

Abstract
The presentation of pathogen-derived peptides on Major Histocompatibility Complex class I (MHCI) molecules is essential for the initiation of adaptive CD8+ T cell immunity, which in turn is critical for effective control of many significant human infections. The identification of immunogenic pathogen-derived epitopes and a detailed understanding of how they are recognized by T cell receptors (TCRs) is essential for the design of effective T cell-based vaccines. Here, we have characterized the T cell recognition and immune responses in mice to two naturally presented influenza A virus (IAV)-derived peptides previously identified from virally infected cells via mass spectrometry. These neuraminidase-derived peptides, NA181-190 (SGPDNGAVAV) and NA181-191 (SGPDNGAVAVL), are completely overlapping with the exception of a one amino acid extension at the carboxy terminus of the longer peptide. This minor peptidic difference results in the induction of two completely independent and non-cross-reactive T cell populations that show distinct functional characteristics after IAV infection of B6 mice. We show that the unique TCR reactivity to the overlapping peptides is present in the naïve repertoire prior to immune expansion in B6 mice. Moreover, we provide a structural explanation underlying the distinct CD8+ T cell reactivities, which reinforces the concept that peptide length is a key determinant of antigen specificity in CD8+ T cell responses.
Introduction
CD8+ T cells are essential for the effective control of intracellular pathogens and tumors. Indeed, increasing evidence suggests that for several important human pathogens, the magnitude of the T cell response is a superior correlate of disease control than antibody levels (1). In order for CD8+ T cells to respond to pathogen challenge, they must be activated via the specific recognition of a pathogen-derived peptide fragment (p) in complex with an MHC class I molecule (MHCI) presented on the surface of an antigen presenting cell. Despite the large number of peptide epitopes that can theoretically bind any given MHCI allomorph, relatively few are able to elicit detectable CD8+ T cell responses. Moreover, responses to these epitopes tend to be highly conserved between MHC-matched individuals. A significant effort has been made over the years toward the identification of immunogenic peptides, to be able to biochemically and functionally identify appropriate candidate epitopes for novel and more efficient vaccines (2). There are a multitude of algorithms that facilitate the in silico prediction of immunogenic peptides, based on established MHCI-peptide binding motifs and in vivo peptide binding assays, which aid in the identification of potential immunogenic epitopes, but can fall short in providing a true reflection of the in vivo situation (3–6). Direct analyses of immunogenicity often involve testing the reactivity of T cells against overlapping peptide panels spanning the entire length of proteins of interest. For those peptides that elicit T cell responses, the minimal epitope is then defined as the shortest immunogenic peptide, with longer forms of the peptide thought to be processed down to the minimal epitope in vivo.
We recently reported the identification of influenza A virus (IAV)-derived peptides using an immunopeptidomics approach that interrogated MHCI bound peptides isolated from the surface of infected cells, demonstrating that all 21 identified peptides were able to elicit a detectable CD8+ T cell response following IAV infection of B6 mice (6). Of these, we found two almost identical peptides (differing by a 1 amino acid extension at the C terminus) derived from the neuraminidase protein (NA181-190 and NA181-191) that bound to H-2Db. These peptides could both be presented via both direct and cross-presentation and following infection of dendritic or lung epithelial cell lines (6). The fact that these highly similar peptides are both naturally processed and presented following IAV infection offered a unique opportunity to dissect the endogenous CD8+ T cell responses to highly similar peptides, to further our understanding of the extent to which similarity in presented peptides was reflected in responding T cell populations after infection.
Here, we describe a comprehensive analysis of the CD8+ T cell populations that recognize the overlapping NA181-190 and NA181-191 peptides in complex with H-2Db (referred to forthwith as H-2DbNA181-190 and H-2DbNA181-191, respectively). We show that the T cells elicited following IAV infection that are specific for each of these peptides are entirely distinct, with no evidence of cross-reactivity. Analysis of naïve epitope-specific T cell frequency, TCRαβ repertoire, and T cell response magnitude and quality, further supported the notion that these populations arise entirely independent of one another. Crystal structures of each of the peptides in complex with H-2Db provide a structural basis for the development and elicitation of distinct T cell populations from almost identical peptides.
Materials and Methods
Mice and virus infections
C57BL/6J mice were housed in a specific pathogen-fee animal facility at the Animal Research Laboratories (ARL) at Monash University. Female C57BL/6J mice aged between 7-12 weeks were infected intranasally (i.n.) with 1000 PFU of PR8 H1N1 influenza A virus (A/Puerto/Rico/8/34). For the procedure mice were anesthetized with 2-3 % isofluorane at a flow rate of 2 L/min before administration of the virus. All animal experimentation was conducted following the Australian National Health and Medical Research Council Code of Practice for the Care and Use of Animals for Scientific Purposes guidelines for housing and care of laboratory animals and performed in accordance with Institutional regulations after pertinent review and approval by the Monash University Animal Ethics Committees.
In vitro peptide stimulation and intracellular cytokine staining
Synthetic SGPDNGAVAV (NA181-190) or SGPDNGAVAVL (NA181-191) peptides used for in vitro stimulation of CD8+ T lymphocytes were purchased from GenScript (Hong Kong) and at a purity of >85%. Lymphocytes from the spleen and bronchoalveolar lavage (BAL) were incubated with 1μM peptide (or no peptide) in a round bottom 96-well plate together with 10 U/ml of IL-2 (Roche) and 1 μg/ml of Golgi-plug (BD Bioscience) and cultured for 5hrs at 37°C and 5% CO2. Following peptide stimulation, cells were washed and stained for CD4, CD8, IFNγ, TNF and IL-2 and analyzed by flow cytometry (7).
Isolation of lymphocytes from spleen and lymph nodes
Mice were euthanized by CO2 asphyxiation, followed by removal of the spleen, as well as the inguinal, brachial, axial and cervical lymph nodes (LN) as indicated. Spleen and LNs were mashed through a sterile 70 μm cell strainer in Hanks Balanced Salt Solution (HBSS) to create a single cell suspension. Red blood cells were lysed according to the manufacturers instruction using RBC lysis buffer (Sigma). Cells were re-suspended in 5 ml of cRPMI for further analysis or surface marker staining.
Harvest of brochoalveolar lavage (BAL)
Mice were euthanized by CO2 asphyxiation and a small incision was made in the trachea, through which a small catheter was inserted. The BAL was harvested by flushing the lung 3x with 1 ml of sterile HBSS, which was retrieved and collected for analysis. The BAL was centrifuges at 1600 rpm for 6 min a 4°C followed by re-suspension in cRPMI to be used for antibody staining.
Tetramer-based magnetic enrichment of virus-specific CD8+ T cells
Spleen and major lymph nodes (auxiliary, brachial, cervical, inguinal and mesenteric) were pooled from naïve mice and stained with H-2DbNA181-190 and H-2DbNA181-191 tetramers coupled to APC or PE. Cells were washed to remove excess tetramer and incubated with anti-PE-and anti-APC-conjugated magnetic microbeads (Miltenyi Biotec) and tetramer-bound cells were enriched over an LS column (Miltenyi Biotec) on a magnetic platform. Enriched cells were stained with a cocktail of conjugated antibodies to identify H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cells (tetramer+ CD8α+ TCRβ+, CD11b-, CD11c-, B220-, F4/80- CD4-). Entire samples were acquired on a BD LSRFortessa X-20 for analysis.
Tetramer staining of epitope specific CD8+ T cells
Tetramers to detect epitope-specific CD8+ lymphocytes were generated via coupling of monomers (made at the University of Melbourne) to streptavidin-labeled APC or PE fluorochromes at a final concentration of 1 μg/μl. Lymphocytes were incubated in 50 μl of MACS buffer containing both H-2DbNA181-190-PE and H-2DbNA180-191-APC tetramers (final dilution 1:100, or as indicated in the individual experiment) and incubated at RT in the dark for 1 hour. Following tetramer staining, cells were stained for CD8α and either analysed or single, live, CD8+ tetramer+ cells individually sorted into a 96-well plate for multipex RT-PCR analysis. Gating strategy for spleen shown in Supplemental Fig. 1A, along with tetramer staining on control spleen and BAL samples stained with all antibodies but not H-2DbNA181-190-PE and H-2DbNA180-191-APC tetramers (Supplemental Fig. 1B) and tetramer staining on CD4+ T cell subset from spleen and BAL (Supplemental Fig. 1C). For analysis of tetramer dissociation kinetics, after tetramer staining, cells were washed and incubated for various times at 37°C with anti-H-2Db/Kb Ab (28-8-6, BD Pharmingen) (25 μg/ml) to prevent tetramer rebinding. Cells were then washed and stained with anti-CD8α-BUV395 Ab for flow cytometric analysis.
Multiplex polymerase chain reaction (PCR) based analysis of paired TCR alpha and beta chains
Paired TCR α and β chain mRNA from single cell sorted tetramer-positive lymphocytes was transcribed into cDNA and amplified according to the multiplex PCR protocol published previously (8, 9). Amplified DNA fragments were sequenced at the Micromon Genomics facility at Monash University. DNA sequences were analyzed using IMGT/V-Quest (10, 11).
