

Abstract
Glioblastoma (GBM) is the most aggressive primary brain tumor. In addition to being genetically heterogeneous, GBMs are also immunologically heterogeneous. However, whether the differences in immune microenvironment are driven by genetic driver mutation is unexplored. By leveraging the versatile RCAS/tv-a somatic gene transfer system, we establish a mouse model for Classical GBM by introducing EGFRvIII expression in Nestin-positive neural stem/progenitor cells in adult mice. Along with our previously published Nf1-silenced and PDGFB-overexpressing models, we investigate the immune microenvironments of the three models of human GBM subtypes by unbiased multiplex profiling. We demonstrate that both the quantity and composition of the microenvironmental myeloid cells are dictated by the genetic driver mutations, closely mimicking what was observed in human GBM subtypes. These myeloid cells express high levels of the immune checkpoint protein PD-L1; however, PD-L1 targeted therapies alone or in combination with irradiation are unable to increase the survival time of tumor-bearing mice regardless of the driver mutations, reflecting the outcomes of recent human trials. Together, these results highlight the critical utility of immunocompetent mouse models for preclinical studies of GBM, making these models indispensable tools for understanding the resistance mechanisms of immune checkpoint blockade in GBM and immune cell-targeting drug discovery.
Keywords: EGFRvIII, GEMM of GBM, glioblastoma, microenvironment, PD-L1
1 |. INTRODUCTION
Glioblastoma (GBM) is the most prevalent and deadly primary brain tumor in adults. The tendency of GBM cells to infiltrate the normal brain parenchyma makes complete surgical resection impossible. In addition, these tumors are remarkably resistant to radiotherapy (RT), chemotherapy, and immunotherapy. Therefore, recurrence is inevitable and inescapably fatal. Despite an aggressive multimodal standard therapy, the median survival time of GBM patients is only ~15 months (Stupp et al., 2005). Our current understanding of the complex biology of GBM is largely derived from studies of genetic and molecular changes within cancer cells. The Cancer Genome Atlas (TCGA) initiative has driven characterization of the genome, epigenome, and transcriptome of human GBM (hGBM) and has provided a higher-resolution picture of frequent alterations, revealing different subtypes with distinct expression signatures (Brennan et al., 2013; Cancer Genome Atlas Research Network, 2008; Verhaak et al., 2010). These studies have propelled the development of robust gene expression-based subtypes of GBM named Proneural (PN), Classical (CL), and Mesenchymal (MES) (Wang et al., 2017).
Integration of multi-dimensional genomic data has also established subtype-specific patterns of somatic mutations and DNA copy number alterations. Although not mutually exclusive, aberrations in gene expression of platelet-derived growth factor receptor alpha (PDGFRA), epidermal growth factor receptor (EGFR), and neurofibro-matosis type I (NF1) were predominantly associated with the PN, CL, and MES subtypes, respectively (Herting et al., 2017). One question that emerges is whether orthotopic delivery of these unique oncogenic driver mutations can produce de facto subtypes of murine GBM in immunocompetent mice? Additionally, although the exact cell-of-origin of GBM is still debatable, recent studies demonstrated that different cells-of-origin can influence not only tumor development and phenotype, but also impact response to therapy (Jiang et al., 2017; Sreedharan et al., 2017). It is therefore critical to exclude the influence of cell-of-origin on the development and therapeutic outcomes of preclinical models in order to attribute the observed phenotype to specific driver mutations. To this end, leveraging the versatile RCAS/tv-a system, we created genetically engineered murine models (GEMMs) wherein Nestin-positive cells were exclusively transformed to generate all three GBM subtypes in immunocompetent mice.
RCAS/tv-a is a somatic cell gene transfer system. It allows the introduction of specific driver mutations, such as ligand overexpression or tumor suppressor deletion, in a cell-type specific manner. The replication competent avian sarcoma leukosis virus (RCAS) only infects transgenic cells expressing the avian Tv-a receptor (Dai et al., 2001; Hambardzumyan, Amankulor, Helmy, Becher, & Holland, 2009). In our studies, Tv-a receptor expression is driven under the control of Nestin promoter (Ntv-a) in mice of pure C57BL/6 background. To model human PN GBM with these Ntv-a mice, we delivered RCAS-PDGFB alone, or in combination with RCAS-shRNA-p53 (Hambardzumyan et al., 2009). For human MES GBM, we chose to codeliver RCAS-shRNA-Nf1, RCAS-shRNA-p53, and RCAS-Cre (to delete Pten) (Herting et al., 2017; Ozawa et al., 2014). We found that PDGFB-overexpressing and Nf1-silenced murine tumors induced extensive expression of specific markers distinguishing these two different subtypes (Herting et al., 2017).
In this report, we aimed to model human CL GBM in Ntv-a mice. First, we generated quadruple transgenic mice by crossbreeding four individual mouse lines: Ntv-a (Holland et al., 2000), Cdkn2a−/− (Serrano et al., 1996), EGFRvIII fl-stop-fl (Zhu et al., 2009), and Ptenfl/fl (Suzuki et al., 2001), all of which are on the C57BL/6 background. These mice were injected with a combination of RCAS-Cre (to induce expression of human EGFRvIII while concurrently deleting Pten), RCAS-shRNA-p53, and in some instances, RCAS-TAZ, which was found highly expressed in CL GBM patients when we analyses TCGA data set. TAZ is a transcriptional coactivator that acts downstram of the Hippo pathway. Translocation of TAZ into the nucleus and subsequent binding to TEAD coactivators on DNA induces de novo transcription of genes that control cell proliferation, tissue renewal and regeneration, and tumor initiation (Zheng & Pan, 2019). A recent report indicates that TAZ promotes GBM cell growth through potentiating EGFR signaling (R. Yang et al., 2016). Characterization of tumors developed in these mice demonstrated over 75% of the tumors are GBM (grade IV), while the rest are grade III anaplastic astrocytoma. We demonstrate that EGFRvIII-expressing murine GBM (mGBM) phenocopies human CL GBM in both gene expression profile and histological characteristics.
We have previously shown that genetic driver mutations can create subtype specific tumor microenvironments. For instance, we demonstrated PDGFB-driven gliomas display more disrupted and leaky vessels compared to Nf1-silenced tumors, while the latter exhibit significantly higher presence of tumor-associated macrophages (TAMs; Herting et al., 2017). Further, by investigating the anatomical ontogeny of these TAMs, we established that in the PDGFB-driven GBM microenvironment the vast majority of the TAMs are bone marrow-derived macrophages (BMDMs) of hematopoietic origin and the rest are intrinsic brain microglia (Z. Chen et al., 2017). This is remarkable because recent studies in various neurological diseases have demonstrated functional distinctions between BMDM and microglia with the former usually promoting pathology while the latter protects the CNS from injury and degeneration (Ajami, Bennett, Krieger, McNagny, & Rossi, 2011; London, Cohen, & Schwartz, 2013; Shemer & Jung, 2015; Varvel et al., 2016). In this report, by leveraging the power of the NanoString PanCancer Immune Profiling panel, we further characterized the immune landscape of each subtype of murine GBM. We demonstrate that both the quantity and composition of the microenvironmental immune cells, especially myeloid cells are dictated by the genetic driver mutations, closely mimicking what was observed in human GBM subtypes. These myeloid cells express high levels of the immune checkpoint protein PD-L1, in a subtype-specific manner. However, PD-L1 targeted therapies either alone or in combination with radiation therapy are unable to increase the survival time of tumor-bearing mice regardless of the driver mutations, reflecting the outcomes of recent human trials (Filley, Henriquez, & Dey, 2017; Lakin, Rulach, Nowicki, & Kurian, 2017).
Together, these models offer an attractive system for interrogations of driver mutation-specific phenomena in GBM. More importantly, because these mGBM are orthotopic brain cancers that develop in immunocompetent mice, they provide an invaluable platform for reliable preclinical trials for novel immunotherapies. One immediate application would be using them to investigate the basis for resistance to immune checkpoint inhibitors in GBM.
2 |. MATERIALS AND METHODS
2.1 |. Mice
Ntv-a;Cdkn2a−/−;Ptenfl/fl, Ntv-a;Cdkn2a−/−;EGFRvIIIfl-stop-fl;Ptenfl/fl, and Cdkn2a−/−;EGFRvIIIfl-stop-fl;Ptenfl/fl mice of both genders (equal distribution) in the age range of 8–16 weeks were used for experiments as previously described (Hambardzumyan et al., 2009; Herting et al., 2017; Zhu et al., 2009). The first strain is a mixed genetic background, while the latter two are in a C57BL/6 background. OT-1/Rag2 mice were purchased from Taconic (TAC 002334). C57BL/6J mice of 8 weeks old used for BMDM isolation were purchased from the Jackson labs (000664). Cx3cr1GFP/WT;Ccr2RFP/WT mice were a gift from Richard Ransohoff (Cook et al., 2001; Jung et al., 2000; Saederup et al., 2010). All animals were housed in a climate-controlled, patho-gen-free facility with access to food and water ad libitum under a 12-hour light/dark cycle. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Emory University (Protocol #2003253, #201700633, and #2001765) and Icahn School of Medicine at Mount Sinai (Protocol #201900619).
2.2 |. Virus generation and tumor induction
DF-1 cells (ATCC, CRL-12203) were purchased and grown at 39°C according to supplier instructions. Cells were transfected with RCAS-PDGFB-HA, RCAS-shRNA-p53-RFP, RCAS-shRNA-Nf1, RCAS-shRNA-Pten-RFP, and RCAS-Cre using a Fugene 6 transfection kit (Roche, 11814443001) according to the manufacturer’s instructions. DF-1 cells (4 × 104) in 1 ul neurobasal medium were stereotactically delivered with a Hamilton syringe equipped with a 30-gauge needle for tumor generation. For generation of EGFRvIII tumors, a mixture of DF-1 cells (4 × 104) that were infected with RCAS-Cre, RCAS-shRNA-p53-RFP, and RCAS-shRNA-Pten-RFP were coinjected at a 1:1:1 ratio in a final volume of 1 ul neurobasal medium. Some mice also received RCAS-TAZ coinjection, as detailed in the figure legends. The target coordinates were aiming at the subventricular zone with the coordinates AP-0.0 mm and right-0.5 mm from bregma; depth-1.5 mm from the dural surface (Franklin & Paxinos, 1997). Mice were continually monitored for signs of tumor burden and were sacrificed upon observation of endpoint symptoms including head tilt, lethargy, seizures, and excessive weight loss.
2.3 |. Generation of EGFRvIII model with WT P53 by targeting stem/progenitor cells ex vivo
Tumors were also generated by orthotopic transplantation of EGFRvIII-expressing neurospheres, which were produced in vitro. Subventricular zones (SVZs) were dissected from P0-P2 Cdkn2a−/−; EGFRvIIIfl-stop-fl;Ptenfl/fl mice and dissociated into single cells with Accutase (Biolegend, 423,201). The cells were cultured in serum free neural stem cell media containing 1× B27 supplement (Life Technologies, 17604044), 20 ng/ml b-FGF (ThermoFisher, PHG0261), 5 μg/ml heparin (Stem Cell Technologies, 7980), 25 ng/ml EGF (Lonza, cc-4017FF) and 1 X penicillin/streptomycin (Genesee, 25512) in DMEM: F12 medium (Gibco, 10565018) (Laks et al., 2009). The cells from individual animals were initially plated in 6-well plates to grow neurospheres over 1–2 weeks. 500,000 cells were then plated in 6-well plates and treated with concentrated adenovirus Cre (Vector Biolabs) at 50–100 MOI, and then cultured in 10 cm dishes for another 5–7 days before they were checked by western blot to confirm EGFRvIII expression and loss of PTEN protein expression. They were passaged for less than 8 passages before being injected into 6-week old C57BL/6 mice to form tumors. About 200,000 cells in 5 μl 1× DPBS were injected per animal using a 23-gauge Hamilton syringe at a position 2.5 mm to the right and 1 mm anterior to the bregma at a depth of 2.5–3 mm.
