

Abstract
Chronic lung diseases are a leading cause of morbidity and mortality across the globe, encompassing a diverse range of conditions from infections with pathogenic microorganisms to underlying genetic disorders. The respiratory tract represents an active interface with the external environment having the primary immune function of resisting pathogen intrusion and maintaining homeostasis in response to the myriad of stimuli encountered within its microenvironment. To perform these vital functions and prevent lung disorders, a chemical and biological cross-talk occurs in the complex milieu of the lung that mediates and regulates the numerous cellular processes contributing to lung health. In this review, we will focus on the role of cross-talk in chronic lung infections, and discuss how different cell types and signaling pathways contribute to the chronicity of infection(s) and prevent effective immune clearance of pathogens. In the lung microenvironment, pathogens have developed the capacity to evade mucosal immunity using different mechanisms or virulence factors, leading to colonization and infection of the host; such mechanisms include the release of soluble and volatile factors, as well as contact dependent (juxtracrine) interactions. We explore the diverse modes of communication between the host and pathogen in the lung tissue milieu in the context of chronic lung infections. Lastly, we review current methods and approaches used to model and study these host-pathogen interactions in vitro, and the role of these technological platforms in advancing our knowledge about chronic lung diseases.
Keywords: pulmonary cross-talk, soluble factors, juxtacrine signaling, volatile signaling, in vitro models
Introduction
Cell-mediated signaling events drive a wide range of physiological functions, ranging from tissue repair and homeostasis to immune response and disease, all of which occur in the complex biological environment of the pulmonary tract (1–3). Within the pulmonary tract, the exchange of cell-derived signals is implicated in the development, progression, and maintenance of infection (both acute and chronic), yet the nature of many signaling interactions remains unknown or unclear in both healthy and pathogenic environments (2–5). The severity and progression of lung infections are frequently examined within the scope of factors such as the pathogen-specific mechanism of infection, patient physiology (e.g., immune and vaccination status, age, transplants, etc.), and microenvironment composition (6–10), with the ultimate goal of understanding the role of various microenvironmental and mechanistic components on the pathogenesis of a singular infection. However, understanding the effects of these components, even within the context of a singular infection, is exceedingly difficult as there is immense variation in disease manifestation and outcome, contributed to in part by the complex signaling environment found in the lung. Chronic pulmonary infections (CPIs), broadly characterized by pathogen persistence and immune evasion as the pathogen adapts to or manipulates its microenvironmental niche, illustrate this difficulty, as CPIs can manifest in a myriad of forms resulting from microbial persistence after medical treatment, external exacerbation of underlying conditions (e.g., latent infection), shifts in internal microenvironmental cues, or potentially other understudied areas of the microenvironment, such as the lung microbiome (6, 10–14). The complexity of these pulmonary cell-signaling environments, coupled with the numerous external variables driving chronic infection, underscores the difficulty in accounting for the simultaneous effects of all of these different factors, including understanding how these host-host, host-pathogen, and pathogen-pathogen communications drive immune response and propagate infection.
The ability to interpret the chemical and biological communication in the lung microenvironment enables us to better comprehend the vital conversations that drive the disease state in chronic infections. However, the translation of these intrinsic signals to actionable immune defense can often confound the host biology, as pathogens have evolved numerous mechanisms to evade host defense systems and confound host communication. Further, this communication, which includes the release of signaling factors (e.g., soluble factor or volatile signaling) and physical interactions (e.g., juxtacrine signaling), acts cumulatively to simultaneously promote many different aspects of the infection ranging from epithelial adhesion and invasion by Burkholderia pseudomallei to secretion of Aspergillus fumigatus growth-promoting volatiles by Pseudomonas aeruginosa (15, 16). Understanding how signaling pathways in the lung microenvironment are shifted and manipulated in response to specific stimuli can elucidate key insight into the infection mechanisms of numerous pathogens, and provide information that can lead to novel diagnostics and treatments.
In this review, we focus on how chemical and biological signaling events in the lung microenvironment unfold during chronic lung infections, including recurrent, persistent, and latent infections with Mycobacterium tuberculosis, Aspergillus fumigatus, Pseudomonas aeruginosa, Streptococcus pneumoniae, Staphylococcus aureus, Cryptococcus neoformans, and Burkholderia pseudomallei. These host-pathogen interactions use analogous modes of cellular signaling and communication in the lung that involves soluble factor paracrine signaling (e.g., cytokines), juxtacrine signaling [e.g., pattern recognition receptors (PRR)], and volatile signaling [e.g., volatile organic compounds (VOC)], oftentimes with overlap of specific signaling mechanisms or components. We highlight similar signaling mechanisms across a spectrum of chronic pulmonary pathogens to create novel comparisons between diseases and to encourage interdisciplinary studies that translate methodologies, perspectives, and understanding for these infections. Further, we describe recent innovative in vitro technologies and platforms, in which the researcher has control over the complexity of the system, and suggest their use to examine the complex communication involved in the lung microenvironment during chronic lung infections.
Infections and Cross-Talk at the Molecular Level
The lung microenvironment is a heterogenous interface consisting of innate immune cells (e.g., alveolar macrophages, neutrophils, monocytes, and dendritic cells), mucous-producing and ciliated epithelial cells, rare immune populations [e.g., mucosal-associated invariant T cells (MAIT cells) (17, 18), innate lymphoid cells (ILCs) (19–21)], commensal and pathogenic microbes, and underlying tissue and vascular networks that are all in constant reciprocal communication (Figure 1). This tissue organization, which supports multiple cell populations with vastly different environmental requirements, must parse through the signaling milieu of soluble, volatile, and physical cues to identify the biological and chemical stimuli needed for proper respiratory function and effective immune defense. However, upon pathogen challenge, the lung microenvironment faces new obstacles in converting these stimuli into a coherent immune response, as oftentimes pulmonary pathogens simultaneously use multiple modes of communication (e.g., physical, soluble) to evade immunity and propagate infection; for example, S. pneumoniae-derived neuraminidase (NanA) cleaves mucins on the epithelial surface to enable pathogen attachment on host glycoconjugates, while subsequently secreting pore-forming toxin pneumolysin to disrupt the epithelium (7, 22, 23). The inability of the host to respond to these pathogen challenges leads to an impaired microenvironment where slight deviations from signaling equilibria can promote a chronic condition (24, 25), highlighting the importance of understanding the signaling landscape in CPIs.
Figure 1.

The lung microenvironment is home to a complex signaling network that interprets factors from various sources simultaneously. Signaling phenomena includes the exchange of soluble, volatile, and physical signals between cells of the host immune system, microbes, and host tissue components at varying spatiotemporal concentrations. Translating this complex quagmire of signals requires a broad perspective to see the role of each type of communication in the context of host-pathogen, pathogen-pathogen, and host-host signaling.
Soluble Factor Paracrine Signaling
Virulence Factors—Toxins
Pathogenic microbes employ a wide range of soluble virulence factor signaling mechanisms to combat the host immune response and persist in vivo, the most notable of which includes the use of host-targeting toxins. These toxins function in the early stages of infection to directly cause damage to the host microenvironment and create a favorable niche for the pathogen to survive. For example, S. pneumoniae makes use of a potent pore-forming toxin, pneumolysin, that binds to membrane-bound cholesterol and perforates the host cell, causing cell death through multiple mechanisms including destruction of DNA and cell cycle arrest (26). Pneumolysin has significant cytolytic effects and acts to induce the complement pathway and a proinflammatory cytokine response, leading to the degradation of barrier function in the host epithelium, pathogen uptake via platelet-activating factor receptor (PAFr)-mediated endocytosis, and pathogen trafficking across the epithelium into the underlying tissue for S. pneumonia dissemination (7, 27–29). Another prominent pathogen-derived toxin is the pore-forming compound α-hemolysin (α-toxin) utilized by S. aureus in pulmonary infection. A-toxin acts via binding to its specific cell receptor, ADAM10, wherein it oligomerizes to form the cytolytic pore and induce host cell lysis (30). The degradation of epithelial barrier integrity enables S. aureus, in a manner similar to S. pneumoniae, to invade into the underlying tissue, or to reside within the epithelial layer itself, where internalized bacteria can survive within a quasi-protective niche (31, 32). This intracellular residence permits the pathogen to enter into a dormant state protected from the host immune response and antibiotic therapy, contributing to its chronicity and enabling recurrent S. aureus infection (32).
The effect of S. aureus-derived α-toxin can be amplified during coinfection as well. Polymicrobial infections with fungal pathogen C. albicans activates the agr quorum sensing system in S. aureus, which leads to enhanced α-toxin secretion and decreased host survival, demonstrating how coinfection not only adds complexity to the lung microenvironment but also promotes increased virulence (33). Further, invasive fungal species, such as A. fumigatus and C. neoformans, are armed with a variety of toxins that impede or interfere with intracellular functions required to mount an effective immune response. A. fumigatus, one of the most successful opportunistic fungal pathogens, has a wide array of immunosuppressive secondary metabolites and toxins at its disposal including trypacidin, endocrocin, and gliotoxin (34–38). These toxins confer protection to A. fumigatus against numerous components of the host immune system. Trypacidin, found to localize to fungal conidia, induces high levels of nitric oxide (NO) and hydrogen peroxide (H2O2) production in bronchial epithelial cells, leading to severe oxidative stress and cell necrosis (34). Similarly, endocrocin, another spore-derived fungal toxin, inhibits neutrophil migration and displays significant virulence in a Drosophila model of infection (35). The most widely characterized A. fumigatus toxin, gliotoxin, has been found to inhibit multiple antimicrobial functions (e.g., superoxide defense) in a wide range of leukocytes, in addition to inducing apoptosis and inhibiting NFκB signaling, providing A. fumigatus with a broad repertoire to defend itself against a range of host immune responses (39–42); for a detailed review of gliotoxin's extensive immunomodulatory effects, refer to (43). The culmination of the effects of this library of toxins enables A. fumigatus to survive continual immune assault and create an environment favorable for colonization and proliferation.
Virulence Factors—Immune Modulators
In addition to toxins, there are other virulence factors used to evade host immune responses and confound the cross-talk required for effective pathogen clearance; these factors are a series of distinct compounds ranging from surface-expressed cell wall components to quorum sensing (QS) molecules used for intermicrobial communication. During infection with M. tuberculosis, the pathogen utilizes alternative strategies to manipulate immune cells in the local tissue environment, such as expression of the type VII secretion system, ESX-1 (44). This secretion system transports potent early secreted antigen target 6 kDa (ESAT-6) and culture filtrate protein (CFP-10) that are essential for phagolysosomal escape into the cytosol and induction of host cell necrosis (44–47). This event will trigger a pro-inflammatory reaction and secretion of MMP9 by epithelial cells and M. tuberculosis-infected macrophages, leading to the recruitment of uninfected macrophages and other immune cells to contain the infection in a granuloma (48, 49), although the arrival of uninfected cells may be exploited by pathogenic mycobacteria to further disseminate the infection (50–52). The extent of virulence, and the capability of the mycobacteria to evade the host immune response, has been directly linked to the presence and function of ESAT-6 and other RD1/ESX-1 derived virulence factors, as avirulent strains lack this secretion system (53–55). The exact mechanism of action of ESAT-6 is still somewhat unclear, yet it has been recently found that the ESX-1 secretion system does not completely depend on ESAT-6 for membrane lysis and that its function is contact dependent, despite ESAT-6 being able to disrupt lysosome membranes at an acidic pH (45, 56).
Recently, QS molecules have been identified as another group of potential virulence factors capable of programming pathogen populations to evade immune responses, as well as to inhibit or manipulate the host immune response directly (57). This mechanism is widely observed in the case of P. aeruginosa, where quorum sensing molecule N-3-oxo-dodecanoyl-L-homoeserine lactone (3O-C12-HSL), which regulates the LasR QS circuit and numerous other virulence factors, acts as a virulence factor itself that inhibits the production of proinflammatory cytokines (e.g., TNF, IL-12) and the proliferation and differentiation of T cells (57, 58). Additionally, 3O-C12-HSL does not only target human cells, in which 3O-C12-HSL often interacts with the intracellular activation protein IQGAP1 (57), but also commensal organisms. In a mixed coculture with opportunistic fungal pathogen C. albicans, 3O-C12-HSL was found to inhibit filamentation of the fungi, indicating that P. aeruginosa can use its QS molecules to influence the host as well as other microbes in its surrounding microenvironment (59). This dual functionality of QS molecules is observed with other P. aeruginosa-derived factors, such as Pseudomonas quinolone signal (PQS), which dysregulates host immunity through manipulation of cytokine production and reduction of antibacterial activity of host immune cells (60, 61). The ability of a pulmonary pathogen to utilize QS molecules for more than one function (i.e., to self-regulate and evade immunity) supports their persistence against constant immune challenge while maintaining the ability to proliferate and survive. Other virulence factors take on a more traditional or singular role in establishing a pathway for infection. C. neoformans, a fungal species often associated with meningitis in immunocompromised patients, is capable of establishing a chronic lung infection in immunocompetent patients through the secretion of host-targeting enzymes (e.g., phospholipases, proteases) that degrade host immune components (e.g., phagocytic compartment membranes, lung surfactant) (62–66). These secreted virulence factors drive the ability of C. neoformans to survive and protect itself from initial immune responses until it adapts to the host environment and takes on phenotypic forms to support its persistence in a dormant state (64, 67, 68). The secretion or release of virulence factors, which can encompass different classes of molecules, mediates various anti-immune behaviors to create favorable niches for persistence; further, the presence of multifunctional molecules, such as QS molecules, adds to the complex milieu present in the lung microenvironment and demonstrates the importance of a broad perspective encompassing not only host-pathogen communication but also signaling amongst bacterial and/or fungal kingdoms.
Cytokines
Pathogen-derived soluble factors contribute partially to the cross-talk events in the lung microenvironment; host-derived signals are also present, often in response to or to defend from pathogen-derived factors. Cytokines are a principle form of secreted soluble factor used to trigger the immune system and induce diverse effector functions upon pathogen challenge; careful regulation of these cytokines is vital, as impaired or excessive cytokine signaling can lead to pathogen survival and persistence, or pathological tissue destruction in the lung, respectively. M. tuberculosis-infected macrophages secrete multiple inflammatory cytokines, including TNF, IL-1β, and IL-6, which induce an early proinflammatory response that promotes immune cell activation and migration and results in the generation of a granuloma. Activated T cells producing IFNγ initiate antimicrobial responses, such as the induction of NO and autophagy, to promote mycobacterial clearance (Figure 2) (69–74). However, disruption of IFNγ signaling due to a shortage of activated effector T cells or an increase in regulatory T cells can lead to pathogen persistence within infected macrophages and increased bacterial burden in the lung (75–78).
Figure 2.

The exchange of soluble factors through paracrine signaling generates context-dependent effects. The secretion of proinflammatory factor IFNγ by T cells induces antimicrobial functions (e.g., autophagy) and signaling (e.g., HIF1α-mediated NO production) in M. tuberculosis-infected macrophages, while increasing the virulence of P. aeruginosa through OprF-binding and downstream induction of virulence factor PA-I. The secretion of IFNγ is decreased by interaction with C. neoformans virulence factor GXM, which decreases secretion of inflammatory factors (TNFα) while increasing secretion of anti-inflammatory factors IL-4 and IL-10. OprF, outer membrane porin F; PA-I, type I P. aeruginosa lectin; HIF1α, hypoxia inducible factor-1 α; NO, nitric oxide; GXM, glucuronoxylomannan.
Contrary to the role of IFNγ in M. tuberculosis infection, there is evidence suggesting that the immune activating effects of IFNγ can lead to an increase in virulence by P. aeruginosa, wherein IFNγ binds to P. aeruginosa surface receptor OprF to induce downstream expression of virulence factor PA-I via its QS signaling network (79) (Figure 2). This unintended response to a host cytokine illustrates that cytokine signaling can have dual, context-dependent outcomes. In addition to intercepting host signaling molecules, pathogens can also manipulate the expression and secretion of host factors to alter the signaling environment in their favor. The C. neoformans-derived virulence factor glucuronoxylomannan (GXM) increases secretion of immunomodulatory factors IL-10 and IL-4 while downregulating pro-inflammatory compounds TNF and IFNγ in stimulated CD4+ T cells, driving a permissive immune response that supports increased yeast cell growth (Figure 2) (80); the manipulation of the secretion ratio of IL-4 and IFNγ induce a range of macrophage phenotypes—ranging from classically activated to alternatively activated and intermediate—that simultaneously contribute to antimicrobial and acquiescent outcomes (81). Similarly, macrophages infected with M. tuberculosis secrete increased levels of VEGF while granuloma-associated macrophages have an increased expression ANG-2; the increased levels of VEGF and ANG-2 correlate with increased vascularization and vessel permeability around the site of infection, respectively, which is subsequently used for pathogen dissemination and host manipulation (82, 83). Chronic pulmonary pathogens can not only usurp the balance of pro- and anti-inflammatory cytokines found in the signaling microenvironment to alter the type of immune response, but also influence the surrounding tissue infrastructure to its own benefit (84).
