

Abstract
Background
There is increasing interest in the use of purified cannabidiol (CBD) as a treatment for a wide range of conditions due to its reported anti-inflammatory, anxiolytic, antiemetic and anticonvulsant properties.
Objective
The objective of this study was to assess the safety, tolerability and pharmacokinetics of a single ascending dose of a new lipid-based oral formulation of CBD in healthy volunteers after a high-fat meal.
Methods
A total of 24 eligible healthy volunteers (aged 18–48 years) were randomised to one of three sequential cohorts (each with six active and two placebo subjects). Cohort 1 received 5 mg/kg CBD or placebo, cohort 2 received 10 mg/kg CBD or placebo (cohort 2), and cohort 3 received 20 mg/kg CBD or placebo. Data relating to adverse events, vital signs, clinical laboratory assessments, 12-lead ECGs, physical examinations and concomitant medications were collected to assess safety and tolerability. Blood samples were collected up to 8 days postdose and plasma was analysed by liquid chromatography and mass spectrometry to assess the pharmacokinetics of the CBD formulation.
Results
CBD was well tolerated in the healthy volunteers (mean age: 24.0 years) treated with a single oral dose of CBD. There were no safety concerns with increasing the dose and the safety profiles of the CBD-treated and placebo-treated subjects were similar. The most frequently reported treatment emergent adverse events (TEAEs) were headache (17%) and diarrhoea (8%). There were no reported serious adverse events (SAEs) and no clinical laboratory findings, vital signs, ECGs or physical examination findings that were reported as TEAEs or were of clinical significance during the study. After a high-fat meal, CBD was detected in plasma samples at 15 min postdose; the median time to maximum plasma concentration (Tmax) was 4 h across all three CBD dose cohorts. The CBD plasma exposure [maximum observed plasma concentration (Cmax) and the area under the concentration–time curve (AUC)] increased in a dose-proportional manner and declined to levels approaching the lower level of quantification by day 8. The terminal elimination half-life was approximately 70 h, suggesting that 2–3 weeks are needed to fully eliminate CBD.
Conclusions
This new CBD formulation demonstrated a favourable safety and tolerability profile in healthy volunteers that was consistent with the profiles reported for other purified CBD products. No severe or serious AEs were observed in this study and there were no safety concerns.
Trial Registration
ACTRN12618001424291. Registered August 2018.
Key Points
| Prescribers should be aware of a dose-proportional increase in cannabidiol exposure for doses between 5 and 20 mg/kg when cannabidiol is administered following a high-fat meal. |
| This new formulation of cannabidiol was generally safe and well tolerated, with the most commonly reported adverse events being headache (17%) and diarrhoea (8%). The formulation is now available in the public domain. |
| Safety and pharmacokinetic data obtained following a high-fat meal were consistent with data from other studies of purified pharmaceutical-grade cannabidiol, strengthening support for the idea of administering CBD with food to maximize bioavailability. |
| There is potential for a substantial variation in cannabidiol bioavailability between doses if the fat content of the associated meal varies significantly. |
Introduction
Cannabidiol (CBD) is a major nonpsychoactive cannabinoid derived from the Cannabis plant that has attracted significant interest due to its anti-inflammatory, anxiolytic, antiemetic and anticonvulsant properties, as demonstrated in preclinical trials [1–3]. Recent phase III clinical trials of purified cannabidiol have reported that this cannabinoid reduces the frequency of seizures in paediatric patients with Dravet syndrome as well as in paediatric and adult patients with Lennox–Gastaut syndrome [4, 5]. The purified formulation of cannabidiol used in those trials was approved by the United States Food and Drug Administration (FDA) for the treatment of these conditions in 2018. Based on promising preclinical data, further trials of cannabidiol for a range of other indications such as anxiety, psychosis, autism spectrum disorder, Rett syndrome, Fragile X syndrome and opioid use disorder (amongst others) are underway.
Cannabidiol does not produce a psychoactive “high”. This is due to inverse agonism at CB1 and CB2 receptors—cannabinoid receptors that are members of the G protein-coupled receptor (GPCR) family present throughout the endocannabinoid system. Interactions with multiple other non-endocannabinoid signalling pathways have also been reported, and are still the subject of debate [6, 7]. These include blocking of the equilibrative nucleoside transporter (ENT) [8], the orphan G protein-coupled receptor GPR55, and the transient receptor potential cation channel subfamily M (melastatin) member 8 (TRPM8) channel [9, 10], in addition to enhancing the activity of the 5-HT1a receptor, the α3 and α1 glycine receptors, peroxisome proliferator-activated receptor γ (PPAR-γ), and the transient receptor potential of ankyrin type 1 (TRPA1) channel [3]. It is suggested that the nonpolar nature of cannabidiol may lead to an interaction mechanism involving the insertion of cannabidiol into the cellular lipid bilayer, thereby impacting membrane fluidity and the functioning of sodium, potassium and calcium channels [11].
Cannabidiol is highly lipophilic (logP > 5) and subject to substantial first-pass metabolism; it has low bioavailability and around one-third of the dose is excreted unchanged in the faeces [12]. As is the case with most other cannabinoids, cannabidiol is metabolised in the liver, primarily by cytochrome P450. The principal metabolites are 7-carboxy-cannabidiol (7-COOHCBD) and 7-hydroxy-cannabidiol (7-OH-CBD), as well as the minor metabolite 6-hydroxy-cannabidiol (6-OHCBD). CBD 6α-hydroxylation is catalysed primarily by CYP3A4 and CYP2C19, while CBD 7-hydroxylation is catalysed mainly by CYP2C19 [13]. Other metabolic pathways include further CYP-mediated pathways (1A2, 2D6 and 2C914) and direct conjugations via uridine 5′-diphospho-glucuronosyltransferase (UGT) enzymes such as UGT1A7, UGT1A9 and UGT2B7 [12].
A recently updated review by Iffland and Grotenhermen [1] of the safety profile and side effects of cannabidiol concluded that it has a low toxicity and a better side-effect profile when it is used to treat epilepsy, psychosis or bipolar disorder. Cannabidiol treatment had fewer side effects than traditional antipsychotics such as amisulpride, and coadministration of cannabidiol with traditional antiepileptic drugs (including clobazam, valproic acid, levetiracetam, felbamate, lamotrigine and zonisamide) reduced the doses of medication required and their associated side effects. The most frequently reported side effects of cannabidiol include somnolence, fatigue, gastrointestinal disturbances (including vomiting and diarrhoea), appetite or weight change, and impaired liver function [1].
