

 LSD1 plays a pivotal role in numerous biological functions.
LSD1 plays a pivotal role in numerous biological functions.
Abstract
LSD1 plays a pivotal role in numerous biological functions. The overexpression of LSD1 is reported to be associated with different malignancies. Over the last decade, LSD1 has emerged as an interesting target for the treatment of acute myeloid leukaemia (AML). Numerous researchers have designed, synthesized, and evaluated various LSD1 inhibitors with diverse chemical architectures. Some of these inhibitors have entered clinical trials and are currently at different phases of clinical evaluation. This comprehensive review enlists recent research developments in LSD1 targeting pharmacophores reported over the last few years.
Introduction
Epigenetic events serve crucial functions in biology, and many researches over the past two decades have established their role in carcinogenesis and tumor progression. DNA methylation and acetylation of histone tails are two of the most studied epigenetic events. Histones form the smallest structural unit of chromatin in the form of eight histones (an octamer consisting of a H3/H4 tetramer and two H2A/H2B dimers) to which 147 bp of DNA is wrapped, and this structural unit is known as nucleosome. The amino-terminal tails of highly-conserved histone proteins protrude out of the nucleosome and are the potential sites of post-translational modifications such as methylation, phosphorylation, sumoylation, ubiquitylation, ADP-ribosylation, biotinylation, glycosylation, and carbonylation including widely studied acetylation by histone acetyltransferases (HATs) and histone deacetylation by histone deacetylases (HDACs).1–3 Thus, histone post-translational modifications serve as regulatory marks, which are critical for the control of transcription and chromatin architecture. Strictly regulated gene expression patterns are important during the process of development and differentiation because diverse cell types have common predecessors.4–6 After the discovery of first histone lysine demethylase in 2004,7 numerous other demethylases have been identified, which control gene expressions and decide cell fate. Therefore, a new class of epigenetic modulators known as histone demethylases has emerged. These demethylases play an important role in developmental processes and in the treatment of various human diseases such as neurological disorders and cancer.8 However, it differs from histone acetylation in the sense that histone acetylation takes place only on lysine (K) residue, whereas methylation occurs on both lysine and arginine (R). Furthermore, unlike histone acetylation, which is generally correlated with active transcription, methylation is linked to both transcriptional activation and repression.7
Discovery of lysine specific demethylase 1 (LSD1/KDM1A)7–9
Modifications of histone are assumed to be part of a ‘code’, which is read by proteins via specific binding domains and thus translated into a functional signal by these proteins. Therefore, depending on how histone modification is being read and translated in a specific context can influence chromatin condensation and poise genes for either transcriptional repression or activation. Albeit, individual histone modifications have been correlated with transcriptional states, which are either active or silenced, but many modifications appear to have several as well as opposing roles, and the combination of these modifications along with their genomic context is seemingly essential for biological output. Nonetheless, the methylation of histones is known since 1960s, but it was not until 2000 that the first histone methyltransferase was identified, and even after its discovery debates were still doing the rounds about whether or not enzymes capable of catalyzing the removal of methylation actually exist. Unlike histone acetylation and phosphorylation, the general conception of histone methylation was that it is irreversible and histone exchange is required for demethylation. It was further supported by studies that the half-life of histone is approximately equal to that of histone methyl marks. In addition, a high thermodynamic stability of N–CH3 bond made the concept of enzymatic removal of methyl mark difficult to believe.2,10 However, with an influx of various research studies citing a change in histone methylation patterns with gene expression in a dynamic and reversible manner indicated that active removal of methylation may occur. It was getting clearer to the scientific community that other than the replication-dependent dilution of histone methylation during cell divisions, alternative mechanisms for methylation removal do exist. The conversion of monomethyl arginine to citrulline by PAD4 (peptidyl arginine deiminase 4), acting as a histone deiminase, also supported the idea of enzymatic cleavage of histone mark.11 In a ground-breaking study in 2004, the first bonafide histone demethylase was identified and reported by Shi et al., demonstrating amine oxidase LSD1/KDM1A to be a part of the C-terminal binding protein 1 (CtBP1) co-repressor complex, which could demethylate lysine 4 on histone H3 in a FAD-dependent reaction but is limited to mono- and dimethylated substrates, H3K4me1/2.7a Later, it was identified that LSD1 also demethylate lysine 9 on histone 3 (H3K9me1/2) and also plays an important role in gene activation, e.g., regulating functions of androgen receptor by the activation of androgen receptor-dependent genes.7b–d Other non-histone proteins such as p53, STAT3, HIF-1α, GfI1, etc. have also been reported to be affected by LSD1;12a however, more research is needed to further establish the role of LSD1 in the demethylation of non-histone proteins. Almost two years later in 2006, the first JmjC domain-containing protein as a histone demethylase was reported, and later the same year, JmjC domain-containing demethylases targeting trimethylation marks were identified. Six families of lysine-specific demethylases (KDMs) have been reported so far; KDM1–KDM6. The KDM1 family comprises three members (LSD1/KDM1A/AOF2, LSD1+8a, and LSD2/KDM1B/AOF1) and is flavin-dependent, while families KDM2-6 are dependent on ketoglutarate and Fe(ii) to remove one to three methyl groups from lysine residues.8,12a LSD1+8a, a spliced form of LSD1, which previously was reported to be restricted to neural tissue involving neurite growth and morphogenesis,12b has also recently been reported to play an important role in neural differentiation in small cell lung cancer.12c LSD2 is the only known mammalian homolog of LSD1 and was discovered in 2009. Similar to LSD1, LSD2 also demethylates H3K4Me1/Me2. LSD2 plays a pivotal role in epigenetic regulation with biological functions distinct from those of LSD1. LSD1 and LSD2 share similarities for their amine oxidase (AO) and SWIRM domains but they differ in their substrate interaction fashion.12d Furthermore, LSD2 contains an N-terminal zinc finger domain (Zn-CW) (residues 50–190), which is absent in LSD1, while LSD1 has an insertion (tower domain, residues 416–515), which is absent in LSD2.12a Although, LSD2 has no known specific inhibitors, but some reported inhibitors of LSD1 have shown some efficacy towards LSD2.22
Mechanism of histone demethylation by LSD1
As discussed earlier, LSD1 demethylates lysine residues in flavin-catalyzed oxidation. The availability of a free electron pair at the methylated lysine residue is crucial for the activity of LSD1 and determines the ability of LSD1 to catalyze the oxidation reaction. An aminium cation intermediate forms after the FAD moiety accepts an electron pair from the nitrogen residue followed by the hydrolysis of the aminium cation intermediate to a carbinolamine intermediate, which spontaneously breaks down to formaldehyde and lysine. The hydrogen peroxide by-product is produced during the oxidation of flavin cofactor with molecular oxygen to regenerate FAD (Fig. 1).13,14a Tranylcypromine inhibits the action of LSD1 irreversibly by forming a covalent FAD adduct via single electron transfer (Fig. 1).14b,c
Fig. 1. (A) Mechanism of histone demethylation by LSD1. (B) Mechanism of inhibition of LSD1 by tranylcypromine (1).

LSD1 and its biological functions12,54–60,67–71
LSD1 regulates various biological functions and has been reported to be over-expressed in various types of cancers, such as breast cancer, prostate cancer, neuroblastoma, acute myeloid leukemia, etc.72a The role of LSD1 in cancer development and progression has been established in these types of cancer; however, there are various mechanisms by which it exhibits its anticancer effects. It has been found that LSD1 is generally associated with protein complexes such as CoREST, NuRD, and CTBP1. LSD1 demethylates methylation marks, which are associated with active transcription states, such as monomethyl- and dimethyl-histone H3 lysine 4 (H3K4me1/2), leading to its activity as a transcription repressor.73,74 The interactions of LSD1 with other proteins depend on substrate specificity, e.g., in the absence of RCOR1 (a transcription repressor complex), LSD1 cannot demethylate nucleosomes.72,75 In addition, LSD1 is also found to be associated with active transcription complexes. It demethylates monomethyl- and dimethyl-histone H3 lysine 9 (H3K9Me1/Me2) when bound to nuclear hormone receptors, such as the androgen receptor (AR)7b–d or oestrogen receptor76 (ER). While H3K9Me1 is associated with active transcription states, H3K9Me2 has been associated with repressed transcription states.72a,b Fig. 2 summarizes various biological functions of LSD1 and its inhibition.
Fig. 2. LSD1 and its biological functions.

