

Abstract
The innate immune system is the first line of defense against bacterial and viral infections. The recognition of pathogen associated molecular patterns (PAMPs) by the RLRs, TLRs, and cGAS leads to the induction of type-I interferons (IFN-I) by activating the transcription factor IRF-3. Although the mechanism of IRF-3 activation has been extensively studied, the structural basis of IRF-3 activation upon phosphorylation remains not fully understood. Here, we determined the crystal structures of phosphorylated human and mouse IRF-3 bound to CBP (cAMP response element binding protein (CREB)-binding protein), which reveal that phosphorylated IRF-3 forms a dimer via pSer386 (pSer379 in mouse IRF-3) and a downstream pLxIS motif. Size-exclusion chromatography and cell-based studies show that mutations of key residues interacting with pSer386 severely impair IRF-3 activation and IFN-β induction. By contrast, phosphorylation of Ser396 within the pLxIS motif of human IRF-3 only plays a moderate role in IRF-3 activation. The mouse IRF-3/CBP complex structure reveals that the mechanism of mouse IRF-3 activation is similar but distinct from human IRF-3. These structural and functional studies reveal the detailed mechanism of IRF-3 activation upon phosphorylation.
Keywords: Innate immunity, interferons, transcription factor, phosphorylation, crystal structure
INTRODUCTION
The innate immune system detects the pathogen-associated molecular patterns (PAMPs) such as nucleic acids, lipopolysaccharide (LPS) through pattern recognition receptors (PRRs), which triggers the induction of a variety of cytokines including type-I interferons (IFN-I) to initiate host defense against pathogens (1–7). Viral or bacterial DNA in cytosol are recognized by cyclic GMP-AMP synthase (cGAS), which catalyzes the synthesis of a cyclic dinucleotide cGAMP. cGAMP binds to the adaptor STING (Stimulator of Interferon Genes) and mediates the recruitment and activation of TBK1 (TANK-Binding Kinase 1) and IRF-3 (5, 8). Activated IRF-3 translocates to the nucleus and initiates the transcription of IFN-β gene with other transcription factors such as NF-κB (7, 9–13). Double-stranded RNA and LPS can be recognized by retinoic acid-inducible protein 1 (RIG-I)-like receptors (RLRs) and Toll-like receptors (TLRs), respectively, to induce the expression of IFN-Is through the adaptors MAVS (mitochondrial antiviral signaling) and TRIF (TIR domain-containing adaptor inducing IFN-β). Interestingly, all these three signaling pathways converge at the recruitment of IRF-3 via a conserved pLxIS (p, hydrophilic residue, x, any residue, S, phosphorylation site) motif within the adaptor proteins (9–13).
The interferon regulatory factor (IRF) family transcription factors contain nine members (IRF-1 through IRF-9). These transcription factors contain a highly conserved N-terminal DNA-binding domain and a relatively divergent C-terminal regulatory domain, suggesting that most members function non-redundantly (14). IRF-3 is a key transcription factor that regulates the expression of type I interferon genes. Under resting conditions, IRF-3 adopts an auto-inhibited conformation and is ubiquitously accumulated in the cytoplasm. Bacterial or viral infection triggers the activation of IRF-3 through various innate immune sensing pathways. Phosphorylated IRF-3 binds to p300/CBP, translocates to the nucleus, and initiates the transcription of IFN-I genes (14–16). Interestingly, IRF-7, which is closely related to IRF-3 in terms of the conserved regulatory domain, also plays an important role in regulating the expression of IFN-Is (17).
The mechanism of IRF-3 activation has been extensively studied, which showed that IRF-3 is activated through phosphorylation of the C-terminal serine-rich repeat (SRR) (16). Previous studies by the Fujita lab showed that the phosphorylation site 1 (residues Ser385, Ser386 of human IRF-3) plays a key role in IRF-3 activation. The phosphorylation of Ser386 induced by viral infection has been detected by a specific antibody. They also observed that the dimerization of IRF-3 was abolished by the mutation of Ser386 (18–20). In addition, the Hiscott lab observed that the phosphorylation site 2, which includes residues Ser396, Ser398, Ser402, Thr404, Ser405 of human IRF-3, plays a critical role in IRF-3 activation. The phosphomimetic mutation of these residues (IRF-3 5D) results in a constitutively active phenotype. Moreover, they observed that the S396D mutation alone induces IFN-I expression (21, 22), suggesting that Ser396 also plays a critical role in IRF-3 activation. Another study by the Harrison lab proposed a two-step phosphorylation and activation model, which suggest that phosphorylation at site 2 leads to the alleviation of IRF-3 auto-inhibition that facilitates the phosphorylation at site 1 and eventually leads to the activation of IRF-3 (23). Later on, studies by the Lin group showed that IRF-3 mutant S386D/S396D bound to CBP forms a stable oligomer (24), suggesting that the phosphorylation of both Ser386 and Ser396 is essential for human IRF-3 activation. Based on these studies, we mutated Ser386 and Ser396 to glutamic acid in a truncated form of IRF-3 (residues 189 to 398) and determined the structure of the phosphomimetic IRF-3 in complex with CBP, which reveals that the phosphomimetic IRF-3 mutant forms a dimer (12). However, the higher affinity between phosphorylated IRF-3/CBP complex than that of the S386/396E mutant indicates that the phosphomimetic mutation does not fully recapitulate the interactions between phosphorylated IRF-3.
To elucidate the exact mechanism of IRF-3 activation upon phosphorylation by TBK1, we co-expressed truncated forms of human and mouse IRF-3 C-terminal domains with a CBP fragment, phosphorylated these complexes with TBK1, and determined the crystal structures of the phosphorylated IRF-3/CBP dimers. These structures reveal the molecular basis of IRF-3 activation upon phosphorylation. Biochemical and functional studies based on the structures show that mutations of the key residues mediating IRF-3 dimerization upon phosphorylation dramatically impair IRF-3 dimerization and IRF-3-mediated signaling.
MATERIALS AND METHODS
Protein expression and purification
The cDNA encoding human IRF-3 (residues 189–398), mouse IRF-3 (residues 184–390) dimerization domains were cloned into a modified pET-28a (+) vector containing an N-terminal His6-SUMO tag with appropriate primers obtained from Integrated DNA Technologies (IDT). SUMO fusion of human CBP (residues 2065 to 2111) was cloned into the pET-22b (+) vector using appropriate primers from IDT. Sequences of all the constructs were confirmed by DNA Sequencing. The plasmid containing human IRF-3 or mouse IRF-3 dimerization domain was co-transformed with CBP plasmid into Escherichia coli BL21 (DE3) cells. The cells were grown on LB agar plates containing both kanamycin and ampicillin. Next day, the cell colonies from the plates were transferred to 6 liters of LB liquid medium in flasks with kanamycin and ampicillin in an incubator shaker at 37 °C under 225 rpm. When OD600 reaches ~1.2, BL21 cells were induced with 0.4 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) overnight at 16 °C. The cells were harvested by centrifugation at 4000 rpm for 10 minutes and then suspended in a 200 mL lysis buffer containing 300 mM NaCl and 50 mM Tris⋅HCl (pH 8.0). The cells were lysed by sonication for 10 minutes with 0.5 sec pulse and 0.5 sec rest and the cell lysate was centrifuged at 16,000 rpm for 30 minutes. The supernatant was loaded onto a Ni2+-NTA column (Qiagen). Then a 200 mL of washing buffer containing 500 mM NaCl, 20 mM Tris⋅HCl, 25 mM Imidazole at pH 7.5 was used to wash non-specific binding proteins off the Ni2+-NTA column. The target proteins were then eluted with 75 mL of elution buffer containing 150 mM NaCl, 20 mM Tris⋅HCl, and 250 mM Imidazole at pH 7.5. The His6-SUMO-tag was cleaved with SUMO protease at a concentration of 10 μg/mL at 4 °C overnight and removed using a Ni2+-NTA column. The target proteins in the flow-through were centrifuged to ~2 mL and further purified by gel-filtration chromatography using a HiLoad 16/60 Superdex 75 column (GE Healthcare) equilibrated with a running buffer containing 150 mM NaCl and 20 mM Tris⋅HCl at pH 7.5. All the mutations were introduced into a full-length human IRF-3 pET-28a (+) plasmid using the QuikChange site-directed mutagenesis kit (Agilent) using proper primers. Sequences of all the plasmids were confirmed by DNA Sequencing. The IRF-3 mutants were expressed and purified the same way as the IRF-3 dimerization domain. Mouse TBK1 (mTBK1) was cloned into the pAcGHLTc vector with an N-terminal GST tag and a His6 tag. The 2 μg plasmid was transfected together with 2.5 μL Baculo-Gold bright linearized baculovirus DNA (BD Biosciences) into sf9 insect cells to generate recombinant baculovirus. The recombinant viruses were amplified for at least two rounds (4–6 days/round) before the large-scale protein expression. The insect cells at a density of 2.5 × 106 cells/mL were infected with the TBK1 recombinant baculovirus and cultured at 27°C and harvested 72 hours post infection by centrifugation at 3000 rpm for 10 minutes. The cells were lysed in a buffer containing 150 mM NaCl, 0.2 M Tris⋅HCl, 1% NP-40, 1 mM PMSF at pH 8.0 in a shaker at 4 °C for 2 hours. The cell lysate was centrifuged at 16,000 rpm for 30 minutes. The GST-TBK1 protein in the supernatant was mixed with 6 mL Ni2+-NTA beads and incubated in a shaker at 4 °C for 2 hours. The beads were then spun down and washed three times using a buffer containing 500 mM NaCl, 20 mM Tris⋅HCl, 25 mM Imidazole at pH 7.5. The target protein was eluted with a buffer containing 150 mM NaCl, 20 mM Tris⋅HCl, and 250 mM Imidazole at pH 7.5. The eluted protein was further purified by gel-filtration chromatography using a HiLoad 16/60 Superdex 200 column.