The present study sought to contribute to the evolving knowledge regarding cannabidiol pharmacokinetics by investigating a new lipid-based oral formulation. Although there are already data regarding cannabidiol pharmacokinetics, there is considerable variation between the pharmacokinetic data obtained in different studies [14]. In addition, the majority of the cannabidiol pharmacokinetic and safety studies undertaken have been in target patient groups, where comorbidities and the coadministration of other drugs (or other cannabinoids) may have affected the pharmacokinetic and safety outcomes [15–17]. There are only a few studies in the literature that have been undertaken in healthy adults in the fed state and with orally administered highly purified cannabidiol, and no study has used ascending doses. The objectives of this study were therefore to investigate the safety, tolerability and pharmacokinetics of this new formulation of cannabidiol when it is administered as a single ascending oral dose (5, 10 or 20 mg/kg) in healthy adult subjects after a high-fat meal, as well as to assess whether dose proportionality was present at these clinically relevant doses. It is important to improve our understanding of cannabidiol pharmacokinetics in order to better inform clinical use, such as optimising dosing regimens and facilitating the development of pharmacokinetic models that permit dose prediction based on gender, age, multiple dosing regimens and comorbidities. The cannabidiol formulation used in this study has not previously been tested and is now available in the public domain.
Methods
Ethics
The study was performed in accordance with the ethical principles in the Declaration of Helsinki and the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use—Good Clinical Practice (ICH-GCP) as well as applicable regulatory requirements. The study protocol and associated documentation were approved by Alfred Hospital Human Research Ethics Committee. Written informed consent was obtained from every subject before any study-related procedures were performed.
Participants
The study subjects included healthy male and female adults aged between 18 and 48 years of age who were in good general health and had a body mass index between 23 and 30 kg/m2. Subjects were required to have negative screening results for drugs of abuse and alcohol and a negative serological test, to agree to use contraception if they had child-bearing potential, and to have clinical laboratory values within normal limits.
Exclusion criteria included positive screening results for recent cannabis use, current or recent regular tobacco use (> 2/week in the previous 2 months), any clinically significant presence of acute illness, alcohol consumption > 4 units/day on average or recreational drug use during the 3 months prior to screening, use of prescription or over-the-counter medications (except for hormonal contraception and simple analgesics) in the 7 days prior to administration, standard blood donation within 30 days of screening, a history of heart disease, pregnancy or lactation, participation in another investigational drug clinical trial within 30 days of screening, allergy to soybeans or excipients used in the study drug or placebo, and the consumption of grapefruit, grapefruit juice or Seville oranges within 1 week of administration or the use of cytochrome P450 (CYP3A) inducers/inhibitors during the period from 2 weeks before administration until completion on day 15.
Study Design
The study was a randomised, double-blinded, placebo-controlled, single-dose escalation study in healthy volunteers. A total of 24 subjects were enrolled at a single site and randomly allocated to three cohorts. A single dose of cannabidiol (cohort 1 = 5 mg/kg, cohort 2 = 10 mg/kg or cohort 3 = 20 mg/kg) or placebo was administered to each subject, with treatment allocated in a 3:1 cannabidiol to placebo ratio. Doses were selected based on safety and toxicology data reported for cannabidiol in previous clinical studies and evidence that this dose range would provide potential therapeutic benefits for managing intractable epilepsy [1, 18, 19]. They reflected a target dose of 10 mg/kg, bracketed above (20 mg/kg) and below (5 mg/kg).
Subjects completed a screening visit between day −28 and day −2 to determine eligibility for inclusion in the study (Fig. 1). Subjects who met the eligibility criteria were admitted to the study site on the evening of day −1, where continued eligibility was confirmed. Subjects were fasted overnight before dosing and received a high-fat breakfast as per the US FDA/Center for Drug Evaluation and Research (CDER) guidance (i.e. fats comprised approximately 50% of the total calorific content of the meal, corresponding to 800–1000 cal) [20], which commenced 30 min before (and was completed 5 min before) dosing with cannabidiol or placebo. Eligible subjects were randomised on day 1 and both subjects and site staff were blinded to the treatment allocated.
Fig. 1.
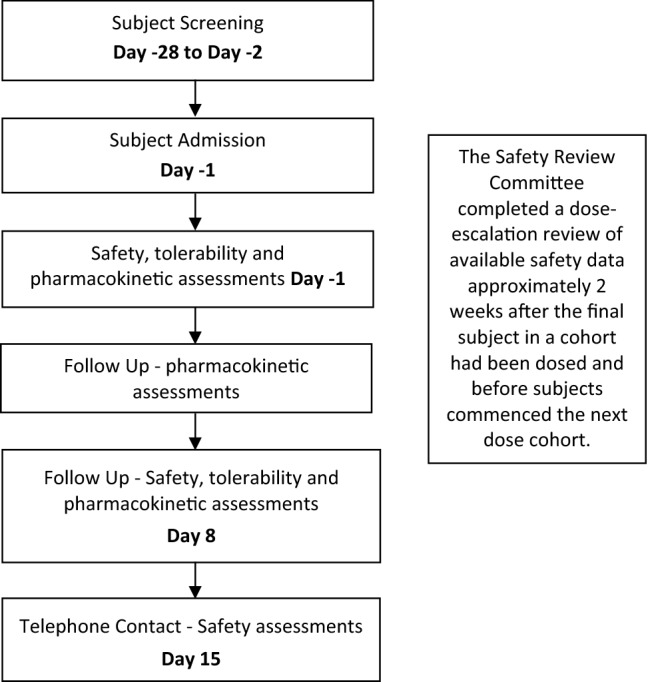
Flow chart of the study design. Safety assessments included AEs, vital signs, physical examination, electrocardiogram (ECG), and routine haematology, blood chemistry and urinalysis clinical laboratory measures
An independent safety review committee (SRC) assessed the safety and tolerability of the study drug in the subjects within each study cohort. The SRC also provided decisions regarding the continuation, modification and termination of the study.