2.4 |. BMDM isolation and polarization
BMDM were isolated from 4 to 12-week-old C57BL/6 mice using a modified established protocol (Weischenfeldt & Porse, 2008). Mice were euthanized via CO2 asphyxiation. The whole, intact femur and tibia were stripped of muscle and collected in sterile Dulbecco’s phosphate-buffered saline (DPBS). Both ends of the bone were cut and the marrow was flushed into a clean petri dish using DPBS supplemented with 4% bovine serum albumin (BSA) (ThermoFisher, 15260037), heparin (STEMCELL, #07980), DNase I (Sigma-Aldrich, 11284932001), and penicillin/streptomycin (Fisher, SV30010). The resulting mixture was briefly triturated and passed through a 70 μm cell strainer. The cells were plated in a 15 cm noncell culture-treated plate for a 6-day differentiation in 15 ml of DMEM (ThermoFisher, 10569010) with 10% fetal bovine serum (FBS; HyClone, SH30396.03) and 40 ng/ml macrophage colony-stimulating factor (M-CSF) (Peprotech, 315–02). An additional 15 ml of media with M-CSF was added after day three. The cells were harvested for experimentation via 10-minute incubation in DPBS with 5 mM EDTA on ice. Cells were plated in 6-well plates in 2 ml of culturing media with M-CSF at a concentration of 20 ng/ml for experimentation. For polarization, a three-hour starvation in fetal bovine serum-deficient media was performed prior to stimulation with 100 ng/ml lipopolysaccharide (LPS) (Sigma Aldrich, L5293) and 40 ng/ml interferon gamma (IFNγ) (Peprotech, 315–05) for M1 polarization or 40 ng/ml IL-4 (Peprotech, 214–14) and 40 ng/ml IL-10 (Peprotech, 210–10) for M2 polarization. Six-hour incubations with the indicated stimulants were performed.
2.5 |. Tumor and cultured cell RNA isolation and analysis
At endpoint, mice were sacrificed with an overdose of ketamine/xylazine and immediately perfused with ice cold Ringer’s solution (Sigma-Aldrich, 96724–100TAB). The brain was extracted, and a piece of tumor was immediately snap-frozen in liquid nitrogen for storage at −80°C. Alternatively, cultured cells were harvested from plates using TRIzol (ThermoFisher, 15596026). RNA was isolated from the frozen tumor pieces or cells with the RNeasy Lipid Tissue Mini Kit (Qiagen, 74804) according to manufacturer’s instructions. RNA quantity was assessed with a NanoDrop 2000 spectrometer, while quality was confirmed via electrophoresis of samples in a 1% bleach gel as previously described (Aranda, LaJoie, & Jorcyk, 2012). RNA was used to generate cDNA with a First Strand Superscript III cDNA synthesis kit (ThermoFisher, 18080051) according to manufacturer’s instructions and with equal amounts of starting RNA. Quantitative-PCR was performed with the validated BioRad PCR primers using SsoAdvanced Universal green Supermix (BioRad, 1725271). Fold-changes in gene expression were determined relative to the M0-polarized group or PDGFB-overexpressing tumors using the 2−ΔΔCt method with β-Actin as a house-keeping gene. The NanoString PanCancer Immune Profiling panel (NanoString) was also utilized to analyze RNA expression in tumor pieces. RNA quality was validated with a BioAnalyzer before assessment with this technique. Gene expression results from NanoString were converted to log2 z-scores prior to analysis.
2.6 |. T cell proliferation assay
Adult OT-1/Rag2 mice were euthanized by asphyxiation. Their spleens were immediately removed and pressed through a 70 μm cell strainer to obtain single cells. The cells were labeled with CellTrace™ CFSE (ThermoFisher, C34554) following manufacturer’s instructions, before they were plated at 50,000 cells per well in a flat-bottom 96-well tissue culture plate. Polarized macrophages or nonpolarized M0 controls were plated thereafter at E:T ratios of 1:1, ½: 1 or ¼: 1. The cells were cultured in complete DMEM (10% FBS, 1× penicillin/streptomycin (Gibco, 15,140,122), 1× l-glutamine (Gibco, 35050061), 1× nonessential amino acid (Gibco, 11,140,050), and DMEM (Gibco 12430054) in the presence of 100 nM of SINNFEKL peptide (Ana-Spec, AS-60193–1) for 4 days at 37°C with a constant supply of 5% CO2. Finally, cells were stained with CD8-PE-Cy5 (BD Pharmingen, 553,034) and examined by flow cytometry.
2.7 |. Tissue samples and pathological appraisal
Human formalin-fixed, paraffin-embedded (FFPE) GBM samples, post-mortem brain specimens, and de-identified clinical information were provided by Emory University. Board-certified neuropathologists graded and diagnosed both the human tumor tissues and murine EGFRvIII samples according to the 2016 World Health Organization Classification of Tumors of the Central Nervous System (Louis et al., 2016). Gene expression profiling to determine the transcriptional subtypes was performed using NanoString nCounter Technology using custom-made probes for 152 genes from the original GBM_2 design (Kaffes et al., 2019).
2.8 |. TCGA analysis
U133 Microarray data for the GBM (TCGA, provisional) dataset were downloaded from cBioPortal (https://www.cbioportal.org) in August, 2019 and sorted into subtypes based on a proprietary key. G-CIMP-positive tumors were excluded from analysis. Data for the same genes analyzed with quantitative-PCR in murine tumors were downloaded. cBioPortal was also used to access GBM patient sample mutational and copy number alteration (CNA) information of 273 GBM samples (Study: GBM multiforme, TCGA Firehose Legacy), data was acquired in April, 2020 (Cerami et al., 2012; Gao et al., 2013).
2.9 |. Clustering analysis
A distance matrix was calculated, and hierarchical clustering was performed on the murine quantitative-PCR data using the factoextra package in R. Principal component analyses on murine tumor and human tumor expression data were also performed using this package. A gene expression heatmap for both sets of data was generated using the heatmap package in R. Clustering of NanoString data was performed using the same packages as well as the nSolver Advanced Analysis plugin.
2.10 |. Tissue processing and immunohistochemistry
Archived FFPE human GBM samples and de-identified clinical information were provided by Emory University. The specimens were sectioned at 5 μm thickness, slide-mounted, and stored at −80°C until use. To process mouse tumor tissues, animals were anesthetized with an overdose of ketamine/xylazine mix and transcardially perfused with ice-cold Ringer’s solution. Brains were removed and processed according to the different applications. For H&E tumor validation and immunohistochemistry staining, brains were fixed in 10% neutral buffered formalin for 72 hr at room temperature (RT), processed in a tissue processor (Leica TP1050), embedded in paraffin, sectioned (5 μm), and slide mounted.
All immunohistochemistry staining was performed on the DISCOVERY XT platform (Ventana Medical Systems). Primary antibodies used in this study include: anti-human EGFR, (1:50, Cell Signaling Technology, D38B1), anti-IBA-1 (1:500, Wako,019–19741), anti-GFAP (1:1,000, Cell Signaling Technology, 3670S), anti-CD31 (1:250, Dianova, DIA-310), anti-phosphorylated Histone 3 (1:400, Millipore, 06–570), anti-CD44 (1:1,000, BD Pharmingen, 550,538), anti-Doub-lecortin (1:400, Santa Cruz, sc-8066), and anti-OLIG2 (1:500, Millipore, AB9610). Digital images of the slides were acquired by using a Nanozoomer 2.0HT whole-slide scanner (Hamamatsu Photonic K.K) and observed offline with NDP view2 software (Hamamatsu). Image analysis was performed using Fiji.
2.11 |. Tumor dissociation and primary cell culturing
Tumor dissociation was performed as previously described. Briefly, tumors were dissected from the brain, minced into pieces <1 mm3, and digested with an enzymatic mixture that includes papain (0.94 mg/ml, Worthington, LS003120), EDTA (0.18 mg/ml, Sigma, E6758), cystine (0.18 mg/ml, Sigma, A8199), and DNase (60 μg/ml, Roche, 11284932001) in 2 ml HBSS (Gibco, 14,175–095). Tumor tissues were kept at 37°C for 30 min with occasional agitation. The digestion was terminated with the addition of 2 ml Ovomucoid (0.7 mg/ml, Worthington, LS003086). Following digestion, single cells were pelleted, resuspended in HBSS, and centrifuged at low speed (84 RCF) for 5 min, before passing through a 70 μm cell strainer.
For tumorsphere cultures, cells were seeded at 5 × 105 cells/ml and grown in Neurocult mouse neurobasal medium (Stem Cell Technologies, 5700) supplemented with 10 ng/ml hEGF (Lonza, cc-4017FF), 20 ng/ml basic-hFGF (ThermoFisher, PHG0261), 1 mg/ml Heparin (Stem Cell Technologies, 7980), and NSC Proliferation Supplements (Stem Cell Technologies, 5701). Fresh medium was added to the cultures every 48 hr. For primary monolayer cultures, neurospheres were dissociated with Accutase (Sigma-Aldrich, A6964) to generate single cells, which were then grown as adherent monolayers on coverslips (Φ = 12 mm) coated with Geltrex (Life Technologies, A14132–01) prepared according to manufacturer’s instructions.
2.12 |. Immunocytofluorescence
Cells grown in monolayer were fixed with 4% paraformaldehyde (PFA, Electron Microscopy Sciences, 15,713–5) for 20 min at RT. They were then washed with PBS, permeabilized with 0.1% Triton, and blocked with 10% normal goat serum and 10% normal donkey serum (Jackson ImmunoResearch). The cells were then incubated in primary antibodies for 2 h at RT. The following antibodies were used at the stated dilutions: rabbit anti-OLIG2 (1:200, Millipore, AB9610); mouse anti-hEGFR (1:100, BioLegend, 352902), and rat anti-CD44 (1:100, BD Pharmingen, 550538). Secondary antibodies conjugated to Alexa-Fluor dyes (488, 555, and 647 nm from Invitrogen) at a dilution of 1:500 in PBS/2% BSA were applied. For nuclear counterstaining, DAPI was used (Sigma). Fluorescent and Differential Interference Contrast (DIC) images were taken on an Olympus FV1000 confocal microscope.
2.13 |. Flow Cytometry
Initial steps of the enzymatic dissociation of the tumors are the same as described above, except 0.5% collagenase D (Sigma, 11088858001) and DNase (Roche, 11284932001) were used in place of papain. Single-cell suspensions were passed through 70 μm cell strainers, centrifuged, and resuspended in 30% Percoll (GE Healthcare, 17-0891-01) solution containing 10% FBS (Hyclone SH30396.03). Cells were separated by centrifugation at 800 g for 15 min at 4°C. The supernatant was carefully removed to get rid of debris and lipids. The cells were then washed in cold PBS and resuspended in RBC lysis buffer (BioLegend, 420301) for 1 min at 37°C. Cells were transferred to an Eppendorf tube and washed once with FACS buffer (DPBS with 0.5% BSA) and blocked with 100 μL of 2× blocking solution (2% FBS, 5% normal rat serum, 5% normal mouse serum, 5% normal rabbit serum, 10 μg/ml anti-FcR [BioLegend, 101319] and 0.2% NaN3 in DPBS) on ice for 30 min. Cells were then stained on ice for 30 minutes and washed with FACS buffer. Antibodies used in this study include: CD45-APC, CD11b-PerCP-Cy5.5, Ly6C-PE-Cy7, Ly6G-V450, PD-L1-BV605, CD3-APC, CD4-APC-Cy7, CD8-PE-Cy5, PD-1-BV605, and rat IgG2b isotype control-BV605 (BioLegend). CX3CR1-GFP and CCR2-RFP were analyzed with detectors B530/30 and YG610/20, respectively. All data were collected on a BD LSR II flow cytometer and analyzed using the FlowJo 10 software (Tree Star Inc.).
2.14 |. Cell cycle analysis
Cell cycle analysis was performed using a Click-iT EdU proliferation assay kit (ThermoFisher, C10636), following manufacturer’s instructions. Briefly, primary murine neurospheres with fewer than 8 passages were dissociated with Accutase to generate single cell suspensions. 1 × 105 cells were seeded in a well of a 12-well tissue culture plate and incubated overnight in the neurosphere medium. At the end of the 23rd hr, the medium was completely replaced with a fresh medium containing 10 μM Edu reagent. Cells were immediately returned to the incubator and grown for one more hour. The cells were lifted with Accutase, fixed with 4% PFA, and permeabilized with saponin, before the EdU was conjugated to a fluorophore with catalysis of a copper compound via a “click chemistry” reaction. The cells were then incubated in a PI staining buffer (200 μg/ml, Sigma, P4864) for 30 min at RT and analyzed on a BD LSR II flow cytometer.
2.15 |. Antibody administration and irradiation of Tumor-Bearing mice
Once tumors developed, mice were administered either a PD-L1 neutralizing antibody (Bio X Cell, BE0101) or its isotype control, rat IgG2b (Bio X Cell, BE0090), via intraperitoneal injection at a dose of 10 mg/kg on the indicated days. X-ray irradiation was administered to the head with the body covered by a lead shield at the indicated doses as previously described (Herting et al., 2017).
2.16 |. Statistical analyses
Graphs were created using GraphPad Prism 8 (GraphPad Software Inc.) or R. Variables from two experimental groups were analyzed using unpaired or paired parametric two-tailed t-tests as appropriate, assuming equal standard deviations. One-way ANOVA was used to compare variables from more than two groups. Kaplan–Meier survival analysis was performed using the log-rank (Mantel-Cox) test and Gehan-Breslow-Wilcoxon test. The details are included in Figure legends. (*) p < .05; (**) p < .01; (***) p < .001; (****) p < .0001; (ns) not significant.
