

Abstract
Since the emergence of SARS-CoV2, to date, no effective antiviral drug has been approved to treat the disease, and no vaccine against SARS-CoV2 is available. Under this scenario, the combination of two HIV-1 protease inhibitors, lopinavir and ritonavir, has attracted attention since they have been previously employed against the SARS-CoV main proteinase (Mpro) and exhibited some signs of effectiveness. Recently, the 3D structure of SARS-CoV2 Mpro was constructed based on the monomeric SARS-CoV Mpro and employed to identify potential approved small inhibitors against SARS-CoV2 Mpro, allowing the selection of 15 drugs among 1903 approved drugs to be employed. In this study, we performed docking of these 15 approved drugs against the recently solved X-ray crystallography structure of SARS-CoV2 Mpro in the monomeric and dimeric states; the latter is the functional state that was determined in a biological context, and these were submitted to molecular dynamics (MD) simulations coupled with the molecular mechanics generalized Born surface area (MM/GBSA) approach to obtain insight into the inhibitory activity of these compounds. Similar studies were performed with lopinavir and ritonavir coupled to monomeric and dimeric SARS-CoV Mpro and SARS-CoV2 Mpro to compare the inhibitory differences. Our study provides the structural and energetic basis of the inhibitory properties of lopinavir and ritonavir on SARS-CoV Mpro and SARS-CoV2 Mpro, allowing us to identify two FDA-approved drugs that can be used against SARS-CoV2 Mpro. This study also demonstrated that drug discovery requires the dimeric state to obtain good results.
Keywords: SARS-CoV2, Proteinase, SARS-CoV, Docking, MD simulations
Graphical abstract
Highlights
-
•
Theoretical methods provide information of the affinity of lopinavir and ritonavir on SARS-CoV and SARS-CoV2 Mpro.
-
•
Binding free energy values suggest that paraziquantel and perampanel can be used against COVID-19.
-
•
Our research demonstrated that drug discovery on SARS-CoV2 Mpro requires the dimeric state to obtain good results.
1. Introduction
In December 2019, another outbreak of acute respiratory disease caused by a novel coronavirus (CoV) was reported in Wuhan, China [1,2]. Analysis of the complete genome of severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) demonstrated that it belongs to betacoronavirus, but it is different from severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East Respiratory coronavirus (MERS-CoV), which caused previous epidemics [1]. This new disease was named coronavirus disease 2019 (COVID-19), previously known as novel coronavirus [2019-nCoV], by the World Health Organization. COVID-19 was first reported in China, and it has now spread quickly to distant nations, including France and the USA. The number of cases within and outside China are increasing abruptly, and no drug has proved to be effective. Therefore, it is crucial to discover and develop drugs to treat the disease. An alternative treatment for COVID-19 is the combination of two HIV-1 protease inhibitors, lopinavir and ritonavir, which was an effective therapy previously used against SARS-CoV [3]. Previous theoretical studies demonstrated that lopinavir and ritonavir form stable complexes with the SARS-CoV main proteinase (SARS-CoV Mpro), with similar affinity [4]. Similar to SARS-CoV Mpro, the main proteinase of SARS-CoV2 (SARS-CoV2 Mpro) exhibits a crucial role in the proteolytic activity of replicase polyproteins, which are indispensable for viral replication. In addition, an alignment of SARS-CoV Mpro and SARS-CoV2 Mpro shows that they share a high percentage of sequence identity (≥95%). Several theoretical studies have been performed to identify inhibitors against SARS-CoV Mpro. Xu et al. constructed a three-dimensional homology model of SARS-CoV2 Mpro based on SARS-CoV Mpro and screened it against 1903 drug inhibitors via protein modeling and virtual screening, highlighting nelfinavir as a potential inhibitor against SARS-CoV2 Mpro [5]. Using X-ray crystallography, the structure of SARS-CoV2 Mpro has recently been solved in complex with the inhibitor N3 (PDB ID: 6LU7), revealing that its structural topology is similar to that of other CoV proteinases. SARS-CoV2 Mpro is built in a homodimer conformation, formed of three domains: domains 1 (residues 8–101) and 2 (residues 102–184) are β-barrels, and domain 3 (residues 201–306) comprises mainly α-helices, and it is connected to domain 2 by an elongated loop region (residues 185–200). The substrate-binding site of SARS-CoV2 Mpro is situated in a cleft between domain 1 and domain 2. Inhibitor N3 was developed using theoretical methods and it can specifically inhibit Mpro from multiple coronaviruses, including SARS-CoV, SARS-CoV2 and MERS-CoV [[6], [7], [8], [9], [10], [11]]. Inhibitor N3 is stabilized at the substrate binding site in an extended conformation by conserved residues (H41 and C145) involved in the catalytic activity of the enzyme, in a similar manner to that observed for other CoV proteinases [4]. The backbone atoms of inhibitor form an antiparallel sheet with some residues of the long strand (residues 155–168), and with residues 189–191 of the loop that connects domain 2 to domain 3.
Proteases are typical targets for drug development because of their recognized enzymatic mechanism, however, many new potential drugs have been ineffective either because of a lack of ligand specificity or because of our incomplete understanding of the conformational state under a biological context of the targeted protease [12]. In order to successfully develop or identify new protease inhibitors, it is necessary to understand important structural features of the protease functions to expand the platform of inhibitor development. Although previous studies have considered the monomeric state of SARS-CoV Mpro or SARS-CoV2 Mpro to search for new inhibitors [5,13] or to understand the molecular basis of inhibitor recognition [4], kinetic studies have indicated that the active form of the SARS CoV main proteinase corresponds to a homodimer [14], suggesting significant conformational differences between the monomer and dimeric states and indicating that drug discovery combining docking and MD simulations should be performed using the homodimeric conformation instead of the monomer. In the present research, the crystallographic dimeric structure of SARS-CoV2 Mpro, the first released structure of this enzyme available at the protein data bank (PDB ID: 6LU7), was docked against 15 Food and Drug Administration (FDA) approved drugs identified in a previous study [5] and then submitted to MD simulations coupled to the MM/GBSA approach to dissect the structural and energetic basis of molecular recognition considering the monomeric and dimeric states. In addition, comparative analysis was performed for dimeric SARS-CoV2 Mpro and SARS-CoV Mpro coupling to lopinavir and ritonavir, which have been shown to be an effective therapy against SARS-CoV Mpro.
2. Methods
2.1. Starting data and preparation systems
Seventeen FDA-approved small drugs (Scheme 1 ), indomethacin (DB00328), naftazone (DB13680), ofloxacin (DB01165), zopiclone (DB01198), sofosbuvir (DB08934), pitavastatin (DB08860), eszopiclone (DB00402), perampanel (DB08883), fenoterol (DB01288), azelastine (DB00972), celecoxib (DB00482), nelfinavir (DB00220), praziquantel (DB01058), ondansetron (DB00904), lemborexant (DB11951), lopinavir (DB01601) and ritonavir (DB00503), were downloaded from DrugBank version 5.0 [15] and optimized at the AM1 level employing Gaussian 09 W [16]. The X-ray crystallography structures of SARS-CoV2 Mpro (PDB ID: 6LU7, 2.16 Å) and SARS-CoV Mpro (PDB ID: 2GX4, 1.93 Å) were used to construct the protein-ligand complexes. PDB structures employed for this research were selected based on their availability (PDB ID: 6LU7, 2.16 Å), high resolution, without mutations and missing residues.
Scheme 1.

2D structure of the compounds used in this research. A) indomethacin, B) naftazone, C) ofloxacin, D) zopiclone, E) lopinavir, F) sofosbuvir, G) pitavastatin, H) eszopiclone, I) ondansetron, J) perampanel, K) fenoterol, L) azelastine, M) ritonavir, N) celecoxib, O) nelfinavir, P) praziquantel, Q) lemborexant, R) Inhibitor N3, S) TG-0205221, T) niclosamide, and U) chloroquine.
