

Abstract
A series of dispiro[indoline-3,2′-pyrrolidine-3′,3″-indolines] was synthesized via a multicomponent polar [3 + 2] cycloaddition (32CA) reaction of isatin derivatives, sarcosine and (E)-3-(2-oxo-2-(pyren-1-yl)ethylidene)indolin-2-one derivatives. The regio- and stereochemistries of the cycloadducts were established on the basis of one-dimensional (1D) (1H-, 13C-, 13C-CRAPT NMR) and two-dimensional (2D) homonuclear and heteronuclear correlation NMR spectrometry experiments (1H–1H gDQFCOSY, 13C–1H-HSQCAD, 13C–1H-HMBCAD, 1H–1H-ROESYAD). The molecular mechanism and regio- and stereoselectivities of the cycloaddition (CA) reaction have been investigated utilizing a density functional theory (DFT) method and were thoroughly explained based on the transition-state stabilities and global/local electrophilicity/nucleophilicity reactivity indices of the reactants.
Introduction
Pyrene is a prominent polynuclear aromatic hydrocarbon and is considered to be one of the most useful skeletons for the construction of fluorogenic chemosensors for a variety of important chemical species.1 In the past several decades, synthesis of pyrrolidine-based heterocycles has been a major source of attraction because they comprise an important class of substances with highly pronounced biological activities, and the pyrrolidine moiety is also the core skeleton of numerous alkaloids.2 Multicomponent [3 + 2] cycloaddition (32CA) reactions are incredibly useful reactions for the synthesis of five-membered heterocycles, usually in a high regio- and stereoselective manner.3 Consequently, polar 32CA of azomethine ylides (generated by the reaction of α-amino acids and carbonyl compounds) to electron-deficient alkenes is considered as one of the most expedient and efficient synthetic protocols for the construction of highly functionalized five-membered heterocycles such as the pyrrolidine ring.4 Moreover, spiropyrrolidine-oxindole derivatives exhibit promising biological applications prospects based on their reported antibiotic, antidiabetic, anticonvulsant, antiviral, antibacterial, anti-inflammatory, antitubercular, and anticancer activities.5 In addition to the selectivity behavior, the understanding of the fundamental principles in the polar 32CA reactions has been gradually developed from prolific studies of the interplay between experimental and theoretical aspects, and it still remains a formidable challenge.6 The steric and electronic effects are two key factors that can impact the regio- and stereoselectivities of these reactions.7 Their regio- and stereochemistries may be controlled either by selecting the appropriate dipole and/or dipolarophile or by conducting the reaction using a catalyst.8
Furthermore, the molecular mechanism and the origins of the regio- and chemoselectivities in 32CA reactions have been theoretically studied based on density functional theory (DFT) reactivity indices9 and molecular electron density theory (MEDT).10
Herein, as a continuation of our research program on 32CA reactions for the synthesis of novel polyfunctionalized spiroheterocycles,11 we report, for the first time, a facile and expeditious protocol for the synthesis of novel pyrene-grafted dispiro[indoline-3,2′-pyrrolidine-3′,3″-indolines] via the 32CA reaction of azomethine ylides (AYs) 3a–d (generated in situ from isatins 1a–d and sarcosine 2) to (E)-3-(2-oxo-2-(pyren-1-yl)ethylidene)indolin-2-ones 4a and 4b. This is followed by extensive theoretical investigations of all possible regio- and stereo-cycloaddition paths using global/local electrophilicity/nucleophilicity reactivity indices and the corresponding transition-state (TS) calculations at the (B3LYP/6-31G(d)) level of theory.
Results and Discussion
Synthesis
The azomethine ylides (AYs) 3a–d were generated in situ from isatin derivatives 1a–d and sarcosine 2 and then trapped with (E)-3-(2-oxo-2-(pyren-1-yl)ethylidene)indolin-2-ones 4a and 4b as dipolarophiles, affording a series of hitherto unknown novel dispiro[indoline-3,2′-pyrrolidine-3′,3″-indolines] bearing pyrene moiety 5a–h (Scheme 1). The reactions were performed under three different conditions to examine the effect of solvent on the reaction time and yields. Different solvents such as toluene (nonpolar), acetonitrile (polar aprotic), and ethanol (polar protic) under reflux conditions were found to exert a substantial influence on the reaction yield. The results of this investigation are shown in Table 1.
Scheme 1. Synthesis of Pyrene-Grafted Dispiro[indoline-3,2′-pyrrolidine-3′,3″-indolines] 5a–h.

Table 1. Synthesis of Pyrene-Grafted Dispiro[indoline-3,2′-pyrrolidine-3′,3″-indolines] 5a–h under Different Conditionsa.
| toluene |
acetonitrile |
ethanol |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| product | R1 | R2 | R3 | time (h) | yield (%) | time (h) | yield (%) | time (h) | yield (%) |
| 5a | H | H | H | 16 | traces | 9 | 65 | 4.5 | 96 |
| 5b | H | H | CH3 | 16 | traces | 9 | 63 | 5 | 91 |
| 5c | CH3 | H | H | 16 | 23 | 9 | 54 | 4 | 89 |
| 5d | CH3 | H | CH3 | 16 | 26 | 9 | 48 | 4.5 | 69 |
| 5e | H | Cl | H | 16 | traces | 12 | 65 | 7 | 95 |
| 5f | H | Cl | CH3 | 16 | traces | 12 | 60 | 7.5 | 82 |
| 5g | CH3 | Cl | H | 16 | 20 | 12 | 61 | 7 | 86 |
| 5h | CH3 | Cl | CH3 | 16 | 23 | 12 | 40 | 7 | 65 |
Reaction conditions: isatins 1a–d (1.2 mmol), sarcosine (2, 1.2 mmol), (E)-3-(2-oxo-2-(pyren-1-yl)ethylidene)indolin-2-ones 4a and 4b (1.0 mmol), solvent (10 mL)/reflux.
As shown in Table 1, the pyrrolidine derivative 5 was obtained as a single regioisomer in good yields (65–96%) with relatively short reaction times (4–7.5 h) when the reactions were carried out in ethanol. Moderate yields (40–65%) were obtained and longer reaction time was observed when acetonitrile was used as a reaction solvent. On the other hand, poor yields (up to 26%) were obtained when the reactions were done in toluene as a reaction medium, even though the reaction time was extended to 16 h. A sensible interpretation of these findings may be attributed to the solvent polarity effect. Thus, as the polarity of the solvent is increased, the azomethine ylide (AY) is increasingly stabilized and consequently increases the percentage of reaction yield. A similar trend is consistently strong with protic solvent because of the strong influence of intermolecular hydrogen bonding. Besides, the electronic demand of the transition states is higher in polar and protic solvents than in nonpolar solvents. In polar protic solvents (e.g., ethanol), the transition state is more relatively stabilized than in aprotic (acetonitrile) and nonpolar (toluene) solvents.12
Regio- and Stereoselectivities
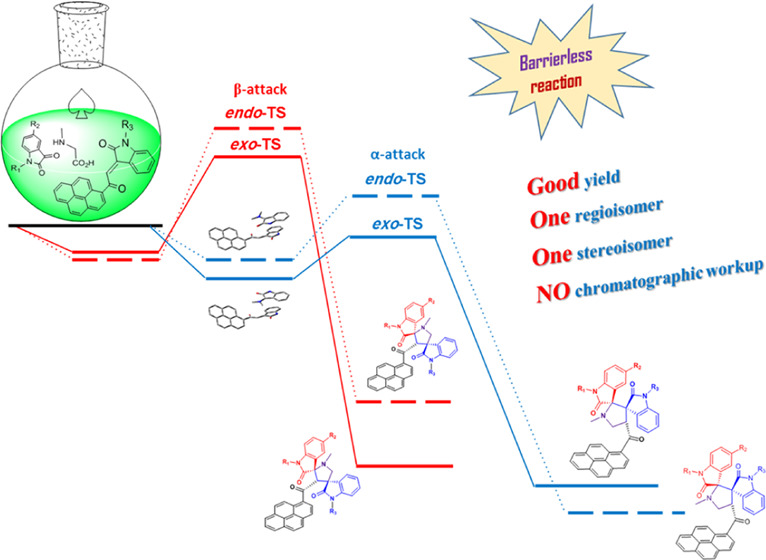
The 32CA reaction of nonstabilized azomethine ylides 3a–d, generated in situ via decarboxylation condensation of isatin derivatives 1a–d with sarcosine (2) to substituted (E)-3-(2-oxo-2-(pyren-1-yl)ethylidene)indolin-2-ones 4a and 4b proceeded in a highly regio- and diastereoselective manner, leads to the exclusive formation of one of four possible diastereomers in all cases. In a surprising twist, and unlike the regiochemical outcome observed previously11d with the β-addition of the same nonstabilized azomethine ylide to (E)-3-aryl-1-(pyren-1-yl)prop-2-en-1-ones as dipolarophiles (Figure 1a), the current cycloaddition (CA) reaction to the (E)-3-(2-oxo-2-(pyren-1-yl)ethylidene)indolin-2-ones demonstrated opposite regioselectivity where the electron-rich carbon atom of the AY reacted with the α-carbon of the enone (Figure 1b). Obviously, a combination of electronic (formation of a highly delocalized enolate anion; Figure 1a) and steric factors (preferential attack at secondary carbon as opposed to a hindered tertiary carbon; Figure 1b) working in concert dictates the preferential formation of one regioisomeric product.
Figure 1.

Comparison of the 32CA reaction of nonstabilized azomethine ylide 3a to (E)-3-aryl-1-(pyren-1-yl)prop-2-en-1-ones (a) versus (E)-3-(2-oxo-2-(pyren-1-yl)ethylidene)indolin-2-ones (b, c).
Although one-step cycloaddition reactions are inherently diastereoselective and the relative stereochemistry of two stereogenic centers are predictable in the above reaction (C4′ and C3′/C3″), one still expects the formation of two diastereomeric products due to the creation of a second spirocenter (C2′/3) during the process (Scheme 1). Interestingly, the exclusive regioselective formation of only one isomer was obtained in all cases. Thus, to fully establish the chemical structures of the products and assign the relative stereochemistry of the stereocenters for the isolated diastereomer in each reaction, extensive one-dimensional (1D) (1H-, 13C-, 13C-CRAPT NMR) and two-dimensional (2D) homonuclear and heteronuclear correlation NMR spectrometry experiments (1H–1H gDQFCOSY, 13C–1H-HSQCAD, 13C–1H-HMBCAD, 1H–1H-ROESYAD) as well as decoupling experiments were conducted in dimethyl sulfoxide (DMSO)-d6 on a selection of compounds (see the Supporting Information (SI)). Hence, using 1,1′-dimethyl-4′-(1-pyrenoyl)-dispiro[indoline-3,2′-pyrrolidine-3′,3″-indoline]-2,2″-dione (5c) as a representative example and a model for the remaining structurally related compounds, the relevant spectra that were used for regiochemical elucidation and stereochemical assignment are shown in Figure 2.
Figure 2.