3 |. RESULTS
3.1 |. Generating adult EGFRvIII-driven tumors from nestin-positive cells using the RCAS/tv-a system
The RCAS/tv-a system has been a valuable tool for studying biology and therapeutic responses of adult PDGFB-overexpressing and NF1-silenced GBM (Hambardzumyan et al., 2009; Herting et al., 2017). To model CL human GBM, we crossbred commercially-available mouse lines to generate Ntv-a;Cdkn2a−/−;EGFRvIII fl-stop-fl;Ptenfl/fl quadruple transgenic mice (Figure 1a). It has been previously documented that EGFRvIII expression alone was not sufficient to generate gliomas when adenovirus-based Cre delivery was used (Zhu et al., 2009). To evaluate the ability of EGFRvIII in inducing adult de novo high-grade gliomas when targeting Nestin-positive cells in the subventricular zone (SVZ), we injected a combination of RCAS-shRNA-p53 and RCAS-Cre into the brain of adult Ntv-a;Cdkn2a−/−;EGFRvIIIfl-stop-fl;Ptenfl/fl mice and monitored tumor formation and survival. The tumor penetrance was nearly 100% with a median survival time of 127 days, indicating the essential requirement of removing these tumor-suppressor genes for tumorigenesis (Figure 1b). Additionally, it was recently demonstrated that TAZ, the cotransactivator of the Hippo pathway, can promote GBM growth by activating the EGFR signaling (R. Yang et al., 2016). Interestingly, when we queried TAZ expression in TCGA database, we found that TAZ expression levels are the highest in CL hGBM among all the three subtypes (Figure 1c). Together, these data prompted us to include the overexpression of TAZ in our RCAS plasmid mix (Figure S1). Remarkably, overexpression of TAZ significantly reduced median survival time (70 vs. 127 days) of the mice compared to those without TAZ (Figure 1b).
FIGURE 1.
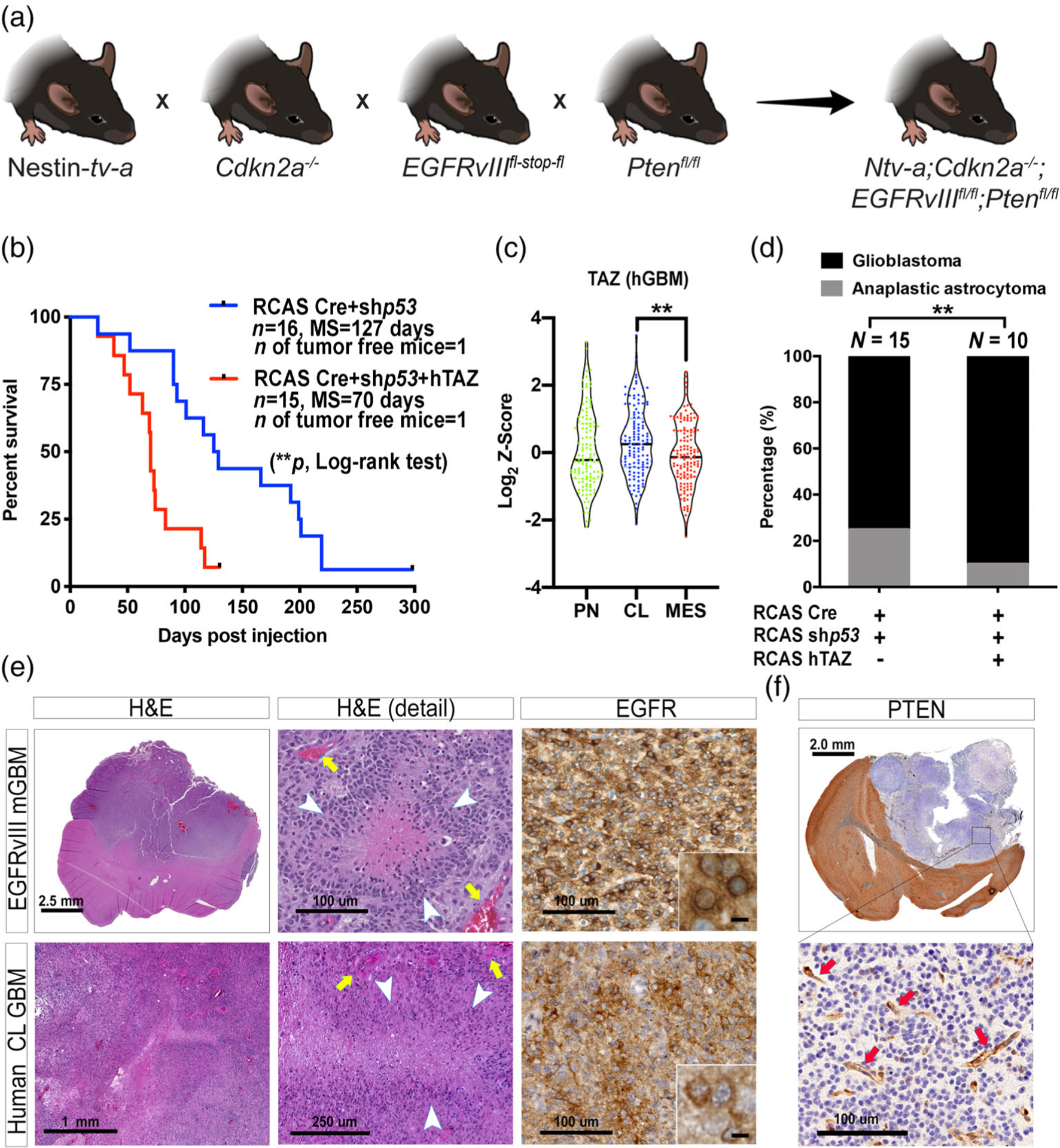
Establishing and characterizing a murine model of CL hGBM. (a) Schematic illustration of the generation of the quadruple transgenic mouse. (b) Survival curves of murine EGFRvIII-expressing tumors with or without hTAZ expression. **p < .01 by log-rank test. (c) TAZ expression in subtypes of human GBM. **p < .01 by one-way ANOVA with Tuckey’s posthoc comparison. (d) Grading of EGFRvIII-expressing tumors with or without hTAZ. **p < .01 by Fisher’s exact test. (e) Hematoxylin and eosin (H&E) staining of CL hGBM and EGFRvIII mGBM; and immunohistochemical analysis of human EGFR in both species. (f) Immunohistochemical analysis of PTEN in EGFRvIII mGBM. Arrows indicate PTEN-positive stromal cells. Scale bars are as indicated, except the ones in inset, which represent 12.5 μm
Histological grading of these EGFRvIII gliomas per the WHO 2016 standard demonstrated that these tumors are a mixture of grade III anaplastic astrocytoma and grade IV GBM, with the majority falling into the latter category (Figure 1d). The proportion of grade IV tumors is further increased by TAZ-overexpression (Figure 1d, p < .01 by Fisher’s test). Similar to CL hGBM, EGFRvIII-expressing mGBM exhibited pseudopalisading necrosis and microvascular proliferation (Figure 1e). IHC analysis for human EGFR showed intense membrane staining in both EGFRvIII mGBM and CL hGBM (Figure 1e). The aggressiveness of this tumor is recapitulated in the murine models and demonstrated by the interdigit structures in the invasive leading edge of the tumor (Figure S2).
To confirm the complete deletion of Pten in these tumors, we stained the tissues to assess the presence of PTEN. Similar to a uniformed distribution of EGFR expression in all tumor cells (Figure 1e), PTEN deletion was also achieved in all of the tumor cells (Figure 1f). PTEN immunopositivity was only visible in the non-neoplastic cells of tumor microenvironment (Figure 1f).
We next evaluated whether EGFRvIII mGBM and CL hGBM exhibit similarities regarding their histological features. Staining for CD44, OLIG2, and DCX displayed similar patterns of expression for CL hGBM and EGFRvIII mGBM (Figure S3). Although, prior published literature indicated that overexpression of the EGFR and mutations of the TP53 tumor suppressor gene are mutually exclusive (Watanabe et al., 1996), later study demonstrated concurrent alterations of EGFR and TP53 were detected and were associated with worse outcome in primary GBM patients (Brennan et al., 2013; Ruano et al., 2009). To better understand the significance of TP53 mutation in GBM cases with altered EGFR, we next evaluated the presence of concomitant mutations of EGFR and TP53 by mining in the TCGA Firehose Legacy dataset that contained information on mutation and CNA of 273 GBM samples. The results demonstrated that nearly 18% of the cases with EGFR mutations (including samples with EGFR mutation and samples that harbor both EGFR mutation and amplification) also harbor TP53 mutation (Figure S4 and Table S1). Since we used the most common EGFR mutation to drive gliomagenesis in our murine model, we next selected patient samples that only had EGFR mutation and excluded samples with EGFR amplification, and further analyzed the samples for TP53 status. Intriguingly, nearly 40% of the samples with EGFR mutation also harbored TP53 mutation (Figure S4c). These data show that EGFR mutations and TP53 mutations coexist in 40% cases of EGFR mutated patients. We modeled this in the current manuscript by using the most common EGFR mutation in GBM, EGFRvIII, in combination with silencing p53.
To assess whether p53 status has an impact on the EGFRvIII-driven tumor microenvironment, we next transformed the neural stem/progenitor cells isolated from Cdkn2a−/−;EGFRvIIIfl-stop-fl;Ptenfl/fl mice with adenoviral-Cre. Resultant cells were then transplanted into recipient C57BL/6 mice. These EGFRvIII-driven p53 WT tumors were compared to tumors generated with the RCAS/tv-a system that were driven with EGFRvIII but harbored silencing of p53. There was no significant difference observed when various stromal and tumor cell-specific protein expressions (pH 3, GFAP, CD31, and IBA1) were evaluated by IHC (Figure S5). These data suggest that the silencing of p53 does not create substantial differences in the EGFRvIII-driven tumor microenvironment. Overall these results indicate that EGFRvIII expression in combination with loss of Pten, silencing of p53, and together with TAZ-overexpression in Nestin-positive cells in the SVZ of adult mice can create tumors with similar neoplastic and non-neoplastic cell compositions to what is observed in CL hGBM; the subtype known to be enriched for EGFRvIII mutation.
3.2 |. Expression profile of murine PDGFB-overexpressing, NF1-silenced, and EGFRvIII-expressing GBMs clusters similarly to their corresponding human subtypes
Previously, we demonstrated the ability to differentiate between PDGFB-overexpressing and Nf1-silenced murine gliomas with an 18-gene signature (Herting et al., 2017). Furthermore, we showed that the expression of these genes was similar when comparing our murine models to their associated human counterparts. To perform a similar analysis but also to include the EGFRvIII murine tumors, we designed a panel of 28 genes. This panel included the aforementioned 18 genes, as well as an additional 10 genes that were suggested to be differentially expressed in EGFRvIII tumors (Nagane et al., 1996; Verhaak et al., 2010; I. Yang, Han, Kaur, Crane, & Parsa, 2010). RNA expression of these genes in murine tumors was analyzed with qPCR (Figure S6). Human expression data of the same genes were obtained from the TCGA U133 microarray database (Figure S7). For murine tumors, a distance matrix based on the gene expression profile was generated (Figure 2a), which showed that tumors with the same driver mutations were more similar to each other compared to tumors generated with different driver mutations. Visualization with hierarchical clustering (Figure S8a) or two-dimensional PCA analysis (Figure S8b) showed that PDGFB-overexpressing, Nf1-silenced, and EGFRvIII tumors clustered based on this 28-gene panel. Furthermore, when this 28-gene panel was analyzed in human samples, they clustered based on subtypes as well albeit with more overlap (Figure S8c). Direct comparison of the expression levels of the genes included in this panel between murine and human tumors by three-dimensional PCA analysis demonstrates striking similarities between the murine models and their associated human counterparts (Figure 2b).
FIGURE 2.
