

Abstract
Protein–protein interactions (PPIs) have pivotal roles in life processes. The studies showed that aberrant PPIs are associated with various diseases, including cancer, infectious diseases, and neurodegenerative diseases. Therefore, targeting PPIs is a direction in treating diseases and an essential strategy for the development of new drugs. In the past few decades, the modulation of PPIs has been recognized as one of the most challenging drug discovery tasks. In recent years, some PPIs modulators have entered clinical studies, some of which been approved for marketing, indicating that the modulators targeting PPIs have broad prospects. Here, we summarize the recent advances in PPIs modulators, including small molecules, peptides, and antibodies, hoping to provide some guidance to the design of novel drugs targeting PPIs in the future.
Subject terms: Target identification, Drug discovery, Target identification, Drug discovery, Target identification
Introduction
PPIs and diseases
Proteins are the basic building blocks of life that are made by amino acids. The amino acids are coded by genes and form the peptides, peptides further form various proteins, and the proteins form the living tissues. Besides, proteins also have a central role in biological processes such as catalyze reactions, transport molecules, immune reactions to the various pathogens, and signal transduction between cells. What is more, the critical biological processes in the cells that directly associate with our health like DNA replication, transcription, translation, and transmembrane signal transduction all rely on the functional specific proteins. The aforementioned biological activities are regulated through protein complexes, which are typically controlled via protein–protein interactions (PPIs).1–3 PPIs in cells form a complicated network which has a term named “interactome”.4,5 The interactome has a significant role in physiological and pathological processes, including signal transduction, cell proliferation, growth, differentiation, and apoptosis, etc.6–8 Therefore, the aberrant PPIs are associated with many human diseases such as cancer, infectious diseases, and neurodegenerative diseases.9–11 Since the classic drug targets are usually enzymes, ion channels, or receptors, the PPIs indicate new potential therapeutic targets.12 In recent years, the PPIs have received increasing attention and became attractive targets.13,14 Recent studies indicate that the PPIs have great potential as an intervention target for novel treatment of refractory diseases, and its regulation is widely regarded as a promising strategy in drug discovery8,15,16 (Table 1).
Table 1.
Summary of some PPI modulators in clinical trials
| PPI | Related disease | Drug | Developer | Status | NCT number | Refs. |
|---|---|---|---|---|---|---|
| Small molecules | ||||||
| MDM2/p53 | Acute myeloid leukemia | Idasanutlin | Roche | Phase III | NCT02545283 | 245 |
| MDM2/p53 | Metastatic melanoma | AMG232 | Amgen | Phase I/II | NCT02110355 | 246 |
| MDM2/p53 | Solid tumor with p53 wild type status | CGM097 | Novartis | Phase I | NCT01760525 | 247 |
| MDM2/p53 | Advanced solid tumor, lymphoma | DS-3032b | Daiichi Sankyo | Phase I | NCT01877382 | 248 |
| MDM2/p53 | Neoplasm malignant | SAR405838 | Sanofi | Phase I | NCT01636479 | 249 |
| Bcl-2/Bax | Chronic lymphocytic leukemia | ABT-199 | AbbVie | Approved in 2016 | – | 250 |
| XIAP/caspase-9 | Relapsed or refractory multiple myeloma | LCL-161 | Novartis | Phase II | NCT01955434 | 251 |
| XIAP/caspase-9 | Recurrent head and neck squamous cell carcinoma | TL32711 | National Cancer Institute | Phase I | NCT03803774 | 252 |
| XIAP/caspase-9 | Solid tumors, lymphoma | ASTX-660 | Astex | Phase I/II | NCT02503423 | 253 |
| XIAP/caspase-9 | Solid cancers | GDC-0917 | Genentech | Phase I | NCT01226277 | 254 |
| PD-1/PD-L1 | Prostatic neoplasms | CA-170 | Astellas | Phase II | NCT01288911 | 255 |
| Gp120/CCR5 | HIV | Maraviroc | Pfizer | Approved in 2007 | – | 256 |
| LFA-1/ICAM-1 | Dry eye | Lifitegrast | Lifelong Vision Foundation | Phase IV | NCT03451396 | 257 |
| Β-catenin/CBP | Liver cirrhosis | RPI-724 | Komagome Hospital | Phase I/II | NCT03620474 | 258 |
| Bromodomain/histone | Cardiovascular diseases | RVX-208 | Resverlogix | Phase III | NCT02586155 | 259 |
| Bromodomain/histone | NUT midline carcinoma | GSK525762 | GSK | Phase I | NCT01587703 | 260 |
| Peptides | ||||||
| MDM2/p53 | Advanced solid tumors, lymphomas | ALRN-6924 | Aileron | Phase I/II | NCT02264613 | 261 |
| Antibodies | ||||||
| CD40/CD40L | Kidney transplantation | Bleselumab | Astellas | Phase II | NCT02921789 | 262 |
| CD40/CD40L | Multiple myeloma | lucatumumab | Novartis | Phase I | NCT00231166 | 263 |
| CD40/CD40L | Relapsed diffuse large B-cell lymphoma | dacetuzumab | Seattle Genetics | Phase II | NCT00435916 | 264 |
| CD40/CD40L | Lupus Nephritis | BI655064 | Boehringer Ingelheim | Phase II | NCT03385564 | 265 |
| CD40/CD40L | Advanced solid tumors | ABBV-428 | AbbVie | Phase I | NCT02955251 | 266 |
| CD40/CD40L | Ulcerative colitis | ABBV-323 | AbbVie | Phase II | NCT03695185 | 267 |
| PD-1/PD-L1 | Non-small lung cancer | Keytruda | Merck Sharp & Dohme | Approved in 2014 | – | 268 |
| PD-1/PD-L1 | Non-small lung cancer | Opdivo | Bristol Myers Squibb | Approved in 2014 | – | 269 |
| PD-1/PD-L1 | Non-small lung cancer | Tecentriq | Roche | Approved in 2016 | – | 198 |
| PD-1/PD-L1 | Merkel cell carcinoma | Bavencio | Merck and Pfizer | Approved in 2017 | – | 270 |
| PD-1/PD-L1 | Non-small lung cancer | Imfinzi | AstraZeneca | Approved in 2017 | – | 199 |
| PD-1/PD-L1 | Unresectable or metastatic melanoma | JS001 | Shanghai Junshi Bioscience | Phase III | NCT03430297 | 271 |
| PD-1/PD-L1 | Advanced/metastatic solid malignancies | IBI308 | Innovent Biologics | Phase I/II | NCT03568539 | 272 |
| PD-1/PD-L1 | Locally advanced or metastatic urothelial bladder cancer | BGB-A317 | BeiGene | Phase II | NCT04004221 | 273 |
Data collected from https://clinicaltrials.gov [last accessed 7th June, 2020]
Challenges in discovering PPIs modulators
The classic small molecule drug discovery approach mainly focuses on the protein–ligand interactions, such as enzymes, ion channels, or receptors, because these proteins typically contain a well-defined ligand-binding site that small molecules can interact with.17 The PPIs modulation through small molecules is generally considered difficult and PPIs were regarded as “undruggable” targets.18,19 It is estimated that there are about 130,000–650,000 types of PPIs in the human interactome.4,8,20 Although the number of protein complexes exceeds that of enzymes and receptors, designing a small molecule to bind to a PPI interface is challenging because of the reasons below. First, the PPIs occur on the interface of a specific domain where two identical or different proteins are in contact. The interface area of the interaction usually reaches 1500–3000 Å2,21 which is larger than that of receptor-ligand contact area (300–1000 Å2),22 and the interface is highly hydrophobic.21 Second, the PPIs interface tends to be flat and contains few grooves or pockets, thus making it difficult for the designed small molecule compounds to bind.23–25 Third, the amino acid residues involved in PPIs are either continuous or discontinuous in their respective protein structures, thus results in high-affinity binding between the proteins, making it difficult for the small molecular compounds to inhibit such high-affinity interaction.26 Forth, compared with traditional drug target enzymes or receptors, PPIs lack endogenous small molecular ligands for reference.26 Besides, compared to traditional small molecule drugs (200–500 Da), drugs acting on PPIs have a higher molecular weight (>400 Da), which makes it challenging to meet the criteria like Lipinski’s “rule of 5”.23,27
Hot-spots
Theoretically, the large binding interfaces are not regarded as the ideal drug targets because it is difficult to find a matching molecule. However, the emergence of “hot-spots” makes the designing drugs for PPIs possible.28 Usually, PPIs happen on several amino acid residues in the interaction regions, having critical roles in the interaction. The regions of the amino acid residues on the PPIs interface that contribute to the binding-free energy are called “hot-spots”.29–31 As the area of PPIs expands, the number of hot-spots increases. The area of all hot-spots is about 600 Å2, usually located at or near the PPIs interface. The hot-spots in the PPIs are identified through a point mutation experiment. Specifically, the amino acid residues on PPI are muted into alanine, and the change of the binding-free energy is measured to determine the residues that contributes significantly to the binding-free energy. Hot-spots have been defined as these sites where alanine mutations cause a significant increase in the binding-free energy of at 2.0 kcal/mol.32 Tryptophan, arginine, and tyrosine are more likely to appear in hot-spots than other amino acids.15,30 Because of the important role of these “hot-spots” amino acids, they are often used to design PPI drugs. Therefore, although the interface of PPIs is relatively large, small molecule drugs only need to act on “hot-spots” to intervene in the PPIs.
Current approaches for the discovery of PPI modulators
Targeting PPIs is challenging because of its unique interface. Compared to the binding pockets of conventional protein targets, the interface of PPIs tends to be flat. Therefore, classic medicinal chemistry methods are less effective for designing and identifying PPIs modulators. Thus, it is necessary to develop more effective approaches for screening the PPI modulators. A wide variety of strategies have been developed to identify hits and leads of PPI modulators in recent years.
High-throughput screening
High-throughput screening (HTS) is a well-established method for discovering classic drug targets. It has been used to identify compounds that target the hot-spots of PPI interfaces.16 Because of the particularity of PPI interface, the compound library used for screening conventional targets may not be suitable for screening PPI modulators. It’s crucial to have a broad compound library to have chemical diversity that may match the PPI target. However, HTS has been proved to be useful in the identification of molecules at the initial stage. For example, it successfully screened out inhibitors against MDM2/p53 interaction.33–35
Fragment-based drug discovery
Fragment-based drug discovery (FBDD) aims to identify molecular fragments from fragment libraries.36 Compared to HTS, FBDD is a better approach for PPIs modulators designing because the PPI interface often consists of discontinuous hot-spots. Surface plasmon resonance (SPR), nuclear magnetic resonance (NMR), X-ray crystallography, and mass spectroscopy (MS) can be utilized for discovery and validation of the fragment hits.37,38 Once the fragment hits are identified, the fragment linking, fragment optimization, and fragment self-assembly can be used to obtain the hits.39 Because the molecular weight of fragments is low and the contact interface is limited, the affinity is relatively low.40 The X-ray crystallography and NMR can provide structural information for the hits optimization. As a result, FBDD is not suitable for the targets with unknown structure. The examples of successful application of FBDD in PPI modulators’ discovery include XIAP/caspase-9,41 Bcl-2/Bax,42 and bromodomains,43 etc.
Structure-based design
Since most PPIs lack endogenous small molecule ligands, it is challenging to rationally design the associate PPI modulators. However, the hot-spots provide important structural information and a basis for the rational design of PPI modulators. At present, there are two design strategies for structure-based design PPI modulators. The first is based on the hot-spots structure. Through bioisosterism and de novo design, the novel small molecule modulators can be obtained.44 For example, during the development of VHL/HIF1α PPI inhibitors, Hyp564 was identified as a crucial amino acid. Through the de novo design targeting the Hyp546, the inhibitors were obtained.45,46 The second is peptidomimetic design which mainly rely on computer modeling and phage display to simulate the secondary structure of the key peptides in PPIs. Furthermore, small molecules were designed or binding peptides were synthesized based on the stable α-helix structure formed by the key peptides.47 The α-helix is the most common identified secondary structure in PPIs.48 At present, many PPI modulators have been successfully developed based on the α-helix structure, including c-Myc/Max,49 Bcl-2/Bax,50 and MDM2/p53.51
Virtual screening
The virtual screening is based on professional application software to screen out hits from compound libraries. One big challenge in developing PPI modulators is to identify the disease-related and druggable PPIs among thousand of available ones. The virtual screening may be useful to locate the binding sites by analyzing the protein surface. It can be classified into both a structure-based approach and a ligand-based approach. The ligand-based approach aims to screen compounds that satisfy the built pharmacophore model. In contrast, the structure-based approach relies on the structural information of the target protein. The virtual screening was successfully applied in the development of PPI modulators including Ubc13/Uevl,52 MDM2/p53,53 and TCF/β-catenin.54
Mechanism of PPIs modulators
The small molecule PPIs modulators can interact not only with protein–protein interface but also with allosteric sites55,56 (Fig. 1). Studies showed the small molecule modulators can either bind to the non-interaction region of the proteins which is named allosteric inhibition or bind to the PPI interface, which is named orthosteric inhibition. Besides PPI inhibition, some modulators can stabilize or even enhance PPI. There are two models to explain the stabling effects: when the modulator binds to the allosteric regulatory site of the protein, it triggers the conformation change of the target protein, thereby enhance the affinity of the target protein to the other protein. In case the modulator binds to the PPI interface, provides more contact sites for the two proteins, the binding force of the two proteins gets enhanced.57 For the PPI with hot-spots, the corresponding ligands can be designed to directly affect PPI. In case the PPI without hot-spots, the PPI can be indirectly regulated through the allosteric mode.58 Specifically, if the PPI hot-spots residues gather together and form appropriate pockets, the orthosteric modulators can be designed and developed based on the pockets structure information to directly influence the associate PPI. If the hot-spots can not form appropriate binding sites, developing the allosteric modulators will be a better choice.59 Most of the small molecules that have been identified to modulate PPIs are inhibitors. The PPI stabilization represents a promising modulation approach since the combination with pre-existing complexes is more advantageous in energy saving compared to the inhibition of complexes formation.60–62 However, the development of PPIs stabilizers has not received sufficient attention as compared to the development of PPIs inhibitors.63
Fig. 1.

Orthosteric and allosteric mechanisms for PPI inhibition and stabilization
Three types of PPIs modulators
Up to date, the PPI modulators can be classified into three categories (Table 2). The first category is the small molecule modulators. Compare to the classic drug targets like enzymes or ion channels, the PPI interface is large, flat, and lacks a suitable size pocket which the small molecules can bind with. What is more, the PPI interface is usually hydrophobic. Therefore, a potent PPI modulator should cover a large surface area and make a large number of hydrophobic contacts. Such a modulator may face pharmacokinetic issues due to its large molecular weight and poor solubility.8 Therefore, the small molecule modulator is more suitable for the tight and narrow PPI interface.44 The second category is an antibody. When targeting a large PPI interface, an alternative other than small molecular compounds is needed to cover the large interface. Although monoclonal antibodies compete with PPIs, because of their large molecular weight, the application of monoclonal antibodies is limited to the extracellular targets. Up to date, the monoclonal antibodies have been successfully used in clinical treatment although they may trigger adverse reactions associated with the immune reactions. The third category is peptides. The peptides are designed based on the structure information of the hot-spots.64 The designed peptides retain the key roles when they bind to proteins, thereby forming a strong affinity with the proteins. Compared with small-molecule PPI modulators and monoclonal antibodies, the molecular weight of peptide is between the two. It has higher target specificity and affinity and is a potential PPI modulator. However, the peptide is susceptible to hydrolysis by various hydrolases in the body, which makes its half-life short.
Table 2.
The advantages and disadvantages of three types of PPI modulators
| Small molecules | Peptides | Antibodies |
|---|---|---|
| Advantages | ||
|
Penetrate cell membrane Oral administration |
Target specificity High affinity |
Strong target specificity High efficiency |
| Disadvantages | ||
| Side effects (low selectivity) |
Short half-life Poor oral administration Unstable physicochemical properties Poor solubility |
Side effects (immunogenicity) Generally act on extracellular targets (huge molecular weight) |
In this review, we summarized the latest advances in PPIs modulators development including the small molecules, peptides, and antibodies. Also, we summarized the up to date some PPIs modulators in clinical trials, hoping to provide some guidance to the design of novel drugs targeting PPIs in the future.
Inhibitors of PPIS
Inhibitors of MDM2/p53 interaction (small molecules, peptides)
The p53 is an important protein that regulates the cell cycle and functions as a tumor suppressor.65 Studies showed ~50% human cancers have alterations in the p53 gene which results in the inactivation of p53 function or loss of p53 expression.66 The mouse double minute 2 (MDM2) is a proto-oncogene and a key negative regulator of p53. A negative feedback loop between MDM2 and p53 has been uncovered as the mechanism of how they regulate each other’s level in the cells (Fig. 2a).67 MDM2 directly binds to and forms a complex with p53, inhibiting the transactivation of p53. Therefore, recovering the impaired the function of p53 by disrupting the MDM2/p53 interaction offers a potential approach for the treatment of cancer.68,69
Fig. 2.

