

Abstract
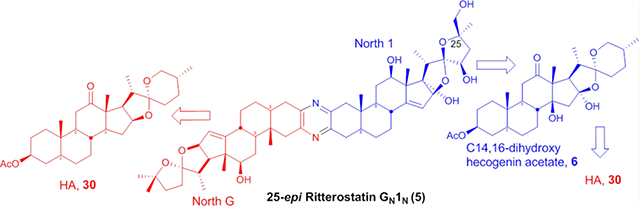
The convergent synthesis of 25-epi ritterostatin GN1N is described for the first time, starting from hecogenin acetate (HA). Stereoselective dihydroxylation employing the chiral ligand (DHQ)2PHAL was used as the key step to introduce the C25 epi-stereocenter on the north 1 segment. The title compound was obtained through a coupling reaction between the C3-keto-azide (cstat North 1) and North G.
Graphical Abstract
INTRODUCTION
Cephalostatin 1 (Cstat 1) was isolated by the Petitt group1 from the marine tubeworm Cephalodiscus gilchristi, collected from the Indian Ocean. It is one of the most potent anticancer molecules reported in the NCI 60 cell line testing (Figure 1).2 Much effort has been devoted to the cephalostatins,3 and very recently a new enantioselective synthesis of this complex molecule has been published.4 A number of congeners have been isolated from marine sources, including cephalostatins 2–195 and ritterazines A–Z.5 The antineoplastic mechanism of the trisdecacyclic pyrazines is only partly known, and the functionality present in these trisdecacyclic steroidal pyrazine bisspiroketals is quite different from that of other anticancer agents, likely indicating a new mechanism of action.6 Beyond in vitro testing, cephalostatin 1 has been shown to be effective in several xenografts including melanoma, sarcoma, leukemia, and in a human mammary carcinoma model.7,8
Figure 1.

IC50 values of Cstat analogs.
Among the series of analogs tested at the NCI (Table 1), 25-epi ritterostatin GN1N 5 exhibited subnanomolar activity (mean GI50 0.48 nM, Table 1 and Figure 1),8 which is 30-fold more potent than ‘natural’ epimer 4. These compounds showed selectivity for CNS, leukemia, and renal cancer cell lines, with ovarian cancer lines being relatively resistant.7,8 As noted previously, this pattern of selectivity is shared with several less potent natural products, including OSW-1, schweinfurthins, and stelletins.7 Several empirical calculations and docking/modeling studies also supported this higher activity.6 In order to move agents of the trisdecacyclic pyrazine class to the clinic, a more efficient route to the north 1 hemisphere was required. This issue is addressed herein via a complete synthesis of 25-epi ritterostatin GN1N in a convergent manner from the readily available hecogenin for both segments, leading to 245 mg of the hybrid molecule.9 In this synthesis, the north G segment could be readily obtained from hecogenin acetate (HA).3
Table 1.
Selected Results from NCI-60 Cell Line Testing (IC50 values in nM)a
| Compd | NSC# | Leukemia HL-60 | Lung A-549 | Colon HT-29 | CNS SF-295 | Breast MCF-7 | Ovary IGROV1 | Melanoma M-14 | Renal A-498 | Prostate PC-3 |
|---|---|---|---|---|---|---|---|---|---|---|
| Cephalostatin 1 (2) n = 9 | 363979 | 0.4 | 0.5 | 1.4 | <0.1 | 0.4 | 4.0 | 1.0 | 1.4 | 0.3 |
| Ritterostatin GN1N (4) n = 3 | 699013 | 2.4 | 9.8 | 79 | 1.3 | 28 | 141 | 11 | >1000 | 7.2 |
| 25-epi Ritterostatin GN1N (5) n = 4 | 720780 | <0.1 | <0.1 | 0.3 | <0.1 | <0.1 | <0.1 | 0.7 | 0.2 | <0.1 |
‘n’ is the number of separate full tests; each test was conducted in triplicate at each point (full details in Supporting Information).
RESULTS AND DISCUSSION
Synthesis of North 1 Segment
The synthesis began from dihydroxyhecogenin acetate 6 derived from crude hecogenin.10 The Δ14 olefin was formed by SOCl2 mediated dehydration of 6 providing the cyclic vinyl ether 7 in 85% yield. The C23 hydroxyl group (8) was introduced via dimethyldioxirane oxidation, followed by reduction of the C16 hydroxyl group using phenylselenol at −45 °C to obtain 9 (Scheme 1).11 Borohydride reduction of 9 at −78 °C gave the C12 alcohol 10 favoring β (β:α, 8:1) and subsequent bis benzoylation, providing 11 in 86% yield over two steps. Regio- and stereoselective [5,6] spiroketal F-ring reductive opening using triethylsilane12 to primary alcohol 12 was followed by sequential iodination to 13, and DBU-assisted E2 elimination ultimately provided the terminal olefin 14 (Scheme 1). These reactions were routinely conducted on >10 g scale with highly reproducible yields (72–88%).
Scheme 1.

Synthesis of Olefin 14
Treatment of terminal olefin 14 with trifluoroacetyl triflate (TFAT) at −40 °C effected spiroketal ring opening,13,12 smoothly providing the expected cyclopentadiene as the C22-OTFA ester 15. Selective deprotection of 15 gave alcohol 16 and its subsequent TBS ether 17 without disturbing the C3-OAc. The C3-keto intermediate 19 was easily obtained in quantitative yield by saponification to 18 followed by oxidation with NMO and catalytic TPAP.13 Reaction of 19 with singlet oxygen led to the stereospecific α−1,4-adduct 20.13 Cleavage of the TBS group at C22 to alcohol 21, followed by Swern oxidation, afforded ketone 22 which was confirmed by X-ray crystallographic analysis (Figure 2).14 Reductive cleavage of the peroxide bond using Zn/AcOH to diol 2312 followed by acid catalyzed cyclization provided olefin 24 (Scheme 2) along with some C3-dimethylacetal 24a which exhibited two additional methoxy acetal singlets at δ 3.08 and 3.15 in addition to the C22-OMe singlet at δ 3.32. This was easily hydrolyzed to 24 by addition of a few drops of water (see Experimental Section).
Figure 2.

X-ray structure of peroxide 22.
Scheme 2.

Preparation of Cstat North 1 Azide, 29
Asymmetric Dihydroxylation
Several ligands were screened for oxy-functionalization of terminal olefin 24 at C25,26 using Sharpless asymmetric dihydroxylation15 (Table 2). As expected, with a reasonable enantiomeric excess (ee) of C25-epi, R-25 (9.5:1 ratio, epi:natural) was obtained when (DHQ)2PHAL AD-mix-α was used, whereas with other ligands the selectivity was poor (Table 2). The C26 primary alcohol of diol 25 was protected as benzoyl ester 26 in 88% yield, followed by acid catalyzed cyclization to spiroketal 2714 in 85% yield (Scheme 2, Figure 3) along with some C3 dimethylacetal [27a, X = (OMe)2]. This material was easily hydrolyzed to the C3 ketone (27) by addition of a few drops of H2O to the reaction flask prior to workup (see Supporting Information).
Table 2.
Asymmetric Dihydroxylation of Olefin 24
 | |
|---|---|
| Ligands screeneda | Compound 25, C25 ratio, epi:natural (R:S) |
| None | 2.2:1 |
| (DHQD)2PHAL | 1:1 |
| (DHQ)2PHAL | 9.5:1 (82%) |
| (DHQD)2PYR | 1:3.8 |
| (DHQ)2PYR | 2:1 |
| DHQD | 1.8:1 |
| DHQ | 2.5:1 |
Conditions: K2OsO4 (2 mol %), ligand (10 mol %), t-BuOH/H2O (1:1), 0 °C, 40 h.
Figure 3.

X-ray structure of 27.
The North 1 coupling partner C3-keto-azide 2916 was prepared by bromination of 27 in THF using phenyltrimethylammonium tribromide (PTAB) in 84% yield (28) followed by azide formation with tetramethylguanidinium azide (TMGA) (29) in 81% yield (Scheme 2). C3-keto-azide 29 of 25-epi Cstat North 1 was thus prepared in 23 steps and 1.6% overall yield from C14,16-dihydroxy hecogenin acetate 6. The North G (30) segment was synthesized according to the literature protocol from HA.3,6
End Game
By coupling aminomethoxime North G 30 with C3-keto-azide of North 1 29 using PVP, Bu2SnCl2 in refluxing benzene provided trisdecacyclic pyrazine 31 in 58% yield (Scheme 3).17 Completion of 25-epi ritterostatin GN1N (5) in nearly quantitative yield simply involved global deprotection with NaOH.
Scheme 3.

Formation of Trisdecacyclic Pyrazine 5
In conclusion, the first synthesis of 25-epi ritterostatin GN1N 5 is reported with excellent stereocontrol. The highly functionalized North 1 hemisphere featured introduction of the C25-epi stereocenter via asymmetric dihydroxylation as the key step. Reactions were performed at multigram scale with reproducible yields.