Sentinel dosing was implemented in the first 2 subjects (1 active and 1 placebo) in each cohort; the remaining subjects in each cohort (5 active and 1 placebo) were dosed when there were—in the opinion of the investigator—no significant safety concerns with the sentinel subjects within at least the first 24 h following administration of the cannabidiol/placebo. Therefore, there were 6 active and 2 placebo subjects per cohort. The occurrence of safety issues within the first 24 h would have resulted in the notification of the SRC, and dosing would have been suspended in the rest of the cohort until a decision to cease or continue dosing was made. However, this was not required post sentinel dosing for any of the three cohorts included in this study. The SRC undertook a dose-escalation review of all available safety data up to day 8, approximately 2 weeks after the final subject in a cohort had been dosed and before the subjects commenced the next dose. All members of the SRC remained blinded for the review of the safety data, as there was no need to unblind them due to safety concerns.
All subjects completed the 3-day study unit confinement period. On day 1, pre- and postdosing safety, tolerability, and pharmacokinetic assessments were performed within 1 h prior to dosing and then at 0.25. 0.5, 1, 2, 4, 8, and 12 h postdose. On day 2, safety, tolerability and pharmacokinetic assessments were performed 24 h and 36 h after dosing. On day 3, safety, tolerability, and pharmacokinetic assessments were performed 48 h after dosing. Subjects were discharged from the study unit on day 3 following collection of the 48-h postdose assessments. Further pharmacokinetic assessments were performed on days 4 and 6, and safety, tolerability and pharmacokinetic assessments were again completed on day 8. All assessments were undertaken at the study unit. Blood sample volumes were: 9 mL for clinical chemistry, 4 mL for haematology, 9 mL for serology and 4 mL for pharmacokinetics. Plasma samples were flash frozen after collection and stored at − 80 °C until analysis.
All adverse events (AEs) and concomitant medications were continually captured for the duration of the study. Adverse events were coded using the Medical Dictionary for Regulatory Activities (MedDRA). Subjects were contacted by telephone on day 15 for final safety assessments.
Study Drug and Formulation
The cannabidiol used for the clinical study was purified from cannabis plant resin using a purification process developed at the Monash Institute of Pharmaceutical Sciences (MIPS). This process comprised dewaxing, normal and reverse-phase flash chromatography and recrystallisation steps to isolate the cannabidiol from plant components, terpenes and other cannabinoids. The purification method was subsequently transferred to a good manufacturing practice (GMP)-certified facility for scale-up manufacturing. The purity of the resulting cannabidiol API (active pharmaceutical ingredient) was confirmed to be > 99.6% using a validated ultra-high-performance liquid chromatography (UHPLC)–UV detection assay.
The formulation of the cannabidiol as outlined in Table 1 was also developed by MIPS and compounded by a sponsor-approved facility. Due to the relatively high oil solubility of cannabidiol, the API was prepared in a lipid-based oral solution (100 g/L) comprising Super Refined soybean oil (to improve oxidative stability) and flavouring agents (to ensure palatability). A placebo was also formulated with the same composition except for cannabidiol. Due to the high purity of the cannabidiol used, the organoleptic properties were comparable for the study and placebo formulations, ensuring that blinding was maintained.
Table 1.
Drug formulation developed by the Monash Institute of Pharmaceutical Sciences (MIPS)
| Ingredient | Concentration (g/L) |
|---|---|
|
Active Cannabidiol (CBD) |
100.00 |
|
Flavours Neotame (sweetener) d-limonene (orange flavour) |
0.10 0.20 |
|
Diluent Maisine CC (glyceryl monolinoleate) Super Refined soybean oil |
10.00 To 1 L |
The cannabidiol formulation and placebo were administered orally via a dispensing syringe. Dosing was based on subject body weight and assigned dose cohort, with the same volume of placebo administered as would have been used for the study drug dose for that cohort. This was to eliminate any effect of lipid dose on tolerance and/or pharmacokinetic outcomes.
The stability of cannabidiol in the developed formulation (100 mg/mL) was evaluated under controlled conditions for up to 12 months at 2–8 °C (refrigerated), 25 °C and 40 °C. When a validated stability-indicating UHPLC-UV detection assay was applied, the cannabidiol concentration in the test formulation was found to remain stable (i.e. it remained within 90–110% of the nominal concentration) when the formulation was stored for up to 12 months at 2–8 °C and 25 °C and for up to 9 months at 40 °C. The stability assessment indicated that the cannabidiol concentration in the formulation would be maintained during the 15-day course of the clinical trial once it was compounded and stored at room temperature.
Bioanalytical Assay of Cannabidiol in Plasma
A UHPLC–tandem mass spectroscopy assay was used for the quantitation of cannabidiol present at concentrations of 0.25–2500 ng/mL in human plasma. Standards were prepared using pooled human plasma and with sodium heparin as the anticoagulant to mirror samples from the clinical study. Prior to analysis, the samples were prepared by solvent protein precipitation and plate-based phospholipid removal to attenuate matrix effects in the plasma that could lead to variability in cannabidiol detection and quantitation. Cannabidiol and deuterated cannabidiol (D3-CBD, used as the internal standard) were monitored using the mass spectrometer with positive ion mode electrospray ionisation. Parent and product ion mass-to-charge ratio (m/z) transitions for CBD and D3-CBD were 315.10 > 193.20 and 318.20 > 196.20, respectively. The assay was validated for linearity, intra- and interassay accuracy and precision, sensitivity, recovery, matrix effects, and sample integrity after multiple freeze–thaw cycles by the Monash University Institute of Pharmaceutical Sciences in accordance with the ICH Harmonised Tripartite Guideline and the US Food and Drug Administration guidelines for bioanalytical method validation [21, 22], and based on predefined acceptable limits for accuracy and precision (± 15% of nominal concentration, ± 20% at the lower limit of quantitation [LLOQ]), carryover effects (≤ 20% of LLOQ) and potential changes during up to three freeze/thaw cycles (± 15% of nominal concentration).