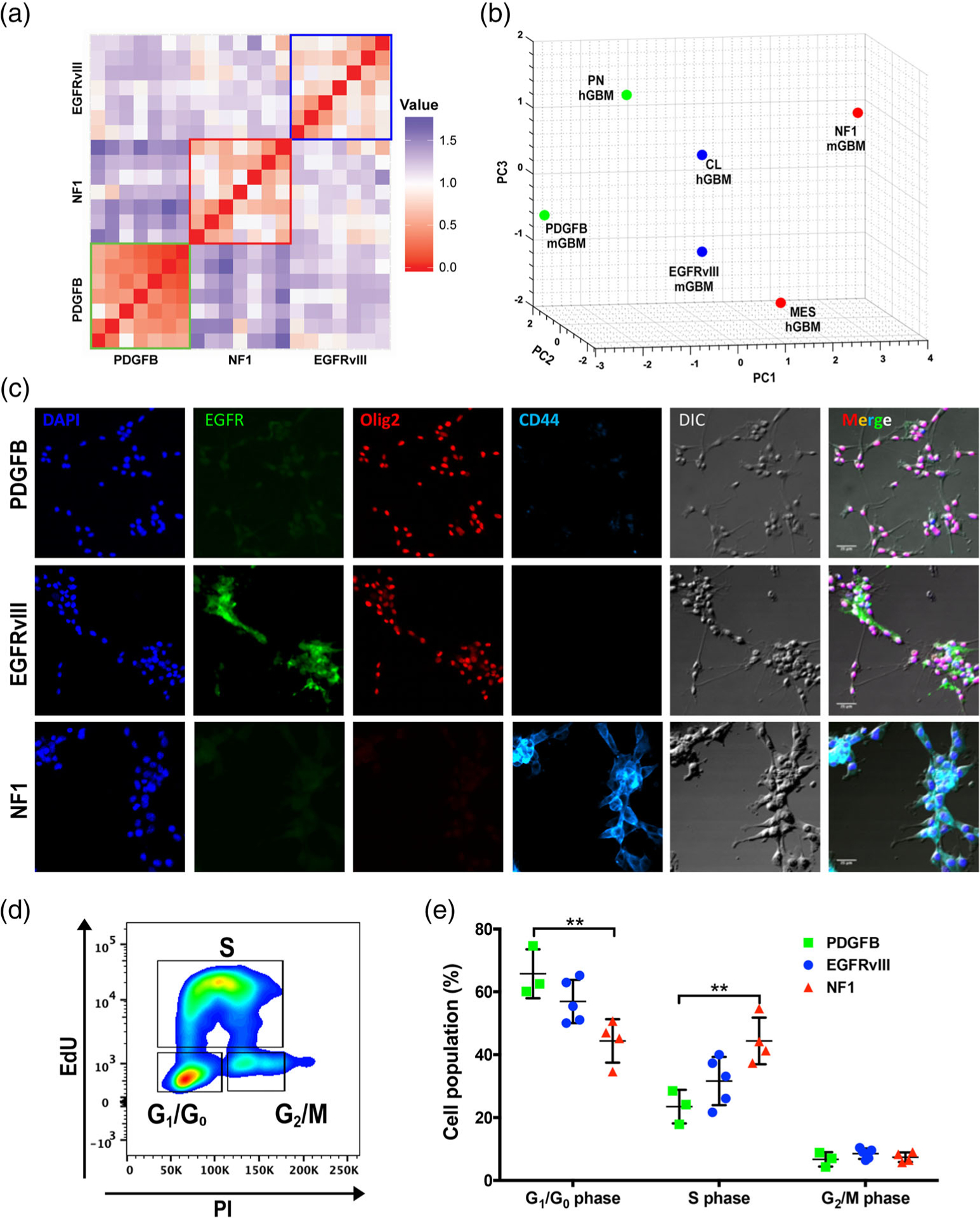
Murine GBM models closely resemble their human counterparts. (a) A distance matrix of murine tumor expression data (N = 7 per group). (b) PCA clustering of murine (N = 7 per group) and human tumor expression data (PN: N = 87, MES: N = 137, and CL: N = 128). (c) Primary cells derived from the three subtypes of murine GBM generated with the RCAS/tv-a system were cultured in vitro and stained for the nuclear marker DAPI as well as the cellular markers EGFR, OLIG2, and CD44. Differential interference contrast microscopy (DIC) was utilized to visualize the morphology of the cells. Scale bars = 25 μm. (d) Flow cytometric plots showing cell-cycle stages as examined by EdU assay. (e) Quantification of the phases of cell cycle. **p < .01 by one-way ANOVA with Tukey’s posthoc test
3.3 |. Genetic driver mutations determine tumor cell expression patterns for various markers, which are preserved in culture
We next assessed whether freshly dissociated tumors can maintain their expression of unique or shared IHC markers when cultured in vitro in serum-free conditions. We derived primary cells from murine tumors of all three subtypes and stained them for CD44, OLIG2, and GFAP. Human EGFR staining was used to ensure cultured cells were neoplastic and expressed their oncogenic driver. Similar to what was observed in vivo, PDGFB-overexpressing and EGFRvIII-expressing glioma cells expressed OLIG2, in contrast to Nf1-silenced tumors (Figure 2c). CD44 staining demonstrated strong positivity in cells derived from Nf1-silenced tumors, less in EGFRvIII-expressing cells, and no expression in PDGFB-overexpressing glioma cells (Figure 2c). We previously demonstrated that significant differences in survival times between PDGFB-driven tumors and Nf1-silenced tumors were attributable to differences in tumor initiation, but not tumor growth kinetics, as measured by longitudinal MRI of the tumors (Herting et al., 2017). We therefore asked whether the proliferation ability of these primary cells differ. An EdU incorporation assay demonstrated a significant difference only exists between PDGFB-overexpressing and Nf1-silenced primary tumor cells (Figure 2d,e). This slower growth rate in PDGFB-overexpressing primary cells could possibly be due to PDGFB-driven gliomas depending more on growth-promoting signals emanating from the tumor microenvironment compared to Nf1-silenced tumors, because depriving this supportive environment in vitro hinders their growth. Together, these data demonstrate primary cells derived from fresh tumors maintain their gene signatures in vitro, and can be used as tools for extensive mechanistic studies.
3.4 |. Driver mutations create unique immune microenvironments
We next asked how these driver genes may affect the immune landscape of each individual GBM subtypes. We chose to use an unbiased approach to examine the immune profile in each of our murine models, covering both the innate and adaptive immune systems. We performed multiplex gene expression profiling using the NanoString PanCancer Immune Profiling panel. This panel included 770 genes associated with 24 different immune cell types. t-Distributed Stochastic Neighbor Embedding (t-SNE) analysis of these samples clearly distinguished the three subtypes with three distinct clusters (Figure 3a). When we collapsed the individual genes into immune function pathways, the heatmap demonstrated that PDGFB tumors cluster tightly together and exhibited a unique signature compared to the other two subtypes, while there is an overlap between Nf1-silenced and EGFRvIII-expressing tumors (Figure 3b). As expected, the activation of all immune cell-related pathways was lowest in PDGFB-overexpressing tumors. When we examined all the 770 genes with an unsupervised hierarchical clustering analysis, we found that the subtypes are tightly grouped together, with each subtype demonstrating uniquely enhanced expression of a collection of genes (Figure 3c). We extracted the genes from each of the three clusters (boxes in Figure 3c), and fed them to the Enrichr program (E. Y. Chen et al., 2013; Kuleshov et al., 2016) for gene ontology analyses. We found that abnormal macrophage physiology is present in both the PDGFB and Nf1 subtypes, whilst abnormal blood vessel morphology uniquely characterized the PDGFB subtype (Figure 3b). Genes that regulate cytokine secretion are enriched in the EGFR subtype, such as those involved in the release of IFNγ and TNFα (Figure 3d).
FIGURE 3.
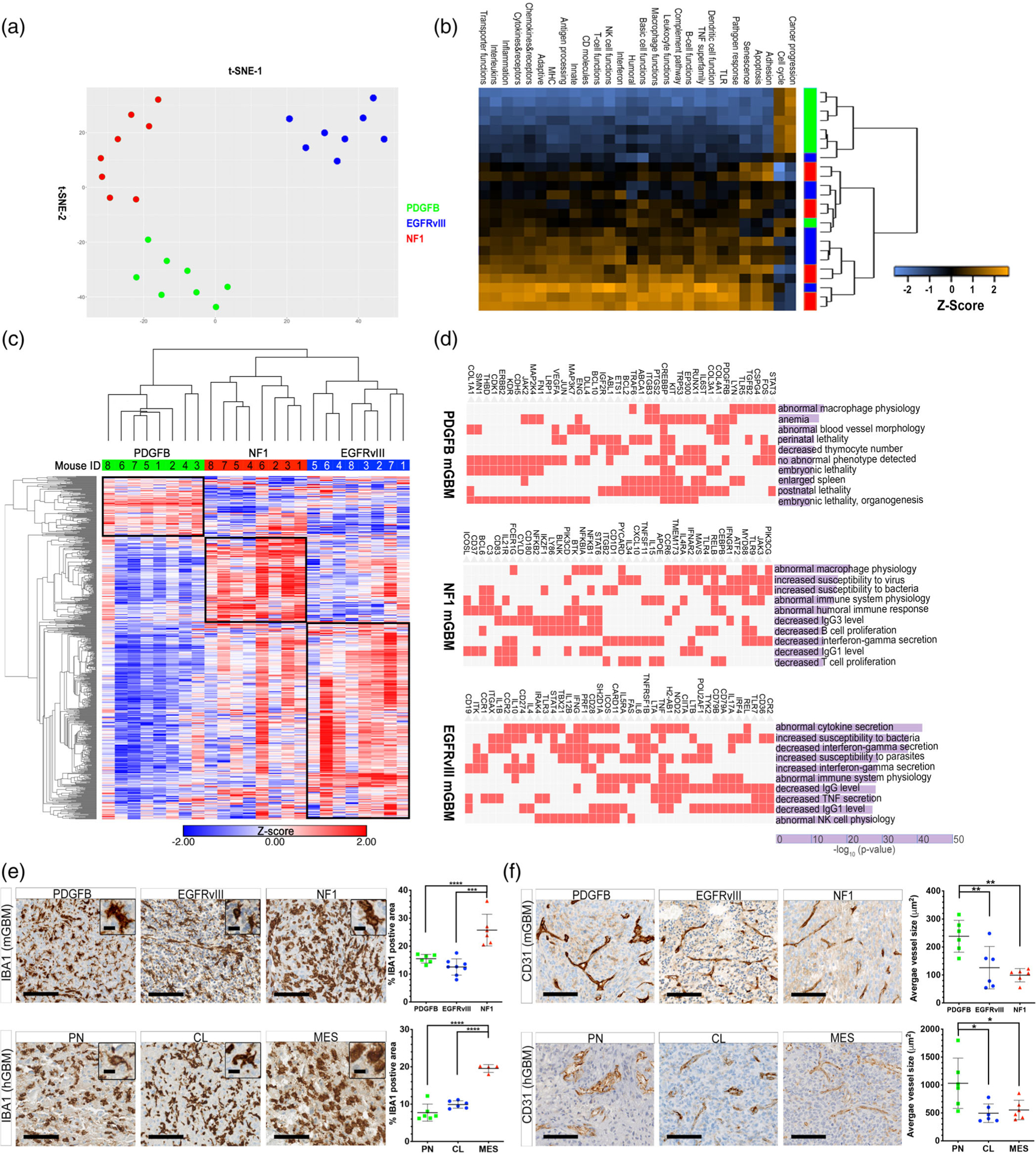
Driver mutation creates unique immune microenvironments in different subtypes of mGBM. (a) t-SNE plot of the NanoString PanCancer Immune Profiling data. (b) Immune pathway function in PDGFB-overexpressing, Nf1-silenced, and EGFRvIII-expressing tumors. (c) Unsupervised hierarchical analysis of NanoString PanCancer Immune Profiling data. (d) Gene Ontology analysis of differentially enriched genes in the three mGBM subtypes. (e) IHC micrographs of IBA1 immunostaining in murine and human GBM subtypes. (f) IHC micrographs of CD31 immunostaining in murine and human GBM subtypes *p < .05, **p < .01, ***p < .001, ****p < .0001, by one-way ANOVA with Tukey’s posthoc test
To further validate these findings, we performed IHC analyses for IBA1, a pan-macrophage marker, and CD31, an endothelial marker that visualizes blood vessels. In agreement with the NanoString results, we found that Nf1 tumors had the highest level of IBA1 immunopositivity, while that of the PDGFB and EGFRvIII tumors are comparable (Figure 3e). This same observation held true for their corresponding human GBM subtypes (Figure 3e). On the contrary, the average size of blood vessels was the highest in PDGFB-overexpressing mGBM and the PN subtype of hGBM, and the vasculature appeared to be deformed (Figure 3f). Together, these results demonstrate that genetic driver mutations create unique immune profiles and microenvironment compositions in these three subtypes of GBM.