EXPERIMENTAL SECTION
General Methods
All reagents purchased were used as received. Tetrahydrofuran (THF) and diethyl ether were distilled from benzophenone ketyl. Benzene, toluene, and methylene chloride (CH2Cl2) were distilled from calcium hydride. Acetonitrile (CH3CN) and methanol were purchased as spectral grade and used without further purification. Sodium sulfate (Na2SO4) was anhydrous. All recrystallization, chromatographic, and workup solvents were distilled. Unless otherwise indicated, all reactions were carried out under a positive pressure of nitrogen in anhydrous solvents, and the reaction flasks were fitted with rubber septa for the introduction of substrates and reagents via syringe. The progress of reactions was monitored by thin layer chromatography (TLC) using silica gel plates. The TLC plates were visualized with a UV lamp (254 nm) and/or with TLC visualizing solutions activated with heat. The two commonly employed TLC visualizing solutions were (i) p-anisaldehyde solution (1350 mL of absolute ethanol, 50 mL of concentrated H2SO4, and 37 mL of p-anisaldehyde) and (ii) iodine.
1H NMR and 13C NMR spectra were recorded on 400–500 MHz spectrometers in CDCl3 solution. Chemical shifts were reported in parts per million (ppm) on the δ scale from an internal standard (NMR descriptions: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad; dd, doublet of doublet; td, triplet of doublet; and/or quint, quintet). Coupling constants, J, are reported in hertz (Hz). Melting points were obtained on a capillary melting point apparatus or automated melting point system and are uncorrected. The high resolution mass measurements were obtained on a LTQ Orbitrap XL mass spectrometer utilizing electrospray ionization (ESI).
C12 Reduction (10) followed by Benzoyl Protection, 11
C23 alcohol (9, 3.1 g, 6.37 mmol) was dissolved in CH2Cl2 and MeOH (1:1, 50 mL). The solution was cooled to −78 °C (dry ice/acetone) under a nitrogen atmosphere. NaBH4 was added into the mixture in three portions in 5-min intervals (200 mg, 5.3 mmol each portion). The solution was maintained at −78 °C for 8 h with vigorous stirring. The temperature was then slowly raised to −20 °C. TLC showed the complete absence of starting material (7:3 EtOAc/Hexane, Rf = 0.3). The reaction was quenched with HCl (2 N, 40 mL). CH2Cl2 was used to extract the reaction mixture (50 mL × 3). The combined organic phase was washed with brine, dried over Na2SO4, and concentrated followed by application of vacuum for 2 h before proceeding to the next step with crude 10. 1H NMR (400 MHz, CDCl3) δ 0.83 (d, J = 6.5, 3H), 0.88 (s, 3H), 1.01 (s, 4H), 1.21 (d, J = 6.7, 3H), 1.28–1.39 (m, 6H), 1.43–1.49 (m, 2H), 1.63–1.75 (m, 6H), 1.80–1.88 (m, 2H), 2.02 (s, 6H), 2.13–2.18 (t, 1H), 2.43 (t, 1H), 3.17 (dd, J = 4.6, 4.6, 1H), 3.48–3.57 (m, 2H), 3.64 (t, 1H), 4.63–4.70 (m, 1H), 4.89 (d, J = 9.5, 1H), 5.37 (s, 1H); LRMS (ESI) m/z calc for C29H45O6 (M+H) 489.32, found 489.27.
To crude 10 in CH2Cl2 (30 mL) were added DMAP (80 mg 0.1 equiv) and pyridine (8 mL) under N2, and the flask was cooled in an ice water bath. To this solution benzoyl chloride (2.2 mL, 19 mmol) was added, and the reaction was stirred for 16 h, until TLC showed starting material disappeared (3:7 EtOAc/Hexane, Rf = 0.5). The solvent was removed under reduced pressure, and the reaction mixture was purified by silica gel column chromatograph by using EtOAc–Hexane to provide the desired product 11 (3:7 EtOAc/Hexane, Rf = 0.5), 3.8 to 3.9 g, yield 86% for two steps. 1H NMR (400 MHz, CDCl3) δ 0.79 (d, J = 6.6, 3H), 0.89 (s, 3H), 0.94 (d, J = 6.7, 3H), 1.01–1.09 (m, 1H), 1.01–1.09 (m, 1H), 1.21 (s, 3H), 1.33–1.43 (m, 3H), 1.46–1.52 (m, 1H), 1.61–1.69 (m, 2H), 1.74–1.84 (m, 3H), 1.88–1.99 (m, 4H), 2.02 (s, 3H), 2.05 (s, 2H), 2.07–2.14 (m, 2H), 2.43 (t, 1H), 3.53 (t, 1H), 3.58–3.62 (m, 1H), 4.63–4.71 (m, 2H), 4.98 (dd, J = 2.1, 2.2, 1H), 5.13 (s, 1H), 5.47 (s, 1H), 7.37–7.56 (m, 6H), 7.99 (d, J = 8.2, 2H), 8.08 (d, J = 8.2, 2H); HRMS (ESI) calcd for C43H53O8 [M+H]+ 697.3740, found 697.3734.
F-Ring Cleavage Compound 12
The C12,23-dibenzoate (11, 10.0 g, 14.35 mmol) and triethylsilane (13.35 g, 114.8 mmol, 8 equiv) were dissolved in CH2Cl2 (100 mL) under N2. BF3·OEt2 (14.5 mL, 114.8 mmol, 8 equiv) was added to the reaction mixture at <5 °C. The reaction was stirred at room temperature for 40 h and then quenched with NaHCO3 (200 mL, ice water bath). The reaction mixture was extracted with CH2Cl2 (50 mL × 3), and the combined organic layer was washed with brine, dried over Na2SO4, and concentrated. The crude mass was subjected to chromatography on silica gel to provide the C26 alcohol (12), 7.21 g, yield 72% (3:7 EtOAc/Hexane, Rf = 0.25). 1H NMR (400 MHz, CDCl3) δ 0.89 (s, 3H), 0.90–0.92 (m, 4H), 0.95 (d, J = 6.5, 3H), 1.24–1.27 (m, 4H), 1.35 (t, 2H), 1.56 (t, 1H), 1.64–1.76 (m, 3H), 1.90–1.97 (m, 8H), 2.02 (s, 3H), 2.04 (s, 2H), 2.08–2.12 (m, 1H), 2.25 (t, 1H), 3.47 (t, 1H), 3.55–3.59 (m, 1H), 4.65–4.70 (m, 4H), 4.84 (dd, J = 1.5, 1.3, 1H), 5.35–5.39 (m, 1H), 5.45 (t, 1H), 7.43 (t, J = 15.0, 4H), 7.55 (t, J = 14.6, 2H), 7.98–8.15 (m, 4H); HRMS (ESI) calcd for C43H55O8 [M+H]+ 699.3897, found 699.3887.
Iodo Compound 13
To the C26 alcohol (12, 5.0 g, 7.15 mmol) in diethyl ether (150 mL) and CH3CN (15 mL) were added triphenylphosphine (4.7 g, 17.9 mmol, 2.5 equiv) and imidazole (2.47 g, 35.8 mmol, 5 equiv) followed by iodine (5.45 g, 21.5 mmol, 3 equiv) at <5 °C. The reaction was stirred at this temperature for 30 min and then at room temperature for an additional 3 h. Aq. NaHSO3 was added to the reaction and stirred for 30 min. The organic solution was separated, and the aqueous phase was extracted with ethyl acetate (25 mL × 3). The combined organic layer was dried over Na2SO4, and removal of the solvent under reduced pressure followed by chromatography on silica gel provided 13 (3:7 EtOAc/Hexane, Rf = 0.65), 5.11 g, yield 88%. 1H NMR (400 MHz, CDCl3) δ 0.84–0.97 (m, 2H), 0.89 (s, 6H), 0.91 (s, 2H), 0.93–0.99 (m, 1H), 1.03 (d, J = 6.5, 3H), 1.26 (s, 3H), 1.36 (t, 2H), 1.56–1.63 (m, 6H), 1.64–1.71 (m, 2H), 1.76–1.86 (m, 1H), 1.88–1.99 (m, 1H), 2.01 (s, 3H), 2.04 (s, 1H), 2.07–2.12 (m, 1H), 2.25 (t, 1H), 3.17 (t, 2H), 3.55 (dd, J = 5.0, 5.1, 1H), 4.67–4.70 (q, 2H), 4.83 (dd, J = 2.2, 2.4, 1H), 5.31–5.35 (m, 1H), 5.46 (t, 1H), 7.41–7.45 (m, 4H), 7.51–7.57 (m, 2H), 8.00–8.06 (m, 4H); 13CNMR (CDCl3, 100 MHz) δ 11.9, 14.1, 16.3, 17.8, 18.3, 19.9, 21.3, 22.5, 25.2, 26.6, 27.2, 28.0, 29.4, 31.2, 31.4, 33.6, 34.0, 34.5, 35.8, 36.4, 37.7, 37.9, 44.0, 51.7, 52.0, 60.0, 73.0, 81.4, 86.5, 87.9, 120.0, 128.3, 129.3, 129.6, 129.8, 130.2, 131.9, 132.0, 132.9, 157.0, 165.6, 165.8, 170.2; LRMS (ESI) calcd for C43H54IO7 [M+H]+ 809.29, found 809.25.
