.2019.05.010. [DOI] [PubMed] [Google Scholar]
- 72.Choi MC, MaruYama T, Chun CH, Park Y. Alleviation of murine osteoarthritis by cartilage-specific deletion of IkappaBzeta. Arthritis Rheumatol. 2018;70:1440–1449. doi: 10.1002/art.40514. [DOI] [PubMed] [Google Scholar]
- 73.Kobayashi H, Chang SH, Mori D, et al. Biphasic regulation of chondrocytes by Rela through induction of anti-apoptotic and catabolic target genes. Nat Commun. 2016;7 doi: 10.1038/ncomms13336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Culley KL, Lessard SG, Green JD, et al. Inducible knockout of CHUK/IKKα in adult chondrocytes reduces progression of cartilage degradation in a surgical model of osteoarthritis. Sci Rep. 2019;9 doi: 10.1038/s41598-019-45334-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ismail HM, Miotla-Zarebska J, Troeberg L, et al. Brief report: JNK-2 controls aggrecan degradation in murine articular cartilage and the development of experimental osteoarthritis. Arthritis Rheumatol. 2016;68:1165–1171. doi: 10.1002/art.39547. [DOI] [PubMed] [Google Scholar]
- 76.Wang Q, Lepus CM, Raghu H, et al. IgE-mediated mast cell activation promotes inflammation and cartilage destruction in osteoarthritis. eLife. 2019;8:390. doi: 10.7554/eLife.39905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Castaño-Betancourt MC, Evans DS, Ramos YFM, et al. Novel genetic variants for cartilage thickness and hip osteoarthritis. PLoS Genet. 2016;12 doi: 10.1371/journal.pgen.1006260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zengini E, Hatzikotoulas K, Tachmazidou I, et al. Genome-wide analyses using UK Biobank data provide insights into the genetic architecture of osteoarthritis. Nat Genet. 2018;50:549–558. doi: 10.1038/s41588-018-0079-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bratus-Neuenschwander A, Castro-Giner F, Frank-Bertoncelj M, et al. Pain-associated transcriptome changes in synovium of knee osteoarthritis patients. Genes (Basel) 2018;9:338. doi: 10.3390/genes9070338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ritter SY, Subbaiah R, Bebek G, et al. Proteomic analysis of synovial fluid from the osteoarthritic knee: comparison with transcriptome analyses of joint tissues. Arthritis Rheum. 2013;65:981–992. doi: 10.1002/art.37823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Buckley MG, Gallagher PJ, Walls AF. Mast cell subpopulations in the synovial tissue of patients with osteoarthritis: selective increase in numbers of tryptase-positive, chymase-negative mast cells. J Pathol. 1998;186:67–74. doi: 10.1002/(SICI)1096-9896(199809)186:1<67::AID-PATH132>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 82.Nasi S, Ea H-K, Chobaz V, van Lent P, Lioté F, So A, et al. Dispensable role of myeloid differentiation primary response gene 88 (MyD88) and MyD88-dependent toll-like receptors (TLRs) in a murine model of osteoarthritis. Joint Bone Spine. 2014;81:320–324. doi: 10.1016/j.jbspin.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 83.Nasi S, Ea H-K, So A, Busso N. Revisiting the role of interleukin-1 pathway in osteoarthritis: interleukin-1α and -1β, and NLRP3 inflammasome are not involved in the pathological features of the murine menisectomy model of osteoarthritis. Front Pharmacol. 2017;8:282. doi: 10.3389/fphar.2017.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.van Lent PLEM, Blom AB, Schelbergen RFP, et al. Active involvement of alarmins S100A8 and S100A9 in the regulation of synovial activation and joint destruction during mouse and human osteoarthritis. Arthritis Rheum. 2012;64:1466–1476. doi: 10.1002/art.34315. [DOI] [PubMed] [Google Scholar]
