

Abstract
Introduction
Plasma markers have been reported to be associated with brain amyloid burden, tau pathology, or neurodegeneration. We aimed to evaluate whether plasma biomarker profiles could predict Alzheimer's disease (AD) pathology and clinical progression in older adults without dementia.
Methods
Cross‐sectional and longitudinal data of participants enrolled in this study were from the Alzheimer's Disease Neuroimaging Initiative (ADNI). Plasma amyloid beta (Aβ)1‐42/Aβ1‐40 ratio was selected as the marker for amyloid pathology, p‐tau181 for tau pathology, and neurofilament light for neurodegeneration. Cut‐offs for these plasma markers were calculated with well‐established positron emission tomography and structural imaging biomarkers as reference. Older adults without dementia were categorized into eight groups at baseline by plasma amyloid/tau/neurodegeneration (A/T/N) cut‐offs. Clinical progression was analyzed using linear mixed‐effects models and Cox proportional hazard models.
Results
A total of 183 participants (97 cognitively normal [CN] subjects and 86 patients with mild cognitive impairment [MCI]; mean age 72.6 years, and 48.1% men) were included. Participants with A+ had significantly higher proportions of apolipoprotein E (APOE) gene ɛ4 carriers than those with A–. Brain atrophy was observed in all groups of CN, whereas cognition decline was obvious in the A+T+N+ group. Compared to A–T–N–, MCI patients with A+T+N+ had faster cognition worsening and faster brain atrophy. In the whole cohort, A+T+N+ and A+T+N– participants were at higher risk of clinical progression.
Discussion
Plasma A/T/N biomarker profiles may predict AD pathology and clinical progression, indicating a potential role for plasma biomarkers in clinical trials. More research is warranted to develop a robust plasma AD framework.
Keywords: Alzheimer's disease, amyloid beta, mild cognitive impairment, neurofilament light, plasma, p‐tau181
1. BACKGROUND
Alzheimer's disease (AD) is characterized by abnormal amyloid deposition, tau aggregates, and neurodegeneration in the brain. 1 Accurate diagnosis and timely intervention at the early preclinical and prodromal stages of AD have become core aims of drug development, the feasibility of which depends heavily on selecting individuals who are at high risk of developing AD. 2 , 3 , 4 Existing clinical routine cannot fulfill accurate diagnosis of preclinical and prodromal AD. The updated 2018 Alzheimer's AT(N) research framework contributes greatly to the definition of AD, by grouping amyloidosis (A), tau pathology (T), and neurodegeneration (N) status using cerebrospinal fluid (CSF), positron emission tomography (PET), and structural imaging biomarkers. 4 , 5 This framework enables researchers to better define preclinical and prodromal AD, and provides prognostic information of clinical progression. 6 , 7 However, the PET technique is difficult to add into clinical routine practice due to high cost and radioactive burden, while magnetic resonance imaging (MRI) can only achieve part of the pathological definition. 3 CSF sampling is relatively invasive and requires skilled operators, so is difficult to use for large‐scale screening. 8 Therefore, inexpensive, readily available, and less invasive plasma markers have gradually become a research focus with a view to filling this gap.