Olefin 14
Iodide (13, 901 mg, 1.13 mmol) was dissolved in CH3CN (25 mL) under an argon atmosphere, and freshly distilled DBU over CaH2 (255 μL, 1.7 mmol, 1.5 equiv) was added to the reaction mixture. The reaction was stirred at 55 °C for 4 h and then at room temperature for 10 h. The solvent was removed under reduced pressure, and the crude mass was purified via silica gel column chromatography using EtOAc–Hexane (3:7 Rf = 0.45) to provide 14, 591 mg, yield 80%. 1H NMR (400 MHz, CDCl3) δ 0.77–0.81 (m, 2H), 0.82 (s, 3H), 0.89 (d, J = 6.4, 3H), 0.94–1.04 (m, 2H), 1.10–1.20 (m, 2H) 1.23 (s, 3H), 1.26–1.34 (m, 4H), 1.37–1.45 (m, 1H), 1.57–1.65 (m, 2H), 1.70 (s, 3H), 1.73–1.86 (m, 2H), 1.88–1.91 (m, 1H), 1.92 (s, 3H), 2.02–2.13 (m, 2H), 2.23 (t, 1H), 2.46 (d, J = 6.4, 2H), 3.53 (dd, J = 5.4, 5.2, 1H), 4.60–4.64 (m, 1H) 4.68 (br, 1H), 4.80 (dd, J = 2.3, 2.3, 1H), 5.37–5.41 (m, 1H), 5.42 (t, 1H), 7.30–7.36 (m, 4H), 7.41–7.47 (m, 2H), 7.95–7.99 (m,4H); 13CNMR (CDCl3, 100 MHz) δ 11.8, 14.1, 16.2, 18.3, 20.6, 21.2, 22.4, 25.2, 26.6, 27.1, 28.0, 29.4, 31.4, 33.6, 34.0, 34.3, 34.5, 35.8, 36.4, 38.1, 39.4, 44.0, 51.7, 52.0, 60.1, 73.0, 73.1, 81.4, 86.4, 87.6, 113.1, 120.1, 128.2, 128.3, 129.3, 129.5, 130.2, 130.3, 132.7, 141.2, 157.0, 165.6, 165.7, 170.2; HRMS (ESI) calcd for C43H53O7 [M+H]+ 681.3792, found 681.3778.
Synthesis of D-Ring Diene 15
The olefin (14, 2.8 g, 4.11 mmol) and 2,6-di-tert-butyl-4-methyl pyridine (3.28 g, 16 mmol, 4 equiv) were dissolved in CH2Cl2 (200 mL) and cooled to −40 °C. Freshly prepared trifluoroacetyl trifluoromethanesulfonate (TFAT, 1.5 mL, 8 mmol, 2 equiv) was added slowly and stirred for 1 h. A second portion of TFAT (1.5 mL, 2 equiv) was added and stirred another 2.5 h, and finally, a third portion of TFAT (0.7 mL, 1 equiv) was added and stirring continued for another 1 h. After the disappearance of starting material shown by TLC (3:7 EtOAc/Hexane, Rf = 0.55), the reaction was quenched with aq. NaHCO3 (100 mL). The organic layer was separated, and the aqueous phase was extracted with CH2Cl2 (15 × 3). The combined organic phase was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The crude product was loaded on a silica gel column and eluted with hexane followed by EtOAc–Hexanes. The 2,6-di-tert-butyl-4-methyl pyridine was recovered first by flushing 1:10 to 1:8, and then desired product 15 was isolated with 1:4 EtOAc–Hexanes respectively. The yield was 70%, 2.23 g. 1H NMR (400 MHz, CDCl3) δ 0.80–0.88 (m, 3H), 0.93 (s, 3H), 0.96 (d, J = 6.5, 3H), 1.10–1.15 (m, 1H), 1.21 (s, 3H), 1.26 (t, 1H), 1.34–1.47 (m, 3H), 1.52 (s, 3H), 1.64–1.75 (m, 2H), 1.78–1.86 (m, 1H), 1.96–2.00 (m, 2H), 2.02 (s, 3H), 2.03–2.09 (m, 1H), 2.14–2.24 (m, 2H), 2.49 (q, 1H), 2.96 (dd, J = 6.6, 6.7, 1H), 4.46 (dd, J = 4.4, 4.4, 1H), 4.63 (br, 1H), 4.67–4.72 (m, 2H), 5.42 (d, J = 10.6, 1H), 5.51 (dd, J = 2.4, 2.4, 1H), 5.88 (s, 1H), 6.15 (s, 1H), 7.38–7.62 (m, 6H), 7.89 (d, J = 8.1, 2H), 8.10 (d, J = 8.2, 2H). 19F NMR (376 MHz) δ −76.43; 13CNMR (CDCl3, 100 MHz) δ 11.9, 13.7, 13.8, 19.2, 20.7, 21.0, 21.8, 27.1, 27.9, 28.9, 32.9, 33.6, 34.5, 35.2, 35.6, 36.8, 44.0, 53.1, 56.6, 60.0, 70.8, 73.0, 76.9, 77.2, 77.5, 79.0, 81.5, 113.1, 114.0, 115.9, 120.6, 125.4, 128.2, 129.2, 130.3, 132.9, 133.1, 140.0, 155.9, 156.2, 156.6, 165.1, 165.2, 170.1, 170.6; LRMS (ESI) calcd for C45H52F3O8 [M+H]+ 777.36, found 777.31.
Synthesis of C22 Alcohol 16
The C22-OTFA (15) was dissolved in THF/MeOH (70 mL, 6:1) and Cs2CO3 (1.3 g, 1 equiv) was added to the reaction and stirred at room temperature. After ensuring the consumption of starting material by TLC (3:7 EtOAc/Hexane, Rf = 0.3), the reaction was diluted by EtOAc (80 mL), followed by water (60 mL). The organic phase was separated, and the aqueous phase was extracted with EtOAc (30 mL × 3). The combined organic phase was washed once with brine (20 mL), dried (Na2SO4), and concentrated. After evaporation of the solvent under reduced pressure, the crude product was purified on a silica gel column using EtOAc–Hexane (1:4) to provide C23 alcohol, 16, 2.38 g in 85% yield. 1H NMR (400 MHz, CDCl3) δ 0.84–0.88 (m, 2H), 0.91 (d, J = 6.4, 3H), 0.93 (s, 3H), 1.05–1.15 (m, 1H), 1.20–1.25 (m, 2H), 1.27 (s, 3H), 1.34–1.50 (m, 3H), 1.65–1.73 (m, 1H), 1.75–1.85 (m, 2H), 2.0–2.07 (m, 9H), 2.13–2.25 (m, 3H), 2.61–2.74 (m, 2H), 3.90 (dd, J = 2.5, 2.4, 1H), 4.46 (dd, J = 4.5, 4.4, 1H), 4.68 (br, 3H), 5.30–5.33 (m, 1H), 5.96 (s, 1H), 6.21 (s, 1H), 7.39–7.47 (m, 4H), 7.52–7.59 (m, 2H), 8.01 (d, J = 8.1, 2H), 8.09 (d, J = 8.2, 2H); LRMS (ESI) calcd for C43H52NaO7 [M+Na]+ 703.36, found 703.33.
