

Abstract
N,N′-Substituted di-isopropyl (NHC-1), benzyl-isopropyl (NHC-2), and adamantyl-isopropyl (NHC-3) benzimidazolium salts react with palladium(II) bromide in pyridine to afford the corresponding trans-dibromidopyridinepalladium(II) complexes Pd-C1, Pd-C2, and Pd-C3 in high yields. A distorted square planar geometry for Pd-C2 and Pd-C3 was confirmed by single-crystal X-ray diffraction. The palladium(II) complexes show a remarkably higher catalytic activity and selectivity, compared to the literature data, in carbonylative Sonogashira coupling reactions of aryl iodides and aryl diiodides with aryl alkynes, alkyl alkynes, and dialkynes. Excellent yields with as low as 0.03 mol % loading of the catalyst were obtained. In the series of benzimidazolium (NHC) precursors, the 1H NMR signals of the α hydrogen show a consistent probing of the N-substituent donor strength. The density functional theory (DFT) quantum mechanical descriptors of the frontier orbitals were calculated. A linear correlation of the calculated absolute softness of the complexes versus the calculated percent buried volume (%Vbur) of their corresponding ligands was obtained. The catalytic activity experimental data are consistent with the hard soft acid base (HSAB)-predicted high affinity of the softest Pd-C3 complex for soft substrates, such as aryl iodides.
Introduction
α,β-Acetylenic carbonyl compounds (ynones) are valuable compounds that have important structural moieties found in agrochemicals and natural products.1 Some ynones have been found to exhibit biological activity of interest such as anticancer or antifungal properties. They are either industrially produced or extracted from different organisms (Figure 1).2−6
Figure 1.

(1) Anticancer cytotoxic uracil analogue, (2) antifungal isolated from fermentation of ischnode benzionum, and (3) antifungal sordaricin analogue.
Ynones are also important precursors for the synthesis of several organic compounds,7−16 including the aromatic heterocycles such as flavones, chromones, quinolones, pyrazoles, furans, and pyrimidines.
The synthesis of ynones in a single step via the carbonylative Sonogashira coupling reaction of terminal alkynes with aryl halides catalyzed by palladium complexes represents a new challenge.16 Different palladium complexes have been designed, synthesized, and evaluated in the catalytic carbonylative Sonogashira coupling reactions.17−24 Nevertheless, important problems remain unsolved, including the high catalyst loading, low catalyst activity, long reaction time, high temperature, and high CO pressure.24−28
In the past two decades, N-heterocyclic carbene (NHC) ligands emerged as versatile and synthetically more accessible molecular species compared to phosphines. Significant advances have been achieved in the applications of their organometallic complexes.29−36 N-heterocyclic carbenes (NHCs) are characterized by their strong σ donation ability, therefore providing high stability to their metal complexes. In the case of palladium, the possibility of formation of the inactive palladium species during the catalytic cycle is significantly diminished.37,38 In addition, NHCs offer the possibility of tuning their electronic and steric properties by modifying their structures depending on the catalytic application.39,40
Since 1970, great efforts were devoted to study the electronic and steric properties of various ligands.41 For instance, Tolman studied the electronic and steric factors of phosphines using the Tolman electronic parameter (TEP) method based on infrared spectral data.42 Later, 1H and 13C NMR spectroscopies were used as probes to study the electronic properties, especially for metal-NHC complexes and their ligand precursors.43 For example, Ganter proposed the use of 1J(C–H) coupling constants in the NMR spectra of N-heterocyclic carbene salts to probe the σ-donating ability of their corresponding NHCs.44 Huynh proposed a method of determination of ligand donor strength based on 13C NMR of palladium(II)-NHC complexes, which probes the electronic effects of both the backbone and N-substituent of the NHC ligands.45−48
We have reported previously new triazole-derived (NHC)palladium(II) complexes and their catalytic activities in different cross-coupling reactions.33 We also reported the high catalytic activity of heteroleptic dibromide- and diiodido-palladium(II)-NHC-pyridine (Br2-Pd-NHC-Py and I2-Pd-NHC-Py) complexes in the carbonylative Sonogashira and carbonylative Suzuki–Miyaura coupling reactions of aryl iodides with different alkynes or boronic acids.49,50
In this work, we report on our latest results giving an insight into the electronic and steric effects in a series of new heteroleptic (NHC)pyridine palladium(II) complexes (Pd-NHC-Py). The approach relies on 1H and 13C NMR analyses and density functional theory (DFT) and “SambVca” (Salerno molecular buried volume) calculations supported by crystal X-ray diffraction data. Adamantyl, benzyl, and isopropyl groups were used as substituents on the benzimidazole to probe the steric and electronic effects on the catalytic efficiency of these complexes in the carbonylative Sonogashira coupling reactions of aryl iodides and aryl diiodides with aryl alkynes, alkyl alkynes, and dialkynes. We also report the one-step synthesis of new compounds, 1-(4-methoxyphenyl)-3-(4-pentylphenyl)prop-2-yn-1-one (3ac), 1-(4-methoxyphenyl)tetradec-2-yn-1-one (5ad), 4-cyclohexyl-1-(4-methoxyphenyl)-but-2-yn-1-one (5ag), and 1,1′-(1,4-phenylene)bis(6-chlorohex-2-yn-1-one) (9a) via the carbonylative Sonogashira coupling reactions using the new (NHC)pyridine palladium(II) complex Pd-C3 as a catalyst.
Results and Discussion
Synthesis of Ligands and Catalysts and Their Molecular Structures
1-Alkyl benzimidazoles (BZ1, BZ2, and BZ3) were prepared in very good yields by the N-alkylation reaction of benzimidazole with alkyl bromides (2-bromopropane, benzyl bromide) in the presence of a base (Schemes 1 and 2). The N-substituted 1-isopropyl-3-alkyl-1H-benzo[d]imidazole-3-ium bromides (NHC-1, NHC-2, and NHC-3) were prepared in good yields by the direct alkylation of 1-alkyl benzimidazoles (BZ1, BZ2, and BZ3) with 2-bromopropane (Scheme 3). The formation of the benzimidazolium bromides was confirmed by the presence of downfield singlet peaks at 7.58 ppm (BZ1), 7.98 ppm (BZ2), and 8.22 ppm (BZ3) in their proton NMR spectra, which are assigned to C-2 protons of the benzimidazole rings. The structures of compounds BZ3, NHC-2, and NHC-3 are depicted in Figure 2. The benzimidazole derivative BZ3 and the benzimidazolium salts NHC-2 and NHC-3 crystallize in the P1̅, C2/c, and P21/n space groups, respectively. Their bond lengths and bond angles are in the normal range compared to similar reported compounds (Table 1).51,52
Scheme 1. Synthesis of Alkyl-1H-benzo[d]imidazole.

Scheme 2. Synthesis of Adamantyl-1H-benzo[d]imidazole (BZ3).

Scheme 3. Synthesis of NHC Ligand Precursors.

Figure 2.

ORTEP diagram of compounds BZ3, NHC-2, and NHC-3 with the atomic numbering scheme. Thermal ellipsoids have been drawn at the 50% probability level for BZ3 and at 30% for NHC-2 and NHC-3.
Table 1. Selected Bond Lengths [Å] and Bond Angles [deg] for Compounds BZ3, NHC-2, and NHC-3.
| BZ3 | NHC-2 | NHC-3 | |||
|---|---|---|---|---|---|
| N(1)–C(1) | 1.304(2) | C(1)–N(1) | 1.328(4) | N(1)–C(1) | 1.328(3) |
| N(1)–C(7) | 1.383(2) | C(1)–N(2) | 1.330(4) | N(1)–C(7) | 1.406(3) |
| N(2)–C(1) | 1.368(2) | C(2)–N(1) | 1.382(4) | N(1)–C(11) | 1.495(3) |
| N(2)–C(2) | 1.3891(19) | C(2)–C(7) | 1.395(5) | N(2)–C(1) | 1.317(3) |
| N(2)–C(8) | 1.4837(18) | C(7)–N(2) | 1.382(4) | N(2)–C(2) | 1.388(3) |
| C(2)–C(7) | 1.405(2) | C(8)–N(1) | 1.485(4) | N(2)–C(8) | 1.478(3) |
| C(11)–N(2) | 1.478(4) | C(2)–C(3) | 1.384(4) | ||
| C(2)–C(7) | 1.392(4) | ||||
| C(1)–N(1)–C(7) | 104.03(14) | N(1)–C(1)–N(2) | 110.2(3) | C(1)–N(1)–C(7) | 107.0(2) |
| C(1)–N(2)–C(2) | 105.31(13) | N(1)–C(2)–C(3) | 132.0(3) | C(1)–N(1)–C(11) | 125.4(2) |
| C(1)–N(2)–C(8) | 126.31(13) | N(1)–C(2)–C(7) | 106.1(3) | C(7)–N(1)–C(11) | 127.5(2) |
| C(2)–N(2)–C(8) | 128.33(12) | C(1)–N(1)–C(2) | 108.7(3) | C(1)–N(2)–C(2) | 108.3(2) |
| N(1)–C(1)–N(2) | 114.99(16) | C(1)–N(1)–C(8) | 126.8(3) | C(1)–N(2)–C(8) | 123.5(2) |
| N(2)–C(2)–C(3) | 133.80(14) | C(2)–N(1)–C(8) | 124.5(3) | C(2)–N(2)–C(8) | 128.0(2) |
| N(2)–C(2)–C(7) | 105.15(14) | C(1)–N(2)–C(7) | 108.1(3) | N(2)–C(1)–N(1) | 111.6(2) |
The palladium(II)-N-heterocyclic carbene pyridine complexes Pd-C1, Pd-C2, and Pd-C3 were prepared in very good yields by reacting palladium(II) bromide with 1.0 equiv of the appropriate ligand precursor (NHC-1, NHC-2, and NHC-3) in an excess amount of pyridine, as shown in Scheme 4. The complexes were purified by passing through a column of silica gel layered with a short pad of celite to afford yellow solids. The disappearance of the acidic C-2 protons in the benzimidazole was used to confirm the formation of the Pd-NHC complexes. Furthermore, the palladation of the NHC ligand precursors is characterized by the appearance of new signals at 154–158 ppm in the 13C NMR spectra of the three complexes assignable to the palladated carbon Pd-C. The X-ray structures of the complexes Pd-C2 and Pd-C3 (Figure 3) were determined from single crystals obtained by slow evaporation of their concentrated solutions in dichloromethane/acetonitrile (8/1) (v/v). The two palladium(II) complexes crystallize in the P21/n and Pbca space groups, respectively. In both compounds, the palladium(II) ion is bonded to the NHC carbon atom, the nitrogen atom of the pyridine ligand, and two trans-bromido anions. The coordination geometry is a distorted square planar. For instance, for compound Pd-C3, the cis-angles are in the range of 87.60(7)–92.21(7)° and the Pd–C, Pd–N, and the average Pd–Br bond lengths are 1.969(2), 2.109(2), and 2.4373(4) Å, respectively (Table 2). These values are in agreement with those reported for similar trans-dibromido(NHC)(pyridine)palladium(II) complexes.53 The dihedral angles between the (NHC) imidazole and the (Pd-C(NHC)-Br1-Br2-Npy) mean planes are 76.5(7) and 86.04(5)° for Pd-C2 and Pd-C3, respectively. The largest value correlates with the higher steric effect of the adamantyl substituent of the benzimidazole in Pd-C3 compared to the benzyl group in Pd-C2. The dihedral angles between the (Pd-C(NHC)-Br1-Br2-Npy) mean plane and the pyridine ligand are 46(1) and 42.2(1)° for Pd-C2 and Pd-C3, respectively.
Scheme 4. Synthesis of Palladium(II)-NHC-pyridine Complexes (Pd-C1, Pd-C2, and Pd-C3).

Figure 3.

