

Abstract
Small Molecular Accurate Recognition Technology (SMART 2.0) has recently been introduced as a NMR-based machine learning tool for the discovery and characterization of natural products. We attempted targeted isolation of sesquiterpene lactones from Eupatorium fortunei with the aid of structural annotation by SMART 2.0 and chemical profiling. Eight germacrene-type (1–7 and 10) and two eudesmane-type sesquiterpene lactones (8 and 9) were isolated from the whole plant of Eupatorium fortunei. With the guidance of the results of the subfractions from E. fortunei obtained by SMART 2.0, their cytotoxic activities were evaluated against five cancer cells (SKOV3, A549, PC3, HEp-2, and MCF-7). Compounds 4 and 8 exhibited IC50 values of 3.9 ± 1.2 and 3.9 ± 0.6 μM against prostate cancer cells, PC3, respectively. Compound 7 showed good cytotoxicity with IC50 values of 5.8 ± 0.1 μM against breast cancer cells, MCF-7. In the present study, the rapid annotation of the mixture of compounds in a fraction by the NMR-based machine learning tool helped the targeted isolation of bioactive compounds from natural products.
Introduction
Natural products have been spotlighted as potential sources providing new insights for the development of therapeutic agents. Multiple chemicals in natural products have hindered the discovery of novel druggable candidates. Recently, natural product chemists have shown interest in the development of new in silico approaches for the rapid annotation of novel compounds and dereplication of the known ones from natural products. In silico methods for spectrometric data generated using nuclear magnetic resonance (NMR) and mass spectrometry (MS), which are the two most popular techniques for characterizing natural products, are being developed.1−4 Although high-resolution MS is the powerful technique for the measurement of tons of metabolites and production of their structural data in a short time, low reproducibility and interpretability have become the weak points for the accurate identification of compounds.5 Meanwhile, NMR spectroscopy guarantees higher reproducibility compared to MS, but it also needs to acquire expertise for the interpretation of 1D and 2D NMR data, which has become a big barrier for novice chemists in the fields of natural products. Recently, Small Molecule Accurate Recognition Technology (SMART 2.0) has been introduced as the NMR-based machine learning study for the structural annotation of compounds in mixtures or unknown compounds.6,7 SMART 2.0 trained on 1H-13C HSQC spectral data of tons of natural products provided the insights for elucidating unknown compounds. We expected this tool to help in simplifying the dereplication step and provide the bypass for the isolation of the targeted compounds in the natural product research.
Eupatorium fortunei Turcz. (Compositae) is widely distributed in Southeast Asian countries, such as Korea, Japan, and China. Eupatorium fortunei has been used as a traditional medicine for the treatment of cold, dropsy, chills, and fevers.8,9 A wide range of secondary metabolites, such as alkaloids,10 phenolic acids,11 benzofuran,12 triterpenoids,13 and sesquiterpenes,14 has been reported as the constituents found in E. fortunei. During the course of discovery of cytotoxic sesquiterpene lactones from Eupatorium species,15 we focused on E. fortunei, which is native to Korea, and especially tried to isolate germacrane-type sesquiterpene lactones having 10-membered monocyclic ring with a cyclic ester structure.16,17 In the present study, we utilized the state-of-art technique, the SMART 2.0 tool, to rapidly discover the targeted compounds in the extract of a natural product and tried NMR spectra-guided isolation of the target compounds, which are expected to have biological activities. Consequently, we succeeded in the isolation of 10 sesquiterpene lactones having potent cytotoxicity from E. fortunei.
Results and Discussion
The experimental steps for the isolation of cytotoxic sesquiterpene lactones from E. fortunei are summarized in Figure 1. For the targeted isolation of the cytotoxic sesquiterpene lactones from E. fortunei, the total extract was fractionated into n-hexane, EtOAc, n-BuOH, and H2O fractions, sequentially. The EtOAc fraction of the subfractions showed the most potent cytotoxic activity against 5 cancer cell lines (A549, MCF-7, HEp-2, SKOV3, and PC3) at a concentration 100 μg/mL (Figure S1). The EtOAc fraction, which was expected to contain the cytotoxic constituents, was subjected to normal column chromatography and fractionated into five subfractions (E1 ∼ 5). Among the subfractions, E1 ∼ 5, E2, and E3, showed the most potent cytotoxic activities of 16.3–32.1 and 15.7–31.6%, respectively, at a concentration of 100 μg/mL against five cancer cell lines and were expected to contain the anticancer compounds of E. fortunei (Figure 2A). The HSQC NMR data of the subfractions E1, E2, and E3, were measured for the introduction into SMART 2.0 analysis (https://smart.ucsd.edu/classic), which resulted in the prediction of the structures of the mixed components in the samples (Figures 2B, S2, and S3). The SMART tool result of low cytotoxicity of subfraction E1 confirmed what was predicted for phenolic compounds and flavonoids (Figure S3). Although the subfraction E2 showed more diverse chemical profiles than E3, both fractions had terpenoid-type components including sesquiterpenes and diterpenes as major proportions of overall profiles. The subfraction E2 showing more complicated profiles was separated to yield seven subfractions (E2–1 ∼ 7), followed by evaluation of the cytotoxic activities against five cancer cells (Figure 2A). Three subfractions E2–4 ∼ 6 showing potent cytotoxic activities except E2–7, showing yellow major spots in the TLC experiment, which were regarded as nonterpenoid-type were studied in the HSQC NMR experiments (Figures 3 and S4). Interestingly, the results predicted by SMART 2.0 analysis showed quite different profiles of four subfractions. The subfractions E2–4, E2–5, and E2–7 showed intricate profiles due to the diverse kinds of chemicals, while terpene lactones including of germacranolide-type occupied the largest portion for E2–6 (Figures 3, S5–S7 and Tables S1–S3). The SMART 2.0 predicted the structures of 24 prenylated sesquiterpene lactones including those of germacranolide-type sesquiterpene lactones with the highest cosine score of 0.88 (Figure 4 and Table S4). Next, we focused on the targeted isolation of the subfractions E2–6 and could isolate seven germacranolide-type sesquiterpene lactones (1–7), 14-hydroxy-8β-[4′-hydroxytigloyloxy]-costunolide (1),18 14-acetoxy-8β-[4′-hydroxytigloyloxy]-costunolide (2),19 14-acetoxy-8β-hydroxy-costunolide (3),20 8β-[4′,5′-dihydroxytigloyloxy]-costunolide (4),21 8β-[4′-hydroxytigloyloxy]-14-oxo-costunolide (5),18 3β-acetoxy-8β-[4′,5′-dihydroxytigloyloxy]-costunolide (6),22 and 2β-hydroxy-8β-[5′-hydroxytigloyloxy]-costunolide (7),23 and two eudesmane-type sesquiterpene lactones 8 and 9, 1β-hydroxy-8β-[4′-hydroxytigloyloxy]-β-cyclocostunolide (8)24 and 1β-hydroxy-8β-[4′-hydroxytigloyloxy]-α-cyclocostunolide (9).25 Interestingly, compounds 9 and 10 contained an exomethylene-γ-lactone and a prenylated ester, which are the same as the seven derivatives with germacranolide-type compounds 1–7.