As mentioned earlier, many non-histone proteins are also target of LSD1 and have variable functional effects. LSD1 regulates methylation levels of DNMT177 and E2F178 during DNA damage. It also represses p53 function by inhibiting the interaction between p53 and 53BP1.79 Furthermore, LSD1 regulates angiogenesis, chromatin remodelling, proliferation, and differentiation of cancer cells by demethylating HIF-1α,80 STAT3,81 and growth factor independent 1 (GFI1).82 Inhibition of LSD1 is reported to target both scaffolding and enzymatic functions of LSD1, and LSD1 inhibitors disrupt (GFI1)/CoREST complex, which is required for cell differentiation.82
Development of LSD1 inhibitors
Because catalytic amine oxidase domain (AOD) of LSD1 requires flavin adenine dinucleotide (FAD) as a cofactor, thus all the members of the LSD1 family are FAD-dependent oxidation enzymes similar to that of monoamine oxidases A and B (MAOs). Owing to the similar homology of LSD1 with MAOs, LSD1 potentially catalyzes their respective oxidation reactions with a mechanism similar to that of MAOs. Due to these observations, known MAO inhibitors (MAOIs) were tested for their inhibitory activity against LSD1. A total of six MAO inhibitors; three non-selective (tranylcypromine/trans-2-phenylcyclopropylamine (TCP/2-PCPA, 1), phenelzine (2), and nialamide (3)) and three selective for either MAOA or MAOB clorgiline (4), deprenyl (5), and pargyline (6), were tested for their ability to inhibit LSD1 activity (Fig. 3).15 On bulk histones, all inhibitors at high concentrations had an inhibitory effect on recombinant LSD1. Although the inhibitory effects of clorgiline, deprenyl, pargyline, and nialamide were minimal, phenelzine and tranylcypromine exhibited a potent inhibitory effect.15 Further screenings suggested that tranylcypromine, phenelzine, and pargyline displayed potent inhibitory activities toward MAOs and LSD1, thus they become the basis for the further development of LSD1 specific inhibitors. Although the LSD1/CoREST complex was not inhibited by pargyline,15 it managed to inhibit LSD1 H3K9 demethylation via the AR complex.7b,16 However, the first attempt to synthesize an LSD1 inhibitor was based on the known mechanisms of action for MAO inhibitors.17 A peptide molecule of 21-residue, propargyl-Lys-derivatized histone 3 tail was synthesized and found to be an irreversible inhibitor of LSD1 by kinetic analysis.17 A peak correlating to the molecular weight of a covalent adduct between the FAD cofactor and inhibitor in the mass spectra of the LSD1/inhibitor mixture17 indicated that irreversible inhibition results from the covalent attachment of the inhibitor to the FAD molecule.13,18 However, with the knowledge of tranylcypromine as a potent LSD1 inhibitor and its ease of synthesis, numerous small molecules based on tranylcypromine as the inhibitors of LSD1 have been developed and are at various levels of clinical development, e.g., GHK2879552 (7) and ORY-1001 (8).
Fig. 3. Structures of MAOs and LSD1 inhibitors.

Due to the prominent role of LSD1 in the development of many types of cancers, LSD1 inhibitors can be divided into two groups:
A. LSD1 inhibitors for the treatment of cancer.
B. LSD1 inhibitors for the treatment of diseases other than cancer.
This review shall discuss developments in the field of LSD1 inhibitors over the past few years, from 2015–2019.
A. LSD1 inhibitors for the treatment of cancer
Tranylcypromine being first in the class of LSD1 inhibitors and due to a high occurrence of nitrogen-based compounds in nature, such as in the form of peptides, pyrimidines, urea, azoles like tri- and tetrazoles, heterocyclics like 6,5-fused and 5,5-fused; small molecules based on these chemical architectures as individual basic pharmacophores, like tranylcypromine or in combination with other pharmacophores, e.g., tranylcypromine with pyrimidine, pyrimidine with triazole, pyrimidine with urea/thiourea, etc. have been extensively used for the development of LSD1 inhibitors.18 Below are some examples of LSD1 inhibitors based on their basic pharmacophores studied over the last five years:
a. Tranylcypromine based inhibitor. Tranylcypromine, a MAOA/B inhibitor, remained a privileged structure from over two decades and is capable of targeting LSD1/KDM1A. Many compounds based on tranylcypromine core have been synthesized (Fig. 4) and some of them are in clinical trials (see LSD1 inhibitors in clinical trial section for more information). Mercurio et al. have reported a novel series of tranylcypromine derivatives. Compound 9b (1S, 2R, LSD1 IC50 = 0.08 μM) obtained from the racemic mixture of 9a, is found to be the most potent LSD1 inhibitor with an excellent in vitro and in vivo profile, which summons further exploration of the molecule. Intriguingly, 9b was more active than its enantiomer 9c (1R, 2S, LSD1 IC50 = 0.22 μM). Compound 9b was orally active and displayed 62% increased survival in the mouse leukemia model.19a Wen et al. have recently reported the synthesis of benzyl-protected tranylcypromine-based sulphonamide 10, having potent LSD1 inhibitory activity and anti-proliferative activity in AML cells.19b Suzuki et al. as an extension of their previously done work have reported a selective inhibitor of LSD1 (11a; LSD1; IC50 = 0.38 μM), which is six times more potent than their previously reported compound 11b (LSD1; IC50 = 2.5 μM) and has displayed comparable anti-proliferative activity, as that of 11a, against neuroblastoma SH-SY5Y cell line having over-expressed LSD1.19c Ganesan et al. have reported a design of fluorinated tranylcypromines (12 LSD1 IC50 = 1.2 μM; 13 LSD1 IC50 = 6.7 μM) as racemic mixtures with potent LSD1 inhibitory and in vitro proliferation inhibitory activities against acute myeloid leukemia (AML) cell lines, MV4-11 and THP-1.20 Yu et al. have developed a series of novel conformationally constrained compounds. These molecules have displayed over 10 000 fold selectivity for LSD1 compared to MAO-A and B, indicating beneficial effects of conformational restriction strategy. The compounds were endowed with potent LSD1 inhibitory activities, and different enantiomers of the same compounds varied in their IC50 values from 270 nM (14a) to 2.2 nM (14b).2114b was able to activate CD86 expression on human MV4-11 AML cells. They further explored structurally restricted molecules and generated a series of spiro compounds with potent activities against LSD1. Compounds based on general structures of 15a and 15b were found to be very potent and selective inhibitors of LSD1.22 Zhang et al. have reported the synthesis and evaluation of their compound CBB3001 (16, LSD1; IC50 = 21.25 μM).
Fig. 4. Examples of tranylcypromine-based LSD1 inhibitors.

This compound is reported to selectively inhibit the growth of human ovarian tetra carcinoma PA-1 and mouse embryonic carcinoma P9 cells by causing down regulation of pluripotent stem cell proteins SOX2 and OCT4.23 Jung et al. have reported the synthesis of compounds 17 and 18 with potent activity against the colony forming activity of cultured leukemic cells.24 Xiong et al. have reported the compounds 19 and 20 containing tranylcypromine along with 6-trifluoroethyl thienopyrimidine and having LSD1 inhibitory activity in nanomolar range with good selectivity over MAO-A/B. The compounds were active against menin-MLL1 PPI cells and MV4-11 cells in low micromolar range and submicromolar range, respectively.25
It is noteworthy to mention that tranylcypromine-based inhibitors have also shown efficacy in diseases other than cancer and have been discussed in detail later in this review. Fig. 4 illustrates examples of some tranylcypromine-based LSD1 inhibitors.
b. Pyrimidine-based inhibitors. In recent years, Liu et al. have worked extensively for the development of potent LSD1 inhibitors and have reported various pyrimidine- and thiourea-based compounds with activities ranging from 0.65 μM to 3.58 μM. Interestingly, thiourea-based compounds were more potent than urea-based compounds. Nonetheless, substitutions to replace thiourea (hydrophilic group) with a chloro or an aryl group led to a decrease in the activity of these compounds.18,26 An optimum activity was achieved with a combination of thiourea, trimethoxy phenyl, and a propargyl group placed at specific positions of pyrimidine to synthesize an orally active LSD1 inhibitor 21 (LSD1 IC50 = 0.65 μM) with potent anti-proliferative (MGC803: IC50 = 4.01 μM; HGC-27: IC50 = 8.92 μM) and anti-metastatic activities in gastric cancer. They further explored the potential of these compounds and synthesized LPE-1 (22, 0.34 μM)27 with improved LSD1 inhibition; however, its effect as an anti-proliferative agent against human oesophageal cancer cell line was almost similar to the previously reported compound 21. A combination of triazole and 6,5-fused pyrimidine was developed and explored for its potent LSD1 inhibitory and anti-proliferative activity. Three compounds viz.23, 24, and 25 were found to possess potent LSD1 inhibitory activities (IC50 = 0.15 μM, 1.19 μM, and 0.5 μM, respectively); however, these compounds were either not tested in detail or had anti-proliferative activity less than or equal to 5-FU (A549 IC50 = 10.34 μM; PC-9 IC50 = 1.99 μM) against A459 (25: IC50 = 10.34 μM) and PC-9 (24: IC50 = 12.72 μM; 25: IC50 = 12.27 μM) cancer cell lines.28 Compound 26 had displayed potent anti-proliferative activity (4-fold more potent than 5-FU against PC-9 cancer cells) but failed to show LSD1 inhibitory activity.28 Further exploring the chemical architecture of these molecules, a reversible LSD1 inhibitor 27 (LSD1 IC50 = 1.72 μM) was synthesized having selectivity toward LSD1 over MAO-A/B.29 Compound 27 displayed the accumulation of H3K4me1/2 and H3K9me2 and also inhibited the migration of A549 cells in a concentration-dependent manner. It further increased the expression of epithelial cell marker E-cadherin and claudin-1 while decreasing the expression of mesenchymal cell marker N-cadherin and upstream transcription factors, Snail and Slug.29 The removal of N-methylpiperazine (28, LSD1 IC50 > 50 μM), a hydrophilic group, led to a decrease in the activity of the compound, indicating the importance of hydrophilic substitution for the activity in this series of molecules.29 Another reversible LSD1 inhibitor based on aryl hydrazine (29, LSD1 IC50 = 0.88 μM) was discovered while exploring new scaffolds for LSD1 inhibitors.30 The docking studies of 29 suggested that it occupies peptide binding region, thus blocking the access of peptide substrate to FAD leading to the inhibition of the demethylase activity of LSD1.30 With continuing efforts to find potent and selective LSD1 inhibitors, a triazole-fused pyrimidine scaffold was discovered. Compound 30 was found to be the most potent molecule of the series with optimum activity in in silico and in vitro assays. Any effort to replace triazole moiety led to decrease in activity pointing out the importance of triazole moiety for hydrogen bonds and strong electrostatic interactions between the compound and LSD1 active site. Compound 30 induced an expression increase of CD11b, a myelomonocytic differentiation marker modulated by KDM1A/LSD1 in a concentration-dependent manner, indicating differential induction of THP-1 cells.31Fig. 5 illustrates various pyrimidine-based LSD1 inhibitors.