Several plasma markers have been reported that could be used as biomarkers or predictors associated with brain amyloid burden, tau pathology, and neurodegeneration. Plasma amyloid beta (Aβ) has a positive correlation with its CSF level and a negative relationship with brain amyloid deposition. 9 In the preclinical or prodromal phase of AD, plasma Aβ1‐42 was observed to be moderately decreased whereas no change was detected in plasma Aβ1‐40 concentration. 9 A longitudinal monocentric cohort reported that individuals with abnormal Aβ PET presented a higher plasma Aβ1‐40/Aβ1‐42 ratio than those with normal Aβ PET. 10 Ovod et al. found a faster turnover rate of Aβ1‐42 compared to Aβ1‐40 and lower levels of Aβ1‐42/Aβ1‐40 in the peripheral blood of amyloid‐positive subjects, which was similar to findings in the CSF. 11 Plasma Aβ1‐42/Aβ1‐40 ratio has also been demonstrated to be able to discriminate between dementia due to AD and dementia not due to AD. 12 Through the immunoprecipitation with mass spectrometry technique, plasma Aβ1‐42/Aβ1‐40 showed high‐precision performance when predicting amyloid burden in the brain. 13 , 14 Plasma phospho‐tau (p‐tau) is currently under consideration to be implemented in clinical practice. It is found that AD patients have higher levels of plasma p‐tau than normal controls, and the combination of plasma and CSF tau could improve diagnostic accuracy. 15 Plasma p‐tau showed similar trajectory with the corresponding CSF p‐tau. 16 In addition, plasma p‐tau and its combination with plasma Aβ were both significantly correlated with tau deposition in the brain. 17 Plasma neurofilament light (NfL) level has been found to be increased in AD and correlated with cerebral hypometabolism, brain atrophy, and cognitive decline, using a large cohort from the Alzheimer's Disease Neuroimaging Initiative (ADNI). 18 , 19 In individuals with positive amyloid pathology, the increased level of plasma NfL correlates with hypometabolism presented by reduced fluorodeoxyglucose (FDG) uptake in AD‐related brain regions. 20 It is therefore suggested that plasma NfL could be used as a predictor to track neurodegeneration in AD. 19
These plasma markers showed favorable capacity of classification concord with PET or CSF biomarkers, which are promising to be implemented in the clinical practice of defining preclinical and prodromal AD. 21 , 22 Unlike the relatively mature AT(N) framework, 4 , 5 plasma research to date has been focused on a single parameter (Aβ1‐42, Aβ1‐42/Aβ1‐40 ratio, t‐tau, p‐tau, NfL, etc) or a purely diagnostic population (mild cognitive impairment [MCI], cognitively normal [CN], or AD dementia). 10 , 19 , 23 Therefore, the primary goal of this study was to investigate whether these plasma amyloid and neurodegeneration marker profiles could be combined to define AD pathology and predict long‐term clinical progression in elderly individuals without dementia.
2. METHODS
2.1. ADNI database
All participants in the cross‐sectional and longitudinal analyses of this study were enrolled from the ADNI. As a public–private project, the ADNI database was launched in 2003 with the primary goal to facilitate research on MCI and AD dementia. 24 It provides clinical and biological assessments such as CSF/blood biological markers; MRI/PET imaging; and demographic, genetic, and neuropsychological information, which can be combined to detect and track disease progression. This multi‐centered research project was approved by institutional review boards at each site and has obtained authorized written informed consent from participants. All data used in the study were downloaded from ADNI in June 2020.
2.2. Participants and cognitive assessments
Individuals from the ADNI database were included in the study if they provided data as follows: (1) plasma Aβ1‐42, Aβ1‐40, p‐tau181, and NfL levels; (2) amyloid PET, tau PET, FDG PET, and structural MRI; and (3) baseline and longitudinal neuropsychological assessments. Patients with AD dementia were diagnosed with a Mini‐Mental State Examination (MMSE) score between 20 and 26, a global Clinical Dementia Rating of 0.5 or 1, and a sum‐of‐boxes Clinical Dementia Rating (CDR‐SB) of 1.0 to 9.0, which met the National Institute of Neurological and Communication Disorders/Alzheimer's Disease and Related Disorders Association criteria. 25 Amnestic MCI subjects were diagnosed with an MMSE score of 24 to 30 and a CDR‐SB score of at least 0.5. Controls with normal cognition were defined as those who had an MMSE score of 24 or higher and a CDR‐SB score of 0 or 0.5.
The general cognition level of participants in the cohort was evaluated by MMSE and the ADAS‐Cog (Alzheimer Disease Assessment Scale‐Cognitive subscale) 11. 26 Clinical information was extracted from the latest merge document “ADNIMERGE.csv.” Longitudinal cognition changes were evaluated by ADNI composite measures for memory, executive functioning, and language domains. All composite cognition scores were downloaded from the document “UW Neuropsych Summary Scores.csv,” and lower scores indicated worse cognition status. Updated information of the ADNI cohort could be searched online (adni.loni.usc.edu).
