

 This review article highlights the recent progress and outlook for small molecule gamma secretase modulators for potential Alzheimer's therapies.
This review article highlights the recent progress and outlook for small molecule gamma secretase modulators for potential Alzheimer's therapies.
Abstract
Alzheimer's disease (AD) is the most common form of progressive neurodegenerative disorder, marked by memory loss and a decline in cognitive function. The major hallmarks of AD are the presence of intracellular neurofibrillary tau tangles (NFTs) composed of hyperphosphorylated tau proteins and extracellular plaques composed of amyloid beta peptides (Aβ). The amyloid (Aβ) cascade hypothesis proposes that the AD pathogenesis is initiated by the accumulation of Aβ peptides in the parenchyma of the brain. An aspartyl intramembranal protease called γ-secretase is responsible for the production of Aβ by the cleavage of the amyloid precursor protein (APP). Clinical studies of γ-secretase inhibitors (GSIs) for AD failed due to the lack of substrate specificity. Therefore, γ-secretase modulators (GSMs) have been developed as potential disease modifying agents to modulate the γ-secretase cleavage activity towards the production of toxic Aβ42 peptides. Following the first-generation ‘nonsteroidal anti-inflammatory drug’ (NSAID) based GSMs, second-generation GSMs (carboxylic acid based NSAID derivatives and non-NSAID derived heterocyclic analogues), as well as natural product-based GSMs, have been developed. In this review, we focus on the recent developments of small molecule-based GSMs that show potential improvements in terms of drug-like properties as well as their current status in human clinical trials and the future perspectives of GSM research.
Introduction
Alzheimer's disease (AD) is the most common form of progressive and chronic neurodegenerative disorder that is usually marked by memory loss and a decline in cognitive function. An estimated 50 million people worldwide are living with AD. In the United States, between 2000 and 2017, deaths from Alzheimer's have increased by 145% affecting 5.8 million people and it is currently ranked as the sixth leading cause of deaths. The estimated cost of care for AD patients is about $290 billion (in 2019) with an expectation to rise to as much as $1.1 trillion by 2050.1 This trend is also seen on a global scale as well. Every year, there are another 10 million new cases and if the trend continues, it is believed to reach 100 million patients by 2050 worldwide if no treatment is found. Therefore, there is an urgent need for disease modifying therapeutics that can delay or prevent AD.2
The most common symptoms of AD are both short- and long-term memory loss, inability of complex thinking, and changes in behavior. In later stages of AD, cognitive decline leads to a loss of function in even the most basic tasks required for living, ultimately leading to death. As of 2019, the FDA has approved five therapeutics for AD, and the drugs fall into two classes:3 a) acetylcholinesterase inhibitors (galantamine, rivastigmine, donepezil, and tacrine) used for mild-to-moderate degree AD patients, and b) a partial N-methyl-d-aspartic acid (NMDA) receptor antagonist (memantine)4 used for moderate-to-severe degree AD patients. In 2013, tacrine was discontinued in the United States due to concerns over safety. The 5th FDA approved therapeutic is a combination drug, ‘memantine + donepezil’ (Namzaric), used to treat patients with a moderate-to-severe degree of AD. All approved treatments are meant to alleviate symptoms but will not slow down the disease progression. Therefore, there exists an urgent need for extremely selective and novel therapeutic approaches with limited to no side effects for the treatment and eventual cure for AD.
Ever since the symptoms were described by the German physician Alois Alzheimer, the major hallmarks of AD are the presence of intracellular neurofibrillary tau tangles (NFTs) composed of hyperphosphorylated tau proteins, and extracellular plaques composed of amyloid beta peptides (Aβ). Under nonpathological conditions, tau is a microtubule-associated protein that is responsible for maintaining neuronal axon stability and aids in transport. In AD, the tau becomes hyperphosphorylated resulting in detachment from microtubules and subsequent aggregation leading to neurotoxicity.5 In addition, the tau spreading is hypothesized to follow a prion-like mechanism in which a specific species of the pathological misfolded protein can recruit and cause the aggregation of normal monomeric tau.6 The amyloid cascade hypothesis proposes that AD pathogenesis is initiated by the accumulation of Aβ peptides in the parenchyma of the brain.7,8 Genetic studies have supported the amyloid hypothesis,9 but the development of anti-Aβ therapeutics remains challenging.10
γ-Secretase complex and activity
The γ-secretase complex (Fig. 1), a macromolecular intramembrane cleaving protease enzyme consisting of an active site in the lipid bilayer of the cellular membrane, falls under the family of intramembrane cleaving proteases. γ-Secretase is a complex enzyme made up of four subunits consisting of presenilin (PS), nicastrin (Nct), anterior pharynx defective-1 (Aph-1), and presenilin enhancer-2 (Pen-2).11 PS includes two genes, PSEN1 (located on chromosome 14 and encodes presenilin-1, PS1) and PSEN2 (located on chromosome 1 and encodes presenilin-2, PS2) and mutations in these two genes are associated with familial AD (FAD).12,13 PS1 and PS2 (molecular weight (M. Wt) ∼50 kDa) are polytopic membrane proteins, and these two proteins are synthesized as single polypeptide chains with nine transmembrane domains (TMDs). PS1 and PS2 undergo autocatalytic endoproteolysis, an internal cleavage event that produces an N-terminal fragment (NTF with 6 TMDs) and a C-terminal fragment (CTF with 3 TMDs).14 PS is the catalytic subunit of γ-secretase.15–17 Nct is a type-I single-pass TM protein with a basic M. Wt of 78 kDa, and a highly glycosylated form resulting in an apparent M. Wt of ∼120 kDa. The Nct protein is responsible for substrate recognition, proper trafficking of the mature complex, stabilization and assembly of PS into the γ-secretase complex, but it is not required for catalytic activity.18,19 Aph-1 is an intramembrane protein with seven TMDs having a M. Wt of ∼30 kDa and is implicated in γ-secretase complex stability, activity and selectivity.20,21 The underlying stability of the γ-secretase complex is associated with a highly conserved GXXXG motif in the TMD-4 of Aph-1, which mediates the protein–protein interactions with PS.22,23 Pen-2, an integral membrane protein having three TMDs with a M. Wt of ∼12 kDa, has been shown to be critical for the maturation of γ-secretase as it promotes endoproteolysis of full-length PS (PS-FL) into PS-NTF and PS-CTF which has been demonstrated by biochemical reconstitution studies.17 Additional studies show that Pen-2 is required for the stability of the γ-secretase complex.20,24–26 Collectively, reports show that mutations in the γ-secretase subunits are associated with AD,12,13 myeloid leukemia27 and familial acne inversa.28
Fig. 1. The cryo-EM structure of the recognition of APP by human γ-secretase at 2.6 Å resolution.32.

The three-dimensional atomic structure of the human γ-secretase assembly by cryo-electron microscopy (cryo-EM) reveals that the active catalytic site is located within the subunit PS (at 4.5, 4.32 and 3.6 Å resolution, respectively)29,30 giving some structural basis for γ-secretase function and regulation. Moreover, recent reports on cryo-EM structures (at 2.7 and 2.6 Å resolution) (Fig. 1) of γ-secretase substrate recognition (Notch and APP, respectively)31,32 reveal that the certain substrate binding specificity of γ-secretase poses a challenge for the design of substrate-specific inhibitors or modulators for AD.
γ-Secretase cleaves within the transmembrane domains (TMDs) of over 90 type-1 membrane proteins of which the amyloid precursor protein (APP) and Notch are the most studied proteins due to their importance in AD and cancer, respectively.33 γ-Secretase cleavage of APP is responsible for the generation of Aβ peptides, and it is considered as the primary therapeutic target in AD. In the brain, Aβ peptides with lengths of 37–43 amino acids are produced through the sequential cleavage of the APP (Fig. 2). The initial β cleavage of the extracellular portion of TM APP by BACE1 (β-secretase) produces a) a soluble ectodomain (ECD) from the cell surface into the lumen, and b) an intracellular membrane bound C-terminal fragment (CTF) C99. Initial proteolysis at the cytoplasm boundary of the membrane bound C-terminal fragment C99 (ε cleavage) by γ-secretase produces another two fragments intracellularly: a) APP intracellular domain (AICD) and b) membrane embedded peptides Aβ49 and Aβ48. If the ε cleavage occurs between amino acids 50 and 49 of the C99, the successive processing steps (ζ and γ cleavages) produce Aβ49 → 46 → 43 → 40 peptides extracellularly. If the cleavage occurs between amino acids 49 and 48, the subsequent cleavage outcomes would be Aβ48 → 45 → 42 peptides. The generated peptides Aβ42 and Aβ40 can be either released into the extracellular domain or further proteolysed resulting in shorter peptides (Aβ39 or 38 from the Aβ42 product line, and Aβ37 from the Aβ40 product line).34,35
Fig. 2. Cartoon representation of the γ-secretase complex processivity of APP.

Among all the Aβ peptides generated, the Aβ40 is the major species36 and mutations in the APP and PS genes may alter the outcome of the processivity by γ-secretase producing a higher ratio of Aβ42 : Aβ40 which is observed in familial Alzheimer's disease (FAD). A shift towards the relative production of longer and hydrophobic Aβ42 is most prone to aggregation and development of neurotoxic oligomers, which in turn form amyloid plaques associated with AD. Therefore, many research groups consider the Aβ42 to be the main delinquent in the pathogenesis of AD. GSMs shift cleavage away from Aβ42 production, without affecting the total Aβ production or processivity of other GS substrates such as Notch, and have become a promising class of compounds.37 However, the above strategies have been pursued over the years in order to develop disease-modifying therapeutics for AD. This review mainly focuses on recent advances of ‘small molecule’ based GSMs pertaining to AD therapeutics. Therefore, in this review, the primary peer-reviewed literature covers from 2016–2020 as there were several review reports that covered these topics prior to 2016.37–41
1) Early γ-secretase inhibitors (GSIs)
Aβ40 and Aβ42, principal fragments produced by the γ-secretase enzyme, aggregate extracellularly leading to the formation of Aβ plaques observed in FAD. Therefore, pharmaceutical companies have been targeting the Aβ for disease-modifying therapeutics or anti-AD drugs. GSIs were the first class of compounds tested to reduce the levels of Aβ formation via inhibition of γ-secretase. Among several clinical GSIs, two compounds, avagacestat (BMS-708163) and semagacestat (LY-450149) (Fig. 3), have advanced into phase-II and phase-III trials, respectively.42–44
Fig. 3. Structures of GSIs that advanced into clinical trials.

However, these two compounds were discontinued from clinical trials due to numerous side effects following treatment. It has been stated that γ-secretase has several substrates, including Notch proteins, which are essential for normal biological functions.45 Notch inhibition by γ-secretase leads to several side effects such as skin cancer and a decrease in the lymphocyte count, which were observed in the semagacestat (LY-450149) phase III trials. Avagacestat (BMS-708163) has also shown similar side effects to those of semagacestat at effective doses.
2) γ-Secretase modulators (GSMs)
First-generation GSMs
With the failure of GSIs in clinical trials, other classes of compounds called γ-secretase modulators (GSMs) were discovered, which can reduce the toxic Aβ while maintaining the total Aβ levels unchanged. The first-generation GSMs were based on nonsteroidal anti-inflammatory drugs (NSAIDs) (Fig. 4) including ibuprofen, indomethacin, sulindac sulfide, flurbiprofen (tarenflurbil), etc.46 Studies show that these NSAID compounds could lower the production of Aβ42 in favor of Aβ38 production as evidenced by both in vitro and in vivo studies.46–48 The role of Aβ38 in AD is unknown, but some reports show that the shorter forms (Aβ38 and Aβ37) are less prone to aggregation and less toxic than Aβ42.34,35,46–48
Fig. 4. Representative structures of first-generation NSAID based GSMs.