ORTEP diagram of compounds Pd-C2 and Pd-C3 with the atomic numbering scheme. Thermal ellipsoids have been drawn at the 30% probability level.
Table 2. Selected Bond Lengths [Å] and Bond Angles [deg] for Complexes Pd-C2 and Pd-C3.
| Pd-C2 | Pd-C3 | ||
|---|---|---|---|
| Pd(1)–C(1) | 1.96(3) | Pd(1)–C(6) | 1.969(2) |
| Pd(1)–N(3) | 2.09(3) | Pd(1)–N(1) | 2.109(2) |
| Pd(1)–Br(1) | 2.420(3) | Pd(1)–Br(2) | 2.4337(4) |
| Pd(1)–Br(2) | 2.425(3) | Pd(1)–Br(1) | 2.4408(3) |
| N(1)–C(1) | 1.33(3) | N(1)–C(5) | 1.331(4) |
| N(1)–C(7) | 1.35(4) | N(1)–C(1) | 1.334(4) |
| N(1)–C(8) | 1.50(4) | N(2)–C(6) | 1.351(3) |
| N(2)–C(1) | 1.33(3) | N(2)–C(7) | 1.410(3) |
| N(2)–C(2) | 1.38(4) | N(2)–C(16) | 1.503(3) |
| N(2)–C(15) | 1.50(4) | N(3)–C(6) | 1.350(3) |
| N(3)–C(22) | 1.35(5) | N(3)–C(12) | 1.390(3) |
| N(3)–C(18) | 1.37(4) | N(3)–C(13) | 1.477(3) |
| C(1)–Pd(1)–N(3) | 179.3(10) | C(6)–Pd(1)–N(1) | 174.01(9) |
| C(1)–Pd(1)–Br(1) | 88.1(7) | C(6)–Pd(1)–Br(2) | 87.60(7) |
| N(3)–Pd(1)–Br(1) | 92.0(7) | N(1)–Pd(1)–Br(2) | 92.21(7) |
| C(1)–Pd(1)–Br(2) | 87.6(7) | C(6)–Pd(1)–Br(1) | 87.78(7) |
| N(3)–Pd(1)–Br(2) | 92.3(7) | N(1)–Pd(1)–Br(1) | 92.35(7) |
| Br(1)–Pd(1)–Br(2) | 175.31(15) | Br(2)–Pd(1)–Br(1) | 175.367(13) |
| C(22)–N(3)–Pd(1) | 124(2) | C(5)–N(1)–Pd(1) | 122.4(2) |
| C(18)–N(3)–Pd(1) | 119(2) | C(1)–N(1)–Pd(1) | 120.0(2) |
| N(1)–C(1)–Pd(1) | 124(2) | N(3)–C(6)–Pd(1) | 118.92(17) |
| N(2)–C(1)–Pd(1) | 128(2) | N(2)–C(6)–Pd(1) | 133.12(17) |
Carbonylative Sonogashira Coupling of 4-Iodoanisole (1a) and Phenylacetylene (2a) by Pd(II)-NHC-Pyridine: Optimization of the Reaction Conditions
There are several reports in the literature on the catalytic activity of Pd(II)-NHC complexes in various cross-coupling reactions, especially the C–C bond coupling.33,36,54,55 However, the literature about the application in carbonylative cross-coupling reactions is still limited, and the systems reported are associated with drawbacks such as high temperature,50 high catalyst loading,31 and long reaction time. In this work, the application of the newly synthesized Pd(II)-NHC-pyridine (Pd-NHC-Py) complexes (Pd-C1, Pd-C2, Pd-C3) was examined in the carbonylative Sonogashira coupling reaction of aryl iodides with aryl and alkyl alkynes. For instance, a model reaction of 4-iodoanisole with phenylacetylene under pressurized carbon monoxide in the presence of a catalytic amount of Pd-NHC-Py (Pd-C3) was adopted (eq 1). The summary of the results of the optimization reactions is included in Table 3. When the reaction was conducted with 0.01 mol % Pd-C3 as a catalyst and triethylamine as a base at 80 °C for 1 h, only 10% of 1-(4-methoxyphenyl)-3-phenylprop-2-yn-1-one (3aa) (Table 3, entry 1) as a product was isolated. The yield of the product 3aa was gradually improved by an increase in temperature (Table 3, entries 1–3). When the reaction time was increased from 1 to 3 h, the isolated yield of the product 3aa has increased from 20 to 55% at 100 °C (Table 3, entry 4). However, an excellent isolated yield (91%) of the alkynone 3aa was obtained at 120 °C (Table 3, entry 5). A complete conversion of 4-iodoanisole and 99% isolated yield product were achieved when the loading of the catalyst was increased to 0.05 mol % at 120 °C. Similarly, when a 0.03 mol % loading of Pd-C3 was used at 120 °C, 99% of the product 3aa was isolated (Table 1, entries 6 and 7). Reducing the temperature from 120 to 100 °C under the optimized conditions (toluene/Et3N/0.030 mol % of Pd-C3/3 h) did not significantly affect the isolated yield (97%) of the alkynone 3aa (Table 3, entry 8). Therefore, a temperature of 100 °C was considered as the optimized temperature for the subsequent reactions. As expected, no product was observed in this reaction when it is conducted in the absence of a base (Table 3, entry 9). Additionally, only 8% of the product was observed with KOH (Table 3, entry 10) and 5% of the product was collected with K2CO3 (Table S1, entry 11) as bases. When toluene was replaced by other solvents such as tetrahydrofuran (THF), dimethylformamide (DMF), and acetonitrile, a significant decrease in the isolated yields was observed (Table 3, entries 12–14). Based on the results of the optimization experiments, the subsequent reactions were carried out using Et3N as a base, toluene as a solvent, and at 100 °C for 3 h. For comparison, commercially available palladium catalysts were also tested (Table 3, entries 17–19).
 |
1 |
Table 3. Optimization of the Carbonylative Sonogashira Coupling Reactions of 4-Iodoanisole (1a) and Phenylacetylene (2a)a.
| entry | NHC-Pd-Py (mol %) | solvent | T (°C) | base | time (h) | yield 3aa (%)b |
|---|---|---|---|---|---|---|
| 1 | Pd-C3 | toluene | 80 | Et3N | 1 | 10 |
| (0.01) | ||||||
| 2 | Pd-C3 | toluene | 100 | Et3N | 1 | 20 |
| (0.01) | ||||||
| 3 | Pd-C3 | toluene | 120 | Et3N | 1 | 52 |
| (0.01) | ||||||
| 4 | Pd-C3 | toluene | 100 | Et3N | 3 | 55 |
| (0.01) | ||||||
| 5 | Pd-C3 | toluene | 120 | Et3N | 3 | 91 |
| (0.01) | ||||||
| 6 | Pd-C3 | toluene | 120 | Et3N | 3 | 99 |
| (0.05) | ||||||
| 7 | Pd-C3 | toluene | 120 | Et3N | 3 | 99 |
| (0.03) | ||||||
| 8 | Pd-C3 | toluene | 100 | Et3N | 3 | 97 |
| (0.03) | ||||||
| 9 | Pd-C3 | toluene | 100 | 3 | 0 | |
| (0.03) | ||||||
| 10 | Pd-C3 | toluene | 100 | KOH | 3 | 8 |
| (0.03) | ||||||
| 11 | Pd-C3 | toluene | 100 | K2CO3 | 3 | 5 |
| (0.03) | ||||||
| 12 | Pd-C3 | THF | 100 | Et3N | 3 | 61 |
| (0.03) | ||||||
| 13 | Pd-C3 | DMF | 100 | Et3N | 3 | 73 |
| (0.03) | ||||||
| 14 | Pd-C3 | CH3CN | 100 | Et3N | 3 | 84 |
| (0.03) | ||||||
| 15 | Pd-C2 | toluene | 100 | Et3N | 3 | 79 |
| (0.03) | ||||||
| 16 | Pd-C1 | toluene | 100 | Et3N | 3 | 85 |
| (0.03) | ||||||
| 17 | Pd(PPh3)2Cl2 | toluene | 100 | Et3N | 3 | 48 |
| (0.03) | ||||||
| 18 | Pd(PhCN)2Cl2 | toluene | 100 | Et3N | 3 | 31 |
| (0.03) | ||||||
| 19 | PdBr2 | toluene | 100 | Et3N | 3 | 22 |
| (0.03) |
Reaction conditions: 4-iodoanisole (1.0 mmol), phenylacetylene (1.5 mmol), base (2.0 mmol), solvent (5 mL), CO (200 psi).
Isolated yield.
The other two Pd catalysts (Pd-C1 and Pd-C2) were also evaluated under the optimized conditions (Et3N/toluene/100 °C/3 h). The efficiencies of these complexes in the carbonylative Sonogashira coupling reactions were clearly less than that for Pd-C3 (Table 4, entries 1–3).
Table 4. Screening of the New NHC-Pd-Py Catalystsa.
| entry | NHC-Pd-Py | yield 3aa (%)b |
|---|---|---|
| 1 | Pd-C1 | 85 |
| 2 | Pd-C2 | 79 |
| 3 | Pd-C3 | 97 |
Reaction conditions: NHC-Pd-Py (0.03 mol %), 4-iodoanisole (1.0 mmol), phenylacetylene (1.5 mmol), Et3N (2.0 mmol), toluene (3 mL), CO (200 psi), 100 °C, 3 h.
Isolated yield.
The evaluation of the complexes Pd-NHC-Py in the carbonylative Sonogashira coupling reactions under optimized conditions (0.03 mol % of Pd-C3, 2.0 equiv of Et3N, 3 mL of toluene, 200 psi CO, 100 °C, 3 h) showed overall high catalytic activities. Particularly, the activity of the complex Pd-C3, functionalized with the adamantyl group as N1-substituent, was remarkable.
To gain insight into this distinguished catalytic activity in the carbonylative Sonogashira coupling reactions, the ligand electronic properties and steric hindrance were investigated.
1H and 13C NMR spectra were used as a probe to study the electronic effects of the N-substituents of the new synthesized ligands. The spectra were recorded in CDCl3. 1H NMR singlet peaks of Hα located on the C2 carbon of the benzimidazolium species were observed (Figure 4). The corresponding chemical shift values are 10.80, 11.35, and 11.51 ppm for the adamantyl (NHC-3)-, isopropyl (NHC-2)-, and benzyl (NHC-1)-substituted benzimidazolium salts, respectively. The downfield shift is consistent with the expected +I effect order of the N-substituents (adamantyl > isopropyl > benzyl). The sensitivity of 1H NMR signals of the hydrogen Hα to the N-substituent donor strengths is clearly demonstrated in this series of benzimidazolium (NHC) precursors in consistence with Huynh results.43
Figure 4.

Chemical shift of Hα of 1-isopropyl-3-alkyl-1H-benzo[d]imidazol-3-ium bromide salts (NHCs) on the 1H NMR scale.
The carbene carbon 13C NMR signals of the three complexes are given in Figure 5. In general, five-membered free NHC carbene carbons are strongly deshielded (>200 ppm),31,56,57 and among the paramagnetic, diamagnetic, and neighboring magnetic anisotropy effects, the first factor is the dominant one in these compounds.43 Therefore, taking into account the N-substituent donor strengths only, the expected chemical shift order would be: Pd-C3 (N-Ad) > Pd-C1 (N-iPr) > Pd-C2 (N-benz). However, the observed order is (Pd-C1 (N-iPr) 160.0 ppm > Pd-C3 (N-Ad) 156.78 ppm > Pd-C2 (N-benz) 154.34 ppm). The expected relative order of Pd-C1 (N-iPr) and Pd-C2 (N-benz) is observed; however, the Pd-C3 (N-Ad) chemical shift is much more upfield than expected only based on N-substituent donor strengths. Clearly, a separate factor inherent to the adamantyl-substituted NHC in Pd-C3 is effective, such as π back-donation. The Pd(II)-(NHC) bonding has been computationally investigated earlier. In one report, it was found that in addition to the major ligand σ-donation component, a minor π-bonding interaction including π acidity and π donation are also effective with a relative major contribution of the former in π-bonding.58,59 NHCs π acceptor ability was probed experimentally based on 77Se NMR chemical shielding in a series of imidazol-2-ylidene selenoureas. The N-adamantyl-substituted derivative was assigned the highest π-accepting ability in this series in comparison to more than 20 other derivatives, including the isopropyl-substituted one.60−62 A significant π back-bonding could explain the upfield shift in Pd-C3 (N-Ad).
Figure 5.

Chemical shift of the carbene carbon in the Pd-NHC-Py complexes on the 13C NMR scale.
To get some insight into the predicted relative reactivity of the three complexes toward substrates that may undergo oxidative addition during the catalytic cycle, quantum mechanical descriptors of the frontier orbitals were calculated using density functional theory (DFT) at the B3LYP/def2-TZVP level. A plot of the frontier orbitals is given in Figure 6. These data were validated by comparing the DFT-calculated bond lengths of Pd-C2 and Pd-C3 complexes with the single-crystal X-ray diffraction data (Table 5).
Figure 6.