2.2. Molecular docking
The seventeen FDA-approved small drugs were docked on monomeric and dimeric SARS-CoV2 Mpro using AutoDock Tools 1.5.6 and AutoDock 4.2 programs [17]. Lopinavir and ritonavir were docked on monomeric and dimeric SARS-CoV Mpro. In the previous docking calculation, hydrogen atoms were added to the ligand, and protein atoms and Kollman and Gasteiger partial charges were assigned for the receptor and ligand, respectively. The grid box was centered on the substrate-binding site of each monomeric subunit with grid points in the x, y and z of 70 × 70 × 70 Å, respectively, with a grid spacing of 0.375 Å. The ligand place was optimized using a Lamarckian genetic algorithm. The protein-ligand conformation with the lowest binding energies was selected as the initial conformer to start MD simulations. The compounds reached the substrate binding site of SARS-CoV Mpro and SARS-CoV2 Mpro, obtaining a root-mean-square deviation (RMSD) of 0.5 Å to 2.0 Å with respect to the co-crystallized compound. About 20 runs were run for each compound and 30 binding poses of the ligand were obtained between compounds and receptor. The docking protocol was validated by reproducing the experimental binding mode of inhibitor N3 and TG-0205221 (scheme) on SARS-CoV2 Mpro (PDB ID: 6LU7) and SARS-CoV Mpro (PDB ID: 2GX4), respectively. By using this methodology, we identified that our docking methodology was able to reproduce the experimental binding mode of both compounds with RMSD values lower than 1.0 Å.
2.3. MD simulations
MD simulations were carried out using the AMBER16 package [18] and the ff14SB force field [19]. The force field of ligands was performed considering AM1-BCC atomic charges and the general Amber force field (GAFF) [20]. Each complex generated through docking was neutralized with 0.10 M NaCl and then solvated using the TIP3P water model [21] in a dodecadic box of 12.0 Å. Previously, MD simulations for each complex were minimized through 1000 steps for the steepest descent and 3000 steps for the conjugate gradient. Then, the systems were heated through 200 ps, the density was equilibrated through 200 ps, and finally, the systems were equilibrated by 600 ps of constant pressure equilibration at 310 K. Once the systems were equilibrated, MD simulations were run for 100 ns with triplicate experiments using an NPT ensemble at 310 K. The electrostatic forces were described by the particle mesh Ewald method [22], and a 10 Å cutoff was chosen for the van der Waals interactions. The SHAKE algorithm [23] was used to constrain bond lengths at their equilibrium values. Temperature and pressure were maintained using the weak-coupling algorithm [24]. The results were analyzed using AmberTools16. Images were built using PyMOL [25].
2.4. Binding free energy and per-residue decomposition calculations
The MM/GBSA [26,27] method was employed to calculate the binding free energy (ΔGbind) values between the receptor and ligand and to calculate per-residue decomposition analysis. To this end, 500 snapshots at time intervals of 100 ps were selected over the equilibrated time, removing all counterions and water molecules with a salt concentration of 0.10 M [28]. ΔGbind and per-residue decomposition calculations were determined as described elsewhere [29], and the ΔGbind values represent the average values of triplicate simulation experiments. Similar experiments were performed using compounds with known experimental affinity to SARS-CoV2 Mpro or SARS-CoV Mpro to validate the ability of MM/GBSA approach to reproduce the experimental binding affinity trend. We observed that the approach was able to reproduce the experimental tendency previously observed for two inhibitors (Chloroquine and niclosamide) of SARS-CoV2 Mpro [30], for which niclosamide showed higher affinity for SARS-CoV2 Mpro in comparison to chloroquine (supplementary material, Table S1). Similarly, TG-0205221, a SARS-CoV Mpro inhibitor, showed a higher affinity for SARS-Cov Mpro (supplementary material, Table S1) in comparison to lopinavir (Table 2), in line with experimental reports [31,32].
Table 2.
Binding free energy components for complexes between ligands and dimeric SARS-CoV2 Mpro and SARS-CoV Mpro systems (in units of kcal/mol).
| System | ΔEvdw | ΔEele | ΔGele,sol | ΔGnpol,sol | DGmmgbsa |
|---|---|---|---|---|---|
| Dimeric SARS-CoV2 Mpro | |||||
| SARS-CoV2sub1-indomethacin | ND | ND | ND | ND | ND |
| SARS-CoV2sub2-indomethacin | −31.7 ± 4.0 | 85.3 ± 11.0 | −68.8 ± 10.0 | −4.1 ± 0.40 | −19.30 ± 3.0 |
| SARS-CoV2sub1-naftazone | −22.17 ± 4.0 | −32.7 ± 11.0 | 40.47 ± 7.0 | −3.0 ± 0.30 | −17.40 ± 3.0 |
| SARS-CoV2sub2-naftazone | −26.38 ± 2.0 | −26.2 ± 5.0 | 36.54 ± 4.0 | −3.2 ± 0.20 | −19.24 ± 3.0 |
| SARS-CoV2sub1-ofloxacin | −32.27 ± 4.0 | −50.4 ± 11.0 | 58.0 ± 11.0 | −4.0 ± 0.40 | −28.67 ± 4.0 |
| SARS-CoV2sub2-ofloxacin | ND | ND | ND | ND | ND |
| SARS-CoV2sub1-zopiclone | −37.88 ± 3.0 | −114.0 ± 10.0 | 130.0 ± 10.0 | −3.9 ± 0.40 | −25.78 ± 3.0 |
| SARS-CoV2sub2-zopiclone | −38.40 ± 3.0 | −117.5 ± 12.0 | 134.6 ± 13.0 | −3.7 ± 0.30 | −25.0 ± 4.0 |
| SARS-CoV2sub1-sofosbuvir | −34.64 ± 5.0 | −20.27 ± 9.0 | 40.0 ± 8.0 | −4.6 ± 0.60 | −19.51 ± 5.0 |
| SARS-CoV2sub2-sofosbuvir | −37.44 ± 5.0 | −20.0 ± 9.0 | 38.1 ± 8.0 | −4.6 ± 0.60 | −23.94 ± 6.0 |
| SARS-CoV2sub1-pitavastatin | −36.5 ± 3.0 | 62.7 ± 22.0 | −44.2 ± 10.0 | −5.0 ± 0.40 | −23.0 ± 5.0 |
| SARS-CoV2sub2-pitavastatin | −40.4 ± 3.0 | 46.9 ± 12.0 | −27.0 ± 6.0 | −4.7 ± 0.30 | −25.20 ± 4.0 |
| SARS-CoV2sub1-eszopiclone | −38.1 ± 4.0 | −123.5 ± 14.0 | 142.2 ± 14.0 | −3.9 ± 0.40 | −23.30 ± 4.0 |
| SARS-CoV2sub2-eszopiclone | −41.1 ± 3.0 | −154.9 ± 21.0 | 172.0 ± 22.0 | −3.9 ± 0.30 | −27.90 ± 3.0 |
| SARS-CoV2sub1-perampanel | −46.2 ± 3.0 | −25.0 ± 4.0 | 39.1 ± 3.0 | −5.6 ± 0.20 | −37.70 ± 3.0 |
| SARS-CoV2sub2-perampanel | −42.6 ± 2.0 | −23.9 ± 4.0 | 39.8 ± 3.0 | −5.0 ± 0.20 | −31.70 ± 2.0 |
| SARS-CoV2sub1-fenoterol | −34.3 ± 4.0 | −120.8 ± 12.0 | 136.6 ± 12.0 | −4.6 ± 0.50 | −23.1 ± 5.0 |
| SARS-CoV2sub2-fenoterol | −33.4 ± 3.0 | −118.8 ± 10.0 | 129.0 ± 10.0 | −4.4 ± 0.20 | −27.60 ± 4.0 |
| SARS-CoV2sub1-azelastine | −34.4 ± 4.0 | −106.7 ± 12.0 | 118.0 ± 12.0 | −3.9 ± 0.40 | −27.0 ± 3.0 |
| SARS-CoV2sub2-azelastine | −39.27 ± 3.0 | −127.7 ± 15.0 | 142.0 ± 15.0 | −4.63 ± 0.30 | −29.6 ± 3.0 |
| SARS-CoV2sub1-celecoxib | −34.42 ± 3.0 | −14.4 ± 3.0 | 30.9 ± 3.0 | −4.7 ± 0.30 | −22.62 ± 3.0 |
| SARS-CoV2sub2-celecoxib | −30.59 ± 4.0 | −20.0 ± 4.0 | 33.0 ± 3.0 | −4.54 ± 0.50 | −22.13 ± 4.0 |
| SARS-CoV2sub1-nelfinavir | −34.42 ± 3.0 | −14.4 ± 3.0 | 30.9 ± 3.0 | −4.7 ± 0.30 | −22.62 ± 3.0 |
| SARS-CoV2sub2-nelfinavir | −48.73 ± 4.0 | −175.06 ± 13.0 | 184.7 ± 10.0 | −5.9 ± 0.30 | −44.99 ± 5.0 |