Fig. 5. Examples of pyrimidine-based LSD1 inhibitors.

c. Piperidine- and morpholine-based inhibitors. Based on their observations and experiments, Song et al. discovered that the addition of the piperidin-4-ylmethyl group can increase the LSD1 inhibitory activity of their previously reported compounds and thus initiated the quest to develop potent LSD1 inhibitors based on piperidine moiety.32 Compounds 31–34 were found to be the most potent compounds amongst the series of piperidine-based analogs. A –CN substitution was found to be critical for the activity of these molecules and any replacement of –CN group diminished the activity. Compounds 31 and 32 exhibited very high selectivity of >160 fold and >640 fold against MAO-B, respectively and inactivity against MAO-A (Ki > 50 μM). 32 increased the cellular level of H3K4me2 in a dose-dependent manner, indicating that it is cell membrane permeable and targets LSD1 as its substrate. These four compounds displayed potent anti-proliferative activity against MV4-11 leukemia cells with EC50 ranging from 280–480 nM, but did not inhibit the growth of normal fibroblast cells.32 Zha et al. have also reported some piperidine-based compounds with compound 35 exhibiting the most potent and reversible activity against LSD1 (IC50 = 4 μM), which can also inhibit the migration of HCT116 and A459 cancer cells.33 Morpholine-based 3-oxoamino-benzsulfonamides (37: LSD1 IC50 = 9.5 μM; 38: LSD1 IC50 = 6.9 μM) have also been reported as a new class of potent and reversible LSD1 inhibitors developed by modifying compound 36 (an analog of SP-2557, see LSD1 inhibitors in the clinical trial section for more details), summoning further exploration.34 Zhao et al. have reported the modification of same parent compound 36 to yield morpholine-based benzohydrazides as potent LSD1 inhibitors. Thus, the synthesized compounds (39; LSD1 IC50 = 0.0014 μM; 40: LSD1 IC50 = 0.0017 μM) exhibited potent LSD1 inhibitory activity, which was 10 times more potent than 36 (LSD1 IC50 = 0.013 μM). These compounds were also endowed with potent anti-proliferative activities against various cancer cell lines by on-targeting histone methylation mediated through LSD1 inhibition.35Fig. 6 illustrates various piperidine- and morpholine-based LSD1 inhibitors.
Fig. 6. Examples of piperidine- and morpholine-based LSD1 inhibitors.

d. 5,5-Fused and 6,5-fused heterocyclic-based inhibitors. Vianello et al. have reported the synthesis of some potent thieno [3,2-b]pyrrole-5-carboxamides as new and reversible inhibitors of LSD1.36 A high throughput screening method was used to screen 34 000 compounds and a hit (41, LSD1 IC50 = 2.9 μM) was identified. Further modifications of this hit generated a series of novel carboxamides with compound 42 (LSD1 IC50 = 0.162 μM) being the most potent amongst the synthetic analogs. In this series, thienopyrrole ring was favored over furopyrrole ring, and a meta substituent on phenyl ring yielded more potent compounds than the ortho substituent. Replacing the amide linkage led to diminished activity. Replacing the phenyl group with any other heterocyclic such as pyridine or pyrimidine had a detrimental effect on the activity of these compounds. Compounds 41 and 42 induced an increase in the mRNA expression of CD14 and CD11B, indicating the ability of compounds to block LSD1/KDM1A and to induce differentiation of THP-1 cells.36 On further exploring the structure of these compounds, 43 (LSD1 = 7.8 nM) with improved LSD inhibitory activity was synthesized. Compound 43 exhibited potent activity to induce the expression of CD14, CD11b, CD86, genes regulated by KDM1A. Both 42 and 43 exhibited anti-clonogenic activity.37 Mai et al. have reported some indole-based potent inhibitors (44, LSD1 IC50 = 0.04 μM; 45, LSD1 IC50 = 0.08 μM) of LSD1.38 On investigating the compounds based on pyrrole and indole, Z-indole-based compounds were found to be more potent than Z-pyrrole-based compounds. These compounds were able to induce the expression of the GFI-1b gene, a differentiation marker in acute promyelocytic leukemia (APL) NB4 cells, indicating the inhibition of LSD1 and were also found active against the APL-NB4 cell line in an anti-proliferative assay. Indole-based compounds were 2- to 4- fold more potent than pyrrole-based compounds for their tumor cell growth inhibitory activity in AML-MV4-11 and APL-NB4 cells lines.38
Xu et al. have reported the design of some novel irreversible inhibitors of LSD1.39 Compounds of this series were first of its kind LSD1 inhibitors capable of irreversibly targeting LSD1, which were not derived from MAO inhibitors. Compound 46 (LSD1 IC50 = 1.23 μM) was found to be the most potent compound with an ability to increase the expression of CD86, a differentiation marker in AML YHP-1 cells having a high expression of LSD1. A benzyl group at N1 and 4-(1-hydroxyethyl)-3-methylenedihydrofuran-2(3H)-one at C3 were found to be crucial for the activity of these compounds.39 Chen et al. have reported two series of orally active compounds based on imidazole and indazole; however, indazole-based compounds were found to be more potent than the imidazole-based compounds.40 Compound 47 (LSD1 IC50 = 0.7 nM) and 48 (LSD1 IC50 = 1 nM) were found to be most potent amongst the synthetics. These compounds had good hERG safety profile and EC50 values of 14 and 8 nM, respectively, against THP-1 cells.40Fig. 7 illustrates the examples of 5,5-fused and 6,5-fused heterocyclic-based LSD1 inhibitors.
Fig. 7. Examples of 5,5-fused and 6,5-fused heterocyclic-based LSD1 inhibitors.

e. Natural compounds and their derivatives as LSD1 inhibitors. Liu et al. have reported a natural compound baicalin (49, LSD1 IC50 = 3.01 μM), which is one of the active ingredients in skullcap, as a potent LSD1 inhibitor.41 The sugar moiety of baicalin is essential for its activity and baicalin without sugar moiety is devoid of LSD1 inhibitory activity. Compound 49 can inhibit the cell migration of MGC-803 cells and can increase the expression of epithelial cell marker, E-cadherin mRNA and can decrease the expression of mesenchymal cell marker, N-cadherin mRNA.41 On further exploring natural products, resveratrol (50, LSD1 IC50 = 15 μM) was found to be an inhibitor of LSD1; therefore, a series of resveratrol derivatives as potent LSD1 inhibitors was generated. Two compounds 51 and 52 were found to be active with an IC50 value of 121 nM and 123 nM against MGC803 cells with a percentage inhibition of LSD1 at 10 μM to be 95.3% and 92.9%, respectively.42 Further exploring the potential of these compounds, compound 53 was synthesized and was reported to have 99.23% inhibition with LSD1 IC50 = 0.283 μM and MV4-11 IC50 = 7.49 μM. Compound 53 was found to be a reversible inhibitor of LSD1 with a fast association and slow dissociation.43
Cheng et al. have reported curcumin (54, LSD1 IC50 = 9.6 μM) based compounds and evaluated them for their LSD1 inhibitory potential. Compounds 55 (LSD1 IC50 = 2.8 μM) and 56 (LSD1 IC50 = 0.8 μM) were found to be endowed with potent LSD1 inhibitory and anti-proliferative activities. The LSD1 inhibitory activity of 56 was nearly 10 fold higher than curcumin. Compound 55 displayed an IC50 value of 4.4 μM against A549 cells, but diminished activity against U87 cells, which have a low level of LSD1 expression.44a
However, it is important to note the promiscuous behavior of natural compounds like curcumin and resveratrol and their potential to act as pan-assay interference compounds (PAINs). More often, natural compounds give false positive results due to their complex structures and higher possibility of interactions with various proteins, thus careful monitoring and analysis of experimental outcomes is of prime importance. Curcumin, in particular, interacts with many biomolecules via non-covalent and covalent bondings. The aromatic and tautomeric structural features of curcumin, along with the flexibility of the linker group, are responsible for non-covalent interactions. The α,β-unsaturated β-diketone moiety interacts covalently with protein thiols via Michael reaction, increasing its chances to act as PAIN.44b–d Resveratrol is also a well-known compound with PAIN potential and has been reported to give false results in an experiment to evaluate the effect of natural products on cellular bilayer modification.44eFig. 8 illustrates the examples of natural compound-based LSD1 inhibitors.