(a) 1H NMR spectrum of (3S,3′R,4′S)-1,1′-dimethyl-4′-(1-pyrenoyl)-dispiro[indoline-3,2′-pyrrolidine-3′,3″-indoline]-2,2″-dione (5c) (DMSO-d6, 400 MHz); (b) 13C NMR spectrum (DMSO-d6, 100 MHz); (c) 1H–1H-gDQFCOSY NMR spectrum; (d) 1H–13C-HSQCAD NMR spectrum; (e) 1H–13C-HMBCAD NMR spectrum; and (f) 1H–1H-ROESYAD NMR spectrum.
Regiochemical Assignment
Analysis of the 13C (Figure 2, spectrum b, showing selected peak picking) and 13C-CRAPT (see the Supporting Information) NMR spectra confirmed the presence of the expected 37 signals (17 aromatic CH’s, 11 aromatic quaternary carbons, 3 carbonyl carbons, 2 quaternary sp3 spirocenters, 1 methylene, 2 methyl, and 1 methine carbons), which is consistent with all carbons being nonequivalent. The three carbonyl carbons of the ketone and the indolinone rings resonate at δ 200.5 (C10), 177.3 (C2″, identified via long-range coupling with N1″–H), and 173.5 (C2) ppm, respectively. The two spirocyclic carbons of the pyrrolidine ring C2′/C3 (δ 78.2 ppm) and C3′/C3″ (δ 61.9 ppm) resonate furthest downfield relative to the remaining carbon atoms of this ring. While the methylene 13C chemical shift (C5′/δ 53.4 ppm) was clearly identified as the only signal with a negative phase in the 1H–13C-HSQCAD spectrum (Figure 2, spectrum d), the attached diastereotopic protons appeared as two apparent triplets [δ 4.64 (app t, J = 8.7 Hz, 1H, C5′–Hb) and 3.56 (app t, J = 8.1 Hz, 1H, C5′–Ha) ppm] and were correlated in the 1H–13C-HSQCAD NMR spectrum by two contours to the corresponding C5′ carbon (Figure 2, spectrum e). The methine proton signal (δ 5.28), which is the most deshielded aliphatic signal in the 1H NMR due to diamagnetic anisotropy, shows strong long-range 1H–13C heteronuclear multiple bond correlations (Figure 2, spectrum e) with the adjacent ketone (C10, δ 201 ppm, 2JCH), methylene carbon C5′H2 (53.4, 2JCH), spirocenter C3′/C3″ carbon (61.9 ppm, 2JCH), as well as the indolinone carbonyl C2″ carbon (δ 177.2 ppm, 3JCH) and C9″ carbon (124.9 ppm, 3JCH) (Figure 2, spectrum f). The N1′ methyl showed strong correlation contours with the C3/C2′ spirocenter (3JCH) and C5′H2 (3JCH), thereby establishing a perfect match between the chemical shift values and the appropriate pyrrolidine ring atoms when used in conjunction with long-range 1H–13C coupling data observed from the methine proton signal. The 1H–13C heteronuclear multiple bond correlation data of N1″–H of the indolinone ring were instrumental for identifying the chemical shifts of all carbon atoms associated with the indolinone five-membered ring (C2″, C3″ C8″, and C9″); meanwhile, the observed ROSEYAD correlation cross-peaks between N1″–H and C7″–H identified the chemical shift of the latter and triggered partial assignment of C4″–C7″ protons. Accordingly, from the 1H–1H-gCOSYAD correlation of C7″–H (off-diagonal cross-peak at δ 5.90/6.42) (Figure 2, spectrum c), the triplet at δ 6.42 was assigned to the adjacent proton C6″–H. The most diagnostic signal to distinguish the regioisomers was that of the methine group (C4′–H) and its corresponding multiplicity. Thus, on first glance and based on observed geminal and vicinal correlations between C4′–H, C5′–Ha, and C5′–Hb in the gDQFCOSY (Figure 2, spectrum c), one immediately infers that the methine and the methylene groups are spin-coupled, clearly supporting the proposed structure 5 (Scheme 1) and ruling against the nonobserved regioisomer 6, in which the same groups are isolated.
Stereochemical Assignment
Although the C4′–H multiplicity was expected to be doublet of doublets (dd) due to the neighboring nonequivalent diastereotopic protons, it appeared as a highly shifted downfield apparent triplet at δ 5.28 ppm (app t, J = 8.3 Hz, 1H, C4′–H) and was correlated to the carbon at δ 53.2 ppm. The three nonequivalent pyrrolidine protons were totally correlated with the same-spin system by 1H–1H-gDQFCOSY (nine-contour square in the aliphatic region (Figure 2, spectrum c)), and the corresponding 13C chemical shifts were also confirmed based on 1H–13C-gHSQCAD cross-peaks (Figure 2, spectrum d). Clearly, the magnetic anisotropic effect impacted the chemical shifts for the pyrrolidine carbons and more so for its protons and outweighed the local atomic environment effect, which warranted the above detailed NMR investigations. Pleasantly, the N1–CH3 (δ 2.77 ppm/1H NMR) group of the indolinone moiety (Figure 2, spectrum a) provided the only entry point that ultimately led to the unambiguous assignment of the correct stereochemistry for the isolated diastereomer. In the ROSEYAD NMR spectrum (Figure 2, spectrum f), there exists a strong correlation cross-peak between the N1–CH3 protons (δ 2.77 ppm) and H-7 (d, δ 6.75 ppm), and identification of the latter triggers the partial assignment of all of the indolinone ring protons C4–H–C7–H. Therefore, from the 1H–1H-gCOSYAD correlation of C7–H (off-diagonal cross-peak at δ 6.75/7.25) (Figure 2, spectrum c), the overlapped triplet at δ 7.25 was assigned to the adjacent proton C6–H. Thus, it appears that the two overlapped signals in the range δ 7.27–7.25 ppm arise from two different indolinone aromatic rings (vide infra). Thus, from the correlation of the δ 7.25 signal, as evident from the contour at δ 7.27/6.36, the triplet at δ 6.36 was tentatively assigned to C5″–H and suggested that the overlapped signals in the range δ 7.27–7.25 comprise a doublet (C4″–H) and a triplet (C6–H). However, this warranted further investigation, which could only be resolved through 1H–1H homonuclear decoupling experiments (vide infra).
It is noted, though, because of the overlap between two signals at δ 7.27–7.25 ppm, 1H–1H-gDQFCOSY cross-peaks were insufficient to totally correlate the C4–H–C7–H spin system and make a clear distinction between signals belonging to the latter and those stemming from the other indolinone C4″–H–C7″–H spin system. Thus, homonuclear decoupling experiments (Figure 3) were conducted to match the chemical shifts of indolinone protons with their respective positions on the indolinone aromatic ring. Hence, irradiation of C5′–Hb (Figure 3, spectrum b), C4′–H (Figure 3, spectrum c), C7″–H (Figure 3, spectrum d), C5″–H (Figure 3, spectrum e), C6″–H (Figure 3, spectrum f), C7–H (Figure 3, spectrum g), C5–H (Figure 3, spectrum h), C6–H (Figure 3, spectrum i), and C4″–H (Figure 3, spectrum j) resulted in the collapse of C4′–H/C5′–Ha into doublets, C5′–Ha/C5′–Hb into doublets, C6″–H into a doublet, C4″–H into a singlet, C7″–H into a singlet, C6–H into a doublet, C7–H into a singlet, and C6–H into a doublet. These studies led to the unambiguous assignment of the chemical shifts for the indolinones and pyrrolidine protons, even for the overlapped signals at δ 7.27–7.25 ppm.
Figure 3.

1H–1H-homonuclear decoupling experiments of 5c: (a) nondecoupled spectrum; (b) irradiation of C5′–Hb; (c) irradiation of C4′–H; (d) irradiation of C7″–H; (e) irradiation of C5″–H; (f) irradiation of C6″–H; (g) irradiation of C7–H; (h) irradiation of C5–H; (i) irradiation of C4″–H; and C6–H; (j) irradiation of C4–H.
Accordingly, the C4″–H–C7″–H protons were matched with δ 7.27, 6.36, 6.42, and 5.90 ppm, respectively, and the C4–H–C7–H protons were assigned to δ 7.58, 7.06, 7.25, and 6.75 ppm, respectively. As expected, each spin system consisted of two triplets and two doublets, providing further support to the above assignments.
Having identified the chemical shifts for each proton in the preceding three independent spin systems, what remained was to examine the ROSEYAD spectrum (Figure 2, spectrum f) for any significant spatial proximity contours that could be used to assign the relative stereochemistry of the three stereocenters. Indeed, three very strong cross-peaks at δ 7.58/3.56, 7.58/5.28, and 5.28/3.56 ppm indicated the close proximity of C4–H, C5′–Ha, and C4′–H to one another (Figure 4), signifying a syn relationship between the indolinone aromatic ring and C4′–H and setting the relative stereochemistry for the C3/C2′ spirocenter and C4′–H. Further evidence to support the syn relationship between C5′–Ha and C4′–H, which was key for stereochemical assignment, was clear in derivative 5h from the large cis coupling constant (C5′–Ha/C4′–H, 3J = 11.1 Hz), while the C5′–Hb and C4′–H trans coupling constant was much smaller (C5′–Hb/C4′–H, 3J = 6.8 Hz) (Figure 4).
Figure 4.

(a) Relevant correlations observed in the 1H–1H-ROSEYAD spectrum of 5c and (b) truncated signals stemming from C4′–H, C5′–Hb, and C5′–Ha of 5h.
The syn geometry of the C4′–H and C3′/C3″ carbonyl was evident from the absence of relevant off-diagonal cross-peaks with the aromatic protons C4″–H–C7″–H. In addition, the syn geometry was expected since it would be retained as a result of the inherently diasteroselective one-step 32CA reaction of an (E)-azomethine ylide ((E)-AY).
32CA Reaction Mechanism
As suggested in Scheme 2, the regio- and stereochemistries for the formation of pyrrolidine derivatives 5a–h rather than the corresponding adducts 6a–h are described by considering the steric factor and the repulsion force. The formation of pyrrolidine derivatives 5a–h proceeds through path A where the nucleophilic carbon of the AY attacked the less hindered carbon of the ethylene derivatives 4a and 4b (α-attack) via “exo”-transition state. This can be explicated by the fact that the corresponding “endo”-transition state would require extra free energy of activation than the exo-transition state due to the electrostatic repulsion between the cis carbonyl groups increasing the free energy of activation.13 On the other hand, concerning path B where the electron-rich carbon atom of the AY attacked the more hindered site of the ethylene derivatives (β-attack), more free energy of activation would be exerted due to the steric effect of the 3° carbon, which could exclude such a path of attack. In addition, the mechanism is illustrated using orbitals (Figures S42).
Scheme 2. Plausible Mechanism for the Formation of Dispiro[indoline-3,2′-pyrrolidine-3′,3″-indolines] 5a–h.

Computational Studies
Energies of Transition-State Structures
As experimentally substantiated, only the isomer of 5a–h could be isolated and 6a–h was not observed at all. It was postulated that regioselectivity curbs the formation of 6a–h (Scheme 2), whereas stereoselectivity restricts the formation of the 5-endo adduct isomer. Therefore, a computational study has been carried out to elucidate the energetics of the reaction paths A and B via exo- and endo-TS for the formation of 5a and 6a, starting from 3a and 4a. Figure 5 represents the computed free-energy profile of the most stable molecular complexes, transition states (exo-TS and endo-TS), unstable intermediates, and the products of reaction paths A and B.
