

Abstract
Splicing of precursor messenger RNA is catalyzed by the spliceosome, a dynamic ribonucleoprotein assembly including five small nuclear (sn)RNAs and >100 proteins. RNA components catalyze the two transesterification reactions, but proteins perform critical roles in assembly and rearrangement. The catalytic core comprises a paired complex of U2 and U6 snRNAs for the major form of the spliceosome and U12 and U6atac snRNAs for the minor variant (∼0.3% of all spliceosomes in higher eukaryotes); the latter shares key catalytic sequence elements and performs identical chemistry. Here we use solution NMR techniques to show that the U12–U6atac snRNA complex of both human and Arabidopsis maintain base-pairing patterns similar to those in the three-helix model of the U2–U6 snRNA complex that position key elements to form the spliceosome’s active site. However, in place of the stacked base pairs at the base of the U6 snRNA intramolecular stem loop and the central junction of the U2–U6 snRNA complex, we see altered geometry in the single-stranded hinge region opposing termini of the snRNAs to enable interaction between the key elements. We then use electrophoretic mobility shift assays and fluorescence assays to show that the protein RBM22, implicated in remodeling the human U2–U6 snRNA complex prior to catalysis, also binds the U12–U6atac snRNA complexes specifically and with similar affinity as to U2–U6 snRNA (a mean Kd for the two methods = 3.4 and 8.0 μM for U2–U6 and U12–U6atac snRNA complexes, respectively), suggesting that RBM22 performs the same role in both spliceosomes.
Introduction
The excision of noncoding intervening sequences (introns) and ligation of flanking coding regions (exons) from precursor messenger (pre-m)RNA is mediated by the spliceosome, a large and dynamic ribonucleoprotein complex found in the nuclei of eukaryotic cells. The vast majority of spliceosomes (99.7%) comprise the snRNAs U1, U2, U4, U5, and U6 in association with at least 100 proteins (actual number depends upon the organism and state of assembly) to form snRNPs.1,2 Pre-mRNA splicing is a critical step in the maturation of pre-mRNA into translatable mRNA transcripts and involves two sequential transesterification reactions catalyzed by RNA components of the spliceosome; thus, the spliceosome is a ribozyme.3
During the first transesterification reaction, the 2′OH of a conserved adenosine residue in the intron, referred to as the branch site due to the branched lariat intermediate it forms, performs a nucleophilic attack at the 5′ splice site, forming a free 3′OH on the exon.4 The free 3′OH attacks the 3′ splice site, joining the two exons while releasing the lariat intron. The transesterification reactions have been previously proposed to involve a two-metal ion center, where one Mg2+ ion activates the nucleophilic 2′OH, while the second Mg2+ ion stabilizes the oxyanion leaving group,5 a mechanism substantiated by the positioning of two metal ions in images derived by cryo-EM.6
Throughout the splicing cycle, snRNPs are delivered to the assembly and the snRNA components undergo dramatic conformational changes. U1 snRNA pairs with the 5′ splice site and U2 snRNA pairs with a specific segment of the intron to form a helix including the bulged branch site. Upon the arrival of the U4/U6/U5 tri-snRNP (B complex), U6 snRNA is unwound from U4 to pair with U2 and the 5′ splice site, and the displaced U4 and U1 snRNPs are released (forming the activated or Bact complex).7 U5 snRNA positions both the 3′ and 5′ splice sites.8 Pairing between U2 and U6 snRNAs results in a multihelix complex that forms the catalytic core of the spliceosome, specifically, a triple helix involving hydrogen bonding between bases of the catalytic AGC triad, a bulged U of the intramolecular stem loop (ISL), and the final GA of the ACAGAGA internal loop, all components of U6 snRNA (B* complex).9−12 Thus, the only snRNAs directly implicated in catalysis belong to the U2–U6 snRNA complex.6,13,14 Each step is reversible and regulated.15
Unlike its counterpart in the self-splicing Group II intron, folding of the spliceosome’s RNA active site requires assistance by the Prp19 complex and associated/related proteins (known as NTC for NineTeen complex), which participates in a number of ATP-dependent remodeling steps.16 However, the only NTC-related protein to contact the U2–U6 snRNA complex in the human spliceosome is RBM22.17 In support of the direct interaction, RBM22 (and Cwc2 in the yeast Saccharomyces cerevisiae(18)) forms photoinduced cross-links with residues in the unpaired 5′ terminus of U6 and ISL region of its respective U6 snRNA;19 residues of Cwc2 in the zinc finger, RNA recognition motif (RRM), and an interdomain connector are involved in the interaction with the RNA;20,21 however, precise interactions in the complex are not yet elucidated. Proximity between RBM22 and the U6 snRNA is observed in cryo-EM images of human spliceosomes in the B22 and C23,24 stages and between Cwc2 and yeast U6 snRNA in the B,25,26 Bact,12,27 B*,28 and C*29,30 stages. Splicing activity is completely inhibited by the deletion of Cwc2 from yeast spliceosomes and restored by supplementation with exogenous Cwc2.19 Thus, Cwc2 and RBM22 are directly involved in facilitating the final remodeling or stabilization of the yeast and human U2–U6 snRNA complex, respectively, into its active conformation.
In this report, we analyze the affinity of RBM22 with its snRNA targets in both the U2–U6 snRNA complex of the major form of the human spliceosome and its U12–U6atac counterpart of the low-abundance (minor) spliceosome. This minor spliceosome is responsible for the splicing of ∼0.3% of introns in a specific (although not fully characterized) subset of higher eukaryote (metazoan) genomes, using the same chemistry, albeit at a somewhat lesser rate than the reaction of the major spliceosome, and has the same intermediates.31,32 However, there are alternative versions of snRNAs accomplishing the function performed by U1, U2, U4, and U6 snRNAs of the major spliceosome (U5 snRNA is identical in both): U12 is the functional analogue of U233 and the sequences of U4atac and U6atac compare to those of U4 and U6 snRNAs of the major spliceosome. There is strong conservation of the AGC triad, ACAGAGA loop, and ISL,34,35 as well as sites proposed to interact with RBM22;19 the principal structural difference is found in a bulged hinge and single-stranded region opposing the termini of both snRNAs in the U12–U6atac complex (Figure 1B) in place of the heterogeneous multihelix central junction observed in the protein-free human U2–U6 snRNA junction. In the absence of protein, the U2–U6 snRNA complex forms a mixture of three-helix and four-helix conformers.36 The two conformers are characterized by different orientations of the stems.37 In addition, the catalytic AGC triad of the U6 snRNA has a different pairing partner in each of the two conformers: in the three-helix conformer in situ, the U6 snRNA AGC catalytic triad forms an intermolecular pairing with U2 snRNA (conformation that is likely to be essential for adopting the active site)13; in the four-helix conformer, it forms an intramolecular pairing in the U6 snRNA ISL.42 In contrast, both the human and Arabidopsis U12–U6atac snRNA complexes adopt only a single conformer in which the catalytic triad (AGC in human, GGC in Arabidopsis) pairs with U12 snRNA and therefore parallels the three-helix conformer of U2–U6 snRNA (Figure 1A), making the minor spliceosome a structurally simpler naturally occurring variant of the major spliceosome for study.
Figure 1.