Figure 1.

Overview of the steps of the target isolation study. E. fortunei crude extract was partitioned successively with n-hexane, EtOAc, n-BuOH, and H2O. The subfractions were obtained from the EtOAc fraction. Subsequently, subfractions were analyzed by HMQC NMR spectroscopy and subjected to MTT assay. HSQC data of fractions were analyzed using SMART analysis. Then, the SMART result was processed with statistical data to target the sesquiterpene lactones.
Figure 2.
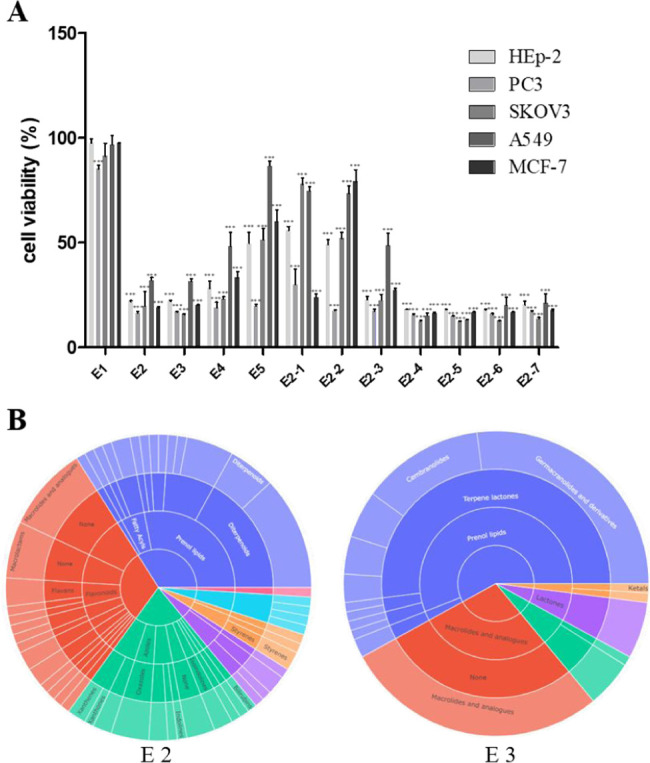
(A) Cytotoxic effect of subfractions (E1 ∼ E5 and E2–1 ∼ E2–7) on human lung cancer A549, breast cancer MCF-7, ovarian cancer SKOV3, laryngeal carcinoma HEp-2, and prostate cancer PC3 cells using MTT assay. Data indicates statistical significance at p < 0.001 compared to the negative control. (B) Various class level classifications of predictive compounds in the SMART tool are shown in the sunburst plot.
Figure 3.

Various class level classifications of predictive compounds in the SMART tool are shown in the sunburst plot (E2–4 ∼ E2–7).
Figure 4.

Structures of sesquiterpene lactones predicted by SMART 2.0.
In the case of subfraction E3, the chemical profile showed that terpenoid-lactones including germacranolide-type compounds occupied the largest portion in the subclass level of the sunburst plot (Figure 3). We expected that the subfraction E3 has a wide range of the germacranolide-type compounds similar to E2 but only 8β-[4′-hydroxytigloyloxy]-costunolide (10)26 was isolated as the major compound (Figure 5). These results indicated that the HSQC signals of compound 10 overwhelmed those of other minor components due to the majority of compound 10 in the subfraction E3. Consequently, we could successfully isolate 10 sesquiterpene lactones, expecting the potent cytotoxic activities using the SMART 2.0 tool. Since compounds 1, 8, and 9 were previously reported about 20–40 years ago, the full assignment of their structures was not determined in the literature. In the present study, the structures of these compounds were fully elucidated from the 1D and 2D NMR spectroscopic data. Among these, the determination of compound 1 is described below.
Figure 5.

Structures of compounds 1–10 isolated from Eupatorium fortunei.