Frontier molecular orbitals at the B3LYP/def2-TZVP level for the complexes Pd-C1, Pd-C2, and Pd-C3.
Table 5. Validation of DFT Calculationsa.
|
Pd-C2 complex |
Pd-C3 complex |
||||
|---|---|---|---|---|---|
| bond length (Å) | DFT calculations | XRD data | bond angle (Å) | DFT calculations | XRD data |
| C2–N1 | 1.352 | 1.330 | C2–N1 | 1.347 | 1.350 |
| C2–N3 | 1.351 | 1.330 | C2–N3 | 1.358 | 1.351 |
| C2–Pd4 | 1.976 | 1.960 | C2–Pd4 | 1.981 | 1.969 |
| Pd4–Br5 | 2.488 | 2.420 | Pd4–Br5 | 2.492 | 2.434 |
| Pd4–Br6 | 2.492 | 2.425 | Pd4–Br6 | 2.493 | 2.440 |
| Pd4–N7 | 2.149 | 2.090 | Pd4–N7 | 2.157 | 2.109 |
| N7–C8 | 1.340 | 1.370 | N7–C8 | 1.340 | 1.334 |
| N7–C9 | 1.340 | 1.350 | N7–C9 | 1.340 | 1.331 |
Comparison between bond lengths of Pd-C2 and Pd-C3 complexes from DFT calculations and XRD data.
These data were validated by comparing the bond lengths of Pd-C2 and Pd-C3 complexes, which were calculated using (DFT). The bond lengths were provided from the coordination data of the XRD structure data of these complexes (Table 5).
Quantum mechanical descriptors of the frontier orbitals were calculated using density functional theory (DFT) at the B3LYP/def2-TZVP level. These data are summarized in Table S1. The calculated ionization potential (IP), electron affinity (EA), absolute electronegativity (χ), absolute hardness (η), and absolute softness (S) of the complexes are given in Table 6. The three complexes show comparable electronegativity (χ) values, while the absolute softness (S) of the complexes increases in the order Pd-C1 < Pd-C2 < Pd-C3. Taking into account the hard soft acid base (HSAB) considerations, the affinity of the softest Pd-C3 complex for soft substrates, such as aryl iodides, is then expected to be relatively higher in consistence with the experimental data. The proposed mechanism for these reactions showed the effect of the hard soft acid base (HSAB) concept in the acceleration of the rate-determining step (Scheme 4). Considerations of the steric effect were approached by evaluation of the percent buried volumes (%Vbur) of the (NHC) ligands in the palladium(II) complexes using SambVca tool,63,64 based on the X-ray data of Pd-C2 and Pd-C3 complexes as well as CSD data available for a diisopropyl(NHC)palladium(II) complex (Figure S43).53 Bondi radii scaled by 1.17, a sphere radius of 3.5 Å, and a Pd-C distance of 2.00 Å were adopted. The ligand percent buried volume increases in the order 28.5 (di-isopropyl (NHC)) < 29.8 (benzyl-isopropyl (NHC)) < 34.1 (adamantyl-isopropyl (NHC)) (Figure 7). A linear correlation of the absolute softness (S) values of Pd-C1 to Pd-C3 was obtained, with an excellent agreement (Figure 8, R2 = 0.989) with the calculated percent ligand volume (%Vbur) of their corresponding ligands. The most active catalyst Pd-C3 bears the greatest S and %Vbur values.
Table 6. Ligand % Buried Volume (%Vbur) and DFT-Calculated Ionization Potential (IP), Electron Affinity (EA), Absolute Electronegativity (χ), Absolute Hardness (η), and Absolute Softness (S) of the Complexes in eVa.

IP = −EHUMO, EA = −ELUMO, χ = (IP + EA)/2, η = (IP – EA)/2, S = (1/2η).
Figure 7.

Molecular structure diagram (B3LYP/def2-TZVP) of Pd-C2 and Pd-C3 complexes.
Figure 8.

DFT-calculated absolute softness (S) versus the calculated percent buried volume (%Vbur) of their corresponding ligands.
Carbonylative Sonogashira Coupling of Aryl Iodides (1) and Aryl Alkynes (2) by Pd-C3
The catalytic activity of Pd-C3 in the carbonylative Sonogashira coupling reactions of various aryl iodides with different aryl alkynes was investigated. We have conducted the reactions under the optimized experimental conditions (0.03 mol % of Pd-C3, 2.0 equiv of Et3N, 3 mL of toluene, 200 psi CO, 100 °C, 3 h). Various aryl iodides were reacted with different functionalized aryl alkynes (eq 2, Table 7). Interesting alkynones were produced in excellent yields starting from various aryl alkynes with different electron-withdrawing and electron-donating substituted aryl iodides and CO. The coupling of 4-iodoanisole “deactivated aryl iodide” with phenylacetylene (Table 7, entry 1) and deactivated substituted phenyl acetylenes (Table 7, entries 2 and 3) afforded the corresponding alkynones in excellent yields (94–97%). The reactions of phenylacetylene with iodobenzene and various activated aryl iodides were also very successful, leading to excellent yields (95–98%) of the corresponding alkynones (Table 7, entries 4 and 5).
 |
2 |
Table 7. Carbonylative Sonogashira Coupling Reactions of Aryl Iodides (1a–c) with Aryl Alkynes (2a–c) Catalyzed by Pd-C3a.

Reaction conditions: Pd-C3 (0.03 mol %), aryl iodide (1.0 mmol), alkyne (1.5 mmol), Et3N (2.0 mmol), toluene (3 mL), CO (200 psi), 100 °C, 3 h.
Isolated yield.
Carbonylative Sonogashira Coupling of 4-Iodoanisole (1a) and 1-Heptyne (4a) by Pd-C3
The catalytic activity of Pd-C3 was also evaluated in the phosphine-free carbonylative Sonogashira coupling reaction of aryl iodides with different alkyl alkynes. For instance, a catalytic amount of Pd-C3 was used in the reaction of 4-iodoanisole with 1-heptyne in the presence of 200 psi of CO (eq 3). The results of the optimization are summarized in (Table 8). Initially, traces of the product [1-(4-methoxyphenyl)oct-2-yn-1-one] were observed with 0.01 mol % loading of Pd-C3 as a catalyst, 2.0 mol equivalent of triethylamine as a base, and toluene as a solvent at temperatures (100–120 °C) for 3–18 h (Table 8, entries 1–4). The isolated yields of [1-(4-methoxyphenyl) oct-2-yn-1-one] (5aa) had improved gradually by increasing the catalyst loading from 0.03 to 0.1 mol % in 18 h at 100 or 120 °C. However, the isolated yields at these levels of the catalyst loading were still relatively low (12–62%) (Table 8, entries 5–7). Improved isolated yields of the product 5aa (50% at 100 °C and 67% at 120 °C) were obtained with increasing the catalyst (Pd-C3) loading to 0.2 mol % in 6 h (Table 8, entries 8 and 9). Excellent yield (98%) of 5aa was successfully achieved at 120 or 100 °C for 18 h (Table 8, entries 10 and 11). Based on the above results, the phosphine-free carbonylative Sonogashira coupling reaction of 4-iodoanisole with 1-heptyne was conducted using 0.2 mol % Pd-C3, 200 psi CO, Et3N as a base, and toluene as a solvent at 100 °C for 18 h. These optimized conditions (Et3N/toluene/100 °C/18 h) were considered for the carbonylative Sonogashira coupling reactions of aryl iodides with different alkyl alkynes.
 |
3 |
Table 8. Carbonylative Sonogashira Coupling Reactions of 4-Iodoanisole (1a) with 1-Heptyne (4a) Catalyzed by Pd-NHC-Py (Pd-C3)a.
| entry | [Pd-C3] (mol %) | T (°C) | time (h) | yield 5aa (%)b |
|---|---|---|---|---|
| 1 | (0.01) | 100 | 3 | traces |
| 2 | (0.01) | 100 | 18 | traces |
| 3 | (0.01) | 120 | 3 | traces |
| 4 | (0.01) | 120 | 18 | traces |
| 5 | (0.03) | 120 | 18 | 12 |
| 6 | (0.1) | 120 | 18 | 62 |
| 7 | (0.1) | 100 | 18 | 46 |
| 8 | (0.2) | 100 | 6 | 50 |
| 9 | (0.2) | 120 | 6 | 67 |
| 10 | (0.2) | 120 | 18 | 98 |
| 11 | (0.2) | 100 | 18 | 98 |
Reaction conditions: 4-iodoanisole (1.0 mmol), 1-heptyne (1.5 mmol), Et3N (2.0 mmol), toluene (3 mL), CO (200 psi).
Isolated yield.
Carbonylative Sonogashira Coupling of Aryl Iodides (1) and Alkyl Alkynes (4) by Pd-C3
Investigation of the carbonylative Sonogashira coupling reactions of aryl iodides with alkyl alkynes has been conducted in the absence of any phosphine with a Pd-C3 loading of 0.20 mol % under optimized conditions (Et3N/toluene/100 °C/18 h) (eq 4). The results of these reactions are summarized in Table 9. For instance, the reaction of iodobenzene with 1-heptyne (Table 9, entry 1), 1-hexyne (Table 9, entry 2), and 3,3-dimethyl-1-butyne (Table 9, entry 3) produced the corresponding alkynones in excellent yields (89–95%). The results showed that the alkynes with linear aliphatic chain, such as 1-heptyne (89%) and 1-dodecyne (87%) (Table 9, entry 4), are relatively less reactive than the alkynes with branched aliphatic chain such as 3,3-dimethyl-1-butyne (Table 9, entry 3) under optimized conditions. This can be explained by the role of the donating methyl group located on the α position to alkyne function, which increases the acidity of the acetylenic proton. Furthermore, a double CO insertion was confirmed when the reaction of 4-iodoanisole was conducted with 3-phenylpropyne to produce (E)-3-benzylidene-5-(4-methoxyphenyl)furan-2(3H)-one (94%) via the formation of alkynone as an intermediate (Table 9, entry 6). No furan was detected when the 3-phenyl-1-propyne was replaced by 3-cyclohexyl-1-propyne in the previous reaction of 4-iodoanisole; however, the expected alkynone was produced in a good isolated yield (67%) (Table 9, entry 7). Various activated aryl iodides were reacted with different alkyl alkynes at a shorter reaction time (6 h). For example, the reactions of 4-nitro-iodobenzene with 1-heptyne (Table 9, entry 8) and 5-phenyl-1-pentyne (Table 9, entry 9) were successful and afforded the expected alkynones in good to excellent isolated yields (76–93%). Similarly, 4-iodoacetophenone reacted efficiently with 1-heptyne to yield 96% of 1-(4-acetylphenyl) oct-2-yn-1-one (Table 9, entry 10).
 |
4 |
Table 9. Carbonylative Sonogashira Coupling Reactions of Iodobenzene (1a–d) with Alkyl Alkynes (4a–h) Catalyzed by Pd-C3a.

