

Abstract
In vivo expansion of adoptively transferred CD8+ T cells is a critical determinant of successful adoptive T cell therapy. Emerging evidence indicates Batf3-dependent conventional type 1 dendritic cells (cDC1s) rarely found within the tumor myeloid compartment are crucial for effector T cell recruitment to the tumor microenvironment. However, the role of cDC1s in expansion of tumor-specific CD8+ T cells remains unclear. Here, we addressed the role of cDC1s and their co-stimulatory molecules, CD40, CD70, and CD80/CD86 in expansion and antitumor efficacy of adoptively transferred in vitro-primed CD8+ T cells recognizing nonmutated tumor-associated self-antigens. We found that Toll-like receptor (TLR)/CD40-mediated expansion and antitumor efficacy of adoptively transferred tumor-specific CD8+ T cells was abrogated in Batf3−/− mice. Further mechanistic studies using mixed bone marrow chimeric mice identified CD40 and CD70 but not CD80/CD86 signaling in cDC1s played a critical role in expansion and antitumor efficacy of adoptively transferred CD8+ T cells. Moreover, induction and activation of cDC1s by administration of Fms-like tyrosine kinase 3 receptor ligand (Flt3L) and TLR/CD40 agonists augmented expansion of adoptively transferred CD8+ T cells, delayed tumor growth and improved survival. These findings reveal a key role for CD40 and CD70 signaling in cDC1s, and have major implications for the design of new vaccination strategies with adoptive T cell therapy.
Keywords: Batf3, dendritic cells, adoptive cell therapy, CD40, CD70, T cells
Introduction
Adoptive cell therapy (ACT) with antigen-specific CD8+ T cells is a promising approach for treating patients with chronic viral infections and various cancers including melanoma (1–4). A growing body of evidence from preclinical and clinical studies has shown that in vivo expansion of adoptively transferred CD8+ T cells is a critical determinant of antitumor efficacy of ACT (5–9). The differentiation status of infused T cells, the duration and type of gamma-chain (γc) cytokine support, preparative lymphodepletion, and intensity of in vivo antigen restimulation are associated with expansion of infused CD8+ T cells (5–11). However, a frequently overlooked consideration is that the role of host antigen-presenting cells (APCs) in the antitumor efficacy of ACT. The tumor microenvironment (TME) is comprised of heterogeneous populations of myeloid-lineage cells that can enhance or dampen function of tumor-specific T cells. These include dendritic cells (DCs), tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs) (12).
Batf3-dependent conventional type 1 DCs (cDC1s) (migratory CD103+ and lymphoid CD8α+ DCs in mice, and CD141+ DCs in humans) excel at cross-presenting tumor antigen to CD8+ T cells, produce IL-12 in response to innate and T cell-derived stimuli, directing the development of Th1-type T cell responses, and play a critical role in antitumor immunity (13–16). Emerging evidence suggests that tumor-residing cDC1s also facilitate trafficking of adoptively transferred T cells by secreting CXCL9/CXCL10, and improve antitumor efficacy of ACT (17). Notably, the secondary expansion of tumor-specific memory CD8+ T cells was not observed in β–catenin-expressing tumors where cDC1s are sparse (17, 18). These findings suggest the involvement of cDC1s in expansion of adoptively transferred tumor-specific T cells; however, the underlying molecular mechanisms and strategy to maximize the engagement of cDC1s in ACT remain elusive.
CD40 is a member of the tumor necrosis factor receptor superfamily, and is broadly expressed on B cells, DCs, monocytes, macrophages, and non-hematopoietic cells (19, 20). The ligand for CD40, CD40L (CD154) is predominantly expressed on activated CD4+ T cells, and is essential for inducing proper ‘licensing’ of DCs to prime CD8+ T cells to become cytotoxic T lymphocytes (CTLs) (21–24). Ligation of CD40L to CD40 on DCs results in secretion of various cytokines including IL-12, and upregulates major histocompatibility complex (MHC) and co-stimulatory molecules such as CD70, CD80 and CD86 for effector T cell functions (19, 25–27). Consequently, administration of agonistic anti-CD40 antibody (Ab) boosts the antitumor activity of naturally occurring T cells as well as adoptively transferred T cells against tumor-associated antigens (28, 29).
While the interaction between CD80/CD86 expressed on APCs and CD28 on T cells serve as an important co-stimulatory signal for the activation of T cells (27), CD70/CD27 axis is critical for effector and memory T cell differentiation of CD8+ T cells, and for reversing tolerance to self-antigen (30–34). Accumulating evidence indicates that CD40-licensed DCs relay the CD4+ T cell help signal by inducing CD70 expression (34, 35). Of note, CD70 expression on DCs is induced by TLR or CD40 although most optimally by combined stimulation (25, 32). Despite the evidence that these co-stimulatory molecules on DCs are associated with antitumor CD8+ T cell immunity, interaction between CD40, CD70 and CD80/CD86 on host cDC1s and effector CD8+ T cells remains to be elucidated.
In this study, we delineate the molecular interactions between cDC1s and adoptively transferred in vitro-activated tumor-specific CD8+ T cells using a preclinical model of melanoma in mixed bone marrow chimeric mice. Our results demonstrate that CD40 and CD70 but not CD80/CD86 signaling in cDC1s plays a crucial role in expansion and therapeutic efficacy of adoptively transferred CD8+ T cells. Moreover, induction and activation of cDC1s by administration of FMS-like tyrosine kinase 3 ligand (Flt3L) and TLR/CD40 agonists enhance expansion of adoptively transferred T cells and improve antitumor efficacy of ACT. These findings suggest that different co-stimulatory signals are required to mediate in vivo expansion of adoptively transferred CD8+ T cells, and induction and activation of cDC1s might have important implications for future adoptive T cell therapy.
Materials and Methods
Mice
All knockout and transgenic mice used in this study were female on a C57BL/6 background. C57BL/6J mice, Pmel-1 T-cell receptor (TCR)-transgenic mice (B6.Cg Thy1a-Tg(TcraTcrb)8Rest/J), Batf3−/− mice (B6.129S(C)-Batf3tm1Kmm/J), CD40−/− mice (B6.129S2-Cd40lgtm1Imx/J), and CD80/CD86−/− mice (B6.129S4-Cd80tm1ShrCd86tm2Shr/J) were purchased from the Jackson Laboratory. CD70−/− mice have been previously described (36, 37). CD27−/− mice were originally generated by Dr. Jannie Borst (Netherlands Cancer Institute) and were backcrossed in the Cao lab with C57BL/6J mice from the Jackson Laboratory. These mice were bred in-house (Roswell Park Comprehensive Cancer Center). CD27−/− mice were backcrossed onto Pmel-1 mice to generate CD27−/− Pmel-1 mice. All mice were 7 to 10 weeks old at the beginning of each experiment and were maintained under specific pathogen-free conditions at the Roswell Park animal facility according to approved institutional guidelines.
Cell lines
The murine B16F10 melanoma cell line (B16) was purchased from the American Type Culture Collection (ATCC) and cultured in in RPMI (Gibco) supplemented with 10% FBS (Sigma), 1% NEAA (Gibco), 2 mM GlutaMAX-1 (Gibco), 100 U/ml penicillin-streptomycin (Gibco), and 55 μM 2-mercaptoethanol (Gibco). Cells were authenticated by morphology, phenotype and growth, and routinely screened for mycoplasma by Mycoalert (Lonza) and were maintained at 37°C in a humidified 5% CO2 atmosphere.
Adoptive T cell therapy
Mice were injected subcutaneously with 5 × 105 B16F10 cells. For adoptive T cell therapy, Pmel-1 splenocytes were cultured with mIL-7 (10 ng/ml; Peprotech) and mIL-15 (10 ng/ml; Peprotech) for 6 days in the presence of 1μM of human (h) gp10025–33 peptide, KVPRNQDWL (GenScript). Mice were treated 12–14 days after tumor inoculation with i.v. adoptive transfer of in vitro-activated 1 × 106 T cells. We injected 15,000 IU recombinant human IL-2 (rhIL-2) (Peprotech, Inc) intraperitoneally once on the day of adoptive transfer and twice a day on the two following days. In some experiments, mice received 500 cGy of sublethal irradiation prior to adoptive T cell transfer to mimic the lymphodepletion. Mice were also vaccinated with 100 μl of saline containing 100 μg of hgp100 peptide, 50 μg of agonistic anti-CD40 Ab (clone FGK4.5, BioXcell), and 50 ug of poly(I:C) (InvivoGen) at the peritumoral site or 50 mg of imiquimod cream 5% (Perrigo) applied on the vaccination sites after adoptive transfer as described before (38, 39). Tumor volumes were calculated by determining the length of short (l) and long (L) diameters (volume = l 2 × L/2). Experimental end points were reached when tumors exceeded 20 mm in diameter or when mice became moribund and showed signs of lateral recumbency, cachexia, lack of response to noxious stimuli, or observable weight loss.
Flow Cytometry
Single cell suspensions from blood, spleen, or tumor were prepared for flow cytometric analysis. Red blood cells in blood were lysed using ACK Lysis Buffer (Life Technologies). Cells were incubated with anti-Fc receptor antibody (clone 2.4G2, BD Biosciences) in Phosphate Buffered Saline (PBS) with 2% fetal bovine serum for 30 mins. Then, surface staining using the following monoclonal antibodies was performed: anti-CD8 (clone 53–6.7, BD Biosciences), anti-CD45 (clone 30-F11, Invitrogen), anti-CD90.1 (clone Ox-7, Biolegend), anti-CD62L (clone MEL-14, Biolegend), anti-CD25 (clone PC61, Biolegend), anti-Ly6C (clone HK1.4, Biolegend), anti-CD11b (clone M1/70, BD Biosciences), anti-CD11c (clone HL3, BD Biosciences), anti-I-Ab (clone AF6–120.1, Biolegend), anti-CD24 (clone M1/69, BD Biosciences), and anti-F4/80 (clone BM8, Biolegend). DAPI or LIVE/DEAD Fixable Near-IR Dead Cell Stain kit (Thermo Fisher)-stained cells were excluded from analysis. Samples were analyzed using LSR II (BD Biosciences) or LSRFortessa (BD Biosciences) with FlowJo software (TreeStar).