Compound 1 was obtained as a white powder and has a molecular formula of C20H26O6, based on HR-ESI-MS analysis (m/z 363.1818 [M + H]+, calcd for 363.1808). The 1H spectral data and HSQC spectrum showed resonances of two methyl protons at δH 1.84 (3H, s, H-5′) and 1.71 (3H, s, H-15); two oxymethine protons at δH 5.84 (1H, d, J = 4.2 Hz, H-8) and 5.19 (1H, t, J = 9.3 Hz, H-6); three olefinic protons at δH 6.77 (1H, t, J = 11.6 Hz, H-3′), 5.15 (1H, dd, J = 12.3, 4.3 H-1), and 4.97 (1H, d, J = 9.9 Hz, H-5); two oxymethylene protons at δH 6.22 (1H, d, J = 3.2 Hz, H-13a) and 5.67 (1H, d, J = 3.2 Hz, H-13b); six methylene groups at δH 4.29 (2H, d, J = 6.0 Hz, H-14a, 4′a), 4.28 (1H, d, J = 3.8 Hz, H-4′b), 3.76 (1H, d, J = 11.9 Hz, H-14a), 3.32 (1H, m, H-3b), 2.51 (1H, m, H-2b), 2.40 (1H, m, H-9b), 2.27 (1H, m, H-2a), 2.20 (1H, m, H-3a), and 2.15 (1H, d, J = 5.7 Hz, H-9a); and one methine proton at δH 3.22 (1H, m, H-7). The 13C NMR and HSQC spectra showed resonance signals for 20 carbons, including two carboxylic carbons at δC 171.8 (C-11) and 167.7 (C-1′); five olefinic quaternary carbons at δC 128.9 (C-2′), 139.0 (C-12), 143.6 (C-4), 138.9 (C-10), and 121.5 (C-11); three olefinic methane carbons at δC 142.9 (C-3′), 134.5 (C-1), and 128.5 (C-5); two oxygenated methine carbons at δC 77.6 (C-6) and 73.5 (C-8); five methylene carbons including two oxygenated methylenes at δC 61.0 (C-4′), 59.8 (C-14), 40.1 (C-9), 39.0 (C-3), and 26.6 (C-2); a methine carbon at δC 53.5 (C-7); and two methyl groups at δC 17.2 (C-15) and 12.8 (C-5′). The COSY data showed correlations between H-1/H-2/H-3, H-5/H-6/H-7/H-8/H-9, and H-3′/H-4′. In addition, the key HMBC correlations between H-2 and C-10, H-3 and C-2/C-4 and C-15, H-3 and C-10, H-5 and C-3, and H-6 and C-8 are also shown. The exomethylene protons (H-13a and 13b) were correlated with a methine carbon (C-7) and a carboxylic carbon (C-11). These results suggested that the backbone of compound 1 is the germacranolide-type sesquiterpene lactone.27 The prenyl group suggested by the additional correlations between H-3′ and C-1′, H-4′ and C-1′/C-2′ was attached to C-8 of the backbone indicated by the signal from H-8 to C-1′. The COSY, HMBC, and ROESY are detailed in Figures 6 and S8–S13. Consequently, compound 1 was identified as 14-hydroxy-8β-[4′-hydroxytigloyloxy]-costunolide, whose H NMR profile was reported in 1987.18
Figure 6.
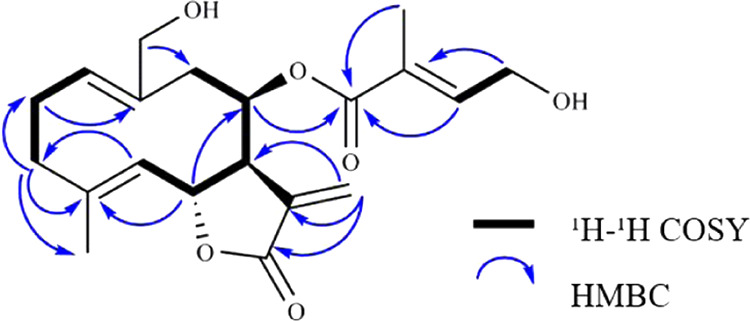
Key 1H-1H COSY and HMBC correlations of 1.
Additionally, compounds 1–10 were evaluated for their anticancer activities against five cell lines A549, MCF-7, HEp-2, SKOV3, and PC3 (Table 1). Compounds 2, 4, and 7–10 showed significant cytotoxic activities against human prostate cancer cell line PC3. Compounds 4 and 8 exhibited IC50 values of 3.9 ± 1.2 and 3.9 ± 0.6 μM, respectively. For human breast cancer cell line MCF-7, among compounds 2, 7, 8, and 10 showing cytotoxic activities, compound 7 exhibited IC50 values of 5.8 ± 0.1 μM. In the case of compound 10, it showed good cytotoxicity against five cancer cell lines (4.3 ± 0.8–20.2 ± 5.0 μM).
Table 1. Cytotoxicity Data of Compounds 1–10 against Five Cell Lines.
| IC50 (μM)a |
|||||
|---|---|---|---|---|---|
| compound | PC3 | SKOV3 | A549 | MCF-7 | HEp-2 |
| 1 | 15.4 ± 1.3 | ≥100 | ≥100 | 13.5 ± 3.5 | 56.3 ± 3.2 |
| 2 | 4.2 ± 1.3 | 32.5 ± 7.4 | 28.3 ± 0.9 | 6.1 ± 1.8 | 20.9 ± 6.2 |
| 3 | 15.2 ± 1.0 | 46.0 ± 6.8 | 52.3 ± 8.0 | 30.0 ± 7.4 | 54.9 ± 3.0 |
| 4 | 3.9 ± 1.2 | 33.8 ± 6.0 | 37.9 ± 7.5 | 12.8 ± 3.3 | 26.4 ± 6.5 |
| 5 | 12.6 ± 4.0 | 31.8 ± 4.6 | 25.9 ± 1.0 | 16.3 ± 3.4 | 25.0 ± 7.1 |
| 6 | 17.4 ± 2.3 | 68.6 ± 2.2 | 61.2 ± 3.3 | 28.9 ± 1.2 | 26.8 ± 1.1 |
| 7 | 8.1 ± 0.3 | 29.4 ± 2.9 | 29.3 ± 7.9 | 5.8 ± 0.1 | 27.6 ± 3.4 |
| 8 | 3.9 ± 0.6 | 36.9 ± 2.5 | 44.8 ± 6.1 | 8.3 ± 0.9 | 22.7 ± 0.3 |
| 9 | 4.7 ± 0.9 | 50.2 ± 6.5 | 45.6 ± 3.6 | 14.6 ± 5.0 | 25.6 ± 1.2 |
| 10 | 4.3 ± 0.8 | 20.2 ± 5.0 | 17.8 ± 3.8 | 6.1 ± 0.3 | 12.6 ± 2.7 |
| docetaxel | 0.6 ± 0.2 | 0.3 ± 0.3 | 1.0 ± 0.4 | 1.4 ± 0.2 | 0.2 ± 0.0 |
The results are IC50 values of compounds against each cancer cell line: PC3 (prostate), SKOV3 (ovarian), A549 (lung), MCF-7 (breast), and HEp-2 (laryngeal); docetaxel was used as a positive control.