| SARS-CoV2sub1-praziquantel | −43.60 ± 4.0 | −18.01 ± 3.0 | 31.3 ± 3.0 | −4.8 ± 0.30 | −35.11 ± 3.7 |
| SARS-CoV2sub2-praziquantel | −43.60 ± 4.0 | −18.01 ± 3.0 | 31.3 ± 3.0 | −4.8 ± 0.30 | −35.11 ± 3.7 |
| SARS-CoV2sub1-ondansetron | −30.0 ± 5.0 | −5.77 ± 5.0 | 20.8 ± 5.0 | −3.4 ± 0.50 | −18.37 ± 5.0 |
| SARS-CoV2sub2-ondansetron | −37.9 ± 5.0 | −15.13 ± 4.0 | 30.0 ± 4.0 | −3.9 ± 0.50 | −26.93 ± 5.0 |
| SARS-CoV2sub1-lemborexant | ND | ND | ND | ND | ND |
| SARS-CoV2sub2-lemborexant | −34.1 ± 4.0 | −5.42 ± 4.0 | 21.6 ± 3.0 | −4.5 ± 0.50 | −22.42 ± 4.0 |
| SARS-CoV2sub1-lopinavir | −49.3 ± 4.0 | −15.2 ± 5.0 | 34.9 ± 4.0 | −6.2 ± 0.44 | −35.80 ± 4.0 |
| SARS-CoV2sub2-lopinavir | −56.9 ± 4.0 | −18.8 ± 6.0 | 40.6 ± 5.0 | −7.2 ± 0.50 | −42.30 ± 4.0 |
| SARS-CoV2sub1-ritonavir | −45.8 ± 5.0 | −10.5 ± 4.0 | 32.5 ± 4.0 | −5.5 ± 0.70 | −29.30 ± 5.0 |
| SARS-CoV2sub2-ritonavir | −47.9 ± 6.0 | −9.0 ± 3.0 | 34.2 ± 5.0 | −5.8 ± 0.80 | −28.50 ± 5.0 |
| Dimeric SARS-CoV2 Mpro | |||||
| SARS-CoVsub1-lopinavir | −38.4 ± 4.0 | −9.5 ± 5.0 | 29.9 ± 5.0 | −4.7 ± 0.44 | −22.70 ± 3.0 |
| SARS-CoVsub2-lopinavir | −43.7 ± 6.0 | −21.7 ± 13.0 | 40.7 ± 13.0 | −5.6 ± 0.80 | −30.30 ± 6.0 |
| SARS-CoVsub1-ritonavir | −49.8 ± 5.0 | −22.6 ± 4.0 | 41.1 ± 4.0 | −6.4 ± 0.60 | −37.70 ± 5.0 |
| SARS-CoVsub2-ritonavir | ND | ND | ND | ND | ND |
3. Results and discussion
3.1. Docking between ligands and monomeric SARS-CoV2 Mpro
Docking studies between ligands and SARS-CoV2 Mpro showed that all ligands: indomethacin (Fig. S1A), naftazone (Fig. S1B), ofloxacin (Fig. S1C), zopiclone (Fig. S1D), sofosbuvir (Fig. S1E), pitavastatin (Fig. S1F), eszopiclone (Fig. S2A), perampanel (Fig. S2B), fenoterol (Fig. S2C), azelastine (Fig. S2D), celecoxib (Fig. S2E), nelfinavir (Fig. S2F), praziquantel (Fig. S3A), ondansetron (Fig. S3B), and lemborexant (Fig. S3C) reached the catalytic binding site of SARS-CoV2 Mpro (supplementary material, Figs. S1–S3 and Table S2). These ligands were mostly stabilized by H41, F140, N142, C145, H163, H164, M165, E166, Q189 and R188 residues through nonpolar interactions. H41, S46, Y54, F140, L141, N142, G143, S144, C145, H163, H164, E166 and D187 established polar interactions through backbone or side chain atoms with some of the compounds: indometachin (Fig. 1 A), naftazone (Fig. S1B), ofloxacin (Fig. S1C), zopiclone (Fig. S1D), sofosbuvir (Fig. S1E), pitavastatin (Fig. S1F), perampanel (Fig. S2B), fenoterol (Fig. S2C), azelastine (Fig. S2D), praziquantel (Fig. S3A), ondansetron (Fig. S3B), lemborexant (Fig. S3C), and ritonavir (Fig. S3E). The residues stabilizing the ligands were mostly distributed between domains 1 (residues 8–101) and 2 (residues 102–184), and the interactions established were similar to those observed in the co-crystallized complex between the SARS-CoV2 Mpro ligand and the inhibitor N3 (PDB ID: 6LU7), highlighting the interactions with conserved residues (H41 and C145) involved in the catalytic activity of the enzyme [33].
Fig. 1.

Binding conformation of complexes between ligands and monomeric SARS-CoV2 Mpro. Maps of the interaction of monomeric SARS-CoV2 Mpro with naftazone (A), zopiclone (B), sofosbuvir (C), pitavastatin (D), eszopiclone (E), and perampanel (F). Each complex corresponds to the most populated conformation obtained thorough MD simulation. The receptor is represented in a green cartoon representation, the interacting residues are depicted in green sticks, and the ligand is shown in a ball and stick representation. The figure was built with PyMOL [25]. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.1.1. Docking of lopinavir or ritonavir with monomeric SARS-CoV2 Mpro and SARS-CoV Mpro
Docking studies show that lopinavir and ritonavir on SARS-CoV2 Mpro and SARS-CoV Mpro reached the catalytic site of both systems (supplementary material, Figs. S3D–G and Table S2). On SARS-CoV2, lopinavir (Fig. S3D) and ritonavir (Fig. S3E) were mostly stabilized by T25, T26, H41, F140, L141, N142, G143, H163, E166, D187, Q189 and R188 residues through nonpolar interactions, whereas ritonavir established polar interactions with the side chain of S46 (Fig. S3E). On SARS-CoV Mpro, lopinavir (Fig. S3F) and ritonavir (Fig. S3G) are mostly stabilized by T25, A46, M49, L141, S144, E166 and Q189 through nonpolar interactions, while ritonavir formed polar interactions with the side chain of Q189 (Fig. S3G).
Comparative analysis between the coupling of lopinavir or ritonavir on SARS-CoV2 Mpro and SARS-CoV Mpro showed that T25, S/A46, Y/M49, L141, S144, E166 and Q189 are present in the stabilization of ligands on SARS-CoV2 Mpro and SARS-CoV Mpro. In addition, these compounds are better stabilized on SARS-CoV2 Mpro than on SARS-CoV Mpro. All these docking-predicted complexes were submitted to MD simulation in the monomeric and dimeric states to validate their stabilization at the catalytic sites of SARS-CoV2 Mpro and SARS-CoV Mpro.
3.2. Convergence of MD simulations
RMSD and radius of gyration (Rg) studies showed that monomeric SARS-CoV2 Mpro and SARS-CoV Mpro in their free and bound states reached equilibrium between 10 and 20 ns with average values that oscillated between 1.6 ± 0.2 and 3.8 ± 0.2 Å for RMSD and 21.9 ± 0.2 and 23.1 ± 0.2 Å for RG (Table S3, supplementary material). Dimeric SARS-CoV2 Mpro and SARS-CoV Mpro in their free and bound states reached equilibrium among 10–30 ns with average values that ranged between 1.5 ± 0.1 and 2.2 ± 0.2 Å for RMSD and 25.8 ± 0.2 and 26.1 ± 0.2 Å for RG (Table S4, supplementary material). Therefore, for further analyses, the first 30 ns were discarded from the 100 ns simulation for each monomer and dimer simulations.
3.3. MD simulations of ligands with monomeric SARS-CoV2 Mpro and SARS-CoV Mpro
MD simulations show that indomethacin, ofloxacin, fenoterol, nelfinavir, praziquantel and ritonavir lost interactions at the catalytic site of SARS-CoV2 Mpro. In contrast, naftazone (Fig. 1A), zopiclone (Fig. 1B), sofosbuvir (Fig. 1C), pitavastatin (Fig. 1D), eszopiclone (Fig. 1E), perampanel (Fig. 1F), azelastine (Fig. 2 A), celecoxib (Fig. 2B), ondansetron (Fig. 2C), and lemborexant (Fig. 2D) maintained interactions with the catalytic site of SARS-CoV2 Mpro. These compounds were mainly stabilized by M49, M165 and Q189 residues through nonpolar interactions. However, S46, G143, S144, H163, M165, C145, E166, P168, D187, T190 and Q192 formed polar interactions with backbone or side chain atoms with some of the compounds, including naftazone (Fig. 1A), zopiclone (Fig. 1B), T190 (Fig. 1C), pitavastatin (Fig. 1D), eszopiclone (Fig. 1E), perampanel (Fig. 1F), azelastine (Fig. 2A), celecoxib (Fig. 2B), ondansetron (Fig. 2C), and lemborexant (Fig. 2D).