The p53/MDM2 interactions and inhibitors. a The p53/MDM2 signaling pathway: MDM2 directly binds to p53 and inhibits its transcriptional activity, causes ubiquitination and proteasomal degradation of p53, and exports p53 out of the nucleus which promotes p53 degradation. b MDM2 (surface)-p53 peptide (green) complex (PDB:1T4F). c The chemical structures of inhibitors of MDM2/p53
The X-ray crystallography disclosed the details of the MDM2/p53 interaction. The interaction between MDM2 and p53 involves four key hydrophobic residues (Phe19, Leu22, Trp23, Leu26) in an α-helix formed by p53 and a small but deep hydrophobic pocket in MDM228 (Fig. 2b). An effective strategy to block their interaction is to design a small molecule compound that mimics the “hot-spots” residue structure of p53, which competes with p53 to bind with MDM2, thereby preventing the inactivation of p53. The peptide-like design, HTS, and structure-based design were adopted as the strategies to screen the MDM2/p53 inhibitors with good drug-like properties.70–72
The imidazoline compounds Nutlins discovered by Vassilev et al.33 through HTS showed strong inhibitory effects against MDM2/p53 interaction (Fig. 2c). As a group of small-molecule inhibitors of MDM2, the Nutlins mimic the effect of p53 peptide segment. The Nutlins bind to the deep hydrophobic pocket in MDM2, therefore block the MDM2/p53 interaction. Studies showed the IC50 of Nutlin-1, Nutlin-2, and Nutlin-3 on MDM2/p53 interaction were 260, 140, and 90 nM, respectively, in vitro.33 Based on the inhibitory dose values, the Nutlin-3 was selected as the lead compound. Roche restructured the Nutlin-3 by substituting the methyl for the 4- and 5-position hydrogen atoms of its imidazole ring, and replaced the cyclomethoxy group at the para position of the benzene ring with a tert-butyl group which prevented the metabolic inactivation of the imidazole ring and the benzene ring.73 Meanwhile, the isopropoxy group was replaced by an ethoxy group to reduce the molecular weight, and the hydrophilic side chain of carbonyl piperazine was replaced by a methylsulfonyl propyl piperazine, therefore obtained the compound RG7112 (Fig. 2c). The homogeneous time-resolved fluorescence (HTRF) assay showed that the compound RG7112 (IC50 = 18 nM) was optimized to be four times more sensitive than that of Nutlin-3.73 RG7112 is the first MDM2 inhibitor entered clinical trials for the treatment of advanced solid tumors.
Using peptides to inhibit PPIs has become a promising way to discover active compounds. Chang et al.74 reported a class of potent MDM2 peptide inhibitors ATSP (Table 3), among which the IC50 value of ATSP-7041 reached 0.9 nM, and the reported Ki values of the ATSP series peptides reached the nanomolar level. The key is that the peptide mimics the key α-helical structure in the p53/MDM2 interaction, thus binding MDM2 competitively with p53. ATSP inhibitors showed a certain biological activity in vivo, which may be related to the good cell membrane permeability produced by the stable α-helix structure. The western blot analysis also showed that the ATSP inhibitors inhibit MDM2 in cells, thereby activating the role of tumor suppressor protein p53.74
Table 3.
Peptide inhibitors of MDM2/p53 interaction reported by Chang et al.
| Name | Sequence | Ki (nM) |
|---|---|---|
| ATSP-1800 | Ac-Gln-Ser-Gln-Gln-Thr-Phe-R8-Asn-Leu-Trp-Arg-Leu- Leu-S5-Gln-Asn-NH2 | 25.9 |
| ATSP-3848 | Ac-Leu-Thr-Phe-Glu-His-Tyr-Trp-Ala-Gln-Leu-Thr-Ser-NH2 | 14.6 |
| ATSP-3900 | Ac-Leu-Thr-Phe-R8-His-Tyr-Trp-Ala-Gln-Leu-S5- Ser-NH2 | 1.0 |
| ATSP-4641 | Ac-Leu-Thr-Phe-R8-Ala-Tyr-Trp-Ala-Gln-Leu-S5- Ser-NH2 | 4.9 |
| ATSP-6935 | Ac-Leu-Thr-Phe-R8-Glu-Tyr-Trp-Ala-Gln-Leu-S5- Ser-NH2 | 1.2 |
| ATSP-7041 | Ac-Leu-Thr-Phe-R8-Glu-Tyr-Trp-Ala-Gln-Cba-S5- Ser-Ala-Ala-NH2 | 0.9 |
| ATSP-7342 | Ac-Leu-Thr-Ala-R8-Glu-Tyr-Trp-Ala-Gln-Cba-S5- Ser-Ala-Ala-NH2 | 536 |
Inhibitors of Bcl-2/Bax interaction (small molecules)
The Bcl-2 family is a key regulator of apoptosis, and it has over twenty members. According to their role in apoptosis, the Bcl-2 family members can be divided into two categories including the anti-apoptotic proteins and the pro-apoptotic proteins (Fig. 3a). The anti-apoptotic proteins include Bcl-2, Bcl-w, Mcl-1, and Bcl-A1. The pro-apoptotic proteins include Bax, Bok and Bak, Bid, Bad, Bmf, Noxa, Puma, Hrk (among them, Bid, Bad, Bmf, Noxa, Puma, and Hrk are BH3-only protein).75,76 Both anti-apoptotic and pro-apoptotic members usually synergize in the form of dimers, having the role of apoptotic switch.77,78 Pro-apoptotic proteins such as Bax and Bad have critical roles in the apoptosis. The functions of these pro-apoptotic proteins are blocked when they bind to the anti-apoptotic proteins like Bcl-2. Therefore, inhibiting the interaction between the pro- and anti-apoptotic proteins prevents the tumor cells from escaping apoptosis.
Fig. 3.

The Bcl-2/Bax interactions and inhibitors. a The Bcl-2 family can be classified into two categories: the anti-apoptosis proteins and pro-apoptosis proteins. The pro-apoptosis proteins can be divided into multi-BH proteins and BH3-only proteins. b The crystal structure of Bcl-2 in complex with Bax BH3 peptide (PDB:2XA0). c The binding modes of ABT-199 binds to Bcl-2 (PDB:6GL8). d The chemical structures of inhibitors of Bcl-2/Bax
The Bcl-2 family members have low homology, but they contain at least one or four conserved Bcl-2 homology (BH) motifs, named BH1, BH2, BH3, and BH4.76 There are two hydrophobic ɑ-helix structures in Bcl-2 which are surrounded by six to seven amphiphilic ɑ-helix structures, of which four amphiphilic ɑ-helix structures form a hydrophobic BH3 “pocket” to interact with Bax (Fig. 3b).79 Compared with the Bax/Bak homodimer, the Bcl-2/Bax homodimer is more stable, which weakens the role of Bax/Bak in inducing cell apoptosis and prevents cell apoptosis. Therefore, the lead compounds should mimic the function of the pro-apoptotic protein domain. The ideal compounds will bind to the hydrophobic pocket on the surface of the anti-apoptotic protein, thereby blocking the anti-apoptotic protein to bind with the BH3 domain and result in the cancer cell apoptosis induction.80,81
Abbott researchers studied the Bcl-XL hydrophobic groove and found that the hydrophobic groove consists of two relatively independent small pockets.82 They used the “SAR by NMR” approach to screen the fragments with BH3 on Bcl-XL, and obtained compound 1 (Kd = 0.30 ± 0.03 mM) and compound 2 (Kd = 4.3 ± 1.6 mM) from the library (Fig. 3d). The researchers used a fragment-based drug design strategy and screened the compounds based on the NMR data. Based on the position and spatial orientation data obtained from the complexes of the Bcl-XL hydrophobic groove-binding pockets with the compound 1 and compound 2, the researchers modified the compound 2’s structure by adding a linking group thereby constructed a highly active new lead compound 3 (IC50 = 36 nM). However, the compound 3 exhibited poor water solubility but high affinity to human serum albumin (HSA). In subsequent structural optimization, the researchers reduced the compound’s affinity to HAS through substituting polar groups at specific sites. It was found that introducing 2-dimethylaminoethyl substituent at the second ligand of compound 3 and substituting piperazine at the first ligand improved its affinity to Bcl-2 protein, and thus compound ABT-737 was obtained (Fig. 3d). The ABT-737 binds to Bcl-XL (Ki < 0.5 nM) and Bcl-2 (Ki < 1 nM), and its IC50 value reaches 35 nM in 10% of human serum. The ABT-737 is not only widely used in the biological studies associated with apoptosis, but also in preclinical studies in lymphoma, small cell lung cancer and chronic lymphoblastic leukemia.83,84
However, the poor oral absorption of ABT-737 significantly limits its clinical application. The ABT-263 (Navitoclax) (Fig. 3d) is the second generation of Bcl-2 anti-apoptotic protein inhibitor based on the structure of ABT-737.85,86 It can bind with Bcl-2 (Ki < 1 nM), Bcl-XL (Ki < 0.5 nM), Bcl-w (Ki < 1 nM), and MCL-1 (Ki = 550 nM). The preclinical studies showed that ABT-263 alone effectively inhibited the small cell lung cancer xenograft tumors growth in mice model. Besides, the ABT-263 also showed synergic effects in inhibiting the solid tumors and blood tumors in combination with other antineoplastic agents.86 However, studies also showed the ABT-263 could temporarily decrease the platelet count.87
The ABT-199 (Venetoclax) (Fig. 3d) is the first small molecule PPI inhibitor approved for marketing. It is a Bcl-2 selective inhibitor based on the structure design of lead compound ABT-263.88 It was approved to be marketed in 2016 for the treatment of chronic lymphoblastic leukemia.89 By studying the complex structure of Bcl-2 protein and small molecule acyl sulfonamide compounds, it was found that the introduction of indole group was beneficial to enhance the binding of drugs to P4 pockets through hydrophobic interaction and resulted in the formation of electrostatic interaction with aspartic acid residues specific to Bcl-2 protein88 (Fig. 3c). The researchers from the Abbvie introduced indole group and azaindole group into the ABT-263 skeleton structure and studied the structure–activity relationship. The studies showed that the ABT-199 had good activity on Bcl-2-dependent hematological cancers.88 The ABT-199 showed a high affinity for Bcl-2 (Ki < 0.01 nM) and a weak affinity for Bcl-XL (Ki = 48 nM). It showed an excellent inhibitory effect on the acute lymphoblastic leukemia cells with high expression of Bcl-2 (EC50 = 8 nM). Compared with the second-generation drug ABT-263, the ABT-199 significantly reduced the damage to the platelets in both in vitro and in vivo studies.
Inhibitors of XIAP/caspase-9 interaction (small molecules)
Inhibitors of apoptosis proteins (IAPs) are an important class of endogenous anti-apoptotic proteins.90 They bind to the caspase or other pro-apoptotic proteins, results in the inhibition of the pro-apoptotic proteins functions and promotes their degradation, thereby regulates the apoptosis.91,92 The IAPs has eight family members: XIAP, c-IAP1, c-IAP2, ML-IAP/Livin, ILP2, NAIP, Bruce/Apollon, and surviving.93 The caspase, a cysteine-containing aspartate proteolytic enzyme, is the main implementer of apoptosis, which induces apoptosis through two pathways. One of which is the death receptor pathway (extrinsic pathway) that mediated through caspase-8. The other one is the mitochondrial pathway (intrinsic pathway), which mediated via cytochrome C/caspase-9 (Fig. 4a).94 The BIR3 domain of the XIAP binds to and inhibits pro-apoptotic caspase-9, thus suspends the apoptosis.95 Interestingly, the endogenous protein inhibitor of the XIAP–caspase-9 interaction exists in the form of Smac (second mitochondria-derived activator of caspase). When the Smac released from the mitochondria, its N-terminal amino acids, alanine–valine–proline–isoleucine (AVPI) bind to the BIR3 domain of XIAP, which makes the XIAP lose the ability to combine with caspase, so as to promote apoptosis.96,97
Fig. 4.

The XIAP/caspase-9 interactions and inhibitors. a The apoptotic pathway. There are two apoptotic pathways: extrinsic and intrinsic. The extrinsic pathway (also known as death receptor) involves the binding of a death receptor ligand to a member of the death receptor family. Active caspase-8 cleaves and activates the executioner caspase-3 and caspase-7, leading to the cell death. The intrinsic pathway (also known as mitochondrial) is mediated by caspase-9. After the mitochondrial membrane is stimulated by apoptosis, it releases cytochrome c and Smac proteins into the cytoplasm. Smac is a pro-apoptotic protein. Cytochrome c combines with Apaf-1 to form a polymer, and promotes pro-caspase-9 to form apoptotic bodies, and then activate caspase-9. The activated caspase-9 can activate other caspases, such as caspase-3, so as to induce apoptosis. b The chemical structures of inhibitors of XIAP/caspas-9
The four amino groups of the AVPI at the N terminus of the Smac protein have a very important role in the binding of XIAP to caspase-9, which competes with the caspase-9 protein for binding to XIAP.98,99 Therefore, the interaction between XIAP and caspase-9 can be inhibited by the Smac protein mimetics that exhibit the similar affinity to XIAP.100 The crystal structure of Smac and XIAP-BIR3 domain revealed that the Val of P2 position and the Ile of P4 position in the Smac formed three hydrogen bonds with the Gly306 and the Thr308 of XIAP-BIR3 domain.99 The 3-position Pro ring bind with the hydrophobic region formed by the Trp323 and Tyr324 of XIAP-BIR3 domain through van der Waals force. Moreover, the Pro ring is essential for maintaining the conformation of AVPI peptide chain, so proline is relatively stable and usually not replaced by other amino acids. Flygare et al.101 discovered the first Smac simulator GDC-0152 through a combination of peptide-like design strategies and high-throughput screening (Fig. 4b). The GDC-0152 binds to XIAP-BIR domain with high affinity by mimicking the structure of the Smac AVPI peptide. Another Smac mimetic GDC-0917 (CUDC-427) (Fig. 4b) has entered phase І clinical trials for the safety evaluation of patients with advanced solid tumors and lymphomas.102 Novartis LCL-161 (Fig. 4b), which is currently progressing rapidly, has entered phase II clinical trials for triple negative breast cancer.103
Inhibitors of Hsp90/Cdc37 interaction (small molecules)
The heat shock protein 90 (Hsp90) is a widely existed, highly conserved molecular chaperone that was discovered in 1962. It is also one of the most abundant proteins in cells. Studies showed the expression of Hsp90 in tumor cells is two to ten times higher than that of normal cells, which indicates it has a very important role in tumor cell growth and survival.104 Studies also showed the Hsp90 participates in the maturation of protein kinases and transcription factors such as Her2, VEGF, mutant p53, CDK4, HIF-1ɑ, Raf -1, Akt, etc. which regulate the cancer cell’s growth and apoptosis signaling pathways.105,106 Hsp90 stabilizes the conformation of the client proteins mentioned above and prevents them from ubiquitination-mediated degradation, thereby stabilizes them to stay in the active form and promotes the tumor growth and metastasis (Fig. 5a).107 Therefore, inhibiting the interaction between the Hsp90 and its client proteins may promote the degradation of the client proteins, and thus results in the inhibition of the tumor growth.108–110
Fig. 5.

The Hsp90/Cdc37 interactions and inhibitors. a Co-chaperone regulation of client protein activation. In the chaperone cycle of Hsp90, the open state Hsp90 firstly combined with HOP through its C terminal. Subsequently, it recruits Hsp40, Hsp70, client protein, and Cdc37 to form a mature complex. After ATP hydrolysis, ADP and mature client protein are released. Hsp90 is converted into an open state and enters the next ATP cycle. b The complex structure DCZ3112 and the N-terminal domain of Hsp90 modeled by molecular docking based on the crystal structure of Hsp90–Cdc37 complex (PDB:2K5B). c The chemical structures of inhibitors of Hsp90/Cdc37
The previous studies also showed that the Hsp90 is a homodimer and each monomer is formed by three highly conserved domains including a N-terminal ATP-binding domain, a middle domain, and a C-terminal dimerization domain.111 The N-terminal domain is an ATP/ADP-binding site that hydrolyzes the ATP in the ATP-binding pocket. The ATP/ADP-binding site acts as a conformational transformation region, regulates the assembly of the Hsp90 involved multi-molecular chaperone complexes.112 The middle domains serve as both nuclear localization sequences and the target protein-binding sites. The middle domains distinguish the different substrate proteins and regulate the activity of specific substrates of molecular chaperones.113 The C-terminal domain is a self-dimerization site of Hsp90, which enhances the interaction of two Hsp90 N-terminal domains.114 The current Hsp90 inhibitors can be classified into three categories: the N-terminal ATP pocket inhibitors, the C-terminal nucleotide site inhibitors, the Hsp90 and chaperone complexes inhibitors. One critical function of Hsp90 is to regulate its client proteins to utilize ATP. The inhibition of such crucial function affects many normal proteins, results in high toxicity.115 Hsp90 inhibitor SNX-5422 developed by Pfizer terminated clinical phase 1 trial in 2011 due to ocular toxicity.116 Therefore, researchers believe that targeting Hsp90 and its molecular chaperones is a new direction for the cancer treatment study.117,118
Among the numerous molecular chaperones of Hsp90, the value of cell division cycle protein 37 (Cdc37) has attracted much attention.110 Many protein kinases (such as EGFR, CDK, Akt) rely on Cdc37 to aggregate onto Hsp90, thus completing the correct folding of the complex’s spatial conformation.110,119 Therefore, inhibiting the interaction of Hsp90 and Cdc37 may deactivates the kinase client proteins, thereby inhibiting the proliferation and growth of tumor cells. In addition, the PPI targeting Hsp90/Cdc37 specifically targets the kinase client protein of Hsp90, thereby improving selectivity and avoiding a series of adverse reactions.