In the present study, we could isolate ten sesquiterpene lactones (1–10), which have been rarely reported in Nature with the guidance of an in silico NMR spectra annotation tool, SMART 2.0. Though SMART 2.0 did not identify the exact structures, the results were quiet reliable to guide the discovery of the targeted compounds with specific moieties among the mixed compounds in the levels of extracts or fractions of natural products.
Experimental Section
General Experimental Procedures
NMR spectra, including 2D NMR (1H-1H COSY, HSQC, and HMBC), were obtained using a Bruker Avance Neo 600 (Bruker, Germany) spectrometer at 600 MHz at the Central Laboratory of Kangwon National University (Chuncheon, Korea). Column chromatography procedures were performed on silica gel Kieselgel 60 (40–60 μm, 230–400 mesh, Merck, Germany) and Sephadex LH-20 gel (18–111 μm, GE Healthcare, Sweden). Thin-layer chromatography (TLC) was performed on precoated silica gel plates (Kieselgel 60 F254 Merck, Germany) and RP-C18 plates (Kieselgel 60 F254s, Merck, Germany). Spots were detected by TLC using UV light or H2SO4-EtOH (v/v) spray followed by heating. Multiple preparative HPLC was performed on a LC-Forte/R system equipped with a YMC UV/Vis detector (YMC, Shimogyo-ku, Japan) using a YMC-Actus Triart C18 column (s-5 μm, 12 nm, 20 × 250 mm2) at a flow rate of 50 mL/min and monitored at 210 nm. Extra high purity solvents were purchased from DaeJung (Sicheung, Korea) for extraction, fractionation, and separation.
Plant Material
The whole plants of E. fortunei were collected from the Medicinal Plant Garden, Seoul National University, Korea in May 2018. The plant was identified by Professor Yong Soo Kwon, the College of Pharmacy of Kangwon National University, and the specimen (KNUEF-01) was deposited in the Herbarium at the College of Pharmacy, Kangwon National University.
SMART 2.0 Analysis
For the HSQC analyses, sample aliquots of 10–20 mg dissolved in 650 μL of methanol-d4 with 0.05 v/v TMS (tetramethylsilane) were ultrasonicated for 10 min and filtered. The dissolved sample was taken into a 5 mm NMR tube. For SMART 2.0 analysis, the measured HSQC spectrum was imported into MestReNova, and the 1H-13C correlation data were converted into a CSV file. The converted CSV file was uploaded to SMART 2.0 (https://smart.ucsd.edu/classic) to obtain the candidate structures. Sunburst was performed using these data. For sunburst, the annotation compounds in the SMART tool were classified based on ClassyFire.28
Extraction and Isolation
The aerial parts of Eupatorium fortunei (10 kg) were extracted three times with 100% MeOH and ultrasonic extraction for 3 h. The crude extract (552.25 g) was dissolved in H2O and partitioned successively with n-hexane (116.53 g), EtOAc (163.68 g), and n-BuOH (56.12 g). A part of the EtOAc fraction (100 g) was chromatographed by RP C18 MPLC with MeOH-H2O (1:3, to 1:0) to obtain subfractions (E1–E5). The subfraction E2 was resolved by silica gel MPLC with hexane–EtOAc (5:1 to 1:5) to give seven fractions. Subfraction E2–6 was separated into six subfractions using a Sephadex LH-20 CC with 70% MeOH. E2-6-3 was resolved by silica gel MPLC with hexane–EtOAc (3:1 to 1:5) to obtain six subfractions containing 1 (99.1 mg) and 2 (134.2 mg). Compounds 4 (4.2 mg) and 5 (2.1 mg) were purified from E2-6-3-3 by multiple preparative HPLC using a gradient solvent system of MeOH-H2O (60%, 10.0 mL/min). Subfraction E2-6-3-4 was further chromatographed on multiple preparative HPLC with an isocratic solvent system of MeOH-H2O (60%, 10.0 mL/min) to afford 3 (7.5 mg), 6 (7.3 mg), 7 (26.6 mg), 8 (10.1 mg), and 9 (3.1 mg). The subfraction E3 was separated by silica gel MPLC with hexane–EtOAc (5:1 to 1:5) and MeOH-CH2Cl2 (1:5 to 1:0) to obtain eleven fractions. Subfraction E3-5 was passed over the Sephadex LH-20 column with 100% MeOH to give five fractions. Compound 10 (29.0 mg) was obtained from E3-5-2 using silica gel MPLC with hexane–EtOAc (5:1 to 0:1).