- 85.Vincent T, Malfait AM. Time to be positive about negative data? Osteoarthritis and cartilage/OARS. Osteoarthritis Research Society. 2017;25:351–353. doi: 10.1016/j.joca.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vincent TL. Mechanoflammation in osteoarthritis pathogenesis. Semin Arthritis Rheum. 2019;49:S36–S38. doi: 10.1016/j.semarthrit.2019.09.018. [DOI] [PubMed] [Google Scholar]
- 87.Gruber J, Vincent TL, Hermansson M, Bolton M, Wait R, Saklatvala J. Induction of interleukin-1 in articular cartilage by explantation and cutting. Arthritis Rheum. 2004;50:2539–2546. doi: 10.1002/art.20369. [DOI] [PubMed] [Google Scholar]
- 88.Watt FE, Ismail HM, Didangelos A, et al. Src and fibroblast growth factor 2 independently regulate signaling and gene expression induced by experimental injury to intact articular cartilage. Arthritis Rheum. 2013;65:397–407. doi: 10.1002/art.37765. [DOI] [PubMed] [Google Scholar]
- 89.Castaño-Betancourt MC, Evans DS, Ramos YF, et al. Novel genetic variants for cartilage thickness and hip osteoarthritis. PLoS Genet. 2016;12 doi: 10.1371/journal.pgen.1006260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yamamoto K, Owen K, Parker AE, et al. Low density lipoprotein receptor-related protein 1 (LRP1)-mediated endocytic clearance of a disintegrin and metalloproteinase with thrombospondin motifs-4 (ADAMTS-4): functional differences of non-catalytic domains of ADAMTS-4 and ADAMTS-5 in LRP1 binding. J Biol Chem. 2014;289:6462–6474. doi: 10.1074/jbc.M113.545376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yamamoto K, Troeberg L, Scilabra SD, et al. LRP-1-mediated endocytosis regulates extracellular activity of ADAMTS-5 in articular cartilage. FASEB J. 2013;27:511–521. doi: 10.1096/fj.12-216671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim J-H, Jeon J, Shin M, et al. Regulation of the catabolic cascade in osteoarthritis by the zinc-ZIP8-MTF1 axis. Cell. 2014;156:730–743. doi: 10.1016/j.cell.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 93.Lee M, Won Y, Shin Y, Kim JH, Chun JS. Reciprocal activation of hypoxia-inducible factor (HIF)-2α and the zinc-ZIP8-MTF1 axis amplifies catabolic signaling in osteoarthritis. Osteoarthritis Cartilage. 2016;24:134–145. doi: 10.1016/j.joca.2015.07.016. [DOI] [PubMed] [Google Scholar]
- 94.Saito T, Fukai A, Mabuchi A, et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat Med. 2010;16:678–686. doi: 10.1038/nm.2146. [DOI] [PubMed] [Google Scholar]
- 95.Buchanan WW. William Hunter (1718–1783) Rheumatology (Oxford) 2003;42:1260–1261. doi: 10.1093/rheumatology/keg003. [DOI] [PubMed] [Google Scholar]
- 96.Dell'accio F, Vincent TL. Joint surface defects: clinical course and cellular response in spontaneous and experimental lesions. Eur Cell Mater. 2010;20:210–217. doi: 10.22203/ecm.v020a17. [DOI] [PubMed] [Google Scholar]
- 97.Leyland KM, Hart DJ, Javaid MK, et al. The natural history of radiographic knee osteoarthritis: a fourteen-year population-based cohort study. Arthritis Rheum. 2012;64:2243–2251. doi: 10.1002/art.34415. [DOI] [PubMed] [Google Scholar]
- 98.Eltawil NM, De Bari C, Achan P, Pitzalis C, Dell'accio F. A novel in vivo murine model of cartilage regeneration. Age and strain-dependent outcome after joint surface injury. Osteoarthritis Cartilage. 2009;17:695–704. doi: 10.1016/j.joca.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rai MF, Hashimoto S, Johnson EE, et al. Heritability of articular cartilage regeneration and its association with ear wound healing in mice. Arthritis Rheum. 2012;64:2300–2310. doi: 10.1002/art.34396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wiegant K, van Roermund PM, Intema F, et al. Sustained clinical and structural benefit after joint distraction in the treatment of severe knee osteoarthritis. Osteoarthritis Cartilage. 2013;21:1660–1667. doi: 10.1016/j.joca.