Chemokines
The manipulation of host signaling by pulmonary pathogens also extends to chemokine signaling, which is a vital process for the recruitment of immune cells to the site of infection. Early in the course of pulmonary infections, the neutrophil-mediated pro-inflammatory response plays an important role in counteracting invading pathogens while also contributing to tissue pathology, making its manipulation an attractive target for pathogens. S. aureus utilizes multiple mechanisms to impede the recruitment of neutrophils to the site of infection by using secreted staphopain (i.e., cysteine proteases) that inactivates CXCR2 on neutrophils and blocks the attractant effects of infection-associated chemoattractants (85); additionally, S. aureus utilizes exoprotein SSL5 to scavenge chemokines and bind to chemokine surface receptors, effectively blocking the ability of neutrophils to respond to receptor-bound chemokines (86). Further, SSL5 is found to act on neutrophils through its capacity as an MMP-inhibitor, blocking potentiation of neutrophil chemokine IL-8 (CXCL8) and preventing neutrophil migration through collagen (87). The modulation of neutrophil recruitment is not limited to S. aureus-derived soluble factors, as S. pneumoniae-derived virulence factor PepO is found to contrastingly increase secretion of chemoattractants CXCL8 and IP-10 (CXCL10) from bronchial cell line BEAS-2B, suggesting that this factor may increase recruitment of neutrophils to the site of infection (88). T cell chemokine CXCL10 has also been associated with Cryptococcus infections; during murine infection with C. gattii, a close pathogenic relative to C. neoformans, an impaired TH1 migration in the lung is correlated with decreased Ip10 expression and poor DC maturation when compared to C. neoformans infection, leading to an attenuated effector T cell response and increased pulmonary infection (89).
Antimicrobial Molecules
Beyond host-host signaling (i.e., paracrine cytokine and chemokine signaling), the role of host-secreted factors directed toward pathogens is an essential functionality in clearing pathogens and preventing chronicity. Host cells utilize an array of armaments to directly kill pathogens, such as reactive oxygen and nitrogen species (ROS, RNS, respectively), cytotoxic molecules (e.g., granzymes, lysozymes, and perforins), antimicrobial peptides (e.g., human cathelicidin, LL-37, defensins, and granulysin), and antimicrobial small molecules [e.g., extracellular vesicles (EVs), miRNA]. ROS play a vital role in the clearance of pathogenic microbes, but must also be tightly controlled to minimize the risk of host-tissue damage via extensive oxidation, as well as host immune suppression at the site of infection. The tight regulation of this defense is evident in the ability of neutrophils to induce different types of ROS signaling based on the size of the microbe it encounters, triggering intracellular ROS against ingested small yeast cells while secreting extracellular ROS against the larger hyphal form of fungi (90). In contrast, pathogens can activate this defense in their favor through toxin secretion, such as P. aeruginosa-derived pyocyanin, which triggers neutrophil apoptosis through inducing ROS and disrupting mitochondrial membrane potential (91). However, ROS-mediated pathogen clearance is not limited to neutrophils, as plasmacytoid DCs are also found to utilize ROS to inhibit the growth of C. neoformans in vitro, although they remain less effective at fungal killing than conventional DCs (92).
In B. pseudomallei infection, bacteremic patients were found to have increased levels of pro-apoptotic compounds granzyme A and B (93); further, stimulating patient-derived monocyte-derived DCs with B. pseudomallei antigens LolC and Hsp60 led to downstream activation of CD8+ T cells and increased granzyme B secretion (94). The increased secretion of granzyme B likely plays a role in patient survival, as NK-cells from deceased patients are characterized by significantly lower levels of granzyme B expression (95). Similarly, control of intracellular M. tuberculosis has been shown to involve CD8+ cytotoxic T cells using the pore-forming protein perforin to enable the entrance of granulysin into infected cells and the subsequent killing of Mycobacterium through osmotic lysis (96). Accordingly, a reduced expression of perforin and granulysin in M. tuberculosis infected tissues have been shown to correlate with disease progression (97, 98).
More recently, work has focused on identifying the antimicrobial effects of other components found in the extracellular communication milieu, such as extracellular vesicles (EVs) or metabolic components. For example, in interactions between A. fumigatus and host immunity, both host and pathogen secrete bioactive EVs that target its respective counterpart and generate proinflammatory and antimicrobial responses (99, 100). Important components of these EVs, such as miRNA, are also involved in mediating intra- and inter-cellular immune mechanisms and responses (101, 102), and are implicated as potential biomarkers of infections such as tuberculosis (103). Further, the metabolic cross-talk between pathogens, commensal organisms, and hosts is believed to be an influential factor in a number of relevant infection models. Indeed, many pulmonary pathogens such as A. fumigatus and P. aeruginosa are known to secrete siderophores that chelate iron from the host for their own use (57, 104, 105). Additionally, nitrogen and sulfur availability is thought to significantly contribute to pathogen survival, where availability of nutrients such as tryptophan and asparagine can modulate pathogen behavior and metabolism, as well as influence host immunity. We refer readers to these excellent recent reviews on the role of metabolic cross-talk in infections (106, 107).
Physical Contact Juxtacrine Signaling
Contact dependent signaling, or juxtacrine signaling, complements soluble factor signaling and is a crucial component of the intercellular signaling microenvironment. Contact-dependent signaling is one of the first modes of communication used to detect pathogens in the lung. Innate immune cells, such as macrophages and dendritic cells, rely on the expression of germline-encoded receptors [e.g., pathogen recognition receptor (PRR)] to sample and detect pathogen-specific molecules [e.g., pathogen associated molecular patterns (PAMPs)]. For example, surface bound receptors toll-like receptor (TLR) and c-type lectin receptor (CLR) detect conserved pathogenic molecules such as lipopolysaccharide (LPS) on the surface of gram-negative bacteria P. aeruginosa and B. pseudomallei, and β-glucans on fungi such as A. fumigatus, respectively (108–110).
Pathogen Recognition Receptors and Associated Molecular Patterns
Toll-Like Receptors (TLRs)
The TLR-signaling axis is present in a wide range of infections, including tuberculosis, where innate immune cells expressing TLR2 are able to detect multiple M. tuberculosis surface components (111), such as LpqH (112), LprG (113), and LprA (114), which impede intracellular signaling pathways and interfere with MHC II antigen processing and presentation. TLR4 detection of intracellular M. tuberculosis modulates intracellular signaling pathways governing the immune response in an antigen-dependent manner. Specifically, TLR2 detection of cell wall components PIM and mannosylated lipoarabinomannan (Man-LAM) result in robust anti-inflammatory responses characterized by decreased secretion of TNF and IL-12p40 and increased secretion of IL-37, respectively (115, 116). At the same time, HSP70-cofactor GrpE activates DCs and leads to their subsequent preferential induction of proinflammatory Th1 cells via TLR4 recognition (117). Additionally, detection of Man-LAM by different macrophage receptors such as the mannose receptor, complement receptors, and DC-SIGN can manipulate induction of proinflammatory responses to reduce local immune cell activation, as well as to inhibit phagosomal maturation inside infected macrophages (118–120).
C-Type Lectin Receptors (CLRs)
In conjunction with TLR pathogen sampling, CLR detection mechanisms also complement the innate immune system's ability to detect a wide range of pathogens. CLRs Dectin-1 and DC-SIGN have been implicated in the recognition, binding, and phagocytosis of A. fumigatus (121, 122), as single nucleotide polymorphisms in genes coding for Dectin-1 and DC-SIGN were associated with a higher risk of invasive pulmonary aspergillosis (123). Further, the absence of Dectin-1 ligand β-1,3-glucan on the surface of A. fumigatus leads to a more efficient complement-mediated antifungal immune response driven by DCs; this is found to be due to improved complement binding in the absence of β-1,3-glucan, and that galactomannan binding via DC-SIGN drives a proinflammatory cytokine response (124). However, A. fumigatus leverages other cell wall components to drive virulence, such as DHN melanin, which is able to block LC3-associated phagocytosis and subsequent fungal killing through impeding of NADPH oxidase from entering the phagosome (125). Host immunity is able to combat the virulence factor DHN melanin through binding via the melanin-sensing C-type lectin receptor (MelLec) found on endothelial cells and myeloid cells, the mutation of which is found to increase the susceptibility of stem-cell transplant patients to aspergillosis and to decrease proinflammatory cytokine secretion in macrophages (126).
The interactions between these PRRs and PAMPs strongly activate immune cells and initiate intra- and intercellular signaling to recruit other immune cells and clear the pathogen; however, this interaction is also vulnerable to manipulation by pathogens through suppression of PRRs or masking of PAMPs to avoid detection (104). Similar to A. fumigatus, C. neoformans utilizes L-DOPA melanin as a defense against oxidative stress from phagocytic immune cells (127–129), yet it relies mostly on its complex capsule to evade detection by host PRRs. C. neoformans contains a heterogenous multi-layered polysaccharide capsule that can block immune detection of fungal PAMPs, such as β-1,3-glucan, while preventing the adherence of complement proteins or antibodies to the surface (130). To further protect itself, C. neoformans can alter the physical characteristics of the capsule to adapt to environmental cues (131, 132), and capsule component glucuronoxylomannan (GXM) can induce L-selectin shedding and decreased TNFR expression on neutrophils, likely impeding their ability to bind to cell surfaces at the site of infection (133).
Epithelial-Pathogen Interactions
Pathogen Adhesion to Epithelium
Pulmonary epithelial cells play multiple roles in maintaining lung immunity, ranging from creating and maintaining a physical mucous barrier to expressing TLRs and initiating immune responses via proinflammatory cytokine secretion (134–138). For a pathogen, epithelial cells often represent the first challenge that must be overcome in order to successfully establish an infection, thereby requiring passage through or destruction of epithelial cells (7, 31, 32). After entrance into the airway, pathogens must surmount the outer layer of the epithelium—containing mucous, surfactant, and scavenging immune cells—as this region is a hostile environment for the establishment of colonies or infections. Therefore, microbes have developed an array of physical methods to pass through the mucous layers and bind to the surface epithelium to prevent their clearance. The pathogen B. pseudomallei expresses genes boaA and boaB, which code for proteins comparable to the Y. enterocolitis adhesin YadA; these adhesin proteins have been found to significantly increase the adhesion of E. coli expressing boaA and boaB to alveolar and bronchial cell lines, and have been implicated the in intracellular survival of the pathogen (139). Further, this adhesive capability of B. pseudomallei can be complemented by the adaptation of a biofilm phenotype, which was found to promote greater adhesion and subsequent invasion and infection by the bacteria (140). Indeed, the ability of B. pseudomallei to adhere to the airway epithelium is directly correlated with its virulence and ability to induce cellular damage (141). The strength of the adhesion of B. pseudomallei to the airway epithelium is also correlated with the expression of genes regulating QS and virulence pathways (142, 143); these correlations underscore the importance of pathogen adhesion for initiating a successful infection. Similarly, C. neoformans utilizes secreted phospholipase B to improve its ability to adhere to pulmonary epithelial cells, likely through interactions between surfactant protein D (SP-D) and polysaccharides on the surface of the C. neoformans capsule (63); additionally, capsule components GXM and MP84 are known to facilitate the binding of different C. neoformans strains to airway epithelial cells (144), enabling C. neoformans to initiate infection at this primary immune barrier (145). SP-D binding is also found to play a role in the immune response against A. fumigatus, wherein SP-D binds specifically to melanin on the fungal conidia and associates with galactomannan and galactosaminogalactan on the fungal cell wall; further, SP-D bound conidia were phagocytosed more readily than un-opsonized conidia, and induced a greater transcription of proinflammatory cytokines (146).
Epithelial Defense Against Pathogens
To counter these various pathogen-driven mechanisms of infection, the host epithelium employs an expanded array of defenses that support its active role as an innate immune component in the airway microenvironment (29, 135–138, 147). These defenses includes the direct secretion of antimicrobial peptide (AMP) classes such as defensins (148–150), cathelicidins (150, 151), lysozymes (152, 153), lactoferrins (152–154), and secretory leukocyte proteinase inhibitor (155, 156), to induce immunity and assist in antimicrobial defense. Some of these AMPs are constitutively secreted, such human β-defensin-1, yet other β-family defensins and AMPs are secreted in response to juxtacrine signaling events, such as TLR activation or NF-κB signaling (135, 136, 148). While the lung epithelium can deploy some defense mechanisms against adherent pulmonary pathogens, once adhered, these pulmonary pathogens can invade into or through the epithelium to establish a protective niche for growth and persistence. Discovery of mechanisms or treatments to decrease this adherence and enable clearance from the lung epithelium prior to pathogen entry can have a significant impact on disease outcome.
Intracellular Cross-Talk
For many pathogens, passage through the epithelial barrier is unknowingly assisted by phagocytic immune cells. Physical detection of pathogens by PRRs can initiate an endocytic process to encapsulate the pathogen within an intracellular vacuole that then uses numerous mechanisms to clear its contents, such as vacuole acidification or fusion with vacuoles containing antimicrobial peptides (e.g., human cathelicidin, LL-37), antimicrobial molecules (e.g., RNS, ROS), or lytic proteins (e.g., cathepsins, phospholipases) (157). During M. tuberculosis infection, activated macrophages release a collection of reactive nitrogen and oxygen species generated via NOX2 into the phagosome to induce oxidative stress and destruction of microbial membranes and intracellular components (157). Induction of autophagy and xenophagy pathways in M. tuberculosis-infected macrophages provides an alternative mechanism of defense against the intracellular pathogen, and modulation of these pathways has been the target of host-directed-therapies (158). This intracellular communication between the host cell and pathogen enables a strong form of self-defense for the cell, as well as a method to clear an acute infection and potentially avoid chronicity. Yet, for many pathogens, including M. tuberculosis, the ability to escape the phagocytic vacuole is the key to survival and persistence and an important mechanism for immune evasion (159–162).
Though not commonly perceived as an intracellular pathogen, S. aureus utilizes several methods to persist within a protective intracellular niche, ranging from secreting factors to block the acidification of the phagosome in leukocytes to disrupting phagosomal membranes for escape into the cytosol in epithelial cells (163, 164). Indeed, S. aureus utilizes its presence inside PMNs to usurp PMN expression of “danger” signals and to manipulate the phenotype of innate immune cells when in coculture, ultimately altering the ability of macrophages to clear S. aureus—infected PMNs and enabling intracellular persistence of the bacteria (165, 166). S. aureus can evade and manipulate PMN cell (i.e., neutrophil) function and antimicrobial effects in different ways, which is reviewed in depth here (165). S. aureus also utilizes the intracellular space of nonprofessional phagocytes, such as epithelial cells, as a protective niche to hide from host immune cells and to diverge into a heterogeneous population of replicating invasive or non-replicating persistent phenotypes depending on its intracellular environment, host cell metabolism, and bacterial colony variance (32, 167).
Once an intracellular pathogen manages to escape from the phagosome, it must rely on additional mechanisms for movement through the cell itself. An important component of this intracellular mobility is actin-based motility, which is utilized by a wide range of pathogens including B. pseudomallei and C. neoformans to manipulate the host cell cytoskeleton for their own movement and eventual dissemination through cell–cell spreading (Figure 3) (168, 169). Intracellular B. pseudomallei that have escaped the phagosome utilize bacterial proteins BimA and BimC to polymerize and remodel actin at its poles, enabling the pathogen to move through the cell to the outer membrane, from which it can protrude out of the cell and infect proximal cells (Figure 3) (168–170). C. neoformans similarly uses cell to cell spreading to infect neighboring cells while evading immune surveillance mechanisms, relying on an actin-dependent method for phagosomal escape and extrusion; however, the host cell utilizes actin flashes, or rapid repolymerizations, to temporarily prevent C. neoformans escape from the phagosome (Figure 3) (171, 172). The importance of actin in the pathogenesis of C. neoformans is also demonstrated by the dependence of macrophages on actin for the uptake and internalization of the extracellular yeast cells, which is significantly decreased in the presence of actin depolymerizing agents (173).