14-Hydroxy-8β-[4′-hydroxytigloyloxy]-costunolide (1)
1H NMR (CD3OD, 600 MHz) δH 6.77 (1H, t, J = 11.6 Hz, H-3′), 6.22 (1H, d, J = 3.2Hz, H-13b), 5.84 (1H, d, J = 4.2 Hz, H-8), 5.67 (1H, d, J = 3.2 Hz, H-13a), 5.19 (1H, t, J = 9.3 Hz, H-6), 5.15 (1H, dd, J = 12.3, 4.3 Hz, H-1), 4.97 (1H, d, J = 9.9 Hz, H-5), 4.29 (2H, d, J = 6.0 Hz, H-14a, 4′b), 4.28 (1H, d, J = 3.8 Hz, H-4′a), 3.76 (1H, d, J = 11.9 Hz, H-14a), 3.32 (1H, m, H-3b), 3.22 (1H, m, H-7), 2.51 (1H, m, H-2b), 2.40 (1H, m, H-9b), 2.27 (1H, m, H-2a), 2.20 (1H, m, H-3a), 2.15 (1H, d, J = 5.7 Hz, H-9a), 1.84 (3H, s, H-5′), 1.71 (3H, s, H-15); 13C NMR (CD3OD, 150 MHz) δC 171.8 (C-11), 167.7 (C-1′), 143.6 (C-4), 142.9 (C-3′), 139.0 (C-12), 138.9 (C-10), 134.5 (C-1), 128.9 (C-2′), 128.5 (C-5), 121.5 (C-11), 77.6 (C-6), 73.5 (C-8), 61.0 (C-4′), 59.8 (C-14), 53.5 (C-7), 40.1 (C-9), 39.0 (C-3), 26.2 (C-2), 17.2 (C-15), 12.8 (C-5′); HREIMS m/z 363.1818 [M + H]+ (calcd for C20H27O6, 363.1808).
14-Acetoxy-8β-[4′-hydroxytigloyloxy]-costunolide (2)
1H NMR (CDCl3, 600 MHz) δH 6.70 (1H, t, J = 5.5 Hz, H-3′), 6.31 (1H, d, J = 2.9 Hz, H-13b), 5.84 (1H, d, J = 4.3 Hz, H-8), 5.62 (1H, d, J = 2.9 Hz, H-13a), 5.19 (1H, dd, J = 11.0, 5.6 Hz, H-1), 5.12 (1H, t, J = 9.3 Hz, H-6), 4.84 (1H, d J = 9.9 Hz, H-5), 4.62 (1H, d, J = 12.4 Hz, H-14), 4.35 (1H, dd, J = 14.9, 6.7 Hz, H-4′b), 4.29 (1H, d, J = 5.03, H-4′a), 4.25 (1H, m, H-14a), 3.24 (1H, dd, J = 14.8, 5.0 Hz, H-9b), 2.95 (1H, m, H-7), 2.42 (1H, m, H-2b), 2.37 (1H, m, H-3b), 2.34 (1H, m, H-2a), 2.22 (1H, m, H-9a), 2.17 (1H, m, H-3a), 1.94 (3H, s, -OAc), 1.82 (3H, s, H-5′), 1.75 (3H, s, H-15); HREIMS m/z 405. 1910 [M + H]+ (calcd for C22H29O7, 405.1913).
14-Acetoxy-8β-hydroxy-costunolide (3)
1H NMR (CD3OD, 600 MHz) δH 6.29 (1H, d, J = 3.4 Hz, H-13b), 5.71 (1H, d, J = 3.4 Hz, H-13a), 5.15 (1H, dd, J = 10.0, 8.4 Hz, H-1), 5.11 (1H, m, H-6), 4.96 (1H, d, J = 10.1 Hz, H-5), 4.51 (1H, d, J = 3.5 Hz, 1H, H-8), 4.14 (1H, d, J = 11.8 Hz, H-14b), 3.88 (1H, d, J = 11.8 Hz, H-14a), 2.95 (1H, m, H-7), 2.87 (1H, dd, J = 14.1 5.1 Hz, H-9b), 2.45 (1H, m, H-9a), 2.42 (1H, m, H-2b), 2.35 (1H, m, H-3b), 2.22 (1H, m, H-2a), 2.19 (1H, m, H-3a), 1.67 (3H, d, J = 1.2 Hz, H-15); 13C NMR (CD3OD, 150 MHz) δC 172.7 (C-12), 143.3 (C-4), 140.4 (C-11), 137.9 (C-10), 134.8 (C-1), 128.3 (C-5), 121.3 (C-13), 77.1 (C-6), 71.7 (C-8), 60.9 (C-14), 54.6 (C-7), 45.2 (C-9), 40.4 (C-3), 26.7 (C-2), 17.3 (C-15); HREIMS m/z 265.1073 [M + H]+ (calcd for C15H21O4, 249.1440).
8β-[4′,5′-Dihydroxytigloyloxy]-costunolide (4)
1H NMR (CD3OD, 600 MHz) δH 6.90 (1H, t, J = 5.9 Hz, H-4′), 6.20 (1H, d, J = 3.3 Hz, H-13b), 5.86 (1H, d, J = 4.0 Hz, H-8), 5.66 (1H, d, J = 3.3 Hz, H-13a), 5.28 (1H, t, J = 9.3 Hz, H-6), 4.97 (1H, m, H-1), 4.90 (1H, d, J = 9.9 Hz, H-5), 4.40 (2H, d, J = 5.9, H-5′), 4.30 (2H, s, H-3′), 2.82 (1H, dd, J = 14.3, 4.8 Hz, H-9b), 2.45 (1H, dd, J = 15.7, 1.4 Hz, H-9a), 2.41 (1H, m, H-2b), 2.38 (1H, m, H-3b), 2.21 (1H, m, H-2a), 2.15 (1H, m, H-3a), 1.80 (3H, s, H-14), 1.53 (3H, s, H-15); 13C NMR (CD3OD, 150 MHz) δC 171.9 (C-11), 167.0, (C-1′), 146.9 (C-3′), 143.6 (C-2′), 138.9 (C-12), 135.50 (C-4), 132.5 (C-10), 131.7 (C-1), 128.6 (C-5), 121.5 (C-13), 77.7 (C-6), 73.9 (C-8), 59.4 (C-5′), 56.9 (C-4′), 53.7 (C-7), 44.7 (C-9), 40.3, (C-3), 27.1 (C-2), 19.5 (C-15), 17.6 (C-14); HREIMS m/z 363.2518 [M + H]+ (calcd for C20H27O6, 363.1808).