Fig. 2.

Binding conformation of complexes between ligands and monomeric SARS-CoV2 Mpro and SARS-CoV Mpro. Diagrams of the interaction of complexes of monomeric SARS-CoV2 Mpro with azelastine (A), celecoxib (B), ondansetron (C) and lemborexant (D) and lopinavir (E). Diagrams of the interaction of monomeric SARS-CoV Mpro with lopinavir (F) and ritonavir (G).
3.3.1. MD simulations of lopinavir or ritonavir with monomeric SARS-CoV2 Mpro and SARS-CoV Mpro
MD simulations showed that ritonavir lost interactions with the catalytic site of SARS-CoV2, whereas lopinavir maintained the interactions with the catalytic site (Fig. 2E). Lopinavir was mostly stabilized by hydrophobic residues (M49, M165 and Q189) similar to those present in the fifteen repositioned compounds (Fig. 1, Fig. 2), while it established polar interactions with the side chain of S46 (Fig. 2E). On SARS-CoV Mpro, lopinavir and ritonavir were mainly stabilized by L27, H41, A46, M49 and C145 through hydrophobic interactions, whereas lopinavir formed polar interactions with backbone atoms of A46 (Fig. 2F), and ritonavir formed polar interactions with the side chain of N142 (Fig. 2G).
Analyses between the coupling of lopinavir or ritonavir on SARS-CoV2 Mpro and SARS-CoV Mpro showed that only hydrophobic contacts with M49 were shared in the stabilization of the fifteen repositioned compounds on SARS-CoV2 Mpro and SARS-CoV Mpro. In addition, the stabilization of these compounds was better on SARS-CoV2 Mpro than on SARS-CoV2 Mpro.
3.3.2. MD simulations of ligands with dimeric SARS-CoV2 Mpro and SARS-CoV Mpro
In contrast, with the observations with monomeric SARS-CoV2 and SARS-CoV Mpro, MD simulations for most of the dimeric systems showed that all the ligands remained on both subunits of the dimer, except for the complexes between indomethacin, ofloxacin and lemborexant with SARS-CoV2 Mpro, in which these compounds only remained at one of the catalytic sites of SARS-CoV2 Mpro. Indomethacin coupled to subunit 2 (Fig. 3 A), naftazone bound to subunits 1 and 2 (Fig. 3B and C), ofloxacin bound to subunit 1 (Fig. 3D), and zopiclone coupled to subunits 1 and 2 (Fig. 3E and F). Sofosbuvir bound at subunits 1 and 2 (Fig. 4 A and B), pitavastatin bound at subunits 1 and 2 (Fig. 4C and D), and eszopiclone bound at subunits 1 and 2 (Fig. 4E and F). Perampanel coupled at subunit 1 or 2 (Fig. 5 A and B), fenoterol bound at subunit 1 or 2 (Fig. 5C and D), and azelastine bound at subunits 1 and 2 (Fig. 5E and F). Celecoxib coupled at subunits 1 and 2 (Fig. 6 A and B), nelfinavir bound at subunits 1 and 2 (Fig. 6C and D), and praziquantel bound at subunits 1 and 2 (Fig. 6E and F). Ondansetron bound at subunits 1 and 2 (Fig. 7 A and B), and lemborexant bound at subunit 2 (Fig. 7C). These compounds were mainly stabilized by L27, H41, M49, N142, C145 and M165 through nonpolar interactions. T25, H41, T45, S46, L141, N142, G143, F140, S144, H163, H164, M165, E166, Q192, and Q189 formed polar interactions with backbone or side chain atoms with some of these compounds: naftazone (Fig. 3B), ofloxacin (Fig. 3D), zopiclone (Fig. 3E and F), sofosbuvir (Fig. 4A and B), pitavastatin (Fig. 4C and D), eszopiclone (Fig. 4E and F), perampanel (Fig. 5A and B), fenoterol (Fig. 5C and D), azelastine (Fig. 5F), celecoxib (Fig. 6A and B), nelfinavir (Fig. 6C and D), praziquantel (Fig. 6E and F), ondansetron (Fig. 7A and B) and lemborexant (Fig. 7C). Comparison of the residues stabilizing these ligands in the monomeric (Fig. 1, Fig. 2) versus dimeric SARS-CoV2 Mpro and SARS-CoV Mpro (Fig. 3, Fig. 7) revealed that the repositioned compounds were better stabilized in the dimeric state than in the monomeric state. In addition, only in the complexes using the dimeric system, the presence of interactions with conserved residues (H41 and C145) involved in the catalytic activity was observed [33].
Fig. 3.

Binding conformation of complexes of ligands with dimeric SARS-CoV2 Mpro. Indomethacin coupled to subunit 2 (A), naftazone bound to subunits 1 (B) and 2 (C), ofloxacin bound to subunit 1 (D), and zopiclone coupled to subunits 1 (E) and 2 (F) of dimeric SARS-CoV2 Mpro.
Fig. 4.

Binding conformation of complexes of ligands with dimeric SARS-CoV2 Mpro. Sofosbuvir bound at subunits 1 (A) and 2 (B), pitavastatin bound at subunits 1 (C) and 2 (D), and eszopiclone bound at subunits 1 (E) and 2 (F) of dimeric SARS-CoV2 Mpro.
Fig. 5.

Binding conformation of complexes of ligands with dimeric SARS-CoV2 Mpro. Perampanel bound at subunits 1 or 2 (A and B), fenoterol bound at subunits 1 or 2 (Fig. C and D), and azelastine bound at subunits 1 and 2 (Fig. E and F) of dimeric SARS-CoV2 Mpro.
Fig. 6.

Binding conformation of complexes of ligands with dimeric SARS-CoV2 Mpro. Celecoxib bound at subunits 1 and 2 (A and B), nelfinavir bound at subunits 1 and 2 (Fig. 6C and D), and praziquantel bound at subunits 1 and 2 (Fig. 6E and F) of dimeric SARS-CoV2 Mpro.
Fig. 7.

Binding conformation of complexes of ligands with dimeric SARS-CoV2 Mpro. Ondansetron bound at subunits 1 and 2 (A and B), lemborexant bound at subunit 2 (C), lopinavir at subunits 1 (D) and 2 (E) and ritonavir at subunit 1 (F) of dimeric SARS-CoV2 Mpro.
3.3.3. MD simulations of lopinavir or ritonavir with dimeric SARS-CoV2 Mpro and SARS-CoV Mpro
MD simulations showed that lopinavir at subunits 1 (Figs. 7D) and 2 (Fig. 7E) and ritonavir at subunits 1 (Figs. 7F) and 2 (Fig. 8 A) were maintained interactions at the catalytic site of SARS-CoV2. Ritonavir and lopinavir were generally stabilized by four residues (M49, M165, L167 and Q189), whereas only lopinavir formed polar interactions with backbone atoms and side chain atoms of T90 and Q189 (Fig. 7E). On SARS-CoV Mpro, lopinavir coupled at subunits 1 (Figs. 8B) and 2 (Fig. 8C) and ritonavir coupled at subunit 1 (Fig. 8D) were mostly stabilized by H41, M49, M165 and Q189 through hydrophobic interactions. Similar nonpolar and polar interactions were observed for the fifteen repositioned compounds (Fig. 3, Fig. 4, Fig. 5, Fig. 6, Fig. 7), except for L167. Comparative analysis of the residues stabilizing ritonavir and lopinavir in the monomeric (Fig. 2) versus dimer SARS-CoV2 Mpro and SARS-CoV Mpro (Fig. 7, Fig. 8) showed that ritonavir is stabilized by similar hydrophobic residues (M49 and M165) in the monomeric and dimeric states, whereas only two residues (H41 and M49) are shared in the stabilization of lopinavir in the monomeric and dimeric SARS-CoV2 Mpro and SARS-CoV Mpro.
Fig. 8.

Binding conformation of complexes of ligands with dimeric SARS-CoV2 and SARS-CoV Mpro. Ritonavir bound at subunit 2 (A) of dimeric SARS-CoV2 Mpro. Lopinavir coupled at subunits 1 (B) and 2 (C) and ritonavir at subunit 1 (D) of dimeric SARS-CoV Mpro.