Statistical Methods
The primary objective of the study was to assess the safety and tolerability of cannabidiol following a single oral dose in healthy volunteers. Primary endpoint data (AEs, vital signs, clinical laboratory assessments, 12-lead ECG data, physical examination and concomitant medications) were summarised in tabular form. Only treatment-emergent AEs (TEAEs) were summarised. All TEAEs were also summarised by System Organ Class (SOC) and Preferred Term (PT), severity, and relationship to cannabidiol. Laboratory data, vital signs and ECGs included a summary of actual values and actual value changes from baseline. Laboratory and ECG data were also categorised by clinical significance.
Data were summarised using descriptive statistics (mean, median, standard deviation, minimum and maximum) or frequency counts and percentages, as appropriate. The baseline was defined as the last available valid, nonmissing assessment before cannabidiol/placebo dosing. An outlier was considered to be any concentration that was a factor of 2 higher or lower than the values measured before and after it, except for Cmax.
The secondary objective of the study was to assess the pharmacokinetics of cannabidiol following a single oral dose in healthy volunteers. Secondary endpoint data included a summary of the plasma cannabidiol concentrations for the three dose cohorts and a summary of the pharmacokinetic parameters for each cannabidiol dose. Individual and mean cannabidiol plasma concentration profiles over time were plotted on linear and semi-logarithmic scales. For each subject who received active treatment, plasma concentration–time data for cannabidiol were used to calculate the following pharmacokinetic parameters by noncompartmental analysis:
Cmax: maximum observed plasma concentration (obtained directly from the data)
Tmax: time to maximum observed concentration (taken directly from the data); if the maximum plasma concentration occurs at more than one time point, the first is chosen
AUClast: area under the plasma concentration versus time curve, calculated using the linear trapezoidal rule from time 0 to the time of the last quantifiable concentration
AUCinf: area under the plasma concentration versus time curve from zero to infinity, calculated as (AUClast + Clast/Kel), where Clast is the last quantifiable cannabidiol concentration
T½: apparent terminal half-life, calculated as T½ = ln(2)/Kel.
Validated liquid chromatography–mass spectrometry bioanalytical methods were used to quantify cannabidiol concentrations. The pharmacokinetic parameters were determined using Phoenix™ WinNonlin® (version 8.0 or higher) software. Dose-related trends in pharmacokinetic parameters were assessed by graphically presenting mean parameters by dose level. The linearity of the dose dependence of Cmax and the AUC parameters was assessed by fitting a power model to these parameters using a generalised linear model approach.
Results
Study Subjects
The 24 subjects enrolled in the study were mostly male (71%) and had a mean age of 24 (min, max: 18, 48 years). The median age in the cannabidiol treatment group (23 years) was slightly lower than that in the placebo group (29.5 years). The characteristics of the treatment and placebo groups were broadly consistent (Table 2), and subjects were randomised in a 3:1 treatment:placebo ratio. Six subjects were administered cannabidiol in each dose cohort (5, 10 and 20 mg/kg).
Table 2.
Subject demographics and baseline characteristics
| Parameter | Cannabidiol dose, mg/kg | |||||
|---|---|---|---|---|---|---|
| 5 (N = 6) | 10 (N = 6) | 20 (N = 6) | Total (N = 18) | Placebo (N = 6) | All subjects (N = 24) | |
| Mean age, years (SD) | 25.8 (5.2) | 25.0 (5.5) | 23.3 (6.6) | 24.7 (5.6) | 31.3 (9.8) | 26.4 (7.2) |
| Gender, n (%) | ||||||
| Male | 4 (67%) | 5 (83%) | 5 (83%) | 14 (78%) | 3 (50%) | 17 (71%) |
| Female | 2 (33%) | 1 (17%) | 1 (17%) | 4 (22%) | 3 (50%) | 7 (29%) |
| Race, n (%) | ||||||
| Asian | – | 1 (17%) | 3 (50%) | 4 (22%) | 2 (33%) | 6 (25%) |
| White | 6 (100%) | 5 (83%) | 3 (50%) | 14 (78%) | 4 (67%) | 18 (75%) |
| Ethnicity, n (%) | ||||||
| Hispanic/Latino | 2 (33%) | 1 (20%) | 1 (33%) | 4 (29%) | – | 4 (22%) |
| Not Hispanic/Latino | 4 (67%) | 5 (83%) | 5 (83%) | 14 (78%) | 6 (100%) | 20 (83%) |
| Mean BMI, kg/m2 (SD) | 23.1 (2.7) | 23.5 (2.1) | 23.4 (4.2) | 23.3 (2.9) | 25.3 (4.0) | 23.8 (3.2) |
BMI body mass index, N number of subjects, SD standard deviation
Safety and Tolerability
Single doses of 5, 10 and 20 mg of cannabidiol per kilogram of body weight were well tolerated. Just under half (42%) of the subjects reported at least one TEAE during the study, and the majority (69%) were related to the treatment (Table 3). A total of 13 TEAEs were reported, and the frequency of TEAEs was fairly similar across the cannabidiol dose groups. Most of the TEAEs (85%) were of mild severity, and the remaining TEAEs were of moderate severity (15%). There were no severe TEAEs.
Table 3.
Summary of treatment emergent adverse events (TEAEs) and serious adverse events (SAEs)
| Cannabidiol dose, mg/kg | ||||||
|---|---|---|---|---|---|---|
| 5 (N = 6) | 10 (N = 6) | 20 (N = 6) | Total (N = 18) | Placebo (N = 6) | All subjects (N = 24) | |
| Subjects | ||||||
| Number with TEAEs | 3 (50%) | 1 (17%) | 3 (50%) | 7 (39%) | 3 (50%) | 10 (42%) |
| Number with related TEAEs | 3 (50%) | – | 3 (50%) | 6 (33%) | 1 (17%) | 7 (29%) |
| Number with moderate or severe TEAEs | 1 (17%) | – | 1 (17%) | 2 (11%) | – | 2 (8%) |
| Number with related and moderate or severe TEAEs | – | – | 1 (17%) | 1 (6%) | – | 1 (4%) |
| Number with SAEs | – | – | – | – | – | 0 (0%) |
| Events | ||||||
| Number of TEAEs | 4 | 1 | 5 | 10 | 3 | 13 |
| Number of related TEAEs | 3 | – | 5 | 8 | 1 | 9 |
| Number of moderate TEAEs | 1 | – | 1 | 2 | – | 2 |
| Number of severe TEAEs | – | – | – | – | – | 0 |
| Number of related and moderate TEAEs | – | – | 1 | 1 | – | 1 |
| Number of related and severe TEAEs | – | – | – | – | – | 0 |
| Number of SAEs | – | – | – | – | – | 0 |
The two moderate TEAEs included an upper respiratory tract infection (20 mg/kg cannabidiol dose cohort) and a headache (5 mg/kg cannabidiol dose cohort); see Table 4. The upper respiratory tract infection was considered to be possibly related to cannabidiol and the headache was considered to be unrelated to cannabidiol. Both events resolved without sequelae. The most frequently reported TEAEs were headache (17%) (cannabidiol group 3/18 versus placebo group 1/6) and diarrhoea (8%) (cannabidiol group 3/18 versus placebo group 0/6). Of these, one of the TEAEs of headache and two of the TEAEs of diarrhoea were assessed as being related to cannabidiol, whereas the remaining three TEAEs were not related to cannabidiol. These TEAEs were of mild severity; only one of the TEAEs of headache was assessed as being of moderate severity.