Analysis of phosphorylated IRF-3 by size-exclusion chromatography (SEC)
Purified full-length human IRF-3 proteins were mixed with GST-mTBK1 in a ratio of 10:1 (w/w) in a 1 mL reaction buffer with 20 mM HEPES pH 7.5, 10 mM MgCl2, 100 mM NaCl, 5 mM ATP, 0.1 mM Na3VO4, 5 mM NaF, 5 mM DTT at 27 °C for ~24 hours. The final concentration of the proteins was about 1 mg/mL. After ~24-hour incubation, the phosphorylated IRF-3 proteins were analyzed using a Superdex 200 (10/300 GL) column eluted with a buffer containing 20 mM Tris⋅HCl and 150 mM NaCl at pH 7.5.
Gradient gel and native gel electrophoresis of phosphorylated IRF-3
Full-length WT IRF-3 and all the mutant proteins were purified using Ni2+-NTA column followed by gel-filtration chromatography as described above. 10 μg of each sample was mixed with 5× loading dye of 250 mM Tris·HCl, pH 6.8, 10% SDS, 30% (v/v) Glycerol, 10 mM DTT, 0.05% (w/v) bromophenol blue and then resolved on 4–20% gradient gels in a buffer containing 25 mM Tris, 192 mM glycine and 0.1% SDS at pH8.4 at 100V for one hour. The gel was stained with Coomassie blue for one hour and destained with a solution containing H2O, methanol, and acetic acid in a ratio of 50/40/10 (v/v/v) until the bands were clearly seen. The gel image was taken using Bio-Rad imager. For native gel electrophoresis, the purified proteins were phosphorylated with GST-mTBK1 using the method described above and each of the phosphorylated proteins with non-phosphorylated wild-type was resolved on 10% native gels running in a buffer containing 25 mM Tris and 192 mM glycine pH 8.4 at 4°C at 100V for 30 minutes. The gels were stained and destained the same way as the gradient gels.
Crystallization, data collection, and structure determination
Purified human IRF-3 (residues 189–398) and mouse IRF3 (residues 184–390) bound to the CBP fragment (residues 2065–2111) were phosphorylated by GST-mTBK1. After 24 hour incubation, the proteins were purified using a HiLoad 16/60 Superdex 75 column eluted with 20 mM Tris⋅HCl and 150 mM NaCl at pH 7.5. The purified phosphorylated proteins were concentrated to a final concentration of ~5 mg/mL. The crystallization screen was performed by hanging drop vapor diffusion technique at 4 °C using Index, Crystal Screen and Crystal Screen 2 reagent kits from Hampton Research. Crystals of human IRF-3 bound to CBP were grown in 0.1 M sodium acetate pH 5.0, 0.2 M MgCl2, ~5% PEG 3350. Crystals of mouse IRF-3 in complex with CBP were grown in 0.2 M ammonium citrate tribasic at pH 7.0 with ~12% PEG 3350. The crystals were flash-frozen in liquid nitrogen in the reservoir solution containing 25% (vol/vol) glycerol. Diffraction data were collected at the Advanced Light Source beamlines 5.0.1 using a Quantum 315R CCD detector. The diffraction data were indexed and integrated with iMosflm and merged with Aimless in the CCP4 package (25). The structures of the pIRF-3/CBP complex were determined by molecular replacement (MR) using the structure of our phosphomimetic IRF-3/CBP complex (PDB ID code 5JEM) as the search model using Phaser in the Phenix package (26). The structures were manually rebuilt using Coot and refined with Phenix. Details of data quality and structure refinement are summarized in Table 1. The structural figures were generated with PyMOL (https://www.pymol.org).
Table 1.
Data collection and refinement statistics for phIRF-3/CBP and pmIRF-3/CBP complexes
| phIRF-3/CBP | pmIRF-3/CBP | |
|---|---|---|
| Data collection | ||
| Space group | C 2 | P 62 |
| Molecules per ASU Cell dimensions | 2 phIRF-3, 2 CBP | 2 pmIRF-3, 2 CBP |
| a, b, c (Å) | 124.01, 68.03, 55.92 | 118.80, 118.80, 72.17 |
| α, β, γ (°) | 90.0, 106.24, 90.0 | 90.0, 90.0, 120.0 |
| Resolution (Å) | 1.68 (1.7 to 1.68)* | 2.23 (2.30 to 2.23) |
| Rmerge | 10.9% (124.3%) | 8.9% (292%) |
| Rpim | 6.8% (79.0%) | 2.7% (86.8%) |
| CC(1/2) (%) | 99.2 (35.2) | 99.9 (38.2) |
| Unique reflections | 48116 | 28422 |
| I / σI | 6.5 (1.0) | 12.6 (1.0) |
| Completeness (%) | 94.7 (90.6) | 99.9 (100.0) |
| Redundancy | 3.4 (3.1) | 12.1 (12.3) |
| Refinement | ||
| Resolution (Å) | 46.93 to 1.68 | 51.44 to 2.23 |
| No. reflections (F > 0) | 48093 | 28389 |
| Rwork / Rfree | 19.1% (22.3%) | 23.1% (25.2%) |
| No. atoms | ||
| Protein | 3670 | 3702 |
| Water | 252 | 14 |
| B-factors (Å2) | ||
| Protein | 24.2 | 91.3 |
| Water | 27.8 | 67.9 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.006 | 0.001 |
| Bond angles (°) | 0.771 | 0.420 |
| Ramachandran plot favored (%) | 97.98 | 95.78 |
| Ramachandran plot outlier (%) | 0.45 | 0.0 |
One crystal was used to collect each of the dataset.
Values in parentheses are for highest-resolution shell.
IFN-β luciferase reporter assays
The cDNA encoding wild-type human IRF-3 was cloned into a pcDNA3.1(−) vector using appropriate primers. Mutants of hIRF-3 were generated using the QuikChange site-directed mutagenesis kit (Agilent). Sequences of the mutants were confirmed by DNA Sequencing. HEK293T cells were plated in CoStar White 96-well plates at 4 × 104 cells per well and each well contains 100 μl DMEM (1×) + GlutaMAX medium (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco). After ~24h incubation at 37°C, the cells were transfected with the IRF-3 plasmids (10 ng per transfection) using Lipofectamine 2000 reagent (Invitrogen) and Opti-MEM medium (Gibco) together with constant amount of IFN-β firefly luciferase reporter plasmids (20 ng per transfection), phRL-TK–Renilla luciferase plasmids (2 ng per transfection) (Promega), and human STING plasmids (0.2 ng per transfection). Transfections with empty pcDNA3.1(−) and WT hIRF-3 with no STING plasmid were used as controls. The cells were incubated for another 24 h to allow the expression of the genes. The half of the cells in the plates were treated with 30 μg/mL cGAMP dissolved in DMEM (1×) + GlutaMAX medium and the other half were treated with the medium only. After ~16 h incubation, the cells were analyzed using the Dual-Glo luciferase reporter assay kit (Promega). Luminescence was quantified with the BioTek Synergy HTX Multi-Mode microplate reader. The relative firefly luciferase activity was normalized by the Renilla luciferase activity. The relative IFN-β reporter fold of induction represents the ratio normalized to control plasmid values with the same treatment.