Protein Expression and purification of H-2Db-peptide complexes
The H-2Db MHCI heavy chain and human β2-microglobulin (β2m) were expressed separately using pET30 vector, and then purified from inclusion bodies in Escherichia coli. Both proteins were resuspended in 8 M urea, 20 mM Tris-HCl pH 8.0, 0.5 mM EDTA and 1 mM DTT. H-2Db and β2m were refolded in complex with either the SGPDNGAVAV (NA181-190) or the SGPDNGAVAVL (NA181-191) peptides (GL Biochem Ltd, Shanghai, China) as described previously (12). Refold buffer containing 3 M Urea, 0.1 M Tris-HCl pH 8.0, 2 mM EDTA, 400 mM L-arginine, 0.5 mM and 5 mM of oxidized/reduced glutathione was chilled at 4°C with continuous stirring. 10 mg of peptide, 10 mg of β2m and 30 mg of H-2Db heavy chain were added to the stirring refolding buffer at 4°C, then dialysed 3 times against 10 L of 10mM Tris-HCl pH 8.0 at 4°C. Proteins were then purified using diethylaminoethyl-cellulose (DEAE-cellulose) and an anion exchange column.
pMHCI Complex Thermal Stability Assay
Thermal stability assays of H-2Db in complex with the two NA peptides were performed in the Real Time Detection instrument (Corbett RotorGene 300) as described previously (13). Each pMHC complexes were tested at two concentrations (1g/l and 0.5g/l) in duplicates, exposed to increasing temperatures in the presence of SYPRO orange (Invitrogen) fluorescent dye. The measurements were made at a rate of 1°C/min from 20°C to 90°C. The fluorescence intensity was monitored with excitation and emission wavelength of 530 nm at 555 nm, respectively. The thermal melt point (Tm) represents the temperature required to unfold 50% protein.
Crystallisation, Data Collection, and Structure Determination
Crystals were made at room temperature via hanging-drop vapour fusion method. A 1:1 drop ratio of pMHC:reservoir solution was used to produce crystals at 5mg/ml of protein and 0.1M sodium citrate pH5.7, 0.2M LiSO4 and 24-28% (w/v) PEG3350 as reservoir solution. The crystals were soaked in a cryoprotectant solution containing the mother liquor solution with20%(w/v) glycerol and then flash frozen in liquid nitrogen. The data was collected on the MX2 beamline at the Australian Synchrotron, Clayton (14). Data were processed using XDS (15) and scaled using Aimless software (16) from the CCP4 suite (17). The structure was determined by molecular replacement using the PHASER (18) program with the H-2Db for the MHC model without the peptide (Protein Data Bank accession number, 3FTG) (19). Manual model building was conducted using the Coot software (20) followed by maximum-likelihood refinement with the Buster program (21). The final models have been validated using the Protein Data Bank validation web site and the final refinement statistics are summarized in SupplementaryTable 1. Coordinates submitted to PDB database, code: 6WZY for H-2DbNA181-190 and 6X00 for H-2DbNA181-191.
All molecular graphics representations were created using PyMol [The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.].
Results
Influenza A virus infection elicits distinct H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cell populations in the spleen and BAL of mice.
Using an unbiased mass spectrometry approach, we previously identified two overlapping overlapping influenza A virus (IAV)-derived peptides; NA181-190 (SGPDNGAVAV) and NA181-191 (SGPDNGAVAVL), which bound to H-2Db and elicited CD8+ T cell responses upon IAV infection of B6 mice (6). To analyse the in vivo response to these highly similar peptides and to determine the extent of cross-reactivity in the responding CD8+ T cell repertoires, we infected B6 mice intranasally (i.n.) with 1000 PFU PR8 IAV and analysed the H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cell responses at the peak of the antiviral CTL response (d10). We previously assessed the response to these peptides by intracellular cytokine staining after short term in vitro peptide restimulation (6). Inherent in this approach is the possibility that the longer NA181-191 peptide may be processed down to the shorter NA181-190 peptide, thus conflating the responding populations. To circumvent this, we stained CD8+ T cell populations from the spleen and lungs (bronchoalveolar lavage; BAL) of infected mice with H-2DbNA181-190 and H-2DbNA181-191 tetramers to allow for a definitive distinction between the two epitope-specific populations. These tetramers had been confirmed to contain the correct length peptide using mass spectrometry prior to use.
At day 10 after infection concurrent staining of cells with H-2DbNA181-190 and H-2DbNA181-191 tetramers showed two distinct epitope-specific CD8+ T cell populations in both the BAL and the spleen of all infected mice analyzed (Fig. 1A). Additionally, both populations were present at similar magnitudes, when quantifying absolute numbers in both tissues (Fig. 1B). To verify that the responding populations were indeed mutually exclusive and not a consequence of preferential tetramer binding, we performed tetramer staining where the concentration of one tetramer was kept constant, while the other was titrated. Titration of an individual tetramer caused a reduction in the percentage of tetramer+ cells as well as a reduced MFI of tetramer staining. Importantly however, even at low concentrations of either tetramer, both H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cell populations remained distinct (Fig. 1C). These findings demonstrate that the H-2DbNA181-190 and H-2DbNA181-191 epitopes elicit mutually exclusive T cell populations, both at the site of infection and in peripheral lymphoid tissue, despite their peptide sequences differing only by the addition of one C-terminal leucine residue in the NA181-191 peptide sequence.
Figure 1: IAV-derived H-2DbNA181-190 and H-2DbNA181-191 epitopes elicit distinct CD8+ T cell responses after PR8 infection.
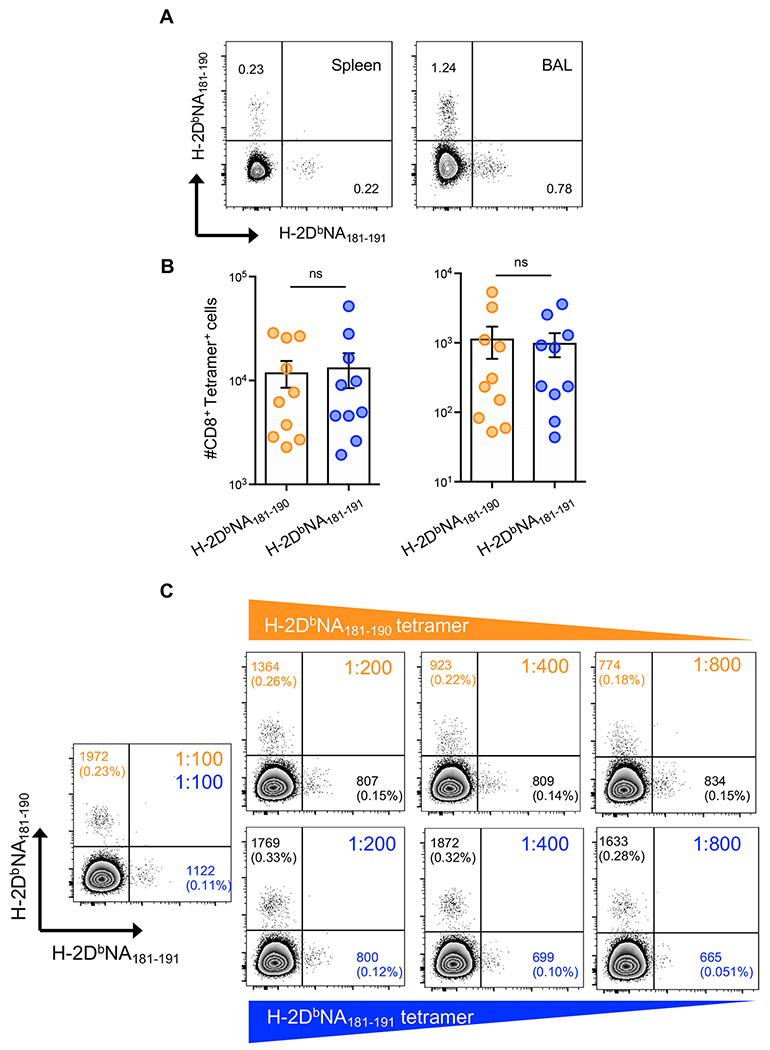
At d10 after intranasal infection with 1000 PFU of PR8 IAV, splenic and BAL-derived CD8+ T cells were stained concurrently with H-2DbNA181-190 and H-2DbNA181-191 tetramers and tetramer binding on live, CD19−, F4/80−, I-Ab-, CD4−, TCRβ+, CD8+, CD44hi lymphocytes from spleen and BAL of infected mice was analysed. A) Dot plots showing H-2DbNA181-190- and H-2DbNA181-191-specific tetramer staining on splenocytes and BAL cells. B) Absolute numbers of H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cells in the spleen (left) and BAL (right) of infected mice. C) Representative dot plots showing concurrent H-2DbNA181-190 and H-2DbNA181-191 tetramer staining with titration of either H-2DbNA181-190 (upper panels) or H-2DbNA181-191 (lower panels) tetramers. Numbers in quadrants represent mean fluorescence intensity of tetramer staining for tetramer+ cells; percentage cells staining with each tetramer shown in parentheses. Data are representative of 2-3 independent experiments (n=4-5 mice/experiment).