Fig. 8. Natural compound-based LSD1 inhibitors.

f. Peptide-based LSD1 inhibitors. Suzuki et al. have reported the synthesis of some peptides, which can inhibit the LSD1 function.45a,b Compound 57a is one of its kind cell-active peptide that works by inhibiting LSD1. LSD1 is known to interact with SNAIL1, a member of the SNAIL/SCRATCH family of transcription factors and to play a role in the expression of the cancer-associated SNAIL1 target gene.45a Compound 57a (LSD1 IC50 = 0.28 μM) inhibits the interaction between LSD1 and SNAIL1 and thus blocks the cancer cell invasion and deactivates LSD1 in a time-dependent manner. It also increases the levels of H3K4Me2 without affecting the levels of H3K4Me1, H3K4Me3, and H3K9Me2.45a Another peptide 57b, having a phenylcyclopropylamine (PCPA) moiety at Lys-4 in the 21-amino acid residue of histone H3, has also been reported.45b A peptide sequence length of 21-amino acid residues is found to be optimum for the activity of peptides of this series.45b Kumarasinghe and Woster have also reported the synthesis of cyclic peptides (58b, 58c) with potent LSD1 inhibitory activity.46a These peptides were developed from 58a (LSD1 IC50 = 2 μM).46b The potency of these peptides was improved through optimization via alanine scanning, and the cell activity against tumor cells was enhanced by lipidation. Fig. 9 illustrates the examples of peptide-based LSD1 inhibitors.
Fig. 9. Peptide-based LSD1 inhibitors.

g. Metal-based LSD1 inhibitors. Despite poor oral bioavailability, metal complexes have been reported that can inhibit some specific enzymes or protein–protein interactions along with epigenetic modulations. In a quest to develop metal-based potent LSD1 inhibitors, Leung et al. have reported first in the class of rhodium(iii)-based metal complexes with LSD1 inhibitory activities. Compound 59 was found to have an LSD1 inhibitory activity of IC50 = 0.04 μM and was able to disrupt LSD1–H3K4me2 interaction in human prostate carcinoma cells. 59 was selective toward LSD1 over other members of the LSD family and MAOA/B and could also increase the expression of differentiation genes, e.g., p21, FOXA2, and BMP2 in PC3 cells, indicating the inhibition of LSD1. Interestingly, compound 59 does not affect the binding of LSD1 with REST or CoREST.47 Inspired from these results, Hu et al. have reported the synthesis of vanadium complexes bearing the tridentate Schiff's base ligand as potent and selective LSD1 inhibitor 60, IC50 = 19.0 μM.48Fig. 10 illustrates the examples of metal-based LSD1 inhibitors.
Fig. 10. Metal-based LSD1 inhibitors.

h. Miscellaneous LSD1 inhibitors. Ogilvie et al. have reported the design and synthesis of potent glycine-based inhibitors and after various chemical modifications, synthesized compound 61 (LSD1, IC50 = 0.083 μM) with potent LSD1 inhibitory activity and greater selectivity over hERG cardiac ion channel. Compound 61 displayed an EC50 value of 0.67 μM against the THP-1 AML cell line. 61 was able to reduce the colony formation of THP-1 and MV-4-11 cells with an up-regulation of cellular differentiation marker CD86.49 Zha et al. have reported the synthesis of potent 5-arylidene barbiturates as selective and reversible LSD1 inhibitors. Compound 62, the most potent compound of the series with an LSD1 activity (IC50 = 0.41 μM), displayed a strong differentiation-inducing effect on LSD1 overexpressed acute promyelocytic leukemia NB4 cells.50 Polyamines as potent LSD1 inhibitors have been reported by various research groups. Woster et al. have reported the synthesis and SAR of some (bis)ureidopropyl and (bis)thioureidopropyldiamine LSD1 inhibitors having a 3-5-3 and 3-6-3 carbon backbone architecture.51 Three of the polyamines 63a, 63b, and 63c have displayed potent LSD1 inhibitory IC50 values of 8 μM, 7 μM, and 5 μM, respectively. These polyamines were capable of increasing the mRNA expression of silenced tumor suppressor genes, such as SFRP2, HCAD, and p16 in the Calu-6 human lung adenocarcinoma line.51 Higuchi et al. have reported the synthesis of polyamine 64 with LSD1 inhibitory activity, Ki = 2.2 μM and an HL-60 IC50 value of 49 μM and have three trans-cyclopentane units along with six stereogenic centers as a part of its chemical architecture.52 Suzuki et al. have reported some small molecules based on H3 peptide that can mimic the γ-turn structure, which is important for the binding of the substrate to the active pocket of LSD1.53 Amongst these compounds, 65 is found to be a potent inhibitor of LSD1 with an IC50 value of 0.622 μM and GI50 value of 5.80 μM in A549 cell lines having over-expressed LSD1. Compound 65 induced the accumulation of histone H3K4me1/2 in a dose-dependent manner along with H3K4me3. The cell cycle analysis indicated the accumulation of cell population in the G1 phase, and increasing concentration of 65 led to the accumulation of cell population in the sub-G1 phase, indicating its apoptosis induction potential.53a Mai et al. have reported quinazoline-based compound 66 and naturally occurring antibiotics used for the treatment of multidrug-resistant bacteria, polymyxin B (67) and E (68), as the inhibitors of LSD1 with a unique binding mode.53b Polymyxin B and polymyxin E (also known as colistin) form a new class of LSD1–CoREST inhibitors, which interact through their circular peptide moieties at the entrance of the H3 tail-binding cleft.53bFig. 11 illustrates the examples of some miscellaneous LSD1 inhibitors.
Fig. 11. Miscellaneous LSD1 inhibitors.

B. LSD1 inhibitors for the treatment of diseases other than cancer
LSD1 inhibitors for viral infections
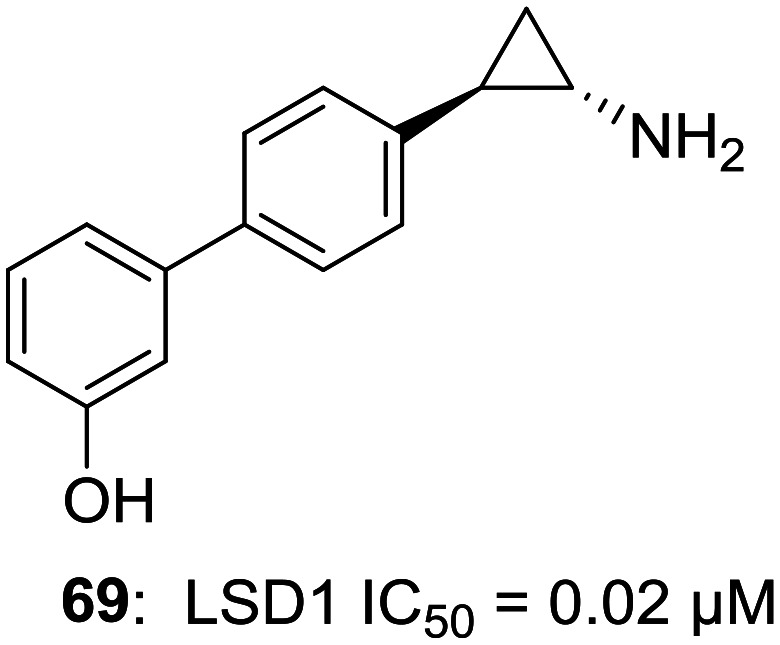
Due to over-expressed LSD1 in various malignancies, the possibility of LSD1 inhibitors as a new class of cancer treatment has drawn a lot of attention. However, efforts have been made to investigate the role of LSD1 inhibitors as a potential treatment for targets other than cancer. There are a growing number of researches which indicate that LSD1 inhibition plays an important role in viral protein expression and viral processes like latency and infection. With not many options for the treatment of viral infections, it becomes even more important to find possible cures. Kristie et al. have worked extensively in the field of establishing links between LSD1/LSD1 inhibitors and viral infections.54–56 Group originally started working with MAO inhibitor tranylcypromine (TCP, 1) as an LSD1 inhibitor and found out that the inhibition of LSD1 blocks the α-herpesviruslytic activity and reactivation from latency. However, the higher IC50 values of MAOIs for LSD1 and broad-spectrum (i.e. non-LSD1) activity eventually led to the discovery of a selective LSD1 inhibitor OG-L002 (69, LSD1 IC50 = 0.02 μM) with potent anti-viral activity against herpes simplex virus (HSV).54
Similar work has been published by other researchers highlighting the role of LSD1 inhibition and its practical implications in infections caused by DNA viruses, e.g., LSD1 inhibition by pargyline (6) in hepatitis B virus (HBV) and the inhibition of LSD1 in human papillomavirus (HPV).57 Intriguingly, LSD1 inhibition works only for the DNA viruses where LSD1 inhibition blocks the transcription of the viral genome. However, the treatment of RNA-based-virus, such as influenza A virus with LSD1 inhibitor, TCP, actually led to a severe attack of influenza A viral infection leading to the discovery of opposing roles of LSD1 depending on the virus type.57,58 Fig. 12 illustrates the example of an LSD1 inhibitor for viral infections.
Fig. 12. LSD1 inhibitor for viral infections.