Western blot
HEK293T cells were transfected with empty pcDNA3.1(−), WT IRF-3 or IRF-3 mutants together with human STING plasmid. The cells were stimulated by 30 μg/mL cGAMP added to the culture media 24h post transfection. After ~16 h incubation, the cells were washed and suspended in PBS and then lysed in 150 mM NaCl, 200 mM Tris⋅HCl (pH 7.5), 1 mM EDTA, and 1% Nonidet P-40 supplemented with one complete EDTA-free protease inhibitor mixture tablet (Roche) and one PhosSTOP phosphatase inhibitor mixture tablet (Roche) for each 10 mL of lysis buffer. The proteins were resolved on 10% SDS-PAGE at 100V for 1.5h and transferred to PVDF membrane in a transfer buffer containing 1x Tris-Glycine + 20% methanol for another 1.5 hours. Then 5% milk in 1xPBST solution was used to block the membrane for 1 hour followed by three rinses with PBST. Next the membrane was incubated with primary antibodies dissolved in 1xPBST with 5% BSA overnight at 4 °C. The membrane was washed three times using PBST next day and further incubated with the corresponding HRP-conjugated secondary antibodies dissolved in PBST. The following antibodies were used in the Western blot experiment: anti–IRF-3 (1:1,000; sc-9082; Santa Cruz) and anti-Actin (1: 4,000; HHF35; Pierce). The proteins were visualized using the Western Lightening Plus ECL (PerkinElmer) according to the manufacturer’s protocol. For the detection of phosphorylated IRF-3, half of the cells transfected with empty pcDNA3.1(−) and WT IRF-3 were stimulated by 30 μg/mL cGAMP and the other half were treated with the medium only. Anti-IRF-3 phospho-Ser386 (1:2,500; ab76493; Abcam) and anti-IRF-3 phospho-Ser396 (1:1,000; 4947S; Cell Signaling) were used.
Immunocytochemistry
HEK293T cells were grown on poly-L-lysine coated coverslips placed in 12-well plates for 24 h and then co-transfected with 20 ng WT IRF-3-HA and 1ng STING plasmids or 20 ng mutant IRF-3-HA and 1ng STING respectively using lipofectamine 2000 reagent mixed with Opti-MEM medium (Gibco). 24 hours post transfection, the medium was replaced with fresh DMEM (1×) + GlutaMAX medium with or without 30 μg/mL cGAMP. After 12 h incubation, cells were washed using PBS, then fixed by 4% paraformaldehyde in PBS for 15 min at room temperature and permeabilized with PBST containing 0.5% Triton X-100 in PBS. Cells were washed and blocked with 5% fetal bovine serum (FBS) in PBST and then incubated with anti-HA tag primary antibody (Cell signaling, 3724; 1:100) overnight. The cells were washed three times with PBS and incubated with Alexa Fluor 488 Goat Anti-Rabbit IgG (Thermo Fisher Scientific, A11034, 1:1000) at room temperature for 1 hour. The coverslips were washed by PBS, mounted on slides with ProLong Gold antifade reagent with 4′−6-diamidino-2-phenylindole (DAPI) and then imaged under Olympus FV1000 fluorescence microscope. The scale bars in the images correspond to 20 μm in length. To quantify the amount of nuclear translocation, 12 IRF-3-HA highly expressed cells or cell clusters were randomly selected in each field. The ROI of nuclear or total IRF-3-HA fluorescence in the same cell or cell cluster was manually drawn. The area integrated fluorescence intensity was calculated by ImageJ (Version 1.51n).
Mass spectrometry
Molecular mass of WT IRF-3 and phosphorylated IRF-3 were determined by MALDI-TOF mass spectrometry using a Bruker Ultraflextreme TOF-TOF mass spectrometer (Protein Chemistry Laboratory, Texas A&M University). The samples were solid phase extracted using Protea LithTip C4 and analyzed using alpha-cyanohydroxycinnamic acid as matrix using the dried drop method. The mass spectrometer was operated in reflector mode and calibrated with angiotensin II, fibrinopeptide, renin substrate and ACTH (18–39 fragment).
Cell culture
HEK293T cells (ATCC, CRL-3216) were cultured in DMEM (1×) + GlutaMAX medium (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco), streptomycin (100 μg/mL) and penicillin (100 U/mL) at 37 °C in a humidified atmosphere containing 5% CO2.
Statistical analysis
Statistical analyses for the luciferase reporter assay and quantification of fluorescence were carried out by Microsoft Excel and Prism respectively. All of the data are presented as mean ± SEM. Two-way ANOVA with Tukey’s multiple comparisons test was used to compare different groups. The statistical significance between the indicated samples and the control is designated as *P < 0.05, **P < 0.01, ***P < 0.001, or NS (P > 0.05).
Data deposition
The atomic coordinates and structural factors of the phosphorylated human and mouse IRF-3/CBP complexes have been deposited in the Worldwide Protein Data Bank, www.wwpdb.org (PDB: 7JFL and 7JFM, respectively).
RESULTS
The molecular basis of human IRF-3 activation upon phosphorylation
To investigate how phosphorylation activates IRF-3, we expressed and purified a truncated human IRF-3 (residues 189 to 398) in complex with a CBP fragment (residues 2065 to 2111) and phosphorylated the complex with TBK1. Mass spectrometry (MS) analysis showed that only one residue of IRF-3 is phosphorylated (Fig. 1A). Next, we crystalized the phosphorylated IRF-3/CBP complex and determined the structure at 1.8 Å resolution (Table 1). Consistent with the MS analysis, the IRF-3/CBP structure showed that only Ser386 was phosphorylated (Fig. 1B to E). Overall, phosphorylated IRF-3/CBP (phIRF-3/CBP) complex forms a dimer through phosphorylated Ser386 and the downstream DLHIS sequence (known as pLxIS motif) (Fig. 1B and C). Superposition of phosphorylated IRF-3/CBP complex and auto-inhibited IRF-3 (PDB: 1QWT) structures reveals a dramatic conformational change of the C-terminal tail upon phosphorylation (Fig. S1A and B). In auto-inhibited IRF-3, the C-terminal tail is folded and blocks the binding of CBP. In the phosphorylated IRF-3/CBP complex, the C-terminal tail of IRF-3 unfolds and interacts with another IRF-3 molecule through extensive hydrogen bonds, hydrophobic interaction, and electrostatic interaction (Fig. 1E, Fig. S1C, D and F). In addition, the IRF-3 dimer is also stabilized via hydrophobic interactions and hydrogen bonds in the central core region of the dimer (Fig. S1E).
Figure 1. Crystal structure of the phosphorylated human IRF-3 bound to CBP.