Tarenflurbil was the first NSAID-based GSM that entered clinical trials and showed salutary effects at higher doses in a phase II trial of a subgroup of patients with mild AD. However, due to weak in vitro potencies (Aβ42 IC50 ∼ 200–300 μM) and poor brain penetration (∼1% brain penetration), it was discontinued from phase III trials.49,50 Ibuprofen, indomethacin, and sulindac sulfide have been withdrawn from trials due to the lack of efficacies resulting from insufficient potencies and poor brain penetration. Following the clinical trial of tarenflurbil, a considerable amount of work was dedicated to the development of GSMs that are more potent, and with enhanced brain penetration. GSM analogues based on NSAIDs are termed as second-generation NSAID-derived GSMs.38 Advances in this class resulted in GSMs with improved preclinical profiles, reducing the levels of Aβ42 production over those of Aβ40.
Second-generation GSMs
The second-generation GSMs can generally be classified into two major classes: a) NSAID-derived carboxylic acid GSMs, and b) heterocyclic non-NSAID-derived GSMs. All these GSMs were shown to bind to the PS subunit of γ-secretase.51–56 However, the lack of structural information on the γ-secretase–GSM complex has made it challenging to define the precise binding sites of these GSMs.
a) NSAID-derived carboxylic acid GSMs
Initial efforts centered on NSAID-derived GSMs with the acetic acid chemotype (Fig. 5).
Fig. 5. Representative structures of second-generation NSAID-derived carboxylic acid GSMs.

Among several reported acid chemotypes, Merck and GSK have developed new compounds by substituting the core aryl ring with piperidine. Optimization of substituents on the nitrogen of piperidine yielded a series of potent piperidine acetic acid analogues such as GSM-1, GSM-2, GSM-10 h, etc. (Fig. 5). These GSMs were reported to reduce Aβ42 while promoting the Aβ38 production and did not exhibit any significant reduction of Aβ40 production or total Aβ levels.57–60
Janssen has developed a GSM, JNJ-40418677, an analogue of flurbiprofen with added substituents on the core aryl ring. Acute treatment of WT mice with JNJ-40418677 showed a reduction of Aβ42 levels in the brain with a concomitant increase in the level of Aβ38 without changing the total Aβ levels in the brain. The increased potency of JNJ-40418677 is attributed to increased lipophilicity, but the safety profile has yet to be determined.61 In addition, itanapraced (CHF 5074) and EVP-0015962 exhibited similar attributes and have been investigated in clinical trials. Chiesi Farmaceutici S.p.A. (an Italian pharmaceutical company) has first reported itanapraced (CHF 5074) (Fig. 5) with improved Aβ42 inhibitory potency (Aβ42 IC50 = 41 μM).62 This compound now has advanced into phase II trials for mild cognitive impairment due to AD (clinical trial identifier: NCT01421056). However, it was found that itanapraced lowered the levels of the soluble CD40 ligand, a marker for microglia activation, but not Aβ42 in plasma or cerebrospinal fluid (CSF); therefore, it is now being termed as a microglia modulator. EVP-0015962, a cyclobutyl group containing analogue of (R)-flurbiprofen, was developed by Forum pharmaceuticals (previously, Envivo). This compound showed an acceptable in vitro potency (Aβ42 IC50 = 67 μM) with a reduction of 22% and 38% with oral doses 10 and 30 mg kg–1, respectively, in addition to reproducible animal efficacy.38,63,64 However, it suffers from poor drug like properties with high lipophilicity, similar to other NSAID GSMs. Phase II clinical trial details were not reported; however, based on the current clinical trial details, it is not active assuming withdrawal from trials. In general, all NSAID-based GSMs lower the production of Aβ42 without affecting the Aβ40 levels while compensating the level of shorter peptides Aβ38 and Aβ37.
Biogen's research on GSMs has been focused on the development of novel carboxylic acid based NSAID like molecules. In 2011, Biogen reported65 a potential preclinical candidate 2 (BIIB042) based on the optimization of their early lead 1 (Fig. 6). Initial lead 1 exhibited good potency (Aβ42 IC50 = 1 μM (in CHO/APP V717F) but suffered from poor brain penetration (0.6 μM after 4 h of dosing at 50 mg kg–1). Further optimization was focused on the flexible and most basic nitrogen center (3° amine). The SAR studies indicated that the introduction of a rigid piperidinyl group and a chiral methyl group at the α-position of the carboxylic acid moiety resulted in the preclinical candidate 2 (BIIB042). Compound 2 exhibited good potency, lowering Aβ42 (Aβ42 IC50 = 170 nM) and increasing Aβ38 (Aβ38 EC50 = 150 nM) levels, and no changes in Aβ40 levels. Recently, Biogen has published66in vivo efficacy studies of compound 2. Initial in vitro potency results of 2 in H4/APP (H4 cells expressing human APP) showed reduced Aβ42 (Aβ42 IC50 = 64 nM) and increased Aβ38 (Aβ38 EC50 = 146 nM) levels with no effect on Aβ40 levels. Similar pharmacodynamic (PD) properties were confirmed in the brains and plasma of mice, rats, and in cynomolgus monkeys after a single oral dose of 2.
Fig. 6. Biogen's NSAID-derived carboxylic acid GSM 1 and an optimized modulator 2 (BIIB042).

In a dose dependent manner, an acute p.o. dose of 2 to CF1-mice (at 3, 10, 30 and 100 mg kg–1 for 4 h) resulted in a significant Aβ42 reduction in both the brain and plasma (60% and 45% in the brain and plasma, respectively, for 100 mg kg–1 dose), increased levels of Aβ38 (∼98% and 82% in the brain and plasma, respectively, for 100 mg kg–1 dose) and a slight decrease in Aβ40 levels (16% and 25% in the brain and plasma respectively for 100 mg kg–1 dose) while brain : plasma ratio (Br : Pl = 0.8) was unchanged for all doses. A similar trend was observed in F344 rats.
In time-dependent studies, a single dose (p.o.) was administered to CF1-mice (10, 30, or 100 mg kg–1, evaluate up to 48 h). For a 10 mg kg–1 dose, the Aβ42 levels decreased by 40% (4 h), Aβ38 increased by ∼50% (16 h), and there was no change in Aβ40 levels. The maximum concentration of 2 in the brain reached 7.5 μM at 4 h postdosing. A similar trend was observed in cynomolgus monkeys when a single 10 mg kg–1 dose of 2 was administered in a 72 h time course-experiment.
Subchronic administration of 2 to 5-month-old Tg2576 mice (3, 10, 30 and 60 mg kg–1 per day doses for 65 days) resulted in a similar trend of Aβ modulation to that for CF1-mice, and Aβ42 levels were significantly reduced (∼50% and ∼80% reduction of soluble and insoluble Aβ42, respectively, at 60 mg kg–1 dose). A chronic dose of 2 to 10 month-old Tg2576 mice (0.3, 1, 10, and 30 mg kg–1 per day for 65 days) resulted in decreased levels of both soluble and insoluble Aβ42 at doses of 10 and 30 mg kg–1 per day. A dose of 2 at 30 mg kg–1 per day showed a significant decrease of parenchymal plaque staining in both the cortex and hippocampus. Collectively, the pharmacodynamic (PD) effects of compound 2 show increased Aβ38 levels and decreased Aβ42 levels. These results are consistent with in vitro and in vivo studies as well as in rodents and non-human primates. Therefore, it can be hypothesized that compound 2 (BIIB042) could be efficacious in modulating the Aβ production in humans. However, additional preclinical safety and tolerability data were not reported before considering compound 2 for clinical trials.
b) Heterocyclic non-NSAID-derived GSMs
Heterocyclic non-NSAID-derived GSMs were further classified into three major classes: Imidazole GSMs, triazole GSMs, and distinct heterocyclic GSMs. E2012, a non-acidic imidazole GSM, was first reported67 by Eisai in 2005 (Fig. 7), and contains the critical arylimidazole moiety. This compound represents the starting point for the generation of several classes of arylimidazole GSMs.
Fig. 7. Structure of Eisai's heterocyclic non-NSAID-derived GSM E2012.
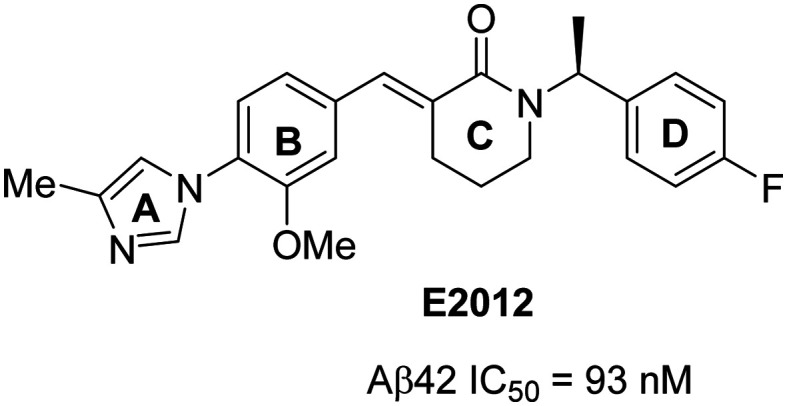
Most of these compounds contain four consecutive rings designated as A (imidazole), B (aryl), C (central core), and D (aromatic). The A and C rings represent the hydrogen bond acceptor (HBA) moieties, while the B and D rings represent aromatic/lipophilic moieties with favorable pharmacophore properties. In general, the non-NSAID derived series of GSMs reduce Aβ42 levels over those of Aβ40, and increase the levels of shorter peptides Aβ37, Aβ38 and Aβ39 while maintaining the total Aβ levels unchanged. The evolution, activity and clinical development profiles of second-generation non-NSAID GSMs have extensively been reviewed in the literature.37–39,41 Therefore, this review focusses on the new GSMs reported by pharmaceutical companies between 2016 and 2019 from primary peer-reviewed literature studies and reviewed based on the classification into imidazole, triazole, and distinct heterocyclic GSMs.
Imidazole GSMs
The University of California at San Diego (UCSD), Albany Molecular Research Inc. (AMRI), and collaborators have described a novel series of aminothiazole derived γ-secretase modulators based on the previously reported56 GSM 3 (Fig. 8). GSM 3 had poor solubility and moderately suppressed Aβ42 formation. Therefore, incorporation of a heterocyclic ring in place of the D ring of compound 3 has generated a new series of compounds with moderate potency and favorable drug-like properties.68,69 The most potent compound70N-ethylpyrazole 4 displayed good in vitro inhibition of Aβ42 (Aβ42 IC50 = 63 nM) and a substantial improvement in solubility (Kin. Sol. pH 7.4 = 4.7 μM). However, it suffers from poor ADME (absorption, distribution, metabolism, and excretion) properties with moderate CYP450 (IC50 = 16 μM for 3A4) and hERG (IC50 = >30 μM) inhibition. Pharmacokinetic (PK) evaluation of compound 4 in mice revealed moderate brain penetration and clearance. Compound 4 has shown improvement in solubility with moderate potency but suffers from high molecular weight and moderate brain penetration. Therefore, there is still more room for improvement of the drug-like properties of this class of compounds before pursuing clinical applications.
Fig. 8. UCSD/AMRI and collaborators' GSMs 3 and 4.

A recent publication from the Bristol-Myers Squibb group reported71 a new tricyclic GSM 6 (Fig. 9), which is the optimized compound based on their previous high-throughput screening (HTS) generated hit 5 (Aβ42 IC50 = 180 nM) (Fig. 9). Initial optimization of 5 resulted in the diamino triazine lead compound 5a (Fig. 9) with improved potency (Aβ42 IC50 = 29 nM).72,73
Fig. 9. Bristol-Myers Squibb's HTS hit 5, triazine lead compound 5a, and tricyclic GSM 6 (top), and optimization strategy for tricyclic analogues (bottom).