Distinct functional properties of H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cells after influenza infection
Given the distinct CD8+ T cell populations responding to each of the H-2DbNA181-190 and H-2DbNA181-191 peptides, we next assessed whether there were any qualitative differences between H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cells after influenza infection with respect to their effector cytokine production. CD8+ T cells were isolated from BAL and spleen and IFNγ, TNF, and IL-2 production assessed after short term in vitro stimulation with NA181-190 and NA181-191peptides. Polyfunctionality (the ability to produce multiple effector molecules) in virus-specific CTLs is a correlate of effective CD8+ T cell immunity (22–24). IAV-specific CTL cytokine production has previously been demonstrated to predominantly occur in an hierarchical manner with the majority of epitope-specific cells producing IFNγ, a subset of those producing TNF, and a subset of the IFNγ+ TNF+ cells producing IL-2 (7). Thus, the proportion of IFNγ+ cells able to produce TNF and/or IL-2 is routinely used as a qualitative indicator of the response, independent of response magnitude. There was a significantly larger proportion of H-2DbNA181-191-specific, compared to H-2DbNA181-190-specific, IFNγ+ splenocytes able to produce multiple cytokines after peptide restimulation (p=0.0058) (Fig. 2A, 2C). This was due to a significantly increased proportion of cells producing TNF (p=0.0046) but no difference in the proportion of IL-2 producers (Fig. 2A, 2C). These differences were not observed in the BAL 10 days after infection (Fig. 2B, 2D). The distinctive cytokine production profiles of the two splenic epitope-specific populations further demonstrate the unique nature of the H-2DbNA181-190- and H-2DbNA181-191-specific CTL populations.
Figure 2: Qualitative analysis of the H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cell cytokine response after IAV infection.
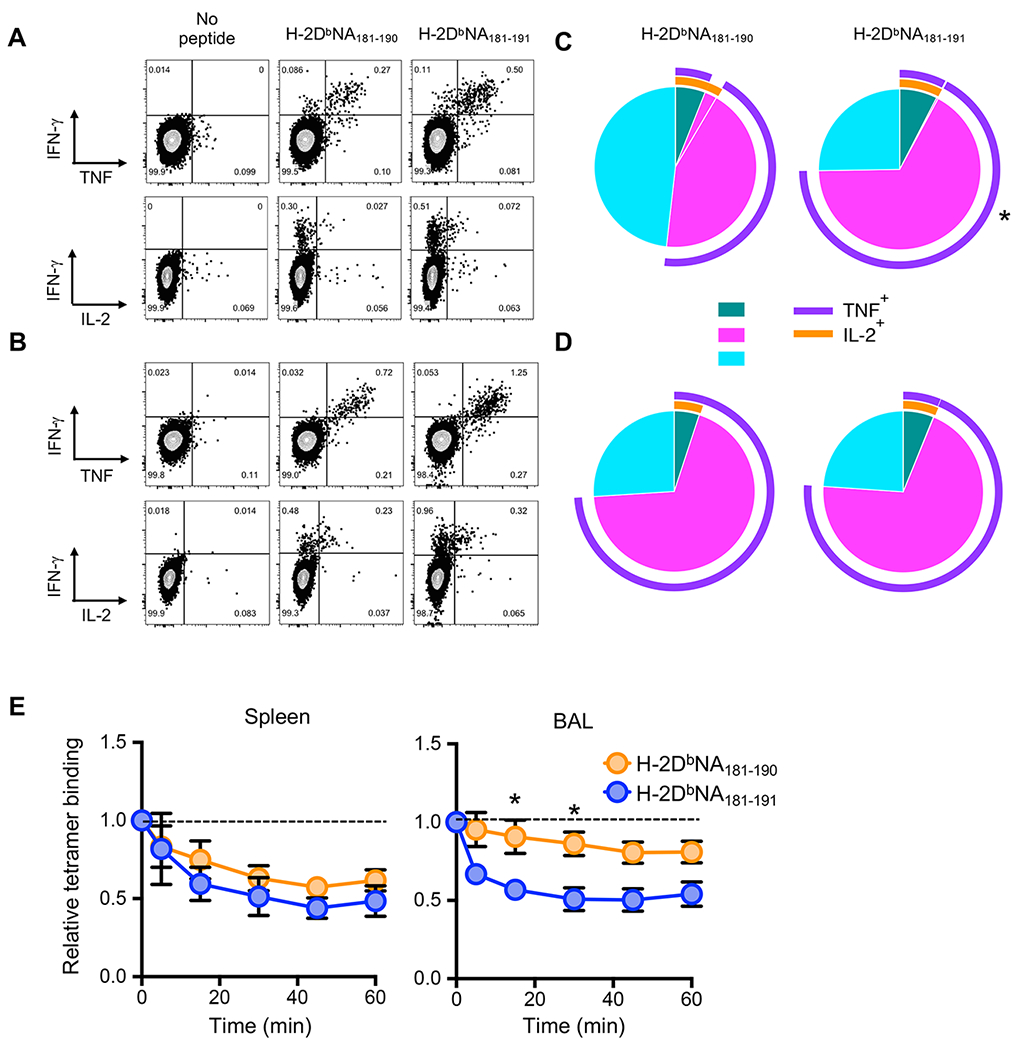
At d10 after intranasal infection with 1000 PFU of PR8 IAV, splenic and BAL-derived cells were enriched for CD8+ T cells. A-D) Cells were stimulated in vitro for 5h with 1 μm NA181-190 or NA181-191 peptides, and stained for CD8, IFNγ, TNF, and IL-2. Representative dot plots showing IFNγ vs TNF staining or IFNγ vs IL-2 staining in live, CD4− CD8+ T cells from A) spleen or B) BAL. Pie charts show the proportion of IFNγ+ cells expressing one (1+), two (2+), or three cytokines (3+), while surrounding arcs indicate which cytokines are produced in addition to IFNγ for CD8+ T cells from C) spleen or D) BAL. Data are representative of 3 independent experiments (n=5 mice/experiment). E) Enriched splenocytes and BAL cells from infected mice were dual stained with H-2DbNA181-190 and H-2DbNA181-191 tetramers and incubated at 37°C for indicated times with anti-H-2Db antibody. Shown is the proportion of total H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cells that remain tetamer-bound after anti-H-2Db antibody addition. Data are representative of 2 independent experiments (n=5 mice/experiment). *p<0.05 using Student’s paired t-test.
To determine whether the distinct H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cell populations display different TCR avidities for their cognate pMHCI complexes, we isolated lymphocytes from spleen and BAL, stained with H-2DbNA181-190 and H-2DbNA181-191 tetramers and assayed the rate of tetramer dissociation, by incubating cells over a time course with an anti-H-2Db blocking (25). While tetramer dissociation was similar on H-2DbNA181-190- and H-2DbNA181-191-specific splenocytes, the H-2DbNA181-191 tetramer dissociated more rapidly than the H-2DbNA181-190 tetramer on BAL cells over a 60 minute interval, despite a tendency (not significant) for H-2DbNA181-191-specific cells to express higher TCRβ levels (Supplemental Fig. 1D). These data suggest that the H-2DbNA181-190-specific CTL population, at least at the site of infection, exhibited a higher avidity for cognate pMHCI than H-2DbNA181-191-specific CTLs (Fig. 2E), again highlighting the unique characteristics of these two CTL populations.
H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cells display distinct TCR repertoire usage after influenza infection
To understand the basis for the unique reactivities of H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cell populations, we single cell sorted H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cells from the same mice on day 10 after influenza infection and performed a paired TCRαβ analysis of the epitope-specific repertoires (Table 1). Detailed analysis of the two TCRαβ repertoires was performed using the TCRdist algorithm (26). Corresponding to the mutually exclusive reactivity of these populations, VJ pairing analysis of the H-2DbNA181-190- and H-2DbNA181-191-specific populations revealed distinct preferences for pairwise gene associations (Fig. 3A). Within the H-2DbNA181-190-specific TCRs, there were preferences for TRBV5-TRBJ1-1/TRAV12-TRAJ5 or TRBV2-TRBJ1-2/TRAV9-2-TRAJ2 combinations, while these were not observed within the H-2DbNA181-191-specific repertoire (Fig. 3A). Analysis of the diversity of TCRs within each of the repertoires (taking into account both similarity as well as identity) shows that TCRs specific for H-2DbNA181-191 were more diverse than those specific for H-2DbNA181-190 (Fig. 3B, upper panel), a finding supported by the fact that the average ‘distance’ across TCRs within a repertoire (i.e. to their nearest neighbors in the repertoire) was greater for H-2DbNA181-191- compared to H-2DbNA181-190-specific TCRs (Fig. 3B, lower panel). Modal CDR3α and β lengths were also different, with H-2DbNA181-190- and H-2DbNA181-191-specific TCRs showing preferred CDR3α lengths of 13 and 14-15aa, respectively, and preferred CDR3β lengths of 13-14 and 14-15aa, respectively. A dissection of TRAV and TRBV usage according to CDR3 length revealed an association between CDR3 length and TRAV or TRBV usage which differed in an epitope dependent manner (Fig. 3C). Analysis of CDR3α and β sequences revealed that H-2DbNA181-190-specific TCRs were characterized by two predominant CDR3α/CDR3β motifs arising from independent TCRs (Fig. 3D); the QVVGQL/SQDRDTEV motif, minor variations of which were observed in all three mice analyzed, and the TGGLSGKL/SQESGDSDY motif, minor variations of which were repeatedly observed in mouse 3. By contrast CD8+ T cells from the same mice but reactive to H-2DbNA181-191 showed completely distinct CDR3 sequences, with less pronounced motif selection (Fig. 3D). Finally, the NNdistance rank score, which provides a measure of the similarity of TCRs used between two epitope specific repertoires, revealed that the distance between the H-2DbNA181-190- and H-2DbNA181-191-specific TCR repertoires was as great as that between as two unrelated IAV-derived epitope-specific repertoires, namely those directed toward DbNP366-374 and DbPA224-233 (Fig. 3E). Collectively, these analyses indicate that, despite the similarity of the peptides being recognized in the context of H-2Db, the TCR repertoires induced by these peptides are as distinct from one another as those recognizing two completely unrelated epitopes.