Reaction conditions: Pd-C3 (0.20 mol %), aryl iodide (1.0 mmol), alkyne (1.5 mmol), Et3N (2.0 mmol), toluene (3 mL), CO (200 psi), 100 °C, 18 h.
Isolated yield.
Time = 6 h.
Carbonylative Sonogashira Coupling of Aryl Diiodides and Dialkynes by Pd-C3
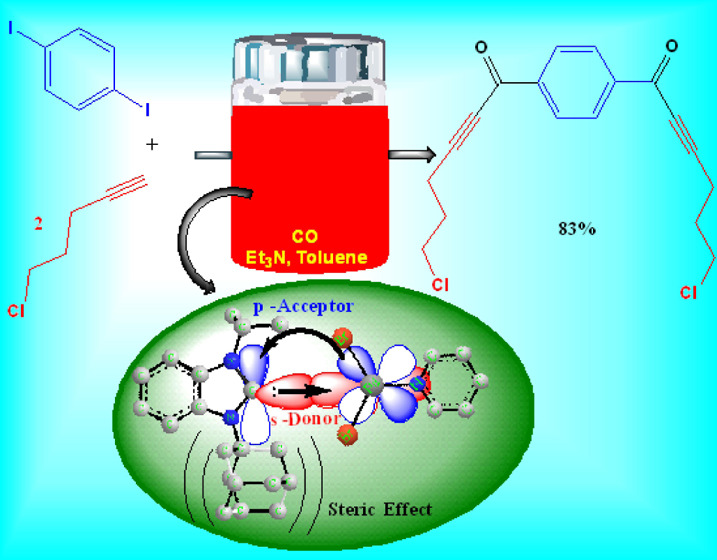
To prove further the high catalytic activity of Pd-C3, we have investigated the carbonylative Sonogashira coupling reactions of aryl diiodides with alkynes and aryl iodides with dialkynes. In fact, these reactions were conducted successfully to give the interesting symmetrical dialkynones that have various medical applications4−6 with very good isolated yields. For instance, 3,3′-(1,4-phenylene)bis[1-(4-nitrophenyl)prop-2-yn-1-one] (7a) (eq 5) and 3,3′-(1,4-phenylene)bis[1-(4-methoxyphenyl)prop-2-yn-1-one] (7b) (eq 6) were obtained via coupling reactions of 1,4-diethynyl benzene with 2 equiv of 4-nitro iodobenzene or 4-iodoanisole in 88% and 82% yields, respectively. Similarly, the coupling reaction of 1,3-diiodobenzene with phenylacetylene gave the expected meta-dialkynone (7c) (eq 7) in a very good isolated yield (78%). Additionally, Pd-C3 was evaluated in the reaction of 1,4-diiodobenzene with different alkyl alkynes such as 5-chloro-1-pentyne (7d) (eq 8), 1-heptyne (7e) (eq 9), and 3,3-dimethyl-1-butyne (4e) (eq 10). It showed a high catalytic performance to produce the corresponding dialkynones with very good isolated yields (77–83%).
 |
5 |
Compared to the catalytic systems reported in the literature (Table 10), Pd-C3 showed the highest catalytic efficiency with lower catalyst loading and shorter reaction time.
Table 10. Comparison of the Activity of Pd-C3 with Previously Published Data in the Carbonylative Sonogashira Coupling Reactions of Aryl Iodides and Alkynes.
| ref | catalyst | co-catalyst/ligand/additive | base | time (h) | yield (%) |
|---|---|---|---|---|---|
| (18) | PdCl2 (6 mol %) | PPh3 (12 mol %) | Et3N | 24 | 46–95 |
| (26) | Pd@MOF (1 mol %) | Et3N | 12 | 62–98 | |
| (50) | Pd-NHC-Py (0.1 mol %) | Et3N | 6 | 88–94 | |
| (65) | Pd(OAc)2 (2 mol %) | tri(2-furyl)- phosphine (4 mol %) | DIPEA | 20 | 62–98 |
| (66) | MCM-41-Pd-PPh2 (4 mol %) | Et3N | 36–72 | 45–91 | |
| (67) | Pd(OAc)2 (3 mol %) | I2 (1.3 mol %) PPh3 (1.3 mol %) | Et3N | 4 | 44–88 |
| this work | Pd-NHC-Py (Pd-C3) (0.03 mol %) | Et3N | 3–18 | 67–98 |
Proposed Mechanism for the Carbonylative Sonogashira Coupling of Aryl Iodides (1) and Alkynes (2) by Pd-C3
The plausible mechanism for the carbonylative Sonogashira coupling reactions that shows the role of the softness and steric effect for complex Pd-C3 is summarized in Scheme 5. The palladium(II) precatalyst (I) undergoes substitutions of the two bromides by the acetylides, obtained by the deprotonation of the alkynes in the presence of the base, to produce the palladium(II) intermediates (II and III) (where L = NHC and Py = Pyridine). The trans–cis isomerization step gives the intermediate (IV), and the reductive elimination of the dialkyne generates the Pd(0) active catalytic species (V). The palladium(II) intermediate [(Ar)(L)Pd(I)(Py)] (VI) is then formed via the oxidative addition of 2-iodophenol to the Pd(0). This step is the rate-determining step19,20,50 that can be faster due to the soft–soft interaction of the softest complex Pd-C3 with soft substrates such as aryl iodides. Therefore, Pd-C3 is more efficient than the relatively harder complexes (Pd-C1 and Pd-C2) (Figure 3). On the other hand, NHCs are known as strong donors, especially in the presence of an adamantyl substituent that has a highly inductive effect +I, leading to a more electron-rich palladium center. The oxidative addition step is enhanced due to the high stability of both the precatalyst (NHC)Pd(II)(Py)Br2 (I) and the proposed active species (NHC)Pd(0)(Py) (V). Further, the acyl palladium intermediate (VII) is formed by the insertion of carbon monoxide (CO) into the Ar–Pd bond. Then, the iodide is substituted by the acetylide to produce the palladium intermediate VIII. Reductive elimination yields the carbonylative Sonogashira product XI with the subsequent regeneration of the Pd(0) catalytic species (V). It is worth noting that the reductive elimination step can be accelerated in the presence of high steric adamantyl substituent more than other substituents “benzyl and isopropyl”.
Scheme 5. Plausible Mechanism for Carbonylative Sonogashira Coupling Reaction Catalyzed by the Pd-NHC-Py Complex.