Further optimization efforts were directed to increase the potency by conformational restriction of triazine derivatives towards the tricyclic analogues (Fig. 9, bottom). Initial cyclization (Hantzsch cyclization) introduced a ring constraint followed by the replacement of triazine by a thiazole heterocycle resulting in compound 6. However, compound 6 (Aβ42 IC50 = 35 nM) and its analogues did not show any improved potency compared to 5a. Compound 6 was profiled in vivo (3xTg mouse model) at doses p.o. of 1, 3, 10, 30, and 100 mg kg–1, resulting in 30%, 40%, and ∼50% Aβ42 reductions in the brain at 10, 30, and 100 mg kg–1 doses, respectively. The measured brain and plasma concentrations (3 h post dose of 30 mg kg–1) were 8900 nM and 8000 nM, respectively, with a Br : Pl ratio of 1.1 indicating significant brain penetration. Compound 6 also exhibited good metabolic stability when incubated with human liver microsomes (rate of metabolism 0.035 nmol min–1 mg–1) and was not a hERG inhibitor. However, it did show some CYP3A4 inhibition (IC50 = 0.7 μM). Thus, the compound suffers from poor drug-like properties with low potency, high M. Wt, and a high clog P value in addition to the CYP3A4 inhibition.
To optimize compound 6 further, another manuscript74 describes the synthesis of a new class of pyrimido[4,5-c]azepine and pyrimido[4,5-c]oxepine based GSMs based on the alternate cyclization strategy. The initial 2-chloropyrimidine derivatives (7, 8, 9, and 10) were synthesized by the Grubbs ring closing metathesis approach, and these derivatives possess a fused bicyclic ring system which is somewhat related to the well-known benzodiazepine drugs that show good brain penetration properties. In addition, the chloroimidazole group has previously demonstrated improved CYP3A4 inhibition compared to methylimidazole. Therefore, the resulting 2-chloropyrimidine derivatives were used in coupling reactions to generate the potent GSMs 7a, 8a, 9a, and 10a, respectively (Fig. 10). The most potent compound of this series, the azepine derivative 7a has shown improved in vitro potency for Aβ42 reduction (IC50 = 5 nM) but failed to show improved in vivo efficacy (at 30 mg kg–1p.o.) which is due to the low brain exposure (data not shown). Therefore, these GSMs suffer from poor in vivo activity and poor drug-like properties with high M. Wt and high clog P values.
Fig. 10. Bristol-Myers Squibb's 2-chloropyrimidine derivatives 7, 8, 9 and 10 (top) and corresponding GSMs 7a, 8a, 9a and 10a (bottom).

A recent publication75 from Bristol-Myers Squibb disclosed further structure–activity relationship (SAR) analysis of 5 and identified the pyrimidine compounds 11 and 12 as new GSMs. An initial change from triazine (C ring of 5) to a pyrimidine core, and then fusion of a carbocyclic ring resulted in the restricted aryl ring (D ring). Further modification at the triazole ring resulted in the chloroimidazole derivative 11 with an improved potency (Aβ42 IC50 = 2 nM) and CYP inhibition profile compared to triazine 5a. However, 11 was reported to be a potent inhibitor of the hERG ion channel (IC50 = 200 nM) with less brain penetration. Therefore, compound 11 was discontinued from further development. Moreover, it is widely stated that the high toxicity of a drug is related to higher lipophilicity (clog P).76 Reducing the lipophilicity is a challenging task, particularly in CNS drugs, because overly polar molecules generally fail to cross the blood–brain barrier and most of the CNS targets prefer lipophilic drugs.77,78 After further optimization, replacing the chloroimidazole group of 11 with methyltriazole, and the phenyl (D ring) group with the 4-fluorophenyl group resulted in the GSM 12 (BMS-932481) (Fig. 11). The potency of 12 (Aβ42 IC50 = 6.6 nM) is slightly less than that of 11, which is a trade-off between potency and lipophilicity.
Fig. 11. Bristol-Myers Squibb's novel bicyclic pyrimidine GSMs 11 and 12 (BMS-932481).

Recent phase-I clinical data for 12 were reported.79 After a single oral dose of 12 at 10–1200 mg, and multiweek daily dosing at 50–200 mg, the analysis of both plasma and CSF samples demonstrated a dose-dependent increase in Aβ37 and Aβ38, and a decrease in Aβ42 and Aβ40, while there was no change in the total Aβ levels. After administration of 12 at 200 mg of daily dose for 24 days, ALT (alanine transaminase) elevations were observed, which was hypothesized to be due to unexpected bio-accumulation of compound 12 (drug) in the liver. Modeling of multiple ascending dose data revealed that a maximum of about ‘25% lowering of Aβ42’ would be achieved at an exposure considered to be free of ALT elevation. Moreover, in the multiple ascending dose (MAD) study, in addition to ALT elevation, several other adverse events were detected, such as elevated bilirubin (for all doses), a decrease in creatinine clearance, etc. Therefore, a sufficient therapeutic index could not be established, and 12 (BMS-932481) was discontinued from clinical trials.
In the search for new GSMs, scientists at Forum Pharmaceuticals considered designing new molecules based on the representative ‘non-NSAID based’ arylimidazole GSM E2012 (Fig. 7). Initially, they envisioned the importance of the central amide hydrogen bond acceptor in E2012 while considering the removal of the cinnamide group. The cinnamide group has been shown to act as a potential Michael acceptor and as an undesirable structural feature.80 Therefore, the design of most potent molecules starts from the amide I (in Fig. 12) with the introduction of a conformational constraint-1 (isosteric amide replacement) into the pendent alkyl chain to generate the oxadiazine II analogues. Then, the introduction of a different conformational constraint-2 (stereoselective synthesis) from the alkyl group (R) to the aryl group (structure II, in Fig. 12) generated novel chiral heteroaryl oxadiazine GSMs 13 and 14 (Fig. 13).81 The only difference between these two structures is the benzofuran in 13vs. benzothiophene in 14.
Fig. 12. Forum Pharmaceutical's molecular design strategy for oxadiazine-based GSMs 13 and 14, etc.

Fig. 13. Forum Pharmaceutical's new oxadiazine-based GSMs 13 and 14.

Compound 14 was shown to be more active (Aβ42 EC50 = 8 nM), but with poor drug-like properties associated with higher clog P (3.7) and a lower CNS-MPO (multi parameter optimization) score (4.1).77 Therefore, compound 13 advanced into further studies. The initial in vitro activity of 13 demonstrated a good potency (Aβ42 EC50 = 35 nM), and great predicted CNS drug-like properties based on a low clog P (2.8) and high CNS-MPO score (5.0). Compound 13 also showed a good metabolic stability (in rat hepatocytes CLint,u = 84 mL min–1 kg–1; and in human hepatocytes CLint,u = 44 mL min–1 kg–1), good aqueous solubility (84 μM in a pH 7.4 buffer), good permeability (PA→B = 10 × 10–6 cm s–1) with a minimal P-gp (P-glycoprotein) efflux potential (efflux ratio = 1.1), minimal cytotoxicity (TC50 > 72 μM in HepG2 cells), and minimal hERG liability (IC50 = 15 μM). The cytochrome P450 (CYP) profiling of compound 13 demonstrated the minimal inhibition against CYP 3A4, 2C19 and 2C8 (IC50 = 25, 17 and >25 μM, respectively) and a moderate inhibition against CYP 2C9, 2D6 and IA2 (IC50 = 8, 2 and 3 μM, respectively). In addition to a good pharmacokinetic (PK) profile in both rats and monkeys, compound 13 has demonstrated a robust in vivo profile in rodents (mouse and rat). A 43% Aβ42 reduction and a 3.2-fold Aβ37 increase was observed after 6 h of post dose (dosed at 30 mg kg–1) of 13 in mice, while 30% Aβ42 reduction and a 2.5-fold Aβ37 increase was observed in rats at the same dose of 13 as that in mice. The excellent results from the potent compound 13 will provide a way for further development of oxadiazine-based analogues towards successful AD therapeutics.
Recent papers82,83 from Janssen Pharmaceuticals described the design and synthesis of a novel series of GSMs in which a ‘cyano attached indole group’ was introduced in place of the classical ‘imidazole heterocycle’ group of the representative GSM E2012 and their previous lead compound 15 (Fig. 14). A high throughput screening (in-house corporate collection of compounds) was performed to evaluate the compounds' ability to increase the production of Aβ38 in vitro. The results showed several hits with the nitrile (cyano) functional group. In a subsequent round of testing, the identified hits were shown to inhibit Aβ42 production with moderate potency. Following further design, synthesis, and SAR analysis of new analogues based on the hits, they identified a novel cyanoindole attached, imidazomorpholine derivative 15a as a potent GSM from the series (Fig. 14).
Fig. 14. One of Janssen Pharmaceutical's previous lead compounds 15 (left), and new imidazomorpholine GSM 15a (right).

The imidazomorpholine 15a was profiled and the results were compared with those of the previous lead 15 (Fig. 14). The in vitro potency of 15a in mice (Aβ42 IC50 = 0.017 μM) was higher than that of 15 (Aβ42 IC50 = 0.049 μM). However, compound 15a was metabolically less stable and less soluble than 15, which may be due to the replacement of the more soluble imidazole group by the less soluble cyano group as observed in all the cyano derivatives in the 15a series (structures not shown in this review). Compound 15a had a higher propensity for the inhibition of CYP450 (CYP3A4 IC50 = >10 μM) (while 15 was an activator), as well as for the inhibition of the hERG channel (hERG IC50 = 5.29 μM) compared to 15 (hERG IC50 = >10 μM). Oral administration of compound 15a in dogs (at 10 mg kg–1p.o.) showed a 37% Aβ42 reduction and 40% Aβ38 increase in the cerebrospinal fluid (CSF) after 8 h post dose, and the maximum plasma concentration (Cmax = 2.4 μM) was reached after 1.6 h post dose. No increase in the liver enzyme levels (ALT and AST) was observed, and no relevant changes were seen in the behavior or appearance. However, due to the replacement of methylimidazole by a cyanoindole group, compound 15a suffered from suboptimal physicochemical properties such as high lipophilicity (clog P = 4.19), high aromatic character and low solubility resulting in poor drug-like properties. Therefore, these compounds were discontinued from further development.
In 2017, researchers at Merck published a paper84 on the discovery of a new class of tetrahydroisoxazole (17 series) GSMs based on Eisai's E2012 (Fig. 7) and their own previous GSMs including 16 (Fig. 15).85–87 To mitigate the potential metabolic activity of the double bond in Merck's 16 and Eisai's E2012, the double bond and the lactam carbonyl groups were replaced by a small heterocyclic fused ring tetrahydrobenzisoxazole as shown in Merck's 17. The tetrahydrobenzisoxazole group not only alleviates the metabolic activity but also locks the side chain conformation of the double bond as shown in 16 and E2012. Their synthetic efforts and SAR studies have generated several compounds with good to excellent selectivity (for Aβ42 reduction) as GSMs. Among the several new analogues, compounds 17a (Aβ42 IC50 = 3.9 × 10–2 μM) and 17b (Aβ42 IC50 = 2.9 × 10–2 μM) (Fig. 16) were shown to be two-fold more active than the representative GSM E2012 (Aβ42 IC50 = 80 nM) and had similar activities to 16 (Aβ42 IC50 = 37 nM). The stereochemistry of the most active enantiomer 17a or 17b was not disclosed in their paper. The incorporation of fluoroaniline in 17a, and difluorobenzaniline in 17b was based on the fact that a) increasing the lipophilicity often results in improved in vitro activity and improved blood–brain barrier permeability of the compounds, and b) controlled pKa would successfully improve the CNS penetration.
Fig. 15. Merck Research Laboratory's oxadiazine 16 and tetrahydrobenzisoxazole analogues 17.