Table 1.
Paired TCR CDR3αβ sequences from immune H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cells
| H-2DbNA181-190-specific TCRs | ||||||||
|---|---|---|---|---|---|---|---|---|
| CDR3α | CDR3β | TRAV | TRAJ | TRBV | TRBJ | M1 | M2 | M3 |
| CALRTQVVGQLTF* | CASSQDRDTEVFF | 12N-3 | 5 | 5 | 1-1 | 15 | ||
| CALRTQVVGQLTF | CASSQDRDTDVFF | 12N-3 | 5 | 5 | 1-1 | 3 | ||
| CADPGTQVVGQLTF | CASGEETDPSGNTLYF | 14-2 | 5 | 13-2 | 1-3 | 2 | ||
| CAMAPLSGSFNKLTF | CASSQDAGDSDYTF | 13-2 | 4 | 2 | 1-2 | 1 | ||
| CALRTQVVGQLTF | CASSQDRDTEVFF | 12N-3 | 5 | 5 | 1-1 | 1 | ||
| CALRTQVVGQLTF | CATTHDTDTEVFF | 12N-3 | 5 | 5 | 1-1 | 1 | ||
| CALRTQVVGQLTF | CASSQDRDTEVFF | 12N-3 | 5 | 5 | 1-1 | 10 | ||
| CALRTQVVGQLTF | CASSLDRDTEVFF | 12N-3 | 5 | 12-1 | 1-1 | 2 | ||
| CALRTQVVGQLTF | CASSQDRDTEVFF | 12N-3 | 5 | 5 | 1-1 | 2 | ||
| CALRTQVVGQFTF | CASSQDRDTEVFF | 12N-3 | 5 | 5 | 1-1 | 1 | ||
| CALRTQVVGQLTF | CASSLDRDTEVFF | 12N-3 | 5 | 12-1 | 1-1 | 1 | ||
| CALRTQDVGQLTF | CASSQDRDTEVFF | 12N-3 | 5 | 5 | 1-1 | 1 | ||
| CALRTQVVGHLTF | CASSQDRDTEVFF | 12N-3 | 5 | 5 | 1-1 | 1 | ||
| CALRTQVVGQLTF | CASSQDRDAEVFF | 12N-3 | 5 | 5 | 1-1 | 1 | ||
| CALIPDTNAYKVIF | CASSDDWGGAGDTQYF | 12D-2 | 30 | 13-1 | 2-5 | 6 | ||
| CVLSAGTGGLSGKLTF | CASSQESGDSDYTF | 9-2 | 2 | 2 | 1-2 | 5 | ||
| CAVSEGTQVVGQLTF | CASSQDNGDSDYTF | 3-1 | 5 | 2 | 1-2 | 3 | ||
| CVLSAGTGGLSGKLTF | CASSQESGDSDYTF | 9-2 | 2 | 2 | 1-2 | 2 | ||
| CAVSGGAGNTGKLIF | CASSQEEGNSDYTF | 9-2 | 37 | 2 | 1-2 | 2 | ||
| CVLSAPNTGGLSGKLTF | CASSQEGGDSDYTF | 9-2 | 2 | 2 | 1-2 | 1 | ||
| CVLSANTGGLSGKLNL | CASSQDNGDSDYTF | 9-2 | 2 | 2 | 1-2 | 1 | ||
| CVLSAGTGGVSGKLTF | CASSQESGDSDYTF | 9-2 | 2 | 2 | 1-2 | 1 | ||
| CVLSAGTGGLSDKLTL | CASSQESGDSDYTF | 9-2 | 2 | 2 | 1-2 | 1 | ||
| CAVRSNYNVLYF | CASSDDRYEQYF | 12D-2 | 21 | 13-3 | 2-7 | 1 | ||
| CAVRMAQVVGQLTF | CASSDAKDSAETLYF | 9-2 | 5 | 13-1 | 2-3 | 1 | ||
| CVLSAGTGGLSGNLTF | CASSFRVSKERLFF | 9-2 | 2 | 15 | 1-4 | 1 | ||
| CALVYTGGLSGKLTF | CASSLLGGYSQNTLYF | 6-7 | 2 | 16 | 2-4 | 1 | ||
| CALANNTNTGKLTF | CAARGTEVFF | 6 | 27 | 19 | 1-1 | 1 | ||
| CALIPDTNAYKVIF | CASSFRVSNERLFF | 12D-2 | 30 | 15 | 1-4 | 1 | ||
| CAVKGGSALGRLHF | CASSDHNSYNSPLYF | 9N-3 | 18 | 13-3 | 1-6 | 1 | ||
| Total | 23 | 19 | 29 | |||||
| H-2DbNA181-191-specific TCRs | ||||||||
| CAAYPGDNSKLIW | CASSPGGITGQLYF | 10 | 38 | 19 | 2-2 | 20 | ||
| CAGYNQGKLIF | CASSGGLGGNTGQLYF | 3-3 | 23 | 13-1 | 2-2 | 4 | ||
| CAVSAMDSNYQLIW | CASSRVRTGGKDTQYF | 9-1 | 33 | 17 | 2-5 | 3 | ||
| CALSALTGSGGKLTL | CTCSEGGAEQFF | 12D-2 | 44 | 1 | 2-1 | 1 | ||
| CAAYPGDNSKLIW | CASSPGGITGQLYF | 10 | 38 | 19 | 2-2 | 1 | ||
| CAMHLSGSFNKLTF | CASSSTGGAETLYF | 13-2 | 4 | 4 | 2-3 | 1 | ||
| CVLSANSGTYQRF | CASSPTKNTEVFF | 9-2 | 13 | 12-1 | 1-1 | 1 | ||
| CALRPTGGSNAKLTF | CASSPWGVEQYF | 6-7 | 42 | 12-2 | 2-7 | 20 | ||
| CAVSHNYAQGLTF | CASSQDPWGSQNTLYF | 9-1 | 26 | 5 | 2-4 | 2 | ||
| CAMSMATGGNNKLTF | CASSFGGKRDTQYF | 16 | 56 | 4 | 2-5 | 1 | ||
| CAVRNSNNRIFF | CASSEGWGYYAEQFF | 3-3 | 31 | 13-1 | 2-1 | 1 | ||
| CALRPTGGSNAKVTF | CASSPWGVEQYF | 6-7 | 42 | 12-2 | 2-7 | 1 | ||
| CAPRGSGGSNAKLTF | CASSLETGDGNTLYF | 13-2 | 42 | 12-2 | 1-3 | 3 | ||
| CAMSLTGFASALTF | CASRDTNSGNTLYF | 6-2 | 35 | 19 | 1-3 | 2 | ||
| CAASPVGDNSKLIW | CASTESMGVSYEQYF | 7-4 | 38 | 13-1 | 2-7 | 1 | ||
| CAASPVGDNSKLIW | CASSINNTGQLYF | 7-4 | 38 | 19 | 2-2 | 1 | ||
| CAASPVGDNSKLIW | CASSFGRGNAEQFF | 7-4 | 38 | 14 | 2-1 | 1 | ||
| CALSEDGSSGNKLIF | CASSFGRGNAEQFF | 12N-3 | 32 | 14 | 2-1 | 1 | ||
| CAPRGFGGSNAKLTF | CASSLETGDGNTLYF | 13-2 | 42 | 12-2 | 1-3 | 1 | ||
| CAFYMGYKLTF | CASGDSYAEQFF | 6-6/6-7 | 9 | 13-1 | 2-1 | 1 | ||
| CAASPVGDNSKLIW | CASTESMGVTYEQYF | 7-4 | 38 | 13-1 | 2-7 | 1 | ||
| CAASPVGDNSKLIW | CASSESLGVYYEQYF | 7-4 | 38 | 13-1 | 2-7 | 1 | ||
| Total | 31 | 25 | 13 | |||||
Motifs colored according to dendrogram groupings (Fig. 3).
Figure 3: Characterization of CD8+ H-2DbNA181-190- and H-2DbNA181-191-specific TCR αβ repertoires after infection.