Role of LSD1 inhibition in inflammation
A recent report indicated the crucial role of LSD1 as an integral part of PKCalpha-LSD1-NFkappaB-cascade, which is important for inflammation development. Therefore, targeting this signal axis could be an effective therapeutic strategy for systemic inflammation.59 Based on these observations, Yuan et al. have investigated and highlighted the importance of LSD1 inhibition in renal inflammation caused by hepatitis B virus (HBV)-associated glomerulonephritis (HBV-GN). The inhibition of LSD1 by tranylcypromine (1) led to the inhibition of TLR-4-related pathway, which is primarily responsible for inflammation in HBV-GN and thus provides evidence for the use of LSD1 inhibitors in HBV-GN.60
Dual inhibitors of LSD1
Studies have revealed an intimate relation between histone deacetylases (HDAC) and LSD1. HDAC1/2 and LSD1 are part of the same cellular complexes viz. CoREST and NuRD co-repressor complex. HDAC1 deacetylates LSD1 at K374, and the inhibition of HDAC by HDAC inhibitors is known to increase H3K4 methylation via transcriptional suppression of histone demethylase. Furthermore, the inhibition of LSD1 increases histone acetylation and enhances the sensitization of cancer cells to HDAC inhibitors. Nonetheless, both; HDACs and LSD1 when over-expressed cause tumor suppressor gene silencing in many cancers; therefore, the simultaneous inhibition of both may prevent tumor growth and its metastasis along with having some synergistic effect. This led Guan et al. to synthesize dual inhibitors of LSD1 and HDAC. Compound 70 (LSD1 IC50 = 1.20 μM; HDAC1 IC50 = 15 nM; HDAC2 IC50 = 23 nM) was found to be the most potent amongst the synthesized analogs.6170 was able to increase methylation of H3K4 and H3K9 and induce apoptosis in MGC-803 cells in a dose-dependent manner. The dose-dependent decrease of mitochondrial membrane potential (MMP) justified the apoptotic nature of 70. Due to the significance of CoREST complex, having HDAC protein, Cole et al. designed a dual inhibitor of LSD1 and HDAC, corin (71) that potentially targets CoREST complex. The IC50 value of 71 for LSD1 was 0.10 μM and for HDAC1 was 0.147 μM against isolated enzymes and LSD1 IC50 = 0.33 μM, HDAC1 IC50 = 0.20 μM for the CoREST ternary complex. Compound 71 was designed based on HDAC inhibitor MS-275 and non-selective LSD1/MAO inhibitor, tranylcypromine. When compared for their anti-proliferative activities, compound 70 (IC50 ∼ 200 nM) displayed 12 fold better anti-proliferative activity against WM983B melanoma cells.62a Milelli et al. have also reported the dual inhibitors of LSD1 and HDAC. Compound 72 was found to be the most potent compound of the series with an IC50 value of 3.45 μM. 72 displayed pronounced cytotoxic effect at 70 μM concentration in MCF cell lines.62b Woster et al. have reported the dual inhibitors of LSD1 and polyamine catabolic enzyme, spermine oxidase (SMOX). The most potent compound 73 exhibited LSD1 IC50 = 50 μM and SMOX IC50 = 25.7 μM with 73 being the most potent SMOX inhibitor reported to date.63Fig. 13 illustrates the examples of LSD1 dual inhibitors.
Fig. 13. Dual inhibitors of LSD1.

LSD1 inhibitors in clinical trials64
A search of LSD1 inhibitors on clinicaltrial.gov by U.S National Library of Medicine yields a report of various clinical trials of LSD1 inhibitors GHK2879552 (7), IMG-7289 (74) and SP-2577 (75), and CC-90011 (77) and INCB059872 on their website. Other than these three, two of the LSD1 inhibitors developed by Oryzon Genomics, ORY1001 (8), and ORY2001 (76) have also entered the clinical trials in Europe and in US, respectively. Except 75 and 77, all others are tranylcypromine-based inhibitors of LSD1. Compound 7 has been developed by GlaxoSmithKline and has entered phase I clinical trial for the treatment of small-cell lung carcinoma (SCLC).65 However, its clinical trial on phase I dose-escalation study in subjects with acute myeloid leukemia (AML) was terminated due to the fact that the risk benefit in relapsed refractory AML did not favor the continuation of the study. Compound 8 is a potent compound with efficacy against leukemia.66 It is also known as Iadademstat/RG6016/RO7051790 and has entered clinical phase IIa trial for the treatment of acute myeloid leukemia (AML) in elderly in combination with azacitidine and also in combination with platinum etoposide for relapsed small-cell lung cancer. IMG-7289/bomedemstat (74) is an orally available irreversible LSD1 inhibitor, which after the successful completion of phase I/IIa trial, has entered phase IIb clinical trials for patients with myelofibrosis. The phase I/IIb report has indicated a safe and well-tolerable profile of IMG-7289. SP-2577 (75), also known as Seclidemstat, has received FDA fast track designation for lead drug candidate and has entered the phase I clinical trial for the treatment of advanced solid tumors for patients with relapsed/refractory Ewing sarcoma. ORY-2001/vafidemstat (76) is the only LSD1 inhibitor, which has an indication also for diseases other than cancer. This dual LSD1/MAOB inhibitor has entered the phase IIa clinical trial for the treatment of mild to moderate Alzheimer diseases. A phase I clinical trial of INCB059872 for the treatment of sickle cell disease was done (NCT03132324);83 however, the trial was terminated as the company decided not to proceed with the trial.84Table 1 summarizes the various LSD1 inhibitors in clinical trials.
Table 1. LSD1 inhibitors in clinical trials.
| Compound name | Chemical structure | Developer company | Clinical trial phase | Indication |
| Tranylcypromine |
|
I/II | AML | |
| 1 | ||||
| GHK2879552 |
|
GlaxoSmithKline | I | Relapsed SCLC |
| 7 | ||||
| ORY1001 |
|
Oryzon Genomics | I/II | AML |
| I | SCLC | |||
| 8 | ||||
| IMG-7289 |
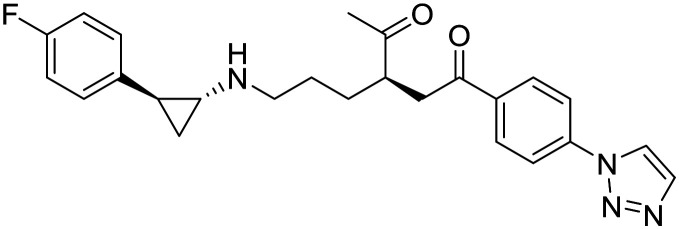
|
Imago BioScience | I | AML |
| II | Myelo-fibrosis | |||
| 74 | ||||
| SP-2577 |
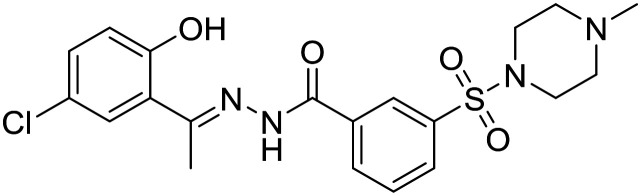
|
Salarius Pharmaceuticals | I | Relapsed Ewing sarcoma |
| 75 | ||||
| ORY2001 |
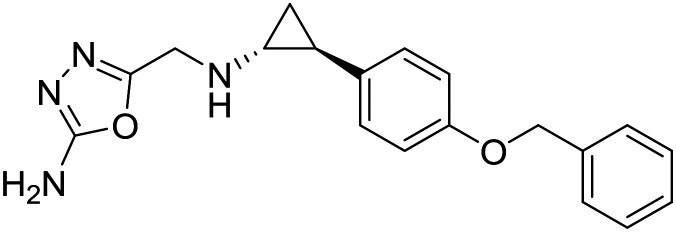
|
Oryzon Genomics | IIa | Mild to moderate Alzheimer |
| I/II | SCLC | |||
| 76 | ||||
| CC-90011 |
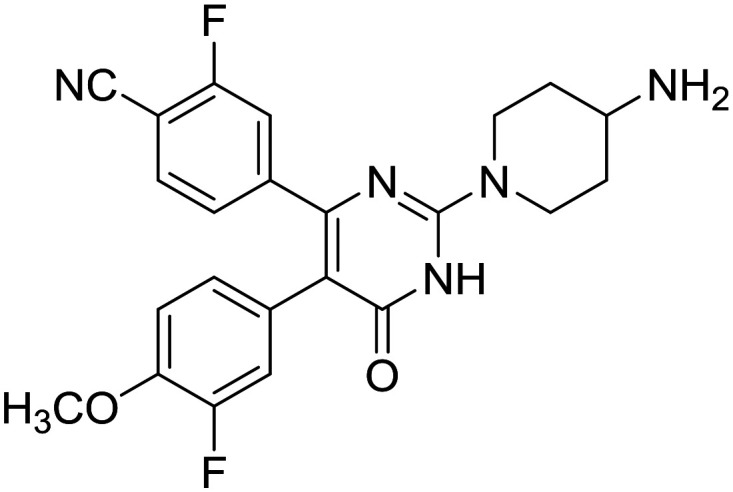
|
Celgene | I | Solid tumor and non-Hodgkin's lymphoma |
| 77 | ||||
| INCB059872 | Unknown | Imago BioScience | I/II | Advanced tumors and hematologic malignancies |
Conclusion
Over the last decade, LSD1 has emerged as a potential therapeutic target particularly for the treatment of various malignancies. As a response to its over-expression in cancers and some other diseases, LSD1 inhibitors have been established as a new class of epigenetic modulators capable of altering the methylation levels of protruding lysine tails of histone proteins. LSD1 performs diverse actions in biology, and targeting LSD1 with a good safety profile is of prime concern. There still does not exist too many LSD1 inhibitors that have entered clinical trials and also no FDA-approved drug has been reported. The scientific community dwells on hopes for the emergence of new potent and selective LSD1 inhibitors that can successfully become approved drugs, and dependent on the trends of LSD1 inhibition and its pharmacological effects; it is merely a matter of time before a potential LSD1 inhibitor-based drug would get an FDA approval.