Figure 5.

Computed free-energy profile of the optimized lowest-energy conformers. In path A, the solid lines connect the intermediates and transition states of the 5a-exo (3S,3′R,4′S) formation path, and the dotted lines connect the intermediates and TS of the 5a-endo (3R,3′R,4′S) formation path. In path B, the solid lines connect the intermediates and transition states of the 6a-exo (3S,3′S,4′R) formation path, and the dotted lines connect the intermediates and TS of the 6a-endo (3R,3′S,4′R) formation path. Relative free energies are given in kcal/mol.
It is obvious from the formation energies (Figure 5) that the nonbonded molecular complex in exo orientation in path A (α attack) is energetically the most favorable orientation, thus confirming the regioselectivity of the reaction. Stereoselectivity can be explained from the activation energies for the formation of 5a and 6a via exo- and endo-TS. The activation energy for the formation of 5a-exo is the lowest among the other stereoisomer 5a-endo as well as 6a-exo/endo (Figure 5). Although energetically, the 5a-endo isomer was found to be more stable than 5a-exo, due to the higher activation energy barrier, it was not formed. Further, as the energy of exo-TS in path A is less than that of the reactants combined (3a + 4a), the formation of 5a-exo appears to be barrierless; however, it is not barrierless for compound 5a-endo or 6a-exo/endo (Figure 5). The effect of substituents on the transition-state stability has been estimated (Figure S41). It is notable that the TS energies varied within 2 kcal/mol for 5b (R1 = CH3), 5c (R3 = CH3), and 5e (R2 = Cl) as examples. Generally, no significant effects of subsituents on the TS have been observed.
The optimized geometries of exo- and endo-TS in paths A and B are given in Figures 6 and 7, respectively. The potential energy surface (PES), as depicted in Figure 8, suggests that the reaction takes place through a one-step two-stage mechanism (Figure 9); first, the 5′···4′ bond is formed and then the 2′···3′ bond is formed.
Figure 6.

Optimized geometries and global electron density transfer (GEDT) at exo-TS (a) and endo-TS (b) in path A. The 5′···4′ and 2′···3′ bond distances are shown with cyan arrows. The green arrow indicates the direction of GEDT.
Figure 7.

Optimized geometries GEDT at exo-TS (a) and endo-TS (b) in path B. The 5′···4′ and 2′···3′ bond distances are shown with cyan arrows. The green arrow indicates the direction of GEDT.
Figure 8.

Potential energy surfaces (PESs) of path A via exo-TS (a) and endo-TS (b) formation reaction as obtained by perturbation of the 5′···4′ (SC2) and 2′···3′ (SC1) bonds. The total electronic energies in the z-axis are given in hartree/particle.
Figure 9.

Potential energy diagram for the 2′···3′ bond length perturbation: (a) the 2′···3′ bond length perturbation in 5a-exo (3S,3′R,4′S) and (b) the 2′···3′ bond length perturbation in 5a-endo (3R,3′R,4′S).
It should be noted at this point that the molecular mechanism of these types of reactions is not well understood. Frequently, the literature is plagued by a confidence in a one-step two-stage mechanism of 32CA,14 and this is regardless of the structure of the cycloaddents (reactants). It is worth mentioning that a body of scientific work challenging this view has been published recently.15 Interestingly, a broad range of mechanisms are now known to occur, proceeding through transition states (TSs) with a range of synchronicities and polarities.16 Consequently, we can suggest that these processes proceed by a one-step mechanism through asynchronous transition states. According to the latest terminology, they should be considered polar but not definitely stepwise processes.
Analysis of the Global/Local Electrophilicity/Nucleophilicity Reactivity Indices
The conceptual density functional theory (CDFT) in the last four decades affords various indices to rationalize and understand the chemical structures.17 The concepts that materialize from this theory have also been extensively used to generate a broad approach to the description of chemical reactivity.18 Accordingly, structures of azomethine ylides 3a–d and ethylene derivatives 4a and 4b in this work were minimized according to the parameters described.
Additionally, theoretical quantitative scales have been shown to be powerful means rationalizing the reactivity of a wide variety of chemical species. These scales have become an appropriate and useful tool as they can be used to justify the electronic aspects of reactivity, selectivity, and their variations induced by field effects due to conformational changes or arising from chemical substitution.
Many global and local reactivity descriptors, defined within the density functional theory (DFT), have been anticipated and shown to be very beneficial in the study and interpretation of reactivity and regioselectivity in polar reactions.19 Well known among these reactivity indices, chemical potential μ, chemical hardness η, and global electrophilicity ω, can be cited.
In this context, the electrophilicity index (ω) has shown to be a powerful theoretical means to predict the electrophilic behavior.20,21 Indeed, ω measures the stabilization in energy when a molecule acquires from the environment a supplementary electronic charge. For this reason, the electrophilicity index takes into account both the propensity of the molecule to acquire an additional electronic charge and the resistance of the system to exchange charge with the environment. The calculated values of μ and η, as well as the electrophilicity index (ω), are presented in Table 2.
Table 2. Global Properties and Global Electrophilicity/Nucleophilicity Indices of Azomethine Ylides 3a–d and Ethylene Derivatives 4a and 4b Involved in 32CA Reactionsa,b,c.
| reactants | μ | η | ω | N |
|---|---|---|---|---|
| Strong Electrophiles | Strong Nucleophiles | |||
| 4a | –4.13 | 2.81 | 3.03 | 5.99 |
| 4b | –4.10 | 2.84 | 2.97 | 6.00 |
| (Z)-3c | –3.36 | 3.21 | 1.76 | 6.55 |
| (Z)-3d | –3.32 | 3.26 | 1.69 | 6.58 |
| (E)-3c | –3.26 | 3.34 | 1.60 | 6.59 |
| (Z)-3a | –3.13 | 3.16 | 1.55 | 6.82 |
| (E)-3d | –3.22 | 3.39 | 1.53 | 6.61 |
| Marginal Electrophiles | ||||
| (Z)-3b | –3.08 | 3.21 | 1.48 | 6.84 |
| (E)-3a | –3.01 | 3.31 | 1.36 | 6.86 |
| (E)-3b | –2.96 | 3.37 | 1.30 | 6.87 |
Electronic chemical potential μ, chemical hardness η, and global electrophilicity/nucleophilicity indices ω and N given in electronvolt. See the text for definitions.
All computations were carried out with the Gaussian 16 suite of programs. Calculations based on the method of DFT were performed using the B3LYP exchange correlation functional, together with the standard 6-31G basis set.
The order of compounds is taken based on the global electrophilicity index ω.
The data summarized in Table 2 reveal that the electronic chemical potentials, μ, of azomethine ylides (AYs) 3a–d (−3.36 < μ < −2.96 eV) are higher than that of ethylene derivatives 4a and 4b, μ = −4.13 and −4.10 eV.
Additionally, the azomethine ylides 3a–d with configuration “E” have an electronic chemical potential higher than its dipole counterparts azomethine ylides 3a–d with configuration “Z”, which means that the electronic flow is again from the E-configuration azomethine ylides 3a–d to (E)-3-(2-oxo-2-(pyren-1-yl)ethylidene)indolin-2-ones 4a and 4b as dipolarophile. Therefore, the ethylene derivatives 4a and 4b act as electrophiles due to the larger value of their ω (2.97 < ω < 3.03) relative to the ω (1.30 < ω < 1.76) values of AYs 3a–d.
It is important to mention that a recent study on the electrophilicity index of reagents involved in CA reactions permitted to develop a unique electrophilicity scale.22a In a study of a set of organic molecules, this scale allowed us to classify these molecules as strong, ω > 1.5 eV, moderate, 0.8 < ω < 1.5 eV, and marginal electrophiles ω < 0.8 eV. Interestingly, this allows us to place our ethylene derivatives 4a and 4b with strong molecules in this scale of electrophilicity, as well as azomethine ylides 3c and d but less than 4a and 4b, while the azomethine ylides 3a and 3b located at the bottom of the electrophilicity scale, which are classified as marginal electrophiles, correspond to good nucleophiles. Moreover, a more systematic study in the same line has used the ω values to classify azomethine ylides and ethylene derivatives on a unique scale.9,19,22,23 On the other hand, the global electron density transfer (GEDT) calculations at TSs have been performed.22b The GEDT value of 0.24e at 5a-exo-TS indicates the polar nature of the reaction (Figures 6 and 7).
Recently, Domingo et al. showed during the study on captodative (CD) ethylenes (Scheme 3), in which the molecule bears more than one functional group with opposite electronic demand, that this type of molecules can behave as good electrophiles and good nucleophiles.24 Therefore, suitable information about the nucleophilicity pattern of reactivity would be desirable. In this sense, the simplest approach to have a different descriptor to give further information about the nucleophilicity has been proposed.24 Based on this idea, an empirical (relative) nucleophilicity index (N) has been introduced, relating the nucleophilicity with the highest occupied molecular orbital (HOMO) energy obtained within the Kohn–Sham scheme.25
Scheme 3. Representative Examples of Captodative (CD) Ethylenes.

A subsequent study involving a wide series of substituted alkenes compounds, as well as substituted aromatic compounds and simple nucleophilic molecules, supported the usefulness of the nucleophilicity N index in the nucleophilicity model.26 In this latter study, such a model permitted to classify organic molecules as strong, N > 3.00 eV, moderate, 2.00 eV < N < 3.00 eV, and marginal nucleophiles, N < 2.00 eV. Therefore, by examining the nucleophilicity descriptor N for these compounds calculated and given in Table 2, we found that the azomethine ylides and ethylene derivatives being classified as a strong nucleophile and azomethine ylide (E)-3b (N = 6.87 eV) represents the best nucleophile of this series. These results are reliable with the expected reactivity pattern (Table 2).
To understand the observed regioselectivity in the cyloaddition reactions, apart from global properties, local parameters of reactivity, including the condensed-to-atom electrophilic and nucleophilic Parr and PY Fukui functions and the local electrophilicity index defined by ωk = ωPk+, are necessary to differentiate the reactive behavior of atoms forming a molecule.27
Several studies have established that the main difference between the regioisomer reaction pathways is essentially related to the orientation of the two asymmetric fragments (dipole and dipolarophile), in which the most favorable regioisomeric reactive channel is that implying the most favorable local electrophilic and nucleophilic interactions.28 Consider, for instance, the interaction between azomethine ylides 3a and 3c, and ethylene derivative 4a, which, according to our classification, will show a global electrophilicity difference Δω = 1.67 and 1.44, respectively. Therefore, an analysis of the local reactivity index in these selected three neutral organic molecules, participating in polar CA reactions, was performed (see the Supporting Information).
From Table 3, it may be seen on the basis of the local descriptor that the highest values of Pk– in azomethine ylides 3a and 3c located at the carbon atom C9 are 0.39 and 0.36, respectively. Furthermore, for the ethylene derivative 4a, C2 carbon represents the electrophilic site with a local electrophilicity value of ωk = 0.34 eV. Therefore, the interaction will take place between the C9 center of azomethine ylides 3a–d and the C2 center of ethylene derivatives 4a and 4b. This result is consistent with the experimental observation, establishing the fact that the CA process between azomethine ylides 3a–d and ethylene derivatives 4a and 4b occurs through interaction between C9–C2ethylene and then C7–C1ethylene, leading to the formation of the pyrrolidine derivatives 5a–h (Scheme 2, Figures 5 and 9).