Conclusions
In this work, remarkably high catalytic activity and selectivity of a series of heteroleptic (NHC)-pyridine palladium(II) complexes derived from a new series of N-substituted N-heterocyclic carbene (NHC) ligands were demonstrated. The scope includes the carbonylative Sonogashira coupling reactions of various aryl iodides and aryl diiodides with a wide range of aryl alkynes, alkyl alkynes, and dialkynes. Excellent isolated yields of the alkynones were produced for the reactions of aryl iodides with both aryl and alkyl alkynes, and very good yields were achieved for the carbonylative coupling of aryl diiodide with aryl alkynes. New compounds including 1-(4-methoxyphenyl)-3-(4-pentylphenyl)prop-2-yn-1-one (3ac), 1-(4-methoxyphenyl) tetradec-2-yn-1-one (5ad), 4-cyclohexyl-1-(4-methoxyphenyl)but-2-yn-1-one (5ag) and 1,1′-(1,4-phenylene)bis(6-chlorohex-2-yn-1-one) (9a) were produced from the carbonylative Sonogashira coupling reactions.
In this series of isopropyl, benzyl, and adamantyl N-substituted (NHC) ligand-based complexes Pd-C1, Pd-C2, and Pd-C3, respectively, the last one showed a significantly higher catalytic activity. The DFT quantum mechanical descriptors of the frontier orbitals for this series of complexes show that Pd-C3 is the softest species, which might be due to the inherent steric and electronic properties of the adamantyl group versus isopropyl and benzyl groups. This is consistent with the highest catalytic activity toward soft substrates observed for this complex.
Experimental Section
Materials and Instrumentation
The carbene ligand precursors and palladium carbene complexes were synthesized using materials that were purchased from Sigma-Aldrich and used as received. Solvents (reagent grade) that were used in the synthesis were distilled and then dried under molecular sieves. Flash column chromatography (packed with 60 F Silica gel from Fluka Chemie AG, Buchs, Switzerland) was used to purify the products; in some cases, appropriate solvents were used for washing the products.
Bruker 400 and 500 MHz NMR devices (Joel 1500 model) were used to collect 1H and 13C NMR spectral data for the products. Tetramethylsilane (TMS) was used as an internal reference, and chemical shifts of the products were recorded in ppm using CDCl3 or DMSO-d6 as the solvent. An electrospray ionization time-of-flight mass spectrometer (ESI-TOF/MS) from Fisher company with ESI scan (0.104–0.499 min, 17 scans) Frag = 180.0 V was used. Elemental analyses were performed on a PerkinElmer Series 11 (CHNS/O) Analyzer 2400 at the Department of Chemistry, King Fahd University of Petroleum and Minerals (KFUPM) in Saudi Arabia. Merck 60 F254 silica gel plates (250 μm layer thickness) were used for thin-layer chromatography (TLC) analyses. A Varian Saturn 2000 GC-MS device (30 m capillary column) was used to analyze the products. Agilent 6890 gas chromatography (GC) was used to monitor the reactions and analyze the products. X-ray data for BZ3, NHC-2, NHC-3, Pd-C2, and Pd-C3 were collected with a Bruker D8 Quest diffractometer (Mo Kα radiation λ = 0.71073 Å) at 298 K using Bruker APEX3 software package.68 Data reduction was performed using SAINT.69 Multiscan absorption correction was performed using SADABS.70 The structures were solved by direct methods with SHELXS using SHELXTL package and refined using full-matrix least-squares procedures on F2 via the program SHELXL-2014.71 Molecular graphics were obtained using ORTEP3.72 All hydrogen atoms, except water hydrogens, were included at calculated positions using a riding model with C–H distances of 0.93 Å for sp2 carbons and 0.96–0.98 Å for sp3 carbons. The isotropic displacement parameters were Uiso(H) = 1.2Ueq(C) and Uiso(H) = 1.5Ueq(C), respectively. The water hydrogen atoms were located on a difference-Fourier map. They were refined isotopically and restrained with DFIX and DANG. The refinement parameters are summarized in Tables 11 and 12.
Table 11. Crystal and Structure Refinement Data for Compounds BZ3, NHC-2, and NHC-3.
| BZ3 | NHC-2 | NHC-3 | |
|---|---|---|---|
| CCDC deposition number | 1948042 | 1948739 | 1949081 |
| empirical formula | C17H20N2 | C17H21BrN2O | C20H27BrN2 |
| formula weight | 252.35 | 349.27 | 375.34 |
| temperature (K) | 298(2) | 296(2) | 296(2) |
| wavelength (Å) | 0.71073 | 0.71073 | 0.71073 |
| crystal system | triclinic | monoclinic | monoclinic |
| space group | P1̅ | C2/c | P21/n |
| unit cell dimensions | |||
| a (Å) | 7.0473(11) | a = 14.9361(11) | a = 10.3429(8) |
| b (Å) | 9.0925(16) | b = 11.3341(9) | b = 16.4241(13) |
| c (Å) | 10.9446(19) | c = 20.2385(16) | c = 10.7998(8) |
| α (deg) | 95.726(6) | α = 90 | α = 90 |
| β (deh) | 103.104(5) | β = 100.857(2) | β = 104.481(2) |
| γ (deg) | 101.050(5) | γ = 90 | γ = 90 |
| volume (Å3) | 662.81(19) | 3364.8(5) | 1776.3(2) Å3 |
| Z | 2 | 8 | 4 |
| calcd density (g/cm3) | 0.075 | 2.444 | 2.316 |
| absorption coefficient (mm–1) | 1.264 | 1.379 | 1.404 |
| F(000) | 272 | 1440 | 784 |
| θ range for data collection (deg) | 2.308–28.858 | 2.381 to 28.415 | 2.382 to 28.345 |
| index ranges | –9 ≤ h ≤ 9, −12 ≤ k ≤ 12, −14 ≤ l ≤ 14 | –19 ≤ h ≤ 19, −15 ≤ k ≤ 15, −27 ≤ l ≤ 27 | –13 ≤ h ≤ 13, −21 ≤ k ≤ 21, −14 ≤ l ≤ 14 |
| reflections collected | 50 526 | 59 457 | 69 444 |
| independent reflections | 3470 [R(int) = 0.0689] | 4222 [R(int) = 0.0592] | 4412 [R(int) = 0.1216] |
| completeness (%) | 99.9 | 99.9 | 99.9 |
| absorption correction | semiempirical from equivalents | semiempirical from equivalents | semiempirical from equivalents |
| max. and min. transmission | 0.989 and 0.858 | 0.628 and 0.520 | 0.934 and 0.608 |
| refinement method | full-matrix least squares on F2 | full-matrix least squares on F2 | full-matrix least squares on F2 |
| data/restraints/parameters | 3470/0/172 | 4222/3/200 | 4412/0/210 |
| goodness of fit on F2 | 1.060 | 1.061 | 1.056 |
| final R indices [I > 2σ(I)] | R1 = 0.0576, wR2 = 0.1398 | R1 = 0.0562, wR2 = 0.1522 | R1 = 0.0492, wR2 = 0.0876 |
| R indices (all data) | R1 = 0.0928, wR2 = 0.1603 | R1 = 0.0877, wR2 = 0.1706 | R1 = 0.1028, wR2 = 0.1046 |
| extinction coefficient | |||
| largest diff. peak and hole | 0.232 and −0.327 e/Å3 | 1.586 and −0.573 e/Å3 | 0.326 and −0.456 e/Å–3 |
Table 12. Crystal and Structure Refinement Data for Complexes Pd-C2 and Pd-C3.
| Pd-C2 | Pd-C3 | |
|---|---|---|
| CCDC deposition number | 1949260 | 1919226 |
| empirical formula | C22H23Br2N3Pd | C25H31Br2N3Pd |
| formula weight | 595.65 | 639.75 |
| temperature (K) | 298(2) | 298(2) |
| wavelength (Å) | 0.71073 | 0.71073 |
| crystal system | monoclinic | orthorhombic |
| space group | P21/n | Pbca |
| unit cell dimensions | ||
| a (Å) | 13.873(5) | 14.7578(5) |
| b (Å) | 10.292(4) | 13.4002(5) |
| c (Å) | 16.064(6) | 25.2603(9) |
| α (deg) | 90 | 90 |
| β (deg) | 96.630(11) | 90 |
| γ (deg) | 90 | 90 |
| volume (Å3) | 2278.3(15) | 4995.4(3) |
| Z | 4 | 8 |
| calcd density (g/cm3) | 1.737 | 1.701 |
| absorption coefficient (mm–1) | 4.334 | 3.960 |
| F(000) | 1168 | 2544 |
| θ range for data collection (deg) | 2.355–28.591 | 3.146–30.120 |
| index ranges | –18 ≤ h ≤ 18, −13 ≤ k ≤ 13, −21 ≤ l ≤ 21 | –18 ≤ h ≤ 20, −18 ≤ k ≤ 18, −34 ≤ l ≤ 35 |
| reflections collected | 36 745 | 68 483 |
| independent reflections | 5713 [R(int) = 0.2071] | 7319 [R(int) = 0.0460] |
| completeness (%) | 99.6 | 99.8 |
| absorption correction | semiempirical from equivalents | semiempirical from equivalents |
| max. and min. transmission | 0.671 and 0.456 | 0.693 and 0.534 |
| refinement method | full-matrix least squares on F2 | full-matrix least squares on F2 |
| data/restraints/parameters | 5713/0/255 | 7319/0/283 |
| goodness of fit on F2 | 1.058 | 1.046 |
| final R indices [I > 2σ(I)] | R1 = 0.1754, wR2 = 0.4189 | R1 = 0.0300, wR2 = 0.0632 |
| R indices (all data) | R1 = 0.2969, wR2 = 0.5053 | R1 = 0.0551, wR2 = 0.0748 |
| extinction coefficient | 0.00213(9) | |
| largest diff. peak and hole | 3.558 and −2.496 e/Å3 | 1.163 and −0.936 e/Å3 |
General Procedures
Palladium N-heterocyclic carbene pyridine complexes (Pd-C1 to Pd-C3) were synthesized based on the modified published procedures.49,50
Synthesis of 1-Alkyl Benzimidazole (BZ1, BZ2, and BZ3)
1-Alkyl benzimidazoles (BZ1–BZ2) were prepared according to modified literature procedures.43,45,74 Benzimidazole (10.0 mmol) was introduced into a clean and dry round-bottom flask with an excess amount of alkyl bromide (12.2 mmol) (2-bromopropane or benzyl bromide). Potassium hydroxide (20.0 mmol) for 2-bromopropane or cesium carbonate (20.0 mmol) with 1 mmol of tetrabutylammonium bromide (TBAB) for benzyl bromide was used as a base. The mixture was dissolved in 100 mL of distilled acetonitrile and stirred for 24 h at 80 °C. The reaction was monitored by TLC using the solvent system ethyl acetate/hexane (ratio = 1/1) until no free benzimidazole was observed. The solvent was then removed by a rotary evaporator. The oily product was purified by extraction twice with 30 mL of ethyl acetate and 20 mL of distilled water. The aqueous layer was separated and washed with ethyl acetate; the separated organic layers were dried.
1-Adamantyl benzimidazole (BZ3) was synthesized via another pathway (Scheme 2). 2-Fluoronitrobenzene (10.0 mmol) was added to 1-adamantylamine (14.5 mmol) and anhydrous potassium fluoride (13.3 mmol) under argon in a 50 mL Schlenk purged and flamed under vacuum. After three vacuum/argon cycles, the mixture was heated and stirred at 180 °C for 48 h and then cooled down to room temperature. Then, dichloromethane was added to the reaction mixture followed by the addition of water. The organic phase was washed with brine and water, dried on sodium sulfate, and concentrated under vacuum. The crude mixture was purified on silica gel using a dichloromethane/hexane mixture (1/5 ratio by volume), affording a yellow-orange solid (0.178 g, 92%) of ortho-nitro-1-adamantylaniline (A) (Scheme 2). The reduction of A was achieved using sodium borohydride to produce ortho-amino adamantyl aniline. The reduction process started by dissolving 10.0 mmol of A in THF, and then 0.25 mmol of Pd/C was added carefully. The resulted slurry was stirred and NaBH4 (25.0 mmol) was added portionwise. The obtained solid was then filtered and extracted three times using a mixture of ethyl acetate and H2O. To fully dry the solid, anhydrous sodium sulfate was added and the solid was filtered off and evaporated to dryness to yield ortho-aminoadamantyl aniline (B) (76%) as a dark oil. Finally, a round-bottom flask was charged with ortho-aminoadamantyl aniline (B) (8.80 mmol), anhydrous THF, trimethyl orthoformate (6.60 mmol), and formic acid (0.880 mmol). The resulting mixture was stirred overnight at room temperature. The solvents were evaporated, and the crude product was purified by column chromatography using hexane/ethyl acetate (7/3) as the eluent to yield N-adamantyl-1H-benzo[d]imidazole (95%).