Conflicts of interest
There is no conflict of interest to declare.
References
- Bird A. Genes Dev. 2002;16:6. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Jenuwein T., Allis C. D. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Margueron R., Trojer P., Reinberg D. Curr. Opin. Genet. Dev. 2005;15:163. doi: 10.1016/j.gde.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Namdar M., Perez G., Ngo L., Marks P. A. Proc. Natl. Acad. Sci. U. S. A. 2010;107:20003. doi: 10.1073/pnas.1013754107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird P. W. Nat. Rev. Cancer. 2003;3:253. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- Yoo C. B., Jones P. A. Nat. Rev. Drug Discovery. 2006;5:37. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- (a) Shi Y., Lan F., Matson C., Mulligan P., Whetstine J. R., Cole P. A., Casero R. A., Shi Y. Cell. 2004;119:941. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]; (b) Metzger E., Wissmann M., Yin N., Muller J. M., Schneider R., Peters A. H. F. M., Gunther T., Buettner R., Schu R. Nature. 2005;437:436. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]; (c) Kahl P., Gullotti L., Heukamp L. C., Wolf S., Friedrichs N., Vorreuther R., Solleder G., Bastian P. J., Ellinger J., Metzger E., Schüle R., Buettner R. Cancer Res. 2006;66:11341. doi: 10.1158/0008-5472.CAN-06-1570. [DOI] [PubMed] [Google Scholar]; (d) Hauser A. T., Bissinger E. M., Metzger E., Repenning A., Bauer U. M., Mai A., Schüle R., Jung M. J. Biomol. Screening. 2012;17:18. doi: 10.1177/1087057111423968. [DOI] [PubMed] [Google Scholar]
- Pedersen M. T., Helin K. Trends Cell Biol. 2010;20:662. doi: 10.1016/j.tcb.2010.08.011. [DOI] [PubMed] [Google Scholar]
- Black J. C., Van Rechem C., Whetstine J. R. Mol. Cell. 2012;48:491. doi: 10.1016/j.molcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister A. J., Schneider R., Kouzarides T. Cell. 2002;109:801. doi: 10.1016/s0092-8674(02)00798-5. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Cell. 2007;128:693. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- (a) Yang G.-J., Lei P.-M., Wong S.-Y., Ma D.-L., Leung C.-H. Molecules. 2018;23:3194. doi: 10.3390/molecules23123194. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Toffolo E., Rusconi F., Paganini L., Tortorici M., Pilotto S., Heise C., Verpelli C., Tedeschi G., Maffioli E., Sala C., Mattevi A., Battaglioli E. J. Neurochem. 2014;128:603. doi: 10.1111/jnc.12457. [DOI] [PubMed] [Google Scholar]; (c) Jotatsu T., Yagishita S., Tajima K., Takahashi F., Mogushi K., Hidayat M., Wirawan A., Ko R., Kanemaru R., Shimada N., Mitani K., Saito T., Takamochi K., Suzuki K., Kohsaka S., Kojima S., Mukae H., Yatera K., Takahashi K. Biochem. Biophys. Rep. 2017;9:86. doi: 10.1016/j.bbrep.2016.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen F., Yang H., Dong Z., Fang J., Wang P., Zhu T., Gong W., Fang R., Shi Y. G., Li Z., Xu Y. Cell Res. 2013;23:306. doi: 10.1038/cr.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavropoulos P., Hoelz A. Expert Opin. Ther. Targets. 2007;11:809. doi: 10.1517/14728222.11.6.809. [DOI] [PubMed] [Google Scholar]
- (a) Maiques-Diaz A., Somervaille T. C. Epigenomics. 2016;8:1103. doi: 10.2217/epi-2016-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Benelkebir H., Hodgkinson C., Duriez P. J., Hayden A. L., Bulleid R. A., Crabb S. J., Packham G., Ganesan A. Bioorg. Med. Chem. 2011;19:3709. doi: 10.1016/j.bmc.2011.02.017. [DOI] [PubMed] [Google Scholar]; (c) Ganesan A. ChemMedChem. 2016;11:1227. doi: 10.1002/cmdc.201500394. [DOI] [PubMed] [Google Scholar]
- Lee M. G., Wynder C., Schmidt D. M., McCafferty D. G., Shiekhattar R. Chem. Biol. 2006;13:563. doi: 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Yang M., Gocke C. B., Luo X., Borek D., Tomchick D. R., Machius M., Otwinowski Z., Yu H. Mol. Cell. 2006;23:377. doi: 10.1016/j.molcel.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Culhane J. C., Szewczuk L. M., Liu X., Da G., Marmorstein R., Cole P. A. J. Am. Chem. Soc. 2006;128:4536. doi: 10.1021/ja0602748. [DOI] [PubMed] [Google Scholar]
- Ma L. Y., Zheng Y. C., Wang S. Q., Wang B., Wang Z. R., Pang L. P., Zhang M., Wang J. W., Ding L., Li J., Wang C., Hu B., Liu Y., Zhang X. D., Wang J. J., Wang Z. J., Zhao W., Liu H. M. J. Med. Chem. 2015;58:1705. doi: 10.1021/acs.jmedchem.5b00037. [DOI] [PubMed] [Google Scholar]
- (a) Vianello P., Botrugno O. A., Cappa A., Dal Zuffo R., Dessanti P., Mai A., Marrocco B., Mattevi A., Meroni G., Minucci S., Stazi G., Thaler F., Trifiró P., Valente S., Villa M., Varasi M., Mercurio C. J. Med. Chem. 2016;59:1501. doi: 10.1021/acs.jmedchem.5b01209. [DOI] [PubMed] [Google Scholar]; (b) Liang L., Wang H., Du Y., Luo B., Meng N., Cen M., Huang P., Wen S. Bioorg. Chem. 2020;99:103808. doi: 10.1016/j.bioorg.2020.103808. [DOI] [PubMed] [Google Scholar]; (c) Khan M. N. A., Tsumoto H., Itoh Y., Ota Y., Suzuki M., Ogasawara D., Nakagawa H., Mizukami T., Miyata N., Suzuki T. MedChemComm. 2015;6:407. [Google Scholar]
- Borrello M. T., Schinor B., Bartels K., Benelkebir H., Pereira S., Al-Jamal W. T., Douglas L., Duriez P. J., Packham G., Haufe G., Ganesan A. Bioorg. Med. Chem. Lett. 2017;27:2099. doi: 10.1016/j.bmcl.2017.03.081. [DOI] [PubMed] [Google Scholar]
- Ji Y. Y., Lin S. D., Wang Y. J., Su M. B., Zhang W., Gunosewoyo H., Yang F., Li J., Tang J., Zhou Y. B., Yu L. F. Eur. J. Med. Chem. 2017;141:101. doi: 10.1016/j.ejmech.2017.09.073. [DOI] [PubMed] [Google Scholar]
- Shi Y., Wu Y.-R., Su M.-B., Shen D.-H., Gunosewoyo H., Yang F., Li J., Tang J., Zhou Y.-B., Yu L.-F. RSC Adv. 2018;8:1666. doi: 10.1039/c7ra13097j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang N., Zhang X., Zhang C., Vo V., Leng F., Saxena L., Yin F., Lu F., Zheng G., Bhowmik P., Zhang H. Bioorg. Med. Chem. 2018;26:1523. doi: 10.1016/j.bmc.2018.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz-Fincke J., Hau M., Barth J., Robaa D., Willmann D., Kürner A., Haas J., Greve G., Haydn T., Fulda S., Lübbert M., Lüdeke S., Berg T., Sippl W., Schüle R., Jung M. Eur. J. Med. Chem. 2018;144:52. doi: 10.1016/j.ejmech.2017.12.001. [DOI] [PubMed] [Google Scholar]
- Wang X., Su M., Li Y., Liu T., Wang Y., Chen Y., Tang L., He Y. P., Ding X., Yu F., Shen J., Li J., Zhou Y., Chen Y. L., Xiong B. Bioorg. Med. Chem. Lett. 2019;29:844. doi: 10.1016/j.bmcl.2019.01.017. [DOI] [PubMed] [Google Scholar]
- Ding L., Wang Z. Z., Sun X. D., Yang J., Ma C. Y., Li W., Liu H. M. Bioorg. Med. Chem. Lett. 2017;27:3521. doi: 10.1016/j.bmcl.2017.05.065. [DOI] [PubMed] [Google Scholar]
- Wang B., Zhao B., Pang L. P., Zhao Y. D., Guo Q., Wang J. W., Zheng Y. C., Zhang X. H., Liu Y., Liu G. Y., Guo W. G., Wang C., Li Z. H., Mao X. J., Yu B., Ma L. Y., Liu H. M. Pharmacol. Res. 2017;122:66. doi: 10.1016/j.phrs.2017.05.025. [DOI] [PubMed] [Google Scholar]