Table 3. Electrophilic and Nucleophilic Parr and PY Fukui Functions for the Most Relevant Heavy Atoms of Azomethine Ylides 3a and 3c and Ethylene Derivative 4a.

It is concluded that the computed electrophilic/nucleophilic Parr functions and Parr–Yang Fukui functions of the reagents involved in CA reactions can explain not only the regioselectivity and chemoselectivity but also the reactivity based on two-center electrophilic–nucleophilic interactions.
Conclusions
In conclusion, an efficient protocol has been developed for the facile synthesis of functionalized dispiro[indoline-3,2′-pyrrolidine-3′,3″-indolines] via a one-pot three-component 32CA of the azomethine ylides with (E)-3-(2-oxo-2-(pyren-1-yl)ethylidene)indolin-2-one derivatives. The pure products were obtained by recrystallization without requiring further purification techniques. The chemical structures of the obtained regioselective products were confirmed on the basis of both 1D and 2D NMR spectroscopy. DFT calculations showed that the formation energies indicate that 32CAs proceed via a one-step two-stage mechanism through asynchronous transition states and the favored reaction path leads to α-adduct path, the most favorable orientation, which well agrees with experimental observations and confirms the regioselectivity of the reaction. The electrophilic/nucleophilic character of a series of azomethine ylides 3a–d and ethylene derivatives 4a and 4b involved in polar CA reactions has been studied. The global electrophilicity pattern of the azomethine ylides and ethylene derivatives involved in the reactions have been quantitatively established in terms of the electrophilicity index ω. The global nucleophilicity values (N) of molecules used in this work have been calculated. Analysis of the reactivity indices based on the frontier molecular orbitals explains the reactivity of these species in polar CA reactions and has been found to correlate well with the experimental results. It is worth mentioning that the obtained data based on experimental, spectral, and theoretical calculations will open a new window in the field of CA reactions.
Experimental Section
All solvents purchased from Sigma-Aldrich are of spectroscopic grade and used without further purifications. Melting points were determined on a Stuart SMP3 melting point apparatus and are uncorrected. NMR spectra were acquired on a Varian NMR instrument (at 400 MHz for 1H, 100 MHz for 13C) and a Bruker Avance III HD NMR spectrometer (600 MHz for 1H, 150 MHz for 13C) in DMSO-d6 solutions, using residual solvent signals as internal standards. Elemental analyses were performed on a Vario EL v2.3 elemental analyzer; the results were found to be in good agreement with the calculated values (±0.3%). The dipolarophiles (E)-3-(2-oxo-2-(pyren-1-yl)ethylidene)indolin-2-ones 4a and 4b were prepared according to the literature procedure.29
Computational Details
Geometry optimizations were performed on 3a, 4a, 5a, and 6a in vacuo with density functional theory (DFT) using B3LYP exchange correlation functional30 and People’s double ζ basis set (6-31G) with added polarization function on the heavy atoms (B3LYP/6-31G(d)). Frequency calculations were performed on the optimized geometries using the same level of theory. The formation of 5′ → 4′ and 2′ → 3′ bonds in both 5a and 6a was probed by bond length perturbation followed by geometry optimization at each point, generating a potential energy surface (PES). Transition-state structures were optimized using the TS guess geometry obtained from PES31 with the QST3 method, as implemented in Gaussian 16. The nature of the TS was confirmed by computing the frequency modes on the optimized TS geometries using the same level of theory. Further, intrinsic reaction coordinate (IRC) scans were performed in both directions starting from each TS (see the Supporting Information). Coordinates for the optimized geometries are given in the Supporting Information (SI). Relative energies were calculated with respect to the sum of the energies of the separated reactants. The energy values were converted to kilocalories per mole from hartree/particle using the conversion factor of 627.509467.
The global electronic properties of azomethine ylides 3a–d and ethylene derivatives 4a and 4b were estimated according to the equations recommended earlier by Parr and Domingo.17,21,24,27 Both quantities of electronic chemical potential μ and chemical hardness η are obtained from one-electron energies of the frontier molecular orbital HOMO and lowest unoccupied molecular orbital (LUMO), εH and εL (Table S1), as μ = (εH + εL)/2 and η = (εL – εH), respectively.
Next, global electrophilicity (ω) is given using electronic chemical potential (μ) and chemical hardness (η) according to the formula
Subsequently, global nucleophilicity (N)27 can be expressed by the following equation
where tetracyanoethylene (TCE) was taken as reference because it presents the lowest HOMO energy in a long series of organic molecules already investigated in polar CA contexts.
Regional Fukui functions for electrophilic (fk–) and nucleophilic (fk) attacks were obtained from a single-point calculation at the optimized structures of the GS of molecules by a method described elsewhere.32
The electrophilic (Pk+), nucleophilic (Pk), and Parr functions were obtained through the analysis of the Mulliken ASD of the radical anion and the radical cation by single-point energy calculations over the optimized neutral geometries using the unrestricted UB3LYP formalism for radical species.27d
Synthetic Procedure
General Procedure for the Synthesis of Dispiro[indoline-3,2′-pyrrolidine-3′,3″-indoline]-2,2″-dione 5a–h
A mixture of isatin 1 (1.2 mmol), sarcosine 2 (107 mg, 1.2 mmol), and chalcone 4 (1 mmol) in absolute ethanol (10 mL) was stirred at reflux for 4–7.5 h and then cooled to room temperature. The solid formed in the reaction mixture was filtered off, washed with n-hexane, and recrystallized from ethanol to obtain the pure cycloadduct 5a–h.
(3S,3′R,4′S)-1′-Methyl-4′-(1-pyrenoyl)-dispiro[indoline-3,2′-pyrrolidine-3′,3″-indoline]-2,2″-dione (5a)
Yield 525 mg (96%) from 176 mg of 1a and 373 mg of 4a; pale yellow crystals, mp 239–240 °C. 1H NMR (DMSO-d6, 600 MHz) δ 10.05 (s, 1H, N1–H), 9.92 (s, 1H, N1″–H), 8.31–8.20 (m, 4Hpyrene), 8.13–8.08 (m, 5Hpyrene), 7.53 (d, J = 6.6, 1H, C4–H), 7.32 (d, J = 6.6 Hz, 1H, C4″–H), 7.12 (t, J = 6.6 Hz, 1H, C6–H), 6.95 (t, J = 6.6 Hz, 1H, C6″–H), 6.54 (d, J = 6.6 Hz, 1H, C5–H), 6.41 (td, J = 7.2, 1.8 Hz, 1H, C5″–H), 6.36 (d, J = 7.2 Hz, 1H, C7–H), 5.90 (d, J = 7.2 Hz, 1H, C7″–H), 5.23 (app t, J = 7.2 Hz, 1H, C4′–H), 4.59 (app t, J = 8.4 Hz, 1H, C5′–Hb), 3.52 (app t, J = 8.4 Hz, 1H, C5′–Ha), 2.14 (s, 3H, N1′–CH3) ppm. 13C NMR (DMSO-d6, 150 MHz) δ 201.1 (q, C10), 177.9 (q, C2″), 175.6 (q, C2), 143.1 (q, C8), 141.9 (q, C8″), 133.2 (q, Cpyrene), 132.5 (q, Cpyrene), 130.9 (q, Cpyrene), 130.3 (CHpyrene), 129.8 (q, C9), 129.7 (CHpyrene), 128.9 (CH, C6), 128.8 (q, Cpyrene), 128.5 (CH, C4″), 128.2 (CH, C6″), 127.5 (CHpyrene), 127.4 (CH, C4), 127.0 (CHpyrene), 126.7 (CHpyrene), 126.4 (2 × CHpyrene), 125.6 (q, C9″), 125.4 (q, Cpyrene), 125.1 (CHpyrene), 124.1 (q, Cpyrene), 124.0 (CHpyrene), 123.6 (q, Cpyrene), 121.7 (C5H), 120.7 (C5″H), 109.5 (C7H), 108.5 (C7″H), 78.7 (C3/C2′), 62.2 (C3′/C3″), 53.8 (C4′H), 53.7 (C5′H2), 35.3 (N1′–CH3) ppm. Anal. Calcd for C36H25N3O3: C, 78.96; H, 4.60; N, 7.67. Found: C, 78.75; H, 4.39; N, 7.49.
(3S,3′R,4′S)-1′,1″-Dimethyl-4′-(1-pyrenoyl)-dispiro[indoline-3,2′-pyrrolidine-3′,3″-indoline]-2,2″-dione (5b)
Yield 511 mg (91%) from 176 mg of 1a and 387 mg of 4b; yellow crystals, mp 227–228 °C. 1H NMR (DMSO-d6, 400 MHz) δ 10.09 (s, 1H, N1–H), 8.43–8.31 (m, 4Hpyrene), 8.28 (d, J = 9.1 Hz, 1Hpyrene), 8.23 (d, J = 8.1 Hz, 1Hpyrene), 8.19–8.05 (m, 3Hpyrene,), 7.39 (dd, J = 7.5, 0.9 Hz, 1H, C4–H), 7.16 (d, J = 8.0 Hz, 1H, C4″–H), 7.03 (t, J = 7.9 Hz, 1H, C6–H), 6.85 (d, J = 8.0 Hz, 1H, C5–H), 6.54 (t, J = 8.4 Hz, 1H, C6″–H), 6.45 (td, J = 7.6, 1.8 Hz, 1H, C5″–H), 6.35 (d, J = 8.2 Hz, 1H, C7–H), 5.94 (d, J = 8.0 Hz, 1H, C7″–H), 5.27 (app t, J = 8.3 Hz, 1H, C4′–H), 4.58 (app t, J = 8.7 Hz, 1H, C5′–Hb), 3.84 (app t, J = 8.1 Hz, 1H, C5′–Ha), 2.40 (s, 3H, N1″–CH3), 2.04 (s, 3H, N1′–CH3) ppm. 13C NMR (DMSO-d6, 100 MHz) δ 202.4 (q, C10), 176.7 (q, C2″), 175.6 (q, C2), 142.5 (q, C8), 142.4 (q, C8″), 132.8 (q, Cpyrene), 131.6 (q, Cpyrene), 130.5 (q, Cpyrene), 129.8 (CHpyrene), 129.7 (q, C9), 129.3 (CHpyrene), 128.5 (CH, C6), 128.4 (q, Cpyrene), 127.8 (CH, C4″), 127.6 (CH, C6″), 127.0 (CHpyrene), 126.7 (CH, C4), 126.5 (CHpyrene), 126.2 (CHpyrene), 125.9 (2 × CHpyrene), 125.0 (q, C9″), 124.7 (q, Cpyrene), 124.2 (CHpyrene), 123.6 (q, Cpyrene), 123.5 (CHpyrene), 123.2 (q, Cpyrene), 121.6 (C5H), 121.0 (C5″H), 109.4 (C7H), 106.9 (C7″H), 78.1 (C3/C2′), 61.2 (C3′/C3″), 54.0 (C4′H), 53.0 (C5′H2), 34.9 (N1′–CH3), 25.4 (N1″–CH3) ppm. Anal. Calcd for C37H27N3O3: C, 79.13; H, 4.85; N, 7.48. Found: C, 78.94; H, 4.91; N, 7.29.