Figure 3.

Pathogens leverage host cytoskeletal components for movement within and between host cells in response to host-initiated physical interactions. Actin-mediated movement of B. pseudomallei is vital for pathogen survival and movement subsequent to phagosomal escape, as proteins BimA and BimC enable actin repolymerization in an Arp2/3-independent manner and the ability to protrude and infect proximal cells. Similarly, actin localization to the phagosome is an important component of C. neoformans phagocytosis and containment, as actin undergoes an Arp2/3-dependent rapid polymerization (“flash”) in response to C. neoformans permeabilization of the phagosome; disruption of the phagosome and C. neoformans extrusion into neighboring cells or extracellularly is an actin-dependent process that results in vacuole formation. Arp2/3, actin-related proteins 2/3; BimA, Burkholderia intracellular motility A; BimC, Burkholderia intracellular motility C.
Volatile Signaling
Whereas there exists a substantial amount of information regarding soluble factor signaling and juxtacrine signaling mechanisms, much less is known regarding volatile signaling. Principally regarded as a potential diagnostic for identifying bacterial species in vivo (174–176), volatile chemicals secreted by microbial species perform multiple functions as both symbiotic and antagonistic factors in complex signaling environments (177–180); indeed, microbial volatile organic compounds (mVOCs) have been described to play an important role in both intra- and inter-kingdom interactions (180). Additionally, these factors can originate from various microbes and kingdoms, and vary based on the microbial makeup of colonies, as well as the morphological form of microbe (i.e., spore vs. filamentous). Secreted mVOCs have been correlated with pathogen identity and even disease severity (181, 182), yet how these mVOCs influence pathogen behavior and host response is still widely unknown. We review mVOCs that target other commensal microbes and mVOCs that target host cells, as well as any downstream effects that result from this volatile cross-talk. However, volatile signaling between pathogens and the host lung is a vastly understudied area that represents a potentially significant wealth of information regarding host-pathogen interactions and as such requires further studies.
Pathogen-Targeted Volatile Signaling
Commensal microbes secrete volatile factors to promote synergetic interactions within an environment, wherein one microbe may secrete a mVOC that is then sequestered and metabolized by another or promote the growth of a neighboring microbe (177, 180). In the widely studied coculture system of P. aeruginosa and A. fumigatus, it has been found that volatile signaling between these two organisms results in stimulation of A. fumigatus growth from P. aeruginosa-derived dimethyl sulfide (Figure 4) (15); this result is contrary to what is observed when these two pathogens are in physical or shared media contact, where P. aeruginosa secretes toxins that inhibit the growth of A. fumigatus (183, 184). These interactions illustrate that the chemical communication between two organisms cannot be solely examined in one context and that growth conditions and the spatial distribution of microbes can yield different pathogen outcomes. This is further evident from the impact of oxygen availability on the A. fumigatus “volatilome,” which generates discernable volatile metabolite profiles dependent on the hypoxic state in the surrounding environment (185). The secretion of specific volatiles can also promote the survival and antibiotic resistance of pathogens, often to the benefit of the rest of the infectious colony. H2S is released by a wide range of pulmonary pathogens, such as Bacillus anthracis, P. aeruginosa, and S. aureus, and inhibition of H2S was found to increase the susceptibility of the bacteria to antibiotics (Figure 4) (186). The cytoprotective role of H2S is believed to result from its ability to control oxidative stress around the bacteria and prevent oxidation and DNA damage (186); this protection not only helps the bacteria resist antibiotic treatment, but also host-based oxidative stress (i.e., ROS). Antibiotic resistance can also be modulated by biogenic ammonia, which has been shown to improve resistance to tetracycline in S. aureus and P. aeruginosa, illustrating the impact of the volatile signaling landscape in the ability of a lung pathogen to survive and persist in a chronic infection (Figure 4) (187). This beneficial effect on growth and resistance is important for infections where there are multiple distal sites of pathogen colonies, as communication between these locations is not limited by the soluble diffusion- or contact-based signaling required for paracrine and juxtacrine signaling, making it more difficult for the host to intercept or impede these signals. However, much is still unknown regarding the detrimental and antagonistic effects of pathogen-derived volatiles on other pathogens in the lungs, as much of the research has focused on either the positive effects of microbial volatile signaling for pathogen survival, or the use of these volatile signals as diagnostics for bacterial colonization and infection (174, 175, 187).
Figure 4.

Volatile signaling in the lung microenvironment supports commensal and pathogenic microbial populations against diverse challenges and stimuli. Pulmonary pathogens S. aureus and P. aeruginosa secrete biogenic ammonia (NH3) and hydrogen sulfide (H2S) which offers protection against pharmacological interventions such as TCN and reactive oxygen species induced by GM, AP, or NA, respectively, to persist in the lung microenvironment. Further, the secretion of DMS from P. aeruginosa has been found to promote A. fumigatus growth, and the bidirectional exchange of volatile factors between the two pathogens leads to increased secretion of proinflammatory factors GM-CSF, IL-8, IL-6, IFNγ, and IL-1β from a lung tissue model of infection. TCN, tetracycline; GM, gentamicin; AP, ampicillin; NA, nalidixic acid; DMS, dimethyl sulfide.
Host-Targeted Volatile Signaling
The effects of volatile communication between microbes and their host counterparts is even less studied due to difficulties associated with subjecting host cells to volatile chemicals in a biomimetic manner. Complex in vitro models can partially solve this difficulty, as cells are not cultured in a traditional submerged manner (188, 189). We discuss some of these in vitro models in subsequent sections. Pathogen-derived volatile signals received by host cells can vary extensively from QS molecules to mVOCs; these factors can then in turn manipulate the behavior of host barriers to infection. One well-characterized volatile compound that performs a range of pathogen functions is 2-amino acetophenone (2-AA), a QS-regulated compound secreted by P. aeruginosa and B. thailandensis (a close nonpathogenic relative of B. pseudomallei) that has been linked to a persistent chronic phenotype in P. aeruginosa (190). Pretreatment with 2-AA in ex vivo murine macrophages led to a decrease in proinflammatory cytokine secretion while modulating innate immune signaling pathways, which can decrease tissue pathology but lead to insufficient clearance of P. aeruginosa in vivo (191). Additionally, 2-AA can disrupt oxidative homeostasis and induce ROS-mediated oxidative stress and apoptosis signaling, which is believed to further aid in the establishment of an environment conducive to chronic infection (192, 193). While the diffusion of these factors into the host tissue has detrimental effects, the influence of pathogen-derived volatiles on the host epithelium and immune cells is much lesser-known. Some cytotoxicity studies have been conducted demonstrating the harmful effects of volatiles such as 1-decanol and 1-octen-3-ol. However, these studies used submerged cell cultures exposed to media containing the mVOC and lacking an air-exposure component, or subjected bacterial cells to the compounds for umu and Ames testing (194, 195). Overall, there is a substantial and immediate need for research in this field that relates the effect of pulmonary pathogen-derived volatiles to human host immunology and disease outcomes, as much of the fundamental characterization of this space is still missing (196).
Modeling Pulmonary Infections and Cross-Talk
The role of cross-talk in pulmonary infections is beginning to be more understood as new analytical methods enable the examination of the cellular secretome (197–199) and novel in vitro lung tissue models facilitate scrutiny of the role of complex microenvironment conditions on lung pathology (178, 189, 200–202). Recapitulating lung tissue in vitro is challenging due to the complexity of the organ, which contains various components such as an air-exposed epithelium, a structurally intrinsic extracellular matrix, a complex vasculature network, and a myriad of immune components (203) (Figure 1), as well as a unique microbiome that contributes to the exchange of chemical signals in this microenvironment (6, 204, 205). The airway itself varies in shape and size, transitioning from the bronchi to bronchioles to the alveoli, reflecting a change in function from a conducting section to a gas exchange section (200, 202, 206), as well as regional differences in cell phenotypes and histology. For example, the small airways are lined with pseudostratified columnar ciliated epithelium with interspersed mucin secreting goblet cells; these features are functionally equipped to generate and move mucus, an important immune defense against extracellular microbes (136, 207, 208). In contrast, the alveoli contain a simple squamous epithelium, which is functionally equipped for the diffusion of gases between the alveoli and the surrounding capillary network (206, 209).
A significant challenge in the development of in vitro lung models is finding a balance between a simple and robust system suited for high throughput examination and a system that more closely recapitulates the full complexity of the human lung microenvironment. Perhaps the most significant power of in vitro systems is that they can be engineered along this spectrum, tailored to a particular level of complexity to answer a specific biological question. This configurability of an in vitro system, including choice of human cell types (e.g., epithelial, endothelial, stromal, immune cells), cell source (e.g., immortalized human cells or primary human cells), ECM components (e.g., collagen, matrigel, laminin), and two- or three-dimensional placement of these components relative to each other, grants the researcher unprecedented control over the microenvironment and the ability to manipulate the in vitro system (200, 210, 211).
Studies utilizing animal models have helped to develop much of our understanding of lung pathology and mechanistic biology. However, there are well-known limitations to animal models in fully elucidating the complex microenvironment and cross-talk between pathogens and the human tissue microenvironment, such as physiological immune system differences and differences in disease susceptibility and development. For example, mice do not become infected with M. tuberculosis in the same way as humans, as they form structurally different granulomas and do not commonly develop necrotizing lesions (212). However, other animal models do offer closer homology to humans for the study of CPIs, such as non-human primates or humanized mice (213–218), although these are more expensive, logistically demanding to use, and raise ethical concerns.
Innovative organotypic and tissue engineered models have incorporated key biological functions (219, 220), extracellular matrix (ECM) and structural components (188, 221, 222), mechanical stimuli (188, 221–225), and placement of pathogens with respect to lung tissue components to mimic in vivo interactions. These in vitro models allow researchers to hone in on specific aspects of the lung physiology and manipulate conditions to better understand the underlying signaling mechanisms governing the pathology of infection. Here, we describe the current landscape of in vitro lung models, with a focus on utilizing these models as a tool for studying the microenvironmental cross-talk in CPIs (Table 1). Many of these models have already elucidated key insights into CPIs, such as the dependence of early M. tuberculosis granuloma formation on ESAT-6 and the disruption of this granuloma formation with inhibition of tissue matrix metalloproteinases (MMPs) (49, 227). However, many models that were not explicitly designed for modeling infection have the potential to be adapted for studying cross-talk in infection. Further, many of these models can be kept in culture for extended periods of time (±4–8 weeks) (220–222, 226, 228), which may enable some investigation into long term host-pathogen interactions similar to that found in chronic infections. While we do not discuss in vitro organoid models here, we refer readers to these sources for lung organoid models (229–232).
Table 1.
Current in vitro microfluidic platforms enable researchers to precisely control the microenvironment through incorporation of various cell types, ECM components, and mechanical stimuli, while maintaining compatibility with a wide range of established readouts.
| In vitro Model Type | Advantages/Disadvantages | Specific examples | Applications for investigating cross-talk in infections |
|---|---|---|---|
| Transwell/cell culture inserts | + Simple and robust + Compatible with standard cell culture equipment + Standard air-liquid interface protocol + No external equipment requirement (e.g., pumps or vacuum for controlled fluid or air flow) + Compatible with established readouts (e.g., microscopy, RNA analysis) + Access to cultures for direct manipulation − Limited customization − Mechanically static − Poorly quantified diffusion gradients |
(189)
|
• Direct contact signaling • Soluble factor signaling • Co-culture with various cell types (e.g., DCs, macrophages, stromal cells) • Air-liquid interface on 3D culture • Cell-permeable inserts can be used for neutrophil/immune cell migration modeling through epithelium |
| Mechanically “breathing” microfluidic model | + Cyclic airflow and mechanical stretching possible + Continuous nutrient/media flow + Customizable + ALI compatible + Compatible with established readouts (e.g., microscopy, RNA analysis) + Control over diffusion gradients − External equipment required (e.g., syringe pump for fluid flow control and vacuum pump for pressure control) − Microfabrication technology may be necessary for additional customization − Difficult to access cultures for direct manipulation |
(188, 221)“Lung-on-a-Chip”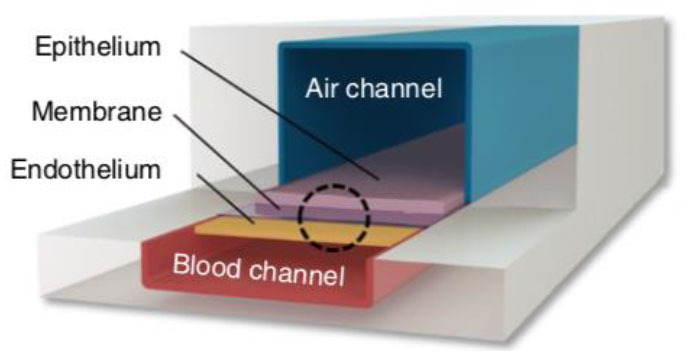
|
• Direct contact signaling • Soluble factor signaling • Investigations involving mechanical stretch • Investigations requiring controlled air or liquid flow • Coculture with multiple cell types (e.g., endothelium) • Porous membrane allows for neutrophil/immune cell migration modeling through epithelium • Control over airflow in ALI model |
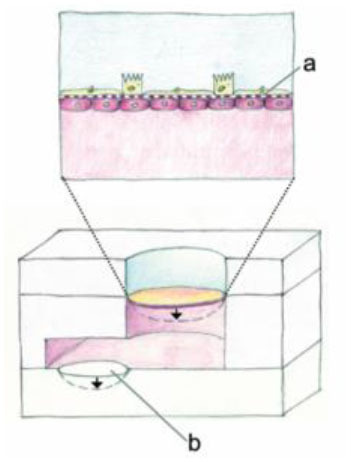
(223)
|
• Direct contact signaling • Soluble factor signaling • Investigations involving mechanical stretch • Investigations requiring controlled air or liquid flow • Coculture with multiple cell types (e.g., endothelium) • Porous membrane allows for neutrophil/immune cell migration modeling through epithelium • Open well for easier access of culture for downstream analysis |
||
| Hydrogel-based microfabricated models | + Customizable + Complexity in tissue components and shape + Multiple signaling modes (i.e., volatile, direct contact, soluble factor, etc.) + ALI compatible + Open systems enable access to cultures for direct manipulation − External equipment may be required if controlled fluid flow or airflow is desired − Microfabrication technology may be necessary for additional customization |
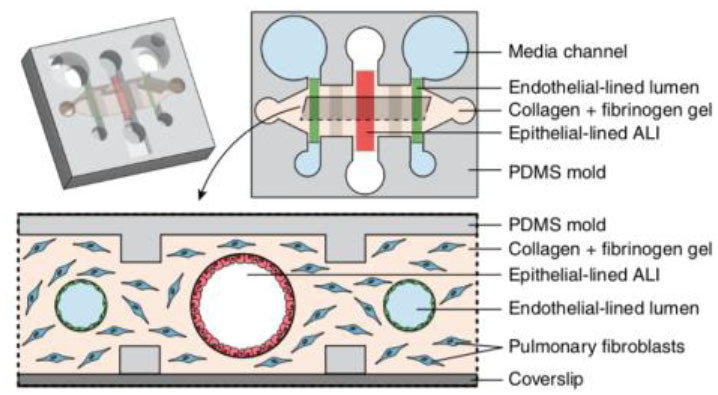
(178)
|
• Volatile signaling coculture experiments • Direct contact signaling • Soluble factor signaling • Investigations involving endothelial, mesenchyme, and epithelial cells • Modular cell culture experiments • Co-infection experiments |
(226)
|
• Co-culture with epithelial and smooth muscle cells • Native ECM interactions (i.e., collagen/matrigel) • Continuous media flow • Air liquid interface • Soluble factor signaling • Direct contact signaling |
Various model types can be adapted for different biological systems, dependent on which components of the lung microenvironment need to be examined.
Transwell Inserts: A Simple, Robust Coculture Model
Advances in culturing techniques have led to an expansion beyond a submerged simple monolayer culture of lung epithelial cells, which fails to replicate tissue-level functionality (233–235). There is much interest and limited success in developing culturing conditions for inducing differentiation of easy to obtain cell lines into various other cellular phenotypes that are found in the airway (219, 220). For example, the establishment of an air liquid interface (ALI), which mimics the native environment of the lung epithelium, can differentiate a small-airway epithelial cell from a cobblestone monolayer culture into a mucociliary phenotype complete with ciliated pseudostratified columnar epithelium (219, 220) and mucin-producing goblet cells (220, 226, 236). Using other epithelial cell lines, an ALI can help create alveolar epithelial models, wherein these cell types can recapitulate alveolar function as measured by the increased surfactant secretion from type II-like alveolar epithelia (188, 237).