In 2004, the researchers resolved the first crystal structure of Hsp90N-Cdc37M which provided a solid structural basis for the design of Hsp90/Cdc37 interaction inhibitors.120 The NMR analysis of the Hsp90N–Cdc37M complex indicated the hydrophobic interaction is the major interaction force between the two proteins.121 The key amino acids of the interface include Met164, Trp193, Ala204, Leu205 of Cdc37M and Ala117, Ala121, Ala124, Ala126, Met130, Phe134 of Hsp90N. The Leu205, the leucine residue of Cdc37, is very important for the formation of Hsp90N–Cdc37M complex. Experiments show that the mutation of Leu205 resulted in the loss or decrease of the binding ability between Hsp90N and Cdc37M.
In 2018, Xie et al.107 first reported the small molecule inhibitor DCZ3112 inhibits the interaction of Hsp90/Cdc37 (Fig. 5c). The DCZ3112 directly binds to the N-terminal domain of Hsp90, inhibits the Hsp90/Cdc37 interaction without affecting the ATPase activity of Hsp90 (Fig. 5b). The DCZ3112 mainly inhibits the proliferation of HER2-positive breast cancer cells and its IC50 value of SK-BR-3 and BT-474 cells was 7.9 and 4.6 μM, respectively. Experiments in SK-BR-3 and BT-474 cells showed that DCZ3112 downregulated the number of Hsp90 client proteins HER2, Akt, RAF-1, CDK4, and CDK6 in a concentration-dependent manner. The in vitro experiments results showed the DCZ3112 has a synergistic effect in inhibiting cell proliferation, inducing G1 arrest, inducing apoptosis, and reducing phosphorylation of Akt and Erk.107
Inhibitors of c-Myc/Max interaction (small molecules)
The c-Myc is a transcription regulator encoded by the proto-oncogene Myc. It is a highly conserved protein with helix structure. It has a critical role in promoting tumorigenesis, maintaining the growth, proliferation and differentiation of tumor cells, angiogenesis and apoptosis.122–124 Aberrant expression of c-Myc has been confirmed in most malignant tumors.125 As a result, c-Myc has become a research hot spot. The c-Myc has a bHLH-ZIP domain, its function depends on the formation of Myc–Max dimer.126 The Myc–Max dimer recognize the CACGTG in E-box sequence on their target DNA and bind to it to activate or enhance the transcription of the regulated genes.126 Therefore, inhibiting the PPI between the c-Myc and Max may inhibit the activation or transcription of oncogenes, indicating an antitumor effect.127,128
Castell et al.129 used the cell-based bimolecular fluorescence complementation (BiFC) to screen small molecules that interfere with c-Myc/Max interaction. Three compounds with good potential were identified from a library of 1990 compounds: MYCMI-6, MYCMI-11 and MYCMI-14 (Fig. 6). MYCMI-6 showed a strong inhibitory effect on c-Myc/Max interaction in both in vitro and cell-based experiments. The surface plasmon resonance (SPR) results showed that MYCMI-6 blocks the c-Myc-driven transcription and MYCMI-6 selectively binds to the bHLH-ZIP domain of c-Myc (Kd = 1.6 ± 0.5 μM). The above results indicate that the MYCMI-6 inhibits the growth of cancer cells in a c-Myc-dependent manner (IC50 = 0.5 μM) and has no effect on normal cells. Also, studies showed MYCMI-6 induces apoptosis, inhibits the proliferation of tumor cells and reduce the microvessel density in the mice xenograft tumors. The validation experiments based on microscale thermophoresis (MST) and SPR showed that MYCMI-6 binds to the bHLH-ZIP domain of c-Myc (Kd = 4.3 μM) and Max (Kd = 3.8 μM), respectively, thus inhibit the interaction between c-Myc/Max proteins.
Fig. 6.
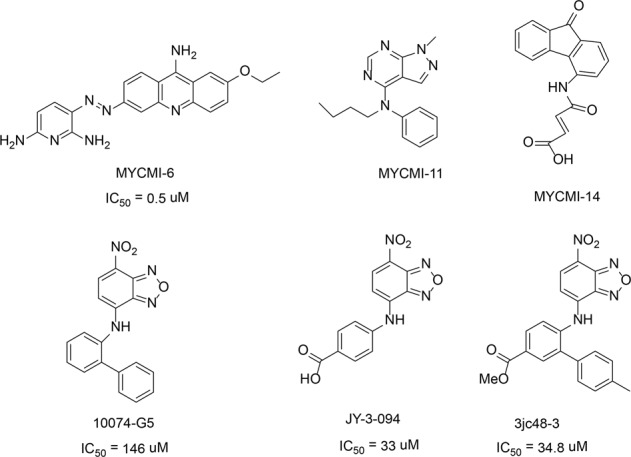
The chemical structures of inhibitors of c-Myc/Max
Chauhan et al.130 discovery that the compound 10074-G5 inhibits the formation of heterodimers between c-Myc/Max (Fig. 6). The nitro and furan rings of 10074-G5 interact with Arg366, Arg367, and Arg372 in the HLH domain, therefore, inhibit the formation of heterodimers between c-Myc and Max.128 The optimization of compound 10074-G5 led to the discovery of compound JY-3-094 (Fig. 6).131 The electrophoretic mobility shift assays (EMSAs) showed the formation of heterodimers between JY-3-094 and c-Myc/Max was five times more active than the 10074-G5 (IC50 = 33 μM vs 146 μM). However, unlike 10074-G5, the JY-3-094 does not inhibit the proliferation of human promyelocytic leukemia (HL60) or Daudi Burkitt lymphoma cell lines because the charged carboxylic acids groups in the molecule impeded the cell entry. By esterifying the carboxylic acid in the JY-3-094 into a series of ester prodrugs, the lower IC50 values were reached in both HL60 and Daudi Burkitt lymphoma cell lines. However, the activity of ester prodrugs is always limited by the activity of carboxylic acid metabolites, so the structural optimization of JY-3-094 continuous. Studies showed the phenyl ring adjacent to aniline in 10074-G5 enhanced the inhibitory effects. The introduction of phenyl ring into the JY-3-094 led to the formation of 3JC48-3, with an IC50 value of 34.8 μM for c-Myc/Max protein inhibitory activity (Fig. 6). Further studies showed that 3JC48-3 inhibits the tumor cell proliferation by inducing cell arrest in the G0/G1 phase. Such a significant increase of c-Myc/Max protein inhibitory effect may be the interaction between phenyl ring with the Phe375/lle381 and Arg378 in the c-Myc/Max.130
Inhibitors of KRAS/PDEδ interaction (small molecules)
Oncogenic RAS is an important antitumor target, and are mutated in about 20–30% of human cancers.132 The RAS family has three members: HRAS, KRAS, and NRAS. The KRAS protein is often mutated in various cancers. Specially, the KRAS mutation has been observed in a large proportion of pancreatic cancers.133 The RAS mutations lead the cells on the hyperactive state for unlimited proliferation. As a molecular switcher, RAS activates the downstream signaling pathways such as MAPK and PI3K-Akt through binding to GTP, thus regulating the growth, proliferation, differentiation, and apoptosis of the cells. If the RAS proteins are continuously activated, it can bind to the downstream effector proteins and transmit signals to the downstream proteins, causing aberrant cell proliferation or tumorigenesis.134 Therefore, RAS proteins can be developed as an important target for the cancer treatment. At present, there are mainly two strategies to inhibiting KRAS. The first strategy is directly targeting the signal pathway of KRAS protein. The second strategy is inhibiting the KRAS membrane association which impairs the localization of KRAS and the signal transduction of tumor proliferation. To carry out its signal transduction function, the RAS proteins need to be recruited to the inner side of the plasma membrane after expression.135 In the process of KRAS relocating to the cell membrane, PDEδ promotes the KRAS protein recruit to the Golgi apparatus135,136 (Fig. 7a). Therefore, through interfering with the interaction between PDEδ and KRAS, the localization of KRAS on the plasma membrane can be inhibited and the signal transduction of carcinogenic RAS can be blocked.137 However, some studies showed that the degree of dependence of KRAS on PDEδ is not yet clear. For example, PDEδ knockout mice are fertile,138 whereas the knockout of KRAS in mice is embryonic lethal,139 indicating KRAS is functional in the absence of PDEδ. Although the relationship between KRAS and PDEδ is vague, blocking KRAS membrane association is a good direction to inhibit the KRAS activity.140
Fig. 7.

The KRAS/PDEδ interactions and inhibitors. a The process of KRAS localization to the plasma membrane. b The chemical structures of inhibitors of KRAS/PDEδ
There are many small molecule compounds that inhibit the interaction between PDEδ and KRAS.137,141–146 In 2018, Chen et al.147 discovered the novel KRAS/PDEδ inhibitors through fragment-based drug design. By applying molecular docking, they found the compound 4 and compound 5 exhibited inhibitory effects on PDEδ and KRAS interaction when the two molecules exist in a specific way (Fig. 7b). The molecular docking model showed that the distance between the benzene ring of the compound 5 and the nitrogen atom of the amide of the compound 4 was 5.3 Å; the distance between the benzene ring of the compound 4 and the nitrogen atom of the imidazole of the compound 5 was 5.0 Å. The above distances are both suitable for using an ether linker to connect the two methylene groups. Therefore, the two series of compound 6 and 7 were obtained (Fig. 7b). A further optimization of the compound 6 and 7’s structure led to the synthesis of compound 8. Compound 8 exhibited a good affinity for PDEδ (Kd = 38 ± 17 nM) (Fig. 7b). The molecular docking data showed the cyclopropyl group in compound 8 forms a hydrophobic interaction with the amino residues Ile129, Val145, and Leu147 in the PDEδ. Compound 8 also exhibited inhibitory effects in Capan-1 cells (IC50 = 8.8 ± 2.4 μM). The RAS family regulates MAPK and PI3K-Akt-mTOR signaling pathways, and studies showed that the compound 5 downregulates the phosphorylation levels of Akt and Erk. In sum, compound 8 induces apoptosis in Capan-1 cells.
Inhibitors of CD40/CD40L interaction (small molecules)
T cells have an important role in the immune system. Their activation requires not only the direct stimulation of foreign antigens, but also the co-stimulus signal transmitted by the interaction of surface molecules.148 The CD40/CD40L pathway is one of the most important co-stimulating pathways in T-cell activation. Because of its critical role in the T-cell activation, the aberrant CD40/CD40L pathway is responsible for various pathological conditions. CD40 is a membrane surface molecule has a key role in B-cell development and activation. It is a surface antigen associated with T cells and B cells function.149 CD40L, a T-cell–B-cell-activating molecule, is widely expressed in the activated T cells, especially CD4+T cells.150
The CD40 and CD40L are a pair of complementary protein molecules. The CD40 is a member of the tumor necrosis factor receptor superfamily and its ligand CD40L (also known as CD154) belongs to the tumor necrosis factor family.151 Both CD40 and CD40L are mainly expressed by T and B cells. As a pair of the membrane proteins, CD40/CD40L participates in various vital physiological processes including B-cell activation, proliferation, differentiation, antibody production, apoptosis, T-cell activation, cytokine production, humoral immunity, cellular immunity, and inflammatory response152 (Fig. 8a). The abnormal expression of CD40/CD40L is closely related to the occurrence and development of inflammatory reaction, autoimmune diseases and immunodeficiency diseases.152–155 Therefore, blocking the interaction between CD40 and CD40L may has great potential to treat the associated diseases.
Fig. 8.

The CD40/CD40L interactions and inhibitors. a CD40/CD40L signal transduction and cellular response. After interacting with CD40L, CD40 recruits and interacts with tumor necrosis factor receptor-associated factor (TRAF) proteins. The activation of CD40-CD40L axis results in cellular events. b The chemical structures of inhibitors of CD40/CD40L
Multiple antibodies that block the interaction of CD40 and CD40L have been tested in preclinical or clinical trials, including: bleselumab, lucatumumab, and dacetuzumab, etc.156 Dacetuzumab is a IgG1 humanized anti-CD40 monoclonal antibody. In the absence of IL-4 and CD40L, dacetuzumab activates the B cells’ proliferation but inhibits the highly differentiated B cells’ proliferation. Besides, dacetuzumab transmits apoptosis signals through caspase-3, mediates antibody-dependent cell-mediated cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP) effects.157 However, most of these antibodies’ trails were terminated due to the severe thrombolysis side effect.158–160 Previous studies demonstrated the thrombolytic side effect may be a feature of antibody treatment.158 What is more, recent studies discovered that the antibody aggregation induced by the mAb Fc domain is also associated with thrombolysis side effect.161 Therefore, to avoid the thrombolysis severe side effect, alternative approaches like using small molecule compounds to block the interaction between CD40 and CD40L need to be developed. Buchwald’s group reported small molecular organic dyes that blocked the interaction between CD40 and CD40L.162,163 Based on these organic dye compounds, they synthesized a series of small molecule compounds that block the interaction between CD40 and CD40L.164 Among them, the IC50 of DRI-C21041, DRI-C21045, and DRI-C25441 were 0.31, 0.17, and 0.36 μM, respectively (Fig. 8b). These compounds also showed inhibitory effects on CD40L-induced B-cell activation, proliferation, and the activation of NF-κB. In addition, they also inhibit the immune response induced by alloantigen.
Inhibitors of Skp2/Skp1 interaction (small molecules)
Ubiquitin-protein degradation system (UPS) is composed of more than 1000 proteins. As the main pathway of protein degradation in cells, UPS has a key role in cell cycle regulation, intracellular signal transduction, gene transcription, metabolic regulation, immune surveillance and other basic cell life processes. The aberrant UPS system is responsible for the occurrence of various diseases.165,166 UPS consists of ubiquitin-activating enzyme 1 (E1), ubiquitin-conjugating enzyme 2 (E2), ubiquitin-protein ligase 3 (E3) and proteasome.167 At present, the E3 has been studied most. Skp1-Cullin 1-F-box (SCF) ubiquitin ligase containing F-box protein is one of the most important ubiquitin ligases and has attracted wide attention.168 SCF is a multi-subunit structure consisting of four parts: Cull, Skp1, Rbx1 and F-box169 (Fig. 9a). As a member of the F-box protein family, S phase kinase-associated protein 2 (Skp2) and Skp1, Cull, and Rbxl constitute E3 ligase, which is involved in the process of catalyzing the transformation of cells from G1 to S phase.170 The overexpression of Skp2 is extremely common in human cancer cells, and Skp2 overexpression promotes cancer invasion and metastasis.171 The interaction between Skp2 and Skp1 is the precondition of the completeness of Skp2–SCF complex and the key to exerting its E3 ligase activity. Therefore, blocking the interaction between Skp2 and Skp1 prevents the formation of Skp2–SCF complex and thus may inhibit the occurrence and development of tumors.
Fig. 9.

The Skp2/Skp1 interactions and inhibitors. a The composition of Skp2–SCF complex. Cullin 1 (Cul1) forms the backbones of ubiquitin ligase complexes. Cul1 is activated by covalent conjugation with NEDD8. The SCF complex consists of the invariable components Rbx1 (RING-finger protein), Cul1 (scaffold protein), and Skp1 (adaptor protein) as well as a variable F-box-protein component, which is responsible for substrate recognition. Skp2 is a member of the F-box protein and is a substrate recognition subunit of the SCF complex. Skp2 can specifically recognize the substrate and mediate its ubiquitination degradation. b The potential-binding pockets on the interface of Skp2–Skp1 complex (PDB:1FQV). c The chemical structures of inhibitors of Skp2/Skp1
The crystal structure of the Skp2–SCF complex shows that Skp2 interacts directly with Skp1 through its F-box domain and indirectly binds to Cul1 and Rbx1.172 Along with the Skp2–Skp1 interface, Chan et al. also reported that Skp2 has 19 “hot-spots” amino acids in contact with Skp1. They classified these key Skp2–Skp1-binding sites into two pocket regions.173 The first region (pocket 1) is near the N terminal of Skp2 and is in the F-box motif which including the amino acid residues of Trp97, Phe109, Glu116, Lys119, and Trp127. The second region (pocket 2) is close to the C terminal of Skp2, formed by a Leu-rich repeat sequence with some residues from the F-box domain (Fig. 9b). Inhibitors bind to one or both of these pockets prevent the formation of Skp2–Skp1 complex.