Table 4.
Summary of treatment-emergent adverse events (TEAEs) by MedDRA term and treatment
| System organ class (MedDRA PT) | Cannabidiol, mg/kg | ||||||
|---|---|---|---|---|---|---|---|
| 5 (N = 6) | 10 (N = 6) | 20 (N = 6) | Total (N = 18) | Placebo (N = 6) | All subjects (N = 24) | ||
| Subjects with at least one TEAE | 3 (50%) | 1 (17%) | 3 (50%) | 7 (39%) | 3 (50%) | 10 (42%) | |
| Infections and infestations | – | – | 1 (17%) | 1 (6%) | – | 1 (4%) | |
| Upper respiratory tract infection | – | – | 1 (17%) | 1 (6%) | – | 1 (4%) | |
| Metabolism and nutrition disorders | 1 (17%) | – | – | 1 (6%) | – | 1 (4%) | |
| Increased appetite | 1 (17%) | – | – | 1 (6%) | – | 1 (4%) | |
| Nervous system disorders | 2 (33%) | 1 (17%) | 1 (17%) | 4 (22%) | 1 (17%) | 5 (21%) | |
| Dizziness postural | 1 (17%) | – | – | 1 (6%) | – | 1 (4%) | |
| Headache | 1 (17%) | 1 (17%) | 1 (17%) | 3 (17%) | 1 (17%) | 4 (17%) | |
| Gastrointestinal disorders | – | – | 3 (50%) | 3 (17%) | – | 3 (13%) | |
| Diarrhoea | – | – | 2 (33%) | 2 (11%) | – | 2 (8%) | |
| Nausea | – | – | 1 (17%) | 1 (6%) | – | 1 (4%) | |
| Skin and subcutaneous tissue disorders | 1 (17%) | – | – | 1 (6%) | – | 1 (4%) | |
| Drug eruption | 1 (17%) | – | – | 1 (6%) | – | 1 (4%) | |
| Musculoskeletal and connective tissue disorders | – | – | – | – | 1 (17%) | 1 (4%) | |
| Back pain | – | – | – | – | 1 (17%) | 1 (4%) | |
| General disorders and administration site conditions | – | – | – | – | 1 (17%) | 1 (4%) | |
| Fatigue | – | – | – | – | 1 (17%) | 1 (4%) | |
MedDRA Medical Dictionary for Regulatory Activities preferred term, SOC system organ class, TEAE treatment emergent adverse event
One sentinel subject in cohort 1 (5 mg/kg cannabidiol) developed a TEAE of drug eruption (allergic drug rash) that required treatment with loratadine. The event was of mild severity, was assessed as probably related to cannabidiol, and resolved without sequelae. As a result of this TEAE, the SRC decided to implement the use of two sentinel subjects in the two remaining dose cohorts (10 and 20 mg/kg cannabidiol). No other TEAEs of allergic drug rash were reported for any other subjects.
There were no SAEs or deaths reported during the study, and there were no subjects who were discontinued from the study due to AEs. No clinical laboratory findings were reported as TEAEs or were of clinical significance. Further, there were no vital signs, ECGs, or physical examination findings that were reported as TEAEs or were of clinical significance during the study.
Overall, cannabidiol was well tolerated in healthy volunteers who were treated with a single oral dose of cannabidiol. There were no safety concerns with increasing the dose, and the safety profiles of the cannabidiol- and placebo-treated subjects were similar.
Pharmacokinetic Results
After oral dosing, cannabidiol was detected in subject plasma samples as early as 15 min postdose in all three cannabidiol dose cohorts, peaked between 2 and 4 h after dosing, and then declined to levels close to the lower limit of quantification (LLOQ) by day 8. The plasma concentration profiles were similar for the subjects within each dose cohort (Figs. 2 and 3). There were no instances of outliers in the pharmacokinetic data.
Fig. 2.

Geometric mean (CV%) plasma cannabidiol concentration–time profile by treatment cohort (linear plot)
Fig. 3.

Geometric mean (CV%) plasma cannabidiol concentration–time profile by treatment cohort (semi-log plot)
The cannabidiol plasma exposure (Cmax and AUC) values increased with increasing dose. The geometric mean (percent geometric coefficient of variance, GeoCV%) Cmax values were 248 (77%), 626 (57%) and 1,003 ng/mL (55%) for the 5, 10 and 20 mg/kg cannabidiol dose cohorts, respectively. Geometric mean (GeoCV%) AUClast values were 1793 (26%), 4,025 (37%) and 7,618 h ng/mL (28%) for the 5, 10 and 20 mg/kg cannabidiol dose cohorts, respectively. Geometric mean (GeoCV%) AUCinf values were 1,905 (25%), 4,227 (37%) and 8,008 h ng/mL (29%) for the 5, 10 and 20 mg/kg cannabidiol dose cohorts, respectively (see Table 5).
Table 5.