3.4. Binding free energy calculations
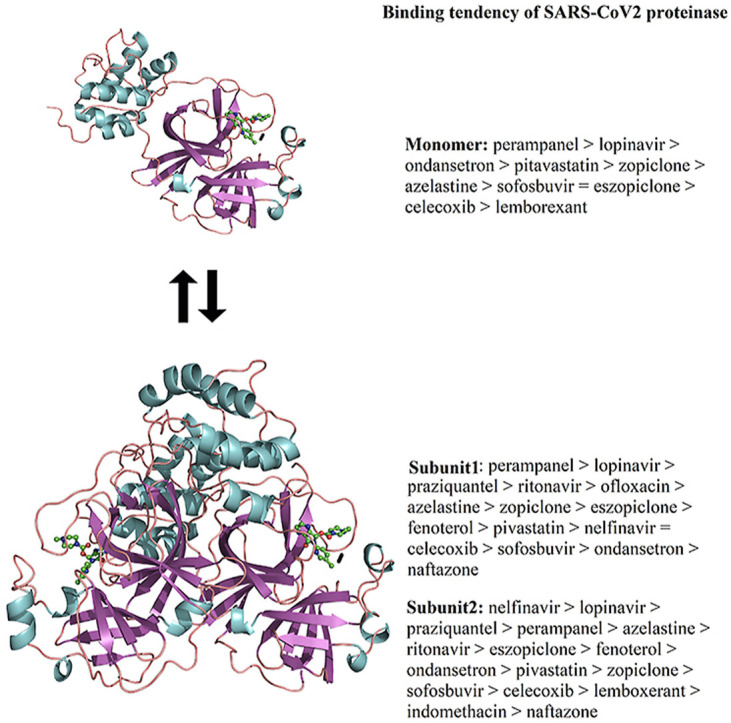
Differences in affinity for the complexes between ligands and monomeric and dimeric SARS-CoV2 Mpro and SARS-CoV Mpro systems were calculated using the MM/GBSA approach, showing that all the bindings are energetically favorable and guided through nonpolar interactions, van der Waals energy (ΔE vdw) and the nonpolar free energy of desolvation (ΔG npol,sol). Binding free energy (ΔGbind) values for the ligands coupled at the monomeric SARS-CoV2 Mpro show the following tendency: perampanel > lopinavir > ondansetron > pitavastatin > zopiclone > azelastine > sofosbuvir = eszopiclone > celecoxib > lemborexant (Table 1 ). However, a higher affinity towards monomeric SARS-CoV Mpro was exhibited by lopinavir than by ritonavir. Comparison of ΔGbind values for the affinity of repositioned compounds with ritonavir or lopinavir shows that perampanel was able to inhibit monomeric SARS-CoV Mpro in a similar manner to lopinavir and ritonavir, which diffuses in the first nanoseconds of MD simulations (see section 3.3.1). Comparisons between the affinity of lopinavir or ritonavir for monomeric SARS-CoV2 Mpro and SARS-CoV Mpro systems showed that these two compounds exhibit a higher affinity by monomeric SARS-CoV Mpro than by SARS-CoV2 Mpro.
Table 1.
Binding free energy components for complexes between ligands and monomeric SARS-CoV2 Mpro and SARS-CoV Mpro systems (in units of kcal/mol).
| System | ΔEvdw | ΔEele | ΔGele,sol | ΔGnpol,sol | DGmmgbsa |
|---|---|---|---|---|---|
| SARS-CoV2 | |||||
| Naftazone | −22.4 ± 2.7 | −5.5 ± 1.0 | 15.8 ± 5.0 | −2.9 ± 0.30 | −15.0 ± 3.0 |
| Zopiclone | −38.0 ± 2.5 | −127.0 ± 11.0 | 141.0 ± 5.0 | −3.9 ± 0.30 | −27.9 ± 3.0 |
| Sofosbuvir | −37.5 ± 4.0 | −22.0 ± 5.0 | 37.0 ± 4.0 | −4.5 ± 0.4 | −27.0 ± 3.0 |
| Pitavastatin | −35.0 ± 5.0 | 6.7 ± 2.0 | 4.9 ± 1.0 | −4.8 ± 0.3 | −28.2 ± 5.0 |
| Eszopiclone | −37.0 ± 2.0 | −109.0 ± 10.0 | 123.0 ± 10.0 | −4.0 ± 0.3 | −27.0 ± 3.0 |
| Perampanel | −39.0 ± 3.0 | −27.0 ± 5.0 | 39.0 ± 5.0 | −4.0 ± 0.2 | −31.0 ± 3.0 |
| Azelastine | −36.0 ± 3.0 | −107.0 ± 15.0 | 120.0 ± 14.0 | −4.3 ± 0.3 | −27.3 ± 3.0 |
| Celecoxib | −31.0 ± 3.0 | −11.0 ± 4.0 | 25.0 ± 3.0 | −4.3 ± 0.4 | −21.3 ± 4.0 |
| Ondansetron | −40.0 ± 2.0 | −16.0 ± 4.0 | 31.0 ± 3.0 | −4.1 ± 0.2 | −29.1 ± 3.0 |
| Lemborexant | −32.0 ± 5.0 | −4.0 ± 3.0 | 19.0 ± 3.0 | −4.2 ± 0.5 | −21.2 ± 5.0 |
| Lopinavir | −44.0 ± 5.0 | −36.0 ± 11.0 | 56.0 ± 10.0 | −5.9 ± 0.5 | −29.9 ± 5.0 |
| SARS-CoV | |||||
| Lopinavir | −51.0 ± 5.0 | −10.0 ± 5.0 | 33.0 ± 4.0 | −7.0 ± 0.6 | −35.0 ± 4.0 |
| Ritonavir | −40.0 ± 6.0 | −9.0 ± 2.0 | 28.0 ± 6.0 | −5.0 ± 0.8 | −26.0 ± 5.0 |
ΔGbind values for the ligands coupled on the first subunit of dimeric SARS-CoV2 Mpro show the following tendency: perampanel > lopinavir > praziquantel > ritonavir > ofloxacin > azelastine > zopiclone > eszopiclone > fenoterol > pitavastatin > nelfinavir = celecoxib > sofosbuvir > ondansetron > naftazone. The ligands coupled at the second subunit showed the following order: nelfinavir > lopinavir > praziquantel > perampanel > azelastine > ritonavir > eszopiclone > fenoterol > ondansetron > pitavastatin > zopiclone > sofosbuvir > celecoxib > lemborexant > indomethacin > naftazone (Table 2 ). Based on this analysis, it is evident that perampanel, and praziquantel can be proposed as anti-COVID-19 clinical drugs, whereas nelfinavir could also exhibit moderate activities against COVID-19. Perampanel is a drug currently employed in epilepsy, with an innovative mechanism of action through AMPA ([2-amino-3- (3-hydroxy-5-methyl-isoxazol-4-yl) propanoic acid) glutamate receptors. Praziquantel is an anthelmintic drug used to treat several sorts of parasitic worm infections, and nelfinavir is a strong HIV1 protease inhibitor used with other antiviral medications to treat HIV. Interestingly, perampanel and praziquantel also exhibit a similar affinity to lopinavir and a higher affinity than ritonavir, both known inhibitors of SARS-CoV Mpro [3]. A comparison of the ΔGbind values of lopinavir and ritonavir on dimeric SARS-CoV Mpro versus SARS-CoV2 Mpro indicated that these compounds exhibit a higher affinity for dimeric SARS-CoV2 Mpro than for SARS-CoV2 Mpro. In addition, a comparison between the monomeric versus dimeric SARS-CoV2 Mpro and SARS-CoV Mpro systems shows that although the employment of the monomeric system allowed us to identify perampanel and lopinavir as good inhibitors of SARS-CoV2, it did not permit to the identification with praziquantel nelfinavir and ritonavir, highlighting the suitability of employing the dimeric system for drug discovery.
3.5. Per-residue free energy decomposition
An analysis of the residues contributing to the ΔGbind values for complexes with monomeric and dimeric SARS-CoV2 Mpro and SARS-CoV Mpro systems resulted in 5–11 residues (Table 3, Table 4, Table 5, Table 6 ). An analysis of the residue stabilizing complexes between ligands and monomeric SARS-CoV2 Mpro and SARS-CoV Mpro systems showed that H41, M49, M165 and Q189 were present in most of the complexes (Table 3), but only H41 and M165 were present for perampanel, the compound with the highest affinity for monomeric SARS-CoV2 (Table 1); instead, it was stabilized by N142, G143, S144 and C145, which together with M165, contributed the most to the ΔGbind value. M49 and M165 were present in the stabilization of lopinavir, the second-best compound, in the monomeric SARS-CoV2 Mpro and SARS-CoV Mpro systems.
Table 3.