Chan et al.173 identified seven compounds that could inhibit the formation of Skp2–Skp1 complex through HTS. Among which, SZL-P1-41 exhibits strong inhibition effects to the Skp2–Skp1 complex formation (Fig. 9c). The molecular docking model shows the SZL-P1-41 binds to pocket 1 rather than pocket 2, which suggests the pocket 1 in the F-box sequence of Skp2 may have a leading role in the Skp2–Skp1 interaction173 (Fig. 9b). The docking model also suggests that the benzothiazole structure of SZL-P1-41 interacts with the Trp97 residue on Skp2 through an aromatic stack and a polar contact; The flavone groups of SZL-P1-41 interact with the Asp98 and Trp127 of Skp2 via hydrogen bonding or hydrophobic interaction; The ethyl group on phenol ring extends into Skp1 region; The piperidine interacts with both Asp98 and Trp127. Both in vitro and in vivo experiments results showed the Skp2 inhibitors could inhibit the Skp2-mediated P27 ubiquitination. The in vivo experiments data also showed the SZL-P1-41 could effectively inhibit the growth of tumors. In addition, the Skp2 inhibitors not only inhibit the formation of Skp2–Skp1 complex, but also reduce the Skp2 E3 ligase activity. The higher doses of SZL-P1-41 also reduces the Skp2 protein expression.
Inhibitors of Keap1/Nrf2 interaction (small molecules, peptides)
The Keap1–Nrf2-ARE signaling pathway is the most important antioxidant stress pathway, which is associated with a variety of oxidative stress-related diseases including cancer, Alzheimer’s disease, Parkinson’s disease, diabetes, and arthritis.174 Under physiological conditions, the Keap1 targets the Nrf2 to initiate the ubiquitin-dependent degradation of protein media. When cells under electrophilic or oxidative stress, the Nrf2 escapes the Keap1-mediated degradation and enters the nucleus, where it mediates the activation of the antioxidant and cytoprotective genes175,176 (Fig. 10a). Therefore, the Nrf2 signaling pathway activators should have therapeutic effect in oxidative stress-induced diseases. Up to date, most of the “Nrf2 activators” are inhibitors of Keap1/Nrf2 interaction which covalently bind with the sulfhydryl groups on the cysteine in Keap1 through oxidation or alkylation. The covalent adduct changes the Keap1 conformation that prevents the Nrf2 interact with Keap1.177 However, the covalent binding is irreversible. As a result, the long term application of the Keap1/Nrf2 inhibitors results in the accumulation of the active Nrf2, which may trigger other problems like cancer178 Therefore, finding non-covalent small molecules that directly interfere with the Keap–Nrf2 interaction, dissociating the two and exerting antioxidant defense effects has become a new therapeutic strategy.179
Fig. 10.

The Keap1/Nrf2 interactions and interactions. a The Keap1–Nrf2-ARE pathway. Under basal conditions, Nrf2 binds to Keap1 and is degraded by proteasomes. Under oxidative stress, Nrf2 escapes the degradation mediated by Keap1 and transfers to nucleus, binding with ARE and Maf protein to initiate the transcription of antioxidative and cytoprotective genes. b The chemical structures of inhibitors of Keap1/Nrf2
In 2006, Hannink’s group analyzed the structure of a complex between the Kelch domain of Keap1 and Nrf2-derrived peptide, thus revealed the binding interface between Nrf2 and Keap1, and determined the key residues of Keap1, including Arg380, Arg415, Arg483, Ser363, Ser508, Ser555, and Ser602, which laid the foundation for the design of Keap1/Nrf2 interaction inhibitors.180
The study of the Keap1/Nrf2 inhibitors begins with the investigation of the polypeptides that inhibits the Keap1/Nrf2 interaction. Up to date, a number of the inhibitive polypeptides have been reported181–184 (Table 4). Hu et al.185 developed a series of fluorescent probes to verify when the peptide chain length is 9 amino acids it has the best activity to inhibit the Keap1/Nrf2 interaction. The peptide inhibitor P1 designed based on the fluorescent probes has a moderate inhibitory activity (IC50 = 3480 ± 920 nM), and its activity increases with the elongation of the polypeptide chain (7–16 amino acids). For instance, the hexadecapeptide P2 (IC50 = 163 ± 11 nM) exhibits the highest activity.181 The acetylation of the N terminus of such a peptide neutralizes the positively charged group at the N terminus, which also greatly changes its electrical property. The nonapeptide P3 was obtained via the modifications as above-mentioned and exhibits great activity (IC50 = 194 ± 49 nM).186 Subsequently, the structure–activity relationship study demonstrated that the heptapeptide also shows activity, such as heptapeptide P4 (IC50 = 8230 ± 262 nM) and P5 (IC50 = 558 ± 53 nM), which exhibits moderate inhibitory activity. The follow-up work focused on acetylation and C18 fatty acid stearic heptapeptide. The compound P6 showed excellent Keap1 inhibitory activity (IC50 = 22 ± 3 nM).182 However, the peptide inhibitor has large molecular weight and poor ability to penetrate the cell membrane. Therefore, it is of great significance to find a class of small size peptide inhibitors with strong membrane permeability. Steel et al. designed and synthesized a number of highly permeable membrane peptides. Among them, the compound P7 induces the expression of heme oxygenase-1 (HO-1) in cells, and inhibits the proinflammatory cytokine-TNF’s expression.187
Table 4.
Some peptide inhibitors of Keap1/Nrf2 interaction
| Name | Sequence | IC50 (nM) |
|---|---|---|
| P1 | H-LDEETGEFL-OH | 3480 ± 920 |
| P2 | H-AFFAQLQLDEETGEFL-OH | 163 ± 11 |
| P3 | Ac-LDEETGEFL-OH | 194 ± 49 |
| P4 | Ac-NAETGEF-OH | 8230 ± 262 |
| P5 | Ac-DAETGEW-OH | 558 ± 53 |
| P6 | St-DPETGEL-OH | 22 ± 3 |
| P7 | YGRKKRRQRRRLQLDEETGEFLPIQ | 24 |
Most of the high binding peptides have poor cell permeability. Subsequently, the high binding peptides’ activity is not ideal. Therefore, screening the small molecule inhibitors have become a hot spot in the study of Keap1/Nrf2 interaction inhibition.188–195 The mile stone of the small molecule inhibitors study is the development of benzothiazepine heterocyclic Keap1–Nrf2 small molecule inhibitors made by GSK through fragment-based drug design.189 After screening 330 fragments via X-ray crystallography, the compounds 9–11 (Fig. 10b) were identified which interact with Arg483, Tyr525, and Ser602, respectively. The binding of the compounds to the amino acids as mentioned above simulating the binding of Nrf2 peptide segment with Keap1, but the binding activity of these three compounds was low (Kd > 1 mM). To improve the compounds’ Keap1-binding activity, various structural modifications were made. Among which, the introduction of methanesulfonamide on the benzene ring of compound 12 significantly increased the compound’s Keap1-binding activity by 20 times (IC50 = 61 μM). When compound 13 was completely introduced, its IC50 strikingly reached 0.27 μM. A series of structural optimizations were performed using compound 13 (Fig. 10b) as the lead: when methyl group is substituted for the chlorine atom on the benzene ring, it can release its potential binding to the sulfonamide center; Introducing an electron-donating group methoxy group at the 7-position of benzotriazole can improve hydrogen bonding and enhance surface bonding; The conversion of benzenesulfonamide ring to seven membered benzothiazide heterocycle can make more space occupied by sulfonamide and benzotriazole sites, and the activity of compound 14 (Fig. 10b) is significantly increased (IC50 = 0.015 μM). These compounds can induce the expression of Nrf2 downstream target protein NQ01 in BEAS-2B cells, and reduce the lung inflammation induced by ozone in animal experiments.
Inhibitors of PD-1/PD-L1 interaction (small molecules, peptides, antibodies)
Studies indicate that the PD-1/PD-L1 signaling pathway has critical role in tumor immune escape and tumor development.196 PD-1 (also known as CD279) is an immunosuppressive receptor belongs to the CD28 superfamily of T-cell regulatory receptors and its natural ligand is PD-L1. Under physiological conditions, PD-1 is mainly expressed in activated immune cells, which promotes the maturation of T lymphocytes, regulates unnecessary or excessive immune response through negative regulation, and maintains immune tolerance. The over activation of PD-1/PD-L1 signaling pathway negatively regulates the function of T cells, which cancels the immune system surveillance function and promotes the escape of tumor cells.197 Therefore, blocking the interaction of PD-1 and PD-L1 maintain the T cells immune function may be a potential strategy for tumor treatment (Fig. 11a). The PD-1/PD-L1 signaling pathway inhibitors include monoclonal antibodies, peptides, and small molecule inhibitors.
Fig. 11.

The PD-1/PD-L1 interactions and inhibitors. a PD-1/PD-L1 signaling pathway. PD-1/PD-L1 interaction causes the phosphorylation of ITIMs and ITSMs in the intracellular domain of PD-1, and then recruits SHP2 to suppress PI3K/Akt, Ras/MAPK/ERK signaling pathway, leading to T-cell exhaustion. b The chemical structures of inhibitors of PD-1/PD-L1
Up to date, there are five monoclonal antibody drugs Pembrolizumab (Keytruda), Opdivo (Nivolumab), Tecentriq (Atezolizumab), Bavencio (Avelumab), and Imfinzi (Durvalumab) have been approved as PD-1/PD-L1 signal pathway inhibitors for the treatment of melanoma, non-small cell lung cancer and other diseases198–202 (Table 5). Pembrolizumab is the first PD-1 inhibitor approved by the FDA for the treatment of advanced or unresectable melanoma, which does not respond to other drugs.203 Pembrolizumab is a highly selective humanized IgG4-κ anti-PD-1 monoclonal antibody, which activates tumor infiltrating lymphocytes (TIL). The combination of PD-1 highly expressed in TIL and PD-L1 expressed in tumor cells is an important factor for tumor immune escape. Pembrolizumab binds to PD-1 on the surface of TIL and inhibits its interaction with PD-L1/2 to activate TIL. Although immunotherapy against PD-1/PD-L1 has been applied in clinic, the use of monoclonal antibodies may affect the proliferation and activation of T cells, thereby trigger severe immune-related adverse reactions, including tissues damage, weaken the function of Fc-immune effect (killing immune cells), etc.204,205
Table 5.
Antibody agents targeting PD-1/PD-L1 approved by FDA
| Name | Diseases | Developer |
|---|---|---|
| Keytruda | Melanoma, non-small cell lung cancer | Merck Sharp & Dohme |
| Opdivo | Melanoma, head and neck cancer | Bristol Myers Squibb |
| Tecentriq | Non-small cell lung cancer, bladder cancer | Roche |
| Bavencio | Merkel cell carcinoma | Merck and Pfizer |
| Imfinzi | Urothelial carcinoma | AstraZeneca |
Compared with monoclonal antibodies, the peptides and small molecule drugs do not have the limitations of monoclonal antibodies as mentioned above.206 Chang et al.207 developed the first hydrolysis-resistant d-peptide antagonists to target the PD-1/PD-L1 pathway by using the mirror-image phage display (Table 6). The optimized compound DPPA-1 binds to PD-L1 at an affinity of 0.51 μM in vitro. The cellular level blockade assay data and tumor-bearing mice experiments results all indicate that the DPPA-1 disrupts the PD-1/PD-L1 interaction under in vivo condition.207 Aurigene developed a small peptide AUNP-12, which is an anti-PD-1 targeted immunotherapy for cancer (the structure of the compound has not been disclosed).208 The AUNP-12 inhibits the binding of PD-1 and PD-L1 under in vitro conditions (IC50 = 0.72 nM), but the time of drug metabolism was short. The animal trials data demonstrated the AUNP-12 has good anti-PD-L1 activity and effectively inhibits the growth and metastasis of tumor cells.
Table 6.
Peptide inhibitors of PD-1/PD-L1 interaction reported by Chang et al.
| Name | Sequence | Kd (μM) |
|---|---|---|
| DPPA-1 | NYSKPTDRQYHF | 0.51 |
| DPPA-2 | KHAHHTHNLRLP | 1.13 |
| DPPA-3 | AAKMDGHLHGGQ | No binding |
| DPPA-4 | TLYQRPSTNLER | 22.0 |
| DPPA-5 | RHTNDYSQFYPK | No binding |
Because the structure of PD-1 and PD-L1 proteins are not available, the development of small molecule PD-1/PD-L1 inhibitors lags far behind the development of antibody drugs. By analyzing the PD-1/PD-L1 complex structure, Zak et al.209 reported that there are three main binding pockets in the contact interface between PD-1 and PD-L1, which provide a rational basis for drug development. With the success of PD-1 monoclonal antibodies and macromolecular biomedical drugs, Bristol Myers Squibb (BMS) conducted an in-depth investigation of small molecule inhibitors of the PD-1/PD-L1 pathway. In 2015, the company disclosed its first patent on a biphenyl immunomodulator. The homogeneous time-resolved fluorescence (HTRF) test results demonstrated that these compounds blocked the interaction between PD-1 and PD-L1. Surprisingly, some of the compounds even reached nanomolar activity. The IC50 of representative compounds 15 and 16 (Fig. 11b) were 18 and 22 nM, respectively.210 In the same year, in another patent disclosed by BMS, additional structural modifications have been made which include the benzene ring in part A of the compound was replaced by 1,4-benzodioxane, and m-cyanobenzene was introduced into the benzene ring of part C through an ether bond. The structural optimization as above-mentioned significantly improved the compound’s PD-1/PD-L1 inhibition activity that their IC50 values reached 0.6–10 nM range. Specifically, the IC50 of representative compounds 17 and 18 (Fig. 11b) were 2.25 and 1.4 nM, respectively.211 To improve the compound’s inhibition activity further, the researchers continued to optimize the structures of this class of compounds. The additional structural modifications include introducing different hydrophilic groups into a part of the hydrophobic biphenyls through a carbon chain, which improved the compounds’ activity further. The representative compounds 19 and 20’s IC50 values were 0.48 and 0.88 nM (Fig. 11b), respectively.212 In 2018, BMS disclosed new compounds with symmetric structures. Compare to other compounds, the new compounds replaced the original groups on the left side with new groups that have the same or similar structures as the ones on the right side based on the original structure, thus forming a compound characterized by “central symmetry”. The activity of this type of compounds is generally less than 1 nM, and the representative compound 21’s IC50 values (Fig. 11b) reaches 0.04 nM.213
Stabilizers of PPIS
Stabilizers of 14-3-3/H+-ATPase (small molecules)
The tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activator protein family (14-3-3 protein) has an important role in PPIs. It is a highly conserved ubiquitous protein family encoded by multiple genes in most organisms.214 There are at least seven highly conserved subtypes of 14-3-3 proteins encoded by different genes in mammals. The 14-3-3 proteins bind to various ligand proteins including kinases, phosphatases, and transmembrane receptors.215 The 14-3-3 proteins regulate more than 500 endogenous molecules’ activity through binding to them.216 Since these endogenous molecules have critical roles in the cell metabolism process, cell cycle modulation, apoptosis, cell differentiation, transcription, signal transduction, and other vital biological events, the intervene of these molecules’ activities may yield severe consequences in the cells.217,218 Specifically, the 14-3-3 proteins also named as “bridge protein” of protein–protein interactions as they bind with transcription factors to form complexes that regulate the expression of associated genes. Due to the important function of 14-3-3 proteins in cells, they have critical roles in various diseases including the nervous system diseases, arthritis, malignant tumors, and infectious diseases etc.218–220
All 14-3-3 proteins have similar tertiary structures and the structure can be divided into three parts: N terminal, conservative core region, and C terminal. Each monomer consists of 9 helium spirals (ɑA~ɑI) located between the N terminal and the C terminal that arranged from an anti-parallel to an L-shaped structure separated by a short loop.221 Under certain conditions, the 14-3-3 proteins can aggregate together in the form of stable homologous/heterodimers that can bind with two ligands simultaneously.222 The dimer formation is a necessary regulatory step for its binding to the ligand protein. The dimer interface consists of αA from one monomer with the combination of and αC and αD from another monomer forming highly conserved facultative grooves. The grooves contain both polar and nonpolar amino acid residues and also contain an intense negative charge. The nonpolar amino acid residues in all subtypes of 14-3-3 proteins mainly distribute along the inner grooves, while the polar amino acid residues are located on the outer surface of the grooves. Such a unique distribution of nonpolar-polar amino acid residues makes the grooves identify the target proteins with common characteristics.
Fusicoccin A, a natural product, is the first reported stabilizer to regulate the interaction between the 14-3-3 protein and its ligand (Fig. 12). Fusicoccin A is a diterpenoid glycoside with a 5-8-5 ring that binds to 14-3-3 receptors. Fusicoccin A stabilizes the complex formed by 14-3-3 protein and plasma membrane ATPase (PMA).223 The crystal structure studies showed that the 14-3-3 dimer forms a complex structure with 52 amino acids at the C terminal of H+-ATPase, and Fusicoccin A fills the gap between the protein–protein interface of the complex.223 The hydrophobic 5-8-5 ring is inserted into the binding channel of the 14-3-3 protein. The bottom of the hydrophobic cavity contains Val153, Phe126, and Met130. The methyl or methoxy substituent is an important condition for contacting the hydrophobic bottom. The 5-8-5 ring has extensive hydrophobic interactions with Pro174, Ile174, Gly178, Leu225, Ile226, and Ile956 of H+-ATPase proteins. In addition, many water-mediated polar interactions were formed between Fusicoccin A and 14-3-3 proteins.