Fig. 16. Merck Research Laboratory's novel tetrahydrobenzisoxazoles 17a and 17b.

The successful synthesis and in vitro activities of 17a and 17b encouraged in vivo profiling in a nontransgenic rat model. Compound 17a significantly reduced cerebrospinal fluid (CSF) Aβ42 by 58% (p = 0.0066, n = 5) while 17b reduced only ∼20% after 3 h of single p.o. dose at 30 mg kg–1 in rats when compared to vehicle-treated rats. Interestingly, the in vitro experiment results showed that compound 17b was the most potent; however, to get an insight into this discrepancy, the rat tissues were collected (after dosing), and the drug levels in the plasma and brain were assessed. The efficacy of compound 17a was determined by its excellent CNS penetration, which provided about six-fold higher exposure compared to 17b. The efflux ratio (ER = 6.8) of 17b was four-fold higher than that of 17a (ER = 1.6), and the higher ER of 17b could be the cause for its low brain penetration property as well as poor in vivo efficacy. Therefore, these promising results would encourage scientists to develop the more efficacious non-NSAID based GSMs. However, to date, there have been no further safety and efficacy data reported for compound 17a, including its future developments towards clinical trials.
A Pfizer group has recently published88 their discovery of a novel series of chromene- and cyclopropyl chromane-derived pyridopyrazine-1,6-dione GSMs for the treatment of AD. Their initial efforts led to the discovery of dihydrobenzofuran-amide based GSM 18 (Fig. 17).89 However, compound 18 was discontinued from further development due to some complications in the process of improving its potency and ADME properties. Further focus on the design and optimization of novel conformationally restricted heterocyclic analogues resulted in the naphthyl derivative 19 (Aβ42 IC50 = 44 nM) with improved potency and polarity.90 Further optimization using ligand-based design strategies such as molecular modeling and conformational analysis resulted in a series of potent compounds with chromene- and cyclopropyl chromane units as a suitable heterocyclic replacement for the naphthyl group that was present in compound 19.
Fig. 17. Pfizer's novel most potent chromane- and cyclopropyl chromane-derived GSMs 20, 21, 22 and 23.

The replacement of the naphthyl group in 19 by the trifluoromethyl chromene group and the introduction of a conformational constraint via insertion of an (S)-methyl group on the methylene linker resulted in compound 20 with improved potency (Aβ42 IC50 = 4.9 nM) and good metabolic stability (CLint,app = 14.3 μL min–1 mg–1).88 The trifluoromethoxy group as an alternative lipophilic and non-metabolizable replacement resulted in compound 21, exhibiting a favorable in vitro profile (Aβ42 IC50 = 6.0 nM, CLint,app = 25.7 μL min–1 mg–1). Further optimization led to the discovery of cyclopropyl chromane derivatives 22 (Aβ42 IC50 = 4.9 nM; CLint,app = 34.5 μL min–1 mg–1) and 23 (Aβ42 IC50 = 5.1 nM; CLint,app = 42.5 μL min–1 mg–1) (Fig. 17) with a trifluoromethyl group and trifluoromethoxy group, respectively, at the 6-position on the chromane ring. The installed gem-dimethyl substitution on the chromene–methylene or cyclopropyl chromane–methylene showed improved lipophilicity, which is an advantage for CNS activity. The opposite stereoisomer of each active compound was less potent. Collectively, compound 22 exhibited the best overall in vitro and in vivo profile. Excellent selectivity was achieved for Notch and hERG, with acceptable physicochemical properties. Compound 22 has also demonstrated favorable in vitro ADME characteristics which include a good microsomal stability (HLM CLint,app = 34.5 μL min–1 mg–1), passive permeability (RRCK Papp, A → B = 5.3 × 10–6 cm s–1), and adequate MDR efflux ratio (MDR ER = 2.5). By further examination of compound 22in vivo, in rat time-course studies at oral doses of 10 and 40 mg kg–1, the Aβ42 levels in the brain were maximally reduced by 40% and 62% with unbound brain concentrations of 11 and 67 nM at the 10 and 40 mg kg–1 doses, respectively. The in vivo efficacy of compound 22 was achieved at significantly lower doses and with lower free drug levels compared to earlier compounds in this series. Therefore, compound 22 has advanced into further developments of in vivo efficacy and safety studies. However, to date, no data were reported regarding the development of clinical trials.
Triazole GSMs
Several efforts have been directed to develop more efficacious GSMs consisting of reduced electron density in the privileged B ring system of E2012 (Fig. 7). This was accomplished by the introduction of a) more electron-withdrawing groups, and b) replacing the C ring (aniline) with heteroaromatic groups (triazine or thiazole groups, etc.) in order to generate more polar B–C ring fragments (see Fig. 18). However, to achieve good potency, the A–B ring sequence seems to be mandatory in all reported non-NSAID based GSMs. Hence, to achieve a good potency without changing the structural elements of the A–B ring system, scientists at Boehringer Ingelheim have come up with an idea of GSMs that lack the imidazole group and the electron-rich aniline moiety. Besides achieving a desirable potency, the modifications would also limit CYP inhibition and the formation of potential reactive metabolites. A recent publication91 has disclosed the discovery of a novel, distinct class of potent and in vivo efficacious GSMs consisting of tetrahydroindazolyl amines (Fig. 19 and 20) as a replacement of the prominent arylimidazole motif.
Fig. 18. Roche's GSM 24 (left), and Boehringer Ingelheim's tetrahydroindazolyl amine hit compounds 25 and 26 (right).

Fig. 19. Structure of Boehringer Ingelheim's compound ent1-27 and its X-ray structure.

Fig. 20. Structures of Boehringer Ingelheim's ent2-27 and representative compound (R)-28 (BI-1408).

The initial hypothesis was that the triazolopyridine group of Roche's GSM 24 (Fig. 18) could be a good probe to identify novel A–B structures.92,93 In the selection process of an amine core for the A ring, they acknowledged the necessity of a hydrogen bond acceptor group as a key pharmacophore along with a conformationally rigid group. The synthesis followed by SAR studies of around 30 analogues identified the two most active compounds: a) triazolopyridine 25 (Aβ42 IC50 = 1.2 μM), which has an indazolyl amine moiety as a bicyclic A–B ring motif, and b) a hydrogenated analogue 26 (Aβ42 IC50 = 1.2 μM) (Fig. 18).
Starting from the more potent compound 26, thorough investigation of tetrahydroindazole substituted triazolopyridines resulted in several new entities; among them, ent1-27 (Aβ42 IC50 = 0.009 μM) and (R)-28 (Aβ42 IC50 = 0.04 μM) were reported to be most the potent compounds (Fig. 19 and 20). Interestingly, the influence of stereochemistry on biological activity revealed that the racemic mixture (racemate) of 27 (Aβ42 IC50 = 0.02 μM) and the other enantiomer ent2-27 (Aβ42 IC50 = 0.194 μM) is less potent than ent1-27 (Fig. 19). Due to the poor metabolic stability (in rodents) of ent1-27, ent2-27 and other potent compounds, the only chiral bicyclic tetrahydroindazolyl amine as the representative compound BI-1408 ((R)-28) has advanced to assess the in vivo activity.91
The initial in vitro profile of the enantiomer BI-1408 ((R)-28) (in H4 cells overexpressing human APPwt, Aβ42 IC50 = 0.04 μM) showed two-fold more activity than its racemate, but they are equipotent in rat primary cortical neurons (Aβ42 IC50 = 0.04 μM). Furthermore, BI-1408 ((R)-28) did not affect the total Aβ concentrations (Aβtotal/Aβ42 = 254) and Notch processing (Aβ42 IC50 = >30 μM) and showed no indication of relevant effects on the CYP3A4 (IC50 = >50 μM) and hERG channel (8% and 39% inhibition at 1 and 10 μM, respectively) parameters. However, it did show some moderate inhibition of CYP2C8 (IC50 = 9.5 μM).
The in vivo PK studies of BI-1408 ((R)-28) in rats demonstrated moderately fast clearance (40 mL min–1 kg–1) when dosed at 1 μmol kg–1 (i.v. bolus dose). However, when administered orally (10 μmol kg–1, p.o. dose) to evaluate the distribution in different tissues, the compound showed no indication of in vivo efflux as evidenced by the ratio of muscle to brain concentrations (CMuscle/CBrain = 0.9). Compound BI-1408 ((R)-28) also showed equal concentration distribution between the brain (CBrain = 2990 nM) and plasma (CPlasma = 2677 nM) indicating favorable results to advance into pharmacodynamic (PD) studies.
In the acute PD measurements of BI-1408 ((R)-28), rats were dosed (p.o.) at 30 mg kg–1, blood samples were collected (after 3 h post dose) to determine the plasma exposure, and the brains were extracted to measure the levels of Aβ42 and Aβtotal. The results indicate that the brain concentration was ∼30 nM, which was estimated from unbound plasma concentration. The brain Aβ42 levels lowered by ∼50% while the Aβtotal levels remained unchanged. Collectively, the successful identification and optimization of the chiral bicyclic BI-1408 ((R)-28) would make it a potential candidate for clinical trials. However, no additional safety and efficacy data were reported.
Recent developments of effective GSMs focused on increased lipophilicity which could enhance the potency, but might reduce their drug-like properties.94 Therefore, a recent report from Takeda Pharmaceutical has disclosed the ligand-lipophilicity efficiency (LLE = pIC50 – log D, a higher LLE represents greater drug-likeness of the compound) method as a drug-likeness guideline in a lead generation and optimization study for the discovery of efficacious GSMs.95 Based on the LLE method, they have discovered a new GSM, 5,6,7,8-tetrahydro[1,2,4]triazolo[4,3′a]pyridine derivative 34. An initial introduction with SAR of various substituents at the 8-position of 29 resulted in several more potent analogues: 30 (trifluoroethyl amide), 31 (2-hydroxypropane-2-yl), and 32 and 33 (spiro compounds). Further optimization of 31 resulted in the most potent compound 34-R (Aβ42 IC50 = 26 nM) while its isomer 34-S showed less potency (Aβ42 IC50 = >10 000 nM) (Fig. 21). The in vivo Aβ42 lowering effect experiments for compounds 30–33 and 34-R (Fig. 21) in wild-type mice (C57BL/6J) resulted in compound 34-R as the most potent when orally administered at doses of 10 mg kg–1 and 30 mg kg–1. Compound 34-R afforded the most positive Aβ42-lowering effect (44% Aβ42 reduction at 3 h after 10 mg kg–1p.o. administration), while compound 30 showed the least positive Aβ42-lowering effect (only 4% Aβ42 reduction at 3 h after 30 mg kg–1p.o. administration) which is most likely due to the much lower in vitro mice microsomal clearance of 34-R (MLM = –7 μL min–1 mg–1).
Fig. 21. Takeda Pharmaceutical's tetrahydro[1,2,4]triazolo[4,3,a]pyridine based GSMs (29–33, 34-R and 34-S) with in vitro Aβ42-lowering effect values (NR = not reported).