Bone Marrow Chimeras
To generate bone marrow chimera or mixed bone marrow chimeras, bone marrow-recipient C57BL/6 mice were irradiated at 1,100 cGy. Radiation was delivered in two equal doses of 550 cGy 3 hours apart. To obtain donor bone marrow (C57BL/6 wild type, Batf3−/−, CD40−/−, CD70−/−, or CD80/CD86−/− mice), femurs and tibiae were harvested, and the bone marrow was flushed out. After washing, a total of 5 × 106 bone marrow cells for bone marrow chimera or 2.5 × 106 cells of each for mixed bone marrow chimera were resuspended in PBS for transfer into mice after radiation. After 8–12 weeks of bone marrow transfer, recipients were used for the experiments.
Statistical Analysis
Statistical analysis was performed using Student’s t-test or Mann-Whitney U test for comparisons between two groups, one-way ANOVA for multiple comparisons, or the Mantel-Cox method (log-rank test) for survival analysis using GraphPad Prism 8.02 (GraphPad Software). The p values for comparison of means were determined by two-tailed t-test and one-way and two-way ANOVA. P <0.05 was considered statistically significant. Data are presented as mean ± SEM.
Results
Vaccination with the cognate antigen and TLR/CD40 agonists enhances expansion and antitumor efficacy of adoptively transferred tumor-associated antigen-specific T cells
To dissect the molecular interactions between host DCs and adoptively transferred tumor-specific CD8+ T cells, we sought to validate a preclinical model of ACT using Pmel-1 TCR transgenic CD8+ T cells that can recognize gp100 expressed on B16 tumor cells (40). Pmel-1 T cells were activated with hgp100 peptide, IL-7, and IL-15 for 6 days in vitro before ACT (38, 39). While previous reports showed that in vivo stimulation of host APCs with a recombinant viral vaccine expressing the cognate antigen augments expansion of infused CD8+ T cells in this preclinical model (7–11, 40), vaccination with defined factors would be ideal for potential clinical application. Combined innate and adaptive stimuli synergistically induce CD8+ T cell expansion in vivo (41). Therefore, we evaluated the potential synergy of agonistic anti-CD40 Ab and TLR7 agonist (imiquimod) in expansion and antitumor efficacy of adoptively transferred Pmel-1 T cells in B16 tumor-bearing C57BL/6 mice in combination with subcutaneous vaccination with hgp100and systemic IL-2 administration (Fig. 1A).
FIGURE 1. Vaccination with the cognate antigen and TLR/CD40 agonists enhances expansion and antitumor efficacy of adoptively transferred tumor-associated antigen-specific T cells.
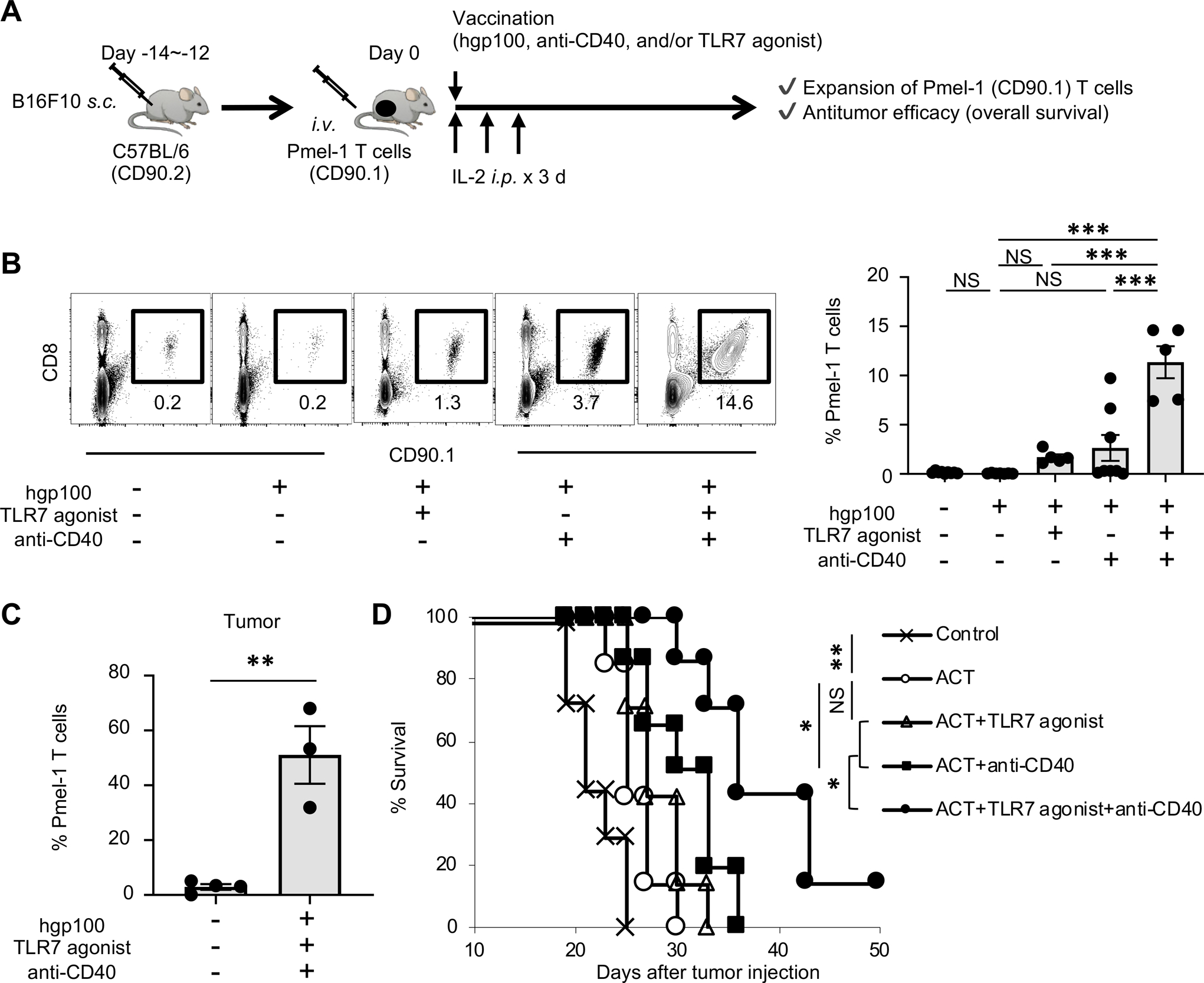
(A) Experimental scheme of adoptive cell therapy (ACT) with vaccination. Vaccination with hgp100 peptide, agonistic anti-CD40 Ab, and imiquimod cream (TLR7 agonist) were given peritumorally.
(B) Frequency of infused Pmel-1 T cells (CD8+ CD90.1+) among total CD45+ cells in spleen of B16 tumor-bearing C57BL/6 mice 7 days after ACT with various combinations of vaccination. Left shows representative flow-cytometric plots gated with CD45+ population from mice in the absence (–) or presence (+) of indicated combinations of regimens; numbers denote percent CD90.1+ CD8+ cells. Right panel shows frequency of Pmel-1 T cells in each group (n = 5–9 mice per group). Data shown are pooled from two independent experiments. NS, not significant, ***P <0.001 by one-way ANOVA with Tukey’s multiple comparisons.
(C) Frequency of infused Pmel-1 T cells (CD8+ CD90.1+) among total CD45+ cells in B16 tumors 7 days after ACT with or without vaccination (hgp100 peptide, agonistic anti-CD40 Ab, and TLR7 agonist) (n = 3–5 mice per group). **P <0.01 by unpaired two-tailed t-test.
(D) Survival curves in B16 tumor-bearing C57BL/6 mice in different treatment groups as indicated (n = 7 mice per group). *P <0.05, **P <0.01 by log-rank (Mantel-Cox) test. Data shown are representative of two independent experiments.
Values are mean ± SEM.
Adoptively transferred Pmel-1 T cells expressing CD90.1 can be distinguished from endogenous T cells expressing CD90.2 in C57BL/6 mice. In order to evaluate the expansion of Pmel-1 T cells, we harvested spleen 7 days after ACT, and analyzed the frequency of CD90.1+ CD8+ cells (Pmel-1 T cells) (Supplementary Fig. 1).
We found that vaccination with hgp100 peptide alone did not affect expansion of Pmel-1 T cells, whereas TLR7 or CD40 stimulation with hgp100 moderately enhanced expansion of Pmel-1 T cells. In contrast, combination of TLR7 and CD40 agonist synergistically expanded Pmel-1 T cells (Fig. 1B) in line with previous studies (41). Phenotypic analysis revealed that activation and effector differentiation of Pmel-1 T cells, characterized by upregulation of CD25 and downregulation of CD62L, respectively, were greatly enhanced by dual TLR7/CD40 stimulation (Supplementary Fig. 2 A, B). Phenotypic analysis of harvested B16 tumors 7 days after ACT confirmed Pmel-1 T cell infiltration into tumors (Fig. 1C). Furthermore, antitumor efficacy of adoptively transferred Pmel-1 T cells was synergistically enhanced by combined TLR7 and CD40 stimulation (Fig. 1D). Together, these findings validate a preclinical model of ACT to investigate the role of host DCs in expansion and antitumor efficacy of adoptively transferred tumor-specific CD8+ T cells.