Fig. 12.

The chemical structures of stabilizers of 14-3-3
Richter et al.224 reported that the pyrrolidone derivatives could stabilize the interaction between 14-3-3 and PMA. Among these, the compound 22 exhibited the highest activity (Fig. 12). The crystal structure of the pyrazole derivatives and 14-3-3/PMA complex showed that the rigid pyrazole part penetrated into the protein–protein interaction interface deeply, therefore enlarged the interface with PMA. Compared with the natural product Fusicoccin A (EC50 = 498 ± 65 nM), the activity of compound 22 was better (EC50 = 33 ± 4 μM). Furthermore, compound 22 showed a good selectivity and has no effect on 14-3-3/C-Raf or 14-3-3/p53 interactions.
Stabilizers of S100 pentamer (small molecules)
The S100 protein was given its name because it is well soluble in 100% saturated ammonium sulfate under neutral conditions.225 Up to date, at least 20 members of the S100 protein family have been identified, including S100A1-A15, S100B, S100P, etc.226 The S100 proteins mainly exist in the forms of homodimers, heterodimers, trimers, and tetramers in the cells.227 Previous studies have shown that the S100 proteins act as a calcium sensor, which regulates many intracellular and extracellular activities in a calcium-dependent manner.228 The binding of calcium ions changes the S100 protein conformation, exposing its binding sites to the target proteins. Therefore, various biological functions of S100 protein can be exerted through regulating the calcium ions under in vivo conditions.229 For example, S100 regulates protein phosphorylation, enzyme activity, cell proliferation, cell differentiation, inflammatory reaction induction, and protects cells from oxidative damage.225,229 Studies showed that the high expression of S100A4 associate with rheumatoid arthritis, kidney fibrosis, and cardiac hypertrophy.230,231 Garrett et al.232 reported several phenothiazines that block the activity of S100A4. One of these compounds, trifluoroperazine inhibits the S100A4 function through stabilizing its inactive pentamer (Fig. 13b). The complex structure study discovered that the trifluoroperazine forms a pentamer complex with S100A4 and the two molecules are in contact with each other at the interface. Further analysis of the complex structure found that trifluoroperazine binds to a hydrophobic patch, which includes the side chains of Ile82, Met85, and Cys86, from one protomer and Phe89 as well as Phe93 from the other (Fig. 13a). The methylated piperazine ring of trifluoroperazine interacts with the protomer of Ser44, Phe45, Leu46, and Gly47. In addition, the carbonyl oxygen atom of the protomer Phe45 forms a hydrogen bond with the nitrogen atom on the piperazine ring.
Fig. 13.

Proteins and small molecule inhibitors of S100 pentamer. a The binding modes of trifluoroperazine binds to S100 (PDB:3KO0). Due to the clarity, only two adjacent S100A4 monomers and their contact interface are shown. b The chemical structure of a stabilizer of S100 pentamer
Stabilizers of influenza nucleoprotein protomers (small molecules)
Influenza virus is the pathogen causing acute infectious disease influenza. Influenza virus nucleoprotein (NP) is its main structural protein and the main component of nucleocapsid.233 The ribonucleoprotein complex is composed of ribonucleoprotein and RNA fragments of virus and three kinds of dependent RNA polymerase PA, PB1, and PB2, which participate in the transcription, replication, and assembly of the virus. As the main structural protein of the virus, nucleoprotein contains many functional domains, such as nuclear localization sequences, RNA-binding domains, NP–NP-binding domains, and PB2-binding domains. All these domains have vital functions that are indispensable components of viral replication. Therefore, inhibiting the nucleoprotein function may have antiviral effects.
Gerritz et al.234 reported a triazole compound that induces the formation of higher-order nuclear protein oligomers, which prevents the nuclear proteins entering the nucleus, thereby inhibiting viral replication. Previous studies showed that the binding sites of the triazole compound might be located in two regions on NP: one in NPY289/N309 region and the other in NPY52 region. Six molecules of compound 23 (Fig. 14) bridge two NP trimers (NP_A, NP_A′, NP_A″ and NP_B, NP_B′, NP_B″) to form a hexamer. The structure analysis on compound 10 and NP protein complex showed that compound 10 located between the interfaces of two trimers and stabilizes the complex.234 The other unique structures of the triazole compounds and NP complex include a hydrophobic pocket formed between two NP monomers by the amino acid residues Tyr289, Phe291, Try296, Tyr52, and Tyr313 on each monomer. The nitro moiety on the aromatic ring of compound 23 forms a Π-Π interaction with Tyr289 on NP_A. The piperazine moiety of compound 23 forms a hydrophobic interaction with Tyr254 on NP_B. Further, the hydroxyl group of the NP_B Ser forms a hydrogen bond with the carbonyl group of compound 23.
Fig. 14.
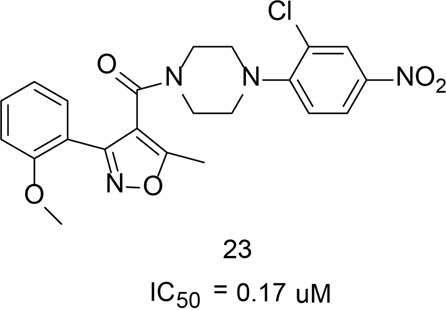
The chemical structures of stabilizers of influenza nucleoprotein
Stabilizers of microtubules (small molecules)
The microtubule is the main component of the cytoskeleton, which is composed by α-tubulin and β-tubulin. Microtubules have a vital role in maintaining cell morphology, cell division, signal transmission, and material transport.235 In the living cells, microtubules aggregate with each other into spindles in the early stages of cell division. The spindles pull chromosomes to move towards the two poles into two daughter cells during mitosis, thereby completing cell proliferation. Under physiological conditions, there is a dynamic balance between the microtubule and tubulin dimer. The microtubule stabilizers stabilize microtubules and promote the multimerization of microtubules, thus block the depolymerization of microtubules, and thereby destroy the dynamic instability of tubulin. Such effect further destroys the rapidly differentiated tumor cells during mitosis, stagnates the cell cycle, and in turn induces the tumor cells to undergo apoptosis.236
Paclitaxel (Fig. 15) is the first approved microtubule stabilizer. Studies showed that the Paclitaxel binds with β-tubulin, promotes the aggregation of microtubulin, stabilizes microtubule structure, hinders the formation of spindles, and leads to cell cycle arrest in G2/M phase.237,238 Zampanolide (Fig. 15) is a 20-membered macrolide isolated from the Tongan marine sponge Fasciospongia rimosa.239 It arrests cells in mitosis and inhibits cell proliferation by stabilizing microtubules. The structural analysis shows that zampanolide induces the disordered curled M-loop into an ordered spiral structure through its side chain.240 The M-loop is composed of eight amino acid residues in the middle region of the tubulin subunits, which maintains the interaction between the microtubule fibrils. The change in the M-loop facilitates the lateral contact between the microtubule fibrils and thus stabilize the microtubules. In 1A9 cells, zampanolide exhibited a IC50 value of 14.3 ± 2.4 nM, which demonstrated itself a potential microtubule stabilizer.241
Fig. 15.
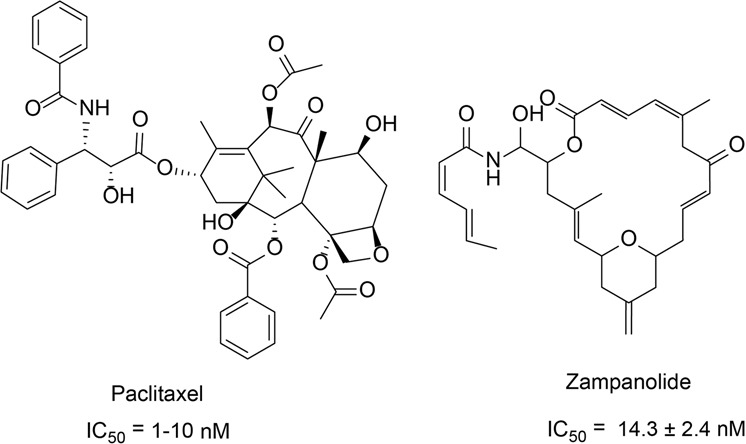
The chemical structures of stabilizers of microtubules
Conclusion
In recent years, new PPIs modulators development has been an attractive goal in preclinical studies.242,243 However, the design of modulators targeting PPIs still faces tremendous challenges. Besides the challenges mentioned previously like the difficult PPI interfaces for the drug design, lack of ligands reference, the ineffectiveness of the classic medicinal chemistry approaches for PPI drug development, lack of guidance rules for the PPI modulators development, the biggest obstacle is the lack of high-resolution PPI proteins structures. Because the medicinal PPI drug design is based on the high-resolution PPI protein structures, more resources should be put into the structural biological studies of the identified PPIs.
Inhibitors and stabilizers are two ways to modulate PPIs. Some of these modulators have been applied in the clinic, some have entered clinical trials, and some have lead compounds that require further structural optimization. Although compounds such as trifluoroperazine and zampanolide exhibit PPIs stabilizing activity, the PPIs stabilizers’ development has not received sufficient attention as compared to the PPIs inhibitors’ development.63 The difficulties for the PPIs stabilizers’ development include the insufficient understanding of PPIs mechanisms, the poor chemical space performance of PPI stabilizers in existing small molecule libraries, and the extreme diversity of the PPIs stabilizers’ molecular structures make it difficult to establish the criteria to guide the design of new PPIs stabilizers. Most of the identified PPI stabilizers are natural products, only a few of them are synthesized through the rational design method. The HTS of the natural products that have PPI stabilization activities may be the direction of finding the lead compounds of PPI stabilizers.
Compared with the traditional small molecule inhibitors, peptides exhibit higher affinity and specificity, making it easier to bind with the target proteins. However, the peptide has two major problems when used as drugs: its instability under in vivo conditions and their poor membrane permeability. Fortunately, new technologies are available now to counter the two problems. To prevent the quick degradation of the peptides after entering the body, the chemical modifications can be applied to improve the stability of the peptides. Regarding the peptides’ poor membrane permeability issue, there is a class of short peptides that have been found in recent years to have the function of penetrating biomembrane and mediate transmembrane transduction of macromolecular substances.244 This brings significant progress to the development of intracellular peptides.
In recent years, remarkable progress has been made in the development of antibodies that regulate PPI, especially the monoclonal antibodies regulate PD-1/PD-L1 interaction. However, due to the high research cost, the instability, and potential severe immunogenic side effects of antibodies, more and more attention has been drawn to the peptides and small molecular inhibitors, especially the small molecular inhibitors. Compared with antibodies, the classic small molecule drugs have advantages such as lower research costs, diverse preparations, oral administration, and better tumor microenvironment penetration.
Decades ago, due to the limited understanding of the PPI properties and very limited available screening techniques by time, the modulation of PPIs has been recognized as one of the most challenging tasks in drug discovery for a long period of time. However, the rapid development of structural biology and the associated methodologies have helped us to understand the PPI properties to a level we could not imagine before. Besides, the rapid development of various high-throughput screening approaches also makes the quick screening of the PPI modulators possible. As a result, great progress has been made in the development of PPI modulators lately. In summary, opportunities and challenges coexist in the discovery of modulators targeting PPIs. In the future, with the emergence of new and better approaches to reveal the structures of protein complexes and the development of structural biology, it is believed that more PPI small molecule modulators will be developed and enter the clinic to benefit the patients.
Acknowledgements
This work was supported by the Key Research and Development Program of Science and Technology Department of Sichuan Province (2019YFS0514), the Chengdu Science and Technology Program (No. 2015-HM01-00463-SF), the State Administration of Traditional Chinese Medicine (JDZX2015210), and the Open Research Fund of Chengdu University of Traditional Chinese Medicine Key Laboratory of Systematic Research of Distinctive Chinese Medicine Resources in Southwest China (2018GZ2011005).
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Haiying Lu, Qiaodan Zhou, Jun He
Contributor Information
Cheng Peng, Email: pengchengchengdu@126.com.
Rongsheng Tong, Email: tongrs@126.com.
Jianyou Shi, Email: shijianyoude@126.com.
References
- 1.Ferrari, S., Pellati, F. & Costi, M. P. Disruption of Protein-Protein Interfaces (Springer, Berlin, 2013).