3.5 |. Driver mutations define myeloid cell composition in GBM
Intrigued by the NanoString results and the IHC investigation of IBA1 staining, we wanted to further examine the myeloid cells in these three subtypes by flow cytometry. BMDM (CD11b+ CD45HiLy6cintLy6gnegCCR2+CX3CR1+), microglia (CD11b+CD45Lo Ly6cnegLy6gnegCCR2negCX3CR1+), and neutrophils (CD11b+CD45+ Ly6c+Ly6g+ CCR2negCX3CR1neg) can be clearly distinguished by the combination of their surface markers, and further confirmed by their chemokine receptor expressions (Figure 4a). We found an increased presence of total myeloid cells in Nf1-silenced murine tumors when compared with the other two subtypes (Figure 4b). Interestingly, in depth analysis of myeloid cell composition in the three subtypes showed marked differences (Figure 4c). While the majority of myeloid cells in PDGFB-overexpressing and EGFRvIII-expressing tumors were monocyte-derived, in Nf1-silenced murine tumors the majority of the myeloid cells were brain-resident microglia (Figure 4c). A similar trend was previously documented in human samples, albeit with limited sample size (Gabrusiewicz et al., 2016). In addition, Nf1-silenced tumors demonstrated increased neutrophil infiltration compared to the other two subtypes (Figure 4c). In summary, our data demonstrate that genetic-driver mutations can dictate not only the number of infiltrating myeloid cells, but also their ontological compositions (Figure 4d).
FIGURE 4.
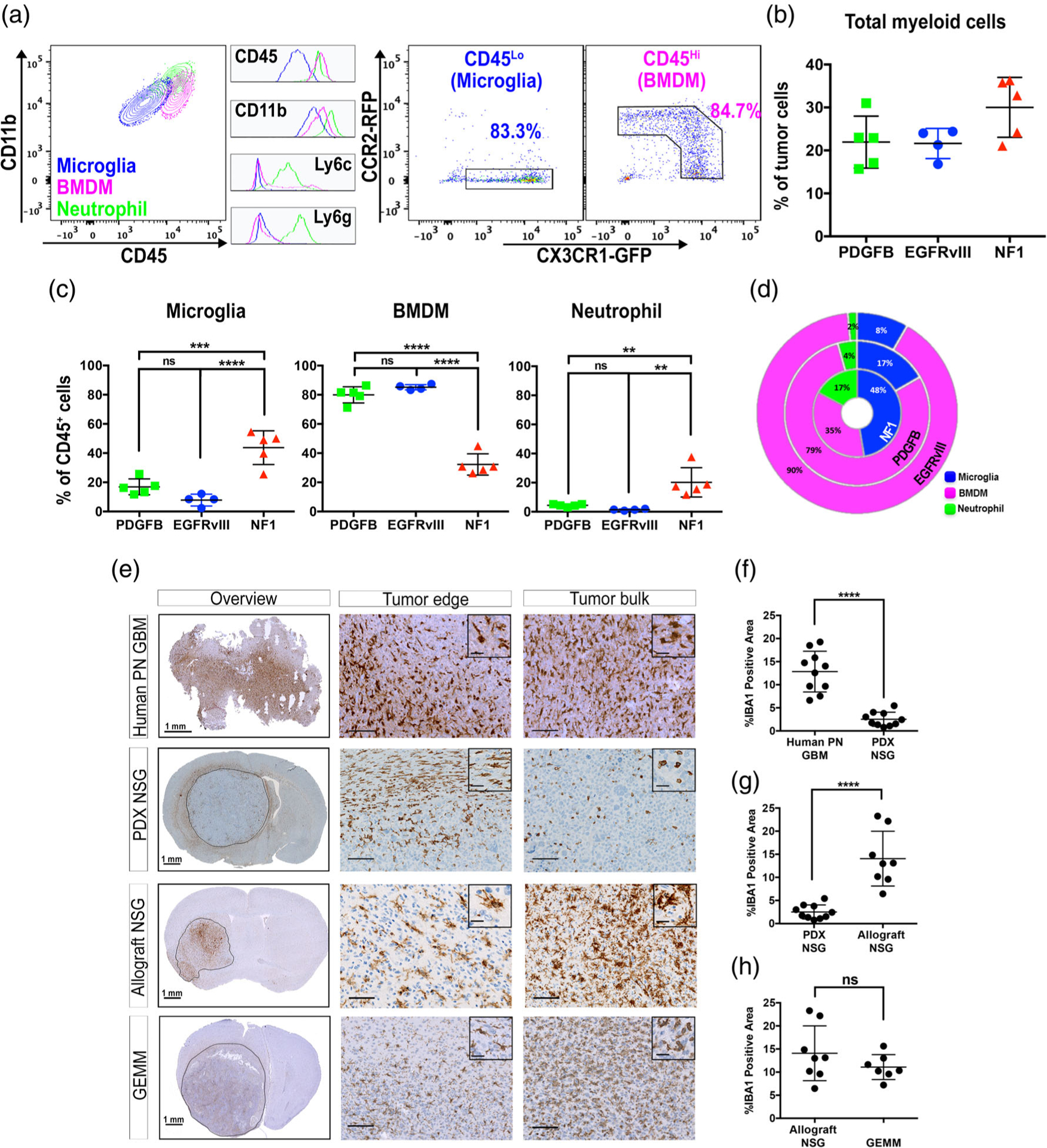
Tumor-associated myeloid cell populations differ significantly between GBM subtypes. Mice were euthanized when their tumor burden reached terminal stage, and their tumors were analyzed by flow cytometry or immunohistochemistry. (a) Representative flow cytometry plots of myeloid cells in mGBM. Histograms of the key surface markers of each population are shown in the middle. (b) Percent of total myeloid cell present in each tumor subtype. (c) Percent of each type myeloid cell present in each tumor subtype. (d) Donut plot demonstrating the differential composition of TAMs in three subtypes of mGBM. (e) Immunohistochemical analyses of TAM infiltration in NSG mice. The tissue sections were stained with IBA1. (f–h) Quantification of IBA1 staining. **p < .01, ***p < .001, ****p < .0001, (ns) not significant, by one-way ANOVA with Tukey’s posthoc test (c) or unpaired t-test (f–h)
3.6 |. PDX models exhibit impaired myeloid cell infiltration
Immunocompromised mice are routinely used for glioma studies, especially when employing patient-derived xenografts (PDX). Recently, there has been increasing use of these models to study TAMs, especially with NOD scid gamma (NSG) mice. These mice carry two mutations on the NOD/ShiLtJ genetic background: severe combined immune deficiency (scid) and a complete null allele of the IL2 receptor common gamma chain (IL2rgnull). The scid mutation occurs in the DNA repair complex protein Prkdc, which renders the mice B- and T-cell deficient. The IL2rgnull mutation prevents cytokine signaling through multiple receptors, resulting in a lack of functional NK cells (Coughlan et al., 2016; Shultz et al., 2005). To determine whether the immune deficiency of PDX models impacts the infiltrative capacity of TAMs, we created PDX in NSG mice using tumor cells derived from patients diagnosed with PN GBM. When we examined TAM infiltration by IHC staining of IBA1, to our surprise, the abundance of TAMs was markedly reduced in PDX compared to human PN GBM (Figure 4e,f). This reduction could be a result of either impaired TAM migration due to immunodeficiency, or incompatibility between chemokines (produced by human tumor cells) and their receptors (present on murine TAMs), or both. To answer this question, we orthotopically transplanted freshly dissociated tumor cells from PDGFB-overexpressing, Nf1-silenced, or EGFRvIII-expressing murine tumors into NSG mice to create allograft models. We found strong TAM infiltration in these mice, as exemplified by IHC staining of IBA1 in a PDGFB-overexpressing allograft NSG mouse (Figure 4e,g), showing no difference from that of the GEMM (Figure 4e,h). To further determine whether the reduction in TAM infiltration in PDX was due to decreased numbers of microglia or BMDM, we evaluated the quantity of both these myeloid cells by FACS analysis. In agreement with the IHC results, there is a significant reduction in total myeloid cells in PDX compared with allografts (Figure S9a), which is a collective result of diminished infiltration of both microglia and BMDM (Figure S9b). We did not observe a difference in neutrophil infiltration between these two models (data not shown). Together, these data suggest that the impaired microglia and BMDM infiltration was not a result of extreme immunodeficiency of the NSG mice, but most probably an effect of species incompatibility between human chemokines and mouse chemokine receptors that are involved in the recruitment of microglia and macrophages to GBM.
3.7 |. Differential PD-L1 expression in murine tumors and polarized macrophages
One of the major functions of TAMs in tumor development is to provide immune suppression via checkpoint engagement. To test whether our murine models behave similarly to hGBM and are suitable for studies to reveal the mechanisms of primary resistance to checkpoint blockade, we first assessed the expression of Pdcd1 (Pd1) and its ligands Pdcd1lg1 (Pdl1) and Pdcd1lg2 (Pdl2) in the NanoString dataset. On the RNA level, the expression of each marker was shown to be highest in EGFRvIII-expressing tumors (Figure 5a). We next evaluated the expression levels of PD-1 in T cells by flow cytometry. First, we did not observe any difference in CD4 or CD8 T cell abundance in the three subtypes of tumors (Figure S10). Interestingly, we found that PDGFB-overexpressing and EGFRvIII-expressing tumors demonstrated similar degree of PD-1 expression on cytotoxic T cells, both significantly higher than that of the NF1 subtype (Figure 5b). PD-1 expression on CD4 T cells is the highest in EGFRvIII tumors among all three subtypes (Figure 5b). In a separate flow cytometry panel, we investigated PD-L1 expression in naïve brain and in PDGFB-overexpressing, Nf1-silenced, and EGFRvIII-expressing tumors as shown in Figure 5c,d. The results demonstrated that TAMs in all three mGBM subtypes expressed high levels of PD-L1, where the highest expression was observed in BMDM in Nf1-silenced tumors (Figure 5e). Although tumor cells also expressed PD-L1, it was at a much lower level compared to TAMs in all three tumor types (Figure 5e).
FIGURE 5.
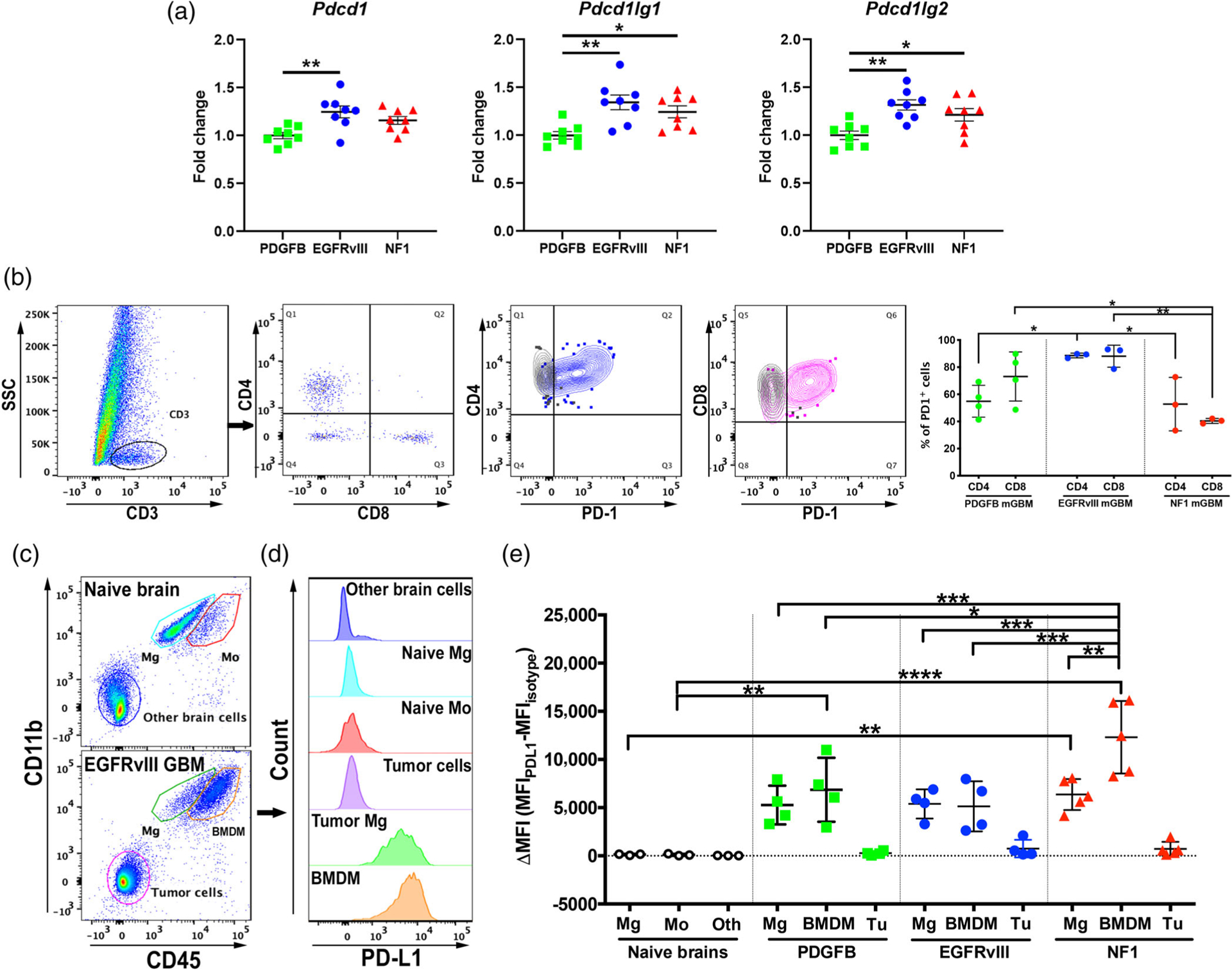
PD-L1 expression is elevated the most in tumor-associated BMDM. (a) Transcription of PD-1 and its ligands are elevated in EGFRvIII mGBM compared to other subtypes. (b) Flow cytometry analysis of PD-1 expression in tumor infiltrating lymphocytes. (c) Gating strategy as represented by CD45 and CD11b (other parameters not shown). Microglia (Mg, CD45LoCD11b+), bone marrow-derived myeloid cells (Mo, CD45HiCD11b+), non-myeloid cells in normal brain, and tumor cells (CD45−CD11b−) are identified in naïve or GBM brain tissues by FACS. (d) Representative off-set overlay histogram of PD-L1 expression in each cell type. (e) Quantification of mean fluorescent intensity (MFI) of PD-L1 compared to isotype control. Mg, microglia. Mo, monocytes. Oth, other normal brain cells. BMDM: bone marrow-derived macrophages. Tu: tumor cells. *p < .05, **p < .01, ***p < .001, by one-way ANOVA with Tukey’s posthoc test
It has been suggested that alternatively activated M2 macrophages are the most immunosuppressive in GBM (Cui et al., 2018). We next sought to address the question whether M1 or M2 genes are most prevalent in our mouse models by mining the NanoString data for expression of specific genes. We employed a panel previously shown to be markers of either M1 or M2 macrophages (Z. Chen & Hambardzumyan, 2018) and generated gene expression heatmaps (Figure 6a). This analysis illustrated that both M1 and M2 genes appear to be concurrently present in all three subtypes, and there is no clear indication of either M1 or M2 dominance in any subtype (Figure 6a).