- Wang S., Zhao L. J., Zheng Y. C., Shen D. D., Miao E. F., Qiao X. P., Zhao L. J., Liu Y., Huang R., Yu B., Liu H. M. Eur. J. Med. Chem. 2017;125:940. doi: 10.1016/j.ejmech.2016.10.021. [DOI] [PubMed] [Google Scholar]
- Wang S., Li Z. R., Suo F. Z., Yuan X. H., Yu B., Liu H. M. Eur. J. Med. Chem. 2019;167:388. doi: 10.1016/j.ejmech.2019.02.039. [DOI] [PubMed] [Google Scholar]
- Li Z. R., Wang S., Yang L., Yuan X. H., Suo F. Z., Yu B., Liu H. M. Eur. J. Med. Chem. 2019;166:432. doi: 10.1016/j.ejmech.2019.01.075. [DOI] [PubMed] [Google Scholar]
- Li Z., Ding L., Li Z., Wang Z., Suo F., Shen D., Zhao T., Sun X., Wang J., Liu Y., Ma L., Zhao B., Geng P., Yu B., Zheng Y., Liu H. Acta Pharm. Sin. B. 2019;9:794. doi: 10.1016/j.apsb.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Zhou C., Yao Y., Wei L., Feng Z., Deng L., Song Y. J. Med. Chem. 2016;59:253. doi: 10.1021/acs.jmedchem.5b01361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi J., Xu S., Zhang L., Bi X., Ren Y., Liu Y. C., Gu Y., Xu Y., Lan F., Zha X. Bioorg. Chem. 2018;78:7. doi: 10.1016/j.bioorg.2018.02.016. [DOI] [PubMed] [Google Scholar]
- Xi J., Xu S., Wu L., Ma T., Liu R., Liu Y. C., Deng D., Gu Y., Zhou J., Lan F., Zha X. Bioorg. Chem. 2017;72:182. doi: 10.1016/j.bioorg.2017.04.006. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Li Y., Wang W. J., Xiang P., Luo X. M., Yang L., Yang S. Y., Zhao Y. L. Bioorg. Med. Chem. Lett. 2016;26:4552. doi: 10.1016/j.bmcl.2015.06.054. [DOI] [PubMed] [Google Scholar]
- Sartori L., Mercurio C., Amigoni F., Cappa A., Fagá G., Fattori R., Legnaghi E., Ciossani G., Mattevi A., Meroni G., Moretti L., Cecatiello V., Pasqualato S., Romussi A., Thaler F., Trifiró P., Villa M., Vultaggio S., Botrugno O. A., Dessanti P., Minucci S., Zagarrí E., Carettoni D., Iuzzolino L., Varasi M., Vianello P. J. Med. Chem. 2017;60:1673. doi: 10.1021/acs.jmedchem.6b01018. [DOI] [PubMed] [Google Scholar]
- Vianello P., Sartori L., Amigoni E., Cappa A., Fagá G., Fattori R., Legnaghi E., Ciossani G., Mattevi A., Meroni G., Moretti L., Cecatiello V., Pasqualato S., Romussi A., Thaler F., Trifiró P., Villa M., Botrugno O. A., Dessanti P., Minucci S., Vultaggio S., Zagarrí E., Varasi M., Mercurio C. J. Med. Chem. 2017;60:1693. doi: 10.1021/acs.jmedchem.6b01019. [DOI] [PubMed] [Google Scholar]
- Rodriguez V., Valente S., Rovida S., Rotili D., Stazi G., Lucidi A., Ciossani G., Mattevi A., Botrugno O. A., Dessanti P., Mercurio C., Vianello P., Minucci S., Varasi M., Mai A. MedChemComm. 2015;6:665. [Google Scholar]
- Liu H. M., Suo F. Z., Li X. B., You Y. H., Lv C. T., Zheng C. X., Zhang G. C., Liu Y. J., Kang W. T., Zheng Y. C., Xu H. W. Eur. J. Med. Chem. 2019;175:357. doi: 10.1016/j.ejmech.2019.04.065. [DOI] [PubMed] [Google Scholar]
- Nie Z., Shi L., Lai C., Severin C., Xu J., Del Rosario J. R., Stansfield R. K., Cho R. W., Kanouni T., Veal J. M., Stafford J. A., Chen Y. K. Bioorg. Med. Chem. Lett. 2019;29:103. doi: 10.1016/j.bmcl.2018.11.001. [DOI] [PubMed] [Google Scholar]
- Zheng Y. C., Shen D. D., Ren M., Liu X. Q., Wang Z. R., Liu Y., Zhang Q. N., Zhao L. J., Zhao L. J., Ma J. L., Yu B., Liu H. M. Bioorg. Chem. 2016;69:129. doi: 10.1016/j.bioorg.2016.10.004. [DOI] [PubMed] [Google Scholar]
- Duan Y. C., Guan Y. Y., Zhai X. Y., Ding L. N., Qin W. P., Shen D. D., Liu X. Q., Sun X. D., Zheng Y. C., Liu H. M. Eur. J. Med. Chem. 2017;126:246. doi: 10.1016/j.ejmech.2016.11.035. [DOI] [PubMed] [Google Scholar]
- Duan Y., Qin W., Suo F., Zhai X., Guan Y., Wang X., Zheng Y., Liu H. Bioorg. Med. Chem. 2018;26:6000. doi: 10.1016/j.bmc.2018.10.037. [DOI] [PubMed] [Google Scholar]
- (a) Wang J., Zhang X., Yan J., Li W., Jiang Q., Wang X., Zhao D., Cheng M. Bioorg. Med. Chem. Lett. 2019;29:126683. doi: 10.1016/j.bmcl.2019.126683. [DOI] [PubMed] [Google Scholar]; (b) Baell J. B. J. Nat. Prod. 2016;79:616. doi: 10.1021/acs.jnatprod.5b00947. [DOI] [PubMed] [Google Scholar]; (c) Baell J. B., Walter M. A. Nature. 2014;513:481. doi: 10.1038/513481a. [DOI] [PubMed] [Google Scholar]; (d) Priyadarsini K. A. Curr. Pharm. Des. 2013;19:2093. doi: 10.2174/138161213805289228. [DOI] [PubMed] [Google Scholar]; (e) Ingólfsson H. I., Thakur P., Herold K. F., Hobart E. A., Ramsey N. B., Periole X., de Jong D. H., Zwama M., Yilmaz D., Hall K., Maretzky T., Hemmings Jr H. C., Blobel C., Marrink S. J., Koçer A., Sack J. T., Andersen O. S. ACS Chem. Biol. 2014;9:1788. doi: 10.1021/cb500086e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Itoh Y., Aihara K., Mellini P., Tojo T., Ota Y., Tsumoto H., Solomon V. R., Zhan P., Suzuki M., Ogasawara D., Shigenaga A., Inokuma T., Nakagawa H., Miyata N., Mizukami T., Otaka A., Suzuki T. J. Med. Chem. 2016;59:1531. doi: 10.1021/acs.jmedchem.5b01323. [DOI] [PubMed] [Google Scholar]; (b) Kakizawa T., Ota Y., Itoh Y., Tsumoto H., Suzuki T. Bioorg. Med. Chem. Lett. 2015;25:1925. doi: 10.1016/j.bmcl.2015.03.030. [DOI] [PubMed] [Google Scholar]
- (a) Kumarasinghe I. R., Woster P. M. Eur. J. Med. Chem. 2018;148:210. doi: 10.1016/j.ejmech.2018.01.098. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kumarasinghe I. R., Woster P. M. ACS Med. Chem. Lett. 2014;5:29. doi: 10.1021/ml4002997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C., Wang W., Liang J. X., Li G., Vellaisamy K., Wong C. Y., Ma D. L., Leung C. H. J. Med. Chem. 2017;60:2597. doi: 10.1021/acs.jmedchem.7b00133. [DOI] [PubMed] [Google Scholar]
- Lu L.-P., Liu J.-H., Cen S.-H., Jiang Y. L., Hu G.-Q. Bioorg. Med. Chem. Lett. 2019;29:681. doi: 10.1016/j.bmcl.2018.10.004. [DOI] [PubMed] [Google Scholar]
- Mould D. P., Alli C., Bremberg U., Cartic S., Jordan A. M., Geitmann M., Maiques-Diaz A., McGonagle A. E., Somervaille T. C. P., Spencer G. J., Turlais F., Ogilvie D. J. Med. Chem. 2017;60:7984. doi: 10.1021/acs.jmedchem.7b00462. [DOI] [PubMed] [Google Scholar]
- Xu S., Zhou C., Liu R., Zhu Q., Xu Y., Lan F., Zha X. Bioorg. Med. Chem. 2018;26:4871. doi: 10.1016/j.bmc.2018.08.026. [DOI] [PubMed] [Google Scholar]
- Nowotarski S. L., Pachaiyappan B., Holshouser S. L., Kutz C. J., Li Y., Huang Y., Sharma S. K., Casero Jr. R. A., Woster P. M. Bioorg. Med. Chem. 2015;23:1601. doi: 10.1016/j.bmc.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umezawa N., Tsuji K., Sato S., Kikuchi M., Watanabe H., Horai Y., Yamaguchi M., Hisamatsu Y., Umehara T., Higuchi T. RSC Adv. 2018;8:36895. doi: 10.1039/c8ra07879c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Ota Y., Miyamura S., Araki M., Itoh Y., Yasuda S., Masuda M., Taniguchi T., Sowa Y., Sakai T., Itami K., Yamaguchi J., Suzuki T. Bioorg. Med. Chem. 2018;26:775. doi: 10.1016/j.bmc.2017.12.045. [DOI] [PubMed] [Google Scholar]; (b) Speranzini V., Rotili D., Ciossani G., Pilotto S., Marrocco B., Forgione M., Lucidi A., Forneris F., Mehdipour P., Velankar S., Mai A., Mattevi A. Sci. Adv. 2016;2:e1601017. doi: 10.1126/sciadv.1601017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y., Vogel J. L., Narayanan A., Peng H., Kristie T. M. Nat. Med. 2009;15:1312. doi: 10.1038/nm.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill J. M., Quenelle D. C., Cardin R. D., Vogel J. L., Clement C., Bravo F. J., Foster T. P., Bosch-Marce M., Raja P., Lee J. S., Bernstein D. I., Krause P. R., Knipe D. M., Kristie T. M. Sci. Transl. Med. 2014;6:265. doi: 10.1126/scitranslmed.3010643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y., Quenelle D., Vogel J. L., Mascaro C., Ortega A., Kristie T. M. mBio. 2013;4:00558. doi: 10.1128/mBio.00558-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwergel C., Stazi G., Mai A., Valente S. Future Sci. OA. 2018;10:1133. doi: 10.4155/fmc-2018-0065. [DOI] [PubMed] [Google Scholar]