Female C57BL/6J mice were infected i.n. with 1000 PFU of PR8 influenza virus and individual H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cells from pooled spleen and lymph nodes were single cell sorted for subsequent multiplexed analysis of paired TCR CDR3α and CDR3β sequences. All clonotypes analyzed have arisen independently of one another (i.e. data is independent of clonal abundance). H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cell repertoires were characterized from the same mice (N=3 mice). (A) Gene pairing structure of H-2DbNA181-190- and H-2DbNA181-191-specific TCRαβ clonotypes. Enrichments or depletions of gene segments relative to background are shown for all labeled genes by up or down arrows where the number of arrowheads reflects the base-2 logarithm of the fold change. (B) Upper panel is a modified version of the Simpson’s Diversity Index, incorporating receptor similarity as well as identity; lower panel is the nearest neighbor distance between each TCRαβ within the repertoire and its nearest 10% of neighbors, averaged across the entire repertoire. Dashed red lines are an estimate of distances obtained from background non-antigen specific TCR repertoires composed of randomly selected TCRαβ chains from bulk sequences (26). (C) Shown are the CDR3α (left panels) and CDR3β length distributions (right panels) for each epitope-specific population, coloured according to gene segment usage. Segment must represent ≥5% of dataset in order to be depicted. (D) TCRdist clustering trees depicting the distance between TCRs within the epitope-specific TCRαβ repertoires. The thickness of the branches is proportional to the number of unique TCRαβ clones represented by those branches. The TCR logo containing the CDR3 sequence is shown on the left with residue height scaled by frequency. The colored bars underneath represent the source of the nucleotides that encode the logo, as depicted in key. The branches are colored according to TCR generation probability as depicted in key, with red being the easiest TCRs to generate via recombination (26). (E) Heat map showing a distance measure between different epitope-specific repertoires, based on the nearest neighbor distance measures within each repertoire. Yellow repertoires are similar to one another, while blue repertoires are distinct. TCR repertoires from 3 mice were analyzed, with n=23, 19, and 29 sequences for H-2DbNA181-190, and n=31, 25, 13 sequences for H-2DbNA181-191.
No cross-reactivity in naïve H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cell precursors prior to influenza infection
While it is clear that H-2DbNA181-190 and H-2DbNA181-191 induce the expansion of unique T cell populations in response to IAV infection, it was possible that the distinctive reactivities emerged as a consequence of selective expansion, and that cross-reactive T cells are present in the naïve repertoire. To determine the extent of unique vs cross-reactivity within the naïve H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cell populations, we performed tetramer-based magnetic enrichment, using the H-2DbNA181-190 and H-2DbNA181-191 tetramers, on spleen and lymph node cells from naïve B6 mice. As was observed in the immune response, both the H-2DbNA181-190- and H-2DbNA181-191-specific naïve populations were uniquely specific and showed no cross-reactivity (Fig. 4A). In addition, the frequency of naïve H-2DbNA181-190- and H-2DbNA181-191-specific precursors was similar, suggesting a similar expansion capacity of H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cell populations in response to influenza infection (Fig. 4B).
Figure 4: Frequency and specificity of naïve H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cell precursor populations.
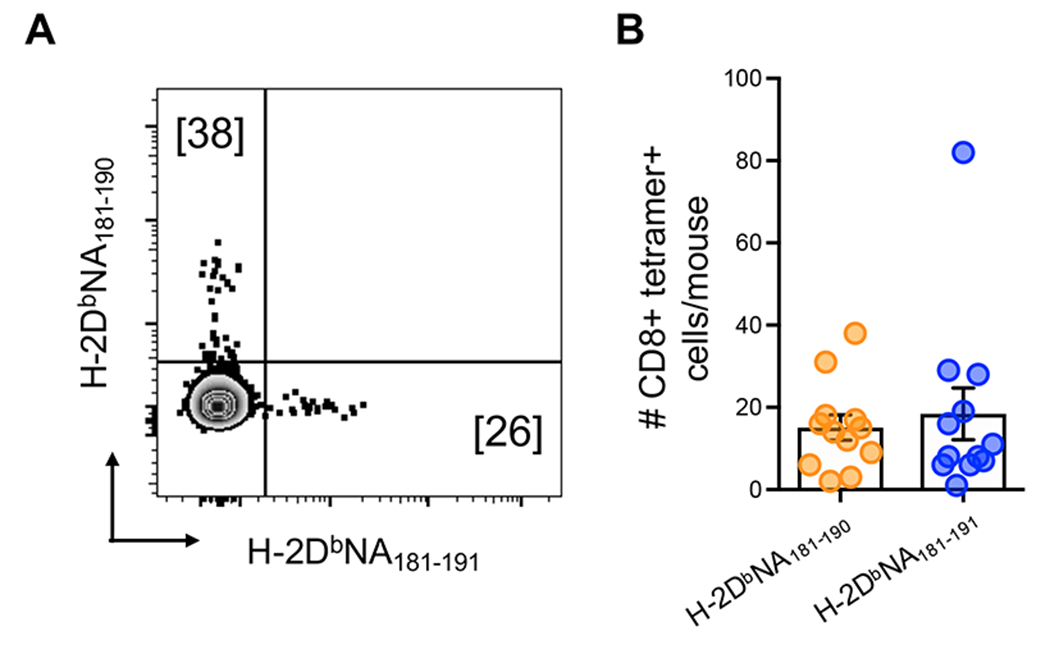
Pooled spleen and major peripheral LN samples from naïve B6 mice were stained with H-2DbNA181-190 and H-2DbNA181-191 tetramers, magnetically enriched for tetramer-binding cells, and then stained with a panel of fluorescently conjugated antibodies for identification of epitope-specific CD8+ T cells. (A) Representative dot plot of live, CD8+ CD3+ CD4− CD11b− CD11c− F4/80− B220− I-Ab− CD44lo H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ lymphocytes. (B) Number of naïve H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cell precursors from the pooled spleen and LNs of individual mice. Data are combined from 3 different experiments (n=4 mice/experiment).
A structural basis for unique reactivities of H-2DbNA181-190- and H-2DbNA181-191-specific CD8+ T cells
Given the lack of cross-reactivity in T cell populations specific for H-2DbNA181-190 and H-2DbNA181-191, and the unique TCR repertoires specific to each peptide, we next sought to explain these findings at a molecular level by determining the structures of the pMHC complexes. We firstly assessed if the additional residue in NA181-191 could destabilize the overall peptide-MHC complex by performing a thermal stability assay. Both the H-2DbNA181-190 and H-2DbNA181-191 pMHCI complexes showed similar stability, with a Tm of ~ 50°C (Fig. 5A), thereby indicating that both peptides were bound equally well by the H-2-Db molecule.
Figure 5: Structure of H-2DbNA181-190 and H-2DbNA181-191 epitopes bound to H-2Db MHC-I.
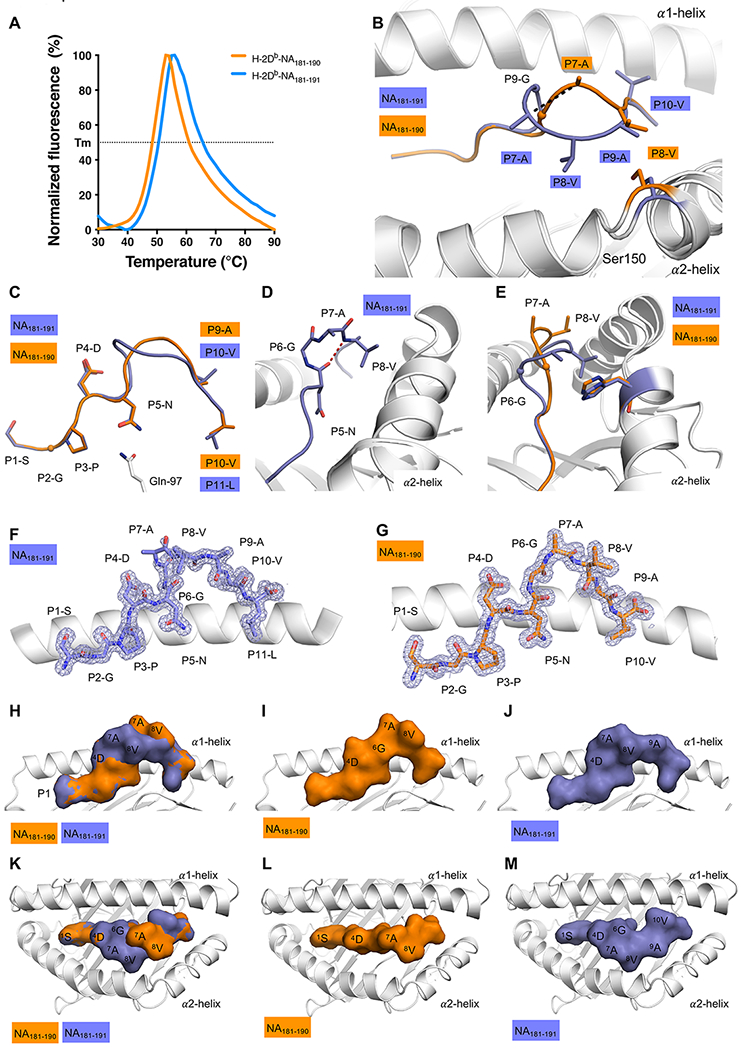
(A) Thermal shift assay data. Normalized Fluorescence (in %) is shown on Y axis, and temperature in °C is shown on the X axis. The data for H-2DbNA181-190 is shown in orange, the data for H-2DbNA181-191 is shown in blue. The calculated thermal melt point (Tm) represents the temperature required to unfold 50% of the protein. All panels (B-M) show H-2Db in white, NA181-190 and NA181-191 peptides in orange and blue, respectively. (B) Superposition of the H-2Db structures presenting the NA181-190 and NA181-191 peptides: the Ser150 of the α2-helix is colored accordingly to the peptide presented by H-2Db. Ser150 is represented as stick and the peptides are in stick and cartoon representation. The maximum displacement observed for the Cα atom of the P7-A represented by black dashed line. (C) Overlay of the peptides represented as stick and cartoon, with only the Gln97 (white stick) of the H-2Db molecule shown. (D) Closer view on the NA181-191 peptide, colored in blue, highlighting the secondary structure 310 helix formed in the central region of the peptide, with the hydrogen bond between P5-N (i) and P8-V (i+3) represented by a red dashed line. (E) Side view of the cleft to show the exposed P7-A and P8-V residues of the NA181-190 peptide (orange), that are partially buried and NA181-191 peptide structure (blue), in contact with His155 (stick colored accordingly to the peptide presented by H-2Db). (F and G) The 2Fo-Fc electron density map contoured at 1 σ (grey mesh) around the (F) NA181-191 peptide, and (G) NA181-190 peptide, presented by the H-2Db molecule (white cartoon). (H to M) Surface presentation of the NA181-190 (I and L) and NA181-191 (J and M) peptide in H-2Db cleft. Overlay of NA181-190 and NA181-191 peptide (H and K). Side view (H, I ,J) and top view (K,L,M). The peptide is shown as surface representation and colored accordingly, the H-2Db cleft is shown in white cartoon.