FIGURE 6.
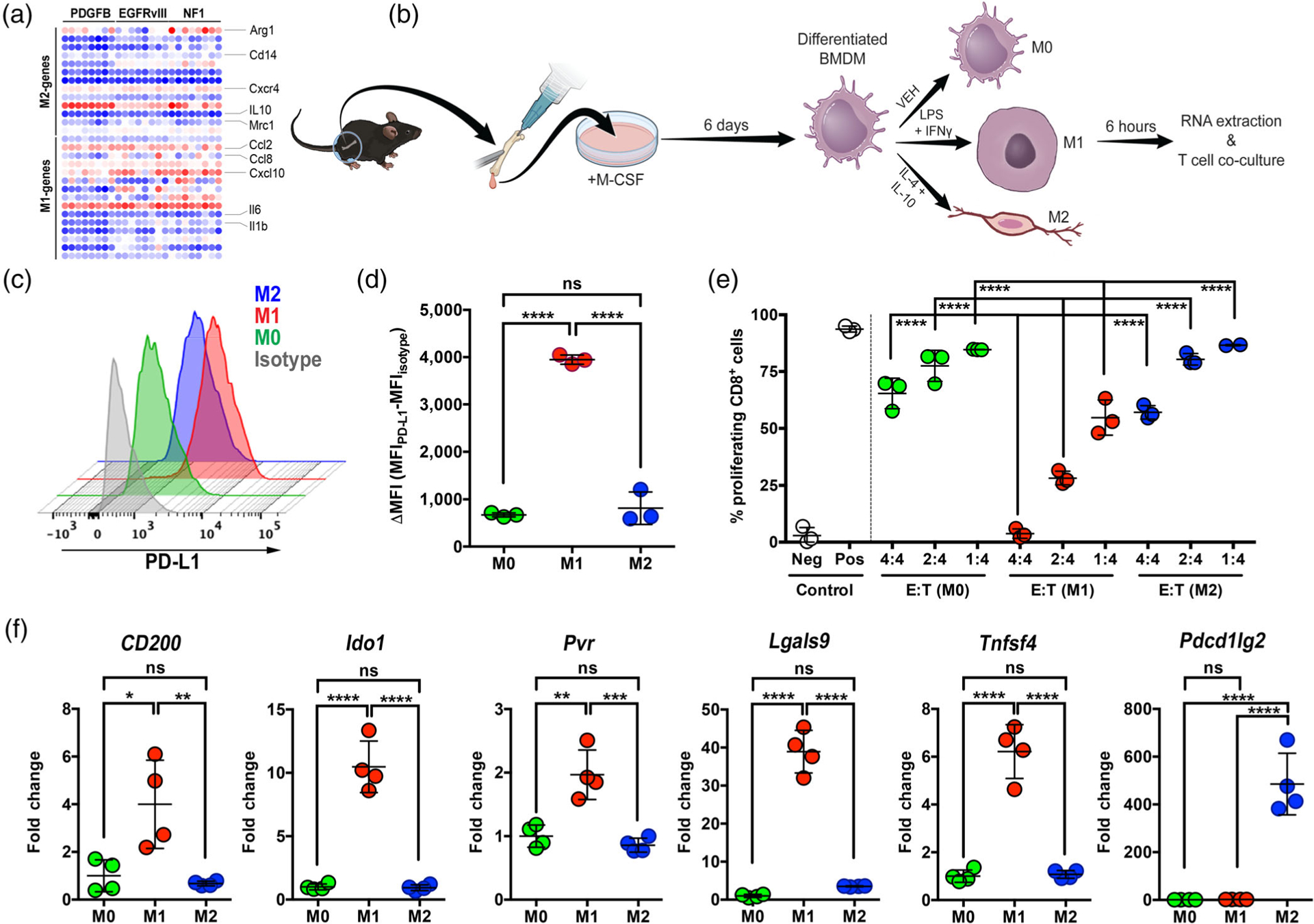
LPS/IFNγ-stimulated (M1) BMDM express high levels of PD-L1 and suppress T cell proliferation in vitro. (a) M1 or M2 differentiated macrophages are equally found in all three subtypes of mGBM. (b) Schematic representation of BMDM isolation and polarization. (c) Representative off-set histograms of PD-L1 expression in polarized BMDM in vitro. (d) Quantification of PD-L1 expression in these polarized cells. (e) BMDM suppress CD8 T-cell proliferation in an E: T (effector: target) ratio-dependent manner when the T cells are stimulated with 100 nM SINNFEKL peptide. (f) M1-polarized BMDM display the highest levels of immunosuppressive markers as examined by qPCR. *p < .05, **p < .01, ***p < .001, ****p < .0001, (ns) not significant, by one-way ANOVA with Tukey’s posthoc test
There is an assumption in the literature that M2-polarized TAMs are immunosuppressive while M1-polarized TAMs are tumoricidal, although no direct evidence has been presented to support these claims. To better understand the connection of M1 and M2 polarization to PD-L1 expression, we used primary BMDM and polarized them to either M1 or M2 phenotypes by stimulation with known classic activation molecules, including LPS and interferon gamma (IFNγ) for M1, as well as IL-4 and IL-10 for M2, as shown in the diagram in Figure 6b. We then stained M0 macrophages (vehicle-stimulated), M1, and M2 with anti-PD-L1 antibody and quantified their expression by flow cytometry (Figure 6c). Quantification of mean fluorescent intensity (MFI) demonstrated that PD-L1 expression was significantly higher in M1 macrophages compared to M0 and M2 (Figure 6d). To determine whether high expression of PD-L1 translated into actual biological function, we performed a T-cell suppression assay to determine OT-1 cell proliferation in the presence of polarized macrophages (Figure S10). The data demonstrate that M1 macrophages most potently suppress the proliferation of stimulated T cells (Figure 6e). We next performed qPCR on M0, M1, and M2 macrophages with a panel of immunosuppressive molecules, and showed that 5 out of 6 tested markers were significantly upregulated in M1 macrophages compared to M0 and M2 (Figure 6f). In total, these results suggest that M1-polarized macrophages may be more immunosuppressive than previously appreciated. Validation of our polarization scheme was performed to ensure M1 and M2 BMDMs indeed fell into each category (Figure S12).
3.8 |. Anti-PD-L1 immunotherapy alone or in combination with RT does not prolong mouse survival in Nf1 and PDGFB-tumor bearing mice
We and others have recently described that TAMs have mixed phenotype and do not fall into either M1 or M2 categories (Z. Chen & Hambardzumyan, 2018). Thus, in addition to our current data demonstrating high PD-L1 expression by TAMs, we decided to test whether blocking PD-L1 would have anti-tumor efficacy alone or in combination with X-ray radiation therapy (RT). PDGFB-overexpressing and Nf1-silenced tumor-bearing mice were treated with PD-L1 neutralizing antibody or antibody isotype control antibody with or without concomitant RT according to the indicated treatment scheme (Figure 7a). We specifically choose PDGFB-overexpressing and Nf1-silenced tumors based on the polarized immune profiles and differential TAM compositions between these two subtypes. PD-L1 neutralizing antibody was administered at the dosage recommended by the manufacturer and based upon previously published results (Wirsching et al., 2019). Treatment of mice bearing PDGFB-overexpressing tumors with PD-L1 neutralization with or without RT yielded no extension in median survival compared to the isotype control group (Figure 7b). As expected, both groups that received RT demonstrated a significant extension in median survival compared to the non-irradiated groups. Administration of PD-L1 neutralizing antibody similarly conferred no survival benefit when combined with radiation in mice bearing Nf1-silenced tumors (Figure 7c). These results are consistent with the majority of previously published literature demonstrating limited efficacy of immune checkpoint blockade in human GBM (Romani, Pistillo, Carosio, Morabito, & Banelli, 2018). In summary, our study produced valuable immunocompetent models that will allow the neuro-immuno-oncology field to determine the primary resistance mechanisms to checkpoint inhibitors in GBM.
FIGURE 7.