(3S,3′R,4′S)-1,1′-Dimethyl-4′-(1-pyrenoyl)-dispiro[indoline-3,2′-pyrrolidine-3′,3″-indoline]-2,2″-dione (5c)
Yield 499 mg (89%) from 193 mg of 1b and 373 mg of 4a; pale yellow, mp 233–234 °C. 1H NMR (DMSO-d6, 400 MHz) δ 9.95 (s, 1H, N1″–H), 8.35–8.33 (m, 1Hpyrene), 8.32–8.31 (m, 2Hpyrene), 8.23 (d, J = 9.1 Hz, 1Hpyrene), 8.15 (d, J = 9.5 Hz, 1Hpyrene), 8.13–8.12 (m, 2Hpyrene), 8.11 (d, J = 2.9 Hz, 1Hpyrene), 8.09 (d, J = 1.7 Hz, 1Hpyrene), 7.58 (dd, J = 7.5, 0.9 Hz, 1H, C4–H), 7.27 (d, J = 7.1 Hz, 1H, C4″–H), 7.25 (td, J = 7.7, 1.2 Hz, 1H, C6–H), 7.06 (td, J = 7.6, 0.9 Hz, 1H, C5–H), 6.75 (d, J = 7.6 Hz, 1H, C7–H), 6.42 (td, J = 7.6, 1.4 Hz, 1H, C6″–H), 6.36 (td, J = 7.6, 1.2 Hz, 1H, C5″–H), 5.90 (dd, J = 7.5, 0.7 Hz, 1H, C7″–H), 5.28 (app t, J = 8.3 Hz, 1H, C4′–H), 4.64 (app t, J = 8.7 Hz, 1H, C5′–Hb), 3.56 (app t, J = 8.1 Hz, 1H, C5′–Ha), 2.77 (s, 3H, N1–CH3), 2.11 (s, 3H, N1′–CH3) ppm. 13C NMR (DMSO-d6, 100 MHz) δ 200.5 (q, C10), 177.3 (q, C2″), 173.5 (q, C2), 144.0 (q, C8), 141.4 (q, C8″), 132.9 (q, Cpyrene), 132.1 (q, Cpyrene), 130.5 (q, Cpyrene), 129.8 (q, C9), 129.5 (CHpyrene), 129.4 (CHpyrene), 128.5 (q, Cpyrene), 128.4 (CH, C6), 127.9 (CH, C4″), 127.7 (CH, C6″), 127.0 (CHpyrene), 126.7 (CH, C4), 126.4 (CHpyrene), 126.3 (CHpyrene), 126.0 (2 × CHpyrene), 124.9 (q, C9″), 124.7 (CHpyrene), 124.1 (q, Cpyrene), 123.7 (q, Cpyrene), 123.6 (CHpyrene), 123.2 (q, Cpyrene), 122.0 (C5H), 120.1 (C5″H), 108.2 (C7H), 108.1 (C7″H), 78.2 (C3/C2′), 61.9 (C3′/C3″), 53.4 (C5′H2), 53.2 (C4′H), 34.9 (N1′–CH3), 25.3 (N1–CH3) ppm. Anal. Calcd for C37H27N3O3: C, 79.13; H, 4.85; N, 7.48. Found: C, 79.02; H, 4.60; N, 7.35.
(3S,3′R,4′S)-1,1′,1″-Trimethyl-4′-(1-pyrenoyl)-dispiro[indoline-3,2′-pyrrolidine-3′,3″-indoline]-2,2″-dione (5d)
Yield 397 mg (69%) from 193 mg of 1b and 387 mg of 4b; pale yellow, mp 249–250 °C. 1H NMR (DMSO-d6, 400 MHz) δ 8.42–8.36 (m, 3Hpyrene), 8.34 (d, J = 12.0 Hz, 1Hpyrene), 8.28 (d, J = 12.0 Hz, 1Hpyrene), 8.19–8.11 (m, 3Hpyrene), 8.07 (d, J = 8.0 Hz, 1Hpyrene), 7.39 (d, J = 8.0 Hz, 1H), 7.21–7.14 (m, 2H), 7.00 (td, J = 8.0, 4.0 Hz, 1H), 6.97–6.87 (m, 2H), 6.74 (d, J = 8.0 Hz, 1H), 6.31 (d, J = 8.0 Hz, 1H), 5.56 (dd, J = 12.0, 8.0 Hz, 1H, C4′–H), 3.88 (dd, J = 8.0, 8.0 Hz, 1H, C5′–Hb), 3.66 (dd, J = 12.0, 8.0 Hz, 1H, C5′–Ha), 2.98 (s, 3H, N1–CH3), 2.51 (s, 3H, N1″–CH3), 2.02 (s, 3H, N1′–CH3) ppm. 13C NMR (DMSO-d6, 100 MHz) δ 202.4 (q, C10), 175.0 (q, C2″), 174.7 (q, C2), 144.1 (q, C8), 143.8 (q, C8″), 132.5 (q, Cpyrene), 132.3 (q, Cpyrene), 130.6 (q, Cpyrene), 130.1 (q, C9), 130.0 (CHpyrene), 129.2 (CHpyrene), 128.7 (2xCH, C4″H & C6H), 127.7 (CHpyrene), 127.2 (CH, C6″), 126.8 (CHpyrene), 126.3 (CH, C4), 126.0 (CHpyrene), 125.6 (CHpyrene), 124.9 (q, C9″), 124.5 (CHpyrene), 124.3 (CHpyrene), 123.7 (q, Cpyrene), 123.6 (CHpyrene), 123.5 (CHpyrene), 123.4 (CHpyrene), 123.1 (q, Cpyrene), 122.3 (C5H), 121.5 (C5″H), 108.4 (C7H), 107.6 (C7″H), 78.0 (C3/C2′), 60.1 (C3′/C3″), 53.8 (C5′H2), 53.3 (C4′H), 35.1 (N1′–CH3), 25.4 (N1″–CH3), 25.3 (N1–CH3) ppm. Anal. Calcd for C38H29N3O3: C, 79.28; H, 5.08; N, 7.30. Found: C, 79.00; H, 4.87; N, 7.10.
(3S,3′R,4′S)-5-Chloro-1′-methyl-3′-(1-pyrenoyl)-dispiro[indoline-3,2′-pyrrolidine-4′,3″-indoline]-2,2″-dione (5e)
Yield 553 mg (95%) from 217 mg of 1c and 373 mg of 4a; pale yellow, mp 229–230 °C. 1H NMR (DMSO-d6, 600 MHz) δ 10.60 (s, 1H, N1–H), 10.34 (s, 1H, N1″–H), 8.45 (d, J = 9.0 Hz, 1Hpyrene), 8.38 (d, J = 7.2 Hz, 2Hpyrene), 8.31–8.28 (m, 2Hpyrene), 8.23 (d, J = 6.6 Hz, 1Hpyrene), 8.15–8.13 (m, 3Hpyrene), 7.44 (d, J = 7.2 Hz, 1H, C4″–H), 7.25 (dd, J = 8.4, 2.4 Hz, 1H, C6–H), 7.18 (d, J = 8.4 Hz, 1H, C4–H), 7.05 (t, J = 6.6 Hz, 1H, C6′–H), 6.89 (t, J = 6.6 Hz, 1H, C5′–H), 6.59 (d, J = 7.2 Hz, 1H, C7–H), 6.44 (d, J = 7.2 Hz, 1H, C7″–H), 5.51 (dd, J = 11.4, 7.6 Hz, 1H, C4′–H), 3.64 (dd, J = 7.6, 7.6 Hz, 1H, C5′–Hb), 3.58 (dd, J = 11.4, 7.6 Hz, 1H, C5′–Ha), 2.01 (s, 3H, N1′–CH3) ppm. 13C NMR (DMSO-d6, 150 MHz) δ 203.2 (q, C10), 177.4 (q, C2″), 177.1 (q, C2), 143.3 (q, C8), 142.3 (q, C8″), 133.1 (q, Cpyrene), 133.0 (q, C9), 131.1 (CHpyrene), 130.5 (q, Cpyrene), 130.1 (CHpyrene), 129.7 (CH, C6), 129.3 (CH, C4″), 129.1 (CH, C6″), 128.5 (C–Cl), 127.6 (CH, C4), 127.2 (CHpyrene), 126.8 (CHpyrene), 126.4 (Cpyrene), 126.2 (q, Cpyrene), 126.07 (CHpyrene), 125.7 (q, C9″), 125.0 (CHpyrene), 124.7 (CHpyrene), 124.6 (q, Cpyrene), 124.4 (CHpyrene), 124.2 (q, Cpyrene), 123.9 (q, Cpyrene), 121.5 (C5″H), 111.4 (C7H), 109.4 (C7″H), 78.3 (C3/C2′), 60.7 (C3′/C3″), 54.5 (C5′H2), 53.9 (C4′H), 35.4 (N1′–CH3) ppm. Anal. Calcd for C36H24ClN3O3: C, 74.29; H, 4.16; N, 7.22; Cl, 6.09. Found: C, 74.05; H, 3.98; N, 7.30; Cl, 6.16.
(3S,3′R,4′S)-5-Chloro-1′,1″-dimethyl-4′-(1-pyrenoyl)-dispiro[indoline-3,2′-pyrrolidine-3′,3″-indoline]-2,2″-dione (5f)
Yield 489 mg (82%) from 217 mg of 1c and 387 mg of 4b; yellow cystals, mp 254–255 °C. 1H NMR (DMSO-d6, 400 MHz) δ 10.59 (br s, 1H, N1–H), 8.36–8.30 (m, 3Hpyrene), 8.28 (d, J = 9.2 Hz, 1Hpyrene), 8.23 (d, J = 9.2 Hz, 1Hpyrene), 8.13–8.05 (m, 3Hpyrene), 8.02 (d, J = 8.0 Hz, 1Hpyrene), 7.44 (d, J = 7.2 Hz, 1H, C4″–H),7.08 (dd, J = 8.4, 2.4 Hz, 1H, C6–H), 7.04 (d, J = 2.4 Hz, 1H, C4–H), 7.00 (t, J = 7.2 Hz, 1H, C6′–H), 6.90 (t, J = 7.2 Hz, 1H, C5′–H), 6.50 (d, J = 8.4 Hz, 1H, C7–H), 6.36 (d, J = 8.0 Hz, 1H, C7″–H), 5.48 (dd, J = 12.0, 8.0 Hz, 1H, C4′–H), 3.77 (dd, J = 8.0, 8.0 Hz, 1H, C5′–Hb), 3.56 (dd, J = 12.0, 8.0 Hz, 1H, C5′–Ha), 2.52 (s, 3H, N1″–CH3), 2.00 (s, 3H, N1′–CH3) ppm. 13C NMR (DMSO-d6, 100 MHz) δ 202.2 (q, C10), 176.4 (q, C2″), 174.8 (q, C2), 143.8 (q, C8), 141.7 (q, C8″), 132.6 (q, Cpyrene), 132.2 (q, C9), 130.6 (CHpyrene), 130.1 (q, Cpyrene), 129.7 (CHpyrene), 129.2 (CH, C6), 128.8 (CH, C4″), 128.7 (CH, C6″), 127.7 (C–Cl), 127.1 (CH, C4), 126.8 (CHpyrene), 126.4 (CHpyrene), 126.2 (Cpyrene), 126.1 (q, Cpyrene), 126.0 (CHpyrene), 125.7 (q, C9″), 125.6 (CHpyrene), 125.0 (CHpyrene), 124.7 (q, Cpyrene), 124.1 (CHpyrene), 124.2 (q, Cpyrene), 123.4 (q, Cpyrene), 121.9 (C5″H), 111.0 (C7H), 107.8 (C7″H), 78.0 (C3/C2′), 60.0 (C3′/C3″), 53.8 (C5′H2), 53.2 (C4′H), 35.1 (N1′–CH3), 25.4 (N1″–CH3) ppm. Anal. Calcd for C37H26ClN3O3: C, 74.55; H, 4.40; N, 7.05; Cl, 5.95. Found: C, 74.39; H, 4.25; N, 6.87; Cl, 5.70.