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 101.Intema F, Van Roermund PM, Marijnissen ACA, et al. Tissue structure modification in knee osteoarthritis by use of joint distraction: an open 1-year pilot study. Ann Rheum Dis. 2011;70:1441–1446. doi: 10.1136/ard.2010.142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.van der Woude JAD, Wiegant K, van Roermund PM, et al. Five-year follow-up of knee joint distraction: clinical benefit and cartilaginous tissue repair in an open uncontrolled prospective study. Cartilage. 2017;8:263–271. doi: 10.1177/1947603516665442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mastbergen SC, Saris DBF, Lafeber FPJG. Functional articular cartilage repair: here, near, or is the best approach not yet clear? Nat Rev Rheumatol. 2013;9:277–290. doi: 10.1038/nrrheum.2013.29. [DOI] [PubMed] [Google Scholar]
- 104.Watt FE, Hamid B, Garriga C, et al. The molecular profile of synovial fluid changes upon joint distraction and is associated with clinical response in knee osteoarthritis. Osteoarthritis Cartilage. 2020;28:324–333. doi: 10.1016/j.joca.2019.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tjörnstrand BA, Egund N, Hagstedt BV. High tibial osteotomy: a seven-year clinical and radiographic follow-up. Clin Orthop Relat Res. 1981;160:124–136. [PubMed] [Google Scholar]
- 106.Koshino T, Wada S, Ara Y, Saito T. Regeneration of degenerated articular cartilage after high tibial valgus osteotomy for medial compartmental osteoarthritis of the knee. Knee. 2003;10:229–236. doi: 10.1016/s0968-0160(03)00005-x. [DOI] [PubMed] [Google Scholar]
- 107.Parker DA, Beatty KT, Giuffre B, Scholes CJ, Coolican MRJ. Articular cartilage changes in patients with osteoarthritis after osteotomy. Am J Sports Med. 2011;39:1039–1045. doi: 10.1177/0363546510392702. [DOI] [PubMed] [Google Scholar]
- 108.Kobayashi H, Saito T, Koshino T. Immunolocalization of carboxy-terminal type II procollagen peptide in regenerated articular cartilage of osteoarthritic knees after reduction of mechanical stress. Osteoarthritis Cartilage. 2002;10:870–878. doi: 10.1053/joca.2002.0839. [DOI] [PubMed] [Google Scholar]
- 109.Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8:235–253. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Valverde-Franco G, Binette JS, Li W, et al. Defects in articular cartilage metabolism and early arthritis in fibroblast growth factor receptor 3 deficient mice. Hum Mol Genet. 2006;15:1783–1792. doi: 10.1093/hmg/ddl100. [DOI] [PubMed] [Google Scholar]
- 111.Tang J, Su N, Zhou S, et al. Fibroblast growth factor receptor 3 inhibits osteoarthritis progression in the knee joints of adult mice. Arthritis Rheumatol. 2016;68:2432–2443. doi: 10.1002/art.39739. [DOI] [PubMed] [Google Scholar]
- 112.Xu W, Xie Y, Wang Q, et al. A novel fibroblast growth factor receptor 1 inhibitor protects against cartilage degradation in a murine model of osteoarthritis. Sci Rep. 2016;6 doi: 10.1038/srep24042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tachmazidou I, Hatzikotoulas K, Southam L, et al. Identification of new therapeutic targets for osteoarthritis through genome-wide analyses of UK Biobank data. Nat Genet. 2019;51:230–236. doi: 10.1038/s41588-018-0327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lohmander LS, Hellot S, Dreher D, et al. Intraarticular sprifermin (recombinant human fibroblast growth factor 18) in knee osteoarthritis: a randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. 2014;66:1820–1831. doi: 10.1002/art.38614. [DOI] [PubMed] [Google Scholar]