CD40 in hematopoietic cells is critical for expansion of Pmel-1 T cells in vivo
Next, we tested a role of CD40 signaling in expansion and antitumor efficacy of adoptively transferred tumor-specific T cells restimulated by hgp100/TLR7/CD40 agonists. To this end, we first evaluated expansion and antitumor efficacy of Pmel-1 T cells in CD40−/− mice. Although activation of host APCs by TLR7 agonist remains intact, expansion and antitumor efficacy of adoptively transferred Pmel-1 T cells were abrogated in CD40−/− mice (Fig. 2A, B).
FIGURE 2. CD40 signaling in hematopoietic cells is critical for expansion of adoptively transferred Pmel-1 T cells.
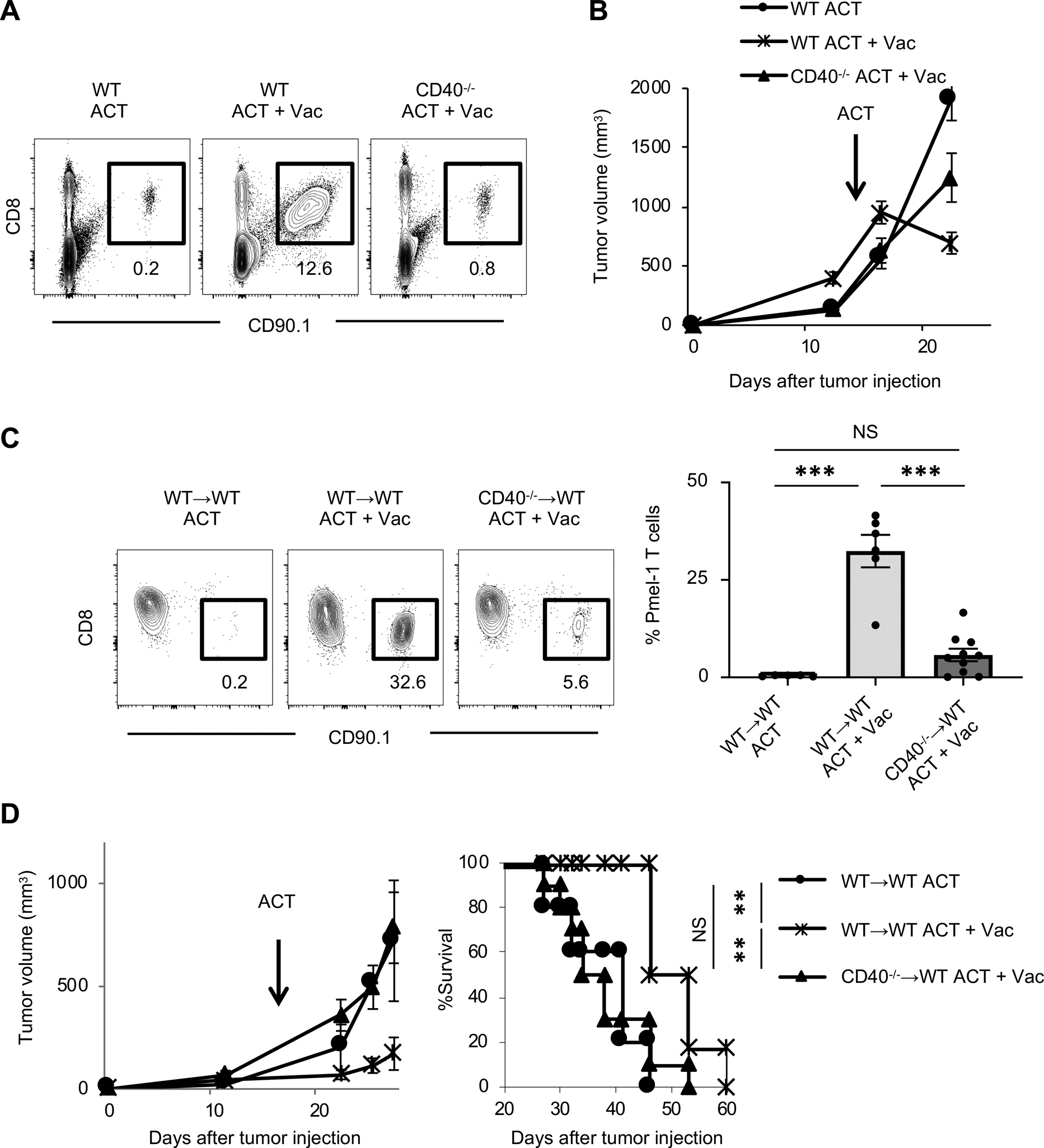
(A) Representative flow-cytometric plots gated with CD45+ cells in spleens of B16 tumor-bearing wild type C57BL/6 (WT) or CD40−/− mice 7 days after ACT with or without vaccination (Vac: hgp100, agonistic anti-CD40 Ab, and TLR7 agonist); numbers denote percent CD90.1+ CD8+ cells (Pmel-1 T cells).
(B) Tumor growth curves in B16 tumor-bearing WT or CD40−/− mice in different treatment groups (n = 7–9 mice per group). Data shown are representative of two independent experiments.
(C) Frequency of infused Pmel-1 T cells (CD8+ CD90.1+) among total CD45+ cells in peripheral blood of B16 tumor-bearing bone marrow chimeric mice (WT→WT or CD40−/−→WT mice) 7 days after ACT ± Vac (hgp100, agonistic anti-CD40 Ab, and TLR7 agonist). Left shows representative flow-cytometric plots gated with CD45+ population from mice in an indicated treatment; numbers denote percent CD90.1+ CD8+ cells (Pmel-1 T cells). Right shows frequency of Pmel-1 T cells in each group (n = 5–10 mice per group). Data shown are pooled from two independent experiments. NS, not significant, ***P <0.001 by one-way ANOVA with Tukey’s multiple comparisons.
(D) Tumor growth and survival curves in B16 tumor-bearing bone marrow chimeric mice (WT→WT or CD40−/−→WT mice) treated with ACT ± Vac (hgp100, agonistic anti-CD40 Ab, and TLR7 agonist) (n = 5–8 mice per group). NS, not significant, **P < 0.01 using log-rank (Mantel-Cox) test. Data shown are from two independent experiments.
Values are mean ± SEM.
CD40 is broadly expressed on non-hematopoietic cells as well as bone marrow-derived immune cells (19). To determine a role of CD40 in hematopoietic lineage cells in expansion and antitumor efficacy of Pmel-1 T cells, we generated bone marrow chimeras where lethally irradiated wild-type (WT) C57BL/6 mice were reconstituted with bone marrow cells from WT or CD40−/− mice. Although we observed moderate expansion of Pmel-1 T cells in mice reconstituted with CD40−/− bone marrow, the degree of expansion was substantially less than in mice reconstituted with WT bone marrow (Fig. 2C). In line with this result, antitumor efficacy of ACT was not observed in mice reconstituted with bone marrow cells from CD40−/− mice (Fig. 2D). These results suggest the critical role of CD40 in hematopoietic cells in expansion of adoptively transferred Pmel-1 T cells, and are in line with a previous study suggesting TLR stimulation alone is not sufficient to overcome peripheral tolerance and induce T cell proliferation (42).
CD40 signaling in cDC1s is required for the expansion of Pmel-1 T cells
We next sought to assess the role of host cDC1s for expansion and antitumor efficacy of antigen-specific CD8+ T cells in the context of ACT, and we treated B16 tumor-bearing Batf3−/− mice with ACT and vaccination. We found that expansion of Pmel-1 T cells was abolished in Batf3−/− mice even with hgp100/TLR7/CD40 stimulation (Fig. 3A). In addition, the frequency of CD62L expressing Pmel-1 T cells remained high in Batf3−/− mice similar to Pmel-1 T cells in WT mice without the vaccination (Supplementary Fig. 3), suggesting failed effector differentiation as well as expansion. Accordingly, adoptively transferred Pmel-1 T cells had no antitumor efficacy in Batf3−/− mice (Fig. 3B).
FIGURE 3. CD40 signaling in Batf3-dependent cells is required for the expansion of Pmel-1 T cells.