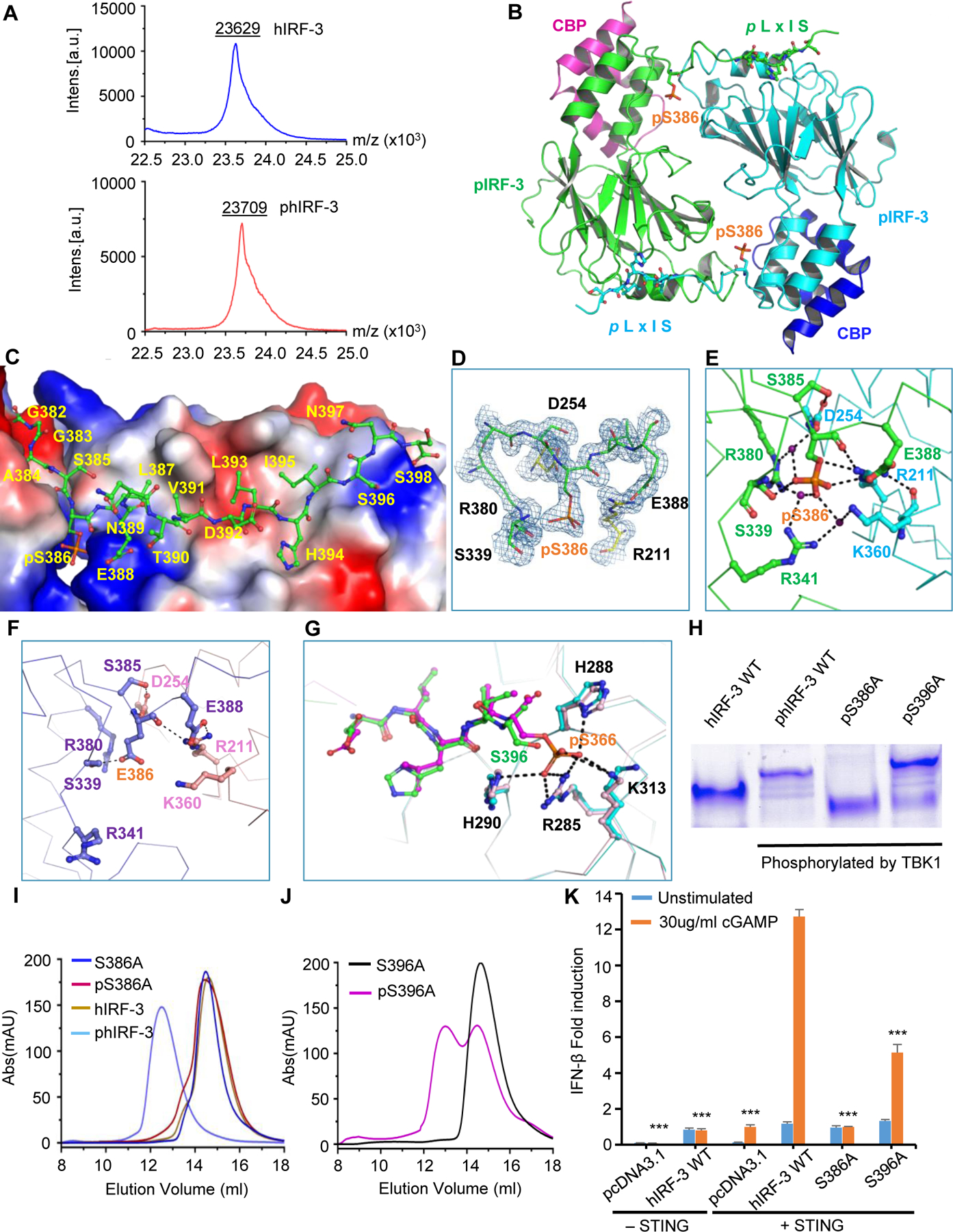
(A) Mass spectrometry analyses of human IRF-3 (residues 189–398) before and after TBK1 phosphorylation. (B) Structure of phosphorylated human IRF-3 (residues 189–398) bound to CBP. Phosphorylated Ser386 and residues of the pLxIS motif are indicated by ball-and-stick models. IRF-3 are shown as green and cyan ribbons. CBP are shown by magenta and blue ribbons. (C) Structure of IRF-3 C-terminal region containing phosphorylated Ser386 and the pLxIS motif. One IRF-3 molecule is shown by green ball-and-stick model. The other IRF-3 in the IRF-3 dimer is shown by the surface representation with positively charged and negatively charged surfaces in blue and red, respectively. (D) 2Fo - Fc map showing pSer386 and its interacting residues contoured at 2.0σ. (E) Interactions between pSer386 and surrounding residues. (F) Interactions between Glu386 and surrounding residues in the phosphomimetic S386/396E IRF-3 dimer (PDB: 5JEM). (G) Superposition of the phIRF-3/CBP dimer and pSTING/IRF-3 complex structures (PDB: 5JEJ). The green and cyan colored ribbons represent IRF-3 in the IRF-3/CBP dimer. The magenta ribbon represents phosphorylated STING and the pink colored ribbon indicates IRF-3 in pSTING/IRF-3 complex. (H) Native gel electrophoresis showing the dimerization state of wild-type IRF-3 and its S386A and S396A mutants upon phosphorylation by TBK1. (I) Size-exclusion chromatography showing how mutation S386A affect the dimerization of phosphorylated IRF-3 as compared to wild-type IRF-3. (J) Size-exclusion chromatography showing how mutation S396A affect the dimerization of phosphorylated IRF-3. (K) IFN-β luciferase reporter assays showing the effects of S386A and S396A mutations on IRF-3 mediated signaling. The data are mean ± s.e.m. and representative of three independent assays. **P < 0.01, ***P < 0.001 values were calculated by comparisons of signals in cells transfected with S386A, S396A mutants and those transfected with wild-type IRF-3.
The phIRF-3/CBP complex structure reveals that pSer386 reaches into a highly positively charged pocket surrounded by residues Arg211, Arg380, Arg341 and Lys360 and interacts with these residues via electrostatic interactions and an extensive network of hydrogen bonds (Fig. 1E). Specifically, pSer386 interacts with Arg211 from another IRF-3 molecule through a network of three hydrogen bonds. In addition, Arg211 also interacts with Lys360 and Glu388 through hydrogen bonds, thus making critical contribution to the formation of IRF-3 dimer. Arg380 forms a hydrogen bond with pSer386 within the same IRF-3 molecule via its side chain guanidinium group. A water molecule forms a network of three hydrogen bonds with Arg380, pSer386 and Asp254, making additional contributions to the interactions between pSer386 and Arg380. In addition, Asp254 of the other IRF-3 molecule interacts with Ser385 upstream pSer386 via two hydrogen bonds. Arg341 is within 4.0 Å from pSer386 and interacts with pSer386 through electrostatic interactions. Moreover, Arg341 also interacts with the phosphate group of pSer386 via a solvent mediated hydrogen bond. Similarly, the sidechain of Lys360 from the other IRF-3 in the dimer is within 4.0 Å from the phosphate group and interacts with pSer386 via electrostatic interaction and a solvent mediated hydrogen bond. In addition, Ser339 forms a hydrogen bond with pSer386 through its side chain hydroxyl group. Ser339 also interacts with the phosphate group of pSer386 via a solvent mediated hydrogen bond through its main-chain amine group. Based on these structural analyses, it is obvious that Arg380, Arg211, Ser339 and Asp254 play key roles in promoting IRF-3 dimerization by interacting with pSer386 though electrostatic interactions and a network of hydrogen bonds (Fig. 1E). Strikingly, every polar atom of pSer386 contributes to one or more hydrogen bonds in the phIRF-3 dimer.
Although the overall structure of the phosphomimetic S386/396E IRF-3/CBP dimer is similar to the phIRF-3/CBP dimer (Fig. S2A), Glu386, which mimics pSer386, contributes much less significantly to IRF-3 dimerization (Fig. 1F). Similar to the phIRF-3/CBP dimer, Arg380 is less than 4 Å from Glu386 and likely interacts with each other via electrostatic interactions. By contrast, the closest distance between the sidechains of Arg211 and Glu386 is over 5 Å and Arg211 forms no hydrogen bonds directly with Glu386. Instead, Arg211 stabilizes the phosphomimetic IRF-3 dimer mainly through its interaction with the sidechain of Glu388. In addition, Arg341 and Lys360 are further away from Glu386 and do not interact with Glu386 directly. Moreover, Lys360, Gln356, Ser351 are not involved in the interaction with any residues downstream of Glu386 in the phosphomimetic dimer (Fig. S2B). Based on these structural comparison, the phosphomimetic dimer does not fully recapitulate the extensive intermolecular interactions observed in the phIRF-3/CBP dimer, explaining why phosphorylated IRF-3 dimer is more stable compared to the phosphomimetic dimer.
In addition to Ser386, Ser396 within the pLxIS motif is also involved in IRF-3 activation and can be phosphorylated by TBK1 (6, 21, 23, 24, 27–29). Western blot showed that both Ser386 and Ser396 are phosphorylated upon cGAMP stimulation in HEK293T cells transfected with wild-type IRF-3 (Fig. S2C). In the phIRF-3/CBP complex structure, the electron density for Ser396 and surrounding residues was well defined. However, we did not observe the phosphorylation of Ser396 in the structure, likely due to the truncation at residue Ser398 that prevents the phosphorylation of Ser396 by TBK1. To investigate how phosphorylation of Ser396 contributes to IRF-3 activation, we superimposed the structure of pSTING/IRF-3 (PDB: 5JEJ) complex over the pIRF-3/CBP complex structure (Fig. 1G). The pLxIS motif of the phosphorylated STING is well aligned with the pIRF-3 pLxIS motif (Fig. 1G). Thus, it is likely that phosphorylated Ser396 could reach into the positively charged pocket surrounded by Arg285, His288, His290 and Lys313 and interact with them through electrostatic interactions in a similar fashion as pSer366 of pSTING (Fig. 1G).
In order to distinguish the roles of Ser386 and Ser396 in IRF-3 activation, we expressed and purified both S386A and S396A mutants of human IRF-3, phosphorylated them by TBK1, and analyzed them by native PAGE. Interestingly, the phosphorylated S386A mutant showed a single lower band, which is indicative of a monomer whereas the phosphorylated S396A mutant exhibited two bands indicating a mixture of both monomer and dimer (Fig. 1H). Consistent with the native gel result, size-exclusion chromatography (SEC) showed that phosphorylated S386A mutant was eluted at the same position as un-phosphorylated S386A and wild type IRF-3, while the phosphorylated S396A mutant showed two peaks, which correspond to a mixture of IRF-3 monomer and dimer (Fig. 1I and J). These results demonstrate that both Ser386 and Ser396 are involved in IRF-3 dimerization but Ser386 plays a more important role in IRF-3 activation compared to Ser396. To further explore how these two residues affect IRF-3 mediated signaling, we conducted IFN-β luciferase reporter assays in cells transfected with STING and IRF-3. We observed that the S386A mutation blocked the IFN-β reporter activation and the S396A mutation reduced the reporter signal by about 50%, demonstrating that both Ser386 and Ser396 are involved in IRF-3 mediated signaling but Ser386 is more crucial (Fig. 1K). Altogether, these extensive structural and functional studies provide critical insights into the detailed mechanism of IRF-3 activation upon phosphorylation.