Key pharmacokinetic parameters for the subjects
| Pharmacokinetic parameters | |||||
|---|---|---|---|---|---|
| Cmax (ng/mL) | Tmax (h) | AUClast (h ng/mL) | AUCinf (h ng/mL) | T1/2 (h) | |
| Cohort 1 (5 mg/kg cannabidiol) | |||||
| N | 6 | 6 | 6 | 6 | 6 |
| Mean | 296 | 4.00 | 1840 | 1951 | 70.3 |
| SD | 173 | 2.19 | 438 | 441 | 7.2 |
| CV (%) | 58 | 55 | 24 | 23 | 10 |
| Median | 296 | 4.00 | 1883 | 1967 | 69.4 |
| Minimum | 97 | 2.00 | 1170 | 1234 | 61.6 |
| Maximum | 466 | 8.00 | 2395 | 2484 | 78.6 |
| GeoMean | 248 | 3.56 | 1793 | 1905 | 70.0 |
| GeoCV% | 77 | 56 | 26 | 25 | 10 |
| Cohort 2 (10 mg/kg cannabidiol) | |||||
| N | 6 | 6 | 6 | 6 | 6 |
| Mean | 704 | 3.67 | 4253 | 4466 | 67.1 |
| SD | 373 | 0.82 | 1605 | 1689 | 14.1 |
| CV (%) | 53 | 22 | 38 | 38 | 21 |
| Median | 598 | 4.00 | 3514 | 3677 | 64.6 |
| Minimum | 293 | 2.00 | 2806 | 3016 | 46.9 |
| Maximum | 1316 | 4.00 | 6599 | 6997 | 89.9 |
| GeoMean | 626 | 3.56 | 4025 | 4227 | 65.8 |
| GeoCV% | 57 | 29 | 37 | 37 | 21 |
| Cohort 3 (20 mg/kg cannabidiol) | |||||
| N | 6 | 6 | 6 | 6 | 6 |
| Mean | 1090 | 4.06 | 7838 | 8248 | 68.9 |
| SD | 372 | 0.16 | 1813 | 1949 | 11.1 |
| CV (%) | 34 | 4 | 23 | 24 | 16 |
| Median | 1180 | 4.00 | 8276 | 8697 | 67.4 |
| Minimum | 359 | 4.00 | 4432 | 4640 | 57.0 |
| Maximum | 1376 | 4.38 | 9595 | 10,216 | 85.2 |
| GeoMean | 1003 | 4.06 | 7618 | 8008 | 68.2 |
| GeoCV% | 55 | 4 | 28 | 29 | 16 |
Pharmacokinetic parameters were determined using Phoenix WinNonlin 8.0
AUClast area under the plasma concentration versus time curve from time zero to the last quantifiable concentration, AUCinf area under the plasma concentration versus time curve extrapolated to infinite time, Cmax maximum observed plasma concentration, CV coefficient of variance, GeoCV geometric mean coefficient of variance, GeoMean geometric mean, Kel apparent terminal elimination rate constant, SD standard deviation, T1/2 apparent terminal elimination half-life, Tmax time of maximum observed plasma concentration
Plasma half-life after cannabidiol dosing was consistent across the dose cohorts, with geometric mean T1/2 values of 70.0, 65.8 and 68.2 h for the 5, 10 and 20 mg/kg cannabidiol doses, respectively. Based on a half-life of approximately 70 h (3 days), an estimated 2–3 weeks would be required for cannabidiol to be fully eliminated (see Table 5).
Dose proportionality for cannabidiol was observed using a power model for the analysis of the pharmacokinetic parameters AUClast, AUCinf and Cmax. The estimated slope [beta] values (95% confidence limits [CLs]) for these three parameters were 1.04 (0.78 to 1.30), 1.04 (0.78 to 1.30) and 1.01 (0.50 to 1.51), respectively. The point estimates for beta for each pharmacokinetic parameter were very close to 1, suggesting that plasma increases in cannabidiol were dose proportional. Goodness of fit (R2) values for the regression models for AUClast, AUCinf and Cmax were 0.82, 0.82 and 0.53 respectively, indicating that the dose-proportionality model accounted for most of the variability in the pharmacokinetic parameters across the dose range (see Table 5).
Discussion
The current study is, to our knowledge, the first ascending dose study of purified cannabidiol in healthy adults in the fed state, and provides a number of new insights into the safety and pharmacokinetics of cannabidiol at clinically relevant doses. The significance of subjects being in the fed rather than the fasted state relates to evidence from two previous studies reporting that a high-fat meal before cannabidiol dosing increases bioavailability compared with fasted conditions. Taylor et al. [23] reported a four- to fivefold increase in Cmax and AUC in healthy volunteers, while Birnbaum et al. [17] reported a 14-fold increase in Cmax and fourfold increase in AUC in adult patients with refractory epilepsy. These findings suggest that ingesting food at the time of cannabidiol dosing may be beneficial as it could maximise bioavailability. The fed state of the subjects in the present study is also likely to have contributed to increased cannabidiol exposure.
The study by Taylor et al. [23] was a phase I, randomised, double-blind, placebo-controlled trial of oral cannabidiol in healthy volunteers, but they investigated a higher range of cannabidiol doses (1500–6000 mg; equivalent to 20–85 mg/kg in a 70 kg adult) than in our study, and the subjects were predominantly in the fasted state. In Taylor et al.’s study, the pharmacokinetic findings for subjects who received a high-fat meal before oral dosing with cannabidiol at 1500 mg (~ 20 mg/kg) were largely comparable to those in the current study. They reported a median Tmax (range) of 3.0 h (1.5–5.0), which was similar to the Tmax of 4.0 (2.0–8.0) h found in our study. The geometric means (GeoCV%) for Cmax and AUCt were 1628 ng/mL (51.4%) and 8347 ng h/mL (34.1%), respectively, for the ~ 20 mg/kg fed dose in the Taylor et al. study [23] versus 1003 ng/mL (55%) and 7618 ng h/mL (28%), respectively, for the 20 mg/kg dose group in the present study. This difference in Cmax may be due to fewer sampling points on day 1 in our study than in Taylor et al.’s study. When plasma was sampled to the steady state (72 h), the mean (%CV) terminal elimination half-life (T1/2,z) in the Taylor et al. study [23], 60.5 h (20.2), was lower than that in the present study (T1/2 ~ 70 h) with steady-state sampling out to 168 h.