Next, to elucidate whether the different length of the peptides resulted in a substantially different peptide landscape presented by the H-2Db molecule, we determined the structures of both NA181-191 and NA181-190 in complex with H-2Db at high resolution (1.55 and 1.50 Å, respectively, Supplemental Table 1 and Fig. 5B–M). The superposition of the two H-2Db-peptide complexes revealed that the overall structure of the H-2Db molecule is conserved with a root mean square deviation (r.m.s.d.) of 0.13 Å of the Cα atoms of the α1α2 domains (residues 1-180 of the H2Db heavy chain). The only notable difference resides in the hinge of the α2 helix (residues 148-152 of the H2Db heavy chain; r.m.s.d. = 0.78 Å) resulting in the cleft of H-2Db being more open when in complex with the longer NA181-191 peptide, compared to the NA181-190 peptide, with a maximum displacement of 1.1 Å for the Ser150 Cα atom (Fig. 5B). While the overall MHC conformation remains similar between the two complexes, the peptide structures are different (r.m.s.d. of 1.98 Å), with a maximum displacement of 4.3 Å for the P7-Ala Cα atom in the central region of the peptides (Fig. 5B). The N-terminal end of the peptides (the 1SGPDN5 motif) and the C-terminal part of the peptide (the 9AV10 and 10VL11 motifs) share the same conformation in both H-2DbNA181-191 and H-2DbNA181-190 (Fig. 5C). The peptides both bind canonically to the H-2Db molecule. Namely, P2 and PΩ anchor into the B and F pockets of H-2Db, as well as P5-Asn acting as a secondary anchor residue and forming a double hydrogen bond with the Gln97 of the H-2Db antigen binding cleft, as previously observed in other peptide-H-2Db structures (12, 19, 27, 28). The extra residue in the NA181-191 is accommodated via the formation of a 310-helix turn (residue P5-P8) in the central region of the peptide (Fig. 5D) with the side chain of P8-Val intercalating itself between the peptide backbone and the H-2Db α2-helix, leading to the opening of the cleft observed in the H-2DbNA181-191 structure (Fig. 5B). Contrary to what is often observed for other long peptides (29), the formation of the 310-helix in the NA181-191 peptide stabilizes the longer peptide which does not bulge out of the cleft more than the NA181-190 peptide (Fig. 5C) but still presents some mobility (Fig. 5F). The central part of the NA181-191 peptide is poorly defined in the electron density map as a result of higher mobility, whereas the NA181-190 peptide adopts a rigid conformation in the cleft of H-2Db, with a very clear electron density map (Fig. 5G). As a result, the two peptides present different exposed surfaces accessible for TCR recognition (Fig. 5H–M). Both peptides exposed the P4-Asp to the surface at their N-terminal region (Fig. 5H, 5K), then a central hydrophobic patch with 7AV8 and 7AVAV10 (for NA181-190 and NA181-191 peptides, respectively) adopting different conformations (Fig. 5H, 5K). The NA181-190 peptide side chains of the 7AV8 motif are fully exposed (Fig. 5E, 5I and 5L), while in the NA181-191 peptide conformation they are partially buried against the His155 of the α2-helix, and their backbone is exposed instead (Fig. 5E, 5J and 5M).
Overall, despite the high sequence identity of the two peptides, the additional residue within the NA181-191 epitope was accommodated within the antigen binding cleft and completely altered the conformation of the NA181-191 peptide compared to that observed for the shorter overlapping NA181-190 peptide. The substantial structural differences between the two pMHCI complexes present an entirely different surface to T cells and thus provide an explanation for the unique T cell reactivities.
Discussion
We have observed that H-2Db molecules bound to two substantially overlapping peptides derived from the IAV NA protein induced entirely distinct populations of CD8+ T cells after viral infection. Both the NA181-190 and NA181-191 peptides were directly identified from H-2Db molecules both after direct IAV infection of cell lines and during cross-presentation. Moreover, reproducible immune responses were observed to both of these epitopes following in vivo IAV infection, demonstrating that both of these peptides are naturally processed and presented following infection. Intriguingly, despite differing only by a single amino acid extension at the carboxy terminus of the peptide, we have demonstrated completely distinct reactivities of responding T cell populations, evidenced by tetramer staining, TCR repertoires and functionality. We found that the unique reactivities of H-2DbNA181-190- and H-2DbNA181-191- specific CD8+ T cells originated in the naïve repertoire, indicating that it was not a product of selective expansion of preferred T cells.
T cell epitope identification is critically important for the ongoing development of immunological treatments for allergies, cancer and chronic virus immunotherapies, and the development of T cell-based vaccines against pathogens such as influenza or malaria. Historically, CD8+ T cell peptide epitopes have been identified by assaying reactivity of T cell clones to overlapping peptide pools of defined length. Once reactivity to a pool is observed the specific reactivity is then honed to a particular peptide and the minimal epitope is defined as the shortest peptide eliciting the most robust response (30, 31). A more targeted approach is the use of T cell epitope prediction tools based on algorithms that have been trained on previously defined epitopes (32), which can reduce the number of peptides that need to be tested experimentally. An assumption in defining minimal T cell epitopes is that MHCI restricted reactivity directed toward particular peptide regions is related. Thus, largely overlapping peptides would be considered to be eliciting the same or similar populations of T cells. Such analyses also provide little insight into which of the overlapping epitopes are naturally processed and presented.
Mass spectrometry-based immunopeptidomics approaches have changed the way we think about T cell epitope discovery. In particular, the direct global analysis of MHCI-presented peptides has yielded the surprising finding that a sizeable proportion of MHCI-bound peptides are longer than the canonical lengths of 8-10 aa (4, 6, 33, 34). Moreover, in several instances - including in the recent analyses of naturally processed and presented Vaccina virus (VacV)-derived peptides presented by mouse H-2b (4) and influenza B-derived peptides presented by HLA-A2 (35) - MHCI molecules have been found to present multiple overlapping peptides. In one case after VacV infection, the H-2Db molecule was found to present five peptides derived from the F5 protein that all had the same amino terminus (aa 279) but differed from each other by an extension of a single amino acid at the carboxy terminal end (4) . How presentation of such similar peptides impacts on their antigenicity and the elicitation of T cell responses will enable a deeper understanding, and potential predictability, of the structure-function relationship for TCR recognition of pMHCI. Moreover, the natural presentation of these heterogenous but related peptide antigens almost certainly imparts an advantage to the host; unravelling the impact of such presentation on TCR breadth and diversity and its role in controlling infection will inform more effective vaccine design and clinical monitoring.