Widespread use of cell culture inserts technologies, such as Transwell inserts, have allowed for multiple monoculture and coculture experiments utilizing an ALI to differentiate epithelial cells into a biomimetic small airway model (219, 220, 238). The Transwell insert, produced by Corning Life Sciences, utilizes a simple and robust design that integrates into standard laboratory equipment. The insert consists of a permeable membrane that sits above the bottom of a standard tissue culture well. The membrane pore size can be chosen from 0.4 to 8.0 μm, with the larger pore sizes often used for cell migration assays, and the smaller pore sizes (typically 0.4 μm) used for coculture, soluble factor signaling, and creating ALI cultures. The inserts can also be used to establish an ALI in lung epithelial models (219, 220, 238) by culturing epithelial cells on the permeable membrane and removing the appropriate amount of media such that the cells are exposed to air on the apical (top) side, while still receiving nutrients through the permeable membrane on the basolateral (bottom) side. For coculture, cells can be cultured on top of the permeable membrane, and a different cell type can be cultured in the well-plate floor or even on the underside of the membrane, allowing the two cell types to communicate via soluble factor signaling through the porous membrane (239, 240). This functionality is particularly useful for studying cross-talk in infections.
Transwell Models for Studying Pathogenic Infection
Due to the ease of use and efficacy of the Transwell cell inserts for establishing an ALI and a mucocilliary differentiated population, Transwell insert models have been utilized in several studies specifically for studying signaling involved in lung infections. The simplest model utilizes a single cell type (e.g., epithelial cells) cultured on the Transwell insert membrane to establish an ALI and a differentiated airway epithelium. The cells are then exposed to a pathogen of interest either by incorporating pathogen conditioned media into the bottom chamber (238) or by introducing the pathogen itself on the apical side of the airway epithelium (241, 242). An example of both these approaches can be seen in a study done by Halldorsson et al. (238) in 2010. This study took advantage of the ability to induce tight junction formation using an ALI produced by a Transwell insert to study the effect of azithromycin treatment for maintaining tight junction integrity during a challenge with live P. aeruginosa and bacterial culture conditioned media. Increasing in complexity, the two-chamber system provided by the Transwell insert also lends itself to coculture with an additional cell type in the bottom chamber, such as endothelial or PMC cells (239, 240).
Building upon these simpler systems, advanced models have also been developed that incorporate a three-dimensional culture into a Transwell insert-based platform. These models often include other tissue features such as an ECM/stromal component (189, 243) and immune components (189). For example, Bhowmick et al. created a 3D chitosan-collagen scaffold polymerized on top of a Transwell insert membrane before culturing human small airway epithelial cells on top of this scaffold and airlifting (exposing to air) the model. When compared to a non-3D scaffold control, they found differences in protein expression in uninfected, H1N1, and H3N3 flu virus-infected epithelial cells (243). These results suggest that the addition of ECM components makes a difference in cellular functions.
Additionally, Nguyen Hoang et al. (189) developed a Transwell insert that incorporates both a stromal component and an immune component within the model. In this model, fibroblasts were suspended in type 1 collagen, forming a 3D stromal layer on top of the Transwell insert membrane. Dendritic cells are then cultured on top of this stromal layer prior to epithelial cell culture and airlifting (189). It was found that the capacity of DC to produce chemokines is regulated by the 3D organotypic model, and that soluble components secreted in their lung tissue model induced CCL18 in DC, a function absent in other culture conditions including conditioned media, suggesting that control over the 3D microenvironment is critical for immune system function – an important consideration when designing models for studying host-pathogen interactions. This lung tissue model has been further adapted to incorporate monocytes and macrophages for the study of early granuloma formation in M. tuberculosis infection (49, 227, 244). These studies enabled visualization and quantification of immune cell clustering in virulent M. tuberculosis infection at the site of infection in the tissue, which is not possible to assess in single-culture systems, thus providing a unique opportunity to study host-pathogen interactions in experimental tissue.
Limitations of Transwell Insert Models
Transwell insert models are versatile and easily integrated into standard cell culture equipment, supporting their widespread use and adaptation. However, on the spectrum of complexity for in vitro models, they are relatively limited in recapitulating the lung tissue microenvironment and function. For example, the membrane separating the compartments is made of a polymeric material and does not fully recapitulate the extracellular matrix environment seen in the lamina propria of lung tissue (226). Therefore, any desired control over 2D or 3D placement of ECM hydrogels or cell types is challenging in Transwell inserts, which are designed for a monolayer or single layer of 3D gel in each compartment. Further, while 96 well Transwell inserts are available, cell insert models tend to require relatively numerous amounts of cells and reagents. This can limit these models to easy-to-obtain cell types, which may not be as biologically relevant as valuable cells, such as primary cells from patients with lung disease. Therefore, meso- or microscale models may be better suited than traditional well-plate culture for applications requiring the use of rare or valuable cell types. Lastly, the diffusion profile of factors between the apical chamber and the basal chamber is poorly characterized, making it difficult to use Transwells for studies where diffusion of signaling factors needs to be tightly controlled (245, 246).
Microfluidic and Microfabricated In vitro Models
Microfluidic and microfabrication techniques have become a useful tool for adding additional control and complexity to in vitro models. Microfluidic systems, which typically contain a channel or network of channels with at least one dimension on the sub-millimeter scale, permit more complex control of fluid and airflow, allowing for dynamic culturing conditions (188, 221, 226, 247). Other systems create dynamic culture conditions by adding modular functionalities and components (i.e., different cell types, microorganisms, etc.) at different experimental timepoints (178, 248). Moreover, the ability to shape hydrogels or create microscale compartments offers spatial control of different cell types that have varying levels of communication with each other from diffusion-based signaling to volatile signaling (178, 188, 221, 226). Finally, microfabrication techniques enable microscale culture dimensions and volumes, thus requiring the use of fewer cells than a traditional macroscale (e.g., well-plate) approach (202, 211, 249, 250).
Dynamic “Breathing” Lung Tissue Models
The microfabricated in vitro lung model (the “lung-on-a-chip”) developed by Huh et al. (188) in 2010 has been an influential model over the past decade. A schematic of a “lung-on-a-chip” type model and other models discussed can be found in Table 1. The model features a thin microfabricated porous membrane separating a top and bottom chamber. Alveolar-like epithelial cells are seeded on top of the porous membrane in the top chamber, where an air–liquid interface is created. Endothelial cells are seeded on the basal side of the membrane in the bottom channel where blood or media is flowed. Because it is a closed system, airflow can be controlled both on the apical ALI and flow rate of blood or media can be controlled in the bottom chamber. To add an additional functionality to the model, the design includes two side chambers on either side of the central channels, which can be put under cyclic vacuum or pressure, thus stretching the membrane that contains the cells in the inner channel. By modulating the pressure on the side chambers, the researchers can induce cyclical mechanical strain, similar to the mechanical stretching that alveolar tissue undergoes during breathing (188). This model is versatile and has found widespread use for studying different pathologies such COPD (221), pulmonary edema (251), and cigarette-induced lung damage (222), and has also been adapted to model the small airway where pseudostratified epithelium and mucin producing cells are observed after ALI exposure (221, 222). Notably, the alveolar model has been used as an infection model, where pathogens are introduced to the apical compartment and the effect of this direct contact is measured in the epithelium and endothelium below (188). Conditioned media or other relevant factors can also be introduced into the bottom compartment as a method for investigating soluble factor signaling.
Other models have also been developed with the goal of modeling cyclical mechanical stretching to mimic breathing in vitro, such as Stucki et al. (223) who developed a lung model that has actuation functionality. Much like Huh et al. (188) this model cultures alveolar epithelial cells on a thin porous membrane where the bottom side of the membrane is lined with endothelial cells. The actuation is brought by another flexible membrane at the bottom of the media chamber on top of a void channel. When negative pressure is applied in the void channel, the flexible membrane at the bottom of the media channel pulls down, thus pulling down on the flexible membrane containing the epithelial cells. This is similar to how the diaphragm decreases the pressure in the air cavity as breathing occurs in vivo. While the concept and materials (i.e., a porous membrane) are similar to the Huh et al. (188) “lung-on-a-chip,” this model differs slightly in geometry of where the actuation occurs, pulling down on the membrane rather than from the side. Further, the upper chamber is open, as it does not have a ceiling, removing the ability to control the apical airflow, but providing pipette access to the culture by users. In fact, human primary pulmonary alveolar cells collected from patients who underwent partial lung resection were utilized in this model, illustrating its applicability for valuable cell populations (223). This model has also been used for applications such as wound healing (224) and has been modified to include microelectronics for impedance measurements (225). Similar to the Huh et al. (188) model, this model can be used for study of cross-talk such as direct or soluble factor signaling, as pathogens can be directly introduced on the apical side or conditioned media can be introduced to the bottom compartment.
Suspended Microfluidic Small Airway Models
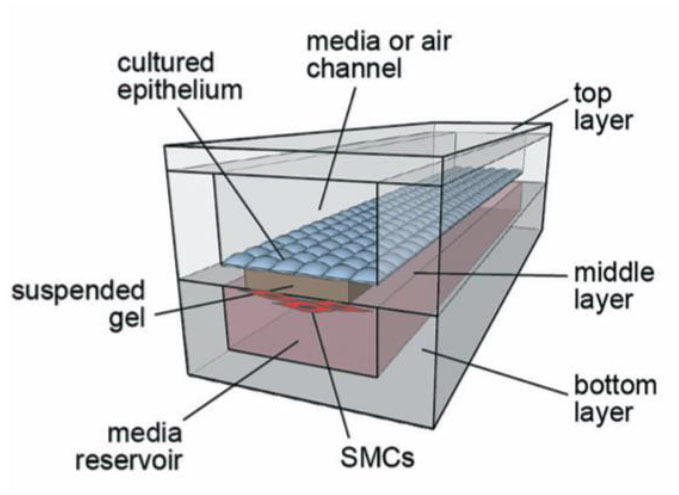
Previously described alveolar models focus on the interaction between epithelial and endothelial cells, as these are the most dominant cell type in that region of the lung. Other small airway microfluidic models, including the “small-airway on a chip” pioneered by Benam et al. (221) in 2016, also looks at the interaction between endothelial and epithelial cells. However, at the level of the small airway, such as the bronchioles, smooth muscle cells are also a relevant component, as the bronchioles will constrict or relax to increase the diameter of the airway, thus regulating airflow to different parts of the lung. Humayun et al. (226) developed a microfabricated small airway model that incorporates smooth muscle cells and epithelial cells. The model looks similar to the small-airway on a chip from Benem et al. (221) in that it contains a top chamber where the epithelial cells are grown, separated by a thin membrane with a bottom chamber that contains a second cell type (in this case smooth muscle cells). However, this model differs in several ways. For one, the cell type in the bottom chamber is smooth muscle cells (SMC) and not endothelial, which enables the researcher to model diseases such as asthma or pulmonary hypertension where SMC are important in disease pathogenesis. Secondly the “membrane” separating the two chambers is a suspended ECM hydrogel, more specifically a mixture of matrigel and collagen. This more closely represents the ECM environment than Transwell inserts or the other in vitro models previously discussed that rely on a bioinert polymer such as polydimethylsiloxane (PDMS). Further, this model does not include the mechanical functionality of the other in vitro models, as it is not a closed microfluidic system. However, the open microfluidic system offers simple fabrication and pipette accessibility. Since this model represents the functionality of the small airway for bronchoconstriction, it is perhaps better suited for studies of diseases where bronchoconstriction is relevant, such as asthma and COPD. However, it can be used as an interesting model to investigate the cross-talk involvement of smooth muscle cells in infections. Alternatively, this model can be altered such that the smooth muscle cells are replaced with a different cell type (e.g., endothelial cells). Further, because it utilizes a 3D suspended ECM, other cell-types could be encapsulated within the ECM, such as stromal cells or immune components, similar to the inclusion of DC in the Transwell 3D model by Nguyen Hoang et al. (189).
Microfluidic Lung Tissue Model for Studying Soluble and Volatile Factor Signaling
Apart from models that incorporate mechanical components to stimulate breathing in vitro, Barkal et al. (178) developed a platform specifically designed for probing multikingdom cell signaling in the lung microenvironment; this model not only allows for investigating direct and soluble factor cross-talk via placing pathogens in direct contact with the host lung tissue model, but also incorporates functionality for investigating volatile signaling. Moreover, this model incorporates a wide range of cellular components, including a three-dimensional collagen-based ECM with suspended fibroblast cells, where channels for the epithelium and endothelium are cast through, with one larger epithelium channel in the middle and two smaller endothelium channels on either side parallel to the epithelial channel. Similar to the Humayun et al. (226) model, the compartments are not separated by an inert plastic or polymer, such as PDMS, but rather a more biologically relevant hydrogel with the addition of suspended fibroblasts. Further, the air liquid interface is uniquely created in a three-dimensional tubular channel, more similar to in vivo bronchial structure (178). The endothelium-lined channels are cast cylindrically in the same manner as the epithelial lined channels. The authors are able to uniquely model a microenvironmental infection by introducing A. fumigatus directly into the apical lung epithelial channel and allowing it to grow out into the surrounding ECM. They measure cytokine concentrations in the media in the side endothelial channels in response to this “infection.” The authors further took advantage of the side endothelial channels and ECM by introducing blood immune components to the endothelial channels. Specifically, neutrophils were introduced via capillary flow into the side channels of the A. fumigatus infected model and migration into the ECM as a result of infection was measured. Finally, the authors designed an insert that clicks into the lung in vitro model chip that can culture two separate fungi or bacteria next to the model, enabling examination of multikingdom volatile signaling by culturing A. fumigatus and P. aeruginosa in this side insert. Cytokine levels again were measured from the endothelial side channels and shown to differ based on what combination of pathogens were included in the multi-kingdom culture. Notably this model offers multiple functionalities for studying pathogens that can contribute to chronic infections and cross-talk amongst these different cell types and kingdoms within a co-infection scenario. Ultimately, this platform, in addition to those described above, can enable researchers to probe very specific aspects of the lung microenvironment for a wide range of conditions and infections in a controlled and versatile manner.
Conclusion
Deciphering the complex signaling phenomena of the lung microenvironment, in the context of CPIs, remains a significant challenge due to the myriad of factors contributing to this signaling milieu. The communication amongst host and pathogen cells through secreted factors, physical contact, and even volatile factors, can drive acute and chronic infections as pathogens evade immunity, but it can also control and impede progression of disease through coordinated immune defenses. Further, accounting for the numerous signaling components and mechanisms occurring simultaneously in the lung in chronic infections, the exchange of signals in this microenvironment becomes extremely convoluted, requiring innovative and creative studies to develop an understanding of this space. The recent advances in in vitro modeling can help facilitate these studies, as these platforms are continually increasing in complexity and adaptability, enabling researchers to utilize these models in conjunction with in vivo studies and to answer specific research queries. Further, the large gap in our understanding surrounding volatile signaling in the lung represents a significant opportunity for fundamental and applied research studies, which can be further facilitated through the use of these novel in vitro platforms. Ultimately, we aimed to highlight similar signaling mechanisms in diverse pulmonary infections to elucidate novel connections and similarities between diseases, as well as to paint a picture of the complexity of this signaling environment during infection.
Author Contributions
SBB and MS conceived of the topic for the manuscript. SBB and AH wrote and organized the manuscript. AT, SB, and MS edited and provided feedback on the manuscript. All authors reviewed and approved the final manuscript.
Conflict of Interest
The authors declare the following potential conflicts of interest in companies pursuing open microfluidic technologies: AT: Stacks to the Future, LLC. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Fang Yun Lim for critical feedback on the manuscript.