- 115.Hochberg MC, Guermazi A, Guehring H, et al. Effect of intra-articular sprifermin vs placebo on femorotibial joint cartilage thickness in patients with osteoarthritis: the FORWARD randomized clinical trial. JAMA. 2019;322:1360–1370. doi: 10.1001/jama.2019.14735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Guhring H, Kraines J, Moreau F, et al. OP0010 cartilage thickness modification with sprifermin in knee osteoarthritis patients translates into symptomatic improvement over placebo in patients at risk of further structural and symptomatic progression: post-hoc analysis of the phase II FORWARD trial. Ann Rheum Dis. 2019;78(suppl 2):70–71. [Google Scholar]
- 117.Dell'accio F, De Bari C, Eltawil NM, Vanhummelen P, Pitzalis C. Identification of the molecular response of articular cartilage to injury, by microarray screening: Wnt-16 expression and signaling after injury and in osteoarthritis. Arthritis Rheum. 2008;58:1410–1421. doi: 10.1002/art.23444. [DOI] [PubMed] [Google Scholar]
- 118.Dell'Accio F, De Bari C, El Tawil NMF, et al. Activation of WNT and BMP signaling in adult human articular cartilage following mechanical injury. Arthritis Res Ther. 2006;8:R139. doi: 10.1186/ar2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Corr M. Wnt-beta-catenin signaling in the pathogenesis of osteoarthritis. Nat Clin Pract Rheumatol. 2008;4:550–556. doi: 10.1038/ncprheum0904. [DOI] [PubMed] [Google Scholar]
- 120.Yasuhara R, Ohta Y, Yuasa T, et al. Roles of β-catenin signaling in phenotypic expression and proliferation of articular cartilage superficial zone cells. Lab Invest. 2011;91:1739–1752. doi: 10.1038/labinvest.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yuasa T, Kondo N, Yasuhara R, et al. Transient activation of Wnt/beta-catenin signaling induces abnormal growth plate closure and articular cartilage thickening in postnatal mice. Am J Pathol. 2009;175:1993–2003. doi: 10.2353/ajpath.2009.081173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kawaguchi H. Regulation of osteoarthritis development by Wnt-beta-catenin signaling through the endochondral ossification process. J Bone Miner Res. 2009;24:8–11. doi: 10.1359/jbmr.081115. [DOI] [PubMed] [Google Scholar]
- 123.Funck-Brentano T, Bouaziz W, Marty C, Geoffroy V, Hay E, Cohen-Solal M. Dkk-1-mediated inhibition of Wnt signaling in bone ameliorates osteoarthritis in mice. Arthritis Rheumatol. 2014;66:3028–3039. doi: 10.1002/art.38799. [DOI] [PubMed] [Google Scholar]
- 124.Lories RJU, Peeters J, Bakker A, et al. Articular cartilage and biomechanical properties of the long bones in Frzb-knockout mice. Arthritis Rheum. 2007;56:4095–4103. doi: 10.1002/art.23137. [DOI] [PubMed] [Google Scholar]
- 125.Nalesso G, Thomas BL, Sherwood JC, et al. WNT16 antagonises excessive canonical WNT activation and protects cartilage in osteoarthritis. Ann Rheum Dis. 2017;76:218–226. doi: 10.1136/annrheumdis-2015-208577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Monteagudo S, Cornelis FMF, Aznar-Lopez C, et al. DOT1L safeguards cartilage homeostasis and protects against osteoarthritis. Nat Commun. 2017;8:15889–15912. doi: 10.1038/ncomms15889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Deshmukh V, Hu H, Barroga C, et al. A small-molecule inhibitor of the Wnt pathway (SM04690) as a potential disease modifying agent for the treatment of osteoarthritis of the knee. Osteoarthritis Cartilage. 2018;26:18–27. doi: 10.1016/j.joca.2017.08.015. [DOI] [PubMed] [Google Scholar]
- 128.Deshmukh V, O'Green AL, Bossard C, et al. Modulation of the Wnt pathway through inhibition of CLK2 and DYRK1A by lorecivivint as a novel, potentially disease-modifying approach for knee osteoarthritis treatment. Osteoarthritis Cartilage. 2019;27:1347–1360. doi: 10.1016/j.joca.2019.05.006. [DOI] [PubMed] [Google Scholar]
- 129.Yazici Y, McAlindon TE, Fleischmann R, et al. A novel Wnt pathway inhibitor, SM04690, for the treatment of moderate to severe osteoarthritis of the knee: results of a 24-week, randomized, controlled, phase 1 study. Osteoarthritis Cartilage. 2017;25:1598–1606. doi: 10.1016/j.joca.2017.07.006. [DOI] [PubMed] [Google Scholar]