- 2.Stelzl U, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 3.Rual JF, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 4.Venkatesan K, et al. An empirical framework for binary interactome mapping. Nat. Methods. 2009;6:83–90. doi: 10.1038/nmeth.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koh GC, Porras P, Aranda B, Hermjakob H, Orchard SE. Analyzing protein-protein interaction networks. J. Proteome Res. 2012;11:2014–2031. doi: 10.1021/pr201211w. [DOI] [PubMed] [Google Scholar]
- 6.Arkin MR, Whitty A. The road less traveled: modulating signal transduction enzymes by inhibiting their protein-protein interactions. Curr. Opin. Chem. Biol. 2009;13:284–290. doi: 10.1016/j.cbpa.2009.05.125. [DOI] [PubMed] [Google Scholar]
- 7.Loregian A, Palu G. Disruption of protein-protein interactions: towards new targets for chemotherapy. J. Cell Physiol. 2005;204:750–762. doi: 10.1002/jcp.20356. [DOI] [PubMed] [Google Scholar]
- 8.Nero TL, Morton CJ, Holien JK, Wielens J, Parker MW. Oncogenic protein interfaces: small molecules, big challenges. Nat. Rev. Cancer. 2014;14:248–262. doi: 10.1038/nrc3690. [DOI] [PubMed] [Google Scholar]
- 9.White AW, Westwell AD, Brahemi G. Protein-protein interactions as targets for small-molecule therapeutics in cancer. Expert Rev. Mol. Med. 2008;10:e8. doi: 10.1017/S1462399408000641. [DOI] [PubMed] [Google Scholar]
- 10.Blazer LL, Neubig RR. Small molecule protein–protein interaction inhibitors as CNS therapeutic agents: current progress and future hurdles. Neuropsychopharmacology. 2008;34:126–141. doi: 10.1038/npp.2008.151. [DOI] [PubMed] [Google Scholar]
- 11.Rosell M, Fernandez-Recio J. Hot-spot analysis for drug discovery targeting protein-protein interactions. Expert Opin. Drug Discov. 2018;13:327–338. doi: 10.1080/17460441.2018.1430763. [DOI] [PubMed] [Google Scholar]
- 12.Milroy LG, Grossmann TN, Hennig S, Brunsveld L, Ottmann C. Modulators of protein-protein interactions. Chem. Rev. 2014;114:4695–4748. doi: 10.1021/cr400698c. [DOI] [PubMed] [Google Scholar]
- 13.Hill TA, Shepherd NE, Diness F, Fairlie DP. Constraining cyclic peptides to mimic protein structure motifs. Angew. Chem. Int. Ed. 2014;53:13020–13041. doi: 10.1002/anie.201401058. [DOI] [PubMed] [Google Scholar]
- 14.Nevola L, Giralt E. Modulating protein-protein interactions: the potential of peptides. Chem. Commun. 2015;51:3302–3315. doi: 10.1039/C4CC08565E. [DOI] [PubMed] [Google Scholar]
- 15.Scott DE, Bayly AR, Abell C, Skidmore J. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016;15:533–550. doi: 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- 16.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 17.Santos R, et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017;16:19–34. doi: 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coyne AG, Scott DE, Abell C. Drugging challenging targets using fragment-based approaches. Curr. Opin. Chem. Biol. 2010;14:299–307. doi: 10.1016/j.cbpa.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 19.Winter A, et al. Biophysical and computational fragment-based approaches to targeting protein-protein interactions: applications in structure-guided drug discovery. Q. Rev. Biophys. 2012;45:383–426. doi: 10.1017/S0033583512000108. [DOI] [PubMed] [Google Scholar]
- 20.Stumpf MPH, et al. Estimating the size of the human interactome. Proc. Natl Acad. Sci. 2008;105:6959–6964. doi: 10.1073/pnas.0708078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith MC, Gestwicki JE. Features of protein-protein interactions that translate into potent inhibitors: topology, surface area and affinity. Expert Rev. Mol. Med. 2012;14:e16. doi: 10.1017/erm.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng AC, et al. Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 2007;25:71–75. doi: 10.1038/nbt1273. [DOI] [PubMed] [Google Scholar]
- 23.Buchwald P. Small-molecule protein-protein interaction inhibitors: therapeutic potential in light of molecular size, chemical space, and ligand binding efficiency considerations. IUBMB Life. 2010;62:724–731. doi: 10.1002/iub.383. [DOI] [PubMed] [Google Scholar]
- 24.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat. Rev. Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 25.Diaz-Eufracio BI, Naveja JJ, Medina-Franco JL. Protein-protein interaction modulators for epigenetic therapies. Adv. Protein Chem. Struct. Biol. 2018;110:65–84. doi: 10.1016/bs.apcsb.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 26.Ivanov AA, Khuri FR, Fu H. Targeting protein-protein interactions as an anticancer strategy. Trends Pharmacol. Sci. 2013;34:393–400. doi: 10.1016/j.tips.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997;23:3–25. doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- 28.Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2009;49:223–241. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geppert T, Hoy B, Wessler S, Schneider G. Context-based identification of protein-protein interfaces and “hot-spot” residues. Chem. Biol. 2011;18:344–353. doi: 10.1016/j.chembiol.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 30.Moreira IS, Fernandes PA, Ramos MJ. Hot spots–a review of the protein-protein interface determinant amino-acid residues. Proteins. 2007;68:803–812. doi: 10.1002/prot.21396. [DOI] [PubMed] [Google Scholar]
- 31.Janin J, Bahadur RP, Chakrabarti P. Protein-protein interaction and quaternary structure. Q. Rev. Biophys. 2008;41:133–180. doi: 10.1017/S0033583508004708. [DOI] [PubMed] [Google Scholar]
- 32.Thorn KS, Bogan AA. ASEdb: a database of alanine mutations and their effects on the free energy of binding in protein interactions. Bioinformatics. 2001;17:284–285. doi: 10.1093/bioinformatics/17.3.284. [DOI] [PubMed] [Google Scholar]
- 33.Vassilev LT, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 34.Grasberger BL, et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J. Med. Chem. 2005;48:909–912. doi: 10.1021/jm049137g. [DOI] [PubMed] [Google Scholar]
- 35.Allen JG, et al. Discovery and optimization of chromenotriazolopyrimidines as potent inhibitors of the mouse double minute 2-tumor protein 53 protein-protein interaction. J. Med Chem. 2009;52:7044–7053. doi: 10.1021/jm900681h. [DOI] [PubMed] [Google Scholar]
- 36.Hajduk PJ, Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat. Rev. Drug Discov. 2007;6:211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 37.Silvestre HL, Blundell TL, Abell C, Ciulli A. Integrated biophysical approach to fragment screening and validation for fragment-based lead discovery. Proc. Natl Acad. Sci. USA. 2013;110:12984–12989. doi: 10.1073/pnas.1304045110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Magee TV. Progress in discovery of small-molecule modulators of protein-protein interactions via fragment screening. Bioorg. Med. Chem. Lett. 2015;25:2461–2468. doi: 10.1016/j.bmcl.2015.04.089. [DOI] [PubMed] [Google Scholar]
- 39.Rees DC, Congreve M, Murray CW, Carr R. Fragment-based lead discovery. Nat. Rev. Drug Disco. 2004;3:660–672. doi: 10.1038/nrd1467. [DOI] [PubMed] [Google Scholar]
- 40.Schuffenhauer A, et al. Library design for fragment based screening. Curr. Top. Med. Chem. 2005;5:751–762. doi: 10.2174/1568026054637700. [DOI] [PubMed] [Google Scholar]
- 41.Wu B, et al. HTS by NMR of combinatorial libraries: a fragment-based approach to ligand discovery. Chem. Biol. 2013;20:19–33. doi: 10.1016/j.chembiol.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lugovskoy AA, et al. A novel approach for characterizing protein ligand complexes: molecular basis for specificity of small-molecule Bcl-2 inhibitors. J. Am. Chem. Soc. 2002;124:1234–1240. doi: 10.1021/ja011239y. [DOI] [PubMed] [Google Scholar]
- 43.Chung CW, Dean AW, Woolven JM, Bamborough P. Fragment-based discovery of bromodomain inhibitors part 1: inhibitor binding modes and implications for lead discovery. J. Med. Chem. 2012;55:576–586. doi: 10.1021/jm201320w. [DOI] [PubMed] [Google Scholar]
- 44.Sheng C, Dong G, Miao Z, Zhang W, Wang W. State-of-the-art strategies for targeting protein-protein interactions by small-molecule inhibitors. Chem. Soc. Rev. 2015;44:8238–8259. doi: 10.1039/C5CS00252D. [DOI] [PubMed] [Google Scholar]
- 45.Buckley DL, et al. Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1alpha. Angew. Chem. Int. Ed. 2012;51:11463–11467. doi: 10.1002/anie.201206231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buckley DL, et al. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J. Am. Chem. Soc. 2012;134:4465–4468. doi: 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mason JM. Design and development of peptides and peptide mimetics as antagonists for therapeutic intervention. Future Med. Chem. 2010;2:1813–1822. doi: 10.4155/fmc.10.259. [DOI] [PubMed] [Google Scholar]
- 48.Bullock BN, Jochim AL, Arora PS. Assessing helical protein interfaces for inhibitor design. J. Am. Chem. Soc. 2011;133:14220–14223. doi: 10.1021/ja206074j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yap JL, et al. Pharmacophore identification of c-Myc inhibitor 10074-G5. Bioorg. Med. Chem. Lett. 2013;23:370–374. doi: 10.1016/j.bmcl.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yin H, et al. Terphenyl-based Bak BH3 α-helical proteomimetics as low-molecular-weight antagonists of Bcl-x L. J. Am. Chem. Soc. 2005;127:10191–10196. doi: 10.1021/ja050122x. [DOI] [PubMed] [Google Scholar]
- 51.Cheng L, Yin H, Farooqi B. p53 α-Helix mimetics antagonize p53/MDM2 interaction and activate p53. Mol. Cancer Therap. 2005;4:1019–1025. doi: 10.1158/1535-7163.MCT-04-0342. [DOI] [PubMed] [Google Scholar]
- 52.Scheper J, et al. Protein-protein interaction antagonists as novel inhibitors of non-canonical polyubiquitylation. PLoS ONE. 2010;5:e11403. doi: 10.1371/journal.pone.0011403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lawrence HR, et al. Identification of a disruptor of the MDM2-p53 protein-protein interaction facilitated by high-throughput in silico docking. Bioorg. Med. Chem. Lett. 2009;19:3756–3759. doi: 10.1016/j.bmcl.2009.04.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tian W, et al. Structure-based discovery of a novel inhibitor targeting the beta-catenin/Tcf4 interaction. Biochemistry. 2012;51:724–731. doi: 10.1021/bi201428h. [DOI] [PubMed] [Google Scholar]
- 55.Lu S, Shen Q, Zhang J. Allosteric methods and their applications: facilitating the discovery of allosteric drugs and the investigation of allosteric mechanisms. Acc. Chem. Res. 2019;52:492–500. doi: 10.1021/acs.accounts.8b00570. [DOI] [PubMed] [Google Scholar]
- 56.Changeux JP. The concept of allosteric modulation: an overview. Drug Discov. Today Technol. 2013;10:e223–e228. doi: 10.1016/j.ddtec.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 57.Petta I, Lievens S, Libert C, Tavernier J, De Bosscher K. Modulation of protein–protein interactions for the development of novel therapeutics. Mol. Ther. 2016;24:707–718. doi: 10.1038/mt.2015.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cossins BP, Lawson ADG. Small molecule targeting of protein-protein interactions through allosteric modulation of dynamics. Molecules. 2015;20:16435–16445. doi: 10.3390/molecules200916435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang N, Lodge JM, Fierke CA, Mapp AK. Dissecting allosteric effects of activator-coactivator complexes using a covalent small molecule ligand. Proc. Natl Acad. Sci. USA. 2014;111:12061–12066. doi: 10.1073/pnas.1406033111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fischer G, Rossmann M, Hyvonen M. Alternative modulation of protein-protein interactions by small molecules. Curr. Opin. Biotechnol. 2015;35:78–85. doi: 10.1016/j.copbio.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thiel P, Kaiser M, Ottmann C. Small-molecule stabilization of protein-protein interactions: an underestimated concept in drug discovery? Angew. Chem. Int Ed. Engl. 2012;51:2012–2018. doi: 10.1002/anie.201107616. [DOI] [PubMed] [Google Scholar]
- 62.Zarzycka B, et al. Stabilization of protein-protein interaction complexes through small molecules. Drug Discov. Today. 2016;21:48–57. doi: 10.1016/j.drudis.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 63.Gestwicki J, P M. Chemical control over protein-protein interactions: beyond inhibitors. Combin. Chem. High. Throughput Screen. 2007;10:667–675. doi: 10.2174/138620707782507296. [DOI] [PubMed] [Google Scholar]
- 64.Jesus Perez de Vega M, M M-M, R G-M. Modulation of protein-protein interactions by stabilizing/mimicking protein secondary structure elements. Curr. Top. Med. Chem. 2007;7:33–62. doi: 10.2174/156802607779318325. [DOI] [PubMed] [Google Scholar]
- 65.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat. Rev. Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 66.Feki A, Irminger-Finger I. Mutational spectrum of p53 mutations in primary breast and ovarian tumors. Crit. Rev. Oncol. Hematol. 2004;52:103–116. doi: 10.1016/j.critrevonc.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 67.Wang X, Jiang X. Mdm2 and MdmX partner to regulate p53. FEBS Lett. 2012;586:1390–1396. doi: 10.1016/j.febslet.2012.02.049. [DOI] [PubMed] [Google Scholar]
- 68.Shangary S, Wang S. Targeting the MDM2-p53 interaction for cancer therapy. Clin. Cancer Res. 2008;14:5318–5324. doi: 10.1158/1078-0432.CCR-07-5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shangary S, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl Acad. Sci. USA. 2008;105:3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thangudu RR, Bryant SH, Panchenko AR, Madej T. Modulating protein-protein interactions with small molecules: the importance of binding hotspots. J. Mol. Biol. 2012;415:443–453. doi: 10.1016/j.jmb.2011.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen L, et al. P53 alpha-helix mimetics antagonize p53/MDM2 interaction and activate p53. Mol. Cancer Therap. 2005;4:1019–1025. doi: 10.1158/1535-7163.MCT-04-0342. [DOI] [PubMed] [Google Scholar]
- 72.Ding K, et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2006;49:3432–3435. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 73.Vu B, et al. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 2013;4:466–469. doi: 10.1021/ml4000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chang YS, et al. Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl Acad. Sci. USA. 2013;110:E3445–E3454. doi: 10.1073/pnas.1303002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li X, Liu R, Fang H. Bcl-2: research progress from target to launched drug. Acta Pharma. Sin. 2018;53:509–517. [Google Scholar]
- 76.Moldoveanu T, Follis AV, Kriwacki RW, Green DR. Many players in BCL-2 family affairs. Trends Biochem. Sci. 2014;39:101–111. doi: 10.1016/j.tibs.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 78.Cory S, Adams JM. Killing cancer cells by flipping the Bcl-2/Bax switch. Cancer Cell. 2005;8:5–6. doi: 10.1016/j.ccr.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 79.Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 80.Vogler M, Dinsdale D, Dyer MJ, Cohen GM. Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell Death Differ. 2009;16:360–367. doi: 10.1038/cdd.2008.137. [DOI] [PubMed] [Google Scholar]
- 81.Billard C. Design of novel BH3 mimetics for the treatment of chronic lymphocytic leukemia. Leukemia. 2012;26:2032–2038. doi: 10.1038/leu.2012.88. [DOI] [PubMed] [Google Scholar]
- 82.Oltersdorf T, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 83.Yu Y, et al. ABT737 induces mitochondrial pathway apoptosis and mitophagy by regulating DRP1-dependent mitochondrial fission in human ovarian cancer cells. Biomed. Pharmacother. 2017;96:22–29. doi: 10.1016/j.biopha.2017.09.111. [DOI] [PubMed] [Google Scholar]
- 84.Paoluzzi L, et al. The BH3-only mimetic ABT-737 synergizes the antineoplastic activity of proteasome inhibitors in lymphoid malignancies. Blood. 2008;112:2906–2916. doi: 10.1182/blood-2007-12-130781. [DOI] [PubMed] [Google Scholar]
- 85.Park CM, et al. Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins. J. Med. Chem. 2008;51:6902–6915. doi: 10.1021/jm800669s. [DOI] [PubMed] [Google Scholar]
- 86.Tse C, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 87.Rudin CM, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin. Cancer Res. 2012;18:3163–3169. doi: 10.1158/1078-0432.CCR-11-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Souers AJ, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 89.Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2018;17:197–223. doi: 10.1038/nrd.2017.227. [DOI] [PubMed] [Google Scholar]
- 90.Cummins JM, et al. X-linked inhibitor of apoptosis protein (XIAP) is a nonredundant modulator of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis in human cancer cells. Cancer Res. 2004;64:3006–3008. doi: 10.1158/0008-5472.CAN-04-0046. [DOI] [PubMed] [Google Scholar]
- 91.Rumble JM, Duckett CS. Diverse functions within the IAP family. J. Cell Sci. 2008;121:3505–3507. doi: 10.1242/jcs.040303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gyrd-Hansen M, Meier P. IAPs: from caspase inhibitors to modulators of NF-κB, inflammation and cancer. Nat. Rev. Cancer. 2010;10:561–574. doi: 10.1038/nrc2889. [DOI] [PubMed] [Google Scholar]
- 93.Hunter AM, LaCasse EC, Korneluk RG. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis. 2007;12:1543–1568. doi: 10.1007/s10495-007-0087-3. [DOI] [PubMed] [Google Scholar]
- 94.Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25:4798–4811. doi: 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- 95.Salvesen GS, Duckett CS. IAP proteins: blocking the road to death’s door. Nat. Rev. Mol. Cell Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 96.Chai J, et al. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature. 2000;406:855–862. doi: 10.1038/35022514. [DOI] [PubMed] [Google Scholar]
- 97.Shiozaki EN, Shi Y. Caspases, IAPs and Smac/DIABLO: mechanisms from structural biology. Trends Biochem. Sci. 2004;29:486–494. doi: 10.1016/j.tibs.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 98.Shiozaki EN, et al. Mechanism of XIAP-mediated inhibition of caspase-9. Mol. Cell. 2003;11:519–527. doi: 10.1016/S1097-2765(03)00054-6. [DOI] [PubMed] [Google Scholar]
- 99.Wu G, et al. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408:1008–1012. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 100.Srinivasula SM, et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature. 2001;410:112–116. doi: 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- 101.Flygare JA, et al. Discovery of a potent small-molecule antagonist of inhibitor of apoptosis (IAP) proteins and clinical candidate for the treatment of cancer (GDC-0152) J. Med. Chem. 2012;55:4101–4113. doi: 10.1021/jm300060k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tolcher AW, et al. A phase I dose-escalation study evaluating the safety tolerability and pharmacokinetics of CUDC-427, a potent, oral, monovalent IAP antagonist, in patients with refractory solid tumors. Clin. Cancer Res. 2016;22:4567–4573. doi: 10.1158/1078-0432.CCR-16-0308. [DOI] [PubMed] [Google Scholar]
- 103.Bardia A, et al. Paclitaxel with inhibitor of apoptosis antagonist, LCL161, for localized triple-negative breast cancer, prospectively stratified by gene signature in a biomarker-driven neoadjuvant trial. J. Clin. Oncol. 2018;36:3126–3133. doi: 10.1200/JCO.2017.74.8392. [DOI] [PubMed] [Google Scholar]
- 104.Isaacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell. 2003;3:213–217. doi: 10.1016/S1535-6108(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 105.Schopf FH, Biebl MM, Buchner J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017;18:345–360. doi: 10.1038/nrm.2017.20. [DOI] [PubMed] [Google Scholar]