The unique CD8+ T cell reactivities observed here in response to H-2DbNA181-190 and H-2DbNA181-191 can be explained by the distinctive topologies of these peptides bound to H-2Db. A key feature of peptide binding to MHCI is that the ends of the MHCI peptide binding cleft are closed, meaning that accommodation of longer peptides is often achieved by maintaining the peptide termini as anchors, and causing the central part of the peptide to protrude out of the cleft (36–38). This was observed for two HIV Nef derived peptides (Nef 138-145 (RYPLTFGW) and Nef 138-147 (RYPLTFGWCF)) bound to HLA-A*24:02, which induced distinct CTL reactivities in HIV+ individuals (39). In these structures, the central residues (P4-7) of Nef 138-147 protruded, resulting in the solvent exposure of a phenylalanine residue. In the current study, instead of bulging out, H-2DbNA181-191 is stabilized inside the H-2Db cleft, resulting in two unique peptide arrangements despite the almost complete identity of the bound peptides. Those unique pMHCI morphologies present an entirely different landscape to the T-cell. Given the unique closed termini of the MHCI binding cleft, as compared to MHCII which is open-ended and can accommodate longer peptides by overhang at either terminus, it is possible that, as a general phenomenon, even highly homologous peptides of different lengths are completely antigenically distinct. This is supported by a study that used a combinatorial peptide library approach to assess patterns of cross-reactivity in TCRs of defined specificity, which revealed that peptide length was the best predictor of TCR cross-reactivity (40). Interestingly, different pMHCI landscapes not only induce distinct TCR repertoires but also allow for distinct recognition strategies adopted by TCRs. For example, structures of TCRs recognizing overlapping 13-mer and 11-mer EBV-derived BZLF1 peptides presented by HLA-B*35:08 and HLA-B*35:01, respectively, revealed completely distinct TCR recognition ‘solutions’. The HLA-B*35:08-13mer-specific TCR perched on top of the rigid bulged peptide, making minimal MHC contacts, while the HLA-B*35:01-11mer-specific TCR flattened the more malleable 11-mer, considerably changing the pMHCI conformation upon binding (38, 41). Recent immunopeptidomics data demonstrating the frequency with which overlapping peptides are naturally processed and presented (4, 6, 35, 42), highlight the possibility of encoding multiple distinct antigenic epitopes in the one peptide sequence, facilitating the targeting of multiple epitopes through a single vaccine construct.
Exceptions to this rule exist, however. Elongated peptides can alternatively be accommodated by MHCI via N- or C-terminal extensions (42–44). A comparison of structures of synthesized 10-, 12-, 14-, and 20-mer overlapping peptides in HLA-B*08:01 revealed that the peptides were accommodated via N terminal overhang and that the 8aa core binding region of all of these peptides showed an almost identical structure in the cleft (45), suggesting that N- or C- terminally extended conformations may be less likely to generate antigenically distinct pMHCI complexes. Importantly, the arginine residue in the HLA-B*0801 A pocket was critical for opening up the N terminus and allowing peptide overhang, suggesting that the ability to bind elongated peptides via maintenance of terminal anchors versus terminal overhang may be HLA allomorph dependent (45). In addition, structural dissimilarity between pMHCI complexes doesn’t always result in distinct TCR reactivities (46, 47). For example, TCR repertoires specific for HLA-A2 bound to either the 26-35 decamer peptide of MART-1, or the 27-35 nonamer peptide were found to be largely cross-reactive, despite considerable structural differences between the two pMHCI complexes (46). Thus, further analysis of T cell reactivities to overlapping peptides is essential to determine the generality of these findings.
We observed here that the distinct T cell specificity for H-2DbNA181-190 and H-2DbNA181-191 epitopes was not a consequence of selective expansion after viral infection but pre-existed in the available naïve repertoire. Combined with the TCRdist Nearest Neighbor analysis, this further indicated that no part of the pMHCI structure was perceived as antigenically similar by the TCR repertoire. Moreover, it indicated that the development of the naïve repertoires was likely driven by selection on distinct self peptide-MHCI complexes in the thymus (48).
In summary, we have identified completely overlapping CD8+ T cell peptides from influenza A virus that are both immunogenic in B6 mice and that induce completely distinct CTL responses after infection. Understanding the extent to which such overlapping epitopes are naturally processed and presented by MHCI, as well as the extent to which they induce uniquely reactive T cell populations has implications for the selection of epitopes in multi-epitope T cell-based vaccines.
Supplementary Material
Key Points.
Overlapping IAV peptides are recognized by completely distinct CD8 T cell populations
Distinct T cell reactivities are reflected in TCR use, function, and naïve repertoire.
The single aa peptide extension drives distinct peptide conformations within H-2Db
Acknowledgments
We thank Dr Phil Bradley (Fred Hutchinson Cancer Research Centre, Seattle) for assistance with TCRdist analysis. We thank the staff at the Monash Animal Research Platform Facility, Monash FlowCore Platform, and the Monash Macromolecular crystallization facility. We thank the staff at the Australian Synchrotron for assistance with data collection.
References
- 1.Tscharke DC, Croft NP, Doherty PC, and La Gruta NL. 2015. Sizing up the key determinants of the CD8(+) T cell response. Nat Rev Immunol 15: 705–716. [DOI] [PubMed] [Google Scholar]
- 2.Zhong W, Reche PA, Lai CC, Reinhold B, and Reinherz EL. 2003. Genome-wide characterization of a viral cytotoxic T lymphocyte epitope repertoire. J Biol Chem 278: 45135–45144. [DOI] [PubMed] [Google Scholar]
- 3.Altuvia Y, and Margalit H. 2004. A structure-based approach for prediction of MHC-binding peptides. Methods 34: 454–459. [DOI] [PubMed] [Google Scholar]
- 4.Croft NP, Smith SA, Pickering J, Sidney J, Peters B, Faridi P, Witney MJ, Sebastian P, Flesch IEA, Heading SL, Sette A, La Gruta NL, Purcell AW, and Tscharke DC. 2019. Most viral peptides displayed by class I MHC on infected cells are immunogenic. Proc Natl Acad Sci U S A 116: 3112–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davenport MP, Ho Shon IA, and Hill AV. 1995. An empirical method for the prediction of T-cell epitopes. Immunogenetics 42: 392–397. [DOI] [PubMed] [Google Scholar]
- 6.Wu T, Guan J, Handel A, Tscharke DC, Sidney J, Sette A, Wakim LM, Sng XYX, Thomas PG, Croft NP, Purcell AW, and La Gruta NL. 2019. Quantification of epitope abundance reveals the effect of direct and cross-presentation on influenza CTL responses. Nat Commun 10: 2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.La Gruta NL, Turner SJ, and Doherty PC. 2004. Hierarchies in cytokine expression profiles for acute and resolving influenza virus-specific CD8+ T cell responses: correlation of cytokine profile and TCR avidity. J Immunol 172: 5553–5560. [DOI] [PubMed] [Google Scholar]
- 8.Cukalac T, Kan WT, Dash P, Guan J, Quinn KM, Gras S, Thomas PG, and La Gruta NL. 2015. Paired TCRalphabeta analysis of virus-specific CD8(+) T cells exposes diversity in a previously defined ‘narrow’ repertoire. Immunol Cell Biol 93: 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dash P, McClaren JL, Oguin TH 3rd, Rothwell W, Todd B, Morris MY, Becksfort J, Reynolds C, Brown SA, Doherty PC, and Thomas PG. 2011. Paired analysis of TCRalpha and TCRbeta chains at the single-cell level in mice. J Clin Invest 121: 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brochet X, Lefranc MP, and Giudicelli V. 2008. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res 36: W503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giudicelli V, Brochet X, and Lefranc MP. 2011. IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb Protoc 2011: 695–715. [DOI] [PubMed] [Google Scholar]
- 12.Gras S, Chadderton J, Del Campo CM, Farenc C, Wiede F, Josephs TM, Sng XYX, Mirams M, Watson KA, Tiganis T, Quinn KM, Rossjohn J, and La Gruta NL. 2016. Reversed T Cell Receptor Docking on a Major Histocompatibility Class I Complex Limits Involvement in the Immune Response. Immunity 45: 749–760. [DOI] [PubMed] [Google Scholar]
- 13.Gras S, Wilmann PG, Chen Z, Halim H, Liu YC, Kjer-Nielsen L, Purcell AW, Burrows SR, McCluskey J, and Rossjohn J. 2012. A structural basis for varied alphabeta TCR usage against an immunodominant EBV antigen restricted to a HLA-B8 molecule. J Immunol 188: 311–321. [DOI] [PubMed] [Google Scholar]
- 14.Aragao D, Aishima J, Cherukuvada H, Clarken R, Clift M, Cowieson NP, Ericsson DJ, Gee CL, Macedo S, Mudie N, Panjikar S, Price JR, Riboldi-Tunnicliffe A, Rostan R, Williamson R, and Caradoc-Davies TT. 2018. MX2: a high-flux undulator microfocus beamline serving both the chemical and macromolecular crystallography communities at the Australian Synchrotron. J Synchrotron Radiat 25: 885–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kabsch W 2010. Xds. Acta Crystallogr D Biol Crystallogr 66: 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evans P 2006. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62: 72–82. [DOI] [PubMed] [Google Scholar]