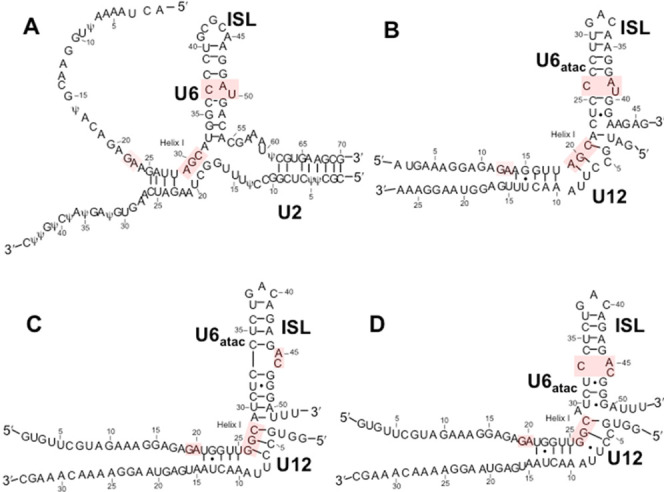
Secondary structural folds for (A) the major spliceosome U2–U6 snRNA complex in its three-helix conformation (note: Ψ modifications replaced by U in the transcribed samples used in these experiments), and folds proposed for (B) the human U12–U6atac snRNA complex; (C) the Arabidopsis minor spliceosome U12–U6atac snRNA complex that maintains five base pairs between the catalytic triad and ISL bulge; and (D) alternate fold for the Arabidopsis snRNA complex (proposed here) that maintains the AY·C (where Y is a pyrimidine) motif seen in human U6 snRNA and proposed for the human U6atac ISL. Lines denoting Watson–Crick base pairs and dots denoting G·U pairs are those proposed in the publications.
If the role of the junction is to assist in positioning stems containing catalytically essential elements into formation, we ask how the open hinged region of the minor spliceosomal snRNA complex achieves this function. As a first step in understanding how the hinged region impacts on the relative placement of critical elements that participate in forming the catalytic site of the U12–U6atac vs those in the U2–U6 snRNA complex, we characterize base-pairing patterns of the U12–U6atac complex of the human, as well as the Arabidopsis (Figure 1C,D), minor spliceosomes, and analyze the geometry within the hinged region to position the catalytically essential elements with respect to that of key elements in the U2–U6 snRNA complex.
Many, but not all, of the snRNP and non-snRNP proteins appear to be the same in both types of spliceosome or are replaced by functional analogues.38 Because of the very low abundance of minor spliceosomes in metazoan cells, far less is known about the protein complement than is known for major spliceosomes, and the presence of RBM22 in the minor spliceosome has not yet been documented.38 We therefore ask whether RBM22 interacts with U12–U6atac analogously to its interaction with U2–U6 snRNA, i.e., whether the different geometry imposed by the difference in the central junction versus hinge impacts on its interaction.
In this report, we show that RBM22 binds to the human U12–U6atac complex with similar affinity as to U2–U6 snRNA. Characterization of these structure–function relationships in the minor spliceosome compared to that of the U2–U6 snRNA complex provides valuable insight into the process and control of pre-mRNA splicing and leads to greater understanding of RNA-mediated catalysis.
Results
Topology of Protein-Free Human and Arabidopsis U12–U6atac snRNA Complexes
Our first goal was to characterize the base-pairing pattern for the human U12–U6atac snRNA complex. We started by analysis of the bimolecular construct representing the human U12–U6atac snRNA complex that includes the key elements required for long-range interactions associated with catalysis, i.e., the ACAGAGA loop, the catalytic AGC triad, and the U6atac ISL (Figure 1B; sequences in the Supporting Information). Analysis was performed by homonuclear and heteronuclear NMR techniques (details in the Experimental Section).39,40 We verified that the U12 and U6atac snRNA strands were fully paired as a homogeneous conformation by migration of the paired complex as a single band, different from that of individual strands, on nondenaturing PAGE (polyacrylamide gel electrophoresis) (Figures S-1 and S-2). A one-dimensional (1D) spectrum of imino protons illustrated peaks of similar areas, consistent with the anticipated number of hydrogen-bonded imino protons undergoing medium-slow exchange (Figure S-3).
1D and two-dimensional (2D) spectra of imino/amino protons displayed line broadening that we attributed to tumbling anisotropy caused by the long single-stranded 5′ region of the full U6atac snRNA strand (originally included because of its equivalence to the U6 snRNA that forms photo-cross-links with the protein RBM22).19 Subsequent NMR studies were carried out using a unimolecular construct in which the 3′ end of the truncated U6atac strand was linked to the U12 snRNA strand by a stable tetraloop (Figure 2A). Folding of the unimolecular U12–U6atac construct into a single conformation was also confirmed by a single band of the anticipated size in nondenaturing PAGE (Figure S-1). Identical chemical shifts were shown by overlaying spectra for the two constructs in 1D and 2D NOESY (nuclear Overhauser effect spectroscopy) and TOCSY (total correlation spectroscopy) spectra, indicating structural equivalence of the areas probed (Figure S-4).
Figure 2.
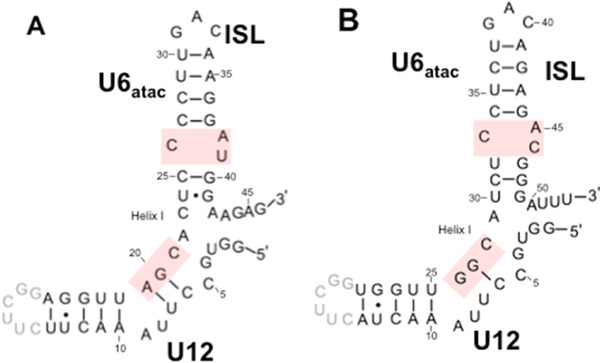
Unimolecular constructs of the (A) human and (B) Arabidopsis U12–U6atac snRNA complexes used for NMR experiments. In each case, a stable UUCG tetraloop (in gray font) was used to connect the 3′ truncated U12 strand and the 5′ end of an abbreviated U6atac strand. The catalytic triad AGC (GGC in Arabidopsis) and ISL bulge elements implicated in folding to form a catalytic complex (highlighted in pink, along with the final two nucleotides of the GGAGAGA loop not included in this construct) are preserved in the unimolecular constructs; numbering of nucleotides for preserved elements in the corresponding snRNAs is the same as in Figure 1B,C/D. Lines denoting Watson–Crick base pairs and dots denoting G·U pairs are those identified in NMR spectra reported here.
Assignments of imino, amino, and adenine H2 protons were made from 2D NOESY spectra of the unimolecular construct (in 95%H2O/5%D2O) by analysis of intra- and internucleotide nOes (Figure 3, top left and bottom panels). Definitive identification of base pairs was aided by the distinctly different 15N chemical shifts of imino protons of uridine versus guanosine in a 2D 15N1H HSQC (heteronuclear single quantum coherence) spectrum (Figure 3, right panel). Sequential nOes were analyzed from 2D NOESY spectra of nonexchangeable protons, assisted by 2D TOCSY, 13C–1H HSQC, and 3D NOESY-HSQC experiments using a uniformly 13C–15N-labeled unimolecular U12–U6atac snRNA construct. Chemical shifts for aromatic, ribose (1′, 2′, and 3′), imino, and amino protons are listed in Table S-1 (SI).
Figure 3.

(Top-left panel) Imino–amino and (bottom-left panel) imino–imino regions of an NOESY spectrum of exchangeable protons of the human U12–U6atac snRNA complex acquired with a mixing time of 150 ms, 10 °C, at 600 MHz (Figure 2A). Base-pairing assignments from these NMR spectra confirm a base-pairing pattern consistent with the fold originally proposed for the human U12–U6atac snRNA complex (Figure 1B). (Bottom-right panel) 1H–15N HSQC spectrum illustrating through-bond interactions between imino 1H and adjoining 15N nuclei was used to confirm assignments of imino protons. Details of the sample preparation and acquisition of NMR spectra are in the Experimental Section.