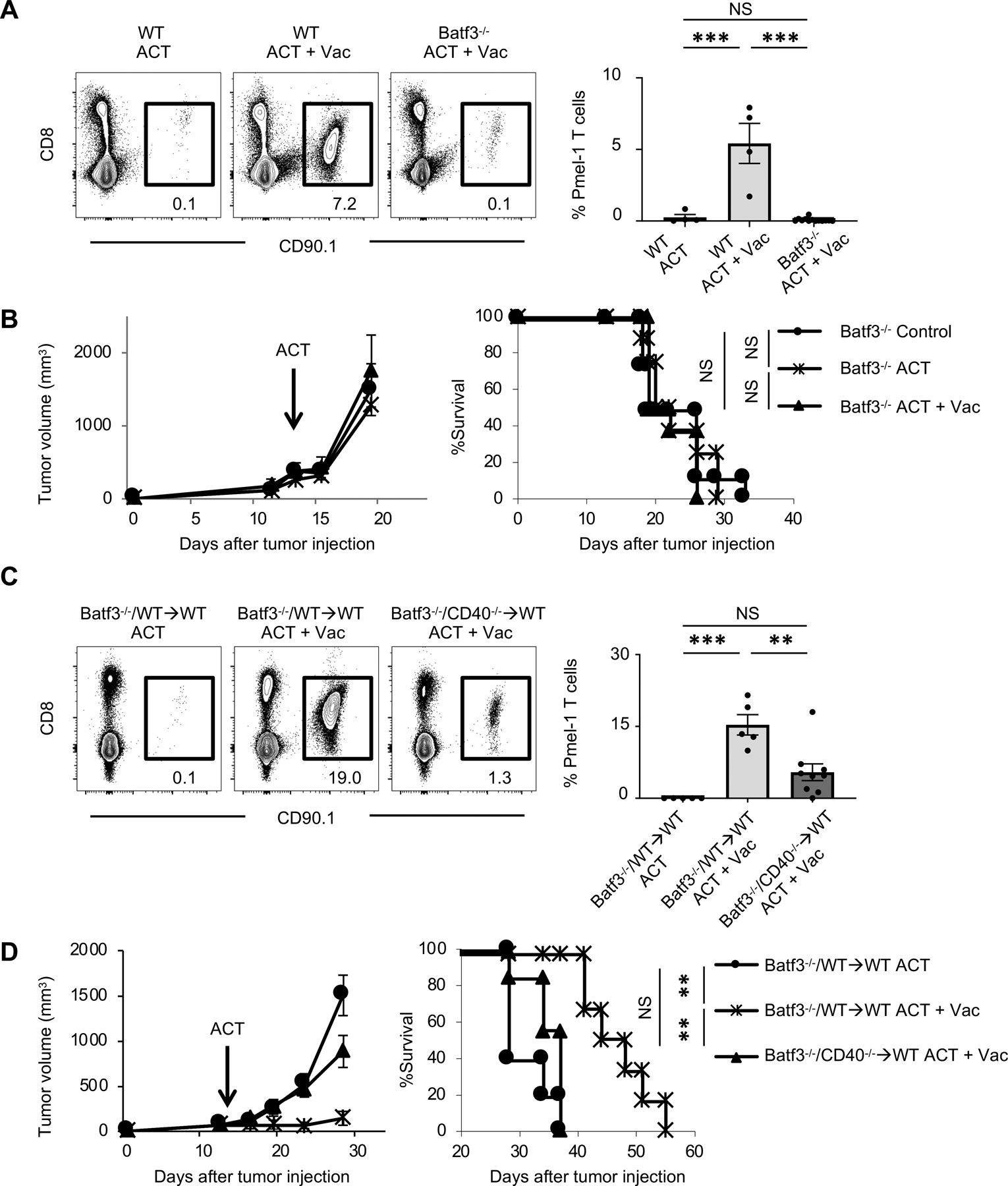
(A) Frequency of infused Pmel-1 T cells (CD8+ CD90.1+) among total CD45+ cells in spleen of B16 tumor-bearing wild type (WT) C57BL/6 or Batf3−/− mice 7 days after ACT ± Vac (hgp100, agonistic anti-CD40 Ab, and TLR7 agonist). Left shows representative flow-cytometric plots gated with CD45+ population from mice in an indicated treatment; numbers denote percent CD90.1+ CD8+ cells (Pmel-1 T cells). Right shows frequency of Pmel-1 T cells in each group (n = 4–13 mice per group). Data shown are pooled from two independent experiments. NS, not significant, ***P <0.001 by one-way ANOVA with Tukey’s multiple comparisons.
(B) Tumor growth and survival curves in B16 tumor-bearing Batf3−/− mice treated with ACT ± Vac. (hgp100, agonistic anti-CD40 Ab, and TLR7 agonist). (n = 5–7 mice per group). Batf3−/− control mice received IL-2 only. NS, not significant by log-rank (Mantel-Cox) test. Data shown are representative of two independent experiments.
(C) Frequency of infused Pmel-1 T cells among total CD45+ cells in spleen of B16 tumor-bearing mixed bone marrow chimera mice (Batf3−/− and WT→WT or Batf3−/− and CD40−/−→WT mice) 7 days after ACT ± Vac (hgp100, agonistic anti-CD40 Ab, and TLR7 agonist). Left shows representative flow-cytometric plots gated with CD45+ population from mice in an indicated treatment; numbers denote percent CD90.1+ CD8+ cells (Pmel-1 T cells). Right shows frequency of Pmel-1 T cells in each group (n = 5–9 mice per group). NS, not significant, **P <0.01, ***P <0.001 by one-way ANOVA with Tukey’s multiple comparisons.
(D) Tumor growth and survival curves in B16 tumor-bearing mixed bone marrow chimera mice (Batf3−/− and WT→WT or Batf3−/− and CD40−/−→WT mice) treated with ACT ± Vac (hgp100, agonistic anti-CD40 Ab, and TLR7 agonist). (n = 5–7 mice per group). NS, not significant, **P <0.01 by log-rank (Mantel-Cox) test. Data shown are from two independent experiments.
Values are mean ± SEM.
Our findings using CD40−/−, bone marrow chimeric CD40−/−, and Batf3−/− mice are suggestive of a key role played by CD40 signaling in Batf3-dependent cells for expansion and antitumor efficacy of ACT. To test this notion, we generated Batf3−/−/CD40−/− or Batf3−/−/WT mixed bone marrow chimeras into WT mice. Unlike Batf3−/−/CD40−/− bone marrow chimera in which the CD40 deficiency is restricted to the Batf3-dependent cells, CD40/CD40L pathway in cDC1s remains intact in the mixed chimeric mice reconstituted with Batf3−/−/WT bone marrow. Although moderate improvement in expansion of Pmel-1 T cells was observed in Batf3−/−/CD40−/− mixed chimera with vaccination compared to Batf3−/−/WT mixed chimera without vaccination, ablation of CD40 expression in Batf3-dependent cells resulted in a significant reduction in the frequency of adoptively transferred Pmel-1 T cells (Fig. 3C). Furthermore, while adoptive transfer of Pmel-1 T cells with vaccination resulted in a significant delay in tumor growth in mice reconstituted with Batf3−/−/WT bone marrow cells, Batf3−/−/CD40−/− reconstituted mice showed rapid tumor growth and poor survival similar to Batf3−/−/WT reconstituted mice without vaccination (Fig. 3D). Collectively, these results revealed a crucial role of CD40 signaling in host Batf3-dependent cells for adoptively transferred CD8+ T cells to expand and mediate antitumor immunity.
Expansion of adoptively transferred T cells is associated with expression of CD70 but not CD80/CD86 in recipient mice
CD70 and CD80/CD86 on DCs are of major importance for T cell co-stimulation (25–27, 32, 43). To investigate the effect of CD70 and CD80/CD86 in recipient mice in expansion of adoptively transferred CD8+ T cells, we first analyzed the proportion of Pmel-1 T cells in B16 tumor-bearing CD70−/− and CD80/CD86−/− mice treated with ACT. Although vaccination facilitated expansion of Pmel-1 T cells in WT, CD70−/−, and CD80/CD86−/− mice, the frequency of infused Pmel-1 T cells was substantially decreased in CD70−/− mice compared to WT mice (Fig. 4A). In contrast, no difference in expansion of Pmel-1 T cells was identified between WT and CD80/CD86−/− mice.
FIGURE 4. CD27-CD70 axis is critical for expansion of adoptively transferred Pmel-1 T cells.
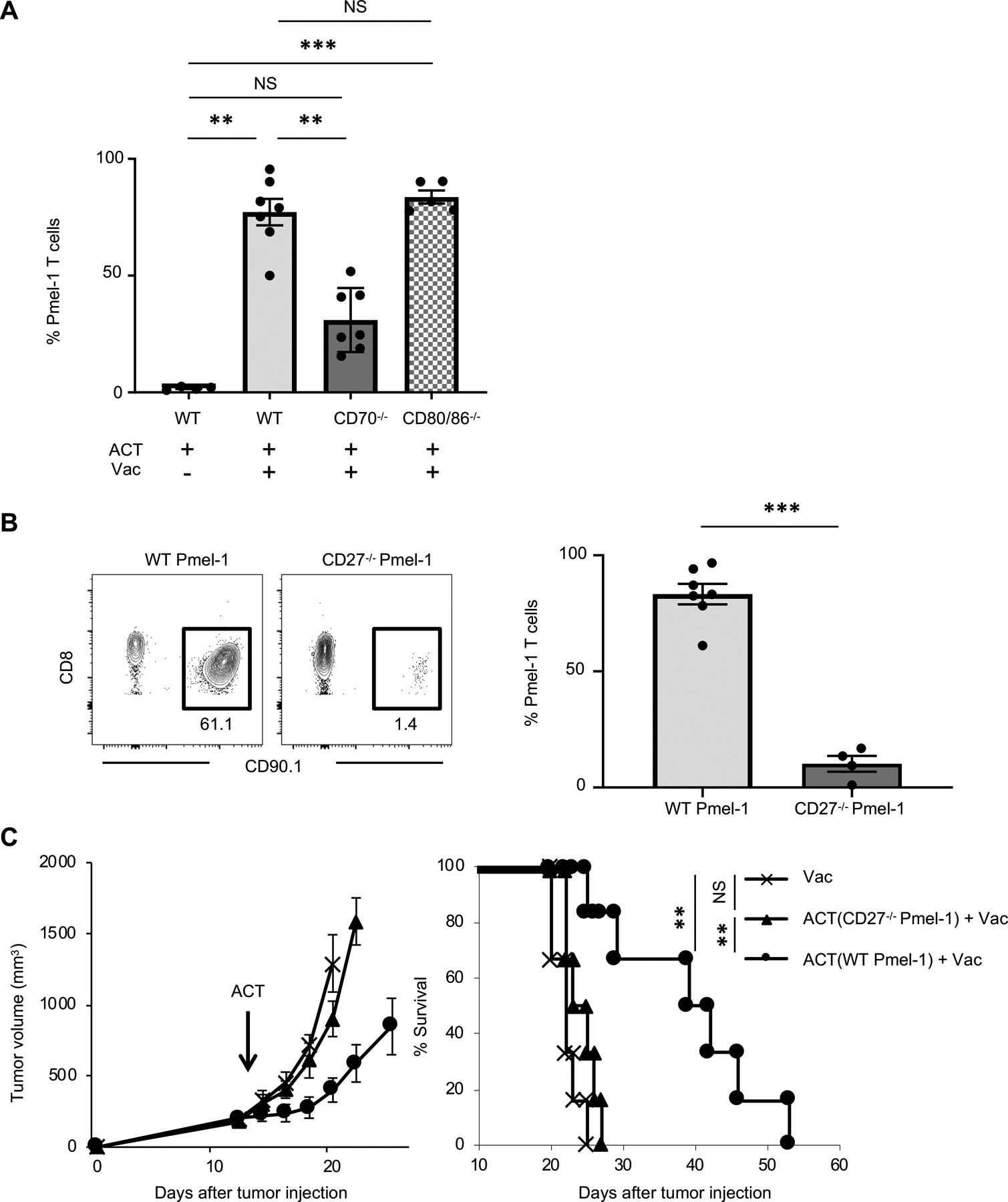
(A) Frequency of infused Pmel-1 T cells (CD8+ CD90.1+) among total CD45+ cells in peripheral blood of B16 tumor-bearing wild type (WT) C57BL/6, CD70−/−, or CD80−/− mice 7 days after ACT ± Vac (hgp100, agonistic anti-CD40 Ab, and TLR7 agonist). (n = 4–7 mice per group). Data shown are pooled from two independent experiments. NS, not significant, **P <0.01, ***P <0.001 by one-way ANOVA with Tukey’s multiple comparisons. Mice were irradiated (500 cGy) before ACT.