Interestingly, Taylor et al. [23] reported a less than proportional increase in exposure to cannabidiol relative to dose from 20 to 90 mg/kg in the fasted state, whereas a proportional relationship was established for doses of 5–20 mg/kg when administered in the fed state in the present study. This finding also contrasts with the current prescribing information for Epidiolex, which indicates a less than dose-proportional pharmacokinetic relationship at doses of 5–20 mg/kg (presumably in the fasted state) [24].
The new cannabidiol formulation used in our study was well tolerated; the most frequently reported TEAEs were headache and diarrhoea, and most were of mild severity and resolved within a short duration (less than 1 day). There were no deaths, SAEs, severe TEAEs or discontinuations in the study. No clinically significant abnormalities were detected in any clinical laboratory parameters, vital signs, physical examinations or ECGs. The safety findings reported here are also similar to those observed in the Taylor et al. study [23] and other clinical trials of cannabidiol reported in the literature. In other studies, the most frequently reported TEAEs for cannabidiol were tiredness/somnolence, diarrhoea, nausea, headache, pyrexia, decreased appetite, vomiting and anaemia [1, 6, 15, 18, 25]. Larger-scale clinical studies and expanded-access programs in patients with epilepsy have also shown that cannabidiol oral formulations appear to be well tolerated [5, 26–29].
Overall, the results of our study are noteworthy as they establish a linear CBD pharmacokinetic profile at an ascending dose range from 5 to 20 mg/kg in fed healthy adults. This dose range (5–20 mg/kg) is relevant for the treatment of patients with Lennox–Gastaut or Dravet syndrome, for which several phase 3 clinical trials have demonstrated a reduction in seizure frequency with cannabidiol doses of up to 20 mg/kg [5, 6, 29]. In addition, the fed state of the subjects in this study better reflects the ketogenic diet commonly used with intractable epilepsy. Our results establish variability parameters for intersubject bioavailability (%CV (AUC)) across the ascending 5–20 mg/kg dose range in fed healthy adults. Published pharmacokinetic studies to date for patients in the fed state [17, 23] have reported a reduction in %CV (AUC), but only when compared to a single fixed dose. Our data will permit later fed-state pharmacokinetic comparisons in the field. Finally, the safety data collated for the clinically relevant dose levels used in this study can inform clinicians on potential AE and SAE considerations relative to the dose range assessed, especially taking into consideration the fed state, which may influence physiological responses and drug absorption. However, it should be noted that the fed versus fasted effect could result in increased variation in bioavailability between doses if the fat content of the associated meal does not remain consistent.
A limitation of this study is that it was performed in healthy volunteers rather than a patient cohort with the potential for drug–drug and drug–disease interactions. Two recent trials have documented clinically significant interactions with commonly prescribed antiepileptic drugs [15, 30], while another study has reported that the metabolism of cannabidiol is significantly altered in patients with moderate or severe hepatic impairment [12]. Further limitations are that only plasma cannabidiol was measured, not its primary metabolites. In addition, only a single dose of cannabidiol was given rather than multiple dosing to reach the steady state. These limitations may impact on the generalisability of the results. Despite this, the data generated in this study under controlled conditions in fed healthy volunteers should help to inform pharmacokinetic models that may facilitate dose adjustments by considering variations in age, sex, disease states and comorbidities in the target patient groups.
Conclusion
The findings of this study contribute to the evolving knowledge of cannabidiol pharmacokinetics and indicate that this new oral lipid-based formulation of cannabidiol is generally safe and well tolerated at all doses studied. No severe or serious AEs were observed and there were no safety concerns. Pharmacokinetic analyses indicate a terminal elimination half-life (T1/2) for cannabidiol of 70 h, with the maximal concentration achieved at approximately 4 h postdose. In addition, a dose-proportional increase in plasma cannabidiol was observed with ascending oral doses of 5–20 mg/kg, implying that the clearance of cannabidiol remained constant over time. This study provides the first pharmacokinetic data for ascending doses of cannabidiol in fed healthy volunteers, strengthening the evidence on which to base decisions regarding the use of cannabidiol in clinical care.
Compliance with Ethical Standards
Funding
This study was supported by the Victorian Department of Health & Human Services (Australia), and this sponsor also provided support for open access to this publication.
Conflict of interest
There is no conflict of interest to declare as the sponsor has no commercial interest and is putting the formulation into the public domain.
Ethical Approval
All procedures in this study were in accordance with the 1964 Helsinki Declaration (and its amendments). The study protocol and amendments were approved by the Alfred Hospital Human Research Ethics Committee—Approval Number: 308/17.
Informed Consent
All subjects provided written informed consent at enrolment.
References
- 1.Iffland K, Grotenhermen F. An update on safety and side effects of cannabidiol: a review of clinical data and relevant animal studies. Cannabis Cannabinoid Res. 2017;2(1):139–154. doi: 10.1089/can.2016.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szaflarski JP, Bebin EM. Cannabis, cannabidiol, and epilepsy—from receptors to clinical response. Epilepsy Behav. 2014;41:277–82. 10.1016/j.yebeh.2014.08.135. [DOI] [PubMed]
- 3.Devinsky O, Cilio MR, Cross H, Fernandez-Ruiz J, French J, Hill C, et al. Cannabidiol: pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia. 2014;55(6):791–802. doi: 10.1111/epi.12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel A, Devinsky O, Cross JH, Villanueva V, Wirrell E, VanLandingham K et al. Cannabidiol (CBD) significantly reduces drop seizure frequency in Lennox–Gastaut syndrome (LGS): results of a multi-center, randomized, double-blind, placebo controlled trial (GWPCARE4) (S21.001). Neurology. 2017;88(Suppl 16).