From NOESY spectra of exchangeable protons, we identified four sharp resonances of imino protons attributed to Watson–Crick base pairs and upfield shifted imino protons attributed to two G·U wobble pairs between U24-G41 and U13-G15 (Figure 2A). We assigned all five A–U, six G–C, and two G·U pairs anticipated for the fold depicted in Figure 1B. Continuous A-type stacking of base pairs was observed by continuous imino–imino and aromatic–anomeric nOes between nucleotides on the 5′ side of the construct, through the ISL hairpin loop; there were breaks in continuity on the 3′ side at the point where the U6atac 3′ and U12 5′ termini separate from the loop and again at the U12 snRNA bulged (“hinge”) region. Base pair interactions defined by exchange-protected imino protons, and the proposed fold, are shown in Figure 2A. This base-pairing pattern suggests a similar sequence and base-pairing environment for each of the critical ion binding regions of the human U12–U6atac complex as seen in the U2–U6 snRNA complex42,41,43 and agrees with the fold originally proposed by Tarn and Steitz.44 To investigate the possibility of deviations from A-type helical parameters in the single-stranded region that may have an impact in the overall folding opposing the 3′ terminus of U6atac and 5′ of U12, we measured the relative intensity of NOESY cross-peaks (Figure 4) between aromatic protons in A–C and C–U steps of the U6atac strand and compared them with cross-peaks of equivalent protons/steps in A-type duplex regions. Using the cytosine H5–H6 distance as a reference, we calculated an increased distance of 4.7 Å for the A22H8–C23H6 step, compared to an equivalent dinucleotide step distance of 3.2 Å in the A-type Helix I of the U12 snRNA segment of this sample. (A table of all distances is in the Supporting Information.) In the single stranded region not constrained by A-type helical parameters, such change could be due to elongation or increased twist45 (see Discussion).
Figure 4.

Aromatic–anomeric (base–H1′) region of an NOESY spectrum of nonexchangeable protons used for analysis of sequential assignments, collected with a mixing time of 250 ms, at 600 MHz, at 25 °C. Nucleotides and connectivities outlined identify base–H1′ nOes involving residues in the single-stranded segment of the human U6atac strand opposing the 5′ and 3′ termini. Decreased intensities of intra- and internucleotide base–H1′ nOes between neighboring nucleotides, relative to equivalent dinucleotide steps in A-type helical regions, suggest that the single-stranded segment opposing the 5′ and 3′ termini adopts a greater elongation between bases. Distances obtained for base–H1′ and base–base nOes in the single-stranded segment of the ISL of human and Arabidopsis U12–U6atac constructs and corresponding steps in duplex regions are included in the text and in Tables S-3 and S-4 of the Supporting Information. Acquisition parameters and sample details are in the Experimental Section, and further annotated spectrum is found in Figure S-5.
To investigate the likelihood that this perturbation of A-type helical parameters is a common feature of U12–U6atac snRNA complexes, we also probed the base-pairing patterns of the U12–U6atac snRNA complex from Arabidopsis. This U12–U6atac snRNA complex provided a unique opportunity because small differences in sequence may have an impact on conformation. The nucleotide sequence AUC (nt 29–31) is found in the U6atac snRNA of the Arabidopsis snRNA complex in the single-stranded region opposing the opening, which may behave differently from ACU (nt 22–24) in the human sequence; in particular, the Arabidopsis U30 in the single-stranded region opposing the 3′ terminus of U6atac and 5′ of U12 (unlike C23 in the equivalent position in the human sequence) allows for an additional A–U Watson–Crick base pair in the ISL, which would promote a shift in base-pairing register by one nucleotide but maintain the five base pairs as in the U2–U6 snRNA complex, as originally postulated by Shukla and Padgett (Figure 1C);33 alternatively, the absence of the additional base pair would result in a fold with the same ISL bulge as in the human U2–U6 snRNA (Figure 1A) and the human U12–U6atac snRNA (Figure 1B); this alternative fold is depicted in Figure 1D.41
As for the human U12–U6atac snRNA complex (Figure 2A), we analyzed the lowest energy fold of the unimolecular Arabidopsis construct shown in Figure 2B by solution NMR. Folding of the unimolecular sample into a single conformation was confirmed by the appearance of a single RNA band on nondenaturing PAGE. Chemical shifts in 1D and 2D spectra for equivalent base pairs were very similar to those of the human U12–U6atac snRNA complex, implying that the substitution of the tetraloop to link the two strands in this construct did not perturb folding patterns. From homonuclear NMR experiments, we assigned exchangeable and nonexchangeable protons. As in the human construct, we assigned five G–C, five A–U, and one G·U pair (there is the potential for three additional G·U pairs, but all three are exchange-broadened beyond detection). These data unequivocally fit a pattern shown in Figure 1D41 in which the base-pairing pattern fully matches the pairing patterns expected for the fold in Figure 1B and not for those anticipated for the fold in Figure 1C.33 If the register were indeed shifted to form the pairing in Figure 1C, we would have expected to observe an additional G·U pair, which, because of mid-helix position, would have been “visible”; however, we see only one G·U pair in Helix I, in the same spectral position and with the same neighbors, as found in the human complex. Chemical shifts for aromatic, ribose (1′, 2′, and 3′), imino, and amino protons for nucleotides in the hinge region of the ISL are listed in Table S-2. We also calculated the distances between sets of protons in this single-stranded C28–A29 and A29–U30 steps and compared the measurements with distances between corresponding protons in stacked regions. The distance for C28H6-A29H8 was 4.6 Å, compared to 3.2 Å for the same protons in the A-type helix step C40H6-A41H8 on the 3′ side of the U6atac hairpin loop, comparable to the increased distance in the analogous region of the human construct. All distance calculations are outlined in SI.
Binding Affinity of NTC-Related Protein RBM22 to Human U2–U6 and U12–U6atac
To quantify and compare the binding affinity of the NTC-related protein RBM22 to RNA constructs representing the U2–U6 snRNA complex of the major spliceosome and the U12–U6atac snRNA complex of the minor spliceosome, we used EMSA and fluorescence assay techniques. Starting with the bimolecular U2–U6 snRNA complex including sequence elements analogous to those in U6 snRNA shown to cross-link with RBM22,19,20 i.e., the single-stranded 5′ terminal region of U6 and the ISL hairpin loop (Figure 1A), we verified complete pairing of strands by migration of a single-shifted band and disappearance of bands corresponding to individual strands on a nondenaturing gel (details in the Experimental Section, Figure S-1). We observed a single band consistent with the size of the full-length RMB22 in nondenaturing PAGE confirming the monomer state of the protein (Figure 5). For each EMSA assay, we mixed 30 μM RNA with protein concentrations ranging from 6 to 42 μM. After electrophoresing on a horizontal two-way nondenaturing polyacrylamide gel (details in the Experimental Section, Figure 5) and staining with EtBr to visualize RNA, we observed a single band for protein-free RNA that migrated toward the anode according to the construct size. Upon adding protein to the RNA sample, we observed a new band migrating at a slower rate (still toward the anode) that increased in intensity with each increase in protein concentration and a concomitant decrease in the intensity of the band representing free RNA. At a concentration ratio of 1.2:1 and 1.4:1 protein/RNA, we noted the absence of the free RNA band and the maximum intensity of the RNA–protein band, suggesting complete binding. As a control for nonspecific interaction, we used an RNA fragment representing U5 snRNA (30 μM, sequence in the Supporting Information) in place of U2–U6 snRNA. There was no observable shift in the protein or RNA bands upon mixing the U5 fragment with RBM22 (Figure S-2), consistent with specificity for the U2–U6–RBM22 interaction. Intensity of each unbound and RBM22-shifted RNA band was quantified by UVP VisionWorks software (uvp.com/visionworks) and plotted data as outlined in Figure 6. Error bars indicate the standard deviation (SD) for three independent experiments.