- 130.Yazici Y, McAlindon TE, Gibofsky A, et al. Lorecivivint, a novel intra-articular CLK/DYRK1A inhibitor and Wnt pathway modulator for treatment of knee osteoarthritis: a phase 2 randomized trial. Arthritis Rheumatol. 2020 doi: 10.1002/art.41315. published online May 20. [DOI] [Google Scholar]
- 131.Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94:1287–1312. doi: 10.1152/physrev.00005.2014. [DOI] [PubMed] [Google Scholar]
- 132.Deng Y, Lu J, Li W, et al. Reciprocal inhibition of YAP/TAZ and NF-κB regulates osteoarthritic cartilage degradation. Nat Commun. 2018;9 doi: 10.1038/s41467-018-07022-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Roelofs AJ, Zupan J, Riemen AHK, et al. Joint morphogenetic cells in the adult mammalian synovium. Nat Commun. 2017;8 doi: 10.1038/ncomms15040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Denk F, Bennett DL, McMahon SB. Nerve growth factor and pain mechanisms. Annu Rev Neurosci. 2017;40:307–325. doi: 10.1146/annurev-neuro-072116-031121. [DOI] [PubMed] [Google Scholar]
- 135.Yang S, Huang Y, Ye Z, Li L, Zhang Y. The efficacy of nerve growth factor antibody for the treatment of osteoarthritis pain and chronic low-back pain: a meta-analysis. Front Pharmacol. 2020 doi: 10.3389/fphar.2020.00817. published online June 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dakin P, DiMartino SJ, Gao H, et al. The efficacy, tolerability, and joint safety of fasinumab in osteoarthritis pain: a phase iib/iii double-blind, placebo-controlled, randomized clinical trial. Arthritis Rheumatol. 2019;40:307. doi: 10.1002/art.41012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tive L, Bello AE, Radin D, et al. Pooled analysis of tanezumab efficacy and safety with subgroup analyses of phase III clinical trials in patients with osteoarthritis pain of the knee or hip. J Pain Res. 2019;12:975–995. doi: 10.2147/JPR.S191297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Schnitzer TJ, Easton R, Pang S, et al. Effect of tanezumab on joint pain, physical function, and patient global assessment of osteoarthritis among patients with osteoarthritis of the hip or knee: a randomized clinical trial. JAMA. 2019;322:37–48. doi: 10.1001/jama.2019.8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Watt FE, Blauwet MB, Fakhoury A, Jacobs H, Smulders R, Lane NE. Tropomyosin-related kinase A (TrkA) inhibition for the treatment of painful knee osteoarthritis: results from a randomized controlled phase 2a trial. Osteoarthritis Cartilage. 2019;27:1590–1598. doi: 10.1016/j.joca.2019.05.029. [DOI] [PubMed] [Google Scholar]
- 140.Krupa E, Jiang G-L, Jan C. Efficacy and safety of intra-articular injection of tropomyosin receptor kinase A inhibitor in painful knee osteoarthritis: a randomized, double-blind and placebo-controlled study. Osteoarthritis Cartilage. 2019;27:1599–1607. doi: 10.1016/j.joca.2019.05.028. [DOI] [PubMed] [Google Scholar]
- 141.Inglis JJ, McNamee KE, Chia S-L, et al. Regulation of pain sensitivity in experimental osteoarthritis by the endogenous peripheral opioid system. Arthritis Rheum. 2008;58:3110–3119. doi: 10.1002/art.23870. [DOI] [PubMed] [Google Scholar]