- 5.Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, et al. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376(21):2011–20. 10.1056/NEJMoa1611618. [DOI] [PubMed]
- 6.Thiele EA, Marsh ED, French JA, Mazurkiewicz-Beldzinska M, Benbadis SR, Joshi C, et al. Cannabidiol in patients with seizures associated with Lennox–Gastaut syndrome (GWPCARE4): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2018;391(10125):1085–1096. doi: 10.1016/S0140-6736(18)30136-3. [DOI] [PubMed] [Google Scholar]
- 7.Tzadok M, Uliel-Siboni S, Linder I, Kramer U, Epstein O, Menascu S, et al. CBD-enriched medical cannabis for intractable pediatric epilepsy: the current Israeli experience. Seizure. 2016;35:41–44. doi: 10.1016/j.seizure.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 8.Carrier EJ, Auchampach JA, Hillard CJ. Inhibition of an equilibrative nucleoside transporter by cannabidiol: a mechanism of cannabinoid immunosuppression. Proc Natl Acad Sci USA. 2006;103(20):7895–7900. doi: 10.1073/pnas.0511232103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Petrocellis L, Vellani V, Schiano-Moriello A, Marini P, Magherini PC, Orlando P, et al. Plant-derived cannabinoids modulate the activity of transient receptor potential channels of ankyrin type-1 and melastatin type-8. J Pharmacol Exp Ther. 2008;325(3):1007–1015. doi: 10.1124/jpet.107.134809. [DOI] [PubMed] [Google Scholar]
- 10.Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153(2):199–215. doi: 10.1038/sj.bjp.0707442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watkins AR. Cannabinoid interactions with ion channels and receptors. Channels (Austin, Tex) 2019;13(1):162–167. doi: 10.1080/19336950.2019.1615824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor L, Crockett J, Tayo B, Morrison G. A phase 1, open-label, parallel-group, single-dose trial of the pharmacokinetics and safety of cannabidiol (CBD) in subjects with mild to severe hepatic impairment. J Clin Pharmacol. 2019;59(8):1110–1119. doi: 10.1002/jcph.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang R, Yamaori S, Takeda S, Yamamoto I, Watanabe K. Identification of cytochrome P450 enzymes responsible for metabolism of cannabidiol by human liver microsomes. Life Sci. 2011;89(5–6):165–170. doi: 10.1016/j.lfs.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 14.Millar SA, Stone NL, Yates AS, O'Sullivan SE. A systematic review on the pharmacokinetics of cannabidiol in humans. Front Pharmacol. 2018;9:1365. 10.3389/fphar.2018.01365. [DOI] [PMC free article] [PubMed]
- 15.Wheless JW, Dlugos D, Miller I, Oh DA, Parikh N, Phillips S, et al. Pharmacokinetics and tolerability of multiple doses of pharmaceutical-grade synthetic cannabidiol in pediatric patients with treatment-resistant epilepsy. CNS Drugs. 2019;33(6):593–604. doi: 10.1007/s40263-019-00624-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sands TT, Rahdari S, Oldham MS, Caminha Nunes E, Tilton N, Cilio MR. Long-term safety, tolerability, and efficacy of cannabidiol in children with refractory epilepsy: results from an expanded access program in the US. CNS Drugs. 2019;33(1):47–60. doi: 10.1007/s40263-018-0589-2. [DOI] [PubMed] [Google Scholar]
- 17.Birnbaum AK, Karanam A, Marino SE, Barkley CM, Remmel RP, Roslawski M, et al. Food effect on pharmacokinetics of cannabidiol oral capsules in adult patients with refractory epilepsy. Epilepsia. 2019;60(8):1586–1592. doi: 10.1111/epi.16093. [DOI] [PubMed] [Google Scholar]
- 18.Bergamaschi MM, Queiroz RH, Zuardi AW, Crippa JA. Safety and side effects of cannabidiol, a Cannabis sativa constituent. Curr Drug Saf. 2011;6(4):237–49. [DOI] [PubMed]
- 19.O'Connell B, Gloss D, Devinsky O. Cannabinoids in treatment-resistant epilepsy: a review. Epilepsy Behav. 2017;70:P341–8. 10.1016/j.yebeh.2016.11.012. [DOI] [PubMed]
- 20.FDA. Guidance for industry: food-effect bioavailability and fed bioequivalence studies. Silver Spring: Center for Drug Evaluation and Research (CDER); 2002.
- 21.FDA. Bioanalytical method validation: guidance for industry. Silver Spring/Rockville: Center for Drug Evaluation and Research (CDER)/Center for Veterinary Medicine (CVM); 2018.
- 22.ICH. Harmonised tripartite guideline. Validation of analytical procedures: text and methodology Q2(R1). Geneva: International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; 2005.
- 23.Taylor L, Gidal B, Blakey G, Tayo B, Morrison G. A phase i, randomized, double-blind, placebo-controlled, single ascending dose, multiple dose, and food effect trial of the safety, tolerability and pharmacokinetics of highly purified cannabidiol in healthy subjects. CNS Drugs. 2018 doi: 10.1007/s40263-018-0578-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greenwich Biosciences. Epidiolex: full prescribing information (FDA approved). Carlsbad: Greenwich Biosciences; 2020.
- 25.World Health Organisation. Cannabidiol. Critical review. Report. Geneva: WHO; 2018.
- 26.Szaflarski JP, Bebin EM, Comi AM, Patel AD, Joshi C, Checketts D, et al. Long-term safety and treatment effects of cannabidiol in children and adults with treatment-resistant epilepsies: expanded access program results. Epilepsia. 2018;59(8):1540–1548. doi: 10.1111/epi.14477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Devinsky O, Marsh E, Friedman D, Thiele E, Laux L, Sullivan J, et al. Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. Lancet Neurol. 2016;15(3):270–278. doi: 10.1016/s1474-4422(15)00379-8. [DOI] [PubMed] [Google Scholar]
- 28.Devinsky O, Verducci C, Thiele EA, Laux LC, Patel AD, Filloux F, et al. Open-label use of highly purified CBD (Epidiolex) in patients with CDKL5 deficiency disorder and Aicardi, Dup15q, and Doose syndromes. Epilepsy Behav. 2018;86:131–7. 10.1016/j.yebeh.2018.05.013. [DOI] [PubMed]
- 29.Devinsky O, Patel AD, Cross JH, Villanueva V, Wirrell EC, Privitera M, et al. Effect of cannabidiol on drop seizures in the Lennox–Gastaut syndrome. N Engl J Med. 2018;378(20):1888–977. 10.1056/NEJMoa1714631. [DOI] [PubMed]
- 30.Morrison G, Crockett J, Blakey G, Sommerville K. A phase 1, open-label, pharmacokinetic trial to investigate possible drug-drug interactions between clobazam, stiripentol, or valproate and cannabidiol in healthy subjects. Clin Pharmacol Drug Dev. 2019 doi: 10.1002/cpdd.665. [DOI] [PMC free article] [PubMed] [Google Scholar]