Footnotes
Funding. This work was supported by grants from the Karolinska Institutet, Stockholm County Council, the Swedish Governmental Agency for Innovation Systems (VINNOVA) under the frame of NordForsk (Project no. 90456, PerAID), the Swedish Research Council under the frame of ERA PerMED (Project 2018-151, PerMIT), the Swedish Lung and Heart Foundation, the National Institutes of Health (NIH R35GM128648), the University of Washington, and by the National Science Foundation Graduate Research Fellowship Program under Grant no. DGE-1256082 (SBB). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
References
- 1.Cooper GM. Chapter 13: Signaling molecules and their receptors. In: The Cell: A Molecular Approach. 2nd ed Sunderland: Sinauer Associates; (2000). [Google Scholar]
- 2.Lee IT Yang CM . Inflammatory signalings involved in airway and pulmonary diseases. Mediators Inflamm. (2013) 2013:791231. 10.1155/2013/791231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Branchett WJ, Lloyd CM. Regulatory cytokine function in the respiratory tract. Mucosal Immunol. (2019) 12:589–600. 10.1038/s41385-019-0158-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Domingo-Gonzalez R, Prince O, Cooper A, Khader SA. Cytokines and chemokines in Mycobacterium tuberculosis infection. Microbiol Spectr. (2016) 4:1–37. 10.1128/microbiolspec.TBTB2-0018-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Werner JL, Steele C. Innate receptors and cellular defense against pulmonary infections. J Immunol. (2014) 193:3842–50. 10.4049/jimmunol.1400978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Dwyer DN, Dickson RP, Moore BB. The lung microbiome, immunity, and the pathogenesis of chronic lung disease. J Immunol. (2016) 196:4839–47. 10.4049/jimmunol.1600279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henriques-Normark B, Tuomanen EI. The pneumococcus: epidemiology, microbiology, and pathogenesis. Cold Spring Harb Perspect Med. (2013) 3:1–16. 10.1101/cshperspect.a010215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belperio J, Palmer SM, Weigt SS. Host-pathogen interactions and chronic lung allograft dysfunction. Ann Am Thorac Soc. (2017) 14:S242–S6. 10.1513/AnnalsATS.201606-464MG [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vila-Córcoles A, Ochoa-Gondar O, Llor C, Hospital I, Rodríguez T, Gómez A. Protective effect of pneumococcal vaccine against death by pneumonia in elderly subjects. Eur Respir J. (2005) 26:1086–91. 10.1183/09031936.05.00030205 [DOI] [PubMed] [Google Scholar]
- 10.Malhotra S, Hayes D, Wozniak DJ. Cystic fibrosis and Pseudomonas aeruginosa: the host-microbe interface. Clin Microbiol Rev. (2019) 32:00138–18. 10.1128/CMR.00138-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris J, Keane J. How tumour necrosis factor blockers interfere with tuberculosis immunity. Clin Exp Immunol. (2010) 161:1–9. 10.1111/j.1365-2249.2010.04146.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Budden KF, Shukla SD, Rehman SF, Bowerman KL, Keely S, Hugenholtz P, et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir Med. (2019) 7:907–20. 10.1016/S2213-2600(18)30510-1 [DOI] [PubMed] [Google Scholar]
- 13.Huffnagle GB, Dickson RP, Lukacs NW. The respiratory tract microbiome and lung inflammation: a two-way street. Mucosal Immunol. (2017) 10:299–306. 10.1038/mi.2016.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ubags NDJ, Marsland BJ. Mechanistic insight into the function of the microbiome in lung diseases. Eur Respir J. (2017) 50:1602467. 10.1183/13993003.02467-2016 [DOI] [PubMed] [Google Scholar]
- 15.Briard B, Heddergott C, Latgé JP. Volatile compounds emitted by pseudomonas aeruginosa stimulate growth of the fungal pathogen Aspergillus fumigatus. MBio. (2016) 7:1–5. 10.1128/mBio.00219-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.David J, Bell RE, Clark GC. Mechanisms of disease: host-pathogen interactions between Burkholderia species and lung epithelial cells. Front Cell Infect Microbiol. (2015) 5:80. 10.3389/fcimb.2015.00080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartmann N, Harriff MJ, McMurtrey CP, Hildebrand WH, Lewinsohn DM, Kronenberg M. Role of MAIT cells in pulmonary bacterial infection. Mol Immunol. (2018) 101:155–9. 10.1016/j.molimm.2018.06.270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meierovics A, Yankelevich WJC, Cowley SC. MAIT cells are critical for optimal mucosal immune responses during in vivo pulmonary bacterial infection. Proc Natl Acad Sci U S A. (2013) 110:3119–28. 10.1073/pnas.1302799110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barlow JL, McKenzie ANJ. Innate lymphoid cells of the lung. Annu Rev Physiol. (2019) 81:429–52. 10.1146/annurev-physiol-020518-114630 [DOI] [PubMed] [Google Scholar]
- 20.Lai DM, Shu Q, Fan J. The origin and role of innate lymphoid cells in the lung. Mil Med Res. (2016) 3:1–10. 10.1186/s40779-016-0093-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ardain A, Porterfield JZ, Kløverpris HN, Leslie A. Type 3 ILCs in lung disease. Front Immunol. (2019) 10:92. 10.3389/fimmu.2019.00092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berry AM, Paton JC. Additive attenuation of virulence of Streptococcus pneumoniae by mutation of the genes encoding pneumolysin and other putative pneumococcal virulence proteins. Infect Immun. (2000) 68:133–40. 10.1128/IAI.68.1.133-140.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Canvin JR, Marvin AP, Sivakumaran M, Paton JC, Boulnois GJ, Andrew PW, et al. The role of pneumolysin and autolysin in the pathology of pneumonia and septicemia in mice infected with a type 2 pneumococcus. J Infect Dis. (1995) 172:119–23. 10.1093/infdis/172.1.119 [DOI] [PubMed] [Google Scholar]
- 24.Cavalcanti YVN, Brelaz MCA, Neves JKDAL, Ferraz JC, Pereira VRA. Role of TNF-alpha, IFN-gamma, and IL-10 in the development of pulmonary tuberculosis. Pulm Med. (2012) 2012:745483. 10.1155/2012/745483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flynn JL, Chan J. Tuberculosis: latency and reactivation MINIREVIEW tuberculosis: latency and reactivation. Infect Immun. (2001) 69:4195–201. 10.1128/IAI.69.7.4195-4201.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rai P, He F, Kwang J, Engelward BP, Chow VTK. Pneumococcal pneumolysin induces DNA damage and cell cycle arrest. Sci Rep. (2016) 6:1–2. 10.1038/srep22972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Pee K, Neuhaus A, D'Imprima E, Mills DJ, Kühlbrandt W, Yildiz Ö. CryoEM structures of membrane pore and prepore complex reveal cytolytic mechanism of Pneumolysin. Elife. (2017) 6:1–22. 10.7554/eLife.23644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tweten RK. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect Immun. (2005) 73:6199–209. 10.1128/IAI.73.10.6199-6209.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thornton JA, Durik-Eder K, Tuomanen EI. Pneumococcal pathogenesis: innate invasion yet organ-specific damage. J Mol Med. (2011) 88:103–7. 10.1007/s00109-009-0578-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berube BJ, Wardenburg JB. Staphylococcus aureus α-toxin: nearly a century of intrigue. Toxins. (2013) 5:1140–66. 10.3390/toxins5061140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soong G, Martin FJ, Chun J, Cohen TS, Ahn DS, Prince A. Staphylococcus aureus protein A mediates invasion across airway epithelial cells through activation of RhoA GTPase signaling and proteolytic activity. J Biol Chem. (2011) 286:35891–8. 10.1074/jbc.M111.295386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palma Medina LM, Becker AK, Michalik S, Yedavally H, Raineri EJM, Hildebrandt P, et al. Metabolic cross-talk between human bronchial epithelial cells and internalized Staphylococcus aureus as a driver for infection. Mol Cell Proteomics. (2019) 18:892–908. 10.1074/mcp.RA118.001138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Todd OA, Fidel PL, Harro JM, Hilliard JJ, Tkaczyk C, Sellman BR, et al. Candida albicans augments Staphylococcus aureus virulence by engaging the staphylococcal agr quorum sensing system. MBio. (2019) 10:1–16. 10.1128/mBio.00910-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gauthier T, Wang X, Dos Santos J, Fysikopoulos A, Tadrist S, Canlet C, et al. Trypacidin, a spore-borne toxin from Aspergillus fumigatus, is cytotoxic to lung cells. PLoS One. (2012) 7:e29906. 10.1371/journal.pone.0029906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berthier E, Lim FY, Deng Q, Guo C-J, Kontoyiannis DP, Wang CCC, et al. Low-volume toolbox for the discovery of immunosuppressive fungal secondary metabolites. PLoS Pathog. (2013) 9:e1003289. 10.1371/journal.ppat.1003289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malekinejad H, Fani F, Hassani-Dizaji S, Shafie-Irannejad V, Fink-Gremmel F. Aspergillus fumigatus toxins cause cytotoxic and apoptotic effects on human T lymphocytes (Jurkat cells). World Mycotoxin J. (2013) 6:65–71. 10.3920/WMJ2012.1481 [DOI] [Google Scholar]
- 37.Kamei K, Watanabe A. Aspergillus mycotoxins and their effect on the host. Med Mycol. (2005) 43:95–9. 10.1080/13693780500051547 [DOI] [PubMed] [Google Scholar]
- 38.Suen YK, Fung KP, Lee CY, Kong SK. Gliotoxin induces apoptosis in cultured macrophages via production of reactive oxygen species and cytochrome c release without mitochondrial depolarization. Free Radic Res. (2001) 35:1–10. 10.1080/10715760100300541 [DOI] [PubMed] [Google Scholar]
- 39.Schlam D, Canton J, Carreño M, Kopinski H, Freeman SA, Grinstein S. Gliotoxin suppresses macrophage immune function by subverting. MBio. (2016) 7:1–15. 10.1128/mBio.02242-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Niide O, Suzuki Y, Yoshimaru T, Inoue T, Takayama T, Ra C. Fungal metabolite gliotoxin blocks mast cell activation by a calcium- and superoxide-dependent mechanism: implications for immunosuppressive activities. Clin Immunol. (2006) 118:108–16. 10.1016/j.clim.2005.08.012 [DOI] [PubMed] [Google Scholar]
- 41.Kupfahl C, Geginat G, Hof H. Gliotoxin-mediated suppression of innate and adaptive immune functions directed against Listeria monocytogenes. Med Mycol. (2006) 44:591–9. 10.1080/13693780600815411 [DOI] [PubMed] [Google Scholar]
- 42.Pahl HL, Krauss B, Schulze-Osthoff K, Decker T, Traenckner EB, Vogt M, et al. The immunosuppressive fungal metabolite gliotoxin specifically inhibits transcription factor NF-κB. J Exp Med. (1996) 183:1829–40. 10.1084/jem.183.4.1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arias M, Santiago L, Vidal-García M, Redrado S, Lanuza P, Comas L, et al. Preparations for invasion: modulation of host lung immunity during pulmonary Aspergillosis by gliotoxin and other fungal secondary metabolites. Front Immunol. (2018) 9:2549. 10.3389/fimmu.2018.02549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, et al. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell. (2007) 129:1287–98. 10.1016/j.cell.2007.05.059 [DOI] [PubMed] [Google Scholar]
- 45.Conrad WH, Osman MM, Shanahan JK, Chu F, Takaki KK, Cameron J, et al. Mycobacterial ESX-1 secretion system mediates host cell lysis through bacterium contact-dependent gross membrane disruptions. Proc Natl Acad Sci U S A. (2017) 114:1371–6. 10.1073/pnas.1620133114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Houben D, Demangel C, van Ingen J, Perez J, Baldeón L, Abdallah AM, et al. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol. (2012) 14:1287–98. 10.1111/j.1462-5822.2012.01799.x [DOI] [PubMed] [Google Scholar]
- 47.Simeone R, Bobard A, Lippmann J, Bitter W, Majlessi L, Brosch R, et al. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog. (2012) 8:e1002507. 10.1371/journal.ppat.1002507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Volkman HE, Pozos TC, Zheng J, Davis JM, Rawls JF, Ramakrishnan L. Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science. (2010) 327:466–9. 10.1126/science.1179663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parasa VR, Muvva JR, Rose JF, Braian C, Brighenti S, Lerm M. Inhibition of tissue matrix metalloproteinases interferes with Mycobacterium tuberculosis-induced granuloma formation and reduces bacterial load in a human lung tissue model. Front Microbiol. (2017) 8:2370. 10.3389/fmicb.2017.02370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ehlers S, Schaible UE. The granuloma in tuberculosis: dynamics of a host-pathogen collusion. Front Immunol. (2012) 3:1–9. 10.3389/fimmu.2012.00411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Silva Miranda M, Breiman A, Allain S, Deknuydt F, Altare F. The tuberculous granuloma: an unsuccessful host defence mechanism providing a safety shelter for the bacteria? Clin Dev Immunol. (2012) 2012:139127. 10.1155/2012/139127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramakrishnan L. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol. (2012) 12:352–66. 10.1038/nri3211 [DOI] [PubMed] [Google Scholar]
- 53.Pym AS, Brodin P, Brosch R, Huerre M, Cole ST. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol. (2002) 46:709–17. 10.1046/j.1365-2958.2002.03237.x [DOI] [PubMed] [Google Scholar]
- 54.Lewis KN, Liao R, Guinn KM, Hickey MJ, Smith S, Behr MA, et al. Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette-Guérin attenuation. J Infect Dis. (2003) 187:117–23. 10.1086/345862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brodin P, Rosenkrands I, Andersen P, Cole ST, Brosch R. ESAT-6 proteins: protective antigens and virulence factors? Trends Microbiol. (2004) 12:500–8. 10.1016/j.tim.2004.09.007 [DOI] [PubMed] [Google Scholar]
- 56.De Leon J, Jiang G, Ma Y, Rubin E, Fortune S, Sun J. Mycobacterium tuberculosis ESAT-6 exhibits a unique membrane-interacting activity that is not found in its ortholog from non-pathogenic Mycobacterium smegmatis. J Biol Chem. (2012) 287:44184–91. 10.1074/jbc.M112.420869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Turkina MV, Vikström E. Bacteria-host crosstalk: sensing of the quorum in the context of Pseudomonas aeruginosa infections. J Innate Immun. (2019) 11:263–79. 10.1159/000494069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hooi DSW, Bycroft BW, Chhabra SR, Williams P, Pritchard DI. Differential immune modulatory activity of Pseudomonas aeruginosa quorum-sensing signal molecules. Infect Immun. (2004) 72:6463–70. 10.1128/IAI.72.11.6463-6470.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hogan DA, Vik Å, Kolter R. A Pseudomonas aeruginosa quorum-sensing molecule influences Candida albicans morphology. Mol Microbiol. (2004) 54:1212–23. 10.1111/j.1365-2958.2004.04349.x [DOI] [PubMed] [Google Scholar]
- 60.Lin J, Cheng J, Wang Y, Shen X. The Pseudomonas quinolone signal (PQS): not just for quorum sensing anymore. Front Cell Infect Microbiol. (2018) 8:230. 10.3389/fcimb.2018.00230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Skindersoe ME, Zeuthen LH, Brix S, Fink LN, Lazenby J, Whittall C, et al. Pseudomonas aeruginosa quorum-sensing signal molecules interfere with dendritic cell-induced T-cell proliferation. FEMS Immunol Med Microbiol. (2009) 55:335–45. 10.1111/j.1574-695X.2008.00533.x [DOI] [PubMed] [Google Scholar]
- 62.Feldmesser M, Kress Y, Novikoff P, Casadevall A. Cryptococcus neoformans is a facultative intracellular pathogen in murine pulmonary infection. Infect Immun. (2000) 68:4225–37. 10.1128/IAI.68.7.4225-4237.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ganendren R, Carter E, Sorrell T, Widmer F, Wright L. Phospholipase B activity enhances adhesion of Cryptococcus neoformans to a human lung epithelial cell line. Microbes Infect. (2006) 8:1006–15. 10.1016/j.micinf.2005.10.018 [DOI] [PubMed] [Google Scholar]