Figure 5.

EMSA studies to quantify the affinity between human RBM22 and the human U12–U6atac snRNA complex (Figure 1B) were performed with 30 μM U12–U6atac and concentrations of RBM22 ranging from 6 to 42 μM. (Top panel) Horizontal nondenaturing PAGE stained with EtBr visualizes bound (red arrow) and unbound RNA (blue arrow), and (bottom panel) the same gel stained with Coomassie Brilliant Blue displays the bound protein complex (red arrow) and free protein (light blue). Further details are in the Experimental Section.
Figure 6.

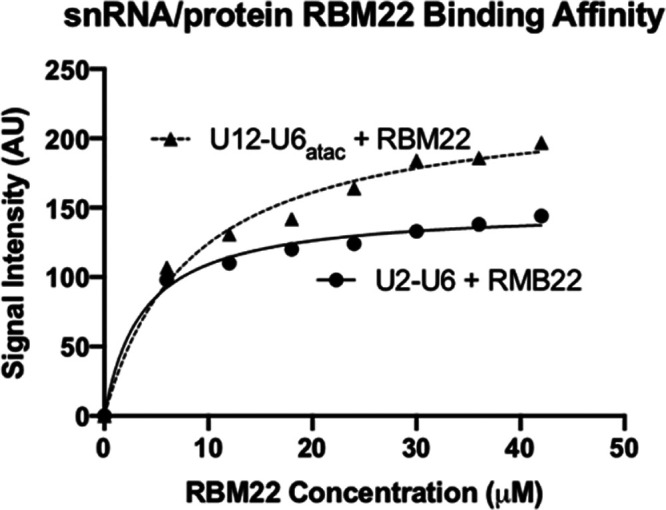
Determination of the dissociation constant Kd for the binding of RBM22 to the U2–U6 snRNA (top) and to the U12–U6atac snRNA (bottom) complex was performed by quantification of the intensity of shifted and unshifted bands from staining by EtBr and then Coomassie Blue (see Figure 1A,B for constructs tested, respectively; see Figure 5 for sample gel). Concentration of the protein added to 30 μM RNA was plotted vs band intensity for staining with EtBr (circles) and Coomassie Blue (triangles) for each RNA complex using GraphPad Prism. The best fit for each set of points was a curve corresponding to a 1:1 stoichiometry. Dissociation constants (Kd) for the binding of U2–U6 snRNA to RBM22 are 3.2 ± 0.5 μM (EtBr)/3.8 ± 0.7 μM (Coomassie), for a mean Kd = 3.5 μM; for the binding of U12–U6atac snRNA to RBM22 are 8.4 ± 0.7 μM (EtBr)/7.9 ± 0.7 μM (Coomassie Blue), for a mean Kd = 8.2 μM. Error bars indicate the SD for three independent experiments.
The same gel was then stained with Coomassie Blue for visualization of protein. We observed RNA-free RBM22 as a single band with minimal migration toward the cathode. For each sample of RNA (30 μM) mixed with increasing aliquots of RBM22 (6–42 μM), the band corresponding to the basic protein (only) disappeared and a band appeared on the anode side in the identical location of the shifted RNA band visualized by EtBr. Only with the final concentrations of 36 and 42 μM (ratios of 1.2:1 and 1.4:1 protein/RNA) did we see evidence of the unbound protein. We quantified the intensity of each Coomassie-stained band with UVP VisionWorks software (uvp.com/visionworks) and plotted the data as before (Figure 6).
We also performed equivalent assays in reverse, i.e., varying RNA concentrations while maintaining a constant protein concentration; however, the quantification of EtBr results was clearer with the concentration of RNA kept low and constant. For each set of quantified bands acquired by the two staining procedures, we plotted band intensity vs protein concentration using GraphPad Prism (details in the Experimental Section; graphpad.com/scientific-software/prism). For the EtBr data, the points were best fit with a line describing a 1:1 stoichiometry and a Kd = 3.2 ± 0.5 μM (Figure 6). For the Coomassie Blue data for the same gel, the best-fit curve indicated a 1:1 stoichiometry and a Kd = 3.8 ± 0.7 μM (also in Figure 6). Thus, independent of the staining method, the RNA–protein interaction was described by similar parameters. We used the same EMSA protocol to test for affinity of RBM22 to the bimolecular U12–U6atac snRNA complex (Figure 1B), an interaction that has not previously been shown. We observed a similar pattern of electrophoretic shifts and specificity as seen with U2–U6, that is, an increase in intensity for a shifted RNA–protein band upon increasing RBM22 concentrations, and a disappearance of the free RNA band. Quantification of intensity and plotting for EtBr and Coomassie Blue staining were performed as above using GraphPad Prism (graphpad.com/scientific-software/prism), revealing Kd = 8.4 ± 0.7 and 7.9 ± 0.7 μM, respectively (Figure 6). Three independent assay experiments were performed for U12–U6atac as with U2–U6, with error bars indicating the SD (Figure 6). Based on these data, we observe that the binding of RBM22 to U2–U6 has an ∼38% greater affinity than to U12–U6atac (while in the same range). Because the sequence elements of the U2–U6 snRNA complex shown to form cross-links are present in the minor spliceosome U12–U6atac snRNA complex, the data suggest that the junction of the U2-U6 snRNA complex (the only region that is markedly different in sequence or topology from the central “hinged” region of the U12-U6atac snRNA complex) may participate in an interaction with the protein.
As a secondary method to probe the binding affinity of RBM22 to the U2–U6 and U12–U6atac snRNA complexes, we measured changes in the intrinsic protein fluorescence derived from endogenous tryptophan upon its binding to the target RNA complexes. Tryptophan fluorescence is strongly influenced by the protein’s environment, and the intensity of endogenous tryptophan can therefore be used to study protein conformational changes such as that resulting from RNA binding. An independent measurement was particularly valuable in this case because results from EMSA can be perturbed (in particular, an underestimate of Kd) as a result of shearing forces as a complex migrates through the gel matrix.46 The RBM22 construct used here contains four tryptophan residues, including one in the RRM that, in the yeast homolog Cwc2, has been shown to interact with U6 snRNA.20 Excitation at 300 nm was utilized to avoid the effect of absorbance by the increasing concentrations of RNA at the optimal tryptophan excitation at 280 nm (Figures S-6 and S-7). As a control to correct for inner filter effects, we used a nonbinding RNA fragment representing U5 snRNA (30 μM, Figure S-8). For each fluorescence assay, we mixed 15–30 μM protein with RNA concentrations ranging from 3 to 42 μM; emission values for each mixture (constant [protein] following incubation with varied [RNA]) reveal a decrease in tryptophan fluorescence intensity upon binding of RMB22 to the U12–U6atac snRNA complex (Figure S-6), up to a total decrease in the fluorescence of 73% upon saturation (∼1.2:1 RNA/protein for each of the RNA complexes). Data for the array of samples for U2–U6 (Figure S-7) and U12–U6atac (using GraphPad Prism) reveal a Kd = 3.1 ± 0.4 and 7.8 ± 0.4 μM, respectively (Figure 7). Error bars indicate the SD for three independent fluorescence assays for both U2–U6 and U12–U6atac. These values are very similar to those obtained by EMSA (varying either RNA or protein and measured by either EtBr or Coomassie Blue), thus minimizing any method-dependent bias in the determination of binding affinity and strengthening the conclusions that RBM22 binds the human U12–U6atac snRNA complex with a modestly lesser affinity than that for the human U2–U6 snRNA complex.