(B) Frequency of infused WT Pmel-1 T cells or CD27−/− Pmel-1 CD8+ T among total CD45+ cells in peripheral blood of B16 tumor-bearing WT mice 7 days after ACT ± Vac (hgp100, agonistic anti-CD40 Ab, and TLR7 agonist). Mice were irradiated (500 cGy) before ACT. Left shows representative flow-cytometric plots gated with CD45+ population from mice in an indicated treatment; numbers denote percent CD90.1+ CD8+ cells (Pmel-1 T cells) (n = 4–7 mice per group). ***P <0.001 by unpaired two-tailed t-test.
(C) Tumor growth and survival curves in B16 tumor-bearing C57BL/6 mice with ACT of WT Pmel-1 T cells or CD27−/− Pmel-1 T cells + Vac (hgp100, agonistic anti-CD40 Ab, and TLR7 agonist). (n = 7 mice per group). **P < 0.01 using log-rank (Mantel-Cox) test. Data shown are representative of two independent experiments.
Values are mean ± SEM.
Given the decreased expansion of Pmel-1 T cells in CD70−/− mice, we further investigated the role of CD27, the ligand of CD70, in Pmel-1 T cells by generating CD27−/− Pmel-1 mice. We adoptively transferred WT Pmel-1 T cells or CD27−/− Pmel-1 T cells to WT C57BL/6 mice, and evaluated their expansion and antitumor efficacy. We found that CD27−/− Pmel-1 T cells failed to expand (Fig. 4B) and had decreased antitumor efficacy compared with WT Pmel-1 T cells (Fig. 4C). Altogether, these results indicate that CD27-CD70 signaling plays a pivotal role in expansion and antitumor efficacy of adoptively transferred Pmel-1 T cells.
CD70 but not CD80/CD86 signaling in Batf3-dependent cells plays a critical role in expansion and antitumor efficacy of infused Pmel-1 T cells
Finally, we sought to determine the role of CD70 and CD80/CD86 in Batf3-dependent cells in expansion and antitumor efficacy of Pmel-1 T cells. To this end, we generated Batf3−/−/CD70−/−, Batf3−/−/CD80/CD86−/−, or Batf3−/−/WT mixed bone marrow chimeras into WT. As in Batf3−/−/CD40−/− bone marrow chimera described above, CD70 or CD80/CD86 deficiency is restricted to Batf-3-dependent cells in Batf3−/−/CD70−/− or Batf3−/−/CD80/CD86−/− mixed bone marrow chimera, while CD70 or CD80/CD86 on cDC1s remains intact in the mixed chimeric mice reconstituted with Batf3−/−/WT bone marrow.
Similar to the results of conventional knock out mice described in Fig. 4A, expansion of infused Pmel-1 T cells was markedly decreased in Batf3−/−/CD70−/− mixed bone marrow chimera but not in Batf3−/−/CD80/CD86−/− mixed bone marrow chimera compared to Batf3−/−/WT mixed bone marrow chimera (Fig. 5A). Furthermore, whereas improved survival was observed in mice reconstituted with Batf3−/−/CD80/CD86−/− bone marrow chimera as in mice reconstituted with Batf3−/−/WT bone marrow, antitumor efficacy of ACT with vaccination was significantly decreased in mice reconstituted with Batf3−/−/CD70−/− bone marrow (Fig. 5B). Taken together, these data suggest CD70 but not CD80/CD86 signaling in host Batf3-dependent cells plays a critical role in expansion and antitumor efficacy of adoptively transferred Pmel-1 T cells.
FIGURE 5. CD70 but not CD80/CD86 signaling in Batf3 dependent cells plays a critical role in expansion and antitumor efficacy of infused Pmel-1 T cells.
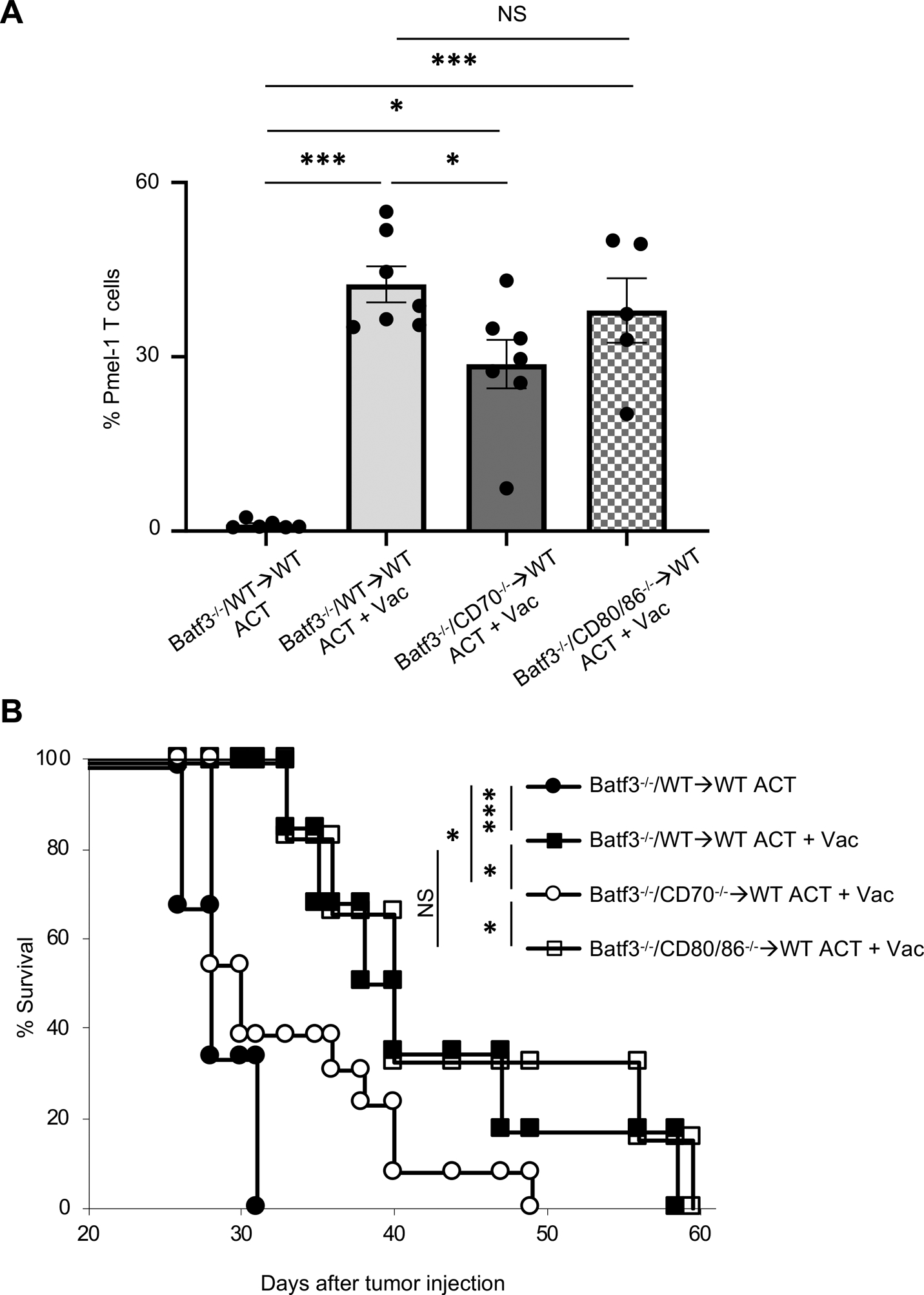
(A) Frequency of infused Pmel-1 T cells (CD8+ CD90.1+) among total CD45+ cells in peripheral blood of B16 tumor-bearing mixed bone marrow chimera mice (Batf3−/− and WT→WT, Batf3−/− and CD70−/−→WT mice, or Batf3−/− and CD80−/−→WT mice) 7 days after ACT ± Vac (hgp100, agonistic anti-CD40 Ab, and TLR7 agonist). (n = 5–7 mice per group). NS, not significant, *P <0.05, ***P <0.001 by one-way ANOVA with Tukey’s multiple comparisons. Mice were irradiated (500 cGy) before ACT.