- 106.Li J, Soroka J, Buchner J. The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta. 2012;1823:624–635. doi: 10.1016/j.bbamcr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 107.Chen X, et al. DCZ3112, a novel Hsp90 inhibitor, exerts potent antitumor activity against HER2-positive breast cancer through disruption of Hsp90-Cdc37 interaction. Cancer Lett. 2018;434:70–80. doi: 10.1016/j.canlet.2018.07.012. [DOI] [PubMed] [Google Scholar]
- 108.Porter JR, Fritz CC, Depew KM. Discovery and development of Hsp90 inhibitors: a promising pathway for cancer therapy. Curr. Opin. Chem. Biol. 2010;14:412–420. doi: 10.1016/j.cbpa.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 109.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin. Cancer Res. 2012;18:64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Taipale M, et al. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell. 2012;150:987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Patel HJ, Modi S, Chiosis G, Taldone T. Advances in the discovery and development of heat-shock protein 90 inhibitors for cancer treatment. Expert Opin. Drug Discov. 2011;6:559–587. doi: 10.1517/17460441.2011.563296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wandinger SK, Richter K, Buchner J. The Hsp90 chaperone machinery. J. Biol. Chem. 2008;283:18473–18477. doi: 10.1074/jbc.R800007200. [DOI] [PubMed] [Google Scholar]
- 113.Hainzl O, Lapina MC, Buchner J, Richter K. The charged linker region is an important regulator of Hsp90 function. J. Biol. Chem. 2009;284:22559–22567. doi: 10.1074/jbc.M109.031658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu. Rev. Biochem. 2006;75:271–294. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- 115.Garg G, Khandelwal A, Blagg BS. Anticancer inhibitors of Hsp90 function: beyond the usual suspects. Adv. Cancer Res. 2016;129:51–88. doi: 10.1016/bs.acr.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rajan A, et al. A phase I study of PF-04929113 (SNX-5422), an orally bioavailable heat shock protein 90 inhibitor, in patients with refractory solid tumor malignancies and lymphomas. Clin. Cancer Res. 2011;17:6831–6839. doi: 10.1158/1078-0432.CCR-11-0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Solit DB, Chiosis G. Development and application of Hsp90 inhibitors. Drug Discov. Today. 2008;13:38–43. doi: 10.1016/j.drudis.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 118.Pearl LH, Prodromou C, Workman P. The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem. J. 2008;410:439–453. doi: 10.1042/BJ20071640. [DOI] [PubMed] [Google Scholar]
- 119.Gray PJ, Prince T, Cheng J, Stevenson MA, Calderwood SK. Targeting the oncogene and kinome chaperone CDC37. Nat. Rev. Cancer. 2008;8:491–495. doi: 10.1038/nrc2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Roe SM, et al. The mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50cdc37. Cell. 2004;116:87–98. doi: 10.1016/S0092-8674(03)01027-4. [DOI] [PubMed] [Google Scholar]
- 121.Sreeramulu S, et al. The human Cdc37.Hsp90 complex studied by heteronuclear NMR spectroscopy. J. Biol. Chem. 2009;284:3885–3896. doi: 10.1074/jbc.M806715200. [DOI] [PubMed] [Google Scholar]
- 122.Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kress TR, Sabo A, Amati B. MYC: connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer. 2015;15:593–607. doi: 10.1038/nrc3984. [DOI] [PubMed] [Google Scholar]
- 124.Sabo A, et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature. 2014;511:488–492. doi: 10.1038/nature13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012;18:5546–5553. doi: 10.1158/1078-0432.CCR-12-0977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nair SK, Burley SK. X-ray structures of Myc-Max and Mad-Max recognizing DNA: molecular bases of regulation by proto-oncogenic transcription factors. Cell. 2003;112:193–205. doi: 10.1016/S0092-8674(02)01284-9. [DOI] [PubMed] [Google Scholar]
- 127.Follis AV, Hammoudeh DI, Wang H, Prochownik EV, Metallo SJ. Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chem. Biol. 2008;15:1149–1155. doi: 10.1016/j.chembiol.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 128.Hammoudeh DI, Follis AV, Prochownik EV, Metallo SJ. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J. Am. Chem. Soc. 2009;131:7390–7401. doi: 10.1021/ja900616b. [DOI] [PubMed] [Google Scholar]
- 129.Castell A, et al. A selective high affinity MYC-binding compound inhibits MYC:MAX interaction and MYC-dependent tumor cell proliferation. Sci. Rep. 2018;8:10064–10081. doi: 10.1038/s41598-018-28107-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chauhan J, et al. Discovery of methyl 4′-methyl-5-(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)-[1,1′-biphenyl]-3-carboxylate, an improved small-molecule inhibitor of c-Myc-max dimerization. Chem. Med. Chem. 2014;9:2274–2285. doi: 10.1002/cmdc.201402189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang H, et al. Disruption of Myc-Max heterodimerization with improved, cell-penetrating analogs of the small molecule 10074-G5. Oncotarget. 2013;4:936–947. doi: 10.18632/oncotarget.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat. Rev. Drug Discov. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kang HM, et al. Inhibitory activity of diarylheptanoids on farnesyl protein transferase. Nat. Prod. Res. 2004;18:295–299. doi: 10.1080/14786410310001620691. [DOI] [PubMed] [Google Scholar]
- 135.Schmick M, et al. KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell. 2014;157:459–471. doi: 10.1016/j.cell.2014.02.051. [DOI] [PubMed] [Google Scholar]
- 136.Chandra A, et al. The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat. Cell Biol. 2011;14:148–158. doi: 10.1038/ncb2394. [DOI] [PubMed] [Google Scholar]
- 137.Zimmermann G, et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–642. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 138.Zhang H, et al. Deletion of PrBP/delta impedes transport of GRK1 and PDE6 catalytic subunits to photoreceptor outer segments. Proc. Natl Acad. Sci. USA. 2007;104:8857–8862. doi: 10.1073/pnas.0701681104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Johnson L, et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997;11:2468–2481. doi: 10.1101/gad.11.19.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cox AD, Der CJ, Philips MR. Targeting RAS membrane association: back to the future for anti-RAS drug discovery? Clin. Cancer Res. 2015;21:1819–1827. doi: 10.1158/1078-0432.CCR-14-3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jiang Y, et al. Structural biology-inspired discovery of novel KRAS-PDEdelta inhibitors. J. Med. Chem. 2017;60:9400–9406. doi: 10.1021/acs.jmedchem.7b01243. [DOI] [PubMed] [Google Scholar]
- 142.Kim J, et al. Low-dielectric-constant polyimide aerogel composite films with low water uptake. Polym. J. 2016;48:829–834. doi: 10.1038/pj.2016.37. [DOI] [Google Scholar]
- 143.Zimmermann G, et al. Structure guided design and kinetic analysis of highly potent benzimidazole inhibitors targeting the PDEdelta prenyl binding site. J. Med. Chem. 2014;57:5435–5448. doi: 10.1021/jm500632s. [DOI] [PubMed] [Google Scholar]
- 144.Murarka S, et al. Development of pyridazinone chemotypes targeting the PDEdelta prenyl binding site. Chemistry. 2017;23:6083–6093. doi: 10.1002/chem.201603222. [DOI] [PubMed] [Google Scholar]
- 145.Martin-Gago P, Fansa EK, Wittinghofer A, Waldmann H. Structure-based development of PDEdelta inhibitors. Biol. Chem. 2017;398:535–545. doi: 10.1515/hsz-2016-0272. [DOI] [PubMed] [Google Scholar]
- 146.Martin-Gago P, et al. A PDE6delta-KRas inhibitor chemotype with up to seven H-bonds and picomolar affinity that prevents efficient inhibitor release by Arl2. Angew. Chem. Int. Ed. 2017;56:2423–2428. doi: 10.1002/anie.201610957. [DOI] [PubMed] [Google Scholar]
- 147.Chen L, Zhuang C, Lu J, Jiang Y, Sheng C. Discovery of novel KRAS-PDEdelta inhibitors by fragment-based drug design. J. Med Chem. 2018;61:2604–2610. doi: 10.1021/acs.jmedchem.8b00057. [DOI] [PubMed] [Google Scholar]
- 148.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Van Kooten C. Immune regulation by CD40-CD40-L interactions - 2 Y2K update. Front. Biosci. 2000;5:880–893. doi: 10.2741/A557. [DOI] [PubMed] [Google Scholar]
- 150.O’Sullivan B, Thomas R. Recent advances on the role of CD40 and dendritic cells in immunity and tolerance. Curr. Opin. Hematol. 2003;10:272–278. doi: 10.1097/00062752-200307000-00004. [DOI] [PubMed] [Google Scholar]
- 151.Meabed MH, Taha GM, Mohamed SO, El-Hadidy KS. Autoimmune thrombocytopenia: flow cytometric determination of platelet-associated CD154/CD40L and CD40 on peripheral blood T and B lymphocytes. Hematology. 2007;12:301–307. doi: 10.1080/10245330701383957. [DOI] [PubMed] [Google Scholar]
- 152.Elgueta R, et al. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009;229:152–172. doi: 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wagner DH, et al. Expression of CD40 identifies a unique pathogenic T cell population in type 1 diabetes. Proc. Natl Acad. Sci. USA. 2002;99:3782–3787. doi: 10.1073/pnas.052247099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pamukcu B, Lip GY, Snezhitskiy V, Shantsila E. The CD40-CD40L system in cardiovascular disease. Ann. Med. 2011;43:331–340. doi: 10.3109/07853890.2010.546362. [DOI] [PubMed] [Google Scholar]
- 155.Senhaji N, Kojok K, Darif Y, Fadainia C, Zaid Y. The contribution of CD40/CD40L axis in inflammatory bowel disease: an update. Front. Immunol. 2015;6:529. doi: 10.3389/fimmu.2015.00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Croft M, Benedict CA, Ware CF. Clinical targeting of the TNF and TNFR superfamilies. Nat. Rev. Drug Discov. 2013;12:147–168. doi: 10.1038/nrd3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Oflazoglu E, et al. Macrophages and Fc-receptor interactions contribute to the antitumour activities of the anti-CD40 antibody SGN-40. Br. J. Cancer. 2009;100:113–117. doi: 10.1038/sj.bjc.6604812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kawai T, Andrews D, Colvin RB, Sachs DH, Cosimi AB. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat. Med. 2000;6:114. doi: 10.1038/72162. [DOI] [PubMed] [Google Scholar]
- 159.Boumpas DT, et al. A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 2003;48:719–727. doi: 10.1002/art.10856. [DOI] [PubMed] [Google Scholar]
- 160.Schulze-Neick I, et al. End-stage heart failure with pulmonary hypertension: levosimendan to evaluate for heart transplantation alone versus combined heart-lung transplantation. Transplantation. 2004;78:1237–1238. doi: 10.1097/01.TP.0000137790.63159.48. [DOI] [PubMed] [Google Scholar]
- 161.Mirabet M, Barrabes JA, Quiroga A, Garcia-Dorado D. Platelet pro-aggregatory effects of CD40L monoclonal antibody. Mol. Immunol. 2008;45:937–944. doi: 10.1016/j.molimm.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 162.Margolles-Clark E, Umland O, Kenyon NS, Ricordi C, Buchwald P. Small-molecule costimulatory blockade: organic dye inhibitors of the CD40–CD154 interaction. J. Mol. Med. 2009;87:1133–1143. doi: 10.1007/s00109-009-0519-3. [DOI] [PubMed] [Google Scholar]
- 163.Margolles-Clark E, Kenyon NS, Ricordi C, Buchwald P. Effective and specific inhibition of the CD40-CD154 costimulatory interaction by a naphthalenesulphonic acid derivative. Chem. Biol. Drug Des. 2010;76:305–313. doi: 10.1111/j.1747-0285.2010.01014.x. [DOI] [PubMed] [Google Scholar]
- 164.Chen J, et al. Small-Molecule Inhibitors of the CD40-CD40L costimulatory protein-protein interaction. J. Med Chem. 2017;60:8906–8922. doi: 10.1021/acs.jmedchem.7b01154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat. Rev. Cancer. 2008;8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. Mol. Cell Biol. 2013;14:369–381. doi: 10.1038/nrm3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chaugule VK, Walden H. Specificity and disease in the ubiquitin system. Biochem. Soc. Trans. 2016;44:212–227. doi: 10.1042/BST20150209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Heo J, Eki R, Abbas T. Deregulation of F-box proteins and its consequence on cancer development, progression and metastasis. Semin. Cancer Biol. 2016;36:33–51. doi: 10.1016/j.semcancer.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Skaar JR, Pagan JK, Pagano M. SCF ubiquitin ligase-targeted therapies. Nat. Rev. Drug Discov. 2014;13:889–903. doi: 10.1038/nrd4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Z H. E3 ubiquitin ligase Skp2 as an attractive target in cancer therapy. Front. Biosci. 2015;20:474–490. doi: 10.2741/4320. [DOI] [PubMed] [Google Scholar]
- 171.Hershko DD. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer. 2008;112:1415–1424. doi: 10.1002/cncr.23317. [DOI] [PubMed] [Google Scholar]
- 172.Zheng N, et al. Structure of the Cul1–Rbx1–Skp1–F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–709. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
- 173.Chan CH, et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell. 2013;154:556–568. doi: 10.1016/j.cell.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tkachev VO, Menshchikova EB, Zenkov NK. Mechanism of the Nrf2/Keap1/ARE signaling system. Biochemistry. 2011;76:407–422. doi: 10.1134/s0006297911040031. [DOI] [PubMed] [Google Scholar]
- 175.Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 2006;38:769–789. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- 176.Padmanabhan B, et al. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol. Cell. 2006;21:689–700. doi: 10.1016/j.molcel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 177.Hong F, Sekhar KR, Freeman ML, Liebler DC. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J. Biol. Chem. 2005;280:31768–31775. doi: 10.1074/jbc.M503346200. [DOI] [PubMed] [Google Scholar]
- 178.Zhang DD. The Nrf2-Keap1-ARE signaling pathway: the regulation and dual function of Nrf2 in cancer. Antioxid. Redox Signal. 2010;13:1623–1626. doi: 10.1089/ars.2010.3301. [DOI] [PubMed] [Google Scholar]
- 179.Magesh S, Chen Y, Hu L. Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med. Res. Rev. 2012;32:687–726. doi: 10.1002/med.21257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lo SC, Li X, Henzl MT, Beamer LJ, Hannink M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006;25:3605–3617. doi: 10.1038/sj.emboj.7601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hancock R, et al. Peptide inhibitors of the Keap1-Nrf2 protein-protein interaction. Free Radic. Biol. Med. 2012;52:444–451. doi: 10.1016/j.freeradbiomed.2011.10.486. [DOI] [PubMed] [Google Scholar]
- 182.Hancock R, Schaap M, Pfister H, Wells G. Peptide inhibitors of the Keap1-Nrf2 protein-protein interaction with improved binding and cellular activity. Org. Biomol. Chem. 2013;11:3553–3557. doi: 10.1039/c3ob40249e. [DOI] [PubMed] [Google Scholar]
- 183.Wells G. Peptide and small molecule inhibitors of the Keap1-Nrf2 protein-protein interaction. Biochem. Soc. Trans. 2015;43:674–679. doi: 10.1042/BST20150051. [DOI] [PubMed] [Google Scholar]
- 184.Georgakopoulos ND, Talapatra SK, Gatliff J, Kozielski F, Wells G. Modified peptide inhibitors of the Keap1-Nrf2 protein-protein interaction incorporating unnatural amino acids. ChemBioChem. 2018;19:1810–1816. doi: 10.1002/cbic.201800170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hu L, et al. Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorg. Med Chem. Lett. 2013;23:3039–3043. doi: 10.1016/j.bmcl.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Inoyama D, et al. Optimization of fluorescently labeled Nrf2 peptide probes and the development of a fluorescence polarization assay for the discovery of inhibitors of Keap1-Nrf2 interaction. J. Biomol. Screen. 2012;17:435–447. doi: 10.1177/1087057111430124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Steel R, Cowan J, Payerne E, O’Connell MA, Searcey M. Anti-inflammatory effect of a cell-penetrating peptide targeting the Nrf2/Keap1 interaction. ACS Med. Chem. Lett. 2012;3:407–410. doi: 10.1021/ml300041g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Jiang CS, et al. Identification of a novel small-molecule Keap1-Nrf2 PPI inhibitor with cytoprotective effects on LPS-induced cardiomyopathy. J. Enzym. Inhib. Med. Chem. 2018;33:833–841. doi: 10.1080/14756366.2018.1461856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Davies TG, et al. Monoacidic inhibitors of the Kelch-like ECH-associated protein 1: nuclear factor erythroid 2-related factor 2 (KEAP1: NRF2) protein–protein interaction with high cell potency identified by fragment-based discovery. J. Med. Chem. 2016;59:3991–4006. doi: 10.1021/acs.jmedchem.6b00228. [DOI] [PubMed] [Google Scholar]
- 190.Jiang ZY, et al. Discovery of potent Keap1-Nrf2 protein-protein interaction inhibitor based on molecular binding determinants analysis. J. Med. Chem. 2014;57:2736–2745. doi: 10.1021/jm5000529. [DOI] [PubMed] [Google Scholar]
- 191.Jiang ZY, et al. Structure–activity and structure–property relationship and exploratory in vivo evaluation of the nanomolar Keap1–Nrf2 protein–protein interaction inhibitor. J. Med. Chem. 2015;58:6410–6421. doi: 10.1021/acs.jmedchem.5b00185. [DOI] [PubMed] [Google Scholar]
- 192.Zhuang C, Narayanapillai S, Zhang W, Sham YY, Xing C. Rapid identification of Keap1–Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J. Med. Chem. 2014;57:1121–1126. doi: 10.1021/jm4017174. [DOI] [PubMed] [Google Scholar]
- 193.Bertrand HC, et al. Design, synthesis, and evaluation of triazole derivatives that induce Nrf2 dependent gene products and inhibit the Keap1–Nrf2 protein–protein interaction. J. Med. Chem. 2015;58:7186–7194. doi: 10.1021/acs.jmedchem.5b00602. [DOI] [PubMed] [Google Scholar]
- 194.Sun HP, et al. Novel protein–protein interaction inhibitor of Nrf2–Keap1 discovered by structure-based virtual screening. Med. Chem. Commun. 2014;5:93–98. doi: 10.1039/C3MD00240C. [DOI] [Google Scholar]
- 195.Marcotte D, et al. Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorg. Med. Chem. 2013;21:4011–4019. doi: 10.1016/j.bmc.2013.04.019. [DOI] [PubMed] [Google Scholar]
- 196.Dermani FK, Samadi P, Rahmani G, Kohlan AK, Najafi R. PD-1/PD-L1 immune checkpoint: potential target for cancer therapy. J. Cell Physiol. 2019;234:1313–1325. doi: 10.1002/jcp.27172. [DOI] [PubMed] [Google Scholar]
- 197.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Socinski MA, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N. Engl. J. Med. 2018;378:2288–2301. doi: 10.1056/NEJMoa1716948. [DOI] [PubMed] [Google Scholar]
- 199.Antonia SJ, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N. Engl. J. Med. 2017;377:1919–1929. doi: 10.1056/NEJMoa1709937. [DOI] [PubMed] [Google Scholar]
- 200.Garon EB, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015;372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 201.Ferris RL, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2016;375:1856–1867. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Dirix LY, et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1b JAVELIN Solid Tumor study. Breast Cancer Res. Treat. 2018;167:671–686. doi: 10.1007/s10549-017-4537-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Mullard A. 2014 FDA drug approvals. Nat. Rev. Drug Discov. 2015;14:77–81. doi: 10.1038/nrd4545. [DOI] [PubMed] [Google Scholar]
- 204.Naidoo J, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol. 2015;26:2375–2391. doi: 10.1093/annonc/mdv383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hwang SJ, et al. Bullous pemphigoid, an autoantibody-mediated disease, is a novel immune-related adverse event in patients treated with anti-programmed cell death 1 antibodies. Melanoma Res. 2016;26:413–416. doi: 10.1097/CMR.0000000000000260. [DOI] [PubMed] [Google Scholar]
- 206.Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug Discov. Today. 2010;15:40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 207.Chang HN, et al. Blocking of the PD-1/PD-L1 interaction by a D-peptide antagonist for cancer immunotherapy. Angew. Chem. 2015;54:11926–11930. doi: 10.1002/ange.201506225. [DOI] [PubMed] [Google Scholar]
- 208.Sasikumar, P. G. N. et al. Immunosuppression modulating compounds. WO2011161699 (2011).