(3S,3′R,4′S)-5-Chloro-1,1′-dimethyl-4′-(1-pyrenoyl)-dispiro[indoline-3,2′-pyrrolidine-3′,3″-indoline]-2,2″-dione (5g)
Yield 512 mg (86%) from 234 mg of 1d and 373 mg of 4a; pale yellow cystals, mp 231–232 °C. 1H NMR (DMSO-d6, 400 MHz) δ 10.05 (br s, 1H, N1″–H), 8.36–8.31 (m, 2Hpyrene), 8.26–8.20 (m, 3Hpyrene), 8.17–8.07 (m, 4Hpyrene), 7.62 (d, J = 2.4 Hz, 1H, C4–H),7.34 (dd, J = 8.4, 2.4 Hz, 1H, C6–H), 7.27 (d, J = 7.6 Hz, 1H, C4–H), 6.82 (d, J = 8.4 Hz, 1H, C7–H), 6.48 (t, J = 7.6 Hz, 1H, C6″–H), 6.42 (t, J = 7.6 Hz, 1H, C5″–H), 5.92 (d, J = 8.0 Hz, 1H, C7″–H), 5.29 (t, J = 8.0 Hz, 1H, C4′–H), 4.58 (t, J = 8.0 Hz, 1H, C5′–Hb), 3.57 (t, J = 8.0 Hz, 1H, C5′–Ha), 2.77 (s, 3H, N1–CH3), 2.13 (s, 3H, N1′–CH3) ppm. 13C NMR (DMSO-d6, 100 MHz) δ 202.8 (q, C10), 176.9 (q, C2″), 175.0 (q, C2), 143.2 (q, C8), 142.8 (q, C8″), 132.7 (q, Cpyrene), 132.6 (q, C9), 130.7 (q, Cpyrene), 130.1 (q, Cpyrene), 129.8 (CHpyrene), 129.4 (CHpyrene) 129.3 (CH, C6), 128.9 (CH, C4″), 128.8 (CH, C6″), 128.1 (q, Cpyrene) 127.2 (CHpyrene), 126.9 (CH, C4), 126.7 (C–Cl), 126.1 (CHpyrene), 125.7 (q, Cpyrene), 125.5 (q, C9″), 125.4 (CHpyrene), 125.1 (CHpyrene), 124.2 (CHpyrene), 124.0 (CHpyrene), 123.9 (q, Cpyrene), 123.8 (CHpyrene), 123.5 (q, Cpyrene), 121.0 (C5″H), 110.1 (C7H), 109.1 (C7″H), 77.9 (C3/C2′), 60.7 (C3′/C3″), 54.1 (C4′H), 53.8 (C5′H2), 35.1 (N1′–CH3), 25.6 (N1–CH3) ppm. Anal. Calcd for C37H26ClN3O3: C, 74.55; H, 4.40; N, 7.05; Cl, 5.95. Found: C, 74.43; H, 4.20; N, 6.90; Cl, 5.81.
(3S,3′R,4′S)-5-Chloro-1,1′,1″-trimethyl-4′-(1-pyrenoyl)-dispiro[indoline-3,2′-pyrrolidine-3′,3″-indoline]-2,2″-dione (5h)
Yield 396 mg (65%) from 234 mg of 1d and 387 mg of 4b; pale yellow crystals, mp 267–268 °C. 1H NMR (DMSO-d6, 400 MHz) δ 8.43–8.37 (m, 3Hpyrene), 8.34 (d, J = 8.0 Hz, 1Hpyrene), 8.29 (d, J = 8.0 Hz, 1Hpyrene), 8.17 (d, J = 4.0 Hz, 1Hpyrene), 8.16–8.15 (m, 1Hpyrene), 8.14 (d, J = 4.0 Hz, 1Hpyrene), 8.09 (d, J = 8.0 Hz, 1Hpyrene), 7.38 (d, J = 7.2 Hz, 1H, C4″–H), 7.27 (dd, J = 8.4, 2.4 Hz, 1H, C6–H), 7.12 (d, J = 2.4 Hz, 1H, C4–H), 7.04 (t, J = 8.0 Hz, 1H, C6″–H), 6.93 (t, J = 7.6 Hz, 1H, C5″–H), 6.79 (d, J = 8.4 Hz, 1H, C7–H), 6.40 (d, J = 8.0 Hz, 1H, C7″–H), 5.57 (dd, J = 11.1, 6.7 Hz, 1H, C4′–H), 3.86 (dd, J = 8.6, 6.8 Hz, 1H, C5′–Hb), 3.66 (dd, J = 11.1, 8.7 Hz, 1H, C5′–Ha), 2.97 (s, 3H, N1–CH3), 2.56 (s, 3H, N1″–CH3), 2.04 (s, 3H, N1′–CH3) ppm. 13C NMR (DMSO-d6, 100 MHz) δ 202.2 (q, C10), 174.7 (q, C2″), 174.6 (q, C2), 143.6 (q, C8), 143.0 (q, C8″), 132.6 (q, Cpyrene), 132.2 (q, Cpyrene), 130.6 (q, Cpyrene), 130.1 (q, C9), 129.9 (CHpyrene), 129.3 (CHpyrene), 129.0 (q, Cpyrene), 128.8 (CH, C6), 127.8 (CH, C4″), 127.2 (CH, C6″), 127.0 (C–Cl), 126.8 (CH, C4), 126.5 (CHpyrene), 126.4 (CHpyrene), 126.1 (2 × CHpyrene), 125.6 (q, C9″), 125.3 (CHpyrene), 124.6 (q, Cpyrene), 124.5 (q, Cpyrene), 123.6 (CHpyrene), 123.5 (q, Cpyrene), 123.4 (CHpyrene), 121.7 (C5″H), 110.1 (C7H), 107.9 (C7″H), 78.0 (C3/C2′), 60.3 (C3′/C3″), 53.7 (C5′H2), 53.4 (C4′H), 35.2 (N1′–CH3), 25.5 (N1″–CH3), 25.3 (N1–CH3) ppm. Anal. Calcd for C38H28ClN3O3: C, 74.81; H, 4.63; N, 6.89; Cl, 5.81. Found: C, 74.66; H, 4.50; N, 6.90; Cl, 5.70.
Acknowledgments
The authors are highly indebted to the Deanship of the Scientific Research (DSR), Umm Al-Qura University, for the financial support through the project number 19-SCI-1-01-0008. S.A.A. is highly indebted to Alexander von Humboldt Foundation (AvH) and Prof. Dr. Karola Rück-Braun, Technical University Berlin (TU-Berlin), Germany, for their continuous help and support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c03510.
NMR spectra of the synthesized compounds; potential energy surfaces (PES) of path B; intrinsic reaction coordinates (IRCs) connecting the reactant and the products via exo/endo-TS; potential energy diagram for the 2′···3′ bond length perturbation in 6a-exo and 6a-endo; coordinates of the optimized geometries; frontier orbital energies (eV) for AYs 3a–d and ethylene derivatives 4a and 4b; effect of substitutions on the energetics of the cycloaddition reaction; Parr function of 3a–c, 4a, and 4b; and condensed-to-atom Fukui functions of 3a, 3c, and 4a (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Weng J.; Mei Q.; Ling Q. Q.; Fan Q.; Huang W. A new colorimetric and fluorescent ratiometric sensor for Hg2+ based on 4-pyren-1-yl-pyrimidine. Tetrahedron 2012, 68, 3129–3134. 10.1016/j.tet.2011.12.071. [DOI] [Google Scholar]
- a Cui C.-B.; Kakeya H.; Osada H. Novel mammalian cell cycle inhibitors, spirotryprostatins A and B, produced by Aspergillus fumigatus, which inhibit mammalian cell cycle at G2/M phase. Tetrahedron 1996, 52, 12651–12666. 10.1016/0040-4020(96)00737-5. [DOI] [Google Scholar]; b Xue J.; Zhang Y.; Wang X.-I.; Fun H. K.; Xu J. H. Photoinduced reactions of 1-acetylisatin with phenylacetylenes. Org. Lett. 2000, 2, 2583–2586. 10.1021/ol000110a. [DOI] [PubMed] [Google Scholar]; c Klumpp D. A.; Yeung K. Y.; Prakash G. K. S.; Olah G. A. Preparation of 3, 3-diaryloxindoles by superacid-induced condensations of isatins and aromatics with a combinatorial approach. J. Org. Chem. 1998, 63, 4481–4484. 10.1021/jo980588g. [DOI] [Google Scholar]
- Padwa A.; Pearson W. H.. Synthetic Applications of 1,3 Dipolar Cycloaddition Chemistry toward Heterocycles and Natural Products; John Wiley & Sons, Inc., 2002. [Google Scholar]
- a Nájera C.; Sansano J. M. Katalytische enantioselektive 1,3-dipolare Cycloaddition von Azomethinyliden und Alkenen: der direkte Weg zu enantiomerenangereicherten mehrfach substituierten Prolinderivaten. Angew. Chem. 2005, 117, 6428–6432. 10.1002/ange.200501074. [DOI] [Google Scholar]; b Gothelf K. V.Cycloaddition Reactions in Organic Synthesis; Wiley-VCH: Weinheim, 2002; pp 211–247. [Google Scholar]; c Gothelf K. V.; Jørgensen K. A. Asymmetric 1, 3-dipolar cycloaddition reactions. Chem. Rev. 1998, 98, 863–910. 10.1021/cr970324e. [DOI] [PubMed] [Google Scholar]; d Lakshmi N. V.; Thirumurugan P.; Perumal P. T. An expedient approach for the synthesis of dispiropyrrolidine bisoxindoles, spiropyrrolidine oxindoles and spiroindane-1,3-diones through 1,3-dipolar cycloaddition reactions. Tetrahedron Lett. 2010, 51, 1064–1068. 10.1016/j.tetlet.2009.12.079. [DOI] [Google Scholar]; e Poornachandran M.; Raghunathan R. Synthesis of spirooxindolo/spiroindano nitro pyrrolizidines through regioselective azomethine ylide cycloaddition reaction. Synth. Commun. 2007, 37, 2507–2517. 10.1080/00397910701462575. [DOI] [Google Scholar]
- a Murugan R.; Anbazhagan S.; Narayanan S. Synthesis and in vivo antidiabetic activity of novel dispiropyrrolidines through [3 + 2] cycloaddition reactions with thiazolidinedione and rhodanine derivatives. Eur. J. Med. Chem. 2009, 44, 3272–3279. 10.1016/j.ejmech.2009.03.035. [DOI] [PubMed] [Google Scholar]; b Karthikeyan K.; Kumar R. S.; Muralidharan D.; Perumal P. T. Diastereoselective synthesis of pyrrolidines via 1, 3-dipolar cycloaddition of a chiral azomethine ylide. Tetrahedron Lett. 2009, 50, 7175–7179. 10.1016/j.tetlet.2009.10.030. [DOI] [Google Scholar]; c Girgis A. S. Regioselective synthesis of dispiro[1H-indene-2,3′-pyrrolidine-2′,3″-[3H]indole]-1,2″(1″H)-diones of potential anti-tumor properties. Eur. J. Med. Chem. 2009, 44, 91–100. 10.1016/j.ejmech.2008.03.013. [DOI] [PubMed] [Google Scholar]; d Kumar R. R.; Peruma S.; Manju S. C.; Bhatt P.; Yogeeswari P.; Sriram D. An atom economic synthesis and antitubercular evaluation of novel spiro-cyclohexanones. Bioorg. Med. Chem. Lett. 2009, 19, 3461–3465. 10.1016/j.bmcl.2009.05.018. [DOI] [PubMed] [Google Scholar]; e Aicher T. D.; Knorr D. C.; Smith H. C. Synthesis of chiral tetrahydropyrrolo[2,1-b]thiazol-5(6H)-ones. Tetrahedron Lett. 1998, 39, 8579–8580. 10.1016/S0040-4039(98)01962-5. [DOI] [Google Scholar]; f Baldwin J. E.; Freeman R. T.; Lowe C.; Schofield C. J.; Lee E. A γ-lactam analogue of the penems possessing antibacterial activity. Tetrahedron 1989, 45, 4537–4550. 10.1016/S0040-4020(01)89088-8. [DOI] [Google Scholar]; g Trapani G.; Franco M.; Latrofa A.; Carotti A.; Cellamare S.; Serra M.; Ghiani C. A.; Tuligi G.; Biggio G.; Liso G. Synthesis and Anticonvulsant Activity of Some 1,2,3,3a-Tetrahydropyrrolo[2,1-b]-benzothiazol-, -thiazol-or -oxazol–1–ones in Rodents. J. Pharm. Pharmacol. 1996, 48, 834–840. 10.1111/j.2042-7158.1996.tb03984.x. [DOI] [PubMed] [Google Scholar]