Per-residue free energy for complexes between ligands and monomeric SARS-CoV2 Mpro and SARS-CoV Mpro (values kcal/mol).
| Residue | Lig2 | Lig4 | Lig5 | Lig6 | Lig7 | Lig8 | Lig10 | Lig11 | Lig14 | Lig15 | Lig16 | Lig16∗ | Lig17∗ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T25 | −0.580 | −1.609 | |||||||||||
| L27 | −0.645 | −0.781 | −0.671 | −1.031 | |||||||||
| H41 | −0.602 | −0.652 | −1.345 | −0.596 | −1.073 | −1.206 | −1.702 | −0.926 | −1.512 | ||||
| T45 | −0.611 | ||||||||||||
| S46/A46 | −0.226 | −0.878 | −0.877 | −1.722 | −1.029 | ||||||||
| M49 | −1.403 | −0.515 | −0.822 | −1.527 | −2.776 | −1.191 | −0.563 | −1.609 | −1.884 | −2.241 | −1.613 | ||
| L50 | −0.906 | 1.191 | |||||||||||
| F140 | −0.912 | ||||||||||||
| L141 | −0.985 | −1.148 | −0.634 | −0.553 | |||||||||
| N142 | −1.186 | −3.120 | −0.945 | −2.735 | −1.371 | −0.631 | |||||||
| G143 | −0.576 | −3.576 | −2.144 | −0.691 | |||||||||
| S144 | −0.673 | −2.172 | −0.933 | −1.154 | |||||||||
| C145 | −2.102 | −2.645 | −2.150 | −2.101 | −0.511 | −0.763 | −0.670 | ||||||
| H163 | −1.514 | −0.692 | −1.437 | −2.590 | |||||||||
| M165 | −1.563 | −2.178 | −2.688 | −2.987 | −1.911 | −2.137 | −1.619 | −1.905 | −3.285 | −1.720 | −1.974 | −1.881 | |
| D166 | −1.819 | −0.761 | −2.215 | ||||||||||
| L167 | −0.946 | −0.876 | |||||||||||
| P168 | −0.837 | −0.590 | |||||||||||
| P169 | |||||||||||||
| H172 | −0.502 | ||||||||||||
| D187 | −0.570 | −1.356 | −0.717 | −0.916 | |||||||||
| R188 | −0.752 | −1.253 | −0.516 | −0.932 | −0.513 | ||||||||
| Q189 | −1.336 | −2.542 | −1.787 | −1.238 | −1.279 | −1.181 | −1.628 | −0.919 | −2.253 | ||||
| T190 | −1.250 | ||||||||||||
| A191 | −1.270 | ||||||||||||
| Q192 | −2.243 |
Indomethacin = lig1, naftazone = lig2, ofloxacin = lig3, zopiclone = lig4, sofosbuvir = lig5, pitavastatin = lig6, eszopiclone = lig7, perampanel = lig8, fenoterol = lig9, azelastine = lig10, celecoxib = lig11, nelfinavir = lig12, praziquantel = lig13, ondansetron = lig14, lemborexant = lig15, lopinavir = lig16, and ritonavir = lig17. ∗Denotes complexes between monomeric SARS-CoV Mpro with lopinavir (lig16∗) and ritonavir (lig17∗).
Table 4.
Per-residue free energy for complexes between ligands and dimeric SARS-CoV2 Mpro (values kcal/mol).
| Residue | Lig1sub2 | Lig2Sub1 | Lig2Sub2 | Lig3Sub1 | Lig4Sub1 | Lig4Sub2 | Lig5Sub1 | Lig5Sub2 | Lig6Sub1 | Lig6Sub2 | Lig7Sub1 | Lig7Sub2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T25 | −0.891 | |||||||||||
| L27 | −0.185 | −0.529 | −0.447 | −0.591 | −0.684 | −0.663 | −0.675 | −0.662 | −0.568 | |||
| H41 | −2.507 | −0.544 | −0.611 | −1.752 | −0.672 | −0.611 | −1.079 | −0.486 | −2.520 | −0.838 | ||
| C44 | −1.719 | |||||||||||
| T45 | −0.586 | −3.105 | ||||||||||
| S46 | −0.578 | −1.761 | ||||||||||
| M49 | −1.110 | −2.972 | −1.486 | −0.874 | −1.452 | −1.543 | −0.860 | |||||
| L50 | −1.449 | |||||||||||
| F140 | −0.610 | −0.992 | −0.871 | −1.352 | −1.256 | −1.284 | ||||||
| L141 | −0.961 | −1.546 | −2.391 | −1.987 | −1.465 | |||||||
| N142 | −1.456 | −1.911 | −1.567 | −1.208 | −0.642 | −0.714 | −0.854 | −2.523 | −0.962 | −0.777 | ||
| G143 | −1.335 | −1.460 | −0.849 | −1.369 | −1.462 | −0.841 | −1.191 | |||||
| S144 | −0.502 | −0.717 | −0.648 | −0.859 | −1.844 | −0.132 | −1.269 | −1.106 | ||||
| C145 | −1.592 | −1.855 | −1.125 | −1.876 | −2.287 | −0.746 | −0.596 | −0.637 | −1.960 | −2.133 | ||
| H163 | −0.466 | −0.925 | −2.322 | −1.626 | −1.403 | −0.709 | −0.191 | −1.465 | −1.622 | |||
| H164 | −1.565 | −1.065 | ||||||||||
| M165 | −1.069 | −0.748 | −0.776 | −1.732 | −1.258 | −2.432 | −0.554 | −2.578 | −1.654 | |||
| D166 | −2.040 | −3.466 | −0.604 | −2.836 | −4.313 | |||||||
| L167 | −0.572 | |||||||||||
| R188 | −1.492 | |||||||||||
| Q189 | −1.521 | −1.129 | −2.417 |
Indomethacin = lig1, naftazone = lig2, ofloxacin = lig3, zopiclone = lig4, sofosbuvir = lig5, pitavastatin = lig6, eszopiclone = lig7. Sub1 and Sub2 denote subunits 1 or 2 of dimeric SARS-CoV2 Mpro.
Table 5.
Per-residue free energy for complexes between ligands and dimeric SARS-CoV2 Mpro (values kcal/mol).
| Residue | Lig8sub1 | Lig8Sub2 | Lig9Sub1 | Lig9Sub2 | Lig10Sub1 | Lig10Sub2 | Lig11Sub1 | Lig11Sub2 | Lig12Sub1 | Lig12Sub2 | Lig13Sub1 | Lig13Sub2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T25 | −1.271 | −0.527 | ||||||||||
| L27 | −0.776 | −0.633 | −0.737 | −1.097 | −0.572 | −1.929 | −0.141 | −0.517 | −0.831 | −0.725 | ||
| H41 | −1.038 | −0.528 | −1.898 | −1.611 | −2.726 | −0.556 | −0.833 | −1.449 | −1.328 | |||
| V42 | −0.622 | |||||||||||
| T45 | −0.544 | |||||||||||
| S46 | −0.531 | |||||||||||
| M49 | −1.669 | −0.776 | −2.057 | −1.981 | −1.216 | −2.325 | −1.516 | |||||
| L50 | −1.151 | −0.761 | ||||||||||
| F140 | −0.849 | −0.918 | −1.214 | −0.627 | −0.611 | |||||||
| L141 | −0.710 | −0.526 | −0.544 | |||||||||
| N142 | −2.929 | −2.445 | −2.018 | −1.194 | −0.795 | −0.735 | −0.929 | |||||
| G143 | −2.328 | −2.454 | −0.792 | −0.650 | −1.684 | −1.672 | ||||||
| S144 | −1.672 | −1.517 | −1.247 | −1.486 | −1.415 | |||||||
| C145 | −1.566 | −2.232 | −1.109 | −0.913 | −1.598 | −0.518 | −3.112 | −2.718 | ||||
| H163 | −1.652 | −0.707 | ||||||||||
| H164 | −0.708 | −0.560 | ||||||||||
| M165 | −2.233 | −1.841 | −1.661 | −3.722 | −1.750 | −1.667 | −1.248 | −3.034 | −1.805 | −1.984 | −1.903 | |
| D166 | −1.017 | −0.950 | −4.882 | −0.849 | ||||||||
| L167 | −0.702 | |||||||||||
| P168 | −0.778 | |||||||||||
| D187 | −1.061 | −1.119 | −1.468 | |||||||||
| R188 | −0.659 | −0.898 | −1.323 | −0.504 | −0.969 | |||||||
| Q189 | −1.367 | −1.248 | −2.608 | −3.590 | −1.936 | −0.556 | ||||||
| T190 | −1.119 | |||||||||||
| A191 | −2.158 | |||||||||||
| Q192 | −0.988 | −1.391 |
Perampanel = lig8, fenosterol = lig9, azelastine = lig10, celecoxib = lig11, nelfinavir = lig12, praziquantel = lig13. Sub1 and Sub2 denote subunits 1 or 2 of dimeric SARS-CoV2 Mpro.