The mechanism of mouse IRF-3 activation is similar but distinct from human IRF-3
Based on the structure of phIRF-3/CBP complex, the C-terminal tail of IRF-3 mediates the IRF-3 dimerization upon phosphorylation. We wondered if a similar mechanism is involved in IRF-3 activation in other mammalian species. First, we aligned the C-terminal sequences of IRF-3 across different species and observed that Ser386 and the pLxIS motif containing Ser396 of hIRF-3 are highly conserved in other species (Fig. 2A). However, Lys381 of mouse IRF-3 and Lys383 of rat IRF-3 replace Glu388 and Asn389 of human IRF-3 (Fig. 2A). To elucidate the mechanism of mouse IRF-3 activation, we phosphorylated mouse IRF-3 (residues 184 to 390) /CBP complex using TBK1 and determined the crystal structure of the phosphorylated mouse IRF-3/CBP complex (pmIRF-3/CBP) (Fig. S3A, Table 1). Similar to hIRF-3, MS analysis showed that only one residue of mIRF-3 is phosphorylated (Fig. S3B). The overall structures of the pmIRF-3/CBP and phIRF-3/CBP complexes are similar (rmsd 1.3 Å, Fig. 2B). We observed that Ser379, which corresponds to Ser386 of human IRF-3, is phosphorylated in the structure (Fig. 2B and C). Due to the replacement of Glu388 and Asn389 of human IRF-3 by Lys381, this region of pmIRF-3 is restructured. Unlike Glu388 in human IRF-3, which contributes to hIRF-3 activation by interacting with Arg211, Lys360 and Gln356, Lys381 of mIRF-3 flips into the solvent and does not interact with any residues nearby (Fig. 2C). However, structures of pSer379 and the pLxIS motif downstream are well preserved. Arg373 is structurally conserved and interacts with pSer379 in a similar fashion as Arg380 in phIRF-3 (Fig. 2C). The side chain of Arg205 adopts a slightly different conformation compared to Arg211 of phIRF-3 and interacts with pSer379 through electrostatic interaction and hydrogen bonds (Fig. 2C). The interactions between Asp247 and pSer379, Ser378 are also well preserved in both mouse and human IRF-3 (Fig. 2C). By contrast, the sidechains of Arg334 and Lys353 move away from pSer379 and do not interact with pSer379 directly (Fig. 2C). These structural comparisons demonstrate that Arg373 and Arg205 of mIRF-3, which correspond to Arg211 and Arg380 of hIRF-3, are critical for the interactions with pSer379 and contribute to the dimerization of mIRF-3 upon phosphorylation.
Figure 2. Comparison of structures of phosphorylated mouse and human IRF-3 bound to CBP.
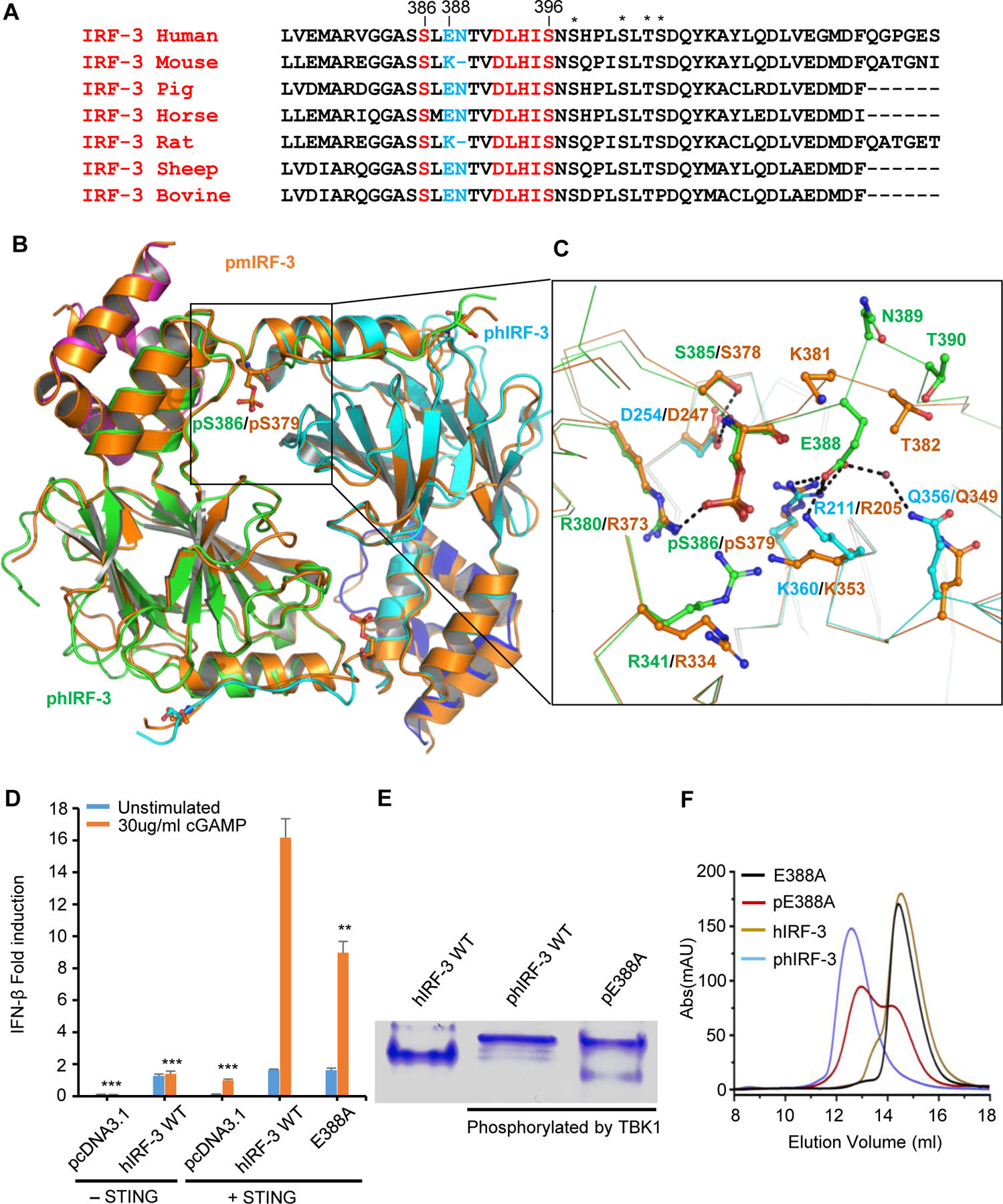
(A) Sequence alignment of C-terminal tail of IRF-3 across different species showing the conserved phosphorylation sites and the pLxIS motif in red. Residues corresponding to Glu388 and Asn389 of human IRF-3 are highlighted in blue. Other potential phosphorylation sites are indicated by the asterisks. (B) Comparison of structures of phosphorylated mouse and human IRF-3/CBP complexes. Phosphorylated mouse IRF-3/CBP is shown by the orange ribbon. Human IRF-3 dimer is colored in green and cyan with CBP bound to hIRF-3 in magenta and blue. Phosphorylated Ser379 of mIRF-3 and Ser386 of hIRF-3 are shown by the ball-and-stick models. (C) Distinct interactions between phosphorylated mouse and human IRF-3. Key residues mediating human and mouse IRF-3 dimerization are colored in green and orange, respectively. Residues interacting with Glu388 of hIRF-3 are in cyan. (D) IFN-β luciferase reporter assays showing the effect of E388A mutation on the signaling mediated by hIRF-3. The data are mean ± s.e.m. and representative of three independent assays. **P < 0.01, ***P < 0.001 values were calculated by comparisons of signals in cells transfected with E388A mutant and those transfected with wild-type IRF-3. (E) Native gel electrophoresis showing the dimerization of wild-type human IRF-3 and its E388A mutant upon phosphorylation. (F) Size-exclusion chromatography showing the effect of E388A mutation on the dimerization of hIRF-3 upon phosphorylation as compared to wild-type IRF-3.
In contrast to mouse IRF-3, Glu388 of human IRF-3 interacts with Arg211, Lys360 and Gln356 through electrostatic interactions and the solvent-mediated hydrogen bond and likely plays additional roles in human IRF-3 activation (Fig. 2C). Indeed, the luciferase reporter assay showed that mutating Glu388 to alanine in human IRF-3 reduced the IFN-β reporter signal by about 45% (Fig. 2D). Furthermore, phosphorylated E388A mutant showed two bands on native gel compared to phosphorylated wild-type IRF-3 (Fig. 2E). In agreement with these results, SEC shows that the E388A mutant of phIRF-3 elutes as two peaks, indicating the dimerization of hIRF-3 was compromised by this mutation (Fig. 2F). Taken together, these structural analyses reveal that mouse IRF-3 is activated in a similar but distinct manner compared to human IRF-3.