2.3. Plasma marker measurements
The ADNI project provides multi‐centered data of plasma Aβ1‐42 and Aβ1‐40 levels, which vary across laboratories. To avoid experimental factors due to different methods for Aβ protein analysis, we only included the latest samples measured by immunoprecipitation in the Bateman Laboratory. 11 Aβ isoforms in human plasma were immunoprecipitated by the anti‐Aβ mid‐domain antibody on the KingFisher (Thermo) automated immunoprecipitation platform, and subsequently handled with Lys‐N protease and liquid chromatography tandem mass spectrometry (LC‐MS/MS). 11 Plasma p‐tau181 and NfL data were both provided by the Clinical Neurochemistry Laboratory, University of Gothenburg, Sweden. Through the Single Molecule array (Simoa) technique, plasma p‐tau181 was analyzed by an in‐house assay using a combination of two monoclonal antibodies (Tau12 and AT270). 27 Plasma NfL concentrations were generated by an in‐house ultra‐sensitive enzyme‐linked immunosorbent assay on a single molecule array platform (Quanterix Corp) in the Clinical Neurochemistry Laboratory of the University of Gothenburg, Sweden. 19 , 28
RESEARCH IN CONTEXT
Systematic review: The authors reviewed literature on plasma biomarkers for Alzheimer's disease (AD) using PubMed and online reports. Although many studies have reported promising markers of AD in plasma, most of them analyzed a single marker or a purely diagnostic group. Relevant publications are appropriately cited.
Interpretation: Our findings indicated that plasma amyloid, tau, and neurodegeneration biomarker profiles could predict AD pathology and clinical progression, supporting potential use of plasma biomarkers in clinical trials.
Future directions: This article proposed that plasma amyloid/tau/neurodegeneration (A/T/N) biomarker profiles could be used as candidate screening tools before applying the standard positron emission tomography or cerebrospinal fluid biomarkers. Replications in other larger cohorts with different populations are required to ascertain robust thresholds of these plasma biomarker profiles.
2.4. Neuroimaging methods
Amyloid PET data were acquired using florbetapir (AV‐45) tracer, and the summary data are regularly updated on ADNI. FreeSurfer (version 4.5.0) was used to segment the native‐space MRI scan for each individual. The mean tracer uptake of selected cortical and reference regions was calculated with the florbetapir scan applied to its corresponding MRI scan. Summary florbetapir standard uptake value ratios (SUVRs) were generated by averaging uptake ratios across four cortical regions (frontal, anterior cingulate, precuneus, and parietal cortex) and then normalizing it by the reference region (whole cerebellum). Brain tau deposit was measured via the flortaucipir (AV‐1451) processing method from the Helen Wills Neuroscience Institute, University of California Berkeley and Lawrence Berkeley National Laboratory. A composite metaROI (region of interest) of bilateral entorhinal, amygdala, fusiform, parahippocampal, inferior, and middle temporal regions were considered for tau PET assessment. 29 FDG PET was used to determine hypometabolic regions that were indicative of AD‐related pathological metabolic change. A set of metaROIs were developed based on literature review of FDG‐PET studies concerning AD and MCI. 23 , 30 The average counts of FDG PET across angular, temporal, and posterior cingulate regions were adopted. Data of structural MRI were obtained by the Siemens Trio scanner and estimates of selected region volume were measured using FreeSurfer software. In this study, we also selected hippocampal atrophy as a reference standard for neurodegenerative marker and calculated the hippocampal volume (HVa) adjusted by the intracranial volume for subsequent analyses. Specific calculation methods have been described in our previous report. 6
2.5. Cut‐offs of imaging biomarkers for classification and diagnostic performance of plasma profiles
Plasma markers were dichotomized by defining cut‐offs that could best discriminate PET or structural MRI abnormal subjects from normal subjects. We selected plasma Aβ1‐42/Aβ1‐40 concentration ratio as the biological marker for brain amyloidosis (A), plasma p‐tau181 level for tau pathology (T), and plasma NfL level for neurodegeneration (N). To optimize concordance with PET/MRI classification, 4 , 5 cut‐offs for plasma A/T/N markers were determined based on Youden index, using receiver operating characteristic (ROC) analyses. For amyloid PET, higher than 1.11 SUVR was considered to have abnormal cortical amyloid deposition. 6 For tau PET, higher than 1.33 SUVR was considered having tau pathology. 29 The well‐acknowledged cut‐off of 6723 mm3 for adjusted hippocampus volume or that of 1.21 for FDG PET were considered to be associated with neurodegeneration (see supporting information). 6 , 23 , 31 Logistic regression model using age, sex, years of education, apolipoprotein E (APOE) genotype was performed to assess the combined diagnostic capability.