- 64.Djordjevic JT. Role of phospholipases in fungal fitness, pathogenicity, and drug development - lessons from Cryptococcus neoformans. Front Microbiol. (2010) 1:125. 10.3389/fmicb.2010.00125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Santangelo RT, Nouri-Sorkhabi MH, Sorrell TC, Cagney M, Chen SCA, Kuchel PW, et al. Biochemical and functional characterisation of secreted phospholipase activities from Cryptococcus neoformans in their naturally occurring state. J Med Microbiol. (1999) 48:731–40. 10.1099/00222615-48-8-731 [DOI] [PubMed] [Google Scholar]
- 66.Garcia-Hermoso D, Janbon G, Dromer F. Epidemiological evidence for dormant Cryptococcus neoformans infection. J Clin Microbiol. (1999) 37:3204–9. 10.1128/JCM.37.10.3204-3209.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hommel B, Mukaremera L, Cordero RJB, Coelho C, Desjardins CA, Sturny-Leclère A, et al. Titan cells formation in Cryptococcus neoformans is finely tuned by environmental conditions and modulated by positive and negative genetic regulators. PLoS Pathog. (2018) 14:e1006982. 10.1371/journal.ppat.1006982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Casadevall A, Coelho C, Alanio A. Mechanisms of Cryptococcus neoformans-mediated host damage. Front Immunol. (2018) 9:855. 10.3389/fimmu.2018.00855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Braverman J, Stanley SA. Nitric oxide modulates macrophage responses to Mycobacterium tuberculosis infection through activation of HIF-1α and repression of NF-κB. J Immunol. (2017) 199:1805–16. 10.4049/jimmunol.1700515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Braverman J, Sogi KM, Benjamin D, Nomura DK, Stanley SA. HIF-1α Is an essential mediator of IFN-γ-dependent immunity to Mycobacterium tuberculosis. J Immunol. (2016) 197:1287–97. 10.4049/jimmunol.1600266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cadwell K. Crosstalk between autophagy and inflammatory signalling pathways: Balancing defence and homeostasis. Nat Rev Immunol. (2016) 16:661–75. 10.1038/nri.2016.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. (2004) 119:753–66. 10.1016/j.cell.2004.11.038 [DOI] [PubMed] [Google Scholar]
- 73.Kim YS, Silwal P, Kim SY, Yoshimori T, Jo EK. Autophagy-activating strategies to promote innate defense against mycobacteria. Exp Mol Med. (2019) 51(12). 10.1038/s12276-019-0290-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Saito S, Nakano M. Nitric oxide production by peritoneal macrophages of Mycobacterium bovis BCG-infected or non-infected mice: regulatory roles of T lymphocytes and cytokines. J Leukoc Biol. (1996) 59:908–15. 10.1002/jlb.59.6.908 [DOI] [PubMed] [Google Scholar]
- 75.Green AM, DiFazio R, Flynn JL. IFN-γ from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J Immunol. (2013) 190:270–7. 10.4049/jimmunol.1200061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cowley SC, Elkins KL. CD4+ T cells mediate IFN-γ-independent control of Mycobacterium tuberculosis infection both in vitro and in vivo. J Immunol. (2003) 171:4689–99. 10.4049/jimmunol.171.9.4689 [DOI] [PubMed] [Google Scholar]
- 77.Shafiani S, Tucker-Heard G, Kariyone A, Takatsu K, Urdahl KB. Pathogen-specific regulatory T cells delay the arrival of effector T cells in the lung during early tuberculosis. J Exp Med. (2010) 207:1409–20. 10.1084/jem.20091885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Scott-Browne JP, Shafiani S, Tucker-Heard G, Ishida-Tsubota K, Fontenot JD, Rudensky AY, et al. Expansion and function of Foxp3-expressing T regulatory cells during tuberculosis. J Exp Med. (2007) 204:2159–69. 10.1084/jem.20062105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu L, Estrada O, Zaborina O, Bains M, Shen L, Kohler JE, et al. Recognition of host immune activation by Pseudomonas aeruginosa. Science. (2005) 309:774–7. 10.1126/science.1112422 [DOI] [PubMed] [Google Scholar]
- 80.Andrade RM, Almeida GM, DosReis GA, Bento CAM. Glucuronoxylomannan of Cryptococcus neoformans exacerbates in vitro yeast cell growth by interleukin 10-dependent inhibition of CD4+ T lymphocyte responses. Cell Immunol. (2003) 222:116–25. 10.1016/S0008-8749(03)00116-3 [DOI] [PubMed] [Google Scholar]
- 81.Arora S, Olszewski MA, Tsang TM, McDonald RA, Toews GB, Huffnagle GB. Effect of cytokine interplay on macrophage polarization during chronic pulmonary infection with Cryptococcus neoformans. Infect Immun. (2011) 79:1915–26. 10.1128/IAI.01270-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oehlers SH, Cronan MR, Beerman RW, Johnson MG, Huang J, Kontos CD, et al. Infection-induced vascular permeability aids mycobacterial growth. J Infect Dis. (2017) 215:813–7. 10.1093/infdis/jiw355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Polena H, Boudou F, Tilleul S, Dubois-Colas N, Lecointe C, Rakotosamimanana V, et al. Mycobacterium tuberculosis exploits the formation of new blood vessels for its dissemination. Sci Rep. (2016) 6:1–11. 10.1038/srep33162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Osherov N, Ben-Ami R. Modulation of host angiogenesis as a microbial survival strategy and therapeutic target. PLoS Pathog. (2016) 12:e1005838 10.1371/journal.ppat.1005838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Laarman AJ, Mijnheer G, Mootz JM, van Rooijen JM, Ruyken M, Malone CL, et al. Staphylococcus aureus Staphopain A inhibits CXCR2-dependent neutrophil activation and chemotaxis. EMBO J. (2012) 31:3607–19. 10.1038/emboj.2012.212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bestebroer J, Van Kessel KPM, Azouagh H, Walenkamp AM, Boer IG, Romijn RA, et al. Staphylococcal SSL5 inhibits leukocyte activation by chemokines and anaphylatoxins. Blood. (2009) 113:328–37. 10.1182/blood-2008-04-153882 [DOI] [PubMed] [Google Scholar]
- 87.Koymans KJ, Bisschop A, Vughs MM, van Kessel KPM, de Haas CJC, van Strijp JAG. Staphylococcal superantigen-like protein 1 and 5 (SSL1 & SSL5) limit neutrophil chemotaxis and migration through MMP-inhibition. Int J Mol Sci. (2016) 17:1–16. 10.3390/ijms17071072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zou J, Zhou L, Hu C, Jing P, Guo X, Liu S, et al. IL-8 and IP-10 expression from human bronchial epithelial cells BEAS-2B are promoted by Streptococcus pneumoniae endopeptidase O (PepO). BMC Microbiol. (2017) 17:187. 10.1186/s12866-017-1081-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Angkasekwinai P, Sringkarin N, Supasorn O, Fungkrajai M, Wang YH, Chayakulkeeree M, et al. Cryptococcus gattii infection dampens Th1 and Th17 responses by attenuating dendritic cell function and pulmonary chemokine expression in the immunocompetent hosts. Infect Immun. (2014) 82:3880–90. 10.1128/IAI.01773-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Warnatsch A, Tsourouktsoglou TD, Branzk N, Wang Q, Reincke S, Herbst S, et al. Reactive oxygen species localization programs inflammation to clear microbes of different size. Immunity. (2017) 46:421–32. 10.1016/j.immuni.2017.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Managò A, Becker KA, Carpinteiro A, Wilker B, Soddemann M, Seitz AP, et al. Pseudomonas aeruginosa pyocyanin induces neutrophil death via mitochondrial reactive oxygen species and mitochondrial acid sphingomyelinase. Antioxidants Redox Signal. (2015) 22:1097–110. 10.1089/ars.2014.5979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hole CR, Leopold Wager CM, Mendiola AS, Wozniak KL, Campuzano A, Lin X, et al. Antifungal activity of plasmacytoid dendritic cells against Cryptococcus neoformans in vitro requires expression of dectin-3 (CLEC4D) and reactive oxygen species. Infect Immun. (2016) 84:2493–504. 10.1128/IAI.00103-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lauw FN, Simpson AJH, Hack CE, Prins JM, Wolbink AM, van Deventer SJH, et al. Soluble granzymes are released during human endotoxemia and in patients with severe infection due to gram-negative bacteria. J Infect Dis. (2000) 182:206–13. 10.1086/315642 [DOI] [PubMed] [Google Scholar]
- 94.Tippayawat P, Pinsiri M, Rinchai D, Riyapa D, Romphruk A, Gan YH, et al. Burkholderia pseudomallei proteins presented by monocyte-derived dendritic cells stimulate human memory T cells in vitro. Infect Immun. (2011) 79:305–13. 10.1128/IAI.00803-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kronsteiner B, Chaichana P, Sumonwiriya M, Jenjaroen K, Chowdhury FR, Chumseng S, et al. Diabetes alters immune response patterns to acute melioidosis in humans. Eur J Immunol. (2019) 49:1092–106. 10.1002/eji.201848037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stenger S, Hanson DA, Teitelbaum R, Dewan P, Niazi KR, Froelich CJ, et al. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science. (1998) 282:121–5. 10.1126/science.282.5386.121 [DOI] [PubMed] [Google Scholar]
- 97.Andersson J, Samarina A, Fink J, Rahman S, Grundström S. Impaired expression of perforin and granulysin in CD8+ T cells at the site of infection in human chronic pulmonary tuberculosis. Infect Immun. (2007) 75:5210–22. 10.1128/IAI.00624-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rahman S, Gudetta B, Fink J, Granath A, Ashenafi S, Aseffa A, et al. Compartmentalization of immune responses in human tuberculosis: Few CD8+ effector T cells but elevated levels of FoxP3+ regulatory T cells in the granulomatous lesions. Am J Pathol. (2009) 174:2211–24. 10.2353/ajpath.2009.080941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Souza JAM, Baltazar L, de M, Carregal VM, Gouveia-Eufrasio L, de Oliveira AG, Dias WG, et al. Characterization of Aspergillus fumigatus extracellular vesicles and their effects on macrophages and neutrophils functions. Front Microbiol. (2019) 10:2008 10.3389/fmicb.2019.02008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shopova IA, Belyaev I, Dasari P, Jahreis S, Stroe MC, Cseresnyes Z, et al. Human neutrophils produce antifungal extracellular vesicles against Aspergillus fumigatus. mBio. (2020) 11:e00596–20. 10.1128/mBio.00596-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Aguilar C, Mano M, Eulalio A. MicroRNAs at the host-bacteria interface: host defense or bacterial offense. Trends Microbiol. (2019) 27:206–18. 10.1016/j.tim.2018.10.011 [DOI] [PubMed] [Google Scholar]
- 102.Alexander M, Hu R, Runtsch MC, Kagele DA, Mosbruger TL, Tolmachova T, et al. Exosome-delivered microRNAs modulate the inflammatory response to endotoxin. Nat Commun. (2015) 6:7321. 10.1038/ncomms8321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Correia CN, Nalpas NC, McLoughlin KE, Browne JA, Gordon SV, MacHugh DE, et al. Circulating microRNAs as potential biomarkers of infectious disease. Front Immunol. (2017) 8:118. 10.3389/fimmu.2017.00118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hernández-Chávez MJ, Pérez-García LA, Niño-Vega GA, Mora-Montes HM. Fungal strategies to evade the host immune recognition. J Fungi. (2017) 3:1–28. 10.3390/jof3040051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Haas H. Fungal siderophore metabolism with a focus on Aspergillus fumigatus. Nat Prod Rep. (2014) 31:1266–76. 10.1039/C4NP00071D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Olive AJ, Sassetti CM. Metabolic crosstalk between host and pathogen: sensing, adapting and competing. Nat Rev Microbiol. (2016) 14:221–34. 10.1038/nrmicro.2016.12 [DOI] [PubMed] [Google Scholar]
- 107.Ren W, Rajendran R, Zhao Y, Tan B, Wu G, Bazer FW, et al. Amino acids as mediators of metabolic cross talk between host and pathogen. Front Immunol. (2018) 9:319. 10.3389/fimmu.2018.00319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Amarsaikhan N, Templeton SP. Co-recognition of β-glucan and chitin and programming of adaptive immunity to Aspergillus fumigatus. Front Microbiol. (2015) 6:344. 10.3389/fmicb.2015.00344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Norris MH, Schweizer HP, Tuanyok A. Structural diversity of Burkholderia pseudomallei lipopolysaccharides affects innate immune signaling. PLoS Negl Trop Dis. (2017) 11:e0005571. 10.1371/journal.pntd.0005571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pier GB. Pseudomonas aeruginosa lipopolysaccharide: a major virulence factor, initiator of inflammation and target for effective immunity. Int J Med Microbiol. (2007) 297:277–95. 10.1016/j.ijmm.2007.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shukla S, Richardson ET, Drage MG, Boom WH, Harding CV. Mycobacterium tuberculosis lipoprotein and lipoglycan binding to toll-like receptor 2 correlates with agonist activity and functional outcomes. Infect Immun. (2018) 86:450–18. 10.1128/IAI.00450-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sánchez A, Espinosa P, García T, Mancilla R. The 19 kDa Mycobacterium tuberculosis lipoprotein (lpqh) induces macrophage apoptosis through extrinsic and intrinsic pathways: a role for the mitochondrial apoptosis-inducing factor. Clin Dev Immunol. (2012) 2012:950503 10.1155/2012/950503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gehring AJ, Dobos KM, Belisle JT, Harding CV, Boom WH. Mycobacterium tuberculosis LprG (Rv1411c): a novel TLR-2 ligand that inhibits human macrophage class II MHC antigen processing. J Immunol. (2004) 173:2660–68. 10.4049/jimmunol.173.4.2660 [DOI] [PubMed] [Google Scholar]
- 114.Pecora ND, Gehring AJ, Canaday DH, Boom WH, Harding CV. Mycobacterium tuberculosis LprA is a lipoprotein agonist of TLR2 that regulates innate immunity and APC function. J Immunol. (2006) 177:422–9. 10.4049/jimmunol.177.1.422 [DOI] [PubMed] [Google Scholar]
- 115.Doz E, Rose S, Court N, Front S, Vasseur V, Charron S, et al. Mycobacterial phosphatidylinositol mannosides negatively regulate host toll-like receptor 4, MyD88-dependent proinflammatory cytokines, and TRIF-dependent co-stimulatory molecule expression. J Biol Chem. (2009) 284:23187–96. 10.1074/jbc.M109.037846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huang Z, Zhao GW, Gao CH, Chi XW, Zeng T, Hu YW, et al. Mannose-capped lipoarabinomannan from Mycobacterium tuberculosis induces IL-37 production via upregulating ERK1/2 and p38 in human type ii alveolar epithelial cells. Int J Clin Exp Med. (2015) 8:7279–87. [PMC free article] [PubMed] [Google Scholar]
- 117.Kim WS, Jung ID, Kim J-S, Kim HM, Kwon KW, Park Y-M, et al. Mycobacterium tuberculosis GrpE, A heat-shock stress responsive chaperone, promotes Th1-biased T cell immune response via TLR4-mediated activation of dendritic cells. Front Cell Infect Microbiol. (2018) 8:95. 10.3389/fcimb.2018.00095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fratti RA, Chua J, Vergne I, Deretic V. Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc Natl Acad Sci U S A. (2003) 100:5437–42. 10.1073/pnas.0737613100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hmama Z, Sendide K, Talal A, Garcia R, Dobos K, Reiner NE. Quantitative analysis of phagolysosome fusion in intact cells: inhibition by mycobacterial lipoarabinomannan and rescue by an 1α,25-dihydroxyvitamin D3-phosphoinositide 3-kinase pathway. J Cell Sci. (2004) 117:2131–9. 10.1242/jcs.01072 [DOI] [PubMed] [Google Scholar]
- 120.Kang PB, Azad AK, Torrelles JB, Kaufman TM, Beharka A, Tibesar E, et al. The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J Exp Med. (2005) 202:987–99. 10.1084/jem.20051239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Serrano-Gómez D, Domínguez-Soto A, Ancochea J, Jimenez-Heffernan JA, Leal JA, Corbí AL. Dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin Mediates binding and internalization of Aspergillus fumigatus conidia by dendritic cells and macrophages. J Immunol. (2004) 173:5635–43. 10.4049/jimmunol.173.9.5635 [DOI] [PubMed] [Google Scholar]