- 17.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, and Wilson KS. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40: 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valkenburg SA, Gras S, Guillonneau C, La Gruta NL, Thomas PG, Purcell AW, Rossjohn J, Doherty PC, Turner SJ, and Kedzierska K. 2010. Protective efficacy of cross-reactive CD8+ T cells recognising mutant viral epitopes depends on peptide-MHC-I structural interactions and T cell activation threshold. PLoS Pathog 6: e1001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emsley P, and Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- 21.Blanc E, Roversi P, Vonrhein C, Flensburg C, Lea SM, and Bricogne G. 2004. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr D Biol Crystallogr 60: 2210–2221. [DOI] [PubMed] [Google Scholar]
- 22.Almeida JR, Price DA, Papagno L, Arkoub ZA, Sauce D, Bornstein E, Asher TE, Samri A, Schnuriger A, Theodorou I, Costagliola D, Rouzioux C, Agut H, Marcelin AG, Douek D, Autran B, and Appay V. 2007. Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J Exp Med 204: 2473–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boyd A, Almeida JR, Darrah PA, Sauce D, Seder RA, Appay V, Gorochov G, and Larsen M. 2015. Pathogen-Specific T Cell Polyfunctionality Is a Correlate of T Cell Efficacy and Immune Protection. PLoS One 10: e0128714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lichterfeld M, Yu XG, Waring MT, Mui SK, Johnston MN, Cohen D, Addo MM, Zaunders J, Alter G, Pae E, Strick D, Allen TM, Rosenberg ES, Walker BD, and Altfeld M. 2004. HIV-1-specific cytotoxicity is preferentially mediated by a subset of CD8(+) T cells producing both interferon-gamma and tumor necrosis factor-alpha. Blood 104: 487–494. [DOI] [PubMed] [Google Scholar]
- 25.La Gruta NL, Doherty PC, and Turner SJ. 2006. A correlation between function and selected measures of T cell avidity in influenza virus-specific CD8+ T cell responses. Eur J Immunol 36: 2951–2959. [DOI] [PubMed] [Google Scholar]
- 26.Dash P, Fiore-Gartland AJ, Hertz T, Wang GC, Sharma S, Souquette A, Crawford JC, Clemens EB, Nguyen THO, Kedzierska K, La Gruta NL, Bradley P, and Thomas PG. 2017. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 547: 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clancy-Thompson E, Devlin CA, Tyler PM, Servos MM, Ali LR, Ventre KS, Bhuiyan MA, Bruck PT, Birnbaum ME, and Dougan SK. 2018. Altered Binding of Tumor Antigenic Peptides to MHC Class I Affects CD8(+) T Cell-Effector Responses. Cancer Immunol Res 6: 1524–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valkenburg SA, Quinones-Parra S, Gras S, Komadina N, McVernon J, Wang Z, Halim H, Iannello P, Cole C, Laurie K, Kelso A, Rossjohn J, Doherty PC, Turner SJ, and Kedzierska K. 2013. Acute emergence and reversion of influenza A virus quasispecies within CD8+ T cell antigenic peptides. Nat Commun 4: 2663. [DOI] [PubMed] [Google Scholar]
- 29.Josephs TM, Grant EJ, and Gras S. 2017. Molecular challenges imposed by MHC-I restricted long epitopes on T cell immunity. Biol Chem 398: 1027–1036. [DOI] [PubMed] [Google Scholar]
- 30.Lewinsohn DA, Winata E, Swarbrick GM, Tanner KE, Cook MS, Null MD, Cansler ME, Sette A, Sidney J, and Lewinsohn DM. 2007. Immunodominant tuberculosis CD8 antigens preferentially restricted by HLA-B. PLoS Pathog 3: 1240–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Powlson J, Wright D, Zeltina A, Giza M, Nielsen M, Rampling T, Venkatrakaman N, Bowden TA, Hill AVS, and Ewer KJ. 2019. Characterization of Antigenic MHC-Class-I-Restricted T Cell Epitopes in the Glycoprotein of Ebolavirus. Cell Rep 29: 2537–2545 e2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paul S, Croft NP, Purcell AW, Tscharke DC, Sette A, Nielsen M, and Peters B. 2019. Benchmarking predictions of MHC class I restricted T cell epitopes. Biorxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hassan C, Chabrol E, Jahn L, Kester MG, de Ru AH, Drijfhout JW, Rossjohn J, Falkenburg JH, Heemskerk MH, Gras S, and van Veelen PA. 2015. Naturally processed non-canonical HLA-A*02:01 presented peptides. J Biol Chem 290: 2593–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rist MJ, Theodossis A, Croft NP, Neller MA, Welland A, Chen Z, Sullivan LC, Burrows JM, Miles JJ, Brennan RM, Gras S, Khanna R, Brooks AG, McCluskey J, Purcell AW, Rossjohn J, and Burrows SR. 2013. HLA peptide length preferences control CD8+ T cell responses. J Immunol 191: 561–571. [DOI] [PubMed] [Google Scholar]
- 35.Koutsakos M, Illing PT, Nguyen THO, Mifsud NA, Crawford JC, Rizzetto S, Eltahla AA, Clemens EB, Sant S, Chua BY, Wong CY, Allen EK, Teng D, Dash P, Boyd DF, Grzelak L, Zeng W, Hurt AC, Barr I, Rockman S, Jackson DC, Kotsimbos TC, Cheng AC, Richards M, Westall GP, Loudovaris T, Mannering SI, Elliott M, Tangye SG, Wakim LM, Rossjohn J, Vijaykrishna D, Luciani F, Thomas PG, Gras S, Purcell AW, and Kedzierska K. 2019. Human CD8(+) T cell cross-reactivity across influenza A, B and C viruses. Nat Immunol 20: 613–625. [DOI] [PubMed] [Google Scholar]
- 36.Guo HC, Jardetzky TS, Garrett TP, Lane WS, Strominger JL, and Wiley DC. 1992. Different length peptides bind to HLA-Aw68 similarly at their ends but bulge out in the middle. Nature 360: 364–366. [DOI] [PubMed] [Google Scholar]
- 37.Liu YC, Chen Z, Burrows SR, Purcell AW, McCluskey J, Rossjohn J, and Gras S. 2012. The energetic basis underpinning T-cell receptor recognition of a super-bulged peptide bound to a major histocompatibility complex class I molecule. J Biol Chem 287: 12267–12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tynan FE, Burrows SR, Buckle AM, Clements CS, Borg NA, Miles JJ, Beddoe T, Whisstock JC, Wilce MC, Silins SL, Burrows JM, Kjer-Nielsen L, Kostenko L, Purcell AW, McCluskey J, and Rossjohn J. 2005. T cell receptor recognition of a ‘super-bulged’ major histocompatibility complex class I-bound peptide. Nat Immunol 6: 1114–1122. [DOI] [PubMed] [Google Scholar]
- 39.Sun X, Fujiwara M, Shi Y, Kuse N, Gatanaga H, Appay V, Gao GF, Oka S, and Takiguchi M. 2014. Superimposed epitopes restricted by the same HLA molecule drive distinct HIV-specific CD8+ T cell repertoires. J Immunol 193: 77–84. [DOI] [PubMed] [Google Scholar]
- 40.Ekeruche-Makinde J, Miles JJ, van den Berg HA, Skowera A, Cole DK, Dolton G, Schauenburg AJ, Tan MP, Pentier JM, Llewellyn-Lacey S, Miles KM, Bulek AM, Clement M, Williams T, Trimby A, Bailey M, Rizkallah P, Rossjohn J, Peakman M, Price DA, Burrows SR, Sewell AK, and Wooldridge L. 2013. Peptide length determines the outcome of TCR/peptide-MHCI engagement. Blood 121: 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tynan FE, Reid HH, Kjer-Nielsen L, Miles JJ, Wilce MC, Kostenko L, Borg NA, Williamson NA, Beddoe T, Purcell AW, Burrows SR, McCluskey J, and Rossjohn J. 2007. A T cell receptor flattens a bulged antigenic peptide presented by a major histocompatibility complex class I molecule. Nat Immunol 8: 268–276. [DOI] [PubMed] [Google Scholar]
- 42.Pymm P, Illing PT, Ramarathinam SH, O’Connor GM, Hughes VA, Hitchen C, Price DA, Ho BK, McVicar DW, Brooks AG, Purcell AW, Rossjohn J, and Vivian JP. 2017. MHC-I peptides get out of the groove and enable a novel mechanism of HIV-1 escape. Nat Struct Mol Biol 24: 387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McMurtrey C, Trolle T, Sansom T, Remesh SG, Kaever T, Bardet W, Jackson K, McLeod R, Sette A, Nielsen M, Zajonc DM, Blader IJ, Peters B, and Hildebrand W. 2016. Toxoplasma gondii peptide ligands open the gate of the HLA class I binding groove. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Remesh SG, Andreatta M, Ying G, Kaever T, Nielsen M, McMurtrey C, Hildebrand W, Peters B, and Zajonc DM. 2017. Unconventional Peptide Presentation by Major Histocompatibility Complex (MHC) Class I Allele HLA-A*02:01: BREAKING CONFINEMENT. J Biol Chem 292: 5262–5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li L, Batliwala M, and Bouvier M. 2019. ERAP1 enzyme-mediated trimming and structural analyses of MHC I-bound precursor peptides yield novel insights into antigen processing and presentation. J Biol Chem 294: 18534–18544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borbulevych OY, Insaidoo FK, Baxter TK, Powell DJ Jr., Johnson LA, Restifo NP, and Baker BM. 2007. Structures of MART-126/27–35 Peptide/HLA-A2 complexes reveal a remarkable disconnect between antigen structural homology and T cell recognition. J Mol Biol 372: 1123–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riley TP, Hellman LM, Gee MH, Mendoza JL, Alonso JA, Foley KC, Nishimura MI, Vander Kooi CW, Garcia KC, and Baker BM. 2018. T cell receptor cross-reactivity expanded by dramatic peptide-MHC adaptability. Nat Chem Biol 14: 934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mandl JN, Monteiro JP, Vrisekoop N, and Germain RN. 2013. T cell-positive selection uses self-ligand binding strength to optimize repertoire recognition of foreign antigens. Immunity 38: 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.