1-Isopropyl-1H-benzo[d]imidazole (BZ1)
Yield = 77%. Sticky brown oil product. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.30 (s, 1H, N2CHbini), 7.69–7.62 (m, 1H, Ar-H), 7.25–7.22 (m, 1H, Ar-H), 7.11–7.08 (m, 2H, Ar-H), 4.42 (sep, 1H, 3J = 6.76 Hz, NCH) 1.40 (d, 6H, 3J = 6.76 Hz, NC(CH3)2); 13C{1H} NMR (125 MHz, CDCl3) δ (ppm): 143.5 (NCN), 139.8, 132.7, 122.1, 121.5, 119.8, 109.7, (Ar-H), 47.2 (NCH), 22.02 [NC(CH3)2]. Anal. calcd for C10H12N2 (160): C, 74.97%; H, 7.55%; N, 17.48%. Found: C, 74.84%; H, 7.23%; N, 17.93%.
1-Benzyl-1H-benzo[d]imidazole (BZ2)
Yield = 87%; light yellow solid; 1H NMR (500 MHz, CDCl3) δ (ppm): 7.98 (s, 1H, N2CHbini), 7.83 (d, 1H, 3J = 7.63 Hz, Ar-H), 7.34–7.24 (m, 6H, Ar-H), 7.18 (d, 2H, 3J = 7.02 Hz, Ar-H), 5.36 (m, 2H, NCH2-Ph); 13C{1H} NMR (125 MHz, CDCl3) δ (ppm); 143.1 (NCN), 135.4, 129.0, 128.3, 127.1, 123.1, 122.3, 120.3, 110.0 (Ar-H), 48.8 (NCH2). Anal. calcd for C14H12N2 (208.26): C, 80.74%; H, 5.81%; N, 13.45%. Found: C, 80.79%; H, 5.63%; N, 13.58%.
1-(Adamantan-1-yl)-1H-benzo[d]imidazole (BZ3)
Yield = 85%; light brown solid; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.22 (s, 1H, N2CHbini), 7.85 (m, 1H, Ar-H), 7.64 (m, 1H, Ar-H), 7.18 (m, 2H, Ar-H), 2.33 (m, 6H, 3CH2), 2.21 (m, 3H, 3CH), 1.81–1.75 (m, 6H, NC 3CH2); 13C{1H} NMR (125 MHz, DMSO-d6) δ (ppm); 140.8 (NCN), 121.6, 121.1, 119.8, 113.9 (Ar-H), 56.6 (NC), 41.13 NC(C)3, 35.45 C(CH)CH2, 28.98 CH(CH2)CH. Anal. calcd for C17H20N2 (252.36): C, 80.91%; H, 7.99%; N, 11.10%. Found: C, 80.73%; H, 7.76%; N, 11.51%.
Synthesis of the Precursors for N-Heterocyclic Carbene Ligands (NHC-1, NHC-2, and NHC-3)
1-Alkyl benzimidazole (BZ1, BZ2, or BZ3) (1.00 mmol) and 2-bromopropane (3.00 mL) were charged into a pressure tube. The mixture was stirred for 48 h at 120 °C and then cooled down to room temperature. For NHC-1 and NHC-3 cases, the filtration was used to collect the products as solid precipitates and then washed several times with diethyl ether (5 mL). The pure product was dried under vacuum. For NHC-2, a brown sticky mixture was obtained. Based on the ionic nature of the product, a careful extraction under fume hood (Caution: the evaporated fumes from the reaction are irritating to the eyes) with 5 mL of water for three times was carried out. The aqueous layer was collected, and water was evaporated under vacuum. The product was obtained as orange crystals.
1,3-Diisopropylbenzimidazole-3-ium Bromide (NHC-1)
Yield = 65%. White solid. 1H NMR (400 MHz, CDCl3) δ (ppm): 11.35 (s, 1H, N2CHbini), 7.80–7.78 (m, 2H, C-H- arom), 7.65–7.63 (m, 2H, C-H arom), 5.19 (sep, 2H, 3J = 6.8 Hz, NCH), 1.85 (d, 12H, 3J = 6.8 Hz, 2(CH3)2); 13C NMR (125 MHz, CDCl3) δ (ppm): 140.7, 130.8, 126. 9, 113.9 (C arom); 52.4 2C(CH3)2, 22.2, 2(CH3)2, GC-MS m/z 283.2 (M+). Anal. calcd for C13H19BrN2, (283.21): C, 55.13%; H, 6.76%; N, 9.89%. Found: C, 55.41; H, 7.02; N, 10.03. ESI-MS: m/z 202.17 [M – Br]+.
1-Benzyl-3-isopropyl-1H-benzo[d]imidazol-3-ium Bromide (NHC-2)
Yield = 85%. Orange crystalline solid. 1H NMR (400 MHz, CDCl3) δ (ppm): 11.51 (s, 1H, N2C-Hbini), 7.78 (d, 1H, 3J = 8.00 Hz, C-H arom), 7.65–7.51 (m, 5H, C-H arom), 7.35–7.29 (m, 3H, C-H-(phenyl)), 5.97 (s, 2H, CH2-Ph), 5.03 (sep, 1H, 3J = 6.4 Hz, NCH), 1.83 (d, 6H, 3J = 6.4 Hz, NC(CH3)2); 13C NMR (125 MHz, CDCl3) δ (ppm): 141.3, 133.0, 131.5, 130.8, 129.3, 129.1, 128.5, 127.3, 127.1, 114.1, 113.6 (C arom), 51.9 (NCH2), 51.4 (NCH2), 22.4 (2(CH3)2). Anal. calcd for C17H19BrN2, (331.26): C, 61.64%; H, 5.78%; N, 8.46%. Found: C, 62.05; H, 5.93; N, 8.80. MS: m/z 331.2 (M+). ESI-MS; m/z 251.2 [M – Br]+.
1-(Adamantan-1-yl)-3-isopropyl-1H-benzo[d]imidazol-3-ium Bromide (NHC-3)
Yield = 88%. Light brown solid. 1H NMR (400 MHz, CDCl3) δ (ppm): 10.82 (s, 1H, N2C-Hbini), 8.03 (d, 1H, 3J = 8 Hz, C-H arom), 7.2 (d, 1H, J = 7.0 Hz, C-H arom), 7.63–7.57 (m, 2H, C-H arom), 5.61 (sep, 1H, 3J = 6.4 Hz, CH), 2.55 (m, 6H, 3CH2adam), 2.41 (m, 3H, 3CHadam), 1.87–1.85 (m, 12H, {3CH2adam} + {2CH3}); 13C NMR (125 MHz, CDCl3) δ (ppm): 140.22, 131.44, 130.49, 126.31, 126.04, 116.77, 114.59 (C arom), 63.02 NC(C)3, 52.86 C(CH3)2, 41.72 C(CH2)3, 35.58 3(CH), 29.54 [3(CH2), C(CH3)2]. Anal. calcd for C20H27BrN2, (375.35): C, 64.00%; H, 7.25%; N, 7.46%. Found: C, 64.321; H, 7.53; N, 7.80. MS: m/z 375 (M+); ESI-MS; m/z 294.2 [M – Br]+.
Synthesis of Palladium N-Heterocyclic Carbene Pyridine Complexes (Pd-C1, Pd-C2, and Pd-C3)
A round-bottom flask (15 mL) was charged by an N-heterocyclic carbene ligand precursor (NHC-1, NHC-2, or NHC-3) (0.50 mmol), palladium(II) bromide (0.50 mmol), potassium carbonate (2.0 mmol), and pyridine (5 mL) (Scheme 4). The reaction mixture was heated to 90 °C for 24 h under stirring. After cooling down to room temperature, a colloidal crude product was diluted with 5 mL of dichloromethane and the mixture was purified by passing through a short silica column covered with a short pad of celite. The flash column was eluted with distilled methanol. The solvents were evaporated using a rotary evaporator. The complexes were washed with ether and then collected and dried at room temperature under vacuum. The complexes were obtained as yellow crystals. A slow crystallization procedure was followed in the preparation of single crystals for these complexes. A saturated solution of the palladium was prepared in dichloromethane/acetonitrile (8/1) (v/v).
(1,3-Diisopropyl-1,3-dihydro-2H-benzo[d]imidazol-2-ylidene)(pyridin-1(2H)-yl) Palladium(II) Bromide (Pd-C1)
Yield = 77%. Yellow solid. 1H NMR (400 MHz, CDCl3) δ (ppm): 9.07–9.06 (m, 2H, C-H- pyr), 7.73–7.70 (m, 1H, C-H- pyr), 7.53–7.52 (m, 2H, C-H arom), 7.31–7.29 (m, 2H, C-H arom), 7.16–7.15 (m, 2H, C-H pyr), 6.27 (sep, 2H, 3J = 7.2 Hz, NCH), 1.73 (d, 12H, 3J = 7.2 Hz, 4CH3), 13C NMR (125 MHz, CDCl3) δ (ppm): 158.1 (Pd-C), 151.6, 136.7, 132.3, 123.3, 121.0, 111.4 (C arom); 53.4 [C(CH3)2], 19.4 (2CH3). Calcd for C18H23Br2N3Pd, (547.63): C, 39.48%; H, 4.23%; N, 7.67%. Found: C, 40.05; H, 4.62; N, 7.94. ESI-MS: m/z 467.12 [M – Br]+.
(1-Benzyl-3-isopropyl-1,3-dihydro-2H-benzo[d]imidazol-2-ylidene)(pyridin-1(2H)-yl) Palladium(II) Bromide (Pd-C2)
Yield = 74%. Yellow crystals. 1H NMR (400 MHz, CDCl3) δ (ppm): 1H NMR (400 MHz, CDCl3) δ (ppm): 9.06–9.05 (m, 2H, C-H pyr), 7.71–7.69 (m, 1H, C-H pyr), 7.48 (m, 2H, C-H arom), 7.45–7.41 (m, 5H, C-H arom), 7.35–7.29 (m, 2H, C-H phenyl), 7.13–7.09 (m, 3H, C-H pyr), 5.87 (s, 1H, CH2-Ph), 5.30 (m, 1H, NCH), 1.67 (m, 6H, NC(CH3)2); 13C NMR (125 MHz, CDCl3) δ (ppm): 154.3 (Pd-C), 153.4, 138.5, 128.8, 128.1, 127.3, 127.1, 125.0, 122.8, 112.3, 110.7 (C arom), 53.5 (NCH2), 51.4 (NCH2), 21.44 [(CH3)2]. Anal. calcd for C22H23Br2N3Pd, (595.68): C, 44.36%; H, 3.39%; N, 7.05%. Found: C, 43.85; H, 4.13; N, 7.48; ESI-MS: m/z 515.11 [M – Br]+.
(1-(Adamantan-1-yl)-3-isopropyl-1,3-dihydro-2H-benzo[d]imidazol-2-ylidene)(pyridin-1(2H)-yl)palladium(II) Bromide (Pd-C3)
Yield = 86%. Light yellow crystals. 1H NMR (400 MHz, CDCl3) δ (ppm): 9.11–9.10 (m, 2H, C-H pyr), 8.87 (d, 1H, 3J = 7.8 Hz, C-H arom), 7.81–7.77 (m, 1H, C-H pyr), 7.64 (d, 1H, 3J = 7.4 Hz, C-H arom), 7.39–7.36 (m, 2H, C-H arom), 7.25–7.18 (m, 2H, C-H- pyr), 7.11 (sep, 1H, 3J = 7 Hz, NCH), 3.24 (m, 6H, 3CH2Adam), 2.43 (m, 3H, 3CHAdam), 1.98 (m, 3H, 3CHAdam), 1.85–1.83 (m, 9H, {3CHAdam + 2CH3}); 13C NMR (125 MHz, CDCl3) δ (ppm): 156.8 (Pd-C), 152.9, 137.8, 135.0, 133.2, 124.6, 122.0, 121.3, 115.7, 112.7 (C arom), 61.8 NC(C)3, 57.2 C(CH3)2, 42.6 C(CH2)3, 36.2 3(CH), 30.3 3(CH2), 20.2 C(CH3)2. Anal. calcd for C25H31Br2N3Pd, (639.77): C, 46.93%; H, 4.88%; N, 6.57%. Found: C, 47.32; H, 5.33; N, 7.13. ESI-MS: m/z 559.09 [M – Br]+.
General Procedure for the Carbonylative Sonogashira Coupling Reactions
Carbonylative Sonogashira coupling reactions were conducted in a 45 mL stainless steel autoclave equipped with a glass liner, a gas inlet valve, and a pressure gauge. Palladium complex (0.030 mol %), aryl iodide (1.0 mmol), alkyne (1.2 mmol), base (2.0 mmol), and a solvent (3 mL) were introduced into the glass liner. The glass liner was then placed carefully in the autoclave. The autoclave was vented three times with carbon monoxide and then pressurized to 200 psi of CO. The mixture was heated to the required temperature and stirred for the required time. After completion of the reaction, the mixture was cooled down to room temperature and the excess of CO was vented slowly and carefully under fume hood. Then, 5 mL of distilled water was added to the mixture. The diluted mixture was extracted three times with 10 mL of ethyl acetate. The solvent was concentrated on a rotary evaporator. Several techniques were used to fully characterize the final carbonyl products, including 1H, 13C NMR, GC, and GC-MS. The spectral data of the alkynones prepared in this study were in full agreement with those reported in the literature.18,29,31,53−55,65−67,73
Analytical and Spectroscopic Data of New Carbonyl Compounds
1-(4-Methoxyphenyl)-3-(4-pentylphenyl)prop-2-yn-1-one (3ac)
Yield = 96%. Light brown solid. 1H NMR (500 MHz, CDCl3) δ (ppm): 8.19 (d, 2H, 3J = 6.6 Hz, CH arom), 7.58 (d, 2H, 3J = 8.24 Hz, CH arom), 7.22 (d, 2H, 3J = 7.38 Hz, C-H arom), 6.98 (d, 2H, 3J = 7.88 Hz, CH arom), (d, 2H, 3J = 7.90 Hz, CH arom), 3.89 (s, 3H, OCH3), 2.64 (t, 2H, 3J = 7.3 Hz, CH2), 1.63 (p, 2H, 3J = 6.1 Hz, CH2), 1.33 (m, 2H, CH2), 0.89 (t, 3H, 3J = 6.1 Hz, CH3); 13C NMR (125 MHz, CDCl3) δ (ppm): 176.7 C=O, 164.4, 146.2, 133.0, 131.9, 130.4, 128.8, 117.4, 113.82 (C arom), 93.0, 86.8, 55.6 (OCH3), 36.0 (CH2), 31.4 (3CH2), 31.3 (CH2), 22.4 (CH2), 13.9 (CH3). IR (ν cm–1): 2362, 2193, 1766, 1626, 1591, 1505, 1459, 1264, 1157. Anal. calcd for C21H22O2, (306.16): C, 82.32%; H, 7.24%; Found: C, 81.82; H, 7.56. MS: m/z 306.2 (M+).
1-(4-Methoxyphenyl)tridec-2-yn-1-one (5ad)
Yield = 87%. Orange solid. 1H NMR (500 MHz, CDCl3) δ (ppm): 8.07 (d, 2H, 3J = 8.25 Hz, C-H arom), 6.91 (d, 2H, 3J = 8.25 Hz, C-H arom), 3.84 (s, 3H, OCH3), 2.45 (t, 2H, 3J = 7.0 Hz, CH2), 1.65–1.62 (m, 2H, CH2), 1.44 (m, 2H, CH2), 1.25 (m, 12H, 6CH2), 0.85 (m, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ (ppm): 176.7 (C = O), 164.1, 138.0, 131.7, 130.2, 116.2, 113.4 (C arom), 95.8, 79.5, 55.3 (OCH3), 31.3 (CH2), 31.7–27.7 (complex of CH2), 22.5 (CH2), 19.0 (CH2), 13.9 (CH3). IR (ν cm–1): 2360, 2200, 1681, 1641, 1599, 1509, 1461, 1258, 1061. Anal. calcd for C20H28O2 (300.44): C, 82.32%; H, 7.24%; Found: C, 81.82; H, 7.56. MS: m/z 300.44 (M+).
4-Cyclohexyl-1-(4-methoxyphenyl)but-2-yn-1-one (5ag)
Yield = 67%. Brown solid. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.09 (d, 2H, 3J = 8.4 Hz, C-H arom), 6.93 (d, 2H, 3J = 8.4 Hz, C-H arom), 3.86 (s, 3H, OCH3), 2.37 (d, 2H, 3J = 6.4 Hz, CH2), 1.88–1.85 (m, 2H, CH2), 1.76-1.65 (m, 3H, “overlap with water from the solvent” C-H), 1.29-1.066 (m, 6H, 3CH2); 13C NMR (125 MHz, CDCl3) δ (ppm): 176.9 (C=O), 164.3, 131.8, 130.4, 113.7 (C arom), 95.0, 80.6, 55.5 (OCH3), 36.9 (CH), 32.8 (CH2), 26.9 (CH), 26.0 (CH2). IR (ν cm–1): 2235, 2200, 1706, 1639, 1599, 1510, 1455, 1257, 1169. Anal. calcd for C17H20O2, (256.2): C, 79.65%; H, 7.86%; Found: C, 80.09; H, 8.11. MS: m/z 256.2 (M+).