8β-[4′-Hydroxytigloyloxy]-14-oxo-costunolide (5)
1H NMR (CDCl3, 600 MHz) δH 9.45 (1H, s, H-14), 6.76 (1H, m, H-3′), 6.62 (1H, m, H-1), 6.46 (1H, t, J = 8.3 Hz, H-8), 6.22 (1H, d, J = 2.8 Hz, H-13b), 5.58 (1H, d, J = 2.8 Hz, H-13a), 5.09 (1H, m, H-6), 5.08 (1H, m, H-5), 4.36 (2H, d, J = 5.7 Hz, H-4′), 2.80 (1H, m, H-9b), 2.52 (1H, m, H-2b), 2.51(1H, m, H-7), 2.41 (1H, m, H-3b), 2.29 (1H, m, H-2a), 2.11 (1H, d, J = 14.2 Hz, H-3a), 1.99 (1H, m, H-9a), 1.95 (3H, s, H-5′), 1.82 (3H, s, H-15); HREIMS m/z 361.2351 [M + H]+ (calcd for C20H25O6, 361.1651).
3β-Acetoxy-8β-[4′,5′-dihydroxytigloyloxy]-costunolide (6)
1H NMR (CDCl3, 600 MHz) δH 6.93 (1H, s, H-3′), 6.32 (1H, s, H-13b), 5.78 (1H, s, H-13a), 5.59 (1H, d, J = 7.4 Hz, H-3), 5.27 (1H, m, H-8), 5.25 (1H, m, H-6), 5.22 (1H, d, J = 10.1 Hz, H-5), 5.08 (1H, s, H-1), 4.45 (2H, t, J = 8.8 Hz, H-4′), 4.39 (2H, d, J = 13.7 Hz, H-5′), 2.98 (1H, s, H-7), 2.72 (2H, d, J = 13.9 Hz, H-2b, 9b), 2.41 (1H, d, J = 13.5 Hz, H-9a), 2.11 (1H, s, H-2a), 2.10 (3H, s, -OAc), 1.90 (3H, s, H-14), 1.79 (3H, s, H-15);HREIMS m/z 421.2561 [M + H]+ (calcd for C22H29O8, 421.1862).
2β-Hydroxy-8β-[5′-hydroxytigloyloxy]-costunolide (7)
1H NMR (CDCl3, 600 MHz) δH 6.39 (1H, q, J = 7.04 Hz, H-3′), 6.33 (1H, d, J = 2.3 Hz, H-13b), 5.85 (1H, d, J = 2.7 Hz, H-8), 5.61 (1H, d, J = 2.3 Hz, H-13a), 5.14 (1H, t, J = 9.1 Hz, H-6), 5.00 (2H, t, J = 11.6 Hz, H-1, 5), 4.75 (1H, dd, J = 15.3, 9.5 Hz, H-2), 4.26 (1H d, J = 12.7 Hz, H-5′), 4.17 (1H, d, J = 12.7 Hz, H-5′), 2.97 (1H, m, H-7), 2.91 (1H, dd, J = 14.3, 4.7 Hz, H-9b), 2.73 (1H, dd, J = 10.9, 5.7 Hz, H-3b), 2.37 (1H, d, J = 14.1 Hz, H-9a), 2.12 (1H, t, J = 10.4 Hz, H-3a), 2.02 (3H, d, J = 7.1 Hz, H-4′), 1.78 (3H, s, H-14), 1.54 (3H, s, H-15); HREIMS m/z 363.2511 [M + H]+ (calcd for C20H27O6, 363.1808).
1β-Hydroxy-8β-[4′-hydroxytigloyloxy]-β-cyclocostunolide (8)
1H NMR (CDCl3, 600 MHz) δH 6.77 (1H, t, J = 5.3 Hz, H-3′), 6.15 (1H, d, J = 2.7 Hz, H-13b), 5.81 (1H, s, H-8), 5.45 (1H, d, J = 2.7 Hz, H-13a), 5.03 (1H, s, H-15b), 4.96 (1H, s, H-15a), 4.54 (1H, t, J = 11.0 Hz, H-6), 4.37 (1H, d, J = 5.8 Hz, H-4′), 3.53 (1H, dd, J = 11.3, 4.3 Hz, H-1), 2.86 (1H, J = 8.7 Hz, H-7), 2.39 (1H, d, J = 10.4 Hz, H-9b), 2.37 (1H, J = 7.3 Hz, H-2b), 2.34 (1H, m, H-3b), 2.27 (1H, d, J = 10.8 Hz, H-5), 2.12 (1H, m, H-3a), 1.63 (1H, m, H-9a), 1.60 (1H, m, H-2a), 1.83 (3H, s, H-5′), 0.97 (3H, s, H-14); HREIMS m/z 363.2534 [M + H]+ (calcd for C20H27O6, 363.1808).
1β-Hydroxy-8β-[4′-hydroxytigloyloxy]-α-cyclocostunolide (9)
1H NMR (CDCl3, 600 MHz) δH 6.78 (1H, s, H-3′), 6.15 (1H, s, H-13b), 5.81 (1H, s, H-8), 5.44 (1H, s, H-13a), 5.36 (1H, s, H-3), 4.45 (1H, m, H-6), 4.37 (2H, s, H-4′), 3.68 (1H, d, J = 8.6 Hz, H-1), 2.82 (1H, m, H-7), 2.37-2.40 (4H, m, H-2a, 2b, 5, 9a), 1.89 (3H, s, H-15), 1.83 (3H, s, H-5′), 1.60 (3H, s, H-14), 1.58 (1H, m, H-9b); HREIMS m/z 363.1801 [M + H]+ (calcd for C20H27O6, 363.1808).