Table 6.
Per-residue free energy for complexes between ligands and dimeric SARS-CoV2 Mpro and SARS-CoV Mpro (values kcal/mol).
| Residue | Lig14sub1 | Lig14Sub2 | Lig15Sub2 | Lig16Sub1 | Lig16Sub2 | Lig17Sub1 | Lig17Sub2 | ∗Lig16Sub1 | ∗Lig16Sub2 | ∗Lig17Sub1 |
|---|---|---|---|---|---|---|---|---|---|---|
| T25 | −0.614 | |||||||||
| T26 | ||||||||||
| L27 | −0.662 | −0.741 | ||||||||
| H41 | −0.613 | −2.124 | −0.890 | −1.138 | −1.425 | −0.757 | −0.723 | |||
| C44 | −0.985 | |||||||||
| S/A∗46 | −0.639 | −0.699 | −0.821 | −0.604 | ||||||
| D48 | −0.848 | |||||||||
| M49 | −2.513 | −2.100 | −2.211 | −1.038 | −2.367 | −1.900 | −2.314 | −1.748 | ||
| L50 | −0.513 | −0.875 | −0.554 | |||||||
| P52 | −0.998 | |||||||||
| L141 | −0.859 | −0.750 | ||||||||
| N142 | −0.654 | |||||||||
| G143 | −0.710 | |||||||||
| C145 | −0.598 | −0.561 | −0.663 | |||||||
| H163 | −2.762 | |||||||||
| M165 | −1.787 | −2.814 | −0.647 | −2.208 | −3.300 | −1.912 | −1.979 | −0.840 | −1.234 | −3.112 |
| D166 | −0.703 | −1.830 | ||||||||
| L167 | −0.700 | −0.525 | −0.850 | −1.533 | ||||||
| P168 | −1.495 | −2.062 | −0.610 | −1.946 | ||||||
| H172 | −0.549 | |||||||||
| D187 | −1.562 | −0.778 | −0.885 | −0.764 | ||||||
| R188 | −0.843 | −0.599 | −0.972 | |||||||
| Q189 | −0.709 | −0.968 | −1.951 | −1.809 | −0.785 | −1.353 | −1.069 | −1.712 | ||
| T190 | −1.138 | −0.782 | ||||||||
| A191 | −1.398 | −0.543 | ||||||||
| Q192 | −0.514 |
Ondansetron = lig14, lemborexant = lig15, lopinavir = lig16, and ritonavir = lig17. ∗denotes complexes between dimeric SARS-CoV Mpro with lopinavir (lig16∗) and ritonavir (lig17∗). Sub1 and Sub2 denote subunit 1 or 2 of dimeric SARS-CoV2 Mpro or SARS-CoV Mpro.
For the dimeric SARS-CoV2 Mpro and SARS-CoV Mpro systems, H41, M49 and M165 were present in the stabilization of almost all the complexes (Table 4, Table 5, Table 6). From these three residues, the energetic contribution of H41 and M49 was only present in one of the subunits for the complex between dimeric SARS-CoV2 Mpro and perampanel (Table 5). H41 was present in both subunits of the complex between SARS-CoV2 Mpro and praziquantel, and M49 was present only in subunit 1 for perampanel (Table 5). H41 and M49 were present in the complexes of dimeric SARS-CoV2 Mpro with nelfinavir (Table 5) and lopinavir (Table 6). M49 was present in the complex of dimeric SARS-CoV2 Mpro with ritonavir (Table 6).
As observed for the complex between perampanel and monomeric SARS-CoV2 Mpro (Table 3), N142, G143, S144 and C145, together with M165, contributed the most to the ΔGbind value (Table 2) on both subunits of dimeric SARS-CoV2 Mpro (Table 5). For praziquantel, the energetic contribution of M49 was only observed for one of the subunits, whereas participation of H41 and M165 was observed for both subunits, and as observed for perampanel, in which N142, G143, S144, C145 and M165 contributed importantly to the ΔGbind value (Table 2). For nelfinavir, the participation of H41, M49 and M165 was seen only in one of the subunits, the one with the higher affinity (Table 2), where it was also observed for the energetic contribution of D187, Q189, T190, A191 and Q192, which contributed importantly to the ΔGbind value (Table 2). Energetic contributions of H41, M49 and M165 residues were observed for complexes of lopinavir with the dimeric SARS-CoV2 Mpro and SARS-CoV Mpro (Table 6). Significant participation of P168, D187, Q189 and T190 was also observed but only for interactions at subunit 2 of the dimeric SARS-CoV2 Mpro in complex with lopinavir (Table 6), whereas Q189 contributed importantly to the ΔGbind value in both subunits of the dimeric SARS-CoV Mpro (Table 6).
Energetic contributions of M49 and M165 were observed for complexes of ritonavir with dimeric SARS-CoV2 Mpro and of H41 M49 and M165 with dimeric SARS-CoV Mpro (Table 6). It was also observed that there was significant participation of P168, Q189 and A191 for interactions of ritonavir at subunit 1 of dimeric SARS-CoV2 Mpro and of D166, L167, P168, and Q189 for ritonavir at subunit 1 of dimeric SARS-CoV Mpro. Overall, this analysis supports the importance of two conserved residues (H41 and C145) [4] in the stabilization of different inhibitors and highlights the importance of other residues (M49, N142, G143, S144, M165, D187, Q189, T190, A191 and Q192) in ligand stabilization. The identification of these 12 hot-spot residues allow to explain the differences in ligand affinity in the monomeric, dimeric and between each subunit of dimer. Although these residues do not form part of the protein-protein interface: domain 1 (residues 10–11 and14), domain 2 (122–127, 137–141, 166, 170 and 172) and domain 3 (280, 283, 285, 286, 290, 298, 299 and 303), they are in close distance of residues forming the protein-protein interface. Therefore, the ligand binding in dimer is impacted not only by the modulating of these key residues, but also by the induced fit binding of ligand.
3.6. Principal component analysis
PCA was performed to provide a quantified estimation of the differences in mobility. To this end, the trace of the diagonalized covariance matrix of the backbone atomic positional fluctuations was determined for the free and bound SARS-CoV2 Mpro and SARS-CoV Mpro in the monomeric (Table S5, supplementary material) and dimeric states (Table 7 and Fig. S4, supplementary material). Based on this analysis, the values for free and bound monomeric SARS-CoV2 Mpro and SARS-CoV Mpro systems suggested that only the binding of sofosbuvir and lopinavir to monomeric SARS-CoV2 Mpro was not coupled to conformational changes of monomeric SARS-CoV2 Mpro. The binding of naftazone, pitavastatin, eszopiclone, perampanel, azelastine, celecoxib, ondansetron and lemborexant was linked to a decrease in the conformational mobility of monomeric SARS-CoV2 Mpro, and the conformational reduction would be coupled to an increase in the ΔGbind value (Table 1), due to an unfavorable entropy component. The binding of zopiclone was coupled to an increase in the conformational mobility, which contributed to a decrease in the ΔGbind value due to a favorable entropy component. The binding of lopinavir and ritonavir was linked to a decrease in the conformational mobility of monomeric SARS-CoV Mpro, which would also be linked to an increase in the ΔGbind value observed in Table 1.
Table 7.