In a recent cellular assay (Neuro2A cells) report,96 compound 34-R reduced both Aβ42 and Aβ40, but increased Aβ37 without affecting the total Aβ levels indicating that compound 34-R would modulate γ-secretase activity by shifting the APP cleavage site. Compound 34-R exhibited no activity against Notch signaling (IC50 = >10 μM). The potent in vivo brain Aβ42 lowering effect of 34-R was confirmed at a dose of 6 mg kg–1 in normal mice with a lower drug exposure level (0.677 μg g–1 in the cortex and 3.307 μg mL–1 in plasma at 3 h after p.o. dosing).
When a chronic 58 day p.o. administration study of 34-R was performed in Tg2576 mice at 10 and 30 mg kg–1 per day doses, a ∼60% reduction in brain soluble Aβ42 and ∼50% reduction in brain insoluble Aβ42 were observed at a 30 mg kg–1 per day dose. Both doses displayed decreased levels of Aβ40 without affecting the total Aβ levels, and also showed a tendency to inhibit plaque deposition. The novel object recognition test (NORT) was performed to assess the effect of 34-R on cognitive functions on the 45th day of the chronic p.o. administration; as a result, the preference ratio was significantly improved to the normal range. In addition, a continuous cognitive improvement was observed up to 7 days after withdrawal from 34-R administration. Therefore, these results suggest that compound 34-R could be described as a potential candidate for disease modifying AD therapy. However, to date, no further pharmacological data were reported.
A recent publication97 from Roche has reported the discovery of potent and novel triazolo-azepine based GSM (S)-37 (Fig. 23), demonstrating the excellent in vitro and in vivo DMPK profiles. Their initial efforts were focused on discovering GSM 36 from 35 (Fig. 22) (Aβ42 IC50 = 143 nM) while addressing the potential drawbacks of compound 35 such as a high content of aromaticity.93,98,99 The trifluorophenyl group in compound 35 was replaced by the trifluoroalkoxy group as shown in compound 36 to enhance the aqueous solubility and to prevent the phototoxicity. However, compound 36 did not give good in vivo toxicology data.
Fig. 23. Roche's optimized and novel triazolo-azepine derivative (S)-37 and triazolopyrimidine derivative 38.

Fig. 22. Roche's structures of compound 35 and alkoxy derivative 36.

Therefore, further optimization of compound 36 led to the discovery of several triazolo-azepine based active compounds in which 38 and (S)-37 have shown superior in vivo profiles (Fig. 23). However, the isomer (S)-37 was selected to undergo entry in human enabling studies due to its excellent in vitro and in vivo DMPK profiles along with in vivo efficacy in a pharmacodynamic mouse model.
The lead optimization of 36 began with the improvement of its in vitro potency (Aβ42 IC50 = 31 nM) and to achieve a maximum tolerable dose (below 100 mg kg–1, oral administration) for toxicology studies. Initial efforts were put to optimize the alkoxy, trifluoromethyl, and oxadiazo groups in 36 which resulted in no superior activity. Therefore, further modifications in the triazolopyridine ring lead to the most potent compounds tetrahydrotriazolopyrimidine derivative 38 and tetrahydrotriazoloazepine derivative (S)-37. The free in vivo IC50 Aβ42 values were evaluated and found to be 3.0 and 2.5 nM, which are in agreement with in vitro potency values of 3.0 and 2.0 nM for compounds 38 and (S)-37, respectively. Pharmacodynamic (PD) studies of these two compounds were performed to determine their efficacy using the double transgenic mice model APP-Swedish (oral administration at 20 mg kg–1 and 30 mg kg–1 for compounds 38 and (S)-37, respectively). They were found to modulate the γ-secretase enzyme by increasing the formation of shorter peptides Aβ37 and Aβ38, while decreasing the pathogenic Aβ42 and also Aβ40, without affecting the total Aβ levels. The PKPD studies of these two compounds showed an excellent correlation, but (S)-37 showed superior activity to that of 38 in dose range finding (DRF) studies in both rodent and nonrodent species. Therefore, (S)-37 was selected for further developments in human enabled studies (GLP) as a highly potent GSM for a potential treatment for AD.
Distinct heterocyclic GSMs
In contrast to NSAID based GSMs, arylimidazole group containing heterocyclic non-NSAID based GSMs were shown to inhibit CYP450 activity in some cases. Recently,100 the Astellas Pharma Inc. has discovered a new class of compounds based on imidazopyridine derivatives as a new class of scaffolds for GSMs. These derivatives lack the strongly basic end groups such as arylimidazole, a common group present in Eisai's E2012 and its analogues. High-throughput screening (HTS) was conducted in the human neuroblastoma cell line SK-N-BE2 using their in-house low molecular weight compound libraries and the inhibition of Aβ42 production was measured. HTS identified a novel compound 39 (pyridinyl imidazopyridine) (Fig. 24), which inhibited the cellular Aβ42 production (IC50 = 7.1 μM) without altering the total Aβ production. Further structural optimization resulted in major alterations in the initial lead structure. Compound 39 has structural similarities to E2012 but lacks the ‘A’ ring moiety. Therefore, strategic optimization and addition of corresponding moieties to the ‘A’ ring resulted in 40 (pyridinyl ethylisoindolinone) as the most potent inhibitor of Aβ42 production (IC50 = 0.39 μM). Compound 40 shows no cytochrome P450 (CYP34A) inhibition and is more metabolically stable (CLint in mice = 611 μL min–1 mg–1).
Fig. 24. Astellas Pharmaceutical's imidazopyridine derivatives 39 (left) and 40 (right).

Further, in vivo demonstration indicated a sustained pharmacokinetic (PK) profile in mice. After oral administration of 30 mg kg–1 dose, the plasma concentration of 40 was 2.5 and 0.96 μM at 1 h and 3 h, respectively, with an acceptable brain to plasma ratio (KPbrain = 0.72). However, 40 did not reduce brain Aβ42 levels due to its low bioavailability with high microsomal clearance. In addition, 40 also possesses 5-fold lower in vitro GSM potency than representative GSM E2012 (Fig. 7), and it suffers from poor drug-like properties measured from the high clog P values and number of aromatic rings.
Recently, Astellas has developed a series of orally active N-ethylpyridine-2-carboxamide derivatives 41 and 42 (Fig. 25).101 The design was based on the optimization of 40 to improve the potency and in vivo efficacy. To block the metabolism of the methylene carbon of isoindolinone of 40 without altering the planar structure, the isoindolinone moiety (A–B ring) of 40 was replaced with a picolinamide (N-ethyl-pyridine-2-carboxamide) moiety to generate compound 41. Further optimization of the benzyl group (D ring) of 41 resulted in the most potent compound 42 with a biphenyl moiety at the D ring.
Fig. 25. Astellas Pharmaceutical's N-ethylpyridine-2-carboxamide derivatives 41 and 42.

These new compounds show significant improvement in the potency and in vivo efficacy. Compound 41 showed less potency (IC50 of Aβ42 = 0.58 μM) than 40 (IC50 = 0.39 μM) and improved metabolic stability (CLint in mice = 324 μL min–1 mg–1) compared to 40 (CLint in mice = 611 μL min–1 mg–1). The linker length optimization in 42 (phenyl to biphenyl) led to an overall improvement in activity (IC50 of Aβ42 = 0.091 μM) and metabolic stability (CLint in mice = 84.7 μL min–1 mg–1). The higher lipophilicity of 42 (ACD/log P = 4.78) vs.41 (ACD/log P = 3.02) correlated positively with the in vitro activity profile. The in vivo Aβ42 reduction studies in mice by oral administration of 42 (at 30 mg kg–1) show 36% and 66% Aβ42 reduction in the brain and plasma, respectively, after 3 h post dose. The brain/plasma ratio of 42 (Kp,brain = 0.45) was lower when compared to that of 40 (Kp,brain = 0.72), but 42 showed undetectable CYP3A4 inhibition. Therefore, compound 42 was considered as a lead candidate for further development of AD therapeutics.
However, from a clinical perspective, it was hypothesized that there is some room for optimization of 42 in terms of its microsomal stability as well as in vitro GSM potency. Therefore, further optimization of 42 (IC50 Aβ42 = 0.091 μM) resulted in several imidazopyridines as novel efficacious GSMs. The most potent, orally active imidazopyridine GSM was found to be 43 (IC50 Aβ42 = 0.029 μM) with a superior activity profile (Fig. 26).102 GSM 43 was shown to induce a robust reduction of Aβ42 levels in the brain, and in the Y-maze test, it showed an excellent efficacy against cognitive deficits in the AD mice model.
Fig. 26. Astellas Pharmaceutical's novel and optimized imidazopyridine derivative 43.

Initial optimization of 42 began with the modification of the biphenyl ring system and the amide group moiety as these two groups are critical for in vitro GSM activity as well as metabolic stability, as shown previously. To maintain the lipophilicity and to block the putative metabolic site, fluorine containing substrates were introduced at the biphenyl ring. The amide group substituent was modified from ethyl to methyl while maintaining the pseudo five-membered ring system through an intramolecular hydrogen bond from the amide hydrogen to the neighboring pyridine nitrogen. It was hypothesized that the intramolecular hydrogen bond is mandatory to fix the direction of the carbonyl group to interact with the γ-secretase complex. The most potent GSM 43 showed the highest in vitro activity (IC50 Aβ42 = 0.029 μM), as well as the highest brain exposure with undetectable cytochrome P450 (CYP) enzyme inhibition. Compound 43 also showed robust brain Aβ42-lowering effects: 42% and 23% at 3 h after oral administration (in rats, n = 2 or 3) at 30 mg kg–1 and 10 mg kg–1, respectively. High brain (1.5 nmol g–1) and plasma (5.7 μM) exposures of 43 were observed when administered at 10 mg kg–1 (3 h after p.o. administration). Finally, to evaluate the effect of GSM 43 on the cognitive functions of AD model (Tg2576) mice, the Y-maze test was conducted, in which cognitive deficits were completely rescued when compound 43 was administered at a low dose of 2 mg kg–1 (p.o.) for 8 days. Therefore, these excellent results suggest that compound 43 has shown an overall better activity than 42 and its analogues. Therefore, it should be considered for further development as a potential candidate for AD therapeutics.
Summary and future perspectives
Ever since the discovery of early non-NSAID imidazole-based GSMs, considerable work has been done in search of disease modifying therapeutics for AD. Following the withdrawal of GSIs from clinical trials due to adverse effects resulting from off-target inhibition and toxicological issues, early NSAID-based acidic GSMs have suffered from poor drug-like properties. This is mainly due to the unsuccessful outcomes in the optimization process of GSMs to achieve optimal potency at nanomolar concentrations. Most importantly, the CNS penetration property of GSMs has been tremendously improved over the years with increased potency associated with increased lipophilicity of the compounds. For example, with the evolution of arylimidazole-based non-NSAID GSMs, major pharmaceutical companies have focused on developing novel GSMs based on the core structure of Eisai's representative GSM E2012. As a result, most of the arylimidazole-based GSMs and their analogues exhibit improved potency and CNS penetration. However, due to high lipophilicity, arylimidazole-based GSMs suffered from CYP450 inhibition and hERG inhibition as well as the nonspecific binding of plasma protein (pp) and brain tissue (bt).
In an effort to mitigate the lipophilicity, the imidazole group was replaced by a cyanoindole group. The resulting GSMs were also shown to be less potent because of poor solubility of the compounds. To further address the lipophilicity issue, the a) central nervous system multiparameter optimization (CNS-MPO, max = 6.0) approach, and b) ligand-lipophilicity efficiency (LLE) method have been used to improve the drug-like properties of a molecule with improved in vitro potency associated in vivo efficacy (higher CNS-MPO and LLE scores represent greater drug-likeness of the compound), and a couple of reports have demonstrated an improvement in terms of the lipophilicity and drug-likeness properties of GSMs. However, regardless of recent advancements in GSM research for AD, there are still several important properties of GSMs such as solubility, metabolic stability, permeability, CYP inhibition, hERG liabilities, and toxicity that need to be further improved prior to human clinical trials. Moreover, whether these GSMs can be developed into PROTAC103–105 that selectively degrades certain γ-secretase complexes remains to be investigated. In addition to the modulation of γ-secretase alone, 1) a combination therapy with different classes of new and most potent GSMs, 2) a combination therapy with GSMs and BACE1-inhibitors, and 3) multitargeted ligands106 for the γ-secretase complex would also be some promising considerations for the development of efficacious therapeutics for AD.
Notes and abbreviations
- AD
Alzheimer's disease
- NFTs
Neurofibrillary tau tangles
- Aβ peptides
Beta (β) amyloid peptides
- APP
Amyloid precursor protein
- GSIs
γ-Secretase inhibitors
- GSMs
γ-Secretase modulators
- NSAID
Nonsteroidal anti-inflammatory drugs
- NMDA
N-Methyl-d-aspartic acid
- CNS
Central nervous system
- PS
Presenilin
- Nct
Nicastrin
- Aph-1
Anterior pharynx defective-1
- Pen-2
Presenilin enhancer-2
- FAD
Familial AD
- TMDs
Transmembrane domains
- NFT
N-Terminal fragment (or amino terminal fragment)
- CTF
Carboxy terminal fragment
- cryo-EM
Cryo-electron microscopy
- BACE1
β-Site APP cleaving enzyme-1
- EDC
Ectodomain
- AICD
APP intracellular domain
- CSF
Cerebrospinal fluid
- CHO cells
Chinese hamster ovary cells
- HBA
Hydrogen bond acceptor
- ADME
Absorption, distribution, metabolism and excretion
- CYP450
Cytochrome P450
- CYP3A4
Cytochrome 3A4
- hERG
Human ether-a-go-go related genes
- HTS
High-throughput screening
- clog P
lipophilicity
- IC50
The half-maximal inhibitory concentration
- EC50
The half-maximal effective concentration
- p.o.
per os (by mouth)
- I.V.
Intravenous
- SAR
Structure–activity relationship
- ALT
Alanine transaminase
- AST
Aspartate transaminase
- MAD
Multiple ascending dose
- CNS-MPO score
CNS multi parameter optimization score
- ER
Efflux ratio
- MDR ER
Multidrug resistance efflux ratio
- LLE
Ligand-lipophilicity efficiency
- NORT
Novel object recognition test
- PK
Pharmacokinetic
- PD
Pharmacodynamic
- ACD/log P
Lipophilicity values calculated by the program developed by ACD Labs
- Log D
Distribution constant
- PROTAC
Proteolysis targeting chimera
Conflicts of interest
LYM is a co-inventor of intellectual property (assay for γ-secretase activity and screening method for γ-secretase inhibitors) licensed to Jiangsu Continental Medical Development.
Acknowledgments
We acknowledge the support of this research from R01NS096275 (YML), RF1AG057593 (YML), the JPB Foundation (YML) and The Cure Alzheimer's Fund (YML), training grant T32 CA062948(GN), Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, the Experimental Therapeutics Center of MSKCC, and the William Randolph Hearst Fund in Experimental Therapeutics.
Biographies