8β-[4′-Hydroxytigloyloxy]-costunolide (10)
1H NMR (CD3OD, 600 MHz) δH 6.80 (1H, td, J = 5.9, 1.3 Hz, H-3′), 6.20 (1H, d, J = 3.3 Hz, H-13b), 5.82 (1H, d, J = 4.1 Hz, H-8), 5.64 (1H, d, J = 3.3 Hz, H-13a), 5.22 (1H, dd, J = 9.7, 8.8 Hz, H-6), 4.97 (1H, dd, J = 11.0, 2.4 Hz, H-1), 4.90 (1H, d, J = 10.0 Hz, H-5), 4.27 (2H, d, J = 5.73 Hz, H-4′), 3.19 (1H, m, H-7), 2.81 (1H, dd J = 14.3, 4.9 Hz, H-9b), 2.45 (1H, m, H-9a), 2.40 (2H, ddt, J = 8.0, 4.7, 4.0 Hz, H-2b, 3b), 2.21 (1H, dd, J = 11.0, 5.5 Hz, H-2a), 2.14 (1H, m, H-3a), 1.83 (3H, d, J = 1.1 Hz, H-5′), 1.79 (3H, d, J = 1.2 Hz, H-15), 1.51 (3H, s, H-14); HREIMS m/z 347.1836 [M + H]+ (calcd for C20H27O5, 347.1858).
Cytotoxicity Assay
The A549 human lung cancer cell line, MCF-7 breast cancer cell line, SKOV3 ovarian cancer cell line, and HEp-2 laryngeal carcinoma and PC3 prostate cancer cell lines were purchased from ATCC (American Type Culture Collection). Cells were cultured in Roswell Park Memorial Institute 1640 medium (RPMI 1640; WelGENE, Deagu, Korea) and Dulbecco’s modified Eagle’s medium (DMEM; WelGENE) containing 10% fetal bovine serum with 1% penicillin/streptomycin (WelGENE) under 5% CO2 conditions at 37 °C. For cell culture, the cells were placed in 96-well plates at 5 × 103 cells/100 μL. After incubation overnight, the cells were treated with samples that were dissolved in various concentrations (1–100 μM) of dimethyl sulfoxide with 1% DMSO as negative control and docetaxel as positive control. After 48 h of incubation, cell viability was evaluated using MTT (Sigma-Aldrich) assay. The MTT solution at a concentration of 0.5 mg/mL was added to each well for 4 h at 37 °C. The absorbance was measured at 490 nm on a SpectraMax i3 multimode microplate reader (Molecular Devices). The IC50 values were calculated using GraphPad Prism software (GraphPad Software Inc., CA) as the average of three cycles of anticancer tests.
Acknowledgments
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (NRF-2018R1C1B6002574 and NRF-2019R1A6A3A13096888).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c03270.
Cytotoxic activities of the fractions and the isolated compounds from E. fortunei; 1H and 13C NMR spectral data of EtOAc subfractions and compound 1 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Tsugawa H.; Ikeda K.; Tanaka W.; Senoo Y.; Arita M.; Arita M. Comprehensive identification of sphingolipid species by in silico retention time and tandem mass spectral library. J. Cheminformatics 2017, 9, 19. 10.1186/s13321-017-0205-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Kuhn M.; Gavin A.; Bork P. Identification of metabolites from tandem mass spectra with a machine learning approach utilizing structural features. Bioinformatics 2020, 36, 1213–1218. 10.1093/bioinformatics/btaa464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacalhau P.; Fernandes L.; Martins M. R.; Candeias F.; Carreiro E. P.; López Ó.; Caldeira A. T.; Totobenazara J.; Guedes R. C.; Burke A. J. In silico, NMR and pharmacological evaluation of an hydroxyoxindole cholinesterase inhibitor. Bioorg. Med. Chem. 2019, 27, 354–363. 10.1016/j.bmc.2018.12.007. [DOI] [PubMed] [Google Scholar]
- Singh D. K.; Sahu A.; Wani A. A.; Bharatam P. V.; Kotimoole C. N.; Batkulwar K. B.; Deshpande A. Y.; Giri S.; Singh S. Stability behaviour of antiretroviral drugs and their combinations. 10: LC-HRMS, LC-MSn, LC-NMR and NMR characterization of fosamprenavir degradation products and in silico determination of their ADMET properties. Eur. J. Pharm. Biopharm. 2019, 142, 165–178. 10.1016/j.ejpb.2019.06.018. [DOI] [PubMed] [Google Scholar]
- Kaufmann A.; Teale P.. Capabilities and Limitations of High-Resolution Mass Spectrometry (HRMS): Time-of-flight and Orbitrap Chemical Analysis of Non-antimicrobial Veterinary Drug Residues in Food; Kay J. F.; Kay J. F.; MacNeil J. D.; Wang J., Eds.; John Wiley & Sons, 2016; p 1. [Google Scholar]
- Zhang C.; Idelbayev Y.; Roberts N.; Tao Y.; Nannapaneni Y.; Duggan B. M.; Min J.; Lin E. C.; Gerwick E. C.; Cottrell G. W. Small molecule accurate recognition technology (smart) to enhance natural products research. Sci. Rep. 2017, 7, 14243 10.1038/s41598-017-13923-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reher R.; Kim H. W.; Zhang C.; Mao H. H.; Wang M.; Nothias L.; Caraballo-Rodriguez A. M.; Glukhov E.; Teke B.; Leao T.; et al. A Convolutional Neural Network-Based Approach for the Rapid Annotation of Molecularly Diverse Natural Products. J. Am. Chem. Soc. 2020, 142, 4114–4120. 10.1021/jacs.9b13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A.; Im M.; Yim N.; Ma J. Y. Reduction of metastatic and angiogenic potency of malignant cancer by Eupatorium fortunei via suppression of MMP-9 activity and VEGF production. Sci. Rep. 2014, 4, 6994 10.1038/srep06994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.; Lee H.; Hwang Y.; Lee J.; Cho W.; Ma J. Y. Eupatorium fortunei and its components increase antiviral immune responses against RNA viruses. Front. Pharmacol. 2017, 8, 511. 10.3389/fphar.2017.00511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K.; Roeder E.; Chen T.; Xiu X. Pyrrolizidine alkaloids from Eupatorium fortunei. Phytochemistry 1992, 31, 2573–2574. 10.1016/0031-9422(92)83336-W. [DOI] [Google Scholar]