Trace of the diagonalized covariance matrix of the backbone atoms for free and bound dimeric SARS-CoV2 and SARS-CoV Mpro systems.
| System | Covariance (nm2) |
|---|---|
| Dimeric SARS-CoV2 Mpro | |
| SARS-CoV2apo | 16.2 |
| SARS-CoV2indomethacin | 20.8 |
| SARS-CoV2naftazone | 15.0 |
| SARS-CoV2ofloxacin | 12.0 |
| SARS-CoV2zopiclone | 16.0 |
| SARS-CoV2sofosbuvir | 16.3 |
| SARS-CoV2pitavastatin | 13.2 |
| SARS-CoV2eszopiclone | 15.3 |
| SARS-CoV2perampanel | 17.4 |
| SARS-CoV2fenoterol | 13.5 |
| SARS-CoV2azelastine | 16.0 |
| SARS-CoV2celecoxib | 18.4 |
| SARS-CoV2nelfinavir | 15.9 |
| SARS-CoV2praziquantel | 14.9 |
| SARS-CoV2ondansetron | 20.9 |
| SARS-CoV2lemborexant | 16.2 |
| SARS-CoV2lopinavir | 15.2 |
| SARS-CoV2ritonavir | 23.4 |
| Dimeric SARS-CoV Mpro | |
| SARS-CoVapo | 21.0 |
| SARS-CoVlopinavir | 20.9 |
| SARS-CoVritonavir | 25.1 |
Analysis of the covariance values for free and bound dimeric SARS-CoV2 Mpro and SARS-CoV Mpro systems indicates that the binding of naftazone, zopiclone, sofosbuvir, eszopiclone, perampanel, azelastine, nelfinavir, praziquantel, lemborexant and lopinavir was not linked to important conformational changes of dimeric SARS-CoV2 Mpro, which means that their coupling with receptors would not impact the affinity observed in Table 2. The binding of ofloxacin, pitavastatin and fenoterol contributed to a decrease in conformational mobility, and the coupling of indomethacin, celecoxib, ondansetron and ritonavir was linked to an increase in dimeric SARS-CoV2 Mpro. However, the binding of lopinavir and ritonavir on dimeric SARS-CoV Mpro was not linked to conformational changes for lopinavir and an increase in the mobility of this receptor, which also means that their coupling on dimeric SARS-CoV Mpro did not impact the affinity observed in Table 2. Overall, this analysis shows that the binding of the best compounds on monomeric SARS-CoV2 Mpro or SARS-CoV Mpro more importantly impacts the ΔGbind value estimated for each ligand due to the conformational changes coupled to the binding, whereas the affinity trends observed for the best compounds on the dimeric systems were not affected.
4. Conclusion
In this research, we first performed the docking of 15 FDA-approved drugs, which were previously identified as potential inhibitors of monomeric SARS-CoV2 Mpro, by employing the recently elucidated crystallographic structure of monomer and dimeric SARS-CoV2 Mpro; then, 100-ns-long MD simulations coupled to the MM/GBSA approach were performed to compare results using both monomeric versus dimeric states, where the latter corresponds to the functional state. Additionally, similar studies were performed, including two known HIV-1 protease inhibitors, lopinavir and ritonavir, which have been previously employed as an effective therapy against SARS-CoV Mpro, to compare the inhibitory differences. Our results identified perampanel (best compound), praziquantel (second best compound) and nelfinavir (third best compound) as potential inhibitors of dimeric SARS-CoV2 Mpro, and these ligands also showed similar inhibitory properties than those of lopinavir and better inhibitory properties than those of ritonavir. Furthermore, comparative analysis of the affinity of lopinavir and ritonavir on SARS-CoV2 Mpro and SARS-CoV Mpro revealed that both compounds showed a higher affinity to SARS-CoV2 Mpro. On the basis of per-residue free energy decomposition, we identified the hot-spot residues (H41, M49, N142, G143, S144, C145, M165, D187, Q189, T190, A191 and Q192) which contribute significantly high to the total binding affinity. Among these residues H41 and C145 are conserved residues. Therefore, these key residues are important for drug binding. This study demonstrates for the first time that the coupling of ligands on dimeric SARS-CoV2 Mpro is linked to differences in the binding affinity in both subunits that may be characteristic of cooperativity. Our study also demonstrates that to obtain more confident drug discovery results, it is better to employ the dimeric state than the monomeric state since ligand binding on the monomer is coupled to conformational changes that contribute to the impact of the ΔGbind value.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The work was supported by grants from CONACYT (CB-A1-S-21278) and SIP/IPN (20201015).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmgm.2020.107762.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G.F., Tan W. N. Engl. J. Med. 2020;382(8):727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu H., Stratton C.W., Tang Y.W. J. Med. Virol. 2020;92(4):401–402. doi: 10.1002/jmv.25678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vastag B. J. Am. Med. Assoc. 2003;290:1695–1696. [Google Scholar]
- 4.Nukoolkarn V., Lee V.S., Malaisree M., Aruksakulwong O., Hannongbua S. J. Theor. Biol. 2008;254(4):861–867. doi: 10.1016/j.jtbi.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu Z., Peng C., Shi Y., Zhu Z., Mu K., Wang X., Zhu W. bioRxiv. 2020 doi: 10.1101/2020.01.27.921627. [DOI] [Google Scholar]
- 6.Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Hilgenfeld R. PLoS Biol. 2005;3(10):e324. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xue X., Yu H., Yang H., Xue F., Wu Z., Shen W., Zhang X.C. J. Virol. 2008;82(5):2515–2527. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ren Z. The newly emerged SARS-like coronavirus HCoV-EMC also has an “Achilles’ heel”: current effective inhibitor targeting a 3C-like protease. Protein Cell. 2013;4:248–250. doi: 10.1007/s13238-013-2841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ren Z., Yan L., Zhang N., Guo Y., Yang C., Lou Z., Rao Z. Protein & cell. 2013;4(4):248. doi: 10.1007/s13238-013-2841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang F., Chen C., Tan W., Yang K., Yang H. Sci. Rep. 2016;6:22677. doi: 10.1038/srep22677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Duan Y. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020:1–5. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 12.Turk B. Nat. Rev. Drug Discov. 2006;5(9):785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 13.Sang P., Tian S.H., Meng Z.H., Yang L.Q. RSC Adv. 2000;10(27):15775–15783. doi: 10.1039/d0ra01899f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graziano V., McGrath W.J., Yang L., Mangel W.F. Biochemistry. 2006;45(49):14632–14641. doi: 10.1021/bi061746y. [DOI] [PubMed] [Google Scholar]
- 15.Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z., Assempour N., Iynkkaran I., Liu Y., Maciejewski A., Gale N., Wilson A., Chin L., Cummings R., Le D., Pon A., Knox C., Wilson M. Nucleic Acids Res. 2018;46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, H. Nakatsuji, Gaussian 09, Revision D. 01. 2009, (Wallingford CT).
- 17.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. J. Comput. Chem. 2009;30:2785e2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Case D.A., Cheatham T.E., Darden T., Gohlke H., Luo R., Merz K.M., Jr., Woods R.J. J. Comput. Chem. 2005;26:1668e1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duan Y., Wu C., Chowdhury S., Lee M.C., Xiong G., Zhang W., Kollman P. J. Comput. Chem. 2003;24:1999e2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 20.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. J. Comput. Chem. 2004;25 doi: 10.1002/jcc.20035. 1157e1174. [DOI] [PubMed] [Google Scholar]
- 21.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. J. Chem. Phys. 1983;79 926e935. [Google Scholar]
- 22.Darden T., York D., Pedersen L. J. Chem. Phys. 1993;98 10089e10092. [Google Scholar]
- 23.Van Gunsteren W.F., Berendsen H.J.C. Mol. Phys. 1977;34:1311e1327. [Google Scholar]
- 24.Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., DiNola A., Haak J.R. J. Chem. Phys. 1984;81 3684e3690. [Google Scholar]
- 25.DeLano W.L. DeLano-Scientific; Palo Alto, CA: 2002. The PyMOL Molecular Graphics System. [Google Scholar]
- 26.Miller B.R., McGee T.D., Swails J.M., Homeyer N., Gohlke H., Roitberg A.E. J. Chem. Theor. Comput. 2012;8:3314e3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 27.Gohlke H., Case D.A. J. Comput. Chem. 2004;25:238. doi: 10.1002/jcc.10379. [DOI] [PubMed] [Google Scholar]
- 28.Onufriev A., Bashford D., Case D.A. Proteins. 2004;55:383. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 29.Bello M., Mendieta-Wejebe J.E., Correa-Basurto J. Biochem. Pharmacol. 2014;90(2):145–158. doi: 10.1016/j.bcp.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 30.Jeon S., Ko M., Lee J., Choi I., Byun S.Y., Park S., Shum D., Kim S. Antimicrob. Agents Chemother. 2020;23(7):64. doi: 10.1128/AAC.00819-20. e00819-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang S., Chen S.J., Hsu M.F., Wu J.D., Tseng C.T.K., Liu Y.F., Chen W.C. J. Med. Chem. 2006;49(16):4971–4980. doi: 10.1021/jm0603926. [DOI] [PubMed] [Google Scholar]
- 32.Wu C.Y., T Jan J., Ma S.H., Kuo C.J., Juan H.F., Cheng Y.S.E., Liang F.S. Proc. Natl. Acad. Sci. Unit. States Am. 2004;101(27):10012–10017. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang C., Wei P., Fan K., Liu Y., Lai L. Biochemistry. 2004;43:4568–4574. doi: 10.1021/bi036022q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.