- 122.Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, et al. Dectin-1 is required for β-glucan recognition and control of fungal infection. Nat Immunol. (2007) 8:31–8. 10.1038/ni1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sainz J, Lupiáñez CB, Segura-Catena J, Vazquez L, Rios R, Oyonarte S, et al. Dectin-1 and DC-SIGN polymorphisms associated with invasive pulmonary Aspergillosis infection. PLoS One. (2012) 7:e32273. 10.1371/journal.pone.0032273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Steger M, Bermejo-Jambrina M, Yordanov T, Wagener J, Brakhage AA, Pittl V, et al. β-1,3-glucan-lacking Aspergillus fumigatus mediates an efficient antifungal immune response by activating complement and dendritic cells. Virulence. (2019) 10:957–69. 10.1080/21505594.2018.1528843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Akoumianaki T, Kyrmizi I, Valsecchi I, Gresnigt MS, Samonis G, Drakos E, et al. Aspergillus cell wall melanin blocks LC3-associated phagocytosis to promote pathogenicity. Cell Host Microbe. (2016) 19:79–90. 10.1016/j.chom.2015.12.002 [DOI] [PubMed] [Google Scholar]
- 126.Stappers MHT, Clark AE, Aimanianda V, Bidula S, Reid DM, Asamaphan P, et al. Recognition of DHN-melanin by a C-type lectin receptor is required for immunity to Aspergillus. Nature. (2018) 555:382–6. 10.26226/morressier.5ac39997d462b8028d89a0bc [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Missall TA, Moran JM, Corbett JA, Lodge JK. Distinct stress responses of two functional laccases in Cryptococcus neoformans are revealed in the absence of the Thiol-Specific Antioxidant Tsa1. Eukaryot Cell. (2005) 4:202–8. 10.1128/EC.4.1.202-208.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Emery HS, Shelburne CP, Bowman JP, Fallon PG, Schulz CA, Jacobson ES. Genetic study of oxygen resistance and melanization in Cryptococcus neoformans. Infect Immun. (1994) 62:5694–7. 10.1128/IAI.62.12.5694-5697.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang Y, Casadevall A. Susceptibility of melanized and nonmelanized Cryptococcus neoformans to nitrogen- and oxygen-derived oxidants. Infect Immun. (1994) 62:3004–7. 10.1128/IAI.62.7.3004-3007.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Campuzano A, Wormley FL. Innate immunity against cryptococcus, from recognition to elimination. J Fungi. (2018) 4:33. 10.3390/jof4010033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.O'Meara TR, Andrew Alspaugh J. The Cryptococcus neoformans capsule: a sword and a shield. Clin Microbiol Rev. (2012) 25:387–408. 10.1128/CMR.00001-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yoneda A, Doering TL. regulation of Cryptococcus neoformans capsule size is mediated at the polymer level. Eukaryot Cell. (2008) 7:546–9. 10.1128/EC.00437-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dong ZM, Murphy JW. Cryptococcal polysaccharides induce L-selectin shedding and tumor necrosis factor receptor loss from the surface of human neutrophils. J Clin Invest. (1996) 97:689–98. 10.1172/JCI118466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dudek M, Puttur F, Arnold-Schrauf C, Kühl AA, Holzmann B, Henriques-Normark B, et al. Lung epithelium and myeloid cells cooperate to clear acute pneumococcal infection. Mucosal Immunol. (2016) 9:1288–302. 10.1038/mi.2015.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.McClure R, Massari P. TLR-dependent human mucosal epithelial cell responses to microbial pathogens. Front Immunol. (2014) 5:386. 10.3389/fimmu.2014.00386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hiemstra PS, McCray PB, Bals R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur Respir J. (2015) 45:1150–62. 10.1183/09031936.00141514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tam A, Wadsworth S, Dorscheid D, Man SF, Sin DD. The airway epithelium: more than just a structural barrier. Ther Adv Respir Dis. (2011) 5:255–73. 10.1177/1753465810396539 [DOI] [PubMed] [Google Scholar]
- 138.Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol. (2015) 16:27–35. 10.1038/ni.3045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Balder R, Lipski S, Lazarus JJ, Grose W, Wooten RM, Hogan RJ, et al. Identification of Burkholderia mallei and Burkholderia pseudomallei adhesins for human respiratory epithelial cells. BMC Microbiol. (2010) 10:250. 10.1186/1471-2180-10-250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kunyanee C, Kamjumphol W, Taweechaisupapong S, Kanthawong S, Wongwajana S, Wongratanacheewin S, et al. Burkholderia pseudomallei biofilm promotes adhesion, internalization and stimulates proinflammatory cytokines in human epithelial A549 cells. PLoS One. (2016) 11:e0160741. 10.1371/journal.pone.0160741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kespichayawattana W, Intachote P, Utaisincharoen P, Sirisinha S. Virulent Burkholderia pseudomallei is more efficient than avirulent Burkholderia thailandensis in invasion of and adherence to cultured human epithelial cells. Microb Pathog. (2004) 36:287–92. 10.1016/j.micpath.2004.01.001 [DOI] [PubMed] [Google Scholar]
- 142.Essex-Lopresti AE, Boddey JA, Thomas R, Smith MP, Hartley MG, Atkins T, et al. A type IV pilin, PilA, contributes to adherence of B. pseudomallei and virulence in vivo. Infect Immun. (2005) 73:1260–4. 10.1128/IAI.73.2.1260-1264.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Srisanga K, Suthapot P, Permsirivisarn P, Govitrapong P, Tungpradabkul S, Wongtrakoongate P. Polyphosphate kinase 1 of Burkholderia pseudomallei controls quorum sensing, RpoS and host cell invasion. J Proteomics. (2019) 194:14–24. 10.1016/j.jprot.2018.12.024 [DOI] [PubMed] [Google Scholar]
- 144.Teixeira PAC, Penha LL, Mendonça-Previato L, Previato JO. Mannoprotein MP84 mediates the adhesion of Cryptococcus neoformans to epithelial lung cells. Front Cell Infect Microbiol. (2014) 4:106. 10.3389/fcimb.2014.00106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Taylor-Smith LM. Cryptococcus-epithelial interactions. J Fungi. (2017) 3:53. 10.3390/jof3040053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wah Wong SS, Rani M, Dodagatta-Marri E, Ibrahim-Granet O, Kishore U, Bayry J, et al. Fungal melanin stimulates surfactant protein D-mediated opsonization of and host immune response to Aspergillus fumigatus spores. J Biol Chem. (2018) 293:4901–12. 10.1074/jbc.M117.815852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ohkuni T, Kojima T, Ogasawara N, Masaki T, Fuchimoto J, Kamekura R, Koizumi J-I, et al. Poly(I:C) reduces expression of JAM-A and induces secretion of IL-8 and TNF-α via distinct NF-κB pathways in human nasal epithelial cells. Toxicol Appl Pharmacol. (2011) 250:29–38. 10.1016/j.taap.2010.09.023 [DOI] [PubMed] [Google Scholar]
- 148.Alekseeva L, Huet D, Féménia F, Mouyna I, Abdelouahab M, Cagna A, et al. Inducible expression of beta defensins by human respiratory epithelial cells exposed to Aspergillus fumigatus organisms. BMC Microbiol. (2009) 9:33. 10.1186/1471-2180-9-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Doss M, White MR, Tecle T, Hartshorn KL. Human defensins and LL-37 in mucosal immunity. J Leukoc Biol. (2010) 87:79–92. 10.1189/jlb.0609382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hielpos MS, Ferrero MC, Fernández AG, Bonetto J, Giambartolomei GH, Fossati CA, et al. CCL20 and beta-defensin 2 production by human lung epithelial cells and macrophages in response to Brucella abortus infection. PLoS One. (2015) 10:e0140408. 10.1371/journal.pone.0140408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bals R, Wang X, Zasloff M, Wilson JM. The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc Natl Acad Sci U S A. (1998) 95:9541–6. 10.1073/pnas.95.16.9541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dubin RF, Robinson SK, Widdicombe JH. Secretion of lactoferrin and lysozyme by cultures of human airway epithelium. Am J Physiol Lung Cell Mol Physiol. (2004) 286:750–5. 10.1152/ajplung.00326.2003 [DOI] [PubMed] [Google Scholar]
- 153.Ganz T. Antimicrobial polypeptides in host defense of the respiratory tract. J Clin Invest. (2002) 109:693–7. 10.1172/JCI0215218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Vargas Buonfiglio LG, Borcherding JA, Frommelt M, et al. Airway surface liquid from smokers promotes bacterial growth and biofilm formation via iron-lactoferrin imbalance. Respir Res. (2018) 19:1–11. 10.1186/s12931-018-0743-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Doumas S, Kolokotronis A, Stefanopoulos P. Anti-inflammatory and antimicrobial roles of secretory leukocyte protease inhibitor. Infect Immun. (2005) 73:1271–4. 10.1128/IAI.73.3.1271-1274.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sallenave JM. The role of secretory leukocyte proteinase inhibitor and elafin (elastase-specific inhibitor/skin-derived antileukoprotease) as alarm antiproteinases in inflammatory lung disease. Respir Res. (2000) 1:87–92. 10.1186/rr18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Weiss G, Schaible UE. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev. (2015) 264:182–203. 10.1111/imr.12266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Rekha RS, Rao Muvva SJ, Wan M, Raqib R, Bergman P, Brighenti S, et al. Phenylbutyrate induces LL-37-dependent autophagy and intracellular killing of Mycobacterium tuberculosis in human macrophages. Autophagy. (2015) 11:1688–99. 10.1080/15548627.2015.1075110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mehta M, Rajmani RS, Singh A. Mycobacterium tuberculosis WhiB3 responds to vacuolar pH-induced changes in mycothiol redox potential to modulate phagosomal maturation and virulence. J Biol Chem. (2016) 291:2888–903. 10.1074/jbc.M115.684597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ehrt S, Schnappinger D. Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell Microbiol. (2009) 11:1170–8. 10.1111/j.1462-5822.2009.01335.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Vandal OH, Nathan CF, Ehrt S. Acid resistance in Mycobacterium tuberculosis. J Bacteriol. (2009) 191:4714–21. 10.1128/JB.00305-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhai W, Wu F, Zhang Y, Fu Y, Liu Z. The immune escape mechanisms of Mycobacterium Tuberculosis. Int J Mol Sci. (2019) 20:340. 10.3390/ijms20020340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Grosz M, Kolter J, Paprotka K, Winkler AC, Schäfer D, Chatterjee SS, et al. Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin α. Cell Microbiol. (2014) 16:451–65. 10.1111/cmi.12233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Moldovan A, Fraunholz MJ. In or out: phagosomal escape of Staphylococcus aureus. Cell Microbiol. (2019) 21:1–9. 10.1111/cmi.12997 [DOI] [PubMed] [Google Scholar]
- 165.Guerra FE, Borgogna TR, Patel DM, Sward EW, Voyich JM. Epic immune battles of history: neutrophils vs. Staphylococcus aureus. Front Cell Infect Microbiol. (2017) 7:286. 10.3389/fcimb.2017.00286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Greenlee-Wacker MC, Rigby KM, Kobayashi SD, Porter AR, DeLeo FR, Nauseef WM. Phagocytosis of Staphylococcus aureus by human neutrophils prevents macrophage efferocytosis and induces programmed necrosis. J Immunol. (2014) 192:4709–17. 10.4049/jimmunol.1302692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rollin G, Tan X, Tros F, Dupuis M, Nassif X, Charbit A, et al. Intracellular survival of Staphylococcus aureus in endothelial cells: a matter of growth or persistence. Front Microbiol. (2017) 8:1354 10.3389/fmicb.2017.01354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Allwood EM, Devenish RJ, Prescott M, Adler B, Boyce JD. Strategies for intracellular survival of Burkholderia pseudomallei. Front Microbiol. (2011) 2:170. 10.3389/fmicb.2011.00170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Srinon V, Chaiwattanarungruengpaisan S, Korbsrisate S, Stevens JM. Burkholderia pseudomallei BimC is required for actin-based motility, intracellular survival, and virulence. Front Cell Infect Microbiol. (2019) 9:63. 10.3389/fcimb.2019.00063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Stevens MP, Stevens JM, Jeng RL, Taylor LA, Wood MW, Hawes P, et al. Identification of a bacterial factor required for actin-based motility of Burkholderia pseudomallei. Mol Microbiol. (2005) 56:40–53. 10.1111/j.1365-2958.2004.04528.x [DOI] [PubMed] [Google Scholar]
- 171.Alvarez M, Casadevall A. Cell-to-cell spread and massive vacuole formation after Cryptococcus neoformans infection of murine macrophages. BMC Immunol. (2007) 8:16. 10.1186/1471-2172-8-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Johnston SA, May RC. The human fungal pathogen Cryptococcus neoformans escapes macrophages by a phagosome emptying mechanism that is inhibited by arp2/3 complex- mediated actin polymerisation. PLoS Pathog. (2010) 6:e1001041. 10.1371/journal.ppat.1001041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Guerra CR, Seabra SH, De Souza W, Rozental S. Cryptococcus neoformans is internalized by receptor-mediated or triggered phagocytosis, dependent on actin recruitment. PLoS One. (2014) 9:e0089250 10.1371/journal.pone.0089250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sethi S, Nanda R, Chakraborty T. Clinical application of volatile organic compound analysis for detecting infectious diseases. Clin Microbiol Rev. (2013) 26:462–75. 10.1128/CMR.00020-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Rees CA, Burklund A, Stefanuto PH, Schwartzman JD, Hill JE. Comprehensive volatile metabolic fingerprinting of bacterial and fungal pathogen groups. J Breath Res. (2018) 12:026001. 10.1088/1752-7163/aa8f7f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lewis JM, Savage RS, Beeching NJ, Beadsworth MBJ, Feasey N, Covington JA. Identifying volatile metabolite signatures for the diagnosis of bacterial respiratory tract infection using electronic nose technology: a pilot study. PLoS One. (2017) 12:e0188879. 10.1371/journal.pone.0188879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rodriguez PA, Rothballer M, Chowdhury SP, Nussbaumer T, Gutjahr C, Falter-Braun P. Systems biology of plant-microbiome interactions. Mol Plant. (2019) 12:804–21. 10.1016/j.molp.2019.05.006 [DOI] [PubMed] [Google Scholar]
- 178.Barkal LJ, Procknow CL, Álvarez-Garciá YR, Niu M, Jiménez-Torres JA, Brockman-Schneider RA, et al. Microbial volatile communication in human organotypic lung models. Nat Commun. (2017) 8:1770. 10.1038/s41467-017-01985-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Schmidt R, Cordovez V, De Boer W, Raaijmakers J, Garbeva P. Volatile affairs in microbial interactions. ISME J. (2015) 9:2329–35. 10.1038/ismej.2015.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Schulz-Bohm K, Martín-Sánchez L, Garbeva P. Microbial volatiles: small molecules with an important role in intra- and inter-kingdom interactions. Front Microbiol. (2017) 8:2484. 10.3389/fmicb.2017.02484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bos LDJ, Sterk PJ, Schultz MJ. Volatile metabolites of pathogens: a systematic review. PLoS Pathog. (2013) 9:e1003311. 10.1371/journal.ppat.1003311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhu J, Jiménez-Díaz J, Bean HD, Daphtary NA, Aliyeva MI, Lundblad LK, et al. Robust detection of P. aeruginosa and S. aureus acute lung infections by secondary electrospray ionization-mass spectrometry (SESI-MS) breathprinting: from initial infection to clearance. J Breath Res. (2013) 7:037106. 10.1088/1752-7155/7/3/037106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Briard B, Bomme P, Lechner BE, Mislin GL, Lair V, Prévost MC, et al. Pseudomonas aeruginosa manipulates redox and iron homeostasis of its microbiota partner Aspergillus fumigatus via phenazines. Sci Rep. (2015) 5:8220. 10.1038/srep08220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Mowat E, Rajendran R, Williams C, McCulloch E, Jones B, Lang S, et al. Pseudomonas aeruginosa and their small diffusible extracellular molecules inhibit Aspergillus fumigatus biofilm formation. FEMS Microbiol Lett. (2010) 313:96–102. 10.1111/j.1574-6968.2010.02130.x [DOI] [PubMed] [Google Scholar]