- 142.von Loga IS, El-Turabi A, Jostins L, et al. Active immunisation targeting nerve growth factor attenuates chronic pain behaviour in murine osteoarthritis. Ann Rheum Dis. 2019;78:672–675. doi: 10.1136/annrheumdis-2018-214489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Knights CB, Gentry C, Bevan S. Partial medial meniscectomy produces osteoarthritis pain-related behaviour in female C57BL/6 mice. Pain. 2012;153:281–292. doi: 10.1016/j.pain.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 144.LaBranche TP, Bendele AM, Omura BC, et al. Nerve growth factor inhibition with tanezumab influences weight-bearing and subsequent cartilage damage in the rat medial meniscal tear model. Ann Rheum Dis. 2017;76:295–302. doi: 10.1136/annrheumdis-2015-208913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.McNamee KE, Burleigh A, Gompels LL, et al. Treatment of murine osteoarthritis with TrkAd5 reveals a pivotal role for nerve growth factor in non-inflammatory joint pain. Pain. 2010;149:386–392. doi: 10.1016/j.pain.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 146.Driscoll C, Chanalaris A, Knights C, et al. nociceptive sensitizers are regulated in damaged joint tissues, including articular cartilage, when osteoarthritic mice display pain behavior. Arthritis Rheumatol. 2016;68:857–867. doi: 10.1002/art.39523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kuttapitiya A, Assi L, Laing K, et al. Microarray analysis of bone marrow lesions in osteoarthritis demonstrates upregulation of genes implicated in osteochondral turnover, neurogenesis and inflammation. Ann Rheum Dis. 2017;76:1764–1773. doi: 10.1136/annrheumdis-2017-211396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sato T, Konomi K, Yamasaki S, et al. Comparative analysis of gene expression profiles in intact and damaged regions of human osteoarthritic cartilage. Arthritis Rheum. 2006;54:808–817. doi: 10.1002/art.21638. [DOI] [PubMed] [Google Scholar]
- 149.Ji Q, Zheng Y, Zhang G, et al. Single-cell RNA-seq analysis reveals the progression of human osteoarthritis. Ann Rheum Dis. 2019;78:100–110. doi: 10.1136/annrheumdis-2017-212863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Obeidat AM, Miller RE, Miller RJ, Malfait AM. The nociceptive innervation of the normal and osteoarthritic mouse knee. Osteoarthritis Cartilage. 2019;27:1669–1679. doi: 10.1016/j.joca.2019.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Walsh DA, McWilliams DF, Turley MJ, et al. Angiogenesis and nerve growth factor at the osteochondral junction in rheumatoid arthritis and osteoarthritis. Rheumatology (Oxford) 2010;49:1852–1861. doi: 10.1093/rheumatology/keq188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhu S, Zhu J, Zhen G, et al. Subchondral bone osteoclasts induce sensory innervation and osteoarthritis pain. J Clin Invest. 2019;129:1076–1093. doi: 10.1172/JCI121561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Vincent TL. Peripheral pain mechanisms in osteoarthritis. Pain. 2020;161:S138–S146. doi: 10.1097/j.pain.0000000000001923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Styrkarsdottir U, Lund SH, Thorleifsson G, et al. Meta-analysis of Icelandic and UK data sets identifies missense variants in SMO, IL11, COL11A1 and 13 more new loci associated with osteoarthritis. Nat Genet. 2018;50:1681–1687. doi: 10.1038/s41588-018-0247-0. [DOI] [PubMed] [Google Scholar]
- 155.Jonsson H. Following the genetic clues towards treatment of hand OA. Nat Rev Rheumatol. 2018;14:503–504. doi: 10.1038/s41584-018-0049-x. [DOI] [PubMed] [Google Scholar]
- 156.Vaysbrot EE, Osani MC, Musetti MC, McAlindon TE, Bannuru RR. Are bisphosphonates efficacious in knee osteoarthritis? A meta-analysis of randomized controlled trials. Osteoarthritis Cartilage. 2018;26:154–164. doi: 10.1016/j.joca.2017.11.013. [DOI] [PubMed] [Google Scholar]
- 157.Xing RL, Zhao LR, Wang PM. Bisphosphonates therapy for osteoarthritis: a meta-analysis of randomized controlled trials. Springerplus. 2016;5 doi: 10.1186/s40064-016-3359-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.van der Worp HB, Howells DW, Sena ES, et al. Can animal models of disease reliably inform human studies? PLoS Med. 2010;7 doi: 10.1371/journal.pmed.1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]