Shekar Mekala
Shekar Mekala obtained his Ph.D. in Organic Chemistry (2012) from the University of Oklahoma, Norman, OK, under the mentorship of Prof. Ronald L. Halterman. He then moved to Upstate New York to pursue his postdoctoral research at Syracuse University, and in the Center for Biotechnology and Interdisciplinary Studies (with Prof. Richard A. Gross) at Rensselaer Polytechnic Institute (RPI), Troy, New York. In 2018, he joined the laboratory of Dr. Yue-Ming Li, as a Research Fellow in the Chemical Biology program at the Memorial Sloan-Kettering Cancer Center, New York City. His current research focusses on the early stage drug discovery, and hit to lead development of small molecule autophagy modulators for cancer and Alzheimer's disease. He is originally from India and obtained a master's degree in chemistry from Osmania University, Hyderabad.

Grady Nelson
Grady Nelson studied under Dr. Venkatram Mereddy at the University of Minnesota: Duluth, where he obtained a Masters in Chemistry in 2013 and a Ph.D. in Integrated Biosciences in 2019. During his time there, he developed small molecule modulators of MCT1 and carried out preclinical studies for the treatment of breast cancer. In 2019, Grady joined Dr. Yueming Li's lab as a postdoctoral fellow. He obtained the Cancer Pharmacology Training grant at the Weill Cornell Medicine. During his fellowship, he continues to work on the development of small molecule modulators with a focus on autophagy.