Mutations of residues interacting with phosphorylated Ser386 and Ser396 impair IRF-3 activation
To investigate how the residues interacting with pSer386 and pSer396 contribute to IRF-3 activation, we generated twelve mutants of full-length hIRF-3, which include mutations R211A, R380A, S339A, R211A/R380A, R211A/R380A/S339A of residues interacting with pSer386, and mutations R285A, H288A, H290A, K313A, R285A/K313A, H290A/K313A, and H288A/H290A/K313A of residues that are likely to interact with pSer396. Each of these mutants was expressed and purified for in vitro phosphorylation (Fig. S3C and D). MS analyses showed that WT full-length IRF-3 can be efficiently phosphorylated by TBK1 (Fig. 3A). Each of the IRF-3 mutants was phosphorylated and analyzed by SEC (Fig. 3B to N). Strikingly, the R211A, R211A/R380A, and R211A/R380A/S339A mutants failed to form dimers upon phosphorylation compared to the wild-type control (Fig. 3B, C, F and G). In addition, mutation R380A severely impaired the dimerization of IRF-3 upon phosphorylation (Fig. 3D). By contrast, mutations S339A, R285A, R285A/K313A, H290A/K313A, and H288A/H290A/K313A have moderate effects on IRF-3 dimer formation (Fig. 3E, H, L, M and N). Moreover, mutations H288A, H290A, and K313A have little effects on IRF-3 dimerization (Fig. 3I, J and K). To further investigate the effect of these mutations on IRF-3 activation, we analyzed these IRF-3 mutants by native gel electrophoresis (Fig. 3O and P). We observed that phosphorylated R211A, R211A/R380A, and R211A/R380A/S339A mutants only showed a single band similar to un-phosphorylated IRF-3, suggesting the activation of IRF-3 was disrupted by these mutations. Phosphorylated R380A mutant appeared as two bands and only a small fraction of this mutant formed dimers, indicating that the activation of IRF-3 was dramatically impaired by this mutation. By contrast, mutations S339A, R285A, R285A/K313A, H290A/K313A, and H288A/H290A/K313A moderately affect the dimerization of IRF-3 upon phosphorylation. Three other mutations H288A, H290A, and K313A individually do not affect the dimerization of IRF-3 (Fig. 3O and P). These results are consistent with the SEC analyses of phosphorylated IRF-3 mutants. Taken together, these studies show that mutations of key residues interacting with pSer386 significantly impair the activation of IRF-3. By contrast, mutations of residues that interact with pSer396 have moderate effects on IRF-3 activation, demonstrating that phosphorylation of Ser386 is more critical for the activation of IRF-3.
Figure 3. Mutations of key residues interacting with pSer386 and pSer396 of hIRF-3 affect IRF-3 dimerization upon phosphorylation.
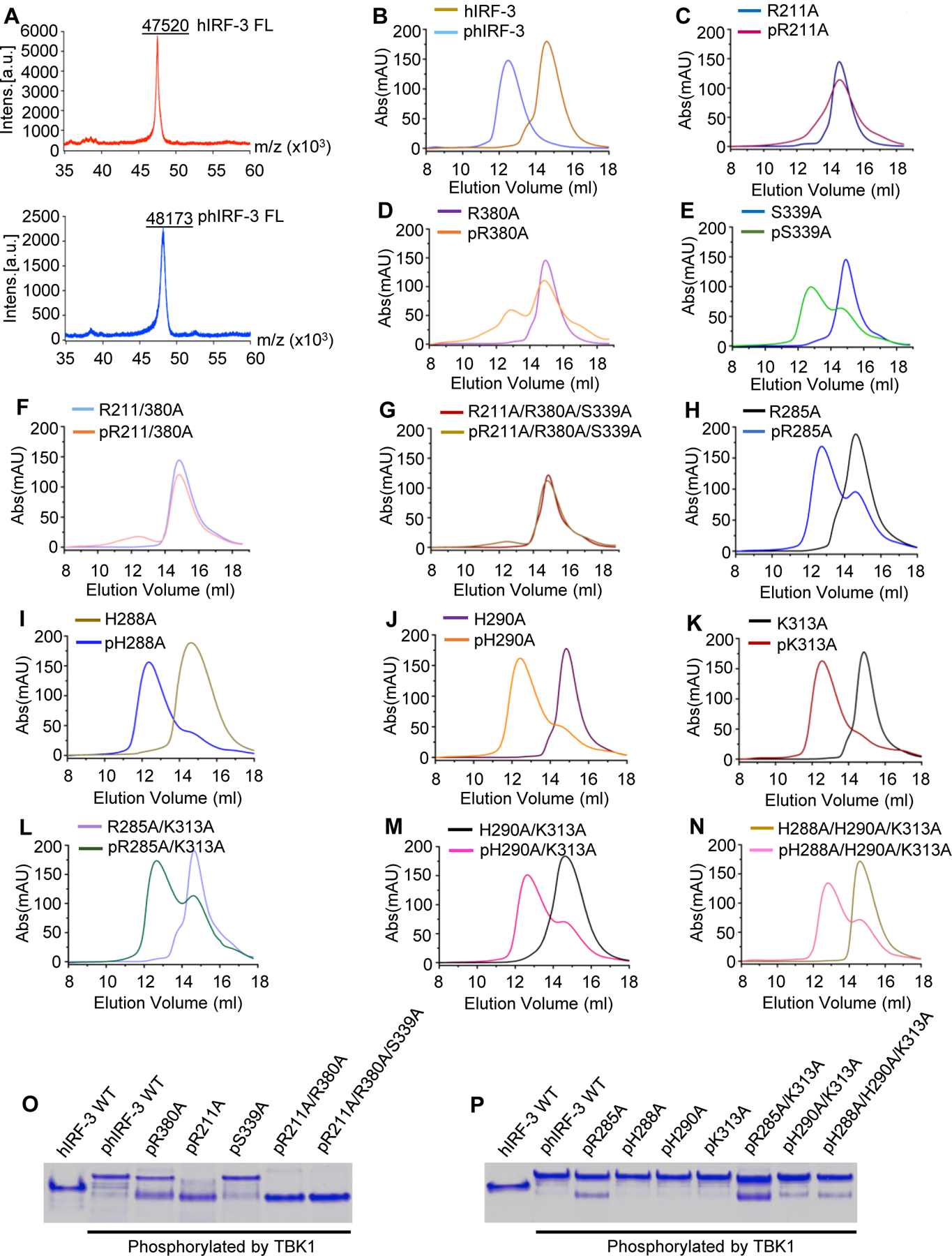
(A) MS analyses of full-length human IRF-3 before and after TBK1 phosphorylation. (B to N) Size-exclusion chromatography analyses showing how mutations of key residues interacting with pSer386 and pSer396 affect IRF-3 dimerization upon phosphorylation. (O and P) Native gel electrophoreses showing how IRF-3 mutations affect its dimerization upon phosphorylation.
Residues mediating IRF-3 dimerization play critical roles in IRF-3 mediated signaling
It has been reported that IRF-3 dimerizes in the cytosol upon phosphorylation and then translocates to the nucleus to initiate the transcription of the IFN-β gene (14, 30–33). To investigate how IRF-3 mediated signaling is affected by mutations of residues involved in IRF-3 dimerization, we conducted IFN-β luciferase reporter assay in cells transfected with full-length IRF-3 mutants. As is shown in Fig. 4A, mutations R211A, R380A, R211A/R380A and R211A/K380A/S339A blocked the induction of the IFN-β reporter, whereas mutation S339A reduced the IFN-β reporter signal by ~50%. As controls, the cells transfected with pcDNA3.1(−) or WT IRF-3 plasmid in the absence of STING plasmid showed almost no signals. The cells co-transfected with pcDNA3.1(−) and STING plasmids also showed very little signals (Fig. 4A). Western blot showed that the expression level of the IRF-3 mutants was similar whereas only a very faint band was observed in the vector control, which corresponds to endogenous IRF-3 (Fig. S3E). Mutations of residues surrounding pSer396, such as mutations R285A, R285A/K313A, H290A/K313A, and H288A/H290A/K313A reduced the reporter signal by 60–70%. By contrast, mutations H288A, H290A, and K313A individually barely impacted the activation of the reporter (Fig. 4B). Similarly, western blot of cells transfected with WT IRF-3 and mutants indicated that these IRF-3 mutants are expressed at similar levels (Fig. S3F). These results demonstrate that the mutations of key residues interacting with pSer386 abrogated IFN-β reporter activation whereas the mutations of residues interacting with pSer396 only partially inhibited IFN-β reporter activation, suggesting that residues mediating IRF-3 dimerization are critical in IRF-3 mediated signaling.