2.6. Statistical analysis
All statistical analyses were carried out using R software (version 3.4.4) and GraphPad (Prism 7.04). ROC curves and logistic regression analyses were conducted by R software using the “OptimalCutpoints” and “pROC” packages. Cut‐off points calculated were used to classify individuals as with abnormal AD pathological changes (+) or without (–) on each predicting variable. Characteristics of the cohort were presented as mean (standard deviation [SD]) or number (percentage [%]) as appropriate. Differences for continuous variables cross the eight plasma A/T/N groups were assessed by the Kruskal‐Wallis test and those for categorical data were evaluated by the chi‐square test or Cochran‐Mantel‐Haenszel test. We then computed linear mixed‐effects models with clinical outcomes to evaluate the longitudinal change in A–T–N–, A+T–N–, A+T+N–, and A+T+N+ individuals. 29 Variables were log transformed and standardized. Unadjusted Kaplan‐Meier plots were constructed to assess the risk of progressing to CDR‐SB > or = 0.5 in CN individuals, and the risk of progressive cognitive deterioration in MCI patients. Progressive cognitive deterioration was defined as: (1) diagnosed as dementia, (2) MMSE score of the last visit below 24, or (3) the decline of MMSE score between the first and last visits of more than three. 32 In addition, we ran univariate and multivariate Cox proportional‐hazards models to predict disease progression. Age, sex, years of education, and APOE genotype were included as covariates in the multivariate model. The statistical significance of all tests was set at a two‐sided P value < .05.
3. RESULTS
3.1. Cut‐offs for plasma A/T/N marker profiles
Participants with eligible plasma and PET/MRI data were enrolled for evaluation of cut‐off points after quality control. Descriptive characteristics of this cohort are described in the supporting information. ROC analyses of PET/MRI‐based abnormal cases versus normal subjects provided statistically optimal cut‐offs for each measure in the plasma (Figure 1 and supporting information). The greatest ROC area under the curve (AUC) for plasma Aβ1‐42/Aβ1‐40 concentration ratio was 0.830 (95% confidence interval [CI] 0.769 to 0.891; sensitivity 68.9% and specificity 87.1%), with a cut‐off value of 0.113 . It improved in logistic regression model analysis using age, sex, years of education, APOE ε4 carrier status as variables (AUC 0.866, Figure 1A; see supporting information). The cut‐off for plasma p‐tau181 was 17.3 pg/mL with an AUC of 0.695 (95% CI 0.606 to 0.783; sensitivity 68.6% and specificity 67.9%). This cut‐off value had a promising negative predictive value of 91.1%, and a logistic regression AUC of 0.716 (Figure 1B; see supporting information). The resultant optimum cut‐offs for plasma NfL level were 32.1 pg/mL based on adjusted hippocampus volume (AUC 0.699, 95% CI 0.67 to 0.728; sensitivity 73.4% and specificity 56.1%) and 31.0 pg/mL on FDG PET (AUC 0.654, 95% CI 0.622 to 0.686; sensitivity 73.7% and specificity 49.9%). Adapting logistic regression models, the AUCs increased to 0.767 (Figure 1C) and 0.706, respectively (see supporting information). We selected the NfL cut‐off level calculated from adjusted hippocampus volume for N due to its better diagnostic performance.
FIGURE 1.
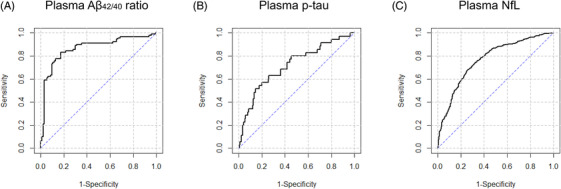
ROC curves for plasma biomarker profiles based on PET or structural imaging. A, ROC curve generated from cut‐off analysis of plasma Aβ1‐42/Aβ1‐40 concentration ratios, based on amyloid PET abnormal versus normal. B, ROC curve generated from cut‐off analysis of plasma p‐tau181 concentration, based on tau PET abnormal versus normal. C, ROC curve generated from cut‐off analysis of plasma NfL level, based on adjusted hippocampus volume abnormal versus normal (by structural imaging MRI). Abbreviations: Aβ, amyloid beta; AUC, area under the ROC curve; NfL, neurofilament light; PET, positron emission tomography; ROC, receiver operating characteristic curve
3.2. Characteristics of the cohort and each group based on plasma A/N classification
A total of 183 older adults without AD dementia (97 CN and 86 MCI) were finally included in the study (see Figure 2 for a flowchart of the screening process). The cohort had mean (SD) age of 72.6 (6.9) years (range from 55.5–88.3 years), 48.1% men, 37.7% APOE ε4 carriers ,and average years of education of 16.5 (2.6). Table 1 summarizes the clinical and demographic characteristics of each plasma A/T/N group and presents statistical differences across the eight groups. Among these, 57 subjects were categorized A–T–N–, 12 A+T–N–, 14 A+T+N–, and 26 A+T+N+ (Table 1).