(B) Survival curves in B16 tumor-bearing mixed bone marrow chimera mice (Batf3−/− and WT→WT, Batf3−/− and CD70−/−→WT mice, Batf3−/− and CD80−/−→WT mice) treated with ACT ± Vac (hgp100, agonistic anti-CD40 Ab, and TLR7 agonist) (n = 6–13 mice per group). Data shown are pooled from two independent experiments. *P < 0.05, ***P < 0.001 using log-rank (Mantel-Cox) test.
Values are mean ± SEM.
Induction and activation of cDC1s enhance expansion of Pmel-1 T cells, and improve antitumor efficacy of ACT
Our findings of an important role of host Batf3-dependent cells in the context of ACT prompted us to hypothesize that induction and activation of cDC1s would augment expansion and antitumor efficacy of adoptively transferred CD8+ T cells. Systemic or in situ administration of Flt3L mobilizes CD103+ DCs expressing TLR3, and TLR3 agonist, poly(I:C), induces activation of CD103+ DCs, enhancing priming of tumor-specific CD8 T cells (15, 44–46). Similar to a TLR7 agonist, poly(I:C) synergizes with agonistic CD40 antibody, and induces type I IFN-mediated expansion of CD8+ T cells in vivo (41). Therefore, we administered Flt3L to mobilize cDC1s, followed by ACT and vaccination with hgp100 and CD40/TLR3 stimulation (Fig. 6A). In situ Flt3L delivery increased both circulating and tumor-residing CD103+ DCs (Fig. 6B, Supplementary Fig, 4A), but did not affect expansion of infused T cells without vaccination (Supplementary Fig, 4B) in B16 tumor-bearing mice. Importantly, in combination with vaccination, expansion of Pmel-1 T cells was substantially enhanced in Flt3L-treated mice compared with control mice receiving PBS (Fig. 6C). We evaluated the antitumor effect of Flt3L, vaccination with hgp100/CD40/TLR3 agonists, ACT alone, any combination of these two, or combination of all three components. Intratumoral Flt3L administration did not alter tumor growth or survival either with vaccination or ACT; however, it did so when combined with both procedures (Fig. 6D). Collectively, these findings indicate that therapeutic efficacy of ACT can be enhanced by induction and activation of cDC1s.
FIGURE 6. Induction and activation of Batf3-dependent CD103+ DCs enhance expansion of Pmel-1 T cells, and improve antitumor efficacy of ACT.
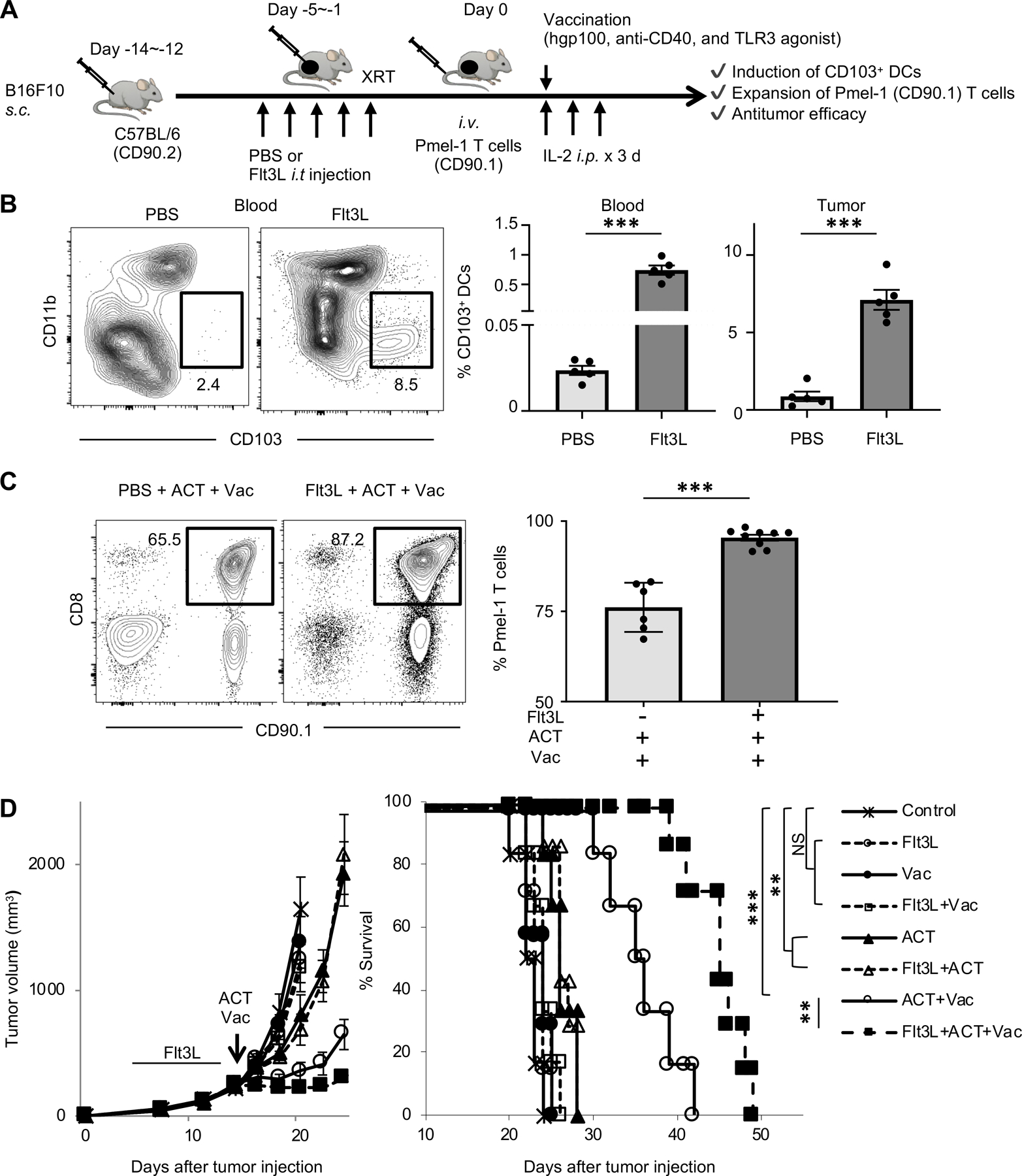
(A) Experimental scheme of ACT with vaccination (Vac) followed by Flt3L injection. Vac with hgp100 peptide, agonistic anti-CD40 Ab, and poly(I:C) (TLR3 agonist) were given peritumorally.
(B) Representative flow-cytometric plots showing CD103 and CD11b expression on dendritic cells (DCs: CD45+ Ly6C− MHC class II+ CD24+ CD11c+); numbers denote percent CD103+ CD11b− cells (CD103+ DCs). Data panels show frequency of CD103+ DCs among total CD45+ cells in peripheral blood (left) and tumors (right) of B16 tumor-bearing C57BL/6 mice treated with PBS or Flt3L (n = 5 mice per group) at one day after the last PBS or Flt3L injection. ***P <0.001 by unpaired two-tailed t-test.
(C) Frequency of infused Pmel-1 T cells (CD8+ CD90.1+) among total CD45+ cells in peripheral blood of B16 tumor-bearing mice in different treatment groups as indicated. Representative flow-cytometric plots gated with CD45+ population; numbers denote percent CD90.1+ CD8+ cells (Pmel-1 T cells) (n = 6–9 mice per group). ***P <0.001 by unpaired two-tailed t-test. Mice were irradiated (500 cGy) before ACT.
(D) Tumor growth and survival curves in B16 tumor-bearing C57BL/6 mice in different treatment groups as indicated (n = 7–9 mice per group). IL-2 was administrated to all groups. Vac with hgp100 peptide, agonistic anti-CD40 Ab, and/or poly(I:C) (TLR3 agonist) were given peritumorally on the day of ACT. **P < 0.01 using log-rank (Mantel-Cox) test. Data shown are representative of two independent experiments.
Values are mean ± SEM.
Discussion
The work described herein highlights the important role of host cDC1s in expansion and antitumor efficacy of adoptively transferred CD8+ T cells targeting nonmutated tumor-associated self-antigen and provides mechanistic insights into the molecular interactions between cDC1s and tumor-specific CD8+ T cells. In agreement with a previous study (17), our study demonstrated that host cDC1s are required for adoptively transferred CD8+ T cells to trigger effective regression of large established, poorly immunogenic tumors. However, our study provides additionally important findings that 1) TLR stimulation of DCs is insufficient, and CD40 signaling is crucial, and 2) CD70- but not CD80/CD86-mediated signaling in cDC1s is critical in expansion and antitumor efficacy of adoptively transferred tumor-specific T cells. Furthermore, gain-of-function experiments indicate that induction and activation of cDC1s by Flt3L and dual TLR/CD40-based vaccination augment expansion of infused T cells, thus enhancing the antitumor efficacy of ACT, suggestive of potential clinical implication of the strategy.