- Shan J., Zhao B., Shan Z., Nie J., Deng R., Xiong R., Tsun A., Pan W., Zhao H., Chen L., Jin Y., Qian Z., Lui K., Liang R., Li D., Sun B., Lavillette D., Xu K., Li B. PLoS Pathog. 2017;13:1006773. doi: 10.1371/journal.ppat.1006773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Nam H. J., Lee W., Yim H. Y., Ahn J. Y., Park S. W., Shin H. R., Yu R., Won K. J., Bae J. S., Kim K. I., Baek S. H. Mol. Cell. 2018;69:398. doi: 10.1016/j.molcel.2018.01.002. [DOI] [PubMed] [Google Scholar]
- Yang Y.-T., Wang X., Zhang Y.-Y., Yuan W.-J. Cell Death Dis. 2019;10:1. doi: 10.1038/s41419-019-1514-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y. C., Ma Y. C., Qin W. P., Ding L. N., Zheng Y. C., Zhu Y. L., Zhai X. Y., Yang J., Ma C. Y., Guan Y. Y. Eur. J. Med. Chem. 2017;140:392. doi: 10.1016/j.ejmech.2017.09.038. [DOI] [PubMed] [Google Scholar]
- (a) Kalin J. H., Wu M., Gomez A. V., Song Y., Das J., Hayward D., Adejola N., Wu M. I., Panova I., Chung H. J., Kim E., Hu Y., Ryu B., Schwabe J. W. R., Mattevi A., Alan R. M., Cole P. A. Nat. Commun. 2018;9:1. doi: 10.1038/s41467-017-02242-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Milelli A., Marchetti C., Turrini E., Catanzaro E., Mazzone R., Tomaselli D., Fimognari C., Tumiatti V., Minarini A. Bioorg. Med. Chem. Lett. 2018;28:1001–1004. doi: 10.1016/j.bmcl.2018.02.034. [DOI] [PubMed] [Google Scholar]
- Holshouser S., Dunworth M., Murray-Stewart T., Peterson Y. K., Burger P., Kirkpatrick J., Chen H. H., Jr Casero R. A., Woster P. M. MedChemComm. 2019;10:778. doi: 10.1039/c8md00610e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y., Liao G., Yu B. J. Hematol. Oncol. 2019;12:129. doi: 10.1186/s13045-019-0811-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer T. M., Besse B., Martinez-Marti A., Trigo J. M., Moreno V., Garrido P., Ferron-Brady G., Wu Y., Park J., Collingwood T., Kruger R. G., Mohammad H. P., Ballas M. S., Dhar A., Govindan R. J. Thorac. Oncol. 2019;14:1828. doi: 10.1016/j.jtho.2019.06.021. [DOI] [PubMed] [Google Scholar]
- Maes T., Mascaró C., Tirapu I., Estiarte A., Ciceri F., Lunardi S., Guibourt N., Perdones A., Lufino M. M. P., Somervaille T. C. P., Wiseman D. H., Duy C., Melnick A., Willekens C., Ortega A., Martinell M., Valls N., Kurz G., Fyfe M., Castro-Palomino J. C., Buesa C. Cancer Cell. 2018;33:495. doi: 10.1016/j.ccell.2018.02.002. [DOI] [PubMed] [Google Scholar]
- Zhang X., Jedrychowski M. P., Chen Y., Serag S., Lavery G. G., Gygi S. P., Spiegelman B. M. Genes Dev. 2016;30:1822. doi: 10.1101/gad.285312.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia S., Karasik E., Gillard B., Williams J., Winchester T., Moser M. T., Smiraglia D. J., Foster B. A. Clin. Epigenet. 2017;9:82. doi: 10.1186/s13148-017-0382-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Saijo K., Skola D., Jin C., Ma Q., Merkurjev D., Glass C. K., Rosenfeld M. G. Proc. Natl. Acad. Sci. U. S. A. 2018;115:E244. doi: 10.1073/pnas.1718759114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosic M., Allen A., Willmann D., Lepper C., Kim J., Duteil D., Schüle R. Nat. Commun. 2018;9:366. doi: 10.1038/s41467-017-02740-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman J. H., Lin B., Schwob J. E. J. Comp. Neurol. 2017;525:3391. doi: 10.1002/cne.24259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Lynch J. T., Harris W. J., Somervaille T. C. P. Expert Opin. Ther. Targets. 2012;16:1239. doi: 10.1517/14728222.2012.722206. [DOI] [PubMed] [Google Scholar]; (b) Diaz A. M., Somervaille T. C. P. Epigenomics. 2016;8:1103. doi: 10.2217/epi-2016-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi M. A., Dong Y., Lane W. S., Speicher D. W., Shiekhattar R. J. Biol. Chem. 2003;278:7234. doi: 10.1074/jbc.M208992200. [DOI] [PubMed] [Google Scholar]
- Shi Y. J., Matson C., Lan F., Iwase S., Baba T., Shi Y. Mol. Cell. 2005;19:857. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Lee M. G., Wynder C., Cooch N., Shiekhattar R. Nature. 2005;437:432. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- Perillo B., Ombra M. N., Bertoni A., Cuozzo C., Sacchetti S., Sasso A., Chiariotti L., Malorni A., Abbondanza C., Avvedimento E. V. Science. 2008;319:202. doi: 10.1126/science.1147674. [DOI] [PubMed] [Google Scholar]
- Wang J., Hevi S., Kurash J. K., Lei H., Gay F., Bajko J., Su H., Sun W., Chang H., Xu G., Gaudet F., Li E., Chen T. Nat. Genet. 2009;41:125. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- Kontaki H., Talianidis I. Mol. Cell. 2010;39:152. doi: 10.1016/j.molcel.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Huang J., Sengupta R., Espejo A. B., Lee M. G., Dorsey J. A., Richter M., Opravil S., Shiekhattar R., Bedford M. T., Jenuwein T., Berger S. L. Nature. 2007;449:105. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- Kim Y., Nam H. J., Lee J., Park D. Y., Kim C., Yu Y. S., Kim D., Park S. W., Bhin J., Hwang D., Lee H., Koh G. Y., Baek S. H. Nat. Commun. 2016;7:10347. doi: 10.1038/ncomms10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Huang J., Dasgupta M., Sears N., Miyagi M., Wang B., Chance M. R., Chen X., Du Y., Wang Y., An L., Wang Q., Lu T., Zhang X., Wang Z., Stark G. R. Proc. Natl. Acad. Sci. U. S. A. 2010;107:21499. doi: 10.1073/pnas.1016147107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Diaz A. M., Spencer G. J., Lynch J. T., Ciceri F., Williams E. L., Amaral F. M. R., Wiseman D. H., Harris W. J., Li Y., Sahoo S., Hitchin J. R., Mould D. P., Fairweather E. E., Waszkowycz B., Jordan A. M., Smith D. L., Somervaille T. C. P. Cell Rep. 2018;22:3641. doi: 10.1016/j.celrep.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Diaz A. M., Lynch J. T., Spencer G. J., Somervaille T. C. P. Mol. Cell. Oncol. 2018;5:e1481813. doi: 10.1080/23723556.2018.1481813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telen M. J., Malik P., Vercellotti G. M. Nat. Rev. Drug Discovery. 2019;18:139. doi: 10.1038/s41573-018-0003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://clinicaltrials.gov/ct2/show/NCT03132324 .