1,1′-(1,4-Phenylene)bis(6-chlorohex-2-yn-1-one) (9a)
Yield = 83%. Off-white solid. 1H NMR (500 MHz, CDCl3) δ (ppm): 7.76 (2, 4H, C-H arom), 3.63 (m, 4H, ClCH2), 2.71 (m, 4H, CH2), 1.76 (m, 4H, CH2); 13C NMR (125 MHz, CDCl3) δ (ppm): 177.1 C=O, 137.9, 130.7 (C arom), 102.5, 94.8, 43.3 (ClCH2), 30.3 (CH2), 16.6 (CH2); IR (ν cm–1): 2359, 2341, 1751, 1647, 1384, 1094, 798. Anal. calcd for C18H16Cl2O2, (335.2): C, 64.49%; H, 4.81%; Found: C, 64.76; H, 5.13. MS: m/z 335.2 (M+).
Analytical and Spectroscopic Data of Known Carbonyl Compounds
1,3-Diphenylprop-2-yn-1-one (3ba)
Yield = 98%; 1H NMR (500 MHz, CDCl3) δ (ppm): 8.23 (d, J = 8.23 Hz, 2H, C-H arom), 7.69 (d, J = 8.23 Hz, 2H, C-H arom), 7.64 (t, J = 7.32 Hz, 1H, C-H arom), 7.47 (m, 5H, C-H arom); 13C NMR (125 MHz, CDCl3) δ (ppm): 178.0, 136.9, 134.1, 134.1, 133.0, 130.8, 129.6, 128.7, 128.7, 128.6, 128.6, 128.4, 120.1, 93.1, 86.9; GC–MS m/z 206 (M+1).
1-(4-Methoxyphenyl)-3-phenylprop-2-yn-1-one (3aa)
Yield = 97%; 1H NMR (500 MHz, CDCl3) δ (ppm): 8.20 (d, J = 8.8 Hz, 2H, C-H arom), 7.67 (d, J = 6.1 Hz, 2H, C-H arom), 7.43 (m, 3H, C-H arom), 6.98 (t, J = 5.8 Hz, 2H, C-H arom), 3.90 (s, 3H, OCH3); 13C NMR (125 MHz, CDCl3) δ (ppm): 176.7, 164.5, 132.9, 132.0, 130.6, 130.5, 130.3, 128.6, 120.4, 113.9, 92.3, 86.9, 55.6; GC–MS m/z 236 (M + 1).
1-Phenylhept-2-yn-1-one (5bb)
Yield = 92%; 1H NMR (500 MHz, CDCl3) δ (ppm): 8.06 (d, J = 7.63 Hz, 2H, C-H arom), 7.51 (t, J = 7.32 Hz, 1H, C-H arom), 7.40 (t, J = 7.02 Hz, 2H, C-H arom), 2.42 (t, J = 7.02 Hz, 2H, CH2) 1.58 (m, 2H, CH2), 1.42 (m, 2H, CH2) 0.88 (t, J = 7.02 Hz, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ (ppm): 178.2, 136.9, 133.5, 129.5, 128.4, 96.9, 79.6, 29.8, 22.0, 18.9, 13.5; GC-MS m/z 186 (M+).
4,4-Dimethyl-1-phenylpent-2-yn-1-one (5bc)
Yield = 95%; 1H NMR (500 MHz, CDCl3) δ (ppm): 8.11 (d, J = 7.02 Hz, 2H, C-H arom), 7.59 (t, J = 7.32 Hz, 1H, C-H arom), 7.49 (t, J = 7.93 Hz, 2H, C-H arom), 1.38 (s, 9H, C(CH3)3); 13C NMR (125 MHz, CDCl3) δ (ppm): 178.4, 137.0, 133.8, 130.5, 128.4, 104.0, 78.1, 30.1; GC-MS m/z 186 (M + 1).
1-(4-Nitrophenyl)-3-phenylprop-2-yn-1-one (3ca)
Yield = 95%; 1H NMR (500 MHz, CDCl3) δ (ppm): 8.29 (d, J = 8.50 Hz, 2H, C-H arom), 8.07 (m, 3H, C-H arom), 7.71 (d, J = 8.50 Hz, 2H, C-H arom), 7.45 (t, J = 6.3 Hz, 2H, C-H arom); 13C NMR (125 MHz, CDCl3) δ (ppm): 177.2, 140.8, 139.9, 133.2, 132.8, 131.1, 129.7, 128.8, 128.5, 128.5, 127.3, 127.3, 118.0, 86.8; GC-MS m/z 251 (M + 1).
3,3′-Benzene-1,3-diylbis(1-phenylprop-2-yn-1-one) (7a)
Yield = 78%; 1H NMR (500 MHz, CDCl3) δ (ppm): 8.23 (d, J = 8.23 Hz, 4H, C-H arom), 8.0 (s, 1H, C-H arom),7.79 (d, J = 7.93 Hz, 2H, C-H arom), 7.66 (t, J = 7.63 Hz, 2H, C-H arom), 7.54 (m, 5H, C-H arom), 13C NMR (125 MHz, CDCl3) δ (ppm): 177.7, 137.0, 136.6, 134.8, 134.8, 134.4, 129.6, 129.3, 128.7, 121.2, 90.7, 87.5 GC-MS m/z 334 (M + 1).
1,1′-Benzene-1,4-diylbisoct-1-yn-3-one (9b)
Yield = 69%; 1H NMR (500 MHz, CDCl3) δ (ppm): 8.20 (s, 4H, C-H arom), 2.43 (t, J = 7.02 Hz, 4H, CH2) 1.58 (m, 4H, CH2), 1.434 (m, 4H, CH2) 0.89 (t, J = 7.02 Hz, 6H, CH3); 13C NMR (125 MHz, CDCl3) δ (ppm): 177.6, 140.5, 129.7, 105.4, 78.1, 29.9, 22.0, 18.9, 13.4; GC-MS m/z 322 (M + 1).
1,1′-(1,4-Phenylene)bis(4,4-dimethylpent-2-yn-1-one) (9c)
Yield = 55%, 1H NMR (500 MHz, CDCl3) δ (ppm): 8.2 (4H, C-H arom), 1.42 (s, 18H, C(CH3)3); 13C NMR (125 MHz, CDCl3) δ (ppm): 177.4, 140.4, 137.7, 129.0, 105.0, 78.0, 77.0, 30.1; GC-MS m/z 294 (M + 1).
Acknowledgments
Saudi Aramco is acknowledged for funding this project under the award number CHEM2432. The authors also acknowledge the Chemistry Department at King Fahd University of Petroleum & Minerals (KFUPM) for support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c02413.
Crystallographic data of the compound BZ3 (CIF)
Crystallographic data of the compound NHC2 (CIF)
Crystallographic data of the compound NHC3 (CIF)
Crystallographic data of the compound Pd-C2 (CIF)
Crystallographic data of the compound Pd-C3 (CIF)
1H NMR spectrum for 3aa; 13C NMR spectrum for 3aa; molecular orbital energy diagram for complexes Pd-C1, Pd-C2, and Pd-C3; steric map of Pd-NHCs-Py complexes; (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Karpov A. S.; Merku E.; Rominger F.; Müller T. J. Concise syntheses of meridianins by carbonylative alkynylation and a four-component pyrimidine synthesis. Angew. Chem., Int. Ed. 2005, 44, 6951. 10.1002/anie.200501703. [DOI] [PubMed] [Google Scholar]
- Whittaker R. E.; Dermenci A.; Dong G. Synthesis of Ynones and Recent Application in Transition-Metal-Catalyzed Reactions. Synthesis 2016, 48, 161–183. 10.1055/s-0035-1560515. [DOI] [Google Scholar]
- Peng S.; Wang Z.; Zhang L.; et al. Streamlined asymmetric α-difunctionalization of ynones. Nat. Commun. 2018, 9, 375 10.1038/s41467-017-02801-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender P. A.; Hardman C. T.; Ho S.; Jeffreys M. S.; Maclaren J. K.; Quiroz R. V.; Ryckbosch S. M.; Shimizu A. J.; Sloane J. L.; Stevens M. C. Scalable synthesis of bryostatin 1 and analogs, adjuvant leads against latent HIV. Science 2017, 358, 218–223. 10.1126/science.aan7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden M. D.; Loy B. A.; Wu X.; Ramirez C. M.; Schrier A. J.; Murray D.; Shimizu A.; Ryckbosch S. M.; Near K. E.; Chun T. W.; Wender P. A.; Zack J. A.; et al. In vivo activation of latent HIV with a synthetic bryostatin analog effects both latent cell “kick” and “kill” in strategy for virus eradication. PLoS Pathog. 2017, 13, e1006575 10.1371/journal.ppat.1006575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh H.; Inoue M. Comprehensive Structure–Activity Relationship Studies of Macrocyclic Natural Products Enabled by Their Total Syntheses. Chem. Rev. 2019, 119, 10002–10031. 10.1021/acs.chemrev.9b00063. [DOI] [PubMed] [Google Scholar]
- Yang Q.; Alper H. Synthesis of Chromones via Palladium-Catalyzed Ligand-Free Cyclocarbonylation of o-Iodophenols with Terminal Acetylenes in Phosphonium Salt Ionic Liquids. J. Org. Chem. 2010, 75, 948–950. 10.1021/jo902210p. [DOI] [PubMed] [Google Scholar]
- Liang B.; Huan M.; You Z.; Xiong Z.; Lu K.; Fathi K.; Chen J.; Yang Z. Pd-Catalyzed Copper-Free Carbonylative Sonogashira Reaction of Aryl Iodides with Alkynes for the Synthesis of Alkynyl Ketones and Flavones by Using Water as a Solvent. J. Org. Chem. 2005, 70, 6097–6100. 10.1021/jo050498t. [DOI] [PubMed] [Google Scholar]
- Arcadi A.; Aschi M.; Marinelli F.; Verdecchia M. Pd-catalyzed regioselective hydroarylation of α-(2-aminoaryl)-α,β-ynones with organoboron derivatives as a tool for the synthesis of quinolines: experimental evidence and quantum-chemical calculations. Tetrahedron 2008, 64, 5354–5361. 10.1016/j.tet.2008.03.015. [DOI] [Google Scholar]
- Kirkham J. D.; Edeson S. J.; Stokes S.; Harrity J. P. Synthesis of Ynone Trifluoroborates toward Functionalized Pyrazoles. Org. Lett. 2012, 14, 5354–5357. 10.1021/ol302418b. [DOI] [PubMed] [Google Scholar]
- Ahmed M. S. M.; Kobayashi K.; Mori A. One-Pot Construction of Pyrazoles and Isoxazoles with Palladium-Catalyzed Four-Component Coupling. Org. Lett. 2005, 7, 4487–4489. 10.1021/ol051841j. [DOI] [PubMed] [Google Scholar]
- Gowrisankar S.; Lee H. S.; Kim J. M.; Kim J. N. Pd-mediated synthesis of 2-arylquinolines and tetrahydropyridines from modified Baylis–Hillman adducts. Tetrahedron Lett. 2008, 49, 1670. 10.1016/j.tetlet.2007.12.13. [DOI] [Google Scholar]
- Bannwarth P.; Valleix A.; Grée D.; Grée R. Flexible Synthesis of Pyrimidines with Chiral Monofluorinated and Difluoromethyl Side Chains. J. Org. Chem. 2009, 74, 4646–4649. 10.1021/jo900674u. [DOI] [PubMed] [Google Scholar]
- She Z.; Niu D.; Chen L.; Gunawan M. A.; Shanja X.; Hersh W. H.; Chen Y. Synthesis of Trisubstituted Isoxazoles by Palladium (II)-Catalyzed Cascade Cyclization–Alkenylation of 2-Alkyn-1-one O-Methyl Oximes. J. Org. Chem. 2012, 77, 3627–3633. 10.1021/jo300090k. [DOI] [PubMed] [Google Scholar]
- Neumann K. T.; Laursen S. R.; Lindhardt A. T.; Bang-Andersen B.; Skrydstrup T. Correction to Palladium-Catalyzed Carbonylative Sonogashira Coupling of Aryl Bromides Using Near Stoichiometric Carbon Monoxide. Org. Lett. 2014, 168, 2216–2219. 10.1021/ol5007289. [DOI] [PubMed] [Google Scholar]
- Taylor C. L.; Bolshan Y. Metal-free synthesis of ynones from acyl chlorides and potassium alkynyl trifluoroborate salts. J. Visualized Exp. 2015, 9, 52401 10.3791/52401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T.; Tanaka M. Carbonylation of organic halides in the presence of terminal acetylenes; novel acetylenic ketone synthesis. J. Chem. Soc., Chem. Commun. 1981, 333 10.1039/c39810000333. [DOI] [Google Scholar]
- Ahmad M. S. M.; Mori A. Carbonylative Sonogashira Coupling of Terminal Alkynes with Aqueous Ammonia. Org. Lett. 2003, 5, 3057. 10.1021/ol035007a. [DOI] [PubMed] [Google Scholar]
- Qi X.; Li-Bing J.; Chong-Liang L.; Rui L.; Xiao-Feng W. palladium-catalyzed One-Pot carbonylative Sonogashira reaction employing formic acid as the CO source. Chem. Asian J. 2015, 10, 1870–1873. 10.1002/asia.201500518. [DOI] [PubMed] [Google Scholar]
- Brennführer A.; Neumann H.; Beller M. Palladium-catalyzed carbonylation reactions of aryl halides and related compounds. Angew. Chem., Int. Ed. 2009, 48, 4114. 10.1002/anie.200900013. [DOI] [PubMed] [Google Scholar]
- Kang S.-K.; Ryu H.-C.; Hong Y.-T. Pd(0)–Cu(I)-catalyzed cross-coupling of alkynylsilanes with triarylantimony(V) diacetates. J. Chem. Soc., Perkin Trans. 1 2001, 1, 736. 10.1039/b007800j. [DOI] [Google Scholar]
- Gadge S. T.; Bhanage B. M. Recent developments in palladium catalysed carbonylation reactions. RSC Adv. 2014, 4, 10367. 10.1039/c3ra46273k. [DOI] [Google Scholar]
- Zhou Z.; Jianying L.; Zhaotao X.; Cai M. Recyclable heterogeneous palladium-catalyzed carbonylative Sonogashira coupling under CO gas-free conditions. Synth. Commun. 2020, 50, 2015–2025. 10.1080/00397911.2020.1762092. [DOI] [Google Scholar]
- Gautam P.; Tiwari N. J.; Bhanage B. M. Aminophosphine Palladium Pincer-Catalyzed Carbonylative Sonogashira and Suzuki–Miyaura Cross-Coupling with High Catalytic Turnovers. ACS Omega 2019, 4, 1560–1574. 10.1021/acsomega.8b02886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genelot M.; Dufaud V.; Djakovitch L. Carbonylative Sonogashira Coupling in the Synthesis of Ynones: A Study of “Boomerang” Phenomena. Adv. Synth. Catal. 2013, 355, 2604–2616. 10.1002/adsc.201300357. [DOI] [Google Scholar]
- Bai C.; Jian S.; Yao X.; Li Y. Carbonylative Sonogashira coupling of terminal alkynes with aryl iodides under atmospheric pressure of CO using Pd(ii)@MOF as the catalyst. Catal. Sci. Technol. 2014, 4, 3261–3267. 10.1039/C4CY00488D. [DOI] [Google Scholar]