Figure 7.

Determination of the dissociation constant Kd for the binding of RBM22 to the U2–U6 snRNA (solid line) and to the U12–U6atac snRNA (dashed line) complex was performed by quantification of intrinsic protein fluorescence derived from endogenous tryptophan upon its binding to the target RNA complexes. For each fluorescence assay, we mixed 15–30 μM protein with RNA concentrations ranging from 3 to 42 μM. The best fit for each set of points was a curve corresponding to a 1:1 stoichiometry. Data for the array of samples for U2–U6 and U12–U6atac (using GraphPad Prism) reveal a Kd = 3.1± 0.4 and 7.8 ± 0.4 μM, respectively. Error bars indicate the SD for three independent experiments.
Discussion
This work addresses questions about the structural features of the U12–U6atac snRNA complex that forms the catalytic center of the minor spliceosome, in comparison with its counterpart in the major spliceosome, and its recognition by the NTC-related protein RBM22 that is postulated to assist in RNA remodeling prior to the first step of splicing. Since all evidence suggests that the major and minor spliceosomes catalyze pre-mRNA splicing by the same mechanism,32 it is expected that they would share certain common features, but base pairing and topological features have not previously been defined. Using bimolecular (Figure 1B) and unimolecular (Figure 2A) constructs representing the human U12–U6atac snRNA complex, results acquired from homonuclear and heteronuclear NMR data confirm the fold originally proposed by Tarn and Steitz (Figure 1B) that positions the catalytic triad and the bulge in the U6 ISL to parallel the spatial context seen in the major spliceosome U2–U6 snRNA complex.44 Two secondary structural folds had been published for the Arabidopsis sequence (Figure 1C44/D41), but here, we identify a base-pairing pattern equivalent to that in the human U12–U6atac snRNA complex.
We noted that the secondary structural folds of both the human and Arabidopsis U12–U6atac snRNA complexes found here display a spacer of four nucleotides between the paired catalytic triad and the bulge in the ISL stem of the U6atac strand opposing the opening between the 5′ and 3′ termini (Figure 1B); this spacing differs from the five-nucleotide spacing observed in the U6 snRNA strand of the human U2–U6 snRNA complex of the major spliceosome opposing the open central junction (Figure 1A) verified in yeast10,27,47 and human22−24 spliceosomes. This difference raises the question of how the minor spliceosome compensates for the decreased number of nucleotides between the catalytic triad and the ISL bulge to create the active site. We then analyzed the distances between nucleotides within this single-stranded region of each U12–U6atac snRNA complex with regard to A-type helical parameters. Internucleotide distances between pairs of protons in individual steps of the U6atac strand are far larger than those in typical A-type helical parameters in both the human and Arabidopsis U12-U6atac snRNA complexes. These increased distances could be the result of either backbone elongation or increased twist. Elongation would relinquish some of the favorable stacking energy associated with an A-type helix, but may enhance flexibility. Alternatively, an increase in rotation would maintain stacking and may facilitate positioning of key catalytic elements. Calculations by Liebl and co-workers suggest that increased rotation, or overwinding of the RNA, is compensated by increased roll (i.e., tilting of the base pair) toward the major groove resulting in reduced helical rise.45 By their calculations, our increased interproton distances would result in a decrease in calculated helical rise (3.2 Å to 2.7 Å). Our data exhibit a similar pattern in the Arabidopsis U12-U6atac snRNA complex as was seen in the human U12-U6atac counterpart, showing a decreased helical rise (3.3 Å to 2.7 Å). We speculate that these changes in helical rise are consistent with the overwinding model described by Liebl et al., but emphasize that our data do not permit us to differentiate unambiguously between elongation or overwinding at this point.
Unlike the Group II intron, in which the RNA sequence contains all components and information necessary for folding into a splicing-competent ribozyme, the snRNA components of the spliceosome do not form a splicing-competent fold in the absence of specific proteins. Evidence implicates the Prp19 complex in this role, where the liaison between the NTC and U2–U6 snRNA in human spliceosomes is RBM22.16,19 RBM22 is the only NTC-related protein to interact specifically with U2–U6 via with cross-linking experiments;19 however, the results do not exclude the possibility of additional contacts that are not clarified in cryo-EM images. Although the inclusion of RBM22 in minor spliceosomes has been predicted,19 its presence has not previously been verified. For this reason, we measured the binding affinity of RBM22 to the U2–U6 and U12–U6atac snRNA complexes by electrophoretic mobility shift assays, in which we monitored the migration of both protein and RNA components in the same gel and by separate fluorescence assays. Results indicate that RBM22 binds the human U2–U6 and human U12–U6atac snRNA complexes specifically with an overall (of all determinations) Kd = 3.4 and 8.0 μM, respectively. Differences between the affinity of RBM22 for the two RNA complexes are quite modest, but the trend is entirely consistent for all determination obtained by several approaches; adding to our confidence, the data represent a real and relevant difference in affinity correlating with additional interactions in the central region.
Conclusions
We have characterized the base-pairing patterns of the U12–U6atac snRNA complex of both human and Arabidopsis minor spliceosomes and characterized the binding properties of the human NTC-related protein RBM22 to constructs representing human U12–U6atac, an interaction that has not previously been demonstrated, and to U2–U6 snRNA complexes from the human major spliceosome. We have shown an equivalent base-pairing context for each of the sequence elements of the human U12–U6atac complex critical for the formation of the catalytic site as found in the U2–U6 snRNA complex, including an analysis of the single-stranded segment opposing termini of the snRNAs, and show that altered geometry may contribute to flexibility of the region allowing for interaction between the catalytically essential elements anticipated for the formation of the active site in the absence of the central junction found in the U2–U6 snRNA complex of the major spliceosome. Binding assay results indicate that RBM22 binds the human U2–U6 and human U12–U6atac snRNA complexes specifically and in the same range for the two RNA complexes with a mean Kd = 3.4 and 8.0 μM, respectively, suggesting that the protein performs the same role in both spliceosomes. The absence of the central junction in the human U12–U6atac snRNA may help explain its somewhat lesser affinity for the protein and the slower rate of catalysis exhibited by the minor spliceosome than its major counterpart, suggesting that this region also forms a recognition site with the protein.
Experimental Section
Formation of snRNA Complexes
All RNA constructs representing snRNA duplexes were transcribed from double-stranded DNA templates (synthesized by IDT, Inc.) using T7 RNA polymerase expressed and purified in the laboratory. The sequence of human U12 snRNA extends from the natural 5′ terminus to a 3′ end truncated beyond any pairing interactions with itself or with U6atac snRNA. Its pairing partner, U6atac snRNA, includes the full natural sequence until its truncation at A27, 13 nucleotides beyond any pairing interactions within the U6atac snRNA ISL (Figure 1A). Thus, upon pairing, all elements anticipated to be double-stranded and/or involved in the formation of a catalytic center were included in each sequence.