Yue-Ming Li
Yue-Ming Li is a Member and Professor of Chemical Biology at the Memorial Sloan-Kettering Cancer Center, and a Professor of Pharmacology and Neuroscience at the Weill Graduate School of Medical Sciences of Cornell University. He received his Ph.D. degree from the University of California, Berkeley and postdoctoral training at Harvard Medical School. Prior to coming to the Memorial Sloan-Kettering Cancer Center in 2002, he was a scientist at Merck Research Laboratories. His lab studies aging-related human disorders, such as Alzheimer's disease and cancer.
References
- Alzheimer's Association, Alzheimer's disease facts and figures, Alzheimer's Dementia, (https://www.alz.org/alzheimers-dementia/what-is-alzheimers).
- World Health Organization, Dementia: key facts, (https://www.who.int/news-room/fact-sheets/detail/dementia).
- Alzheimer's Association, FDA approved treatments alzheimer's, (https://www.alz.org/media/documents/fda-approved-treatments-alzheimers-ts.pdf).
- Briggs R., Kennelly S. P., O'Neill D. Clin. Med. 2016;16:247–253. doi: 10.7861/clinmedicine.16-3-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C., Lee V. M., Trojanowski J. Q. Nat. Rev. Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Sanders D. W., Kaufman S. K., DeVos S. L., Sharma A. M., Mirbaha H., Li A. M., Barker S. J., Foley A. C., Thorpe J. R., Serpell L. C., Miller T. M., Grinberg L. T., Seeley W. W., Diamond M. I. Neuron. 2014;82:1271–1288. doi: 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. A., Higgins G. A. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hardy J., Selkoe D. J. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J., Hardy J. EMBO Mol. Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J., Hou J., Ping J., Cai D. Mol. Neurodegener. 2018;13:64. doi: 10.1186/s13024-018-0299-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertsik N., Chiu D., Li Y.-M. Front. Aging Neurosci. 2014;6:342. doi: 10.3389/fnagi.2014.00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy-Lahad E., Wasco W., Poorkaj P., Romano D. M., Oshima J., Pettingell W. H., Yu C. E., Jondro P. D., Schmidt S. D., Wang K., Crowley A. C., Fu Y.-H., Guenette S. Y., Galas D., Nemens E., Wijsman E. M., Bird T. D., Schellenberg G. D., Tanzi R. E. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Sherrington R., Rogaev E. I., Liang Y., Rogaeva E. A., Levesque G., Ikeda M., Chi H., Lin C., Li G., Holman K., Tsuda T., Mar L., Foncin J. F., Bruni A. C., Montesi M. P., Sorbi S., Rainero I., Pinessi L., Nee L., Chumakov I., Pollen D., Brookes A., Sanseau P., Polinsky R. J., Wasco W., Dasilva H. A. R., Haines J. L., Pericakvance M. A., Tanzi R. E., Roses A. D., Fraser P. E., Rommens J. M., Stgeorge-hyslop P. H. Nature. 1995;375:754–760. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- Thinakaran G., Borchelt D. R., Lee M. K., Slunt H. H., Spitzer L., Kim G., Ratovitsky T., Davenport F., Nordstedt C., Seeger M., Hardy J., Levey A. I., Gandy S. E., Jenkins N. A., Copeland N. G., Price D. L., Sisodia S. S. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- Li Y.-M., Xu M., Lai M. T., Huang Q., Castro J. L., DiMuzio-Mower J., Harrison T., Lellis C., Nadin A., Neduvelil J. G., Register R. B., Sardana M. K., Shearman M. S., Smith A. L., Shi X. P., Yin K. C., Shafer J. A., Gardell S. J. Nature. 2000;405:689–694. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- Esler W. P., Kimberly W. T., Ostaszewski B. L., Diehl T. S., Moore C. L., Tsai J. Y., Rahmati T., Xia W., Selkoe D. J., Wolfe M. S. Nat. Cell Biol. 2000;2:428–434. doi: 10.1038/35017062. [DOI] [PubMed] [Google Scholar]
- Ahn K., Shelton C. C., Tian Y., Zhang X., Gilchrist M. L., Sisodia S. S., Li Y.-M. Proc. Natl. Acad. Sci. U. S. A. 2010;107:21435–21440. doi: 10.1073/pnas.1013246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herreman A., Van Gassen G., Bentahir M., Nyabi O., Craessaerts K., Mueller U., Annaert W., De Strooper B. J. Cell Sci. 2003;116:1127–1136. doi: 10.1242/jcs.00292. [DOI] [PubMed] [Google Scholar]
- Li T., Ma G., Cai H., Price D. L., Wong P. C. J. Neurosci. 2003;23:3272–3277. doi: 10.1523/JNEUROSCI.23-08-03272.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasugi N., Tomita T., Hayashi I., Tsuruoka M., Niimura M., Takahashi Y., Thinakaran G., Iwatsubo T. Nature. 2003;422:438–441. doi: 10.1038/nature01506. [DOI] [PubMed] [Google Scholar]
- Lee S.-F., Shah S., Li H. Q., Yu C., Han W. P., Yu G. J. Biol. Chem. 2002;277:45013–45019. doi: 10.1074/jbc.M208164200. [DOI] [PubMed] [Google Scholar]
- Niimura M., Isoo N., Takasugi N., Tsuruoka M., Ui-Tei K., Saigo K., Morohashi Y., Tomita T., Iwatsubo T. J. Biol. Chem. 2005;280:12967–12975. doi: 10.1074/jbc.M409829200. [DOI] [PubMed] [Google Scholar]
- Lee S.-F., Shah S., Yu C., Wigley W. C., Li H., Lim M., Pedersen K., Han W. P., Thomas P., Lundkvist J., Hao Y. H., Yu G. J. Biol. Chem. 2004;279:4144–4152. doi: 10.1074/jbc.M309745200. [DOI] [PubMed] [Google Scholar]
- Shiraishi H., Sai X., Wang H. Q., Maeda Y., Kurono Y., Nishimura M., Yanagisawa K., Komano H. J. Neurochem. 2004;90:1402–1413. doi: 10.1111/j.1471-4159.2004.02597.x. [DOI] [PubMed] [Google Scholar]
- Steiner H., Winkler E., Edbauer D., Prokop S., Basset G., Yamasaki A., Kostka M., Haass C. J. Biol. Chem. 2002;277:39062–39065. doi: 10.1074/jbc.C200469200. [DOI] [PubMed] [Google Scholar]
- Wolfe M. S. Biochim. Biophys. Acta, Biomembr. 2020;1862:183016. doi: 10.1016/j.bbamem.2019.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinakis A., Lobry C., Abdel-Wahab O., Oh P., Haeno H., Buonamici S., van De Walle I., Cathelin S., Trimarchi T., Araldi E., Liu C., Ibrahim S., Beran M., Zavadil J., Efstratiadis A., Taghon T., Michor F., Levine R. L., Aifantis I. Nature. 2011;473:230–233. doi: 10.1038/nature09999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B. X., Yang W., Wen W., Sun J., Su B., Liu B., Ma D. L., Lv D., Wen Y. R., Qu T., Chen M., Sun M. A., Shen Y., Zhang X. Science. 2010;330:1065. doi: 10.1126/science.1196284. [DOI] [PubMed] [Google Scholar]
- Bai X. C., Yan C., Yang G., Lu P., Ma D., Sun L., Zhou R., Scheres S. H. W., Shi Y. Nature. 2015;525:212–217. doi: 10.1038/nature14892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P., Bai X. C., Ma D., Xie T., Yan C., Sun L., Yang G., Zhao Y., Zhou R., Scheres S. H. W., Shi Y. Nature. 2014;512:166–170. doi: 10.1038/nature13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G. H., Zhou R., Zhou Q., Guo X. F., Yan C. Y., Ke M., Lei J. L., Shi Y. G. Nature. 2019;565:192–197. doi: 10.1038/s41586-018-0813-8. [DOI] [PubMed] [Google Scholar]
- Zhou R., Yang G. H., Guo X. F., Zhou Q., Lei J. L., Shi Y. G. Science. 2019;363:eaaw0930. doi: 10.1126/science.aaw0930. [DOI] [PubMed] [Google Scholar]
- Haapasalo A., Kovacs D. M. J. Alzheimer's Dis. 2011;25:3–28. doi: 10.3233/JAD-2011-101065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takami M., Nagashima Y., Sano Y., Ishihara S., Morishima-Kawashima M., Funamoto S., Ihara Y. J. Neurosci. 2009;29:13042–13052. doi: 10.1523/JNEUROSCI.2362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M. P., Hickman L. J., Eckman C. B., Uljon S. N., Wang R., Golde T. E. J. Biol. Chem. 1999;274:11914–11923. doi: 10.1074/jbc.274.17.11914. [DOI] [PubMed] [Google Scholar]
- Wang R., Sweeney D., Gandy S. E., Sisodia S. S. J. Biol. Chem. 1996;271:31894–31902. doi: 10.1074/jbc.271.50.31894. [DOI] [PubMed] [Google Scholar]
- Crump C. J., Johnson D. S., Li Y.-M. Biochemistry. 2013;52:3197–3216. doi: 10.1021/bi400377p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehlrich D., Berthelot D. J.-C., Gijsen H. J. M. J. Med. Chem. 2011;54:669–698. doi: 10.1021/jm101168r. [DOI] [PubMed] [Google Scholar]
- Bursavich M. G., Harrison B. A., Blain J. F. J. Med. Chem. 2016;59:7389–7409. doi: 10.1021/acs.jmedchem.5b01960. [DOI] [PubMed] [Google Scholar]
- Pettersson M., Kauffman G. W., Ende C. W. A., Patel N. C., Stiff C., Tran T. P., Johnson D. S. Expert Opin. Ther. Pat. 2011;21:205–226. doi: 10.1517/13543776.2011.547479. [DOI] [PubMed] [Google Scholar]
- Golde T. E., Koo E. H., Felsenstein K. M., Osborne B. A., Miele L. Biochim. Biophys. Acta, Biomembr. 2013;1828:2898–2907. doi: 10.1016/j.bbamem.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coric V., van Dyck C. H., Salloway S., Andreasen N., Brody M., Richter R. W., Soininen H., Thein S., Shiovitz T., Pilcher G., Colby S., Rollin L., Dockens R., Pachai C., Portelius E., Andreasson U., Blennow K., Soares H., Albright C., Feldman H. H., Berman R. M. Arch. Neurol. 2012;69:1430–1440. doi: 10.1001/archneurol.2012.2194. [DOI] [PubMed] [Google Scholar]
- Doody R. S., Aisen P. S., Iwatsubo T. N. Engl. J. Med. 2013;369:1661. doi: 10.1056/NEJMc1310845. [DOI] [PubMed] [Google Scholar]
- Doody R. S., Raman R., Farlow M., Iwatsubo T., Vellas B., Joffe S., Kieburtz K., He F., Sun X., Thomas R. G., Aisen P. S., Siemers E., Sethuraman G., Mohs R. N. Engl. J. Med. 2013;369:341–350. doi: 10.1056/NEJMoa1210951. [DOI] [PubMed] [Google Scholar]
- Jurisch-Yaksi N., Sannerud R., Annaert W. Biochim. Biophys.Biochim. Biophys. ActaActa. 2013;1828:2815–2827. doi: 10.1016/j.bbamem.2013.04.016. [DOI] [PubMed] [Google Scholar]
- Weggen S., Eriksen J. L., Das P., Sagi S. A., Wang R., Pietrzik C. U., Findlay K. A., Smith T. E., Murphy M. P., Bulter T., Kang D. E., Marquez-Sterling N., Golde T. E., Koo E. H. Nature. 2001;414:212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- Kuperstein I., Broersen K., Benilova I., Rozenski J., Jonckheere W., Debulpaep M., Vandersteen A., Segers-Nolten I., Van Der Werf K., Subramaniam V., Braeken D., Callewaert G., Bartic C., D'Hooge R., Martins I. C., Rousseau F., Schymkowitz J., De Strooper B. EMBO J. 2010;29:3408–3420. doi: 10.1038/emboj.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Onstead L., Randle S., Price R., Smithson L., Zwizinski C., Dickson D. W., Golde T., McGowan E. J. Neurosci. 2007;27:627–633. doi: 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen J. L., Sagi S. A., Smith T. E., Weggen S., Das P., McLendon D. C., Ozols V. V., Jessing K. W., Zavitz K. H., Koo E. H., Golde T. E. J. Clin. Invest. 2003;112:440–449. doi: 10.1172/JCI18162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock G. K., Black S. E., Hendrix S. B., Zavitz K. H., Swabb E. A., Laughlin M. A. Lancet Neurol. 2008;7:483–493. doi: 10.1016/S1474-4422(08)70090-5. [DOI] [PubMed] [Google Scholar]
- Pozdnyakov N., Murrey H. E., Crump C. J., Pettersson M., Ballard T. E., Am Ende C. W., Ahn K., Li Y.-M., Bales K. R., Johnson D. S. J. Biol. Chem. 2013;288:9710–9720. doi: 10.1074/jbc.M112.398602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohki Y., Higo T., Uemura K., Shimada N., Osawa S., Berezovska O., Yokoshima S., Fukuyama T., Tomita T., Iwatsubo T. EMBO J. 2011;30:4815–4824. doi: 10.1038/emboj.2011.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump C. J., Fish B. A., Castro S. V., Chau D. M., Gertsik N., Ahn K., Stiff C., Pozdnyakov N., Bales K. R., Johnson D. S., Li Y.-M. ACS Chem. Neurosci. 2011;2:705–710. doi: 10.1021/cn200098p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebke A., Luebbers T., Fukumori A., Shirotani K., Haass C., Baumann K., Steiner H. J. Biol. Chem. 2011;286:37181–37186. doi: 10.1074/jbc.C111.276972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumpertz T., Rennhack A., Ness J., Baches S., Pietrzik C. U., Bulic B., Weggen S. PLoS One. 2012;7:e30484. doi: 10.1371/journal.pone.0030484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kounnas M. Z., Danks A. M., Cheng S., Tyree C., Ackerman E., Zhang X., Ahn K., Nguyen P., Comer D., Mao L., Yu C., Pleynet D., Digregorio P. J., Velicelebi G., Stauderman K. A., Comer W. T., Mobley W. C., Li Y.-M., Sisodia S. S., Tanzi R. E., Wagner S. L. Neuron. 2010;67:769–780. doi: 10.1016/j.neuron.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oborski C. E., Iyer R., Maguire B. A., Bora G., Atchison K., Pozdnyakov N., Wood K., Parker C., Subashi T. A., Pettersson M., Johnson D. S., Bales K. R. Neurosci. Med. 2012;3:149–161. [Google Scholar]
- Mitani Y., Yarimizu J., Akashiba H., Shitaka Y., Ni K., Matsuoka N. J. Neurochem. 2013;125:465–472. doi: 10.1111/jnc.12125. [DOI] [PubMed] [Google Scholar]