FIGURE 2.
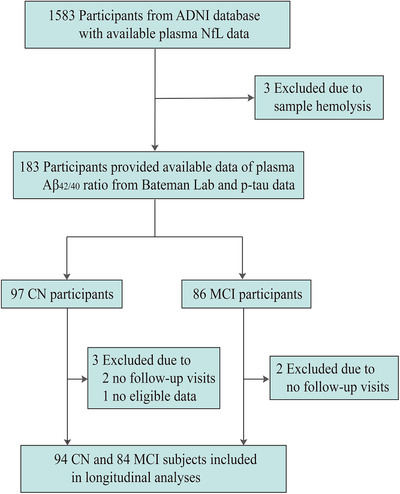
Flowchart. Abbreviations: ADNI, Alzheimer's Disease Neuroimaging Initiative; CN, cognitively normal; MCI, mild cognitive impairment; NfL, neurofilament light
TABLE 1.
Descriptive features of the 183 participants included in this study
| A–T–N– | A–T+N– | A–T–N+ | A–T+N+ | A+T‐N– | A+T+N– | A+T–N+ | A+T+N+ | P | |
|---|---|---|---|---|---|---|---|---|---|
| CN | |||||||||
| N | 30 (30.9) | 7 (7.2) | 15 (15.5) | 7 (7.2) | 6 (6.2) | 5 (5.2) | 12 (12.4) | 15 (15.5) | — |
| Age, years | 69.7 (5.9) | 72.4 (8.3) | 76.3 (3.7) | 76.0 (4.6) | 69.3 (3.1) | 73.3 (4.1) | 77.1 (5.6) | 78.0 (5.7) | < .001 |
| Sex, male | 15 (50) | 3 (42.9) | 5 (33.3) | 2 (28.6) | 2 (33.3) | 1 (20.0) | 8 (66.7) | 8 (53.3) | .526 |
| Education, years | 17.1 (2.5) | 16.9 (2.3) | 16.3 (2.8) | 16.9 (2.8) | 17.0 (1.7) | 15.8 (1.9) | 15.6 (2.6) | 16.9 (2.9) | .744 |
| APOE ɛ4 carriers | 5 (16.7) | 4 (57.1) | 3 (20.0) | 2 (28.6) | 4 (66.7) | 4 (80.0) | 5 (41.7) | 4 (26.7) | .030 |
| ADAS‐cog 11 score | 5.3 (2.4) | 3.1 (2.9) | 6.7 (2.8) | 5.4 (2.1) | 4.3 (2.0) | 7.0 (5.3) | 7.4 (2.5) | 6.7 (2.6) | .026 |
| MMSE score | 28.8 (1.3) | 29.4 (0.5) | 29.5 (1.1) | 28.9 (1.6) | 29.2 (0.8) | 28.4 (1.1) | 28.2 (1.7) | 29.4 (0.7) | .064 |
| Progressors a | 0 | 0 | 5 (33.3) | 1 (14.3) | 0 | 2 (40.0) | 5 (41.7) | 6 (40.0) | — |
| MCI | |||||||||
| N | 27 (31.4) | 1 (1.2) | 14 (16.3) | 9 (10.5) | 6 (7.0) | 9 (10.5) | 9 (10.5) | 11 (12.8) | — |
| Age, years | 67.7 (6.0) | 62 | 73.7 (7.3) | 75.0 (7.4) | 71.2 (7.9) | 67.7 (5.7) | 76.9 (8.2) | 73.0 (6.3) | .009 |
| Sex, male | 14 (51.9) | 0 | 8 (57.1) | 4 (44.4) | 3 (50.0) | 6 (66.7) | 5 (55.6) | 4 (36.4) | .858 |
| Education, years | 16.4 (2.4) | 13 | 16.3 (3.0) | 15.6 (2.4) | 18.7 (1.6) | 17.9 (1.6) | 15.9 (3.1) | 15.1 (2.8) | .078 |
| APOE ɛ4 carriers | 8 (29.6) | 0 | 3 (21.4) | 3 (