To minimize line broadening in NMR experiments most likely associated with the long string of single-stranded nucleotide regions in the unpaired 5′ region of U6atac, we also designed a unimolecular U12–U6atac snRNA construct that includes the key regions of the native complex in a “chimeric” construct in which the 3′ end of the truncated U6atac strand was linked to the 5′ region of the U12 strand by a stable UUCG tetraloop (Figure 2A). For studies of base-pairing patterns in the U12–U6atac snRNA complex of the plant Arabidopsis, we created a similar unimolecular construct as above, from the native sequence of the Arabidopsis U12 and U6atac sequences (Figure 2B).
For the construct representing the human U2–U6 snRNA complex, we used a U2 snRNA sequence truncated at C45 in Helix III and the full native sequence for U6 snRNA. All sequences are specified in the Supporting Information.
Transcribed RNA was purified by denaturing PAGE. The desired band (identified by UV shadowing) was electroeluted, precipitated with ethanol, exchanged with the preferred buffer for NMR or binding experiments (conditions specified with the description of experiments and in legends) using an Amicon Ultra-4 filter (MW cutoff 3 kDa; Merck Millipore, Ltd.), dried, and resuspended to the final concentration used for solution NMR studies in purified H2O or D2O, or for binding experiments in H2O.
For heteronuclear NMR experiments, uniformly 13C–15N-labeled samples for human unimolecular U12–U6atac snRNAs were transcribed with 13C–15N-labeled NTPs (Cambridge Isotope Laboratories, Inc.) and purified as above.
To analyze pairing and folding, U12–U6atac snRNA samples were heated to 70 °C for 3 min in 10 mM NaPi, 50 mM NaCl, pH 6.5, and cooled at room temperature for 30–45 min. Aliquots were loaded onto a 20% nondenaturing gel and electrophoresed at 100 V for 4 h at 4 °C; the gels were then stained with ethidium bromide (EtBr). For the bimolecular constructs, equimolar amounts of U12 and U6atac strands were paired by the above protocol and pairing was verified by the shifting of bands of individual strands to a single band. For both unimolecular and bimolecular constructs, the formation of a homogeneous fold was confirmed by the appearance of a single band analyzed by nondenaturing PAGE (Figures S-1 and S-2). The number of peaks observable in the imino region of 1D 1H NMR spectra for each snRNA construct was commensurate with all anticipated Watson–Crick and non-Watson–Crick pairs, consistent with a single conformation for each construct.
Acquisition of NMR Spectra
To analyze base-pairing patterns of Arabidopsis and human U12–U6atac snRNA samples, homonuclear and heteronuclear NMR spectra were acquired on a Bruker Avance III 600 MHz spectrometer (Hunter College of CUNY, New York, NY). For the examination of exchangeable protons, the samples were exchanged into 10 mM NaPi, pH 6.5, 50 mM NaCl, dried, and suspended in 95% H2O/5% D2O; for spectra of nonexchangeable protons, the dried samples were suspended in 99.996% D2O. The final RNA concentrations were in the range of 0.2–0.5 mM in 10 mM NaPi, 50 mM NaCl, pH 6.5. The NOESY spectrum (Figure 3, left panel) was acquired with an ∼0.5 mM RNA sample in 10 mM NaPi, 50 mM NaCl, 95% H2O, 5% D2O. The 13C–15N-labeled sample (Figure 3, bottom-right panel) was ∼0.2 mM RNA in 10 mM NaPi, 50 mM NaCl, 95% H2O, 5% D2O. The NOESY spectrum of nonexchangeable protons was acquired with an ∼0.5 mM RNA sample in 10 mM NaPi, 50 mM NaCl, in 99.996% D2O.
One-dimensional spectra of exchangeable 1H were acquired with a 3-9-19 WATERGATE pulse sequence for water suppression. Chemical shift assignments were obtained from two-dimensional NOESY spectra of exchangeable and nonexchangeable experiments. Quadrature detection was achieved using the States-TPPI method.39 The spectra were processed and assigned using Bruker Topspin and NMRFAM Sparky software, by the use of the cytosine H5–H6 vector as a known distance in the bases.40 The distances were calibrated using the mean H5–H6 vector from four cytosine bases. The spectra were apodized using a Gaussian function, and zero filling was performed in both dimensions.
13C–1H-HSQC, 1H–15N HSQC, 13C–1H NOESY-HSQC, and HCCH-TOCSY experiments of a 13C–15N-enriched RNA sample were apodized using a Gaussian function, and zero filling was performed in both dimensions. Two-dimensional 1H–15N HSQC spectra were acquired with a 3-9-19 WATERGATE pulse sequence for water suppression. Spectra were processed and assigned using Bruker Topspin and NMRFAM Sparky software.40
Expression and Purification of RBM22
Escherichia coli cells (Rosetta-2) were transformed with a pETM11 vector bearing the full sequence for human RBM22 (pre-mRNA-splicing factor RBM22, UniProtKB Q9NW64) protein (plasmid was a gift from Prof. Reinhard Lührmann, Max Plank Institute for Biophysical Chemistry). For protein expression, the cells were grown at 37 °C in LB media containing 50 μg/mL kanamycin until 0.6 OD at 600 nm was achieved. Media was adjusted with 200 μM IPTG for induction, and the cells were grown overnight (>16 h) at 17 °C while shaking at a rate of 225 rpm.
Purification was adapted from the protocol of Rasche et al.19 The cells were harvested by centrifugation at 15 000g for 30 min, and the cell pellets were stored at −70 °C. For purification, cell pellets from 2 L of cell growth were resuspended in 30 mL IEX Buffer-A (50 mM HEPES-NaOH pH 7.5, 2 mM β-mercaptoethanol) containing 2 μL RNase I (Ambion) and lysed by sonication with a Sonic Dismembrator (Fisher Scientific) and centrifuged at 15 000g for 1 h. The lysate supernatant fraction was filtered at 0.22 μm and loaded onto an ÄKTA Purifier System provided with a HisTrap SP HP 5 mL column for ion-exchange chromatography. Proteins were eluted with a gradient using 50 mM HEPES-NaOH pH 7.5, 2 mM β-mercaptoethanol, 1 M NaCl. Fractions containing proteins were pooled and loaded on the same system provided with a HisTrap HP 5 mL column for immobilized metal affinity chromatography (IMAC). The protein was eluted with a gradient using 50 mM Tris pH 8.5, 500 mM NaCl, 1 M imidazole. Pooled fractions containing the protein were dialyzed against the working buffer (20 mM phosphate buffer, pH 6.5, 100 mM NaCl, 1 mM DTT, 5% glycerol). Protein purity and integrity were tested by sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and migrated as a single band, with size expected for its molecular mass of 50 264 Da. The protein also ran as a single band on nondenaturing PAGE, at a migration rate consistent with monomer formation.
Measurement of Affinity of Human U2–U6 and U12–U6atac to Protein RBM22
Electrophoretic mobility assay (EMSA) techniques and fluorescence assays were used to quantify the affinity between bimolecular U2–U6 and U12–U6atac RNA constructs and protein RBM22. Horizontal PAGE (5% 19:1 acrylamide/bis-acrylamide in 0.2× MOPS/histidine, pH 6.5) with wells located in the center of the gel allowed for migration of anionic and cationic biomolecules or complexes toward the anode and cathode, respectively.48 Analysis of protein–RNA affinity using EMSA was performed using 30 μM RNA human U12–U6atac or human U2–U6 snRNA complexes, or nonspecific RNA, and RBM22 (6–42 μM) for ratios of 0.2:1–1.4:1 (total volume 15 μL per sample). Gels were electrophoresed at 100 V in a Bio-Rad Mini-Sub Cell System, with running buffer (25 mM histidine, 30 mM MOPS, pH 6.5), at 4 °C for 1.5 h.