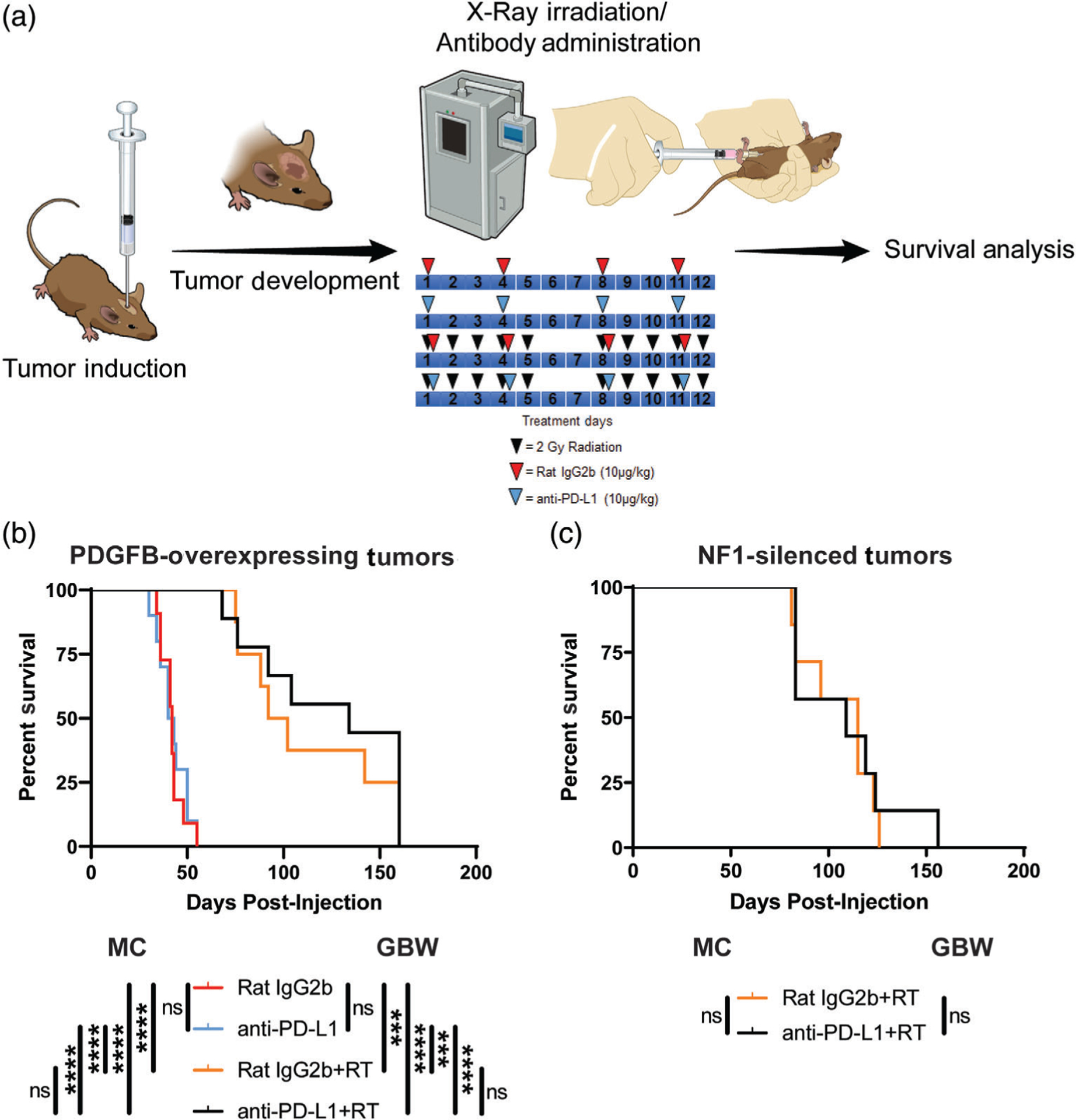
Murine PDGFB-overexpressing and Nf1-silenced tumors do not respond to PD-L1 neutralizing therapy. (a) Schematic illustration of the experimental protocol. (b) Survival curves comparing mice bearing PDGFB-overexpressing tumors treated with isotype control antibody (N = 11), PD-L1 neutralizing antibody (N = 10), isotype control antibody plus x-ray irradiation (N = 8), and PD-L1 neutralizing antibody plus X-ray irradiation (N = 9). (c) Survival curves comparing mice bearing NF1-silenced tumors treated with isotype control antibody plus X-ray irradiation (N = 7) and PD-L1 neutralizing antibody plus X-ray irradiation (N = 6). Mantel-Cox (MC) and Gehan-Breslow-Wilcoxon (GBW) tests, (ns) not significant, ***p < .001, ****p < .0001. RT, X-ray radiation therapy
4 |. DISCUSSION
There has been active investigation of human GBM subtypes, as well as the degree of heterogeneity within the subtypes. Various correlative data have revealed increased TAM-related gene expression and immune infiltration in MES hGBM compared to PN and CL hGBM. In recent years, increased understanding and appreciation of TAMs has resulted in many TAM-centered studies, some of which have utilized PDX models of GBM. In the current study, we made interesting observations that genetic driver mutations create unique immune microenvironments, especially with respect to myeloid cells. More specifically, we found that genetic drivers found in GBM dictate the quantity, compositions, and profiles of myeloid cells and also the immune microenvironment in general. Remarkably, we found that this TAM-rich microenvironment was not recapitulated in PDX models in NSG mice. To answer the question whether these genetically modified mouse models derived by various oncological drivers respond to anti-PD-L1 therapy similarly to human patients we treated the mice with anti-PD-L1 antibodies with or without irradiation. No difference in survival was found among these subtypes of murine GBM treated with anti-PD-L1 antibodies, comparable to what has been observed in human studies (D. A. Reardon et al., 2020). In light of growing evidence of the significant involvement of myeloid cells in suppressing T cell functions in GBM, the establishment of these models serves as an important step toward the mechanistic understanding of the resistance to checkpoint inhibitors in clinical trials for recurrent GBM. Indeed, we revealed that M1, but not M2, in vitro polarized TAMs exhibit immunosuppressive potential through their elevated expression of PD-L1 and other immunosuppressive molecules, suggesting that altering the phenotype of the plastic TAMs in GBM microenvironment may enhance the sensitivity of immune checkpoint treatment.
Elegant work performed by several groups using GEMM demonstrated that different cells-of-origin, even when the same driver mutations are used, could create distinct phenotypes of GBM, directly influencing the expression subtypes and response to therapy (Jiang et al., 2017; Ozawa et al., 2014; Sreedharan et al., 2017). We have previously demonstrated that targeting Nestin-positive cells by either Nf1-silencing or PDGFB-overexpression creates tumors that have significant differences in TAM presence. To have a broader coverage of the heterogeneity of hGBM, we have modified a previously published EGFRvIII-driven model in this study, ensuring the same cell-of-origin, Nestin-positive neural stem/progenitor cells, are used for tumor generation. This allowed for any differences in myeloid composition, immune profile, and therapeutic responses to be attributed directly to driver mutations.
Utilizing gene expression data from the TCGA and the Gene Expression Omnibus databases, several studies have demonstrated enrichment of immune response-related gene expression, especially of TAM genes, in the MES subtype of GBM compared to the other subtypes (Engler et al., 2012), suggesting TAMs could play a subtype-specific role in GBM. In agreement with these findings, we have previously shown increased TAM infiltration in human MES tumors compared to PN and CL tumors by quantification of IBA1 with IHC (Kaffes et al., 2019). Similarly, we showed that Nf1-silenced murine tumors have increased TAM presence compared to PDGFB-overexpressing and EGFRvIII tumors. Interestingly, the overall expression of AIF1 (the gene encoding for the IBA1 protein) is increased in MES hGBM when compared to the other subtypes, but that high expression correlates with worse survival in PN and better survival in MES, with no effect on CL hGBM patients (Kaffes et al., 2019). This suggests that it is not just the number of TAMs that plays a key role in tumorigenesis, but rather their expression profile and/or ontogeny. Indeed, we found that Nf1-silenced mGBM exhibits a significantly higher portion of resident brain microglia; in contrast to PDGFB and EGFRvIII tumors, where the majority of these TAMs are BMDM/macrophages (Figure 4). Additionally, we demonstrated that Nf1-silenced tumors showed increased neutrophil infiltration compared to the other subtypes. Using an unbiased approach, we further demonstrated that driver mutations create distinct immune microenvironments beyond that of the TAMs. Despite having close similarity in terms of myeloid number and composition, PDGFB and EGFRvIII tumors exhibit distinct overall immune expression profiles. These data suggest that driver mutations are critical determinants of immune heterogeneity in GBM when the same cell-of-origin is employed.
Our newly generated EGFRvIII-driven mGBM model closely resembles human CL GBM, which is known to be enriched for tumors that harbor EGFRvIII mutations. Histologically, murine EGFRvIII-driven tumors exhibit critical features of human CL GBM, including microvascular proliferation and pseudopalisading necrosis. IHC evaluation of different proteins of astrocytic and oligodendroglial lineages showed similar expression patterns in human CL and murine EGFRvIII tumors. Comparison of a subtype-related 28-gene panel demonstrated striking similarities in PDGFB-overexpressing, Nf1-silenced, and EGFRvIII-expressing tumors with their PN, MES, and CL counterparts, respectively. Furthermore, freshly dissociated primary cells from all three subtypes can be cultured in serum-free conditions and used for in vitro studies and subsequent orthotopic implantation. These models will allow investigators to dissect the roles of these individual mutations in glioma growth and response to therapy, and thereby improve our ability to develop better precision therapeutics, particularly novel immunotherapies.
Since PDX models are the gold standard for cancer signaling studies, we decided to evaluate myeloid infiltration in PDX models. However, our results indicated PDX models have significant limitations for the use in myeloid studies, as we demonstrated that they have markedly impaired infiltration of both microglia and BMDM (Figure 4). Our results suggest impairment of myeloid cell recruitment is not caused by the extreme immunodeficiency of NSG mice, because murine allografts do not show the same impairment. Rather, impaired myeloid cell infiltration is most likely due to species incompatibilities (human tumor vs. mouse stroma) of various chemokines and their receptors involved in microglia and BMDM recruitment. Our results indicate that for myeloid cell or immunotherapy studies, PDX models should be utilized with caution.
Our study also demonstrated expression of PD-L1 in tumors from all three subtypes, with myeloid cell expression at much higher levels than tumor cells, regardless of subtype. BMDM in Nf1-silenced tumors were shown to have the highest PD-L1 expression levels. We also demonstrated that M1, but not M2-polarized BMDM, express PD-L1 protein with high abundance. Furthermore, in vitro polarized M1-macrophages significantly inhibit the proliferation of T cells compared to M2-polarized macrophages. Although these data are in contrast to other studies suggesting that GBM can induce M2-like macrophages with increased PD-L1 expression (Gabrusiewicz et al., 2018; Wen et al., 2018), our study used canonical procedures to unequivocally polarize the BMDM to an M1 or M2 phenotype. Importantly, our results raised caution for conversion of M2 macrophages into M1 macrophages as a viable GBM therapy, since this switch could lead to upregulation of multiple immunosuppressive pathways.
Traditionally, PD-L1 expression levels quantified by IHC have been employed in solid tumors for response prediction and stratification of patients to anti-PD-L1 therapy. Recent findings show that PD-1/PD-L1 inhibitors appear to have activity in a subset of individuals who do not meet the IHC bioassay cutoff (Diggs & Hsueh, 2017). Furthermore, it is suggested that several additional factors could be involved in the response to anti-PD-1/PD-L1 antibody therapy (Diggs & Hsueh, 2017). While higher expression of PD-L1 at both the RNA and protein levels is correlated with worse outcome in GBM patients (Nduom et al., 2016), current data from randomized trials demonstrate that PD-L1 expression is not a suitable biomarker for predicting response to anti-PD-1 therapy (Platten & Reardon, 2018). Taking advantage of these GEMM models, we investigated how they may differentially respond to PD-L1 blockade alone or in combination with radiation therapy. Our results indicated no significant efficacy with PD-L1 blockade in PDGFB-overexpressing and Nf1-silenced tumor-bearing mice. These data are in agreement with the results from clinical trials showing limited effectiveness of anti-PD-L1 therapy in hGBM (David A. Reardon et al., 2019). The lack of efficacy can be attributed to possible PD-L1 independent immunosuppression in GBM. Additionally, GBMs are notoriously “cold” tumors in terms of infiltrating cytotoxic T lymphocytes (Chongsathidkiet et al., 2018). Indeed, in our murine models, we found that on average T lymphocytes only account for less than 3% of the total CD45+ cells (Figure S10). This inefficacy can also be partially explained by the inability of the systemically delivered antibody to pass through the blood brain barrier (Filley et al., 2017). These possibilities are easily addressable by using Pd1−/− or Pdl1−/− mice in combination with the RCAS/tv-a system, which will allow selective deletion of PD-1 or PD-L1 in neoplastic cells, non-neoplastic cells, or both. Most recently, neoadjuvant PD-1 inhibition (administered prior to surgical resection) in patients with GBM provided some preliminary evidence for potential clinical benefit (Cloughesy et al., 2019; Schalper et al., 2019; Zhao et al., 2019); however, mechanistic determination of how neoadjuvant anti-PD-1 therapy provides superior outcomes to adjuvant therapy remains to be characterized. Adequately designed prospective trials are needed to assess the treatment effects (Berghoff & Preusser, 2019). The immunocompetent preclinical models described in this study provide a valuable platform for dissecting the primary resistance mechanisms of checkpoint blockade in GBM, and will serve as an indispensable tool for the development and validation of novel immunotherapies.
In conclusion, our results show that these driver-mutation induced GEMMs of GBM are a valuable tool for mechanistic studies of immune functions and immunotherapy in GBM. Different oncogenic driver genes create unique immune microenvironmental characteristics in each individual subtype of GBM; a phenomenon that is not recapitulated in patient derived xenograft models. Remarkably, we demonstrated that immune checkpoint molecules are enriched in all subtypes of mGBM, but their response to checkpoint blockade therapy is nonexistent. Future studies focusing on how to substantially improve such immunotherapies will benefit from the availability of these models.
Supplementary Material
ACKNOWLEDGMENTS
We would like to acknowledge Emory Children’s Flow Cytometry Core, Integrated Cellular Imaging Core, Integrated Genomics Core, and Winship Pathology Core Lab, and Winship Cancer Animal Models Core for their services. We extend our thanks to Mr. David R. Schumick for generating illustrations. We thank Dr. Robert C. Castellino for discussions. We are grateful to Dr. Maria Castro and Dr. Mahmoud Alghamri for technical advice with the immunosuppression assay, and Mr. Kai Nie and Ms. Colleen Mosley for technical assistance, and Dr. Alex Chen for providing EGFRvIII cell lines. This work was supported by NIH/NINDS R01 NS100864 and from the Aflac Cancer and Blood Disorders Center start-up funds for Dolores Hambardzumyan, and NIH/NINDS R01 NS100967 for Renee D. Read. PSTP Training Grant 4T32GM008602-20 and NIH/NINDS 1F31NS106887 provided funding for Cameron J. Herting and NIH/NCI 1F31CA232531 provided funding for James L. Ross.