Figure 4. Mutations of residues interacting with pSer386 and pSer396 of hIRF-3 affect IRF-3 mediated signaling and nuclear localization.
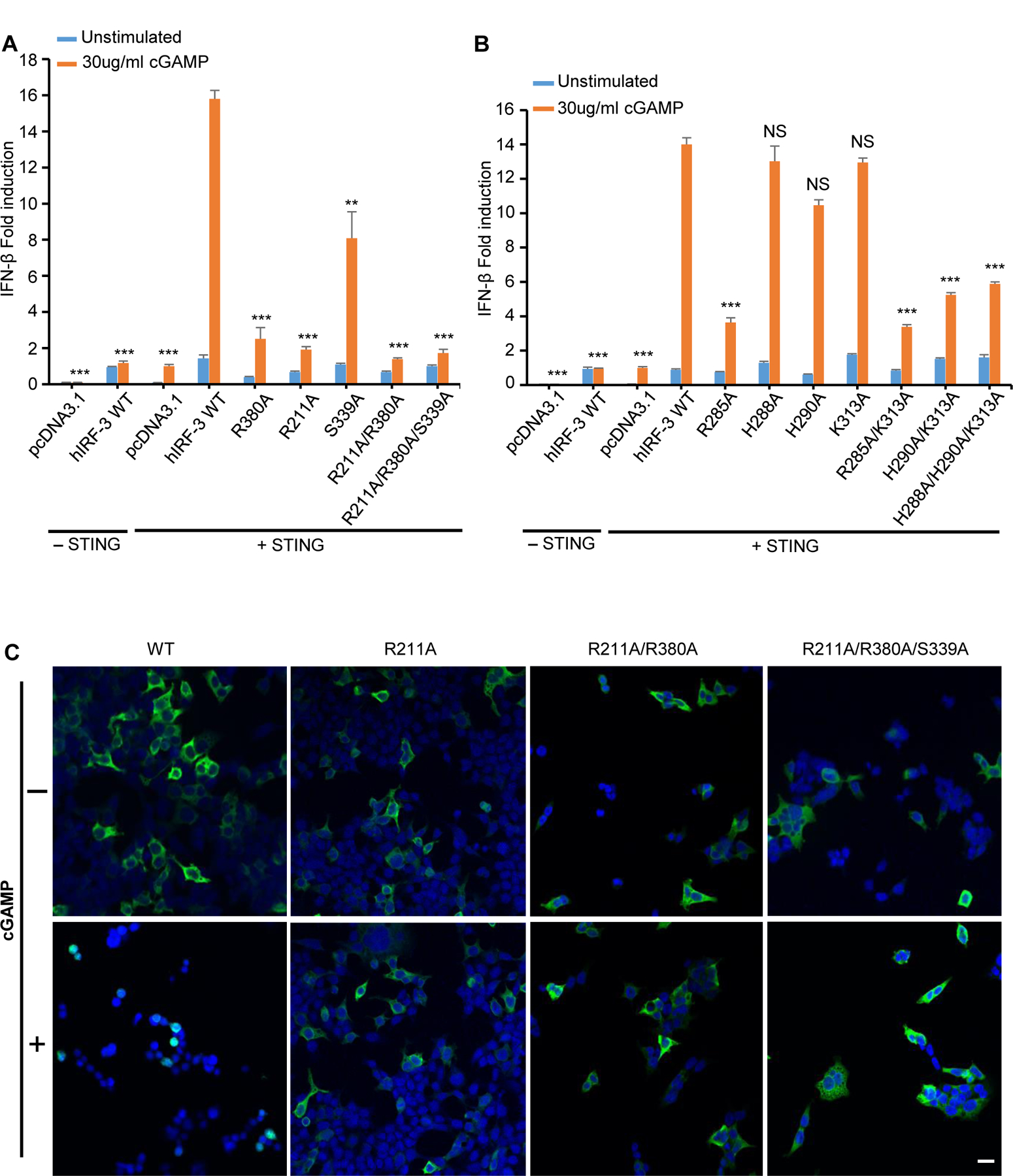
(A and B) IFN-β luciferase reporter assays showing mutations of residues interacting with pSer386 and pSer396 affect IRF-3 mediated signaling. The data are mean ± s.e.m. and representative of three independent assays. **P < 0.01, ***P < 0.001, NS (P > 0.05) values were calculated by comparisons of signals in cells transfected with IRF-3 mutants and those transfected with wild-type IRF-3. (C) Confocal microscopy of HEK293T cells transfected with IRF-3 mutants and STING upon cGAMP stimulation. Scale bars, 20 μm.
To investigate how these mutations affect the subcellular localization of IRF-3, we conducted confocal microscopy analyses of cells transfected with the IRF-3 mutants (Fig. 4C, Fig. S4A). We observed that WT IRF-3 efficiently translocated to the nuclei after cGAMP treatment, whereas the R211A/R380A, R211A/R380A/S339A, R211A, and R380A mutants are mostly localized in the cytosol. The S339A, R285A, R285A/K313A, H290A/K313A and H288A/H290A/K313A mutants partially entered the nuclei. By contrast, the H288A, H290A, and K313A mutants behaved similarly as WT IRF-3 (Fig. 4C, Fig. S4A). We used ImageJ to quantify the amount of nuclear translocation of IRF-3 in the cells and observed that mutations R211A, R380A, R211A/R380A, R211A/R380A/S339A greatly impaired the amount of IRF-3 translocated to the nucleus after cGAMP treatment whereas mutations H288A, H290A, K313A individually did not affect IRF-3 nuclear translocation (Fig. S4B). These results demonstrate that the mutations that affect IRF-3 dimerization impair IRF-3 mediated signaling and nuclear translocation of IRF-3.
DISCUSSION
IRF-3 is the key transcription factor regulating the expression of type I IFNs in response to various pathogens. Here, we determined the crystal structures of phosphorylated human and mouse IRF-3/CBP complexes, which reveal that IRF-3 forms a dimer upon phosphorylation. Compared to auto-inhibited IRF-3, the C-terminal tail of IRF-3 undergoes a dramatic conformational change upon phosphorylation, extending to the binding surface on another IRF-3 molecule and mediates the dimerization of IRF-3. Phosphorylated Ser386 interacts with several residues in a positively charged pocket through extensive electrostatic interaction and hydrogen bonds. Cell-based studies combined with in vitro phosphorylation assays demonstrate that mutations of Ser386 and the residues surrounding pSer386 abrogate IRF-3 dimerization, block its translocation to the nuclei and abolish IRF-3 mediated signaling. By contrast, phosphorylation of Ser396 within the pLxIS motif likely plays a moderate role in IRF-3 activation. Mutations of Ser396 or residues that may interact with pSer396 only partially impair IRF-3 activation and signaling. Moreover, the structural analyses reveal that Glu388 plays additional roles in the activation of human IRF-3. These structural and functional studies established the molecular basis of IRF-3 activation upon phosphorylation.
In previous studies, Ser386 and the adjacent Ser385 (Ser378 and Ser379 in mouse) were considered as two important phosphorylation sites and were identified as critical residues for IRF-3 mediated signaling (18, 19, 34). However, the structures of human and mouse IRF-3 dimer clearly show that only Ser386 is phosphorylated. The phIRF-3/CBP structure reveals that Ser385 interacts with Asp254 via hydrogen bonds to stabilize the IRF-3 dimer. Phosphorylated Ser386 reaches into a large positively charged pocket formed between two molecules of IRF-3 and contributes significantly to IRF-3 dimerization. Mutations of Ser386 and residues interacting with pSer386 dramatically impair IRF-3 dimerization, nuclear translocation and signaling. These results suggest that phosphorylation of Ser386, but not Ser385, is essential for IRF-3 mediated signaling. Consistent with these results, two previous studies showed that mutation of Ser385 to aspartic acid impairs IRF-3 dimerization and no phosphorylated Ser385 was detected with specific antibodies (20, 24).
Although Ser396 (Ser388 in mouse IRF-3) within the pLxIS motif is not phosphorylated in the human and mouse IRF-3 dimer structures presented here, these residues can be phosphorylated in vivo (Fig. S2C). Mutations of Ser396 and key residues that may interact with pSer396 impair IRF-3 dimerization and its functions, suggesting that phosphorylation of Ser396 also plays an important role in IRF-3 activation. Our previous studies demonstrated that the adaptors STING, MAVS, and TRIF employ the conserved pLxIS motif to recruit IRF-3 upon phosphorylation (6, 12). Interestingly, the phosphorylated pLxIS motifs from the adaptors or IRF-3 itself bind to the same surface on IRF-3, suggesting that the pLxIS motif of IRF-3 itself has two functions. First, the pLxIS motif of pIRF-3 occupies the pLxIS motif-binding surface of another IRF-3 molecule, displacing the adaptors and allowing pIRF-3 to dissociate from the adaptors. Second, the phosphorylated pLxIS motif works together with the phosphorylated Ser386 to facilitate IRF-3 dimerization, thus promoting IRF-3 activation.