- Pham T. N.; Pham H. D.; Dang D. K.; Duong T. T.; Le T. P. Q.; Nguyen Q. D.; Nguyen Tien D. Anticyanobacterial phenolic constituents from the aerial parts of Eupatorium fortunei Turcz. Nat. Prod. Res. 2019, 33, 1345–1348. 10.1080/14786419.2018.1476511. [DOI] [PubMed] [Google Scholar]
- Jiang H. X.; Liu Q.; Gao K. Benzofuran derivatives from Eupatorium fortunei. Nat. Prod. Res. 2008, 22, 937–941. 10.1080/14786410701642888. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Li Y.; Pan J.; Gao K. Terpenoids from Eupatorium fortunei TURCZ. Helv. Chim. Acta 2006, 89, 558–566. 10.1002/hlca.200690059. [DOI] [Google Scholar]
- Chen Y.; Jiang H.; Gao K. One novel nortriterpene and other constituents from Eupatorium fortunei Turcz. Biochem. Syst. Ecol. 2013, 47, 1–4. 10.1016/j.bse.2012.10.008. [DOI] [Google Scholar]
- Picman A. K. Biological activities of sesquiterpene lactones. Biochem. Syst. Ecol. 1986, 14, 255–281. 10.1016/0305-1978(86)90101-8. [DOI] [Google Scholar]
- Kupchan S. M.; Maruyama M.; Hemingway R.; Hemingway J.; Shibuya S.; Fujita T.; Cradwick P.; Hardy A.; Sim G. Tumor inhibitors. LXVII. Eupacunin, a novel antileukemic sesquiterpene lactone from Eupatorium cuneifolium. J. Am. Chem. Soc. 1971, 93, 4914–4916. 10.1021/ja00748a048. [DOI] [PubMed] [Google Scholar]
- Kupchan S. M.; Fujita T.; Maruyama M.; Britton R. W. Tumor inhibitors. LXXXIV. Isolation and structural elucidation of eupaserrin and deacetyleupaserrin, new antileukemic sesquiterpene lactones from Eupatorium semiserratum. J. Org. Chem. 1973, 38, 1260–1264. 10.1021/jo00947a002. [DOI] [PubMed] [Google Scholar]
- Zdero C.; Bohlmann F.; Schmeda-Hirschmann G. Beyerene derivatives and other terpenoids from Stevia aristata. Phytochemistry 1987, 26, 463–466. 10.1016/S0031-9422(00)81433-8. [DOI] [Google Scholar]
- Spring O.; Zipper R.; Conrad J.; Vogler B.; Klaiber I.; Da Costa F. B. Sesquiterpene lactones from glandular trichomes of Viguiera radula (Heliantheae; Asteraceae). Phytochemistry 2003, 62, 1185–1189. 10.1016/S0031-9422(02)00747-1. [DOI] [PubMed] [Google Scholar]
- Herz W.; Kumar N. Sesquiterpene lactones from Helianthus grosseserratus. Phytochemistry 1981, 20, 99–104. 10.1016/0031-9422(81)85225-9. [DOI] [Google Scholar]
- Jolad S. D.; Hoffmann J. J.; Bates R. B.; Calders S.; McLaughlin S. Caudatol, a guaianolide from Pericome caudata. Phytochemistry 1990, 29, 3024–3028. 10.1016/0031-9422(90)87128-H. [DOI] [Google Scholar]
- Takahashi T.; Eto H.; Ichimura T.; Murae T. Hiyodorilactones A and B, new tumor inhibitory germacradienolides from Eupatorium sachalinense Makino. Chem. Lett. 1978, 7, 1345–1348. 10.1246/cl.1978.1345. [DOI] [Google Scholar]
- Herz W.; De Groote R. Desacetyleupaserrin and nevadensin from Helianthus pumilus. Phytochemistry 1977, 16, 1307–1308. 10.1016/S0031-9422(00)94387-5. [DOI] [Google Scholar]
- Hernández L. R.; De Riscala E. C.; Catalán C. A.; Díaz J. G.; Herz W. Sesquiterpene lactones and other constituents of Stevia maimarensis and Synedrellopsis grisebachii. Phytochemistry 1996, 42, 681–684. 10.1016/0031-9422(95)00942-6. [DOI] [Google Scholar]
- Hernández L. R.; Catalán C. A.; Cerda-García-Rojas C. M.; Joseph-Nathan P. Sesquiterpene lactones from Stevia breviaristata. Phytochemistry 1994, 37, 1331–1335. 10.1016/S0031-9422(00)90408-4. [DOI] [Google Scholar]
- Bohlmann F.; Schmeda-Hirschmann G.; Jakupovic J. Heliangolides and germacrolides from Disynaphia multicrenulata. Phytochemistry 1984, 23, 1435–1437. 10.1016/S0031-9422(00)80481-1. [DOI] [Google Scholar]
- Buděšínský M.; Šaman D.. Carbon-13 NMR spectra of sesquiterpene lactones. In Annual Reports on NMR Spectroscopy; Elsevier, 1995; pp 231–475. [Google Scholar]
- Feunang Y. D.; Eisner R.; Knox C.; Chepelev L.; Hastings J.; Owen G.; Fahy E.; Steinbeck C.; Subramanian S.; Bolton E. ClassyFire: automated chemical classification with a comprehensive, computable taxonomy. J. Cheminformatics 2016, 8, 61. 10.1186/s13321-016-0174-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.