- a Bentabed-Ababsa G.; Derdour A.; Roisnel T.; Saez J. A.; Perez P.; Chamorro E.; Domingo L. R.; Mongin F. A combined experimental and theoretical study of the polar [3 + 2] cycloaddition of electrophilically activated carbonyl ylides with aldehydes and imines. J. Org. Chem. 2009, 74, 2120–2133. 10.1021/jo8027104. [DOI] [PubMed] [Google Scholar]; b Nacereddine A. K.; Yahia W.; Bouacha S.; Djerourou A. A theoretical investigation of the regio- and stereoselectivities of the 1,3-dipolar cycloaddition of C-diethoxyphosphoryl-N-methylnitrone with substituted alkenes. Tetrahedron Lett. 2010, 51, 2617–2621. 10.1016/j.tetlet.2010.03.025. [DOI] [Google Scholar]; c Benchouk W.; Mekelleche S. M.; Aurell M. J.; Domingo L. R. Understanding the regio-and chemoselective polar [3 + 2] cycloaddition of the Padwa carbonyl ylides with α-methylene ketones. A DFT study. Tetrahedron 2009, 65, 4644–4651. 10.1016/j.tet.2009.04.033. [DOI] [Google Scholar]; d Merino P.; Revuelta J.; Tejero T.; Chiacchio U.; Rescifinab A.; Romeoc G. A DFT study on the 1,3-dipolar cycloaddition reactions of C-(methoxycarbonyl)-N-methyl nitrone with methyl acrylate and vinyl acetate. Tetrahedron 2003, 59, 3581–3592. 10.1016/S0040-4020(03)00547-7. [DOI] [Google Scholar]
- Bakthadoss M.; Sivakumar N. Novel Regio- and Stereoselective Synthesis of Functionalized 3-Spiropyrrolidines and 3-Spiropyrrolizidines Using the Baylis-Hillman Adducts Derived from Nitroolefins. Synlett. 2009, 1014–1018. 10.1055/s-0028-1088206. [DOI] [Google Scholar]
- a Yan X.-X.; Peng Q.; Zhang Y.; Zhang K.; Hong W.; Hou X.-L.; Wu Y.-D. A Highly Enantio- And Diastereoselective Cu-catalyzed 1,3-dipolar Cycloaddition of Azomethine Ylides With Nitroalkenes. Angew. Chem., Int. Ed. 2006, 45, 1979–1983. 10.1002/anie.200503672. [DOI] [PubMed] [Google Scholar]; b Li W.; Shi M. Brønsted acid TfOH-mediated [3 + 2] cycloaddition reactions of diarylvinylidenecyclo-propanes with nitriles. J. Org. Chem. 2008, 73, 4151–4154. 10.1021/jo800390c. [DOI] [PubMed] [Google Scholar]; c Tsubogo T.; Saito S.; Seki K.; Yamashita Y.; Kobayashi S. Development of catalytic asymmetric 1, 4-addition and [3 + 2] cycloaddition reactions using chiral calcium complexes. J. Am. Chem. Soc. 2008, 130, 13321–13332. 10.1021/ja8032058. [DOI] [PubMed] [Google Scholar]; d Yamashita Y.; Guo X.-X.; Takashita R.; Kobayashi S. Chiral silver amide-catalyzed enantioselective [3 + 2] cycloaddition of α-aminophosphonates with olefins. J. Am. Chem. Soc. 2010, 132, 3262–3263. 10.1021/ja100101n. [DOI] [PubMed] [Google Scholar]; e Ghandi M.; Yari A.; Rezaei S. J. T.; Taheri A. Synthesis of novel spiropyrrolidine/pyrrolizine-oxindole scaffolds through 1,3-dipolar cycloadditions. Tetrahedron Lett. 2009, 50, 4724–4726. 10.1016/j.tetlet.2009.06.033. [DOI] [Google Scholar]
- a Domingo L. R.; Aurell M. J.; Perez P.; Contreras R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels–Alder reactions. Tetrahedron 2002, 58, 4417–4423. 10.1016/S0040-4020(02)00410-6. [DOI] [Google Scholar]; b Pérez P.; Domingo L. R.; Aurell M. J.; Contreras R. Quantitative characterization of the global electrophilicity pattern of some reagents involved in 1,3-dipolar cycloaddition reactions. Tetrahedron 2003, 59, 3117–3125. 10.1016/S0040-4020(03)00374-0. [DOI] [Google Scholar]; c Arroyo P.; Picher M. T.; Domingo L. R. The domino reaction between 4, 6-dinitrobenzofuroxan and cyclopentadiene. Insights on the nature of the molecular mechanism. J. Mol. Struct.: THEOCHEM 2004, 709, 45–52. 10.1016/j.theochem.2003.10.072. [DOI] [Google Scholar]; d Arroyo P.; Picher M. T.; Domingo L. R.; Terrier F. A DFT study of the polar diels–alder reaction between 4-aza-6-nitrobenzofuroxan and cyclopentadiene. Tetrahedron 2005, 61, 7359–7365. 10.1016/j.tet.2005.05.080. [DOI] [Google Scholar]
- Domingo L. R. Molecular electron density theory: A Modern View of Reactivity in Organic Chemistry. Molecules 2016, 21, 1319 10.3390/molecules21101319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hussein E. M.; Abdel-Monem M. I. Regioselective synthesis and anti-inflammatory activity of novel dispiro[pyrazolidine-4,3′-pyrrolidine-2′,3″-indoline]-2″,3,5-triones. Arkivoc 2011, 10, 85–98. 10.3998/ark.5550190.0012.a07. [DOI] [Google Scholar]; b Hussein E. M.; Ahmed S. A.; El Guesmi N.; Khairou K. S. 1,3-Dipolar cycloaddition approach to novel dispiro[pyrazolidine-4,3′-pyrrolizidine-2′,3″-indoline]-2″,3,5-triones. J. Chem. Res. 2017, 41, 346–351. 10.3184/174751917X14951017434315. [DOI] [Google Scholar]; c Hussein E. M.; Ahmed S. A.; Althagafi I. I. A convenient regioselective synthesis of novel spirooxindolinopyrrolizidines incorporating the pyrene moiety through [3 + 2]-cycloaddition reaction. Heterocycl. Commun. 2017, 23, 379–384. 10.1515/hc-2017-0036. [DOI] [Google Scholar]; d Hussein E. M.; Moussa Z.; El Guesmi N.; Ahmed S. A. Facile access to regio- and stereoselective synthesis of highly functionalized spiro[indoline-3,2′-pyrrolidines] incorporating a pyrene moiety: experimental, photophysical and theoretical approach. RSC Adv. 2018, 8, 24116–24127. 10.1039/C8RA04312D. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Hussein E. M.; Moussa Z.; Ahmed S. A. Exclusive regioselective 1,3-dipolar cycloaddition of 9-diazo-9H-fluorene and diphenyldiazomethane to 2-arylideneindane-1,3-diones: new approach toward effective synthesis of novel spiropyrazole derivatives. Monatsh. Chem. 2018, 149, 2021–2030. 10.1007/s00706-018-2249-0. [DOI] [Google Scholar]
- Vogel P.; Houk K. N.. Organic Chemistry: Theory, Reactivity and Mechanisms in Modern Synthesis; Wiley-VCH Verlag GmbH & Co. KGaA: Germany, 2019. [Google Scholar]
- a Kumar R. R.; Perumal S.; Senthilkumar P.; Yogeeswari P.; Sriram D. A facile synthesis and antimycobacterial evaluation of novel spiro-pyrido-pyrrolizines and pyrrolidines. Eur. J. Med. Chem. 2009, 44, 3821–3829. 10.1016/j.ejmech.2009.05.010. [DOI] [PubMed] [Google Scholar]; b He J.; Ouyang G.; Yuan Z.; Tong R.; Shi J.; Ouyang L. A Facile Synthesis of Functionalized Dispirooxindole Derivatives via a Three-Component 1,3-Dipolar Cycloaddition Reaction. Molecules 2013, 18, 5142–5154. 10.3390/molecules18055142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisgen R.1,3-Dipolar Cycloaddition Chemistry; Wiley: New York, 1984. [Google Scholar]
- a Jasiński R. In the searching for zwitterionic intermediates on reaction paths of [3 + 2] cycloaddition reactions between 2,2,4,4-tetramethyl-3-thiocyclobutanone S-methylide and polymerizable olefins. RSC Adv. 2015, 5, 101045 10.1039/C5RA20747A. [DOI] [Google Scholar]; b Jasiński R. Competition between the one-step and two-step, zwitterionic mechanisms in the [2 + 3] cycloaddition of gem-dinitroethene with (Z)-C,N-diphenylnitrone: a DFT computational study. Tetrahedron 2013, 69, 927–932. 10.1016/j.tet.2012.10.095. [DOI] [Google Scholar]; c Jasiński R. A stepwise, zwitterionic mechanism for the 1,3-dipolar cycloaddition between (Z)-C-4-methoxyphenyl-N-phenylnitrone and gem-chloronitroethene catalysed by 1-butyl-3-methylimidazolium ionic liquid cations. Tetrahedron Lett. 2015, 56, 532–535. 10.1016/j.tetlet.2014.12.007. [DOI] [Google Scholar]; d Darù A.; Roca-López D.; Tejero T.; Merino P. Revealing stepwise mechanisms in dipolar cycloaddition reactions: Computational study of the reaction between nitrones and isocyanates. J. Org. Chem. 2016, 81, 673–680. 10.1021/acs.joc.5b02645. [DOI] [PubMed] [Google Scholar]; e Khlebnikov A. F.; Koneva A. S.; Virtseva A. A.; Yufit D. S.; Mlostoń G.; Heimgartner H. Concerted vs. non-concerted 1,3-dipolar cycloadditions of azomethine ylides to electron-deficient dialkyl-2,3-dicyanobut-2-enedioates. Helv. Chim. Acta 2014, 97, 453–470. 10.1002/hlca.201300405. [DOI] [Google Scholar]