- 185.Rees CA, Stefanuto PH, Beattie SR, Bultman KM, Cramer RA, Hill JE. Sniffing out the hypoxia volatile metabolic signature of Aspergillus fumigatus. J Breath Res. (2017) 11:036003. 10.1088/1752-7163/aa7b3e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Shatalin K, Shatalina E., Mironov A., Nudler E. H2S: a universal defense against antibiotics in bacteria. Science. (2011) 334:986–90. 10.1126/science.1209855 [DOI] [PubMed] [Google Scholar]
- 187.Bernier SP, Létoffé S, Delepierre M, Ghigo JM. Biogenic ammonia modifies antibiotic resistance at a distance in physically separated bacteria. Mol Microbiol. (2011) 81:705–16. 10.1111/j.1365-2958.2011.07724.x [DOI] [PubMed] [Google Scholar]
- 188.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Yuan Hsin H, Ingber DE. Reconstituting organ-level lung functions on a chip. Science. (2010) 328:1662–8. 10.1126/science.1188302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Nguyen Hoang AT, Chen P, Juarez J, Sachamitr P, Billing B, Bosnjak L, et al. Dendritic cell functional properties in a three-dimensional tissue model of human lung mucosa. Am J Physiol Lung Cell Mol Physiol. (2012) 302:226–37. 10.1152/ajplung.00059.2011 [DOI] [PubMed] [Google Scholar]
- 190.Kesarwani M, Hazan R, He J, Que YA, Apidianakis Y, Lesic B, et al. A quorum sensing regulated small volatile molecule reduces acute virulence and promotes chronic infection phenotypes. PLoS Pathog. (2011) 7:e1002192. 10.1371/journal.ppat.1002192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bandyopadhaya A, Kesarwani M, Que YA, He J, Padfield K, Tompkins R, et al. The quorum sensing volatile molecule 2-amino acetophenon modulates host immune responses in a manner that promotes life with unwanted guests. PLoS Pathog. (2012) 8:e1003024. 10.1371/journal.ppat.1003024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Tzika AA, Constantinou C, Bandyopadhaya A, Psychogios N, Lee S, Mindrinos M, et al. A small volatile bacterial molecule triggers mitochondrial dysfunction in murine skeletal muscle. PLoS One. (2013) 8:e0074528. 10.1371/journal.pone.0074528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bandyopadhaya A, Constantinou C, Psychogios N, Ueki R, Yasuhara S, Martyn JA, et al. Bacterial-excreted small volatile molecule 2-aminoacetophenone induces oxidative stress and apoptosis in murine skeletal muscle. Int J Mol Med. (2016) 37:867–78. 10.3892/ijmm.2016.2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kreja L, Seidel HJ. On the cytotoxicity of some microbial volatile organic compounds as studied in the human lung cell line A549. Chemosphere. (2002) 49:105–10. 10.1016/S0045-6535(02)00159-5 [DOI] [PubMed] [Google Scholar]
- 195.Nakajima D, Ishii R, Kageyama S, Onji Y, Mineki S, Morooka N, et al. Genotoxicity of microbial volatile organic compounds. J Heal Sci. (2006) 52:148–53. 10.1248/jhs.52.148 [DOI] [Google Scholar]
- 196.Bennett JW, Inamdar AA. Are some fungal volatile organic compounds (VOCs) mycotoxins? Toxins (Basel). (2015) 7:3785–804. 10.3390/toxins7093785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Mariappan V, Vellasamy KM, Thimma JS, Hashim OH, Vadivelu J. Identification of immunogenic proteins from Burkholderia cepacia secretome using proteomic analysis. Vaccine. (2010) 28:1318–24. 10.1016/j.vaccine.2009.11.027 [DOI] [PubMed] [Google Scholar]
- 198.Zheng J, Ren X, Wei C, Yang J, Hu Y, Liu L, et al. Analysis of the secretome and identification of novel constituents from culture filtrate of bacillus calmette-guérin using high-resolution mass spectrometry. Mol Cell Proteomics. (2013) 12:2081–95. 10.1074/mcp.M113.027318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Zhao Y, Jamaluddin M, Zhang Y, Sun H, Ivanciuc T, Garofalo RP, et al. Systematic analysis of cell-type differences in the epithelial secretome reveals insights into the pathogenesis of respiratory syncytial virus-induced lower respiratory tract infections. J Immunol. (2017) 198:3345–64. 10.4049/jimmunol.1601291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Miller AJ, Spence JR. In vitro models to study human lung development, disease and homeostasis. Physiology. (2017) 32:246–60. 10.1152/physiol.00041.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Shambat SM, Chen P, Nguyen Hoang AT, Bergsten H, Vandenesch F, Siemens N, et al. Modelling staphylococcal pneumonia in a human 3D lung tissue model system delineates toxin-mediated pathology. Dis Model Mech. (2015) 8:1413–25. 10.1242/dmm.021923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Barkal LJ, Berthier E, Theberge AB, Keller NP, Beebe DJ. Multikingdom microscale models. PLoS Pathog. (2017) 13:e1006424. 10.1371/journal.ppat.1006424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Torrelles JB, Schlesinger LS. Integrating lung physiology, immunology, and tuberculosis. Trends Microbiol. (2017) 25:688–97. 10.1016/j.tim.2017.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Garg N, Wang M, Hyde E, da Silva RR, Melnik AV, Protsyuk I, et al. Three-dimensional microbiome and metabolome cartography of a diseased human lung. Cell Host Microbe. (2017) 22:705–16.e4. 10.1016/j.chom.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Vandeplassche E, Tavernier S, Coenye T, Crabbé A. Influence of the lung microbiome on antibiotic susceptibility of cystic fibrosis pathogens. Eur Respir Rev. (2019) 28:190041. 10.1183/16000617.0041-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Hsia CCW, Hyde DM, Weibel EE. Lung structure and the intrinsic challenges of gas exchange. Compr Physiol. (2016) 6:827–95. 10.1002/cphy.c150028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Voynow JA, Mengr BKR. Mucins, mucus, and sputum. Chest. (2009) 135:505–12. 10.1378/chest.08-0412 [DOI] [PubMed] [Google Scholar]
- 208.Ma J, Rubin BK, Voynow JA. Mucins, mucus, and goblet cells. Chest. (2018) 154:169–76. 10.1016/j.chest.2017.11.008 [DOI] [PubMed] [Google Scholar]
- 209.Shi W, Bellusci S, Warburton D. Lung development and adult lung diseases. Chest. (2007) 132:651–6. 10.1378/chest.06-2663 [DOI] [PubMed] [Google Scholar]
- 210.Kimura H, Yoshizumi M, Ishii H, Oishi K, Ryo A. Cytokine production and signaling pathways in respiratory virus infection. Front Microbiol. (2013) 4:276. 10.3389/fmicb.2013.00276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat Biotechnol. (2014) 32:760–72. 10.1038/nbt.2989 [DOI] [PubMed] [Google Scholar]
- 212.Zhan L, Tang J, Sun M, Qin C. Animal models for tuberculosis in translational and precision medicine. Front Microbiol. (2017) 8:717. 10.3389/fmicb.2017.00717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Perlman RL. Mouse models of human disease: an evolutionary perspective. Evol Med Public Health. (2016) 2016:170–6. 10.1093/emph/eow014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Masopust D, Sivula CP, Jameson SC. Of mice, dirty mice, and men: using mice to understand human immunology. J Immunol. (2017) 199:383–8. 10.4049/jimmunol.1700453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Foreman TW, Mehra S, Lackner AA, Kaushal D. Translational research in the nonhuman primate model of tuberculosis. ILAR J. (2017) 58:151–9. 10.1093/ilar/ilx015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kaushal D, Mehra S, Didier PJ, Lackner AA. The non-human primate model of tuberculosis. J Med Primatol. (2012) 41:191–201. 10.1111/j.1600-0684.2012.00536.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Scanga CA, Flynn JL. Modeling tuberculosis in nonhuman primates. Cold Spring Harb Perspect Med. (2014) 4:a018564 10.1101/cshperspect.a018564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Yong KSM, Her Z, Chen Q. Humanized mice as unique tools for human-specific studies. Arch Immunol Ther Exp (Warsz). (2018) 66:245–66. 10.1007/s00005-018-0506-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Halldorsson S, Asgrimsson V, Axelsson I, Gudmundsson GH, Steinarsdottir M, Baldursson O, et al. Differentiation potential of a basal epithelial cell line established from human bronchial explant. Vitr Cell Dev Biol Anim. (2007) 43:283–9. 10.1007/s11626-007-9050-4 [DOI] [PubMed] [Google Scholar]
- 220.Rayner RE, Makena P, Prasad GL, Cormet-Boyaka E. Optimization of normal human bronchial epithelial (NHBE) cell 3D cultures for in vitro lung model studies. Sci Rep. (2019) 9:2–11. 10.1038/s41598-018-36735-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Benam KH, Villenave R, Lucchesi C, Varone A, Hubeau C, Lee HH, et al. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat Methods. (2016) 13:151–7. 10.1038/nmeth.3697 [DOI] [PubMed] [Google Scholar]
- 222.Benam KH, Novak R, Nawroth J, Hirano-Kobayashi M, Ferrante TC, Choe Y, et al. Matched-comparative modeling of normal and diseased human airway responses using a microengineered breathing lung chip. Cell Syst. (2016) 3:456–66.e4. 10.1016/j.cels.2016.10.003 [DOI] [PubMed] [Google Scholar]
- 223.Stucki AO, Stucki JD, Hall SR, Felder M, Mermoud Y, Schmid RA, et al. A lung-on-a-chip array with an integrated bio-inspired respiration mechanism. Lab Chip. (2015) 15:1302–10. 10.1039/C4LC01252F [DOI] [PubMed] [Google Scholar]
- 224.Felder M, Trueeb B, Stucki AO, Borcard S, Stucki JD, Schnyder B, Geiser T, et al. Impaired wound healing of alveolar lung epithelial cells in a breathing lung-on-a-chip. Front Bioeng Biotechnol. (2019) 7:3. 10.3389/fbioe.2019.00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Mermoud Y, Felder M, Stucki JD, Stucki AO, Guenat OT. Microimpedance tomography system to monitor cell activity and membrane movements in a breathing lung-on-chip. Sensors Actuat B Chem. (2018) 255:3647–53. 10.1016/j.snb.2017.09.192 [DOI] [Google Scholar]
- 226.Humayun M, Chow CW, Young EWK. Microfluidic lung airway-on-a-chip with arrayable suspended gels for studying epithelial and smooth muscle cell interactions. Lab Chip. (2018) 18:1298–309. 10.1039/C7LC01357D [DOI] [PubMed] [Google Scholar]
- 227.Parasa VR, Rahman MJ, Hoang ATN, Svensson M, Brighenti S, Lerm M. Modeling Mycobacterium tuberculosis early granuloma formation in experimental human lung tissue. Dis Model Mech. (2014) 7:281–8. 10.1242/dmm.013854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Wiszniewski L, Jornot L, Dudez T, Pagano A, Rochat T, Lacroix JS, et al. Long-term cultures of polarized airway epithelial cells from patients with cystic fibrosis. Am J Respir Cell Mol Biol. (2006) 34:39–48. 10.1165/rcmb.2005-0161OC [DOI] [PubMed] [Google Scholar]
- 229.Paolicelli G, Luca De A, Jose SS, Antonini M, Teloni I, Fric J, et al. Using lung organoids to investigation epithelial barrier complexity and IL-7 signaling during repiratory infection. Front Immunol. (2019) 10:323. 10.3389/fimmu.2019.00323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Barkauskas CE, Chung MI, Fioret B, Gao X, Katsura H, Hogan BLM. Lung organoids: current uses and future promise. Devolopment. (2017) 144:986–97. 10.1242/dev.140103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Gkatzis K, Taghizadeh S, Huh D, Stainier DYR, Bellusci S. Use of three-dimensional organoids and lung-on-a-chip methods to study lung development, regeneration and disease. Eur Respir J. (2018) 52:1800876. 10.1183/13993003.00876-2018 [DOI] [PubMed] [Google Scholar]
- 232.Sachs N, Papaspyropoulos A, Zomer-van Ommen DD, Heo I, Böttinger L, Klay D, et al. Long-term expanding human airway organoids for disease modeling. EMBO J. (2019) 38:1–20. 10.15252/embj.2018100300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Pampaloni F, Reynaud EG, Stelzer EHK. The third dimension bridges the gap between cell culture and live tissue. Nat Rev Mol Cell Biol. (2007) 8:839–45. 10.1038/nrm2236 [DOI] [PubMed] [Google Scholar]
- 234.Ravi M, Paramesh V, Kaviya SR, Anuradha E, Paul Solomon FD. 3D cell culture systems: advantages and applications. J Cell Physiol. (2015) 230:16–26. 10.1002/jcp.24683 [DOI] [PubMed] [Google Scholar]
- 235.Barrila J, Radtke AL, Crabbé A, Sarker SF, Herbst-Kralovetz MM, Ott CM, et al. Organotypic 3D cell culture models: Using the rotating wall vessel to study host-pathogen interactions. Nat Rev Microbiol. (2010) 8:791–801. 10.1038/nrmicro2423 [DOI] [PubMed] [Google Scholar]
- 236.Faber SC, McCullough SD. Through the looking glass: in vitro models for inhalation toxicology and interindividual variability in the airway. Appl Vitr Toxicol. (2018) 4:115–28. 10.1089/aivt.2018.0002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Ren H, Birch NP, Suresh V. An optimised human cell culture model for alveolar epithelial transport. PLoS One. (2016) 11:e0165225. 10.1371/journal.pone.0165225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Halldorsson S, Gudjonsson T, Gottfredsson M, Singh PK, Gudmundsson GH, Baldursson O. Azithromycin maintains airway epithelial integrity during Pseudomonas aeruginosa infection. Am J Respir Cell Mol Biol. (2010) 42:62–8. 10.1165/rcmb.2008-0357OC [DOI] [PubMed] [Google Scholar]
- 239.Yonker LM, Mou H, Chu KK, Pazos MA, Leung H, Cui D, et al. Development of a primary human co-culture model of inflamed airway mucosa. Sci Rep. (2017) 7:8182. 10.1038/s41598-017-08567-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Hermanns MI, Unger RE, Kehe K, Peters K, Kirkpatrick CJ. Lung epithelial cell lines in coculture with human pulmonary microvascular endothelial cells: development of an alveolo-capillary barrier in vitro. Lab Investig. (2004) 84:736–52. 10.1038/labinvest.3700081 [DOI] [PubMed] [Google Scholar]
- 241.Imundo L, Barascht J, Prince A, Al-awqatit Q. Cystic fibrosis epithelial cells have. Spring. (1995) 92:3019–23. 10.1073/pnas.92.7.3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Duff C, Murphy PG, Callaghan M, McClean S. Differences in invasion and translocation of Burkholderia cepacia complex species in polarised lung epithelial cells in vitro. Microb Pathog. (2006) 41:183–92. 10.1016/j.micpath.2006.07.005 [DOI] [PubMed] [Google Scholar]
- 243.Bhowmick R, Derakhshan T, Liang Y, Ritchey J, Liu L, Gappa-Fahlenkamp H. A three-dimensional human tissue-engineered lung model to study influenza A infection. Tissue Eng Part A. (2018) 24:1468–80. 10.1089/ten.tea.2017.0449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Braian C, Svensson M, Brighenti S, Lerm M, Parasa VR. A 3D human lung tissue model for functional studies on Mycobacterium tuberculosis infection. J Vis Exp. (2015) 2015:1–9. 10.3791/53084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Keenan TM, Folch A. Biomolecular gradients in cell culture systems. Lab Chip. (2007) 8:34–57. 10.1039/B711887B [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Lauffenburger DA, Zigmond SH. Chemotactic factor concentration gradients in chemotaxis assay systems. J Immunol Methods. (1981) 40:45–60. 10.1016/0022-1759(81)90079-X [DOI] [PubMed] [Google Scholar]
- 247.Huh D, Fujioka H, Tung YC, Futai N, Paine R, 3rd, Grotberg JB, et al. Acoustically detectable cellular-level lung injury induced by fluid mechanical stresses in microfluidic airway systems. Proc Natl Acad Sci U S A. (2007) 104:18886–91. 10.1073/pnas.0610868104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Yu J, Berthier E, Craig A, de Groot, Sparks S, Ingram PN, Jarrard DF, et al. Reconfigurable open microfluidics for studying the spatiotemporal dynamics of paracrine signalling. Nat Biomed Eng. (2019) 3:830–41. 10.1038/s41551-019-0421-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Sackmann EK, Fulton AL, Beebe DJ. The present and future role of microfluidics in biomedical research. Nature. (2014) 507:181–9. 10.1038/nature13118 [DOI] [PubMed] [Google Scholar]
- 250.Kimura H, Sakai Y, Fujii T. Organ/body-on-a-chip based on microfluidic technology for drug discovery. Drug Metab Pharmacokinet. (2018) 33:43–8. 10.1016/j.dmpk.2017.11.003 [DOI] [PubMed] [Google Scholar]
- 251.Huh D, Leslie DC, Matthews BD, Fraser JP, Jurek S, Hamilton GA, et al. A human disease model of drug toxicity-induced pulmonary edema in a lung-on-a-chip microdevice. Sci Transl Med. (2012) 4:159ra147 10.1126/scitranslmed.3004249 [DOI] [PMC free article] [PubMed] [Google Scholar]