Formation of C-22 OTBS Ether 17
To the C22 alcohol (16, 2.36 g, 3.47 mmol) in CH2Cl2 (35 mL) were added 2,6-lutidine (2.1 mL, 18 mmol, 5.0 equiv) followed by TBSOTf (4 equiv) in three portions under N2 at room temperature, and the reaction was stirred for 3 h. The reaction mixture was quenched by adding an aq. NaHCO3 solution (20 mL). The aqueous phase was extracted with CH2Cl2 (15 mL × 3), and the combined organic phases were dried over Na2SO4 and concentrated under reduced pressure. The crude product was subjected to chromatography on silica gel to provide 17 (3:7 EtOAc/Hexane, Rf = 0.65), 2.21 g in 81% yield. 1H NMR (400 MHz, CDCl3) δ 0.85 (s, 6H), 0.88 (br, 12H), 0.90–0.95 (m, 5H), 1.04–1.12 (m, 1H), 1.19–1.24 (m, 1H), 1.28 (s, 3H), 1.33–1.42 (m, 3H), 1.44–1.50 (m, 2H), 1.62–1.71 (m, 2H), 1.74–1.80 (m, 1H), 1.97–2.00 (m, 4H), 2.16–2.23 (m, 2H), 2.49 (s, 4H), 2.61–2.70 (m, 1H), 2.72–2.80 (m, 1H), 3.99 (dd, J = 1.4, 1.5, 1H), 4.40 (dd, J = 4.4, 4.2, 1H), 4.66 (br, 2H), 5.29 (d, J = 10.1, 1H), 5.93 (s, 1H), 6.21 (s, 1H), 6.91 (d, J = 7.6, 1H), 7.35–7.56 (m, 6H), 7.98 (d, J = 7.8, 2H), 8.08 (d, J = 8.1, 2H; 13CNMR (CDCl3, 100 MHz) δ −4.6, −3.8, −3.6, 12.1, 12.8, 17.9, 18.3, 20.3, 21.3, 22.0, 24.1, 25.6, 26.0, 27.1, 27.3, 28.1, 29.1, 33.7, 34.6, 35.7, 36.2, 36.4, 36.9, 44.2, 53.2, 56.8, 73.3, 75.1, 78.3, 80.0, 113.0, 120.2, 121.1, 125.6, 128.2, 128.4, 129.3, 129.6, 130.5, 132.7, 132.9, 136.7, 141.7, 154.6, 157.3, 158.5, 165.6, 166.4, 170.5; LRMS (ESI) calcd for C49H67O7Si [M+H]+ 795.46, found 795.41.
Synthesis of C-3 Alcohol 18
C3-acetyl compound (17, 2.21 g, 2.78 mmol) was dissolved in THF/MeOH/H2O (1:1:1, 30 mL), K2CO3 (4.28 g, 30 mmol) was added in one portion, and the reaction mixture was stirred for 70 h at room temperature. After the absence of starting material was ensured from TLC (3:7 EtOAc/Hexane, Rf = 0.2), the mixture was diluted with EtOAc (60 mL) followed by H2O (30 mL). The organic phase was separated, and the aqueous phase was extracted with EtOAc (20 mL × 3). The combined organic phase was dried over Na2SO4, the solvent was evaporated under reduced pressure, and the resulting crude product was subjected to chromatography on silica gel using EtOAc–Hexane to provide the C3-OH, 18 (3:7 EtOAc/Hexane, Rf = 0.2), 1.99 g and yield 95%. 1H NMR (400 MHz, CDCl3) δ −0.23 (s, 3H), −0.07 (s, 3H), 0.79–0.81(m, 1H), 0.86 (s, 9H), 0.91 (s, 3H), 0.92–0.95 (m, 3H), 1.02–1.17 (m, 3H), 1.21–1.25 (m, 2H), 1.28 (s, 3H), 1.31–1.36 (m, 1H), 1.41–1.44 (m, 3H), 1.57–1.71 (m, 3H), 1.75–1.82 (m, 1H), 1.97–2.04 (m, 4H), 2.17–2.27 (m, 2H), 2.64–2.70 (m, 1H), 2.75–2.80 (m, 1H), 3.56–3.65 (m, 1H), 4.01(dd, J = 1.5, 1.5, 1H), 4.40 (dd, J = 4.4, 4.2, 1H), 4.67 (br, 2H), 5.30 (d, J = 10.7, 1H), 5.94 (t, J = 4.1, 1H), 6.22 (d, J = 2.2, 1H), 7.38–7.59 (m, 6H), 7.99 (d, J = 8.1, 2H), 8.10 (d, J = 7.9, 2H); 13CNMR (CDCl3, 100 MHz) δ −4.5, −3.8, −0.03, 12.3, 12.8, 14.2, 18.9, 20.4, 21.0, 22.2, 26.1, 27.4, 28.3, 29.2, 31.2, 34.7, 35.8, 36.2, 36.5, 37.2, 37.8, 44.5, 53.4, 56.9, 60.4, 71.0, 75.2, 78.4, 80.2, 113.0, 121.1, 125.6, 128.2, 128.5, 129.4, 129.6, 130.5, 130.6, 132.8, 133.0, 141.9, 154.6, 158.6, 165.7, 166.5; LRMS (ESI) calcd for C47H65O6Si [M+H]+ 753.45, found 753.41.
Synthesis of C3 Ketone 19
The C3 alcohol (18, 781 mg, 1.04 mmol) was dissolved in CH2Cl2 (10 mL) under N2, and NMO (240 mg, 2 mmol, 2.0 equiv) followed by tetrapropylammonium perruthenate (TPAP, 17.5 mg, 0.05 mmol) was added at room temperature. The mixture was stirred for 3 h. After the disappearance of starting material was ensured by TLC (3:7 EtOAc/Hexane, Rf = 0.55), the solvent was removed and the crude product was subjected to flash chromatography on a short silica gel column to provide C3 ketone 19, 744 mg in 99% yield. 1H NMR (400 MHz, CDCl3) δ −0.24 (s, 3H), −0.07 (s, 3H), 0.85 (s, 12H), 0.93 (d, J = 6.5, 2H), 1.05 (s, 3H), 1.31 (s, 3H), 1.36–1.45 (m, 6H), 1.87–1.95 (m, 2H), 1.98–2.11 (m, 4H), 2.19–2.31 (m, 5H), 2.64–2.71 (m, 1H), 2.79 (t, 1H), 4.01 (d, J = 7.4, 1H), 4.40 (dd, J = 4.5, 4.2, 1H), 4.66 (d, J = 5.3, 2H), 5.31 (dd, J = 2.7, 2.8, 1H), 5.94 (s, 1H), 6.21 (s, 1H), 7.33–7.53 (m, 6H), 7.98 (d, J = 8.2, 2H), 8.09 (d, J = 8.1, 2H); 13CNMR (CDCl3, 100 MHz) δ −4.5, −3.8, 11.3, 12.7, 18.3, 20.3, 22.1, 26.1, 27.5, 28.4, 28.8, 34.5, 35.8, 36.2, 36.4, 37.7, 38.4, 44.3, 46.0, 52.8, 56.9, 75.0, 78.3, 79.7, 113.0, 121.5, 125.5, 128.2, 128.4, 129.3, 129.5, 130.4, 132.7, 133.0, 141.7, 154.0, 158.8, 165.4, 166.2, 210.4; LRMS (ESI) calcd for C47H63O6Si [M+H]+ 751.43, found 751.40.
Peroxide Formation (20) Followed by Deprotection of C22-OTBS (21)
The diene (19, 500 mg, 0.64 mmol) in CH2Cl2 (40 mL) was cooled to −78 °C. 5,10,15,20-Tetraphenyl-21H,23H-porphine (TPP) (0.1 mol %) was added, and oxygen was bubbled into the solution via a balloon with photoactivation (GE Sunlamp 300 W) at a distance of approximately 8 to 9 in. from the reaction flask. The reaction was stirred with irradiation at −78 °C for 2 h. After the disappearance of starting material was ensured by TLC (3:7 EtOAc/Hexane, Rf = 0.4), solvent was removed and the crude peroxide (20) was directly used in the next step after drying for 1 h under high vacuum.
In the second step, crude product 20 was dissolved in THF (7 mL) under N2 and TBAF (tetrabutylammonium fluoride, 1.0 M in THF, 1.5 mL) was added at room temperature. The reaction was stirred for 3 h and then was quenched with brine and extracted with ethyl acetate (10 mL × 3). The combined organics were dried over Na2SO4, the solvent evaporated under reduced pressure, and the crude product was subjected to column chromatography on neutral alumina using EtOAc–Hexane (3:7 EtOAc/Hexane, Rf = 0.22) to provide C22 alcohol (21), 263 mg, 74% for two steps. 1H NMR (400 MHz, CDCl3) δ 0.85 (s, 4H), 0.90–0.96 (m, 3H) 1.04–1.08 (t, 3H), 1.13 (s, 3H), 1.20–1.29 (m, 3H), 1.62–1.69 (m, 1H), 1.74–1.80 (m, 3H), 1.83 (s, 3H), 1.88–1.96 (m, 3H), 2.02–2.11 (m, 4H), 2.24–2.33 (m, 1H), 2.35–2.42 (m, 1H), 3.37 (s, 1H), 4.47 (d, J = 5.2, 2H), 5.22 (d, J = 8.9, 1H), 5.48 (dd, J = 4.4, 5.9, 1H), 6.25 (d, J = 5.6, 1H), 6.44 (d, J = 5.4, 1H), 7.18–7.27 (m, 4H), 7.33–7.43 (m, 2H), 7.80 (d, J = 8.1, 2H), 7.87 (d, J = 8.3, 2H); 13CNMR (CDCl3, 100 MHz) δ 10.6, 10.7, 11.6, 13.4, 14.0, 20.8, 22.2, 22.3, 26.4, 26.8, 28.5, 33.3, 35.0, 35.4, 35.6, 37.5, 37.9, 44.0, 44.9, 45.4, 60.1, 63.3, 72.0, 73.4, 73.6, 97.5, 98.2, 113.1, 128.1, 128.2, 129.2, 129.5, 129.9, 130.0, 132.0, 132.8, 135.9, 141.1, 165.3, 165.5, 170.8, 210.8; LRMS (ESI) calcd for C41H49O8 [M +H]+ 669.34, found 669.29.
Synthesis of C-22 Ketone 2214
A flame-dried flask was charged with DMSO (118 μL, 1.65 mmol) in CH2Cl2 (8 mL) and cooled to −78 °C under an argon atmosphere. TFAA (175 μL, 1.24 mmol) was added to the solution dropwise and stirred for 45 min. To the reaction was added C22-alcohol (21, 263 mg, 0.413 mmol) in CH2Cl2 (6 mL) via syringe over 4 min, and the reaction was stirred another 50 min at −78 °C. Diisopropylethyl amine (456 μL) was added to the reaction, and the temperature was allowed to warm to −30 °C over 1.5 h. TLC (3:7 EtOAc/Hexane, Rf = 0.5) showed no starting material remaining, and the reaction was quenched by adding brine. The organic layer was separated, and the aqueous phase was extracted with CH2Cl2 (10 mL × 3). The combined organics were dried over Na2SO4 and concentrated, and the crude product was subjected to flash column chromatography on neutral Al2O3 by using EtOAc–Hexane to provide C3,22-diketone (22), 187 mg, yield 68%. An X-ray structure was obtained which confirmed the singlet oxygen Diels–Alder reaction proceeded with the desired stereochemistry.14 However, due to the potential instability of the peroxide, this material was normally used immediately. 1H NMR (400 MHz, CDCl3) δ 1.01–1.23 (m, 10H), 1.28–1.34 (m, 1H), 1.50–1.56 (m, 1H), 1.64–1.73 (m, 2H), 1.78–1.87 (m, 3H), 1.94–2.09 (m, 5H), 2.35 (s, 7H), 3.05–3.14 (m, 1H), 4.32 (s, 1H), 4.38 (s, 1H), 5.04 (dd, J = 3.6, 3.3, 1H), 5.40 (dd, J = 4.7, 4.6, 1H), 6.15 (d, J = 5.8, 1H), 6.41 (d, J = 6.0, 1H), 7.12–7.19 (m, 4H), 7.28–7.34 (m, 2H), 7.63 (d, J = 8.2, 2H), 7.68 (d, J = 7.9, 2H); LRMS (ESI) calcd for C41H47O8 [M+H]+ 667.32, found 667.28.
Synthesis of Diol 23
To the C3,22-diketone (22) in CH2Cl2 (6 mL) were added activated Zn powder18 (260 mg, 4 mmol) followed by AcOH (50 μL). The mixture was stirred at roon temperature for 2 h. After the disappearance of starting material from TLC (1:1 EtOAc/Hexane, Rf = 0.3), NaHCO3 (aq. 10 mL) was added to quench the reaction. The organic layer was separated, and the aqueous phase was extracted with CH2Cl2 (10 mL × 3). Combined organics were dried over anhydrous Na2SO4, concentrated under reduced pressure, and purified by chromatography on silica gel to provide 23, 196 mg, yield 71%. 1H NMR (400 MHz, CDCl3) δ 1.00 (s, 3H), 1.04 (s, 2H), 1.17 (t, 3H), 1.22 (d, J = 6.5, 3H), 1.34–1.41 (m, 1H), 1.62–1.68 (m, 1H), 1.72 (s, 3H), 1.80–1.86 (m, 3H), 1.95 (s, 3H), 1.98–2.07 (m, 2H), 2.12–2.27 (m, 3H), 2.45–2.57 (m, 2H), 3.02 (q, 1H), 3.78 (s, 1H), 4.75 (s, 1H), 4.81 (d, J = 10.7, 2H), 5.42 (dd, J = 3.8, 3.6, 1H), 5.73 (d, J = 5.8, 1H), 6.03–6.07 (m, 1H), 6.28 (d, J = 6.0, 1H), 7.34–7.38 (m, 4H), 7.46–7.52 (m, 2H), 7.92 (d, J = 8.2, 2H), 7.98 (d, J = 8.1, 2H); 13CNMR (CDCl3, 100 MHz) δ 10.8, 12.2, 14.2, 14.8, 21.0, 22.1, 26.0, 26.9, 28.7, 34.6, 35.9, 37.8, 37.9, 38.7, 44.4, 45.2, 46.1, 54.5, 60.3, 71.1, 75.7, 87.3, 88.6, 114.8, 128.4, 128.5, 128.9, 129.6, 129.7, 130.6, 132.9, 135.5, 137.7, 138.4, 139.6, 165.4, 165.6, 170.9, 211.2, 213.0; LRMS (ESI) calcd for C41H49O8 [M+H]+ 669.34, found 669.30.
Synthesis of Methoxy Ketal 24
To the C14,16 diol (23, 196 mg, 0.29 mmol) dissolved in dry MeOH (5 mL) was added camphor sulfonic acid (CSA, 6.8 mg, 0.03 mmol), and the reaction was stirred for 4 to 5 h at room temperature. After the disappearance of starting material was ensured from TLC (3:7 EtOAc/Hexane, Rf = 0.45), the solvent was evaporated under reduced pressure and CH2Cl2 (30 mL) was added to the reaction. The mixture was washed with aq. NaHCO3 (5 mL) and brine (10 mL), dried over Na2SO4, and concentrated. The crude product was subjected to chromatography (EtOAc–Hexane) on silica gel to provide 24, 181 mg in 75% yield. Data for C3-ketone, 24: 1H NMR (400 MHz, CDCl3) δ 0.82–0.86 (m, 1H), 0.88–0.94 (m, 3H), 1.01 (s, 3H), 1.13 (d, J = 6.8, 3H), 1.20–1.30 (m, 3H), 1.37 (s, 3H), 1.40–1.47 (m, 1H), 1.52–1.63 (m, 1H), 1.76 (s, 3H), 1.85–1.92 (m, 1H), 1.95–2.01 (m, 2H), 2.04–2.10 (m, 1H), 2.17–2.22 (m, 3H), 2.24–2.30 (m, 2H), 2.39–2.43 (m, 1H), 2.63–2.68 (q, 1H), 3.32 (s, 3H), 4.70 (d, J = 8.0, 2H), 5.10 (t, J = 3.6, 1H), 5.23 (dd, J = 4.4, 4.0, 1H), 5.68 (dd, J = 4.6, 4.8, 1H), 7.38–7.44 (m, 4H), 7.49–7.54 (m, 2H), 7.97 (d, J = 8.2, 2H), 8.02 (d, J = 8.0, 2H); 13CNMR (CDCl3, 100 MHz) δ 8.5, 11.1, 15.0, 22.0, 27.4, 28.1, 33.6, 35.9, 37.7, 38.3, 42.7, 44.2, 45.8, 48.8, 51.8, 53.9, 69.8, 74.9, 91.4, 93.7, 109.8, 113.4, 119.9, 128.1, 128.2, 129.4, 129.6, 130.3, 131.0, 132.6, 141.2, 152.9, 165.5, 165.7, 210.9; LRMS (ESI) calcd for C42H51O8 [M+H]+ 683.35, found 683.31.
Data for C3-Dimethylketal (24a)
Note: The C3-dimethylketal, 24a, was easily converted to C3 ketone 24 by stirring (1–2 h) with a few drops of water in CH2Cl2/MeOH and then subjected to the workup procedure above. 1H NMR (400 MHz, CDCl3) δ 0.81 (s, 3H), 0.85–0.89 (m, 1H), 1.04–1.08 (m, 1H), 1.14 (d, J = 6.8, 3H), 1.19–1.28 (m, 6H), 1.35 (s, 3H), 1.43–1.48 (m, 1H), 1.60–1.63 (m, 1H), 1.77 (s, 3H), 1.80–1.85 (m, 1H), 1.92–1.97 (m, 2H), 2.02 (s, 2H), 2.40–2.45 (m, 2H), 2.55 (s, 1H), 2.63–2.68 (q, 1H), 3.08 (s, 3H), 3.15 (s, 3H), 3.32 (s, 3H), 4.70 (t, J = 3.5, 1H), 4.73 (d, J = 7.1, 2H), 5.10 (t, 1H), 5.24 (dd, J = 5.1, 5.0, 1H), 5.69 (dd, J = 5.0, 4.8, 1H), 7.38–7.44 (m, 4H), 7.50–7.54 (m, 2H), 7.98 (d, J = 8.2, 2H), 8.03 (d, J = 7.9, 2H); 13CNMR (CDCl3, 100 MHz) δ 8.5, 11.3, 14.1, 14.9, 20.9, 22.0, 27.2, 27.6, 28.1, 28.3, 33.8, 34.6, 35.2, 36.1, 38.3, 41.8, 42.7, 47.3, 47.4, 48.8, 52.1, 53.9, 60.3, 69.8, 75.2, 91.4, 93.8, 99.9, 109.8, 113.4, 119.4, 128.1, 128.2, 129.4, 129.5, 130.3, 131.0, 132.5, 132.6, 141.2, 153.5, 165.5, 165.7; LRMS (ESI) calcd for C44H57O9 [M+H]+ 729.40, found 729.37.
Asymmetric Dihydroxylation 25
The olefin 24 (190 mg 0.278 mmol) was mixed with K2OsO4 (2 mg, 0.0056 mmol 2 mol %), (DHQ)2PHAL (20 mg, 0.026 mmol, 5 equiv relative to osmium), and AD mix-α (390 mg). The reaction flask was placed in a 0 °C cooling bath, and tert-butyl alcohol–water (16 mL, 1:1) was added to the flask while stirring was continued for 40 h at this temperature. The reaction was monitored by TLC (1:1 EtOAc/Hexane, Rf = 0.2) by quenching (aq. NaHSO3) reaction mixture aliquots. After the disappearance of starting material was ensured, the reaction was quenched using aq. NaHSO3 (10 mL) and stirred at room temperature for 1 h. The reaction mixture was extracted with EtOAc (15 mL) and CH2Cl2 (20 mL × 3), and the combined organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated. The crude product was chromatographed on silica gel using EtOAc–Hexane followed by 2% MeOH in CHCl3 to provide product 25, 164 mg and yield 82% (the selectivity is 9.5:1, with C25 epi-natural favoring the ‘R’ configuration at C25, whereas in another case the selectivity is 3.8:1, C25 natural-epi when the ligand was (DHQD)2PYR and Na2CO3, K3FeCN6 (see Table 1 in the manuscript)). The selectivity is 9.5:1, with C25 epi-natural favoring the ‘R’ configuration at C25. Data for C3-keto compound, 25: 1H NMR (400 MHz, CDCl3) δ 0.82–0.88 (m, 1H), 1.01 (s, 3H), 1.13 (d, J = 6.9, 3H), 1.15 (s, 2H), 1.17–1.20 (m, 1H), 1.22–1.24 (m, 2H), 1.24 (s, 3H), 1.25–1.26 (m, 1H), 1.27–1.31 (m, 1H), 1.36 (s, 3H), 1.42–1.49 (m, 1H), 1.79–1.90 (m, 1H), 1.92–1.99 (m, 1H), 2.03 (s, 2H), 2.06–2.21 (m, 2H), 2.25–2.32 (m, 2H), 2.52 (s, 1H), 2.63 (q, 1H), 3.32 (s, 3H), 3.15 (s, 3H), 3.39 (s, 2H), 4.72 (t, J = 1.8, 1H), 5.03 (s, 1H), 5.21 (dd, J = 5.2, 4.9, 1H), 5.78 (d, J = 9.7, 1H), 7.40–7.44 (m, 4H), 7.52–7.56 (m, 2H), 7.98 (d, J = 8.2, 2H), 8.02 (d, J = 8.4, 2H); LRMS (ESI) calcd for C42H53O10 [M+H]+ 717.36, found 717.33.
Data for C3-Dimethylacetal (OMe)2 Compound, 25a
1H NMR (400 MHz, CDCl3) δ 0.81 (s, 3H), 0.85–0.91 (m, 1H), 1.04–1.10 (m, 1H), 1.12 (d, J = 6.9, 3H), 1.16 (s, 2H), 1.20–1.23 (m, 2H), 1.24–1.28 (m, 7H), 1.34 (s, 3H), 1.54–1.63 (m, 1H), 1.76–1.86 (m, 2H), 1.89–1.97 (m, 2H), 2.04 (s, 1H), 2.10–2.18 (m, 1H), 2.38 (br, 1H), 2.53 (s, 1H), 2.64 (q, 1H), 2.73 (br, 1H), 3.08 (s, 3H), 3.16 (s, 3H), 3.32 (s, 3H), 3.40 (s, 2H), 4.73 (t, 1H), 5.03 (s, 1H), 5.21 (dd, J = 5.1, 5.0, 1H), 5.78 (d, J = 10.8, 1H), 7.40–7.44 (m, 4H), 7.51–7.55 (m, 2H), 7.98 (d, J = 7.6, 2H), 8.03 (d, J = 7.9, 2H); 13CNMR (CDCl3, 100 MHz) δ 8.6, 11.3, 14.1, 14.7, 23.4, 27.2, 27.6, 28.1, 28.3, 29.6, 33.7, 34.6, 35.1, 36.1, 37.5, 41.8, 42.9, 47.3, 47.4, 48.8, 52.1, 53.8, 68.9, 70.1, 71.7, 75.3, 91.5, 93.8, 100.0, 109.9, 119.3, 128.1, 128.2, 129.4, 129.6, 130.1, 131.0, 132.5, 132.9, 153.5, 165.7, 166.2; LRMS (ESI) calcd for C44H59O11 [M+H]+ 763.40, found 763.37.
Synthesis of Benzoate 26
To the diol (25, 164 mg, 0.25 mmol) in CH2Cl2 (10 mL) were added pyridine (2.5 mL) and DMAP (5 mg, 0.025 mmol, 0.1 equiv) followed by benzoyl chloride (50 μL, 0.4 mmol, 1.5 equiv) under N2 at room temperature, and the reaction was stirred for 16 h. After the disappearance of starting material was ensured via TLC (3:7 EtOAc/Hexane, Rf = 0.45), the solvent was removed under reduced pressure, and the crude product was purified by column chromatography on silica gel using EtOAc–Hexane to provide 26, 0.123 g in 88% yield. 1H NMR (400 MHz, CDCl3) δ 0.77–0.84 (m, 3H), 0.98 (s, 3H), 1.12 (d, J = 6.5, 3H), 1.21 (t, 5H), 1.26 (s, 4H), 1.32 (s, 3H), 1.37–1.45 (m, 1H), 1.83–1.88 (m, 1H), 1.91–1.97 (m, 3H), 2.02–2.07 (m, 1H), 2.14–2.28 (m, 4H), 2.53 (br, 1H), 2.62 (q, 1H), 3.30 (s, 3H), 4.19 (d, J = 4.2, 1H), 4.70 (t, J = 2.8, 1H), 5.00 (t, J = 3.9, 1H), 5.18 (dd, J = 5.5, 5.0, 1H), 5.87 (d, J = 8.6, 1H), 7.34–7.42 (m, 6H), 7.46–7.54 (m, 3H), 7.94–8.02 (m, 6H); 13CNMR (CDCl3, 100 MHz) δ 8.5, 11.1, 14.1, 20.9, 23.8, 27.3, 28.0, 33.5, 35.9, 37.7, 37.8, 38.0, 44.2, 45.8, 48.8, 51.8, 53.4, 53.8, 60.3, 68.4, 70.7, 71.6, 74.9, 91.4, 93.7, 109.9, 119.8, 128.1, 128.2, 128.3, 129.4, 129.5, 129.6, 129.7, 129.8, 130.1, 130.9, 132.6, 132.8, 133.0, 152.8, 165.7, 165.8, 166.2, 171.1, 211.0; LRMS (ESI) calcd for C49H57O11 [M+H]+ 821.39, found 821.35.
Synthesis of Spiroketal 2714
To 26 in CH2Cl2/MeOH (1:1, 10 mL) was added camphor sulfonic acid (CSA, 6 mg) at room temperature, and the reaction was stirred for 4 h. H2O (0.5 mL) was added, and the reaction was stirred for another 1 h (to hydrolyze the C3-dimethylketal, 27a, to C3-ketone 27). After removal of the solvent, the reaction was diluted by adding CH2Cl2 (10 mL) and H2O (5 mL) and extracted with CH2Cl2 (10 mL × 3). The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified on silica gel column chromatography to provide 2714 (3:7 EtOAc/Hexane, Rf = 0.55), 0.101 g and yield 85%. 1H NMR (400 MHz, CDCl3) δ 0.87–0.94 (m, 1H), 1.02 (s, 3H), 1.11 (d, J = 6.4, 3H), 1.23 (t, 3H), 1.30–1.36 (m, 3H), 1.38 (s, 3H), 1.42 (s, 3H), 1.46–1.51 (m, 1H), 1.81–1.83 (m, 1H), 1.89–1.95 (m, 2H), 2.04–2.13 (m, 3H), 2.19–2.23 (m, 1H), 2.25–2.30 (m, 2H), 2.33–2.37 (m, 1H), 2.60–2.65 (q, 1H), 4.33 (d, J = 11.0, 1H), 4.50 (d, J = 11.2, 1H), 4.82 (d, J = 2.2, 1H), 5.19 (dd, J = 4.9, 4.8, 1H), 5.38 (d, J = 7.7, 1H), 5.41 (t, J = 3.8, 1H), 7.37–7.45 (m, 7H), 7.50–7.56 (m, 2H), 7.95–8.0 (m, 3H), 8.06–8.10 (m, 3H); 13CNMR (CDCl3, 100 MHz) δ 8.3, 11.1, 14.1, 14.8, 21.0, 25.1, 27.3, 28.2, 28.4, 33.8, 35.9, 37.8, 37.9, 38.2, 44.3, 45.5, 45.9, 51.7, 54.7, 60.3, 70.1, 72.9, 75.5, 79.6, 89.3, 93.3, 116.4, 121.2, 128.2, 128.3, 128.4, 128.5, 129.3, 129.4, 129.7, 130.0, 130.6, 132.8, 132.9, 133.3, 152.6, 165.7, 166.3, 211.1; LRMS (ESI) calcd for C48H53O10 [M+H]+ 789.36, found 789.32.
Synthesis of α-Bromoketone 28
C3-ketone (27, 148 mg, 0.188 mmol) was dissolved in dry THF (3 mL) under N2 and was cooled to −5 °C. Phenyltrimethylammonium tribromide (PTAB, 70.5 mg, 0.188 mmol, 1 equiv) was added to the reaction in one portion while stirring. In about 5 min, the orange solution turned cloudy and then turned to a light red color. The reaction mixture was stirred for an additional 15 min and then quenched with aq. NaHSO3. The reaction was diluted and extracted with EtOAc (10 mL × 3). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The crude product was chromatographed on silica gel using EtOAc–Hexane to provide C2-bromide 28 (3:7 EtOAc/Hexane, Rf = 0.6), 142 mg in 84 yield %. 1H NMR (400 MHz, CDCl3) δ 0.93–1.00 (m, 1H), 1.11 (s, 8H), 1.24 (t, 1H), 1.30–1.36 (m, 1H), 1.38 (s, 3H), 1.42 (s, 3H), 1.44–1.50 (m, 1H), 1.57–1.66 (m, 1H), 1.74–1.87 (m, 2H), 2.04–2.11 (m, 1H), 2.29–2.38 (m, 2H), 2.39–2.44 (m, 2H), 2.54 (dd, J = 6.2, 6.4, 1H), 2.61 (q, 1H), 2.79 (br, 1H), 4.33 (d, J = 11.0, 1H), 4.59 (d, J = 10.9, 1H), 4.67 (dd, J = 6.0, 6.2, 1H), 4.80 (d, J = 2.3, 1H), 5.19 (dd, J = 5.1, 5.1, 1H), 5.38 (d, J = 7.3, 1H), 5.42 (t, 1H), 7.40–7.46 (m, 7H), 7.52–7.57 (m, 3H), 7.95–7.99 (m, 3H), 8.07–8.09 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 8.3, 11.8, 14.1, 14.8, 21.0, 25.1, 27.3, 27.6, 28.2, 33.4, 38.2, 39.0, 43.5, 45.6, 45.7, 50.8, 51.3, 53.7, 54.7, 60.3, 70.0, 73.0, 75.2, 79.6, 89.3, 93.2, 116.4, 121.5, 128.2, 128.3, 128.5, 129.3, 129.4, 129.7, 130.0, 130.5, 132.9, 133.3, 133.4, 152.0, 165.7, 166.1, 163.3, 200.4; LRMS (ESI) calcd for C48H52BrO10 [M+H]+ 867.27, found 867.23.
Synthesis of α-Azidoketone 29
C2-bromide (28, 142 mg, 0.164 mmol) was dissolved in dry nitromethane (12 mL, freshly distilled) under N2 and cooled in an ice–water bath. A clear solution of tetramethyl guanidinium azide (TMGA, 103 mg, 0.66 mmol, 4.0 equiv) in nitromethane (3 mL) was added to the reaction mixture dropwise. After 5 min, the cooling bath was removed and the reaction was stirred at room temperature for 14 h. The reaction was quenched by adding brine, diluted, and extracted with EtOAc (15 mL × 3). The combined organics were dried (Na2SO4) and concentrated under reduced pressure at room temperature. The crude product was subjected to chromatography on silica gel using EtOAc–Hexane to provide azide 29 (3:7 EtOAc/Hexane, Rf = 0.5) (C25 epi:natural, >9.5:1) favoring C25-epi, 110 mg and a yield of 81%. 1H NMR (400 MHz, CDCl3) δ 0.85–0.93 (m, 1H), 0.94–1.11 (s, 2H), 1.12 (s, 6H), 1.26 (t, 1H), 1.31–1.36 (m, 2H), 1.38 (s, 4H), 1.43 (s, 3H), 1.46–1.60 (m, 2H), 1.84–1.91 (m, 1H), 2.04–2.13 (m, 1H), 2.19–2.27 (m, 1H), 2.30 (d, J = 4.2, 1H), 2.32–2.40 (m, 2H), 2.58–2.67 (q, 1H), 2.75 (br, 1H), 3.93 (dd, J = 6.3, 6.1, 1H), 4.33 (d, J = 10.9, 1H), 4.50 (d, J = 11.0, 1H), 4.80 (d, J = 2.2, 1H), 5.19 (dd, J = 5.2, 5.0, 1H), 5.38 (t, 1H), 5.42 (s, 1H), 7.40–7.45 (m, 7H), 7.52–7.58 (m, 3H), 7.95–8.01 (m, 3H), 8.08–8.10 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 8.3, 12.1, 14.1, 14.8, 20.9, 25.1, 27.3, 27.6, 28.2, 33.3, 37.1, 38.2, 43.3, 44.8, 45.6, 46.8, 51.4, 54.7, 60.3, 63.5, 70.0, 73.0, 75.2, 79.6, 89.3, 93.2, 116.4, 121.5, 128.2, 128.3, 128.4, 128.6, 129.3, 129.4, 129.5, 129.7, 130.0, 130.5, 132.8, 132.9, 133.3, 152.0, 165.7, 166.1, 166.2, 171.0, 204.5; LRMS (ESI) calcd for C48H52N3O10 [M+H]+ 830.36, found 830.33.
Pyrazine Formation – C12, 12′, 23, and 26-Tetra-O-benzoyl Protected 25-epi Ritterostatin GN1N (31)
To the mixture of North G aminomethoxime 30 (17 mg, 0.029 mmol, 1.2 equiv) and North 1 azidoketone 29 (20 mg, 0.024 mmol, 1.0 equiv) in benzene (8 mL) were added polyvinylpyridine (PVP, 37 mg) and dichlorodibutylstannane (catalytic) at room temperature. The reaction flask was heated to 90 °C using a Dean–Stark trap and continued for 4 h at this temperature under N2. TLC (3:7 EtOAc/Hexane, Rf = 0.4) showed the nonpolar ketoazide was almost absent. The reaction was filtered through a short plug of Celite, the solvent was evaporated under reduced pressure, and the crude mass was purified by silica gel column chromatography using EtOAc–Hexane to provide 31 (3:7 EtOAc/Hexane, Rf = 0.4), 18.3 mg in 58% yield. 1H NMR (400 MHz, CDCl3): δ 0.83 (d, J = 6.7, 5H), 0.95 (d, J = 6.4, 2H), 1.11 (s, 9H), 1.25 (s, 1H), 1.26 (s, 3H), 1.34 (s, 3H), 1.39 (s, 3H), 1.42 (s, 7H), 1.65–1.74 (m, 8H), 1.92–2.01 (m, 6H), 2.02–2.07 (m, 1H), 2.10–2.20 (m, 3H), 2.32–2.40 (m, 2H), 2.46–2.64(m, 7H), 2.75–2.87 (m, 3H), 4.33 (d, J = 11.0, 1H), 4.49 (d, J = 11.0, 1H), 4.64 (dd, J = 4.4, 4.7, 1H), 4.82 (s, 1H), 4.96 (d, J = 8.2, 1H), 5.20 (dd, J = 5.2, 4.8, 1H), 5.39 (t, 1H), 5.44 (d, J = 2.1, 1H), 5.49 (d, J = 2.2, 1H), 7.40–7.46 (m, 8H), 7.52–7.56 (m, 4H), 7.95–8.05 (m, 6H), 8.09 (d, J = 8.1, 2H); 13C NMR (CDCl3, 100 MHz) δ 8.3, 11.6, 11.7, 14.0, 14.6, 14.8, 25.1, 26.4, 26.9, 27.1, 27.6, 27.8, 28.3, 29.1, 29.9, 33.1, 33.7, 34.9, 35.0, 35.8, 35.9, 37.1, 38.1, 41.1, 41.2, 41.3, 45.5, 51.4, 51.7, 54.6, 55.8, 70.1, 72.8, 75.7, 79.5, 81.3, 81.9, 84.1, 89.4, 93.3, 108.6, 116.3, 117.2, 121.0, 121.3, 128.2, 128.3, 128.5, 129.3, 129.4, 129.7, 130.1, 130.6, 130.7, 132.7, 132.8, 132.9, 133.2, 148.1, 148.2, 148.3, 148.4, 152.7, 155.7, 165.3, 166.1, 166.2; HRMS (ESI) calcd for C82H93N2O13 [M+H]+ 1313.6677, found 1313.6703.
Global Deprotection of All OBz Groups – Synthesis of 25-epi Ritterostatin GN1N, (5)9
C-12, 12′, 23, 26-Tetra-O-benzoyl protected 25-epi ritterostatin GN1N (31, 15 mg, 0.0114 mmol) was dissolved in 1.5 mL of THF/MeOH/H2O (2:2:1). NaOH (5.0 mg, 10 equiv) was added, and the reaction was stirred for 12 h at room temperature. The solvent was evaporated under reduced pressure, and the crude product was diluted and extracted with EtOAc (5 mL × 2) and CH2Cl2 (5 mL × 4). Combined organics were dried over Na2SO4 and concentrated, and the crude product was purified by flash chromatography on silica gel with 3% MeOH in CH2Cl2 to provide 5 as a colorless solid (10.1 mg, 98%), mp 222–225 °C. Rf = 0.25 (2% MeOH/EtOAc). 1H NMR (400 MHz, CDCl3): δ 0.83 (s, 3H), 0.85 (s, 3H), 0.87 (s, 1H), 0.95 (s, 1H), 0.97 (s, 1H), 0.99 (br, 2H), 1.06 (s, 6H), 1.09 (d, J = 6.8, 3H), 1.15 (s, 4H), 1.18 (s, 3H), 1.19 (s, 3H), 1.23–1.28 (t, 2H), 1.50–1.65 (m, 5H), 1.68–1.75 (m, 3H), 1.80–1.88 (m, 3H), 1.90–1.95 (m, 3H), 1.97–2.10 (m, 3H), 2.31–2.41 (m 2H), 2.43–2.48 (m, 2H), 2.49–2.55 (m, 4H), 2.59 (d, J = 5.6, 1H), 2.60 (s, 1H), 2.63 (s, 1H), 2.78 (d, J = 4.8, 1H), 2.82 (d, J = 5.6, 1H), 2.85 (s, 1H), 2.90 (s, 1H), 3.24 (dd, J = 16.0, 7.8, 1H), 3.32 (m, 1H), 3.55 (d, J = 12.0, 1H), 3.62 (d, J = 12.0, 1H), 3.82–3.86 (m, 1H), 3.99 (br, 1H), 4.18 (m, 1H), 4.69 (s, 1H), 4.81 (s, 1H), 4.93 (d, J = 8.2, 1H), 5.35 (s, 1H), 5.39 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 8.5, 11.6, 11.7, 11.9, 13.3, 13.9, 14.0, 20.6, 22.5, 24.4, 25.2, 27.7, 27.8, 27.9, 28.0, 28.2, 28.4, 28.7, 28.9, 29.1, 29.6, 29.8, 30.0, 31.5, 34.5, 35.9, 36.0, 37.1, 41.2, 45.3, 45.4, 52.0, 52.7, 54.7, 55.7, 67.9, 70.8, 75.0, 78.6, 81.9, 82.6, 84.2, 91.7, 92.7, 116.0, 117.4, 119.8, 120.6, 147.9, 148.3, 148.5, 148.6, 153.1, 156.9; HRMS (ESI) calcd for C54H77N2O9 [M+H]+ 897.5629, found 897.5615.
Supplementary Material
ACKNOWLEDGMENTS
This research work was supported by the NIH (CA 60548) and in part by the Intramural Research Program of NIH, National Cancer Institute, Center for Cancer Research. We sincerely acknowledge Dr. Douglas Lantrip for technical support from the lab and Dr. Hui Cao for his initial experiments on north 1 synthesis and thank Dr. Karl Wood and Arlene Rothwell of Purdue University for mass spectral data.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
Copies of all NMR (1H, 13C and 19F) spectra, X-ray data, and NCI 60 cell line full data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) Pettit GR; Inoue M; Kamano Y; Herald DL; Arm C; Dufresne C; Christie ND; Schmidt JM; Doubek DL; Krupa TS J. Am. Chem. Soc 1988, 110, 2006–2007. [Google Scholar]; (b) Fukuzawa S; Matsunaga S; Fusetani N J. Org. Chem 1995, 60, 608–614. [DOI] [PubMed] [Google Scholar]
- (2). NCI-60 data are available on the web at http://dtp.nci.nih.gov. Cephalostatin 1 is NSC-363979.
- (3).(a) Jeong JU; Sutton SC; Kim S; Fuchs PL J. Am. Chem. Soc 1995, 117, 10157–10158. [Google Scholar]; (b) LaCour TG; Guo C; Bhandaru S; Fuchs PL; Boyd MR J. Am. Chem. Soc 1998, 120, 692–707. [Google Scholar]; (c) Jeong JU; Guo C; Fuchs PL J. Am. Chem. Soc 1999, 121, 2071–2084. [Google Scholar]; (d) Kim S; Sutton SC; Guo C; LaCour TG; Fuchs PL J. Am. Chem. Soc 1999, 121, 2056–2070. [Google Scholar]
- (4).(a) Fortner KC; Kato D; Tanaka Y; Shair MD J. Am. Chem. Soc 2010, 132, 275–280. [DOI] [PubMed] [Google Scholar]; (b) Shi Y; Jia L; Xiao Q; Lan Q; Tang X; Wang D; Li M; Ji Y; Zhou T; Tian W Chem.—Asian J 2011, 6, 786–796. [DOI] [PubMed] [Google Scholar]
- (5).(a) Moser R J. Nat. Prod 2008, 71, 487–491. [DOI] [PubMed] [Google Scholar]; (b) Flessner T; Jautelat R; Scholz U; Winterfeldt E Fortschr. Chem. Org. Naturst 2004, 87, 1–80. [DOI] [PubMed] [Google Scholar]; (c) Lee S; LaCour TG; Fuchs PL Chem. Rev 2009, 109, 2275–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).LaCour TG Ph.D thesis, Purdue University, 2001. [Google Scholar]
- (7).(a) Burgett AW; Poulsen TB; Wangkanont K; Anderson DR; Kikuchi C; Shimada K; Okubo S; Fortner KC; Mimaki Y; Kuroda M; Murphy JP; Schwalb DJ; Petrella EC; Cornella-Taracido I; Schirle M; Tallarico JA; Shair MD Nat. Chem. Biol 2011, 7, 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schwarzenberg KV; Vollmar AM Cancer Lett. 2013, 332, 295–303. [DOI] [PubMed] [Google Scholar]
- (8).Shoemaker RH Nat. Rev. Cancer 2006, 6, 813–823. [DOI] [PubMed] [Google Scholar]
- (9).(a) Kanduluru AK; LaClair JJ; Fuchs PL Org. Lett 2011, 13, 5334–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Unpublished results with Prof. Peng Huang (MD Anderson). [Google Scholar]
- (10).Lee JS; Cao H; Fuchs PL J. Org. Chem 2007, 72, 5820–5823. [DOI] [PubMed] [Google Scholar]
- (11).Lee JS; Fuchs PL Org. Lett 2003, 5, 2247–2250. [DOI] [PubMed] [Google Scholar]
- (12).Lee JS; Fuchs PL J. Am. Chem. Soc 2005, 127, 13122–13123. [DOI] [PubMed] [Google Scholar]
- (13).Lee JS; Fuchs PL Org. Lett 2003, 5, 3619–3622. [DOI] [PubMed] [Google Scholar]
- (14).X-ray structural information for compounds 22 and 27 can be obtained from the Cambridge Crystallographic Data Centre (see Supporting Information).
- (15).(a) Crispino GA; Jeong KS; Kolb HC; Wang ZM; Xu D; Sharpless KB J. Org. Chem 1993, 58, 3785–3786. [Google Scholar]; (b) Kolb HC; VanNieuwenhze MS Chem. Rev 1994, 94, 2483–2547. [Google Scholar]; (c) Lee S; Jamieson D; Fuchs PL Org. Lett 2009, 11, 5–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lee JS; Fuchs PL Org. Lett 2002, 4, 317–318. [DOI] [PubMed] [Google Scholar]
- (17).(a) Guo C; Bhandaru S; Fuchs PL; Boyd MR J. Am. Chem. Soc 1996, 118, 10672–10673. [Google Scholar]; (b) Li W; LaCour TG; Fuchs PL J. Am. Chem. Soc 2002, 124, 4548–4549. [DOI] [PubMed] [Google Scholar]
- (18). The zinc powder was activated in HCl (2 N) solution via Ultrasound vibration for 1 h. The powder was filtered and washed with HCl (2 N), water, and acetone, and then dried in a vacuum desiccator for several hours prior to use.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.