Funding information
NIH/NCI, Grant/Award Number: 1F31CA232531; PSTP Training Grant, Grant/Award Number: 4T32GM008602-20; NIH/NINDS, Grant/Award Numbers: 1F31NS106887, R01 NS100967, R01 NS100864
Footnotes
CONFLICT OF INTEREST
The authors have no relevant competing interests to disclose.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
REFERENCES
- Ajami B, Bennett JL, Krieger C, McNagny KM, & Rossi FM (2011). Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nature Neuroscience, 14(9), 1142–1149. 10.1038/nn.2887 [DOI] [PubMed] [Google Scholar]
- Aranda PS, LaJoie DM, & Jorcyk CL (2012). Bleach gel: A simple aga-rose gel for analyzing RNA quality. Electrophoresis, 33(2), 366–369. 10.1002/elps.201100335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghoff AS, & Preusser M (2019). Does neoadjuvant anti-PD1 therapy improve glioblastoma outcome? Nature Reviews. Neurology, 15(6), 314–315. 10.1038/s41582-019-0178-0 [DOI] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, … Network TR (2013). The somatic genomic landscape of glioblastoma. Cell, 155(2), 462–477. 10.1016/j.cell.2013.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature, 455(7216), 1061–1068. 10.1038/nature07385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, … Schultz N (2012). The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discovery, 2(5), 401–404. 10.1158/2159-8290.cd-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, … Ma’ayan A (2013). Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics, 14, 128 10.1186/1471-2105-14-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Feng X, Herting CJ, Alvarez Garcia V, Nie K, Pong WW, … Hambardzumyan D (2017). Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Research, 77, 2266–2278. 10.1158/0008-5472.can-16-2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, & Hambardzumyan D (2018). Immune Microenvironment in Glioblastoma Subtypes. Frontiers in Immunology, 9, 1004 10.3389/fimmu.2018.01004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, … Fecci PE (2018). Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nature Medicine, 24(9), 1459–1468. 10.1038/s41591-018-0135-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, … Prins RM (2019). Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nature Medicine, 25(3), 477–486. 10.1038/s41591-018-0337-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DN, Chen SC, Sullivan LM, Manfra DJ, Wiekowski MT, Prosser DM, … Lira SA (2001). Generation and analysis of mice lacking the chemokine fractalkine. Molecular and Cellular Biology, 21(9), 3159–3165. 10.1128/MCB.21.9.3159-3165.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlan AM, Harmon C, Whelan S, O’Brien EC, O’Reilly VP, Crotty P, … Little MA (2016). Myeloid engraftment in humanized mice: Impact of granulocyte-Colony stimulating factor treatment and transgenic mouse strain. Stem Cells and Development, 25(7), 530–541. 10.1089/scd.2015.0289 [DOI] [PubMed] [Google Scholar]
- Cui X, Morales RT, Qian W, Wang H, Gagner JP, Dolgalev I, … Chen W (2018). Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials, 161, 164–178. 10.1016/j.biomaterials.2018.01.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, & Holland EC (2001). PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligo-astrocytomas from neural progenitors and astrocytes in vivo. Genes & Development, 15(15), 1913–1925. 10.1101/gad.903001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggs LP, & Hsueh EC (2017). Utility of PD-L1 immunohistochemistry assays for predicting PD-1/PD-L1 inhibitor response. Biomarker Research, 5, 12 10.1186/s40364-017-0093-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler JR, Robinson AE, Smirnov I, Hodgson JG, Berger MS, Gupta N, … Phillips JJ (2012). Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PLoS One, 7(8), e43339 10.1371/journal.pone.0043339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filley AC, Henriquez M, & Dey M (2017). Recurrent glioma clinical trial, CheckMate-143: The game is not over yet. Oncotarget, 8(53), 91779–91794. 10.18632/oncotarget.21586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, & Paxinos G (1997). The mouse brain in stereotaxic coordinates. San Diego: Academic Press. [Google Scholar]
- Gabrusiewicz K, Li X, Wei J, Hashimoto Y, Marisetty AL, Ott M, … Heimberger AB (2018). Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology, 7(4), e1412909 10.1080/2162402X.2017.1412909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, … Heimberger AB (2016). Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight, 1(2), 85841 10.1172/jci.insight.85841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, … Schultz N (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science Signaling, 6(269), pl1. 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, & Holland EC (2009). Modeling adult Gliomas using RCAS/t-va technology. Translational Oncology, 2(2), 89–95. 10.1593/tlo.09100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herting CJ, Chen Z, Pitter KL, Szulzewsky F, Kaffes I, Kaluzova M, … Hambardzumyan D (2017). Genetic driver mutations define the expression signature and microenvironmental composition of high-grade gliomas. Glia, 65(12), 1914–1926. 10.1002/glia.23203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, & Fuller GN (2000). Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nature Genetics, 25 (1), 55–57. 10.1038/75596 [DOI] [PubMed] [Google Scholar]
- Jiang Y, Marinescu VD, Xie Y, Jarvius M, Maturi NP, Haglund C, … Uhrbom L (2017). Glioblastoma cell malignancy and drug sensitivity are affected by the cell of origin. Cell Reports, 18(4), 977–990. 10.1016/j.celrep.2017.01.003 [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, & Littman DR (2000). Analysis of fractalkine receptor CX(3) CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Molecular and Cellular Biology, 20(11), 4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaffes I, Szulzewsky F, Chen Z, Herting CJ, Gabanic B, Velazquez Vega JE, … Hambardzumyan D (2019). Human Mesenchymal glioblastomas are characterized by an increased immune cell presence compared to proneural and classical tumors. Oncoimmunology, 8(11), e1655360 10.1080/2162402X.2019.1655360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, … Ma’ayan A (2016). Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research, 44(W1), W90–W97. 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakin N, Rulach R, Nowicki S, & Kurian KM (2017). Current advances in checkpoint inhibitors: Lessons from non-central nervous system cancers and potential for Glioblastoma. Frontiers in Oncology, 7, 141 10.3389/fonc.2017.00141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laks DR, Masterman-Smith M, Visnyei K, Angenieux B, Orozco NM, Foran I, … Kornblum HI (2009). Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells, 27(4), 980–987. 10.1002/stem.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- London A, Cohen M, & Schwartz M (2013). Microglia and monocyte-derived macrophages: Functionally distinct populations that act in concert in CNS plasticity and repair. Frontiers in Cellular Neuroscience, 7, 34 10.3389/fncel.2013.00034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, … Ellison DW (2016). The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathologica, 131(6), 803–820. 10.1007/s00401-016-1545-1 [DOI] [PubMed] [Google Scholar]
- Nagane M, Coufal F, Lin H, Bogler O, Cavenee WK, & Huang HJ (1996). A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Research, 56(21), 5079–5086. [PubMed] [Google Scholar]
- Nduom EK, Wei J, Yaghi NK, Huang N, Kong LY, Gabrusiewicz K, … Heimberger AB (2016). PD-L1 expression and prognostic impact in glioblastoma. Neuro-Oncology, 18(2), 195–205. 10.1093/neuonc/nov172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, … Holland EC (2014). Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell, 26 (2), 288–300. 10.1016/j.ccr.2014.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platten M, & Reardon DA (2018). Concepts for immunotherapies in Gliomas. Seminars in Neurology, 38(1), 62–72. 10.1055/s-0037-1620274 [DOI] [PubMed] [Google Scholar]
- Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, … Weller M (2020). Effect of Nivolumab vs Bevacizumab in patients with recurrent Glioblastoma: The CheckMate 143 phase 3 randomized clinical trial. JAMA Oncology (Epub ahead of print). 10.1001/jamaoncol.2020.1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon DA, Kaley TJ, Dietrich J, Clarke JL, Dunn G, Lim M, … Venhaus RR (2019). Phase II study to evaluate safety and efficacy of MEDI4736 (durvalumab) + radiotherapy in patients with newly diagnosed unmethylated MGMT glioblastoma (new unmeth GBM). Journal of Clinical Oncology, 37(15_suppl), 2032–2032. 10.1200/JCO.2019.37.15_suppl.2032 [DOI] [Google Scholar]
- Romani M, Pistillo MP, Carosio R, Morabito A, & Banelli B (2018). Immune checkpoints and innovative therapies in Glioblastoma. Frontiers in Oncology, 8, 464 10.3389/fonc.2018.00464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruano Y, Ribalta T, de Lope AR, Campos-Martin Y, Fiano C, Perez-Magan E, … Melendez B (2009). Worse outcome in primary glioblastoma multiforme with concurrent epidermal growth factor receptor and p53 alteration. American Journal of Clinical Pathology, 131(2), 257–263. 10.1309/AJCP64YBDVCTIRWV [DOI] [PubMed] [Google Scholar]
- Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL, … Charo IF (2010). Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One, 5(10), e13693 10.1371/journal.pone.0013693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, Lopez-Janeiro A, Porciuncula A, Idoate MA, … Melero I (2019). Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nature Medicine, 25(3), 470–476. 10.1038/s41591-018-0339-5 [DOI] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, & DePinho RA (1996). Role of the INK4a locus in tumor suppression and cell mortality. Cell, 85(1), 27–37. 10.1016/s0092-8674(00)81079-x [DOI] [PubMed] [Google Scholar]
- Shemer A, & Jung S (2015). Differential roles of resident microglia and infiltrating monocytes in murine CNS autoimmunity. Seminars in Immunopathology, 37(6), 613–623. 10.1007/s00281-015-0519-z [DOI] [PubMed] [Google Scholar]
- Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, … Handgretinger R (2005). Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. Journal of Immunology, 174(10), 6477–6489. 10.4049/jimmunol.174.10.6477 [DOI] [PubMed] [Google Scholar]
- Sreedharan S, Maturi NP, Xie Y, Sundstrom A, Jarvius M, Libard S, … Uhrbom L (2017). Mouse models of pediatric Supratentorial high-grade Glioma reveal how cell-of-origin influences tumor development and phenotype. Cancer Research, 77(3), 802–812. 10.1158/0008-5472.CAN-16-2482 [DOI] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, … Mirimanoff RO (2005). Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England Journal of Medicine, 352(10), 987–996. 10.1056/NEJMoa043330 [DOI] [PubMed] [Google Scholar]
- Suzuki A, Yamaguchi MT, Ohteki T, Sasaki T, Kaisho T, Kimura Y, … Mak TW (2001). T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity, 14(5), 523–534. 10.1016/s1074-7613(01)00134-0 [DOI] [PubMed] [Google Scholar]
- Varvel NH, Neher JJ, Bosch A, Wang W, Ransohoff RM, Miller RJ, & Dingledine R (2016). Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proceedings of the National Academy of Sciences of the United States of America, 113(38), E5665–E5674. 10.1073/pnas.1604263113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, … Hayes DN (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell, 17(1), 98–110. 10.1016/j.ccr.2009.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Hu X, Muller F, Kim H, Squatrito M, & Millelsen T (2017). Tumor evolution of glioma intrinsic gene expression subtype associates with immunological changes in the microenvironment. Cancer Cell, 32(1), 42–56. https://www.ncbi.nlm.nih.gov/pubmed/28697342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Tachibana O, Sata K, Yonekawa Y, Kleihues P, & Ohgaki H (1996). Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathology, 6(3), 217–223; discussion 223–214. 10.1111/j.1750-3639.1996.tb00848.x [DOI] [PubMed] [Google Scholar]
- Weischenfeldt J, & Porse B (2008). Bone marrow-derived macrophages (BMM): Isolation and applications. CSH Protocols, 3(12), 1–6. 10.1101/pdb.prot5080 [DOI] [PubMed] [Google Scholar]
- Wen ZF, Liu H, Gao R, Zhou M, Ma J, Zhang Y, … Wang LX (2018). Tumor cell-released autophagosomes (TRAPs) promote immunosuppression through induction of M2-like macrophages with increased expression of PD-L1. Journal for Immunotherapy of Cancer, 6 (1), 151 10.1186/s40425-018-0452-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirsching HG, Zhang H, Szulzewsky F, Arora S, Grandi P, Cimino PJ, … Holland EC (2019). Arming oHSV with ULBP3 drives abscopal immunity in lymphocyte-depleted glioblastoma. JCI Insight, 4 (13), 128217 10.1172/jci.insight.128217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang I, Han SJ, Kaur G, Crane C, & Parsa AT (2010). The role of microglia in central nervous system immunity and glioma immunology. Journal of Clinical Neuroscience, 17(1), 6–10. 10.1016/j.jocn.2009.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Wu Y, Zou J, Zhou J, Wang M, Hao X, & Cui H (2016). The Hippo transducer TAZ promotes cell proliferation and tumor formation of glioblastoma cells through EGFR pathway. Oncotarget, 7(24), 36255–36265. 10.18632/oncotarget.9199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, … Rabadan R (2019). Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nature Medicine, 25(3), 462–469. 10.1038/s41591-019-0349-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, & Pan D (2019). The hippo signaling pathway in development and disease. Developmental Cell, 50(3), 264–282. 10.1016/j.devcel.2019.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Acquaviva J, Ramachandran P, Boskovitz A, Woolfenden S, Pfannl R, … Charest A (2009). Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proceedings of the National Academy of Sciences of the United States of America, 106(8), 2712–2716. 10.1073/pnas.0813314106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.