- 209.Zak KM, et al. Structure of the complex of human programmed death 1, PD-1, and its ligand PD-L1. Structure. 2015;23:2341–2348. doi: 10.1016/j.str.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Chupak, L. S. & Zheng, X. Compounds useful as immunomodulators. WO2015034820A1 (2015).
- 211.Chupak, S. et al. Preparation of substituted 2,4-dihydroxybenzylamines as immunomodulators. WO2015160641A2 (2015).
- 212.Yeung, K.-S. et al. Compounds useful as immunomodulators. WO2017066227 (2017).
- 213.Yeung, K.-S. et al. Compounds useful as immunomodulators. US patent WO2018044963A1 (2018).
- 214.Aitken A. 14-3-3 proteins: a historic overview. Semin. Cancer Biol. 2006;16:162–172. doi: 10.1016/j.semcancer.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 215.Fu H, Subramanian RR, Masters SC. 14-3-3 Proteins: structure, function, and regulation. Annu Rev. Pharm. Toxicol. 2000;40:617–647. doi: 10.1146/annurev.pharmtox.40.1.617. [DOI] [PubMed] [Google Scholar]
- 216.Ottmann C. Small-molecule modulators of 14-3-3 protein-protein interactions. Bioorg. Med Chem. 2013;21:4058–4062. doi: 10.1016/j.bmc.2012.11.028. [DOI] [PubMed] [Google Scholar]
- 217.Xiaowen Y, et al. Structural basis for protein–protein interactions in the 14-3-3 protein family. Proc. Natl Acad. Sci. USA. 2006;103:17237–17242. doi: 10.1073/pnas.0605779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Hermeking H, Benzinger A. 14-3-3 proteins in cell cycle regulation. Semin. Cancer Biol. 2006;16:183–192. doi: 10.1016/j.semcancer.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 219.Berg D, Holzmann C, Riess O. 14-3-3 proteins in the nervous system. Nat. Rev. Neurosci. 2003;4:752–762. doi: 10.1038/nrn1197. [DOI] [PubMed] [Google Scholar]
- 220.Ottmann C, et al. Phosphorylation-independent interaction between 14-3-3 and exoenzyme S: from structure to pathogenesis. EMBO J. 2007;26:902–913. doi: 10.1038/sj.emboj.7601530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Cau Y, Valensin D, Mori M, Draghi S, Botta M. Structure, function, involvement in diseases and targeting of 14-3-3 proteins: an update. Curr. Med. Chem. 2018;25:5–21. doi: 10.2174/0929867324666170426095015. [DOI] [PubMed] [Google Scholar]
- 222.Hawech P. 14-3-3 proteins—an update. Cell Res. 2005;15:228–236. doi: 10.1038/sj.cr.7290291. [DOI] [PubMed] [Google Scholar]
- 223.Ottmann C, et al. Structure of a 14-3-3 coordinated hexamer of the plant plasma membrane H+-ATPase by combining X-ray crystallography and electron cryomicroscopy. Mol. Cell. 2007;25:427–440. doi: 10.1016/j.molcel.2006.12.017. [DOI] [PubMed] [Google Scholar]
- 224.Richter A, Rose R, Hedberg C, Waldmann H, Ottmann C. An optimised small-molecule stabiliser of the 14-3-3-PMA2 protein-protein interaction. Chem. Eur. J. 2012;18:6520–6527. doi: 10.1002/chem.201103761. [DOI] [PubMed] [Google Scholar]
- 225.Sedaghat F, Notopoulos A. S100 protein family and its application in clinical practice. Hippokratia. 2008;12:198–204. [PMC free article] [PubMed] [Google Scholar]
- 226.Marenholz I, Lovering RC, Heizmann CW. An update of the S100 nomenclature. Biochim Biophys. Acta. 2006;1763:1282–1283. doi: 10.1016/j.bbamcr.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 227.Kuberappa PH, Bagalad BS, Ananthaneni A, Kiresur MA, Srinivas GV. Certainty of S100 from physiology to pathology. J. Clin. Diagn. Res. 2016;10:ZE10–ZE15. doi: 10.7860/JCDR/2016/17949.8022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Malashkevich VN, et al. Structure of Ca2+-bound S100A4 and its interaction with peptides derived from nonmuscle myosin-IIA. Biochemistry. 2008;47:5111–5126. doi: 10.1021/bi702537s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Donato R, et al. Functions of S100 Proteins. Curr. Mol. Med. 2013;13:24–57. doi: 10.2174/156652413804486214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Schneider M, Hansen JL, Sheikh SP. S100A4: a common mediator of epithelial-mesenchymal transition, fibrosis and regeneration in diseases? J. Mol. Chem. 2008;86:507–522. doi: 10.1007/s00109-007-0301-3. [DOI] [PubMed] [Google Scholar]
- 231.Grigorian M, Ambartsumian N, Lukanidin E. Metastasis-inducing S100A4 protein: implication in non-malignant human pathologies. Curr. Mol. Med. 2008;8:492–496. doi: 10.2174/156652408785747942. [DOI] [PubMed] [Google Scholar]
- 232.Garrett SC, et al. A biosensor of S100A4 metastasis factor activation: inhibitor screening and cellular activation dynamics. Biochemistry. 2008;47:986–996. doi: 10.1021/bi7021624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Portela A, Digard P. The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. J. Gen. Virol. 2002;83:723–734. doi: 10.1099/0022-1317-83-4-723. [DOI] [PubMed] [Google Scholar]
- 234.Gerritza SW, et al. Inhibition of influenza virus replication via small molecules that induce the formation of higher-order nucleoprotein oligomers. Proc. Natl Acad. Sci. USA. 2011;108:15366–15371. doi: 10.1073/pnas.1107906108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Parker AL, Kavallaris M, McCarroll JA. Microtubules and their role in cellular stress in cancer. Front. Oncol. 2014;4:153–172. doi: 10.3389/fonc.2014.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 237.Alves RC, Fernandes RP, Eloy JO, Salgado HRN, Chorilli M. Characteristics, properties and analytical methods of paclitaxel: a review. Crit. Rev. Anal. Chem. 2018;48:110–118. doi: 10.1080/10408347.2017.1416283. [DOI] [PubMed] [Google Scholar]
- 238.Alushin GM, et al. High-resolution microtubule structures reveal the structural transitions in alphabeta-tubulin upon GTP hydrolysis. Cell. 2014;157:1117–1129. doi: 10.1016/j.cell.2014.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Field JJ, et al. Microtubule-stabilizing activity of zampanolide, a potent macrolide isolated from the Tongan marine sponge Cacospongia mycofijiensis. J. Med. Chem. 2009;52:7328–7332. doi: 10.1021/jm901249g. [DOI] [PubMed] [Google Scholar]
- 240.Prota AE, et al. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science. 2013;339:587–590. doi: 10.1126/science.1230582. [DOI] [PubMed] [Google Scholar]
- 241.Field JJ, et al. Zampanolide, a microtubule-stabilizing agent, is active in resistant cancer cells and inhibits cell migration. Int. J. Mol. Sci. 2017;18:971–989. doi: 10.3390/ijms18050971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Brown J, Horrocks MH. A sticky situation: aberrant protein-protein interactions in Parkinson’s disease. Semin. Cell Dev. Biol. 2018;99:65–77. doi: 10.1016/j.semcdb.2018.05.006. [DOI] [PubMed] [Google Scholar]
- 243.Ballatore C, et al. Modulation of protein-protein interactions as a therapeutic strategy for the treatment of neurodegenerative tauopathies. Curr. Top. Med. Chem. 2011;11:317–330. doi: 10.2174/156802611794072605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Philippe G, et al. Development of cell-penetrating peptide-based drug leads to inhibit MDMX:p53 and MDM2:p53 interactions. Biopolymers. 2016;106:853–863. doi: 10.1002/bip.22893. [DOI] [PubMed] [Google Scholar]
- 245.Lehmann C, Friess T, Birzele F, Kiialainen A, Dangl M. Superior anti-tumor activity of the MDM2 antagonist idasanutlin and the Bcl-2 inhibitor venetoclax in p53 wild-type acute myeloid leukemia models. J. Hematol. Oncol. 2016;9:50. doi: 10.1186/s13045-016-0280-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Sun D, et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2–p53 inhibitor in clinical development. J. Med. Chem. 2014;57:1454–1472. doi: 10.1021/jm401753e. [DOI] [PubMed] [Google Scholar]
- 247.Holzer P, et al. Discovery of a dihydroisoquinolinone derivative (NVP-CGM097): a highly potent and selective MDM2 inhibitor undergoing phase 1 clinical trials in p53wt tumors. J. Med. Chem. 2015;58:6348–6358. doi: 10.1021/acs.jmedchem.5b00810. [DOI] [PubMed] [Google Scholar]
- 248.Viktor A, et al. Reactivating TP53 signaling by the novel MDM2 inhibitor DS-3032b as a therapeutic option for high-risk neuroblastoma. Oncotarget. 2017;9:2304–2319. doi: 10.18632/oncotarget.23409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.De Weger V, et al. A first-in-human (FIH) safety and pharmacological study of SAR405838, a novel HDM2 antagonist, in patients with solid malignancies. Eur. J. Cancer. 2014;50:121–122. doi: 10.1016/S0959-8049(14)70504-0. [DOI] [Google Scholar]
- 250.Korycka-Wolowiec A, Wolowiec D, Kubiak-Mlonka A, Robak T. Venetoclax in the treatment of chronic lymphocytic leukemia. Expert Opin. Drug Metab. Toxicol. 2019;15:353–366. doi: 10.1080/17425255.2019.1606211. [DOI] [PubMed] [Google Scholar]
- 251.West AC, et al. The SMAC mimetic, LCL-161, reduces survival in aggressive MYC-driven lymphoma while promoting susceptibility to endotoxic shock. Oncogenesis. 2016;5:e216. doi: 10.1038/oncsis.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Benetatos CA, et al. Birinapant (TL32711), a bivalent SMAC mimetic, targets TRAF2-associated cIAPs, abrogates TNF-induced NF-kappaB activation, and is active in patient-derived xenograft models. Mol. Cancer Therap. 2014;13:867–879. doi: 10.1158/1535-7163.MCT-13-0798. [DOI] [PubMed] [Google Scholar]
- 253.Ward GA, et al. ASTX660, a novel non-peptidomimetic antagonist of cIAP1/2 and XIAP, potently induces TNFalpha-dependent apoptosis in cancer cell lines and inhibits tumor growth. Mol. Cancer Therap. 2018;17:1381–1391. doi: 10.1158/1535-7163.MCT-17-0848. [DOI] [PubMed] [Google Scholar]
- 254.Wong H, et al. Learning and confirming with preclinical studies: modeling and simulation in the discovery of GDC-0917, an inhibitor of apoptosis proteins antagonist. Drug Metab. Dispos. 2013;41:2104–2113. doi: 10.1124/dmd.113.053926. [DOI] [PubMed] [Google Scholar]
- 255.Musielak B, et al. CA-170 – a potent small-molecule PD-L1 inhibitor or not? Molecules. 2019;24:2804. doi: 10.3390/molecules24152804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Dorr P, et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005;49:4721–4732. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Perez VL, Pflugfelder SC, Zhang S, Shojaei A, Haque R. Lifitegrast, a novel integrin antagonist for treatment of dry eye disease. Ocul. Surf. 2016;14:207–215. doi: 10.1016/j.jtos.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 258.Kimura K, et al. Safety, tolerability, and preliminary efficacy of the anti-fibrotic small molecule PRI-724, a CBP/beta-catenin inhibitor, in patients with hepatitis C virus-related cirrhosis: a single-center, open-label, dose escalation phase 1 trial. EBioMedicine. 2017;23:79–87. doi: 10.1016/j.ebiom.2017.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Bailey D, et al. RVX-208: a small molecule that increases apolipoprotein A-I and high-density lipoprotein cholesterol in vitro and in vivo. J. Am. Coll. Cardiol. 2010;55:2580–2589. doi: 10.1016/j.jacc.2010.02.035. [DOI] [PubMed] [Google Scholar]
- 260.Mirguet O, et al. Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J. Med. Chem. 2013;56:7501–7515. doi: 10.1021/jm401088k. [DOI] [PubMed] [Google Scholar]
- 261.Carvajal LA, et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 2018;10:eaao3003. doi: 10.1126/scitranslmed.aao3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Kumar MSA, et al. Pharmacokinetics and safety profile of bleselumab (ASKP1240) in patients with moderate-to-severe plaque psoriasis: results from a phase 2A, randomized, double-blind, placebo-controlled, sequential, multiple-dose escalation study. Clin. Therap. 2017;39:e68. doi: 10.1016/j.clinthera.2017.05.208. [DOI] [Google Scholar]
- 263.Byrd JC, et al. Phase I study of the anti-CD40 humanized monoclonal antibody lucatumumab (HCD122) in relapsed chronic lymphocytic leukemia. Leuk. Lymphoma. 2012;53:2136–2142. doi: 10.3109/10428194.2012.681655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Lapalombella R, et al. The humanized CD40 antibody SGN-40 demonstrates pre-clinical activity that is enhanced by lenalidomide in chronic lymphocytic leukaemia. Br. J. Haematol. 2009;144:848–855. doi: 10.1111/j.1365-2141.2008.07548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Albach FN, et al. Safety, pharmacokinetics and pharmacodynamics of single rising doses of BI 655064, an antagonistic anti-CD40 antibody in healthy subjects: a potential novel treatment for autoimmune diseases. Eur. J. Clin. Pharmacol. 2018;74:161–169. doi: 10.1007/s00228-017-2362-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Ye S, et al. A bispecific molecule targeting CD40 and tumor antigen mesothelin enhances tumor-specific. Immun. Cancer Immunol. Res. 2019;7:1864–1875. doi: 10.1158/2326-6066.CIR-18-0805. [DOI] [PubMed] [Google Scholar]
- 267.Argiriadi MA, et al. CD40/anti-CD40 antibody complexes which illustrate agonist and antagonist structural switches. BMC Mol. Cell Biol. 2019;20:29. doi: 10.1186/s12860-019-0213-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Reck M, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non–small-cell lung cancer. N. Engl. J. Med. 2016;375:1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 269.Borghaei H, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 2015;373:1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Boyerinas B, et al. Antibody-dependent cellular cytotoxicity activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor. Cells Cancer Immunol. Res. 2015;3:1148–1157. doi: 10.1158/2326-6066.CIR-15-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Xu R, et al. Recombinant humanised anti-PD-1 monoclonal antibody (JS001) treatment for patients with refractory or metastatic nasopharyngeal carcinoma: preliminary results of an open-label, phase 1b/2, clinical study. Lancet Oncol. 2017;18:S1. doi: 10.1016/S1470-2045(17)30757-X. [DOI] [Google Scholar]
- 272.Hoy SM. Sintilimab: first global approval. Drugs. 2019;79:341–346. doi: 10.1007/s40265-019-1066-z. [DOI] [PubMed] [Google Scholar]
- 273.Zhang T, et al. Abstract 2226: anti-human PD-1 antibody BGB-A317 exhibits potent immune cell activation. Cancer Res. 2016;76:2226. [Google Scholar]