- Ibrahim M. B.; Malik I.; Hussain S. M. S.; Fazal A.; Fettouhi M.; Ali B. E. Synthesis, crystal structures and catalytic activities of new palladium(II)–bis(oxazoline) complexes. Transition Met. Chem. 2016, 41, 739–749. 10.1007/s11243-016-0072-1. [DOI] [Google Scholar]
- He M.; Justin R. S.; Jeffrey W. B. Highly Enantioselective Azadiene Diels–Alder Reactions Catalyzed by Chiral N-Heterocyclic Carbenes. J. Am. Chem. Soc. 2006, 128, 8418–8420. 10.1021/ja062707c. [DOI] [PubMed] [Google Scholar]
- Aktaş A.; Barut D.; Ruya C.; Parham K.; Yetkin T.; Muhittin G.; Gülçinc A. İ. Novel morpholine liganded Pd-based N-heterocyclic carbene complexes: Synthesis, characterization, crystal structure, antidiabetic and anticholinergic properties. Polyhedron 2019, 159, 345–354. 10.1016/j.poly.2018.11.048. [DOI] [Google Scholar]
- Kumar A.; Yuan D.; Huynh H. V. Stereoelectronic Profiling of Expanded-Ring N-Heterocyclic Carbenes. Inorg. Chem. 2019, 58, 7545–7553. 10.1021/acs.inorgchem.9b00786. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Liu J.; Xia C. Aryl-palladium-NHC complex: efficient phosphine-free catalyst precursors for the carbonylation of aryl iodides with amines or alkynes. Org. Biomol. Chem. 2014, 12, 9702–9706. 10.1039/C4OB01878H. [DOI] [PubMed] [Google Scholar]
- Nguyen V. H.; Ibrahim M. B.; Mansour W. W.; El Ali B. M.; Huynh H. V. Postmodification Approach to Charge-Tagged 1,2,4-Triazole-Derived NHC Palladium (II) Complexes and Their Applications. Organometallics 2017, 36, 2345–2353. 10.1021/acs.organomet.7b00329. [DOI] [Google Scholar]
- Kostyukovich A. Y.; Tsedilin A. M.; Sushchenko E. D.; Eremin D. B.; Kashin A. S.; Topchiy M. A.; Asachenko A. F.; Nechaevbc M. S.; Ananikov V. P. In situ transformations of Pd/NHC complexes with N-heterocyclic carbene ligands of different nature into colloidal Pd nanoparticles. Inorg. Chem. Front. 2019, 6, 482–492. 10.1039/C8QI01095A. [DOI] [Google Scholar]
- Tao W.; Wang X.; Ito S.; Nozaki K. Palladium complexes bearing an N-heterocyclic carbene–sulfonamide ligand for cooligomerization of ethylene and polar monomers. J. Polym. Sci., Part A: Polym. Chem. 2019, 57, 474–477. 10.1002/pola.29270. [DOI] [Google Scholar]
- Rajabi F.; Thiel W. R. An Efficient Palladium N-Heterocyclic Carbene Catalyst Allowing the Suzuki–Miyaura Cross-Coupling of Aryl Chlorides and Arylboronic Acids at Room Temperature in Aqueous Solution. Adv. Synth. Catal. 2014, 356, 1873–1877. 10.1002/adsc.201300841. [DOI] [Google Scholar]
- Wang T.; Xie H.; Liu L.; Zhao W. X. N-heterocyclic carbene-palladium (II) complexes with benzoxazole or benzothiazole ligands: Synthesis, characterization, and application to Suzuki–Miyaura cross-coupling reaction. J. Organomet. Chem. 2016, 804, 73–79. 10.1016/j.jorganchem.2015.12.039. [DOI] [Google Scholar]
- Ma X.; Hangxiang W.; Chen W. N-Heterocyclic Carbene-Stabilized Palladium Complexes as Organometallic Catalysts for Bioorthogonal Cross-Coupling Reactions. J. Org. Chem. 2014, 79, 8652–8658. 10.1021/jo5014228. [DOI] [PubMed] [Google Scholar]
- Hopkinson M. N.; Richter C. M.; et al. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. 10.1038/nature13384. [DOI] [PubMed] [Google Scholar]
- Nelson D. J.; Nolan S. P. Quantifying and understanding the electronic properties of N-heterocyclic carbenes. Chem. Soc. Rev 2013, 42, 6723–6753. 10.1039/c3cs60146c. [DOI] [PubMed] [Google Scholar]
- Tolman C. A. Phosphorus ligand exchange equilibriums on zerovalent nickel. Dominant role for steric effects. J. Am. Chem. Soc. 1970, 92, 2953–2956. 10.1021/ja00713a006. [DOI] [Google Scholar]
- Tolman C. A. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev. 1977, 77, 313–348. 10.1021/cr60307a002. [DOI] [Google Scholar]
- Huynh H. V.; Han Y.; Jothibasu R.; Yang J. A. 13C NMR Spectroscopic Determination of Ligand Donor Strengths Using N-Heterocyclic Carbene Complexes of Palladium (II). Organometallics 2009, 28, 5395–5404. 10.1021/om900667d. [DOI] [Google Scholar]
- Verlinden K.; Buhl H.; Frank W.; Ganter C. Determining the Ligand Properties of N-Heterocyclic Carbenes from 77Se NMR Parameters. Eur. J. Inorg. Chem. 2015, 201, 2416–2425. 10.1002/ejic.201500174. [DOI] [Google Scholar]
- Teng Q.; Huynh H. V. Determining the Electron-Donating Properties of Bidentate Ligands by 13C NMR Spectroscopy. Inorg. Chem. 2014, 53, 10964–10973. 10.1021/ic501325j. [DOI] [PubMed] [Google Scholar]
- Teng Q.; Huynh H. V. A unified ligand electronic parameter based on 13C NMR spectroscopy of N-heterocyclic carbene complexes. Dalton Trans. 2017, 46, 614–627. 10.1039/C6DT04222H. [DOI] [PubMed] [Google Scholar]
- Schmid T. E.; Jones D. C.; Songis O.; Diebolt O.; Furst M. R. L.; et al. Mixed phosphine/N-heterocyclic carbene palladium complexes: synthesis, characterization and catalytic use in aqueous Suzuki–Miyaura reactions. Dalton Trans. 2013, 42, 7345–7353. 10.1039/c2dt32858e. [DOI] [PubMed] [Google Scholar]
- Aktaş A.; Erdemir F.; Celepci D. B.; Gök Y.; Aygün M. Mixed phosphine/N-heterocyclic carbene–palladium complexes: synthesis, characterization, crystal structure and application in the Sonogashira reaction in aqueous media. Transit Metal Chem. 2019, 44, 229–236. 10.1007/s11243-018-0286-5. [DOI] [Google Scholar]
- Ibrahim M. B.; Malik I.; Mansour W.; Sharif M.; Fettouhi M.; El Ali B. Efficient N-heterocyclic carbene palladium (II) catalysts for carbonylative Suzuki-Miyaura coupling reactions leading to aryl ketones and diketones. J. Organomet. Chem. 2018, 859, 44–51. 10.1016/j.jorganchem.2018.01.028. [DOI] [Google Scholar]
- Ibrahim M. B.; Malik I.; Mansour W.; Sharif M.; Fettouhi M.; El Ali B. Novel (N-heterocyclic carbene)Pd(pyridine)Br2 complexes for carbonylative Sonogashira coupling reactions: Catalytic efficiency and scope for arylalkynes, alkylalkynes and dialkynes. Appl. Organomet. Chem. 2018, 32, e4280 10.1002/aoc.4280. [DOI] [Google Scholar]
- Williamson C.; Storey J. M. D.; Harrison W. T. A. 1-[(S)-1-Phenylethyl]-1H- benzimidazole. Acta Crystallogr., Sect. E: Struct. Rep. Online 2007, 63, o3482 10.1107/S1600536807029637. [DOI] [Google Scholar]
- Haque R. A.; Haziz U. F. M.; Abdullah A. A. A.; Shaheeda N.; Razali M. R. New non-functionalized and nitrile-functionalized benzimidazolium salts and their silver(I) complexes: Synthesis, crystal structures and antibacterial studies. Polyhedron 2016, 109, 208–217. 10.1016/j.poly.2016.01.039. [DOI] [Google Scholar]
- Han Y.; Huynh H. V.; Tan G. K. Syntheses and Characterizations of Pd(II) Complexes Incorporating a N-Heterocyclic Carbene and Aromatic N-Heterocycles. Organometallics 2007, 26, 6447–6452. 10.1021/om700753d. [DOI] [Google Scholar]
- Chartoire A.; Boreux A.; Martin A. R.; Nolan S. P. Solvent-free arylamination catalysed by [Pd(NHC)] complexes. RSC Adv. 2013, 3, 3840–3843. 10.1039/c3ra40386f. [DOI] [Google Scholar]
- Natte K.; Chen J.; Neumann H.; Beller M.; Wu X. Palladium-catalyzed oxidative carbonylative coupling of arylboronic acids with terminal alkynes to alkynones. Org. Biomol. Chem. 2014, 12, 5590–5593. 10.1039/C4OB01128G. [DOI] [PubMed] [Google Scholar]
- Huynh H. V. Electronic Properties of N-Heterocyclic Carbenes and Their Experimental Determination. Chem. Rev. 2018, 118, 9457–9492. 10.1021/acs.chemrev.8b00067. [DOI] [PubMed] [Google Scholar]
- Tapu D.; Dixon D. A.; Roe C. 13C NMR Spectroscopy of “Arduengo-Type” carbenes and Their Derivatives. Chem. Rev. 2009, 109, 3385–3407. 10.1021/cr800521g. [DOI] [PubMed] [Google Scholar]
- Slattery J.; Thatcher R. J.; Shi Q.; Douthwaite R. E. Comparison of donor properties of N-heterocyclic carbenes and N-donors containing the 1H-pyridin-(2E)-ylidene motif. Pure Appl. Chem. 2010, 82, 1663–1671. 10.1351/PAC-CON-09-11-10. [DOI] [Google Scholar]
- Jacobsen H.; Correa A.; Costabile C.; Cavallo L. π-Acidity and π-basicity of N-heterocyclic carbene ligands. A computational assessment. J. Organomet. Chem. 2006, 691, 4350–4358. 10.1016/j.jorganchem.2006.01.026. [DOI] [Google Scholar]
- Jacobsen H.; Correa A.; Poater A.; Costabile C.; Cavallo L. Understanding the M(NHC) (NHC=N-heterocyclic carbene) bond. Coord. Chem. Rev. 2009, 253, 687–703. 10.1016/j.ccr.2008.06.006. [DOI] [Google Scholar]
- Vummaleti S. V. C.; Nelson D. J.; Poater A.; Gómez-Suárez A.; Cordes D. B.; Slawin A. M. Z.; Nolan S. P.; Cavallo L. What can NMR spectroscopy of selenoureas and phosphinidenes teach us about the π-accepting abilities of N-heterocyclic carbenes?. Chem. Sci. 2015, 6, 1895–1904. 10.1039/C4SC03264K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson R. G. Chemical hardness, and density functional theory. J. Chem. Sci. 2005, 117, 369–377. 10.1007/BF02708340. [DOI] [Google Scholar]
- Poater A.; Cosenza B.; Correa A.; Giudice S.; Ragone F.; Scarano V.; Cavallo L. SambVca: A Web Application for the Calculation of the Buried Volume of N-Heterocyclic Carbene Ligands. Eur. J. Inorg. Chem. 2009, 1759–1766. 10.1002/ejic.200801160. [DOI] [Google Scholar]
- Falivene L.; Cao Z.; Petta A.; Serra L.; Poater A.; Oliva R.; Scarano V.; Cavallo L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11, 872–879. 10.1038/s41557-019-0319-5. [DOI] [PubMed] [Google Scholar]
- Wu X. F.; Jiao H.; Neumann H.; Beller M. Palladium-Catalyzed Carbonylative Coupling of Aryl Iodides and Benzyl Acetylenes to 3-Alkylidenefuran-2-ones under Mild Conditions and Its Density Functional Theory Modeling. Chem. - Eur. J. 2012, 18, 16177 10.1002/chem.201202138. [DOI] [PubMed] [Google Scholar]
- Hao W. Y.; Sha J. C.; Sheng S. R.; Cai M. Z. The first heterogeneous carbonylative Sonogashira coupling reaction catalyzed by MCM-41-supported bidentate phosphine palladium(0) complex. J. Mol. Catal. A: Chem. 2009, 298, 94–98. 10.1016/j.molcata.2008.09.031. [DOI] [Google Scholar]
- Lyu X.; Sun G.; Zhou Y.; et al. Palladium-catalyzed carbonylative Sonogashira cross-coupling for the synthesis of alkynones with formic acid as the CO source. Monatsh. Chem. 2019, 150, 309–315. 10.1007/s00706-018-2331-7. [DOI] [Google Scholar]
- APEX3; Bruker AXS Inc.: Madison, Wisconsin, USA, 2017.
- SAINT; Bruker AXS Inc.: Madison, USA, 2017.
- Sheldrick G. M.SADABS; Bruker AXS Inc.: Madison, Wisconsin, USA, 2017.
- Sheldrick G. M. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrugia L.J. ORTEP-3 for Windows - a version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. 10.1107/S0021889897003117. [DOI] [Google Scholar]
- Wu X.-F.; Neumann H.; Beller M. A General and Convenient Palladium-Catalyzed Carbonylative Sonogashira Coupling of Aryl Bromides. Chem. - Eur. J. 2010, 16, 12104–12107. 10.1002/chem.201001864. [DOI] [PubMed] [Google Scholar]
- Mansour W.; Fettouhi M.; El Ali B. Novel and efficient bridged bis(N-heterocyclic carbene) palladium(II) catalysts for selective carbonylative Suzuki–Miyaura coupling reactions to biaryl ketones and biaryl diketones. Appl. Organomet. Chem. 2020, 34, 5636 10.1002/aoc.5636. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.