The structures of the phosphorylated IRF-3 dimer and the adaptors bound to IRF-3 reveal that Arg285 (Arg278 in mouse) is a key residue interacting with the phosphorylated serine residue within the pLxIS motif. Mutation of Arg285 to alanine moderately impairs IRF-3 activation. These results explain why the R285Q mutation impaired IFN responses to HSV-1 infection and the R285E mutation in either HEK293 cells or fibroblasts of IRF-3-deficient mice showed much less activity upon NDV infection compared to WT IRF-3 (35, 36). Besides Ser386 and the pLxIS motif, Glu388 of human IRF-3 also plays a role in human IRF-3 activation. Size-exclusion chromatography and cell-based studies confirm the contribution of Glu388 in IRF-3 dimerization and IRF-3 mediated signaling. Interestingly, Glu388 is highly conserved in various species except for mouse and rat. The replacement of two residues Glu388 and Asn389 in human by Lys381 in mouse IRF-3 restructures this region and likely reduces the intermolecular interactions of mouse IRF-3 upon phosphorylation that may affect the kinetics of IFN-I induction by mice. In summary, these extensive structural and functional studies provide critical insights into the molecular basis of IRF-3 activation upon phosphorylation.
Supplementary Material
KEY POINTS.
The crystal structures of phosphorylated IRF-3/CBP complexes have been determined.
Both Ser386 and Ser396 are involved in IRF-3 activation but Ser386 is more crucial.
The mechanism of mouse IRF-3 activation is similar but distinct from human IRF-3.
ACKNOWLEDGMENTS
We would like to thank The Berkeley Center for Structural Biology to help us collect X-ray diffraction data. We would also like to acknowledge the members of the Li lab for help with revising the manuscript and critical feedback.
This research was supported in part by the Welch Foundation (Grants A-1931-20170325 to P.L.) and National Institute of Health (Grants R01 AI145287 to P.L.).
REFERENCES
- 1.Paludan SR, and Bowie AG. 2013. Immune sensing of DNA. Immunity 38: 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhat N, and Fitzgerald KA. 2014. Recognition of cytosolic DNA by cGAS and other STING-dependent sensors. Eur J Immunol 44: 634–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burdette DL, and Vance RE. 2013. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol 14: 19–26. [DOI] [PubMed] [Google Scholar]
- 4.Barbalat R, Ewald SE, Mouchess ML, and Barton GM. 2011. Nucleic Acid Recognition by the Innate Immune System. Annual Review of Immunology , Vol 29 29: 185–214. [DOI] [PubMed] [Google Scholar]
- 5.Shu C, Li X, and Li P. 2014. The mechanism of double-stranded DNA sensing through the cGAS-STING pathway. Cytokine Growth Factor Rev 25: 641–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, and Chen ZJ. 2015. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347: aaa2630. [DOI] [PubMed] [Google Scholar]
- 7.Takahasi K, Suzuki NN, Horiuchi M, Mori M, Suhara W, Okabe Y, Fukuhara Y, Terasawa H, Akira S, Fujita T, and Inagaki F. 2003. X-ray crystal structure of IRF-3 and its functional implications. Nature structural biology 10: 922–927. [DOI] [PubMed] [Google Scholar]
- 8.Shu C, Yi G, Watts T, Kao CC, and Li P. 2012. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat Struct Mol Biol 19: 722–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun L, Wu J, Du F, Chen X, and Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao TS, and Fitzgerald KA. 2013. The cGAS-STING Pathway for DNA Sensing. Molecular cell 51: 135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hiscott J, and Lin RT. 2005. IRF-3 releases its inhibitions. Structure 13: 1235–1236. [DOI] [PubMed] [Google Scholar]
- 12.Zhao B, Shu C, Gao X, Sankaran B, Du F, Shelton CL, Herr AB, Ji JY, and Li P. 2016. Structural basis for concerted recruitment and activation of IRF-3 by innate immune adaptor proteins. Proc Natl Acad Sci U S A 113: E3403–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, Zuo X, Kao CC, Herr AB, and Li P. 2013. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 39: 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yanai H, Negishi H, and Taniguchi T. 2012. The IRF family of transcription factors Inception, impact and implications in oncogenesis. Oncoimmunology 1: 1376–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin BY, Liu C, Srinath H, Lam SS, Correia JJ, Derynck R, and Lin K. 2005. Crystal structure of IRF-3 in complex with CBP. Structure 13: 1269–1277. [DOI] [PubMed] [Google Scholar]
- 16.Qin BY, Liu C, Lam SS, Srinath H, Delston R, Correia JJ, Derynck R, and Lin K. 2003. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol 10: 913–921. [DOI] [PubMed] [Google Scholar]
- 17.Ning S, Pagano JS, and Barber GN. 2011. IRF7: activation, regulation, modification and function. Genes Immun 12: 399–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoneyama M, Suhara W, and Fujita T. 2002. Control of IRF-3 activation by phosphorylation. J Interf Cytok Res 22: 73–76. [DOI] [PubMed] [Google Scholar]
- 19.Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, and Fujita T. 1998. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. Embo Journal 17: 1087–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mori M, Yoneyama M, Ito T, Takahashi K, Inagaki F, and Fujita T. 2004. Identification of Ser386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. The Journal of biological chemistry 279: 9698–9702. [DOI] [PubMed] [Google Scholar]
- 21.Servant MJ, Grandvaux N, tenOever BR, Duguay D, Lin R, and Hiscott J. 2003. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. The Journal of biological chemistry 278: 9441–9447. [DOI] [PubMed] [Google Scholar]
- 22.Lin R, Mamane Y, and Hiscott J. 1999. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Molecular and cellular biology 19: 2465–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Panne D, McWhirter SM, Maniatis T, and Harrison SC. 2007. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. The Journal of biological chemistry 282: 22816–22822. [DOI] [PubMed] [Google Scholar]
- 24.Chen W, Srinath H, Lam SS, Schiffer CA, Royer WE Jr., and Lin K. 2008. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. J Mol Biol 379: 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Powell HR, Battye TGG, Kontogiannis L, Johnson O, and Leslie AGW. 2017. Integrating macromolecular X-ray diffraction data with the graphical user interface iMosflm. Nat Protoc 12: 1310–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanaka Y, and Chen ZJ. 2012. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 5: ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu J, and Chen ZJ. 2014. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol 32: 461–488. [DOI] [PubMed] [Google Scholar]
- 29.Lin RT, Heylbroeck C, Pitha PM, and Hiscott J. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Molecular and cellular biology 18: 2986–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hiscott J, Pitha P, Genin P, Nguyen H, Heylbroeck C, Mamane Y, Algarte M, and Lin RT. 1999. Triggering the interferon response: The role of IRF-3 transcription factor. J Interf Cytok Res 19: 1–13. [DOI] [PubMed] [Google Scholar]
- 31.Servant MJ, Grandvaux N, and Hiscott J. 2002. Multiple signaling pathways leading to the activation of interferon regulatory factor 3. Biochem Pharmacol 64: 985–992. [DOI] [PubMed] [Google Scholar]
- 32.Taniguchi T, Ogasawara K, Takaoka A, and Tanaka N. 2001. IRF family of transcription factors as regulators of host defense. Annual review of immunology 19: 623–655. [DOI] [PubMed] [Google Scholar]
- 33.Tamura T, Yanai H, Savitsky D, and Taniguchi T. 2008. The IRF family transcription factors in immunity and oncogenesis. Annual review of immunology 26: 535–584. [DOI] [PubMed] [Google Scholar]
- 34.Suhara W, Yoneyama M, Iwamura T, Yoshimura S, Tamura K, Namiki H, Aimoto S, and Fujita T. 2000. Analyses of virus-induced homomeric and heteromeric protein associations between IRF-3 and coactivator CBP/p300. J Biochem-Tokyo 128: 301–307. [DOI] [PubMed] [Google Scholar]
- 35.Andersen LL, Mork N, Reinert LS, Kofod-Olsen E, Narita R, Jorgensen SE, Skipper KA, Honing K, Gad HH, Ostergaard L, Orntoft TF, Hornung V, Paludan SR, Mikkelsen JG, Fujita T, Christiansen M, Hartmann R, and Mogensen TH. 2015. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J Exp Med 212: 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen W, Lam SS, Srinath H, Jiang Z, Correia JJ, Schiffer CA, Fitzgerald KA, Lin K, and Royer WE Jr. 2008. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nat Struct Mol Biol 15: 1213–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.