DCs are crucial in the induction of T cell immunity as well as in peripheral T cell tolerance (47, 48). Interaction of naive T cells with resting, antigen-presenting DCs results in antigen-specific T cell tolerance, whereas activated DCs induce priming (47, 48). We tested in vivo vaccination strategy comprised of the cognate antigen (hpg100), CD40 and TLR7 agonists based on a previous report (49). Intriguingly, although cDC1s express TLR3 not TLR7 (50), vaccination with hgp100/TLR7 agonist enhanced expansion and antitumor efficacy of Pmel-1 T cells compared to control mice receiving hgp100 alone, and synergize with CD40 stimulation (Fig. 1). In this regard, a TLR7-based adjuvant can stimulate multiple DC subsets (51, 52), and both TLR3 and TLR7 agonists are potent inducers of type I IFN (53). Moreover, TLR3 and TLR7 agonists similarly synergize with CD40 agonist to produce the highest level of CD8+ T cell expansion in a type I IFN-dependent manner (41). Therefore, a possible explanation for cDC1-dependent expansion of Pmel-1 T cells by CD40/TLR7 stimulation is that host type I IFN production induced by TLR7 contributes to the activation of cDC1s. Although we did not directly test this hypothesis, previous studies have shown the critical role of type I IFN signaling on cDC1s for CD8+ T-cell response (54, 55). Notably, vaccination with TLR3/CD40 agonists was also found to induce high levels of CD8+ T cell expansion (Fig. 6C).
Activation of DCs through CD40 cross-linking maintain high levels of MHC class II expression, promote upregulation of co-stimulatory molecules such as CD70, CD80, and CD86, facilitate cytokine (e.g. IL-12) production, and break CD8+ T cell tolerance (19, 25, 56). Our finding of a critical role of CD40 signaling in cDC1s in expansion and antitumor efficacy of Pmel-1 T cells might be particularly relevant to tumors expressing non-mutated self-antigen such as B16 melanoma that can grow normally in Pmel-1 TCR transgenic mice (40). A significant body of evidence has indicated that help signal from CD40-licensed APCs is transmitted to CD8+ T cells via CD27-CD70 interactions (25, 26, 30–35). Our findings using CD70−/− mice and CD27−/− Pmel-1 T cells are in agreement with this, and further demonstrated that CD70 expressed on cDC1s is critical in triggering effective regression of established tumors. Further detailed studies are required to determine whether adoptively transferred CD8+ T cells are restimulated in the TME or in secondary lymphoid organs (SLOs) such as tumor-draining lymph nodes (TdLN) although previous studies suggest that both could be sites of T cell expansion (17, 57).
The limitation of our model using B16 tumors and Pmel-1 T cells is that antitumor efficacy of ACT targeting self-antigens requires vaccination to overcome peripheral tolerance and trigger regression of large established B16 tumors (40). Although our results and others (40, 49) suggest potential clinical utility of the in vivo restimulation in the setting of ACT targeting non-mutated self-antigens and shared tumor-associated antigens such as NY-ESO-1 (58), current clinical ACT protocols do not include vaccination (2, 59). Therefore, the role of cDC1s in expansion of adoptively transferred tumor-infiltrating lymphocytes (2) or neoantigen-specific T cells (59) in current clinical ACT protocols where vaccination is not used remains to be determined. In this regard, Hanada et al., recently developed a novel mouse model for ACT targeting neoantigens where tumor regression can be observed without vaccination (60). Antitumor efficacy of ACT was still markedly enhanced by in vivo antigen restimulation in this model, emphasizing the importance of vaccination with ACT targeting neoantigens as well as nonmutated tumor-associated self-antigens.
Studies reported here and by others indicate that cDC1s play a central role in antitumor T-cell responses (14–16, 18, 55). While rare in blood and tumors, cDC1s can be mobilized by systemic or intratumoral administration of Flt3L in preclinical models and humans (44–46, 61). It has also been shown in animal models and patients that induction and activation of cDC1s facilitate priming of CD8+ T cells, thus enhancing responses to anti-PD-1/PD-L1 therapy (44–46). Our findings further suggest that outcome of ACT can be improved by induction and activation of cDC1s, which has considerable implications for the design of new vaccination strategies and adoptive immunotherapies.
In conclusion, our data demonstrates that CD40 and CD70 signaling in cDC1s is critical for expansion and antitumor efficacy of adoptively transferred CD8+ T cells recognizing nonmutated tumor-associated self-antigens. Induction and activation of cDC1s might be a useful therapeutic strategy to enhance the antitumor efficacy of ACT.
Supplementary Material
Key points.
cDC1s are needed for expansion of in vivo restimulated infused T cells in mice.
CD40 and CD70 signaling in cDC1s is critical in expansion of infused T cells.
Induction and activation of cDC1s enhance efficacy of adoptive T cell therapy.
Acknowledgments
We acknowledge the Division of Laboratory Animal Resources and Flow and Image Cytometry (Roswell Park) for technical assistance. The authors thank Dr. Kunle Odunsi and members of Center for Immunotherapy at Roswell Park for their valuable discussions and intellectual input.
Grant support: This work was supported by Roswell Park Comprehensive Cancer Center and its National Cancer Institute (NCI) award, P30CA016056 involving the use of Roswell Park’s Flow and Image Cytometry, and the Onsite Supply Center. F.I was supported by Roswell Park Alliance Foundation, the Melanoma Research Alliance, the Sarcoma Foundation of America, and NIH/NCI K08CA197966. X.C. was supported by NIH R01HL135325. T.Y. was supported by Astellas Foundation for Research on Metabolic Disorders, and the Nakatomi Foundation. T.O. was supported by Uehara Memorial Foundation.
Footnotes
Disclosures: Tibor Keler and Henry Marsh are employees of and hold stock and options in Celldex Therapeutics which has clinical stage development programs for Flt3L (CDX-301) and an anti-CD40 mAb (CDX-1140). The other authors have no conflicts of interest.
References
- 1.Maus MV, Fraietta JA, Levine BL, Kalos M, Zhao Y, and June CH. 2014. Adoptive immunotherapy for cancer or viruses. Annu Rev Immunol 32: 189–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, Morton KE, Laurencot CM, Steinberg SM, White DE, and Dudley ME. 2011. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin Cancer Res 17: 4550–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, and Riddell SR. 1995. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med 333: 1038–1044. [DOI] [PubMed] [Google Scholar]
- 4.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, and June CH. 2013. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368: 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dudley ME, Gross CA, Langhan MM, Garcia MR, Sherry RM, Yang JC, Phan GQ, Kammula US, Hughes MS, Citrin DE, Restifo NP, Wunderlich JR, Prieto PA, Hong JJ, Langan RC, Zlott DA, Morton KE, White DE, Laurencot CM, and Rosenberg SA. 2010. CD8+ enriched “young” tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin Cancer Res 16: 6122–6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Restifo NP, Dudley ME, and Rosenberg SA. 2012. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 12: 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klebanoff CA, Gattinoni L, Palmer DC, Muranski P, Ji Y, Hinrichs CS, Borman ZA, Kerkar SP, Scott CD, Finkelstein SE, Rosenberg SA, and Restifo NP. 2011. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clin Cancer Res 17: 5343–5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, Waldmann TA, and Restifo NP. 2005. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A 102: 9571–9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, Wrzesinski C, Boni A, Cassard L, Garvin LM, Paulos CM, Muranski P, and Restifo NP. 2009. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med 15: 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, Finkelstein SE, Theoret MR, Rosenberg SA, and Restifo NP. 2005. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest 115: 1616–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM, Surh CD, Rosenberg SA, and Restifo NP. 2005. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. The Journal of experimental medicine 202: 907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gabrilovich DI, Ostrand-Rosenberg S, and Bronte V. 2012. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12: 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shortman K, and Heath WR. 2010. The CD8+ dendritic cell subset. Immunol Rev 234: 18–31. [DOI] [PubMed] [Google Scholar]
- 14.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, and Murphy KM. 2008. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322: 1097–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, Amigorena S, Van’t Veer LJ, Sperling AI, Wolf DM, and Krummel MF. 2014. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26: 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, and Krummel MF. 2016. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 30: 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spranger S, Dai D, Horton B, and Gajewski TF. 2017. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 31: 711–723.e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spranger S, Bao R, and Gajewski TF. 2015. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523: 231–235. [DOI] [PubMed] [Google Scholar]
- 19.Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, and Noelle RJ. 2009. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev 229: 152–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yellin MJ, Sinning J, Covey LR, Sherman W, Lee JJ, Glickman-Nir E, Sippel KC, Rogers J, Cleary AM, Parker M, and et al. 1994. T lymphocyte T cell-B cell-activating molecule/CD40-L molecules induce normal B cells or chronic lymphocytic leukemia B cells to express CD80 (B7/BB-1) and enhance their costimulatory activity. J Immunol 153: 666–674. [PubMed] [Google Scholar]