Gels were first stained with ethidium bromide and visualized and photographed for RNA migration by transillumination at 305 nm. The gels were then washed in 200 mL H2O fixed in 100 mL of 10% acetic acid/50% methanol for 1 h, followed by staining with 100 mL of 0.05% Coomassie Brilliant Blue R-250 in 10% acetic acid overnight, then destained with 100 mL of 10% acetic acid for 2 h, and washed in 200 mL H2O overnight. The gels were then visualized, and the bands were quantified with a UVP GelDoc-It Imaging System equipped with a Gel HR Camera. Band intensity was plotted using GraphPad Prism for the calculation of dissociation constants (Figure 6).
Affinity measurements were also made by monitoring the changes in the fluorescence of endogenous tryptophan of RBM22 upon mixing with RNA. Assays were performed using 15–30 μM RBM22 and 3–42 μM RNA human U12–U6atac or human U2–U6 snRNA complexes so that each independent sample represents a molar ratio of 0.2:1–1.4:1 (RNA/protein). Fluorescence assays were performed using a NanoDrop 3300 Fluorospectrometer (Thermoscientific) with an excitation at 300 nm and detection of emission at 350 nm (Figure S-3). Fluorescence intensity was plotted using GraphPad Prism for the calculation of dissociation constants. As a control for nonspecific interaction, an RNA fragment representing U5 snRNA (30 μM, sequence in Supporting Information) was used in place of U2–U6 and U12–U6atac snRNAs for EMSA and fluorescence experiments.
Acknowledgments
The authors thank Lenura Ziyadinova, Anjelica Gangaram, Nazir Jalili, and NamHee Kim for their technical assistance with the preparation of RNA and protein samples and EMSA experiments, and the NMR facility at the Hunter College of CUNY and Dr. Matthew Devany for guidance with NMR experiments.
Glossary
Abbreviations Used
- EMSA
electrophoretic mobility shift assay
- EtBr
ethidium bromide
- HSQC
heteronuclear single quantum coherence
- IMAC
immobilized metal affinity chromatography
- NMR
nuclear magnetic resonance
- NOESY
nuclear Overhauser effect spectroscopy
- NTC
NineTeen complex
- PAGE
polyacrylamide gel electrophoresis
- TOCSY
total correlation spectroscopy
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c01674.
Nucleotide sequences for constructs: pairing and folding of RNA samples (Figures S-1–S-8 and Tables S-1–S-4) (PDF)
Accession Codes
RBM22, UniProtKB Q9NW64
The authors acknowledge financial support for this research from the PSC-CUNY grant program (award ENHC-46-84).
The authors declare no competing financial interest.
Supplementary Material
References
- Will C. L.; Lührmann R. Spliceosome structure and function. Cold Spring Harbor Perspect. Biol. 2011, 3, a003707 10.1101/cshperspect.a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papasaikas P.; Válcarcel J. The Spliceosome: The ultimate RNA chaperone and sculptor. Trends Biochem. Sci. 2016, 41, 33–45. 10.1016/j.tibs.2015.11.003. [DOI] [PubMed] [Google Scholar]
- Fica S. M.; Mefford M. S.; Piccirilli J. A.; Staley J. P. Evidence for a group II intron-like catalytic triplex in the spliceosome. Nat. Struct. Mol. Biol. 2014, 21, 464–471. 10.1038/nsmb.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M. J.; Query C. C.; Sharp P. A.. Splicing of Precursors to mRNA by the Spliceosome. In The RNA World, Gesteland R. F.; Atkins J. F., Eds.; Cold Spring Harbor Laboratory Press, Cold Spring Harbor: New York, 1993, pp 303–357. [Google Scholar]
- Steitz T. A.; Steitz J. A. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 6498–6502. 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Vielle A.; Espinosa S.; Zhao R. RNAs in the spliceosome: Insight from cryoEM structures. Wiley Interdiscip. Rev.: RNA 2019, e1523 10.1002/wrna.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agafonov D. E.; Kastner B.; Dybkov O.; Hofele R. V.; Liu W. T.; Urlaub H.; Lührmann R.; Stark H. Molecular Architecture of the human U4/U6.U5 tri-snRNP. Science 2016, 351, 1416–1420. 10.1126/science.aad2085. [DOI] [PubMed] [Google Scholar]
- Fabrizio P.; Abelson J. Thiophosphates in yeast U6 snRNA specifically affect pre-mRNA splicing in vitro. Nucleic Acids Res. 1992, 20, 3659–3664. 10.1093/nar/20.14.3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating K. S.; Toor N.; Perlman P. S.; Pyle A. M. A structural analysis of the group II intron active site and implications for the spliceosome. RNA 2010, 16, 1–9. 10.1261/rna.1791310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fica S. M.; Tuttle N.; Novak T.; Li N. S.; Lu J.; Koodathingal P.; Dai Q.; Staley J. P.; Piccirilli J. A. RNA catalyses nuclear pre-mRNA splicing. Nature 2013, 503, 229–234. 10.1038/nature12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C.; Hang J.; Wan R.; Huang M.; Wong C. C.; Shi Y. Structure of a yeast spliceosome at 3.6-angstrom resolution. Science 2015, 349, 1182–1191. 10.1126/science.aac7629. [DOI] [PubMed] [Google Scholar]
- Rauhut R.; Fabrizio P.; Dybkov O.; Hartmuth K.; Pena V.; Chari A.; Kumar V.; Lee C. T.; Urlaub H.; Kastner B.; Stark H.; Lührmann R. Molecular architecture of the Saccharomyces cerevisiae activated spliceosome. Science 2016, 353, 1399–1405. 10.1126/science.aag1906. [DOI] [PubMed] [Google Scholar]
- Madhani H. D.; Guthrie C. A novel base-pairing interaction between U2 and U6 snRNAs suggests a mechanism for the catalytic activation of the spliceosome. Cell 1992, 71, 803–817. 10.1016/0092-8674(92)90556-R. [DOI] [PubMed] [Google Scholar]
- McPheeters D. S.; Abelson J. Mutational analysis of the yeast U2 snRNA suggests a structural similarity to the catalytic core of group I introns. Cell 1992, 71, 819–831. 10.1016/0092-8674(92)90557-S. [DOI] [PubMed] [Google Scholar]
- Hoskins A. A.; Friedman L. J.; Gallagher S. S.; Crawford D. J.; Anderson E. G.; Wombacher R.; Ramirez N.; Cornish V. W.; Gelles J.; Moore M. J. Ordered and dynamic assembly of single spliceosomes. Science 2011, 331, 1289–1295. 10.1126/science.1198830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves de Almeida R.; O’Keefe R. T. The NineTeen Complex (NTC) and NTC-associated proteins as targets for spliceosomal ATPase action during pre-mRNA splicing. RNA Biol. 2015, 12, 109–114. 10.1080/15476286.2015.1008926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrail J. C.; Krause A.; O’Keefe R. T. The RNA binding protein Cwc2 interacts directly with the U6 snRNA to link the nineteen complex to the spliceosome during pre-mRNA splicing. Nucleic Acids Res. 2009, 37, 4205–4217. 10.1093/nar/gkp341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Kooi C. W.; Ren L.; Xu P.; Ohi M. D.; Gould K. L.; Chazin W. J. The Prp19 WD40 domain contains a conserved protein interaction region essential for its function. Structure 2010, 18, 584–593. 10.1016/j.str.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasche N.; Dybkov O.; Schmitzová J.; Akyildiz B.; Fabrizio P.; Lührmann R. Cwc2 and its human homologue RBM22 promote an active conformation of the spliceosome catalytic centre. EMBO J. 2012, 31, 1591–1604. 10.1038/emboj.2011.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitzová J.; Rasche N.; Dybkov O.; Kramer K.; Fabrizio P.; Urlaub H.; Lührmann R.; Pena V. Crystal structure of Cwc2 reveals a novel architecture of a multipartite RNA-binding protein. EMBO J. 2012, 31, 2222–2234. 10.1038/emboj.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P.; Lu G.; Yan C.; Wang L.; Li W.; Yin P. Structure of the mRNA splicing complex component Cwc2: insights into RNA recognition. Biochem J. 2012, 441, 591–597. 10.1042/BJ20111385. [DOI] [PubMed] [Google Scholar]