- a Domingo L. R.; Ríos-Gutiérrez M.; Pérez P. A new model for C–C bond formation processes derived from the molecular electron density theory in the study of the mechanism of [3 + 2] cycloaddition reactions of carbenoid nitrile ylides with electron-deficient ethylenes. Tetrahedron 2016, 72, 1524–1532. 10.1016/j.tet.2016.01.061. [DOI] [Google Scholar]; b Domingo L. R. Theoretical study of the 1,3-dipolar cycloaddition reactions of azomethine ylides. A DFT study of reaction between trifluoromethyl thiomethyl azomethine ylide and acronitrile. J. Org. Chem. 1999, 64, 3922–3929. 10.1021/jo9822683. [DOI] [Google Scholar]; c Jasiński R.; Jasińska E.; Dresler E. A DFT computational study of the molecular mechanism of [3 + 2] cycloaddition reactions between nitroethene and benzonitrile N-oxides. J. Mol. Model. 2017, 23, 13 10.1007/s00894-016-3185-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo L. R.; Ríos-Gutiérrez M.; Pérez P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748 10.3390/molecules21060748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chattaraj P. K.Chemical Reactivity Theory: A Density Functional View; CRC Press, 2009. [Google Scholar]; b Parr R. G.; Yang W.. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, 1989. [Google Scholar]; c De Proft F.; Geerlings P. Conceptual and computational DFT in the study of aromaticity. Chem. Rev. 2001, 101, 1451–1464. 10.1021/cr9903205. [DOI] [PubMed] [Google Scholar]; d Geerlings P.; De Proft F.; Langenaeker W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1873. 10.1021/cr990029p. [DOI] [PubMed] [Google Scholar]; e Geerlings P.; Fias S.; Boisdenghien Z.; De Proft F. Conceptual DFT: chemistry from the linear response function. Chem. Soc. Rev. 2014, 43, 4989–5008. 10.1039/c3cs60456j. [DOI] [PubMed] [Google Scholar]; f Frau J.; Glossman-Mitnik D. Conceptual DFT descriptors of amino acids with potential corrosion inhibition properties calculated with the latest minnesota density functionals. Front. Chem. 2017, 5, 16 10.3389/fchem.2017.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Parr R. G.; Pearson R. G. Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. 10.1021/ja00364a005. [DOI] [Google Scholar]; b Pérez P.; Domingo L. R.; Aizman A.. The Electrophilicity Index in Organic Chemistry. In Theoretical Aspects of Chemical Reactivity; Toro-Labbe A., Ed.; Elsevier: Amsterdam, 2007; Vol. 9, pp 139–201. [Google Scholar]; c Pérez P.; Domingo L. R.; Duque-Noreña M.; Chamorro E. A condensed-to-atom nucleophilicity index. An application to the director effects on the electrophilic aromatic substitutions. J. Mol. Struct.: THEOCHEM 2009, 895, 86–91. 10.1016/j.theochem.2008.10.014. [DOI] [Google Scholar]
- Maynard A. T.; Huang M.; Rice W. G.; Covell D. G. Reactivity of the HIV-1 nucleocapsid protein p7 zinc finger domains from the perspective of density-functional theory. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 11578–11583. 10.1073/pnas.95.20.11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parr R. G.; von Szentpaly L.; Liu S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. 10.1021/ja983494x. [DOI] [Google Scholar]
- a Domingo L. R.; Aurell M. J.; Perez P.; Contreras R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels–Alder reactions. Tetrahedron 2002, 58, 4417–4423. 10.1016/S0040-4020(02)00410-6. [DOI] [Google Scholar]; b Domingo L. R. A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. 10.1039/C4RA04280H. [DOI] [Google Scholar]
- a Steglenko D. V.; Kletsky M. E.; Kurbatov S. V.; Tatarov A. V.; Minkin V. I.; Goumont R.; Terrier F. A theoretical and experimental study of the polar Diels–Alder cycloaddition of cyclopentadiene with nitrobenzodifuroxan. J. Phys. Org. Chem. 2009, 22, 298–307. 10.1002/poc.1469. [DOI] [Google Scholar]; b Lakhdar S.; Terrier F.; Vichard D.; Berionni G.; El Guesmi N.; Goumont R.; Boubaker T. The Diels–Alder Reaction of 4, 6-Dinitrobenzofuroxan with 1-Trimethylsilyloxybuta-1, 3-diene: A Case Example of a Stepwise Cycloaddition. Chem. – Eur. J. 2010, 16, 5681–5690. 10.1002/chem.200903008. [DOI] [PubMed] [Google Scholar]
- a Domingo L. R.; Chamorro E.; Pérez P. Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J. Org. Chem. 2008, 73, 4615–4624. 10.1021/jo800572a. [DOI] [PubMed] [Google Scholar]; b Domingo L. R.; Pérez P. The nucleophilicity N index in organic chemistry. Org. Biomol. Chem. 2011, 9, 7168–7175. 10.1039/c1ob05856h. [DOI] [PubMed] [Google Scholar]; c Chattaraj P. K.; Duleya S.; Domingo L. R. Understanding local electrophilicity/nucleophilicity activation through a single reactivity difference index. Org. Biomol. Chem. 2012, 10, 2855–2861. 10.1039/c2ob06943a. [DOI] [PubMed] [Google Scholar]
- Kohn W.; Sham L. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. 10.1103/PhysRev.140.A1133. [DOI] [Google Scholar]
- Jaramillo P.; Domingo L. R.; Hamorro C. E.; Pérez P. A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J. Mol. Struct.: THEOCHEM 2008, 865, 68–72. 10.1016/j.theochem.2008.06.022. [DOI] [Google Scholar]
- a Pérez P.; Toro-Labbé A.; Aizman A.; Contreras R. Comparison between experimental and theoretical scales of electrophilicity in benzhydryl cations. J. Org. Chem. 2002, 67, 4747–4752. 10.1021/jo020255q. [DOI] [PubMed] [Google Scholar]; b Domingo L. R.; Aurell M. J.; Perez P.; Contreras R. Quantitative Characterization of the Local Electrophilicity of Organic Molecules. Understanding the Regioselectivity on Diels–Alder Reactions. J. Phys. Chem. A 2002, 106, 6871–6875. 10.1021/jp020715j. [DOI] [Google Scholar]; c Chattaraj P. K.; Maiti B.; Sarkar U. Philicity: a unified treatment of chemical reactivity and selectivity. J. Phys. Chem. A 2003, 107, 4973–4975. 10.1021/jp034707u. [DOI] [Google Scholar]; d Domingo L. R.; Pérez P.; Sáez J. A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. 10.1039/C2RA22886F. [DOI] [Google Scholar]
- a Ríos-Gutiérrez M.; Domingo L. R.; Esseffar M.; Oubella A.; Ait Itto M. Y. Unveiling the Different Chemical Reactivity of Diphenyl Nitrilimine and Phenyl Nitrile Oxide in [3 + 2] Cycloaddition Reactions with (R)-Carvone through the molecular electron density theory. Molecules 2020, 25, 1085 10.3390/molecules25051085. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Domingo L. R.; Ríos-Gutiérrez M.; Pérez P. A molecular electron density theory study of the role of the copper metalation of azomethine ylides in [3 + 2] cycloaddition reactions. J. Org. Chem. 2018, 83, 10959–10973. 10.1021/acs.joc.8b01605. [DOI] [PubMed] [Google Scholar]; c Ríos-Gutiérrez M.; Nasri L.; Nacereddine A. K.; Djerourou A.; Domingo L. R. A molecular electron density theory study of the [3 + 2] cycloaddition reaction between an azomethine imine and electron deficient ethylenes. J. Phys. Org. Chem. 2018, 31, e3830 10.1002/poc.3830. [DOI] [Google Scholar]; d Domingo L. R.; Ríos-Gutiérrez M.; Pérez P. A molecular electron density theory study of the reactivity and selectivities in [3 + 2] cycloaddition reactions of C,N-dialkyl nitrones with ethylene derivatives. J. Org. Chem. 2018, 83, 2182–2197. 10.1021/acs.joc.7b03093. [DOI] [PubMed] [Google Scholar]; e Nasri L.; Ríos-Gutiérrez M.; Khorief Nacereddine A.; Djerourou A.; Domingo L. R. A molecular electron density theory study of [3 + 2] cycloaddition reactions of chiral azomethine ylides with β-nitrostyrene. Theor. Chem. Acc. 2017, 136, 104 10.1007/s00214-017-2133-8. [DOI] [Google Scholar]
- Lindwall H. G.; Maclennan J. S. A Condensation of acetophenone with isatin by the Knoevenagel method. J. Am. Chem. Soc. 1932, 54, 4739–4744. 10.1021/ja01351a034. [DOI] [Google Scholar]
- a Miehlich B.; Savin A.; Stoll H.; Preuss H. Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. 10.1016/0009-2614(89)87234-3. [DOI] [Google Scholar]; b Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Banerji B.; Chandrasekhar K.; Killi S. K.; Pramanik S. K.; Uttam P.; Sen S.; Maiti N. C. Silver-catalysed azide–alkyne cycloaddition (AgAAC): assessing the mechanism by density functional theory calculations. R. Soc. Open Sci. 2016, 3, 160090 10.1098/rsos.160090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Contreras R.; Fuentealba P.; Galván M.; Pérez P. A direct evaluation of regional Fukui functions in molecules. Chem. Phys. Lett. 1999, 304, 405–413. 10.1016/S0009-2614(99)00325-5. [DOI] [Google Scholar]; b Fuentealba P.; Pérez P.; Contreras R. On the condensed Fukui function. J. Chem. Phys. 2000, 113, 2544–2551. 10.1063/1.1305879. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.