- 21.Kelleher M, and Beverley PC. 2001. Lipopolysaccharide modulation of dendritic cells is insufficient to mature dendritic cells to generate CTLs from naive polyclonal CD8+ T cells in vitro, whereas CD40 ligation is essential. J Immunol 167: 6247–6255. [DOI] [PubMed] [Google Scholar]
- 22.Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, and Heath WR. 1998. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 393: 478–480. [DOI] [PubMed] [Google Scholar]
- 23.Brossart P, Grunebach F, Stuhler G, Reichardt VL, Mohle R, Kanz L, and Brugger W. 1998. Generation of functional human dendritic cells from adherent peripheral blood monocytes by CD40 ligation in the absence of granulocyte-macrophage colony-stimulating factor. Blood 92: 4238–4247. [PubMed] [Google Scholar]
- 24.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, and Melief CJ. 1998. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 393: 480–483. [DOI] [PubMed] [Google Scholar]
- 25.Bullock TN, and Yagita H. 2005. Induction of CD70 on dendritic cells through CD40 or TLR stimulation contributes to the development of CD8+ T cell responses in the absence of CD4+ T cells. J Immunol 174: 710–717. [DOI] [PubMed] [Google Scholar]
- 26.Tesselaar K, Xiao Y, Arens R, van Schijndel GM, Schuurhuis DH, Mebius RE, Borst J, and van Lier RA. 2003. Expression of the murine CD27 ligand CD70 in vitro and in vivo. J Immunol 170: 33–40. [DOI] [PubMed] [Google Scholar]
- 27.Lenschow DJ, Walunas TL, and Bluestone JA. 1996. CD28/B7 system of T cell costimulation. Annu Rev Immunol 14: 233–258. [DOI] [PubMed] [Google Scholar]
- 28.Liu C, Lewis CM, Lou Y, Xu C, Peng W, Yang Y, Gelbard AH, Lizee G, Zhou D, Overwijk WW, and Hwu P. 2012. Agonistic antibody to CD40 boosts the antitumor activity of adoptively transferred T cells in vivo. J Immunother 35: 276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sotomayor EM, Borrello I, Tubb E, Rattis FM, Bien H, Lu Z, Fein S, Schoenberger S, and Levitsky HI. 1999. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat Med 5: 780–787. [DOI] [PubMed] [Google Scholar]
- 30.Hendriks J, Gravestein LA, Tesselaar K, van Lier RA, Schumacher TN, and Borst J. 2000. CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol 1: 433–440. [DOI] [PubMed] [Google Scholar]
- 31.Keller AM, Schildknecht A, Xiao Y, van den Broek M, and Borst J. 2008. Expression of costimulatory ligand CD70 on steady-state dendritic cells breaks CD8+ T cell tolerance and permits effective immunity. Immunity 29: 934–946. [DOI] [PubMed] [Google Scholar]
- 32.Sanchez PJ, McWilliams JA, Haluszczak C, Yagita H, and Kedl RM. 2007. Combined TLR/CD40 stimulation mediates potent cellular immunity by regulating dendritic cell expression of CD70 in vivo. J Immunol 178: 1564–1572. [DOI] [PubMed] [Google Scholar]
- 33.French RR, Taraban VY, Crowther GR, Rowley TF, Gray JC, Johnson PW, Tutt AL, Al-Shamkhani A, and Glennie MJ. 2007. Eradication of lymphoma by CD8 T cells following anti-CD40 monoclonal antibody therapy is critically dependent on CD27 costimulation. Blood 109: 4810–4815. [DOI] [PubMed] [Google Scholar]
- 34.Taraban VY, Rowley TF, and Al-Shamkhani A. 2004. Cutting edge: a critical role for CD70 in CD8 T cell priming by CD40-licensed APCs. J Immunol 173: 6542–6546. [DOI] [PubMed] [Google Scholar]
- 35.Feau S, Garcia Z, Arens R, Yagita H, Borst J, and Schoenberger SP. 2012. The CD4(+) T-cell help signal is transmitted from APC to CD8(+) T-cells via CD27-CD70 interactions. Nature communications 3: 948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Neill RE, Du W, Mohammadpour H, Alqassim E, Qiu J, Chen G, McCarthy PL, Lee KP, and Cao X. 2017. T Cell-Derived CD70 Delivers an Immune Checkpoint Function in Inflammatory T Cell Responses. J Immunol 199: 3700–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leigh ND, O’Neill RE, Du W, Chen C, Qiu J, Ashwell JD, McCarthy PL, Chen GL, and Cao X. 2017. Host-Derived CD70 Suppresses Murine Graft-versus-Host Disease by Limiting Donor T Cell Expansion and Effector Function. J Immunol 199: 336–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saito H, Okita K, Chang AE, and Ito F. 2016. Adoptive Transfer of CD8+ T Cells Generated from Induced Pluripotent Stem Cells Triggers Regressions of Large Tumors Along with Immunological Memory. Cancer research 76: 3473–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamauchi T, Hoki T, Oba T, Saito H, Attwood K, Sabel MS, Chang AE, Odunsi K, and Ito F. 2020. CX3CR1-CD8+ T cells are critical in antitumor efficacy, but functionally suppressed in the tumor microenvironment. JCI insight 5: e133920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang LN, Feigenbaum L, Kruisbeek AM, Rosenberg SA, and Restifo NP. 2003. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. The Journal of experimental medicine 198: 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ahonen CL, Doxsee CL, McGurran SM, Riter TR, Wade WF, Barth RJ, Vasilakos JP, Noelle RJ, and Kedl RM. 2004. Combined TLR and CD40 triggering induces potent CD8+ T cell expansion with variable dependence on type I IFN. The Journal of experimental medicine 199: 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamilton-Williams EE, Lang A, Benke D, Davey GM, Wiesmuller KH, and Kurts C. 2005. Cutting edge: TLR ligands are not sufficient to break cross-tolerance to self-antigens. J Immunol 174: 1159–1163. [DOI] [PubMed] [Google Scholar]
- 43.Shahinian A, Pfeffer K, Lee KP, Kundig TM, Kishihara K, Wakeham A, Kawai K, Ohashi PS, Thompson CB, and Mak TW. 1993. Differential T cell costimulatory requirements in CD28-deficient mice. Science (New York, N.Y.) 261: 609–612. [DOI] [PubMed] [Google Scholar]
- 44.Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, Chakarov S, Rivera C, Hogstad B, Bosenberg M, Hashimoto D, Gnjatic S, Bhardwaj N, Palucka AK, Brown BD, Brody J, Ginhoux F, and Merad M. 2016. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 44: 924–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hammerich L, Marron TU, Upadhyay R, Svensson-Arvelund J, Dhainaut M, Hussein S, Zhan Y, Ostrowski D, Yellin M, Marsh H, Salazar AM, Rahman AH, Brown BD, Merad M, and Brody JD. 2019. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat Med. [DOI] [PubMed] [Google Scholar]
- 46.Sanchez-Paulete AR, Cueto FJ, Martinez-Lopez M, Labiano S, Morales-Kastresana A, Rodriguez-Ruiz ME, Jure-Kunkel M, Azpilikueta A, Aznar MA, Quetglas JI, Sancho D, and Melero I. 2016. Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov 6: 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, Ravetch JV, Steinman RM, and Nussenzweig MC. 2001. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. The Journal of experimental medicine 194: 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steinman RM 2007. Lasker Basic Medical Research Award. Dendritic cells: versatile controllers of the immune system. Nat Med 13: 1155–1159. [DOI] [PubMed] [Google Scholar]
- 49.Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang XF, Dorta-Estremera SM, Greeley NR, Nitti G, Peng W, Liu C, Lou Y, Wang Z, Ma W, Rabinovich B, Sowell RT, Schluns KS, Davis RE, Hwu P, and Overwijk WW. 2013. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat Med 19: 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Merad M, Sathe P, Helft J, Miller J, and Mortha A. 2013. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 31: 563–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oh JZ, Kurche JS, Burchill MA, and Kedl RM. 2011. TLR7 enables cross-presentation by multiple dendritic cell subsets through a type I IFN-dependent pathway. Blood 118: 3028–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kastenmüller K, Wille-Reece U, Lindsay RW, Trager LR, Darrah PA, Flynn BJ, Becker MR, Udey MC, Clausen BE, Igyarto BZ, Kaplan DH, Kastenmüller W, Germain RN, and Seder RA. 2011. Protective T cell immunity in mice following protein-TLR7/8 agonist-conjugate immunization requires aggregation, type I IFN, and multiple DC subsets. J Clin Invest 121: 1782–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uematsu S, and Akira S. 2007. Toll-like receptors and Type I interferons. J Biol Chem 282: 15319–15323. [DOI] [PubMed] [Google Scholar]
- 54.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, and Schreiber RD. 2011. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. The Journal of experimental medicine 208: 1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, and Gajewski TF. 2011. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. The Journal of experimental medicine 208: 2005–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Diehl L, den Boer AT, Schoenberger SP, van der Voort EI, Schumacher TN, Melief CJ, Offringa R, and Toes RE. 1999. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat Med 5: 774–779. [DOI] [PubMed] [Google Scholar]
- 57.Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, DuPage M, Tammela T, Kerper NR, Farago AF, Robbins R, Crowley DM, Bronson RT, and Jacks T. 2015. Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-tumor T Cell Responses. Immunity 43: 579–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Odunsi K, Jungbluth AA, Stockert E, Qian F, Gnjatic S, Tammela J, Intengan M, Beck A, Keitz B, Santiago D, Williamson B, Scanlan MJ, Ritter G, Chen YT, Driscoll D, Sood A, Lele S, and Old LJ. 2003. NY-ESO-1 and LAGE-1 cancer-testis antigens are potential targets for immunotherapy in epithelial ovarian cancer. Cancer research 63: 6076–6083. [PubMed] [Google Scholar]
- 59.Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, Kriley IR, and Rosenberg SA. 2016. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med 375: 2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hanada KI, Yu Z, Chappell GR, Park AS, and Restifo NP. 2019. An effective mouse model for adoptive cancer immunotherapy targeting neoantigens. JCI insight 4: e124405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Breton G, Lee J, Zhou YJ, Schreiber JJ, Keler T, Puhr S, Anandasabapathy N, Schlesinger S, Caskey M, Liu K, and Nussenzweig MC. 2015. Circulating precursors of human CD1c+ and CD141+ dendritic cells. The Journal of experimental medicine 212: 401–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.