- Bertram K.; Agafonov D. E.; Liu W. T.; Dybkov O.; Will C. L.; Hartmuth K.; Urlaub H.; Kastner B.; Stark H.; Lührmann R. Cryo-EM structure of a human spliceosome activated for step 2 of splicing. Nature 2017, 542, 318–323. 10.1038/nature21079. [DOI] [PubMed] [Google Scholar]
- Galej W. P.; Wilkinson M. E.; Fica S. M.; Oubridge C.; Newman A. J.; Nagai K. Cryo-EM structure of the spliceosome immediately after branching. Nature 2016, 537, 197–201. 10.1038/nature19316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Yan C.; Hang J.; Finci L. I.; Lei J.; Shi Y. An Atomic Structure of the Human Spliceosome. Cell 2017, 169, 918–929. 10.1016/j.cell.2017.04.033. [DOI] [PubMed] [Google Scholar]
- Plaschka C.; Lin P. C.; Nagai K. Structure of a pre-catalytic spliceosome. Nature 2017, 546, 617–621. 10.1038/nature22799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai R.; Wan R.; Yan C.; Lei J.; Shi Y. Structures of the fully assembled Saccharomyces cerevisiae spliceosome before activation. Science 2018, 360, 1423–1429. 10.1126/science.aau0325. [DOI] [PubMed] [Google Scholar]
- Yan C.; Wan R.; Bai R.; Huang G.; Shi Y. Structure of a yeast activated spliceosome at 3.5 A resolution. Science 2016, 353, 904–911. 10.1126/science.aag0291. [DOI] [PubMed] [Google Scholar]
- Wan R.; Bai R.; Yan C.; Lei J.; Shi Y. Structures of the catalytically activated yeast spliceosome reveal the mechanism of branching. Cell 2019, 177, 339–351. 10.1016/j.cell.2019.02.006. [DOI] [PubMed] [Google Scholar]
- Wan R.; Yan C.; Bai R.; Huang G.; Shi Y. Structure of a yeast catalytic step I spliceosome at 3.4 Å resolution. Science 2016, 353, 895–904. 10.1126/science.aag2235. [DOI] [PubMed] [Google Scholar]
- Fica S. M.; Oubridge C.; Galej W. P.; Wilkinson M. E.; Bai X. C.; Newman A. J.; Nagai K. Structure of a spliceosome remodelled for exon ligation. Nature 2017, 542, 377–380. 10.1038/nature21078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarn W. Y.; Steitz J. A. Pre-mRNA splicing: the discovery of a new spliceosome doubles the challenge. Trends Biochem. Sci. 1997, 22, 132–137. 10.1016/S0968-0004(97)01018-9. [DOI] [PubMed] [Google Scholar]
- Turunen J. J.; Niemelä E. H.; Verma B.; Frilander M. J. The significant other: splicing by the minor spliceosome. Wiley Interdiscip. Rev.: RNA 2013, 4, 61–76. 10.1002/wrna.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla G. C.; Padgett R. A. Conservation of functional features of U6atac and U12 snRNAs between vertebrates and higher plants. RNA 1999, 5, 525–538. 10.1017/S1355838299982213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incorvaia R.; Padgett R. A. Base pairing with U6atac snRNA is required for 5′ splice site activation of U12-dependent introns in vivo. RNA 1998, 4, 709–718. 10.1017/S1355838298980207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otake L. R.; Scambrova P.; Hashimoto C.; Steitz J. A. The divergent U12-type spliceosome is required for pre-mRNA splicing and is essential for development in Drosophila. Mol. Cell 2002, 9, 439–446. 10.1016/S1097-2765(02)00441-0. [DOI] [PubMed] [Google Scholar]
- Zhao C.; Bachu R.; Popović M.; Devany M.; Brenowitz M.; Schlatterer J. C.; Greenbaum N. L. Conformational heterogeneity of the protein-free human spliceosomal U2-U6 snRNA complex. RNA 2013, 19, 561–573. 10.1261/rna.038265.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu H.; Perea W.; Greenbaum N. L. Role of the central junction in folding topology of the protein-free human U2-U6 snRNA complex. RNA 2020, 26, 836–850. 10.1261/rna.073379.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sashital D. G.; Cornilescu G.; Butcher S. E. U2-U6 RNA folding reveals a group II intron-like domain and a four-helix junction. Nat. Struct. Mol. Biol. 2004, 11, 1237–1242. 10.1038/nsmb863. [DOI] [PubMed] [Google Scholar]
- Will C. L.; Lührmann R. Splicing of a rare class of introns by the U12-dependent spliceosome. Biol. Chem. 2005, 386, 713–724. 10.1515/BC.2005.084. [DOI] [PubMed] [Google Scholar]
- Marion D.; Ikura M.; Tschudin R.; Bax A. Rapid recording of 2D NMR-spectra without phase cycling - application to the study of hydrogen-exchange in proteins. J. Mag. Res. 1989, 85, 393–399. 10.1016/0022-2364(89)90152-2. [DOI] [Google Scholar]
- Lee W.; Tonelli M.; Markley J. L. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. 10.1093/bioinformatics/btu830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppler A.; Nikstad L. J.; Allmann A. M.; Brow D. A.; Butcher S. E. Metal binding and base ionization in the U6 RNA intramolecular stem-loop structure. Nat. Struct. Biol. 2002, 9, 431–435. 10.1038/nsb800. [DOI] [PubMed] [Google Scholar]
- Burke J. E.; Sashital D. G.; Zuo X.; Wang Y. X.; Butcher S. E. Structure of the yeast U2/U6 snRNA complex. RNA 2012, 18, 673–683. 10.1261/rna.031138.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarn W. Y.; Steitz J. A. Highly diverged U4 and U6 small nuclear RNAs required for splicing rare AT-AC introns. Science 1996, 273, 1824–1832. 10.1126/science.273.5283.1824. [DOI] [PubMed] [Google Scholar]
- Liebl K.; Drsata T.; Lankas F.; Lipfert J.; Zacharias M. Explaining the striking difference in twist-stretch coupling between DNA and RNA: A comparative molecular dynamics analysis. Nucleic Acids Res. 2015, 43, 10143–10156. 10.1093/nar/gkv1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman L. M.; Fried M. G. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2007, 2, 1849–1861. 10.1038/nprot.2007.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anokhina M.; Bessonov S.; Miao Z.; Westhof E.; Hartmuth K.; Lührmann R. RNA structure analysis of human spliceosomes reveals a compact 3D arrangement of snRNAs at the catalytic core. EMBO J. 2013, 32, 2804–2818. 10.1038/emboj.2013.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea W.; Greenbaum N. L. Label-free horizontal EMSA for analysis of protein-RNA interactions. Anal. Biochem. 2020, 599, 113736. 10.1016/j.ab.2020.113736. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.