- Hussain I., Harrison D. C., Hawkins J., Chapman T., Marshall I., Facci L., Ahmed S., Brackenborough K., Skaper S. D., Mead T. L., Smith B. B., Giblin G. M., Hall A., Gonzalez M. I., Richardson J. C. Neurodegener. Dis. 2011;8:15–24. doi: 10.1159/000313903. [DOI] [PubMed] [Google Scholar]
- Hawkins J., Harrison D. C., Ahmed S., Davis R. P., Chapman T., Marshall I., Smith B., Mead T. L., Medhurst A., Giblin G. M., Hall A., Gonzalez M. I., Richardson J., Hussain I. Neurodegener. Dis. 2011;8:455–464. doi: 10.1159/000324511. [DOI] [PubMed] [Google Scholar]
- Van Broeck B., Chen J. M., Treton G., Desmidt M., Hopf C., Ramsden N., Karran E., Mercken M., Rowley A. Br. J. Pharmacol. 2011;163:375–389. doi: 10.1111/j.1476-5381.2011.01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretto I., Radaelli S., Parini C., Zandi M., Raveglia L. F., Dondio G., Fontanella L., Misiano P., Bigogno C., Rizzi A., Riccardi B., Biscaioli M., Marchetti S., Puccini P., Catinella S., Rondelli I., Cenacchi V., Bolzoni P. T., Caruso P., Villetti G., Facchinetti F., Del Giudice E., Moretto N., Imbimbo B. P. J. Med. Chem. 2005;48:5705–5720. doi: 10.1021/jm0502541. [DOI] [PubMed] [Google Scholar]
- Rogers K., Felsenstein K. M., Hrdlicka L., Tu Z., Albayya F., Lee W., Hopp S., Miller M. J., Spaulding D., Yang Z., Hodgdon H., Nolan S., Wen M., Costa D., Blain J. F., Freeman E., De Strooper B., Vulsteke V., Scrocchi L., Zetterberg H., Portelius E., Hutter-Paier B., Havas D., Ahlijanian M., Flood D., Leventhal L., Shapiro G., Patzke H., Chesworth R., Koenig G. Mol. Neurodegener. 2012;7:61. doi: 10.1186/1750-1326-7-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro G. and Chesworth R., US Pat., WO2009086277A1, 2009.
- Xin Z. L., Peng H. R., Zhang A., Talreja T., Kumaravel G., Xu L., Rohde E., Jung M. Y., Shackett M. N., Kocisko D., Chollate S., Dunah A. W., Snodgrass-Belt P. A., Arnold H. M., Taveras A. G., Rhodes K. J., Scannevin R. H. Bioorg. Med. Chem. Lett. 2011;21:7277–7280. doi: 10.1016/j.bmcl.2011.10.047. [DOI] [PubMed] [Google Scholar]
- Scannevin R. H., Chollate S., Brennan M. S., Snodgrass-Belt P. A., Peng H., Xu L., Jung M. Y., Bussiere T., Arastu M. F., Talreja T., Xin Z., Dunstan R. W., Fahrer D., Rohde E., Dunah A. W., Wang J., Kumaravel G., Taveras A. G., Moore Arnold H., Rhodes K. J. Neuropharmacology. 2016;103:57–68. doi: 10.1016/j.neuropharm.2015.12.006. [DOI] [PubMed] [Google Scholar]
- Kimura T., Kawano K., Doi E., Kitazawa N., Shin K., Miyagawa T., Kaneko T., Ito K., Takaishi M., Sasaki T. and Hagiwara H., USA Pat., WO2005115990A1, 2005.
- Wagner S. L., Zhang C., Cheng S., Nguyen P., Zhang X., Rynearson K. D., Wang R., Li Y., Sisodia S. S., Mobley W. C., Tanzi R. E. Biochemistry. 2014;53:702–713. doi: 10.1021/bi401537v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S. L., Rynearson K. D., Duddy S. K., Zhang C., Nguyen P. D., Becker A., Vo U., Masliah D., Monte L., Klee J. B., Echmalian C. M., Xia W., Quinti L., Johnson G., Lin J. H., Kim D. Y., Mobley W. C., Rissman R. A., Tanzi R. E. J. Pharmacol. Exp. Ther. 2017;362:31–44. doi: 10.1124/jpet.117.240861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rynearson K. D., Buckle R. N., Barnes K. D., Herr R. J., Mayhew N. J., Paquette W. D., Sakwa S. A., Nguyen P. D., Johnson G., Tanzi R. E., Wagner S. L. Bioorg. Med. Chem. Lett. 2016;26:3928–3937. doi: 10.1016/j.bmcl.2016.07.011. [DOI] [PubMed] [Google Scholar]
- Shi J. L., Zuev D., Xu L., Lentz K. A., Grace J. E., Toyn J. H., Olson R. E., Macor J. E., Thompson L. A. Bioorg. Med. Chem. Lett. 2016;26:1498–1502. doi: 10.1016/j.bmcl.2015.06.020. [DOI] [PubMed] [Google Scholar]
- Marcin L. R., Thompson III L. A., Boy K. M., Guernon J. M., Higgins M. A., Shi J., Wu Y.-J., Zhang Y. and Macor J. E., US Pat., WO2010083141A1, 2010.
- Gillman K. W., Starrett J. E., Parker M. F., Xie K., Bronson J. J., Marcin L. R., McElhone K. E., Bergstrom C. P., Mate R. A., Williams R., Meredith J. E., Burton C. R., Barten D. M., Toyn J. H., Roberts S. B., Lentz K. A., Houston J. G., Zaczek R., Albright C. F., Decicco C. P., Macor J. E., Olson R. E. ACS Med. Chem. Lett. 2010;1:120–124. doi: 10.1021/ml1000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. J., Zhang Y., Toyn J. H., Macor J. E., Thompson L. A. Bioorg. Med. Chem. Lett. 2016;26:1554–1557. doi: 10.1016/j.bmcl.2016.02.016. [DOI] [PubMed] [Google Scholar]
- Boy K. M., Guernon J. M., Zuev D. S., Xu L., Zhang Y., Shi J., Marcin L. R., Higgins M. A., Wu Y. J., Krishnananthan S., Li J., Trehan A., Smith D., Toyn J. H., Meredith J. E., Burton C. R., Kimura S. R., Zvyaga T., Zhuo X., Lentz K. A., Grace J. E., Denton R., Morrison J. S., Mathur A., Albright C. F., Ahlijanian M. K., Olson R. E., Thompson L. A., Macor J. E. ACS Med. Chem. Lett. 2019;10:312–317. doi: 10.1021/acsmedchemlett.8b00541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J. D., Blagg J., Price D. A., Bailey S., Decrescenzo G. A., Devraj R. V., Ellsworth E., Fobian Y. M., Gibbs M. E., Gilles R. W., Greene N., Huang E., Krieger-Burke T., Loesel J., Wager T., Whiteley L., Zhang Y. Bioorg. Med. Chem. Lett. 2008;18:4872–4875. doi: 10.1016/j.bmcl.2008.07.071. [DOI] [PubMed] [Google Scholar]
- Wager T. T., Hou X., Verhoest P. R., Villalobos A. ACS Chem. Neurosci. 2010;1:435–449. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager T. T., Chandrasekaran R. Y., Hou X., Troutman M. D., Verhoest P. R., Villalobos A., Will Y. ACS Chem. Neurosci. 2010;1:420–434. doi: 10.1021/cn100007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares H. D., Gasior M., Toyn J. H., Wang J. S., Hong Q., Berisha F., Furlong M. T., Raybon J., Lentz K. A., Sweeney F., Zheng N., Akinsanya B., Berman R. M., Thompson L. A., Olson R. E., Morrison J., Drexler D. M., Macor J. E., Albright C. F., Ahlijanian M. K., AbuTarif M. J. Pharmacol. Exp. Ther. 2016;358:138–150. doi: 10.1124/jpet.116.232256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R. F. Chem.-Biol. Interact. 2001;135–136:53–64. doi: 10.1016/s0009-2797(01)00170-3. [DOI] [PubMed] [Google Scholar]
- Bursavich M. G., Harrison B. A., Acharya R., Costa D. E., Freeman E. A., Hodgdon H. E., Hrdlicka L. A., Jin H., Kapadnis S., Moffit J. S., Murphy D. A., Nolan S., Patzke H., Tang C., Wen M., Koenig G., Blain J. F., Burnett D. A. J. Med. Chem. 2017;60:2383–2400. doi: 10.1021/acs.jmedchem.6b01620. [DOI] [PubMed] [Google Scholar]
- Velter A. I., Bischoff F. P., Berthelot D., De Cleyn M., Oehlrich D., Jaroskova L., Macdonald G., Minne G., Pieters S., Rombouts F., Van Brandt S., Van Roosbroeck Y., Surkyn M., Trabanco A. A., Tresadern G., Wu T., Borghys H., Mercken M., Masungi C., Gijsen H. Bioorg. Med. Chem. Lett. 2014;24:5805–5813. doi: 10.1016/j.bmcl.2014.10.024. [DOI] [PubMed] [Google Scholar]
- Bischoff F. P., Velter A. I., Minne G., Pieters S., Berthelot D., De Cleyn M., Gijsen H. J. M., Macdonald G., Surkyn M., Van Brandt S., Van Roosbroeck Y., Zavattaro C., Mercken M., Austin N., Dhuyvetter D., Borghys H., Ahmad I., Samanta S. K. Bioorg. Med. Chem. Lett. 2019;29:1737–1745. doi: 10.1016/j.bmcl.2019.05.023. [DOI] [PubMed] [Google Scholar]
- Zhao Z., Pissarnitski D. A., Huang X., Palani A., Zhu Z., Greenlee W. J., Hyde L. A., Song L., Terracina G., Zhang L., Parker E. M. ACS Med. Chem. Lett. 2017;8:1002–1006. doi: 10.1021/acsmedchemlett.7b00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z. Y., Asberom T., Bara T., Bennett C., Burnett D., Chu I., Clader J., Cohen-Williams M., Cole D., Czarniecki M., Durkin J., Gallo G., Greenlee W., Josien H., Huang X., Hyde L., Jones N., Kazakevich I., Li H., Liu X., Lee J., Maccoss M., Mandal M. B., McCracken T., Nomeir A., Mazzola R., Palani A., Parker E. M., Pissarnitski D. A., Qin J., Song L., Terracina G., Vicarel M., Voigt J., Xu R., Zhang L., Zhang Q., Zhao Z., Zhu X., Zhu Z. J. Med. Chem. 2012;55:489–502. doi: 10.1021/jm201407j. [DOI] [PubMed] [Google Scholar]
- Huang X., Zhou W., Liu X., Li H., Sun G., Mandal M., Vicarel M., Zhu X., Bennett C., McCraken T., Pissarnitski D., Zhao Z., Cole D., Gallo G., Zhu Z., Palani A., Aslanian R., Clader J., Czarniecki M., Greenlee W., Burnett D., Cohen-Williams M., Hyde L., Song L., Zhang L., Chu I., Buevich A. ACS Med. Chem. Lett. 2012;3:931–935. doi: 10.1021/ml300209g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenlee W., Pissarnitski D., Zhao Z. and Zhu Z., US Pat., WO2013066740A1, 2012.
- Pettersson M., Johnson D. S., Rankic D. A., Kauffman G. W., Am Ende C. W., Butler T. W., Boscoe B., Evrard E., Helal C. J., Humphrey J. M., Stepan A. F., Stiff C. M., Yang E., Xie L., Bales K. R., Hajos-Korcsok E., Jenkinson S., Pettersen B., Pustilnik L. R., Ramirez D. S., Steyn S. J., Wood K. M., Verhoest P. R. MedChemComm. 2017;8:730–743. doi: 10.1039/c6md00406g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson M., Johnson D. S., Subramanyam C., Bales K. R., Ende C. W. A., Fish B. A., Green M. E., Kauffman G. W., Lira R., Mullins P. B., Navaratnam T., Sakya S. M., Stiff C. M., Tran T. P., Vetelino B. C., Xie L., Zhang L., Pustilnik L. R., Wood K. M., O'Donnell C. J. Bioorg. Med. Chem. Lett. 2012;22:2906–2911. doi: 10.1016/j.bmcl.2012.02.059. [DOI] [PubMed] [Google Scholar]
- Pettersson M., Johnson D. S., Subramanyam C., Bales K. R., Ende C. W. A., Fish B. A., Green M. E., Kauffman G. W., Mullins P. B., Navaratnam T., Sakya S. M., Stiff C. M., Tran T. P., Xie L., Zhang L., Pustilnik L. R., Vetelino B. C., Wood K. M., Pozdnyakov N., Verhoest P. R., O'Donnell C. J. J. Med. Chem. 2014;57:1046–1062. doi: 10.1021/jm401782h. [DOI] [PubMed] [Google Scholar]
- Gerlach K., Hobson S., Eickmeier C., Gross U., Braun C., Sieger P., Garneau M., Hoerer S., Heine N. Bioorg. Med. Chem. 2018;26:3227–3241. doi: 10.1016/j.bmc.2018.04.053. [DOI] [PubMed] [Google Scholar]
- Baumann K., Flohr A., Goetschi E., Green L., Jolidon S., Knust H., Limberg A., Luebbers T. and Thomas A., US Pat., WO2011101304A2, 2011.
- Baumann K., Green L., Limberg A., Luebbers T. and Thomas A., US Pat., WO2012116965A1, 2012.
- Gijsen H. J., Mercken M. Int. J. Alzheimer's Dis. 2012;2012:295207. doi: 10.1155/2012/295207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai T., Hoashi Y., Tomata Y., Morimoto S., Nakamura M., Watanabe T., Igari T., Koike T. Bioorg. Med. Chem. Lett. 2015;25:4245–4249. doi: 10.1016/j.bmcl.2015.07.101. [DOI] [PubMed] [Google Scholar]
- Murakami K., Watanabe T., Koike T., Kamata M., Igari T., Kondo S. Brain Res. 2016;1633:73–86. doi: 10.1016/j.brainres.2015.12.016. [DOI] [PubMed] [Google Scholar]
- Ratni H., Alker A., Bartels B., Bissantz C., Chen W., Gerlach I., Limberg A., Lu M., Neidhart W., Pichereau S., Reutlinger M., Rodriguez-Sarmiento R.-M., Jakob-Roetne R., Schmitt G., Zhang E., Baumann K. ACS Med. Chem. Lett. 2020;11:1257–1268. doi: 10.1021/acsmedchemlett.0c00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels B., Baumann K., Galley G., Jaeschke G., Jakob-Roetne R., Limberg A., Neidhart W., Ratni H. and Rodriguez-Sarmiento R. M., US Pat., WO2017097728A1, 2017.
- Baumann K., Flohr A., Goetschi E., Green L., Jolidon S., Knust H., Limberg A., Luebbers T. and Thomas A., US Pat., US20110201605A1, 2011.
- Sekioka R., Honjo E., Honda S., Fuji H., Akashiba H., Mitani Y., Yamasaki S. Bioorg. Med. Chem. 2018;26:435–442. doi: 10.1016/j.bmc.2017.11.049. [DOI] [PubMed] [Google Scholar]
- Sekioka R., Honda S., Honjo E., Suzuki T., Akashiba H., Mitani Y., Yamasaki S. Bioorg. Med. Chem. 2020;28:115132. doi: 10.1016/j.bmc.2019.115132. [DOI] [PubMed] [Google Scholar]
- Sekioka R., Honda S., Akashiba H., Yarimizu J., Mitani Y., Yamasaki S. Bioorg. Med. Chem. 2020;28:115455. doi: 10.1016/j.bmc.2020.115455. [DOI] [PubMed] [Google Scholar]
- Deshaies R. J. Nature. 2020;580:329–338. doi: 10.1038/s41586-020-2168-1. [DOI] [PubMed] [Google Scholar]
- Sun X., Gao H., Yang Y., He M., Wu Y., Song Y., Tong Y., Rao Y. Signal Transduction Targeted Ther. 2019;4:64. doi: 10.1038/s41392-019-0101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira M., Calabrese M. F., Bullock A. N., Crews C. M. Nat. Rev. Drug Discovery. 2019;18:949–963. doi: 10.1038/s41573-019-0047-y. [DOI] [PubMed] [Google Scholar]
- Blaikie L., Kay G., Lin P. K. T. MedChemComm. 2019;10:2052–2072. doi: 10.1039/c9md00337a. [DOI] [PMC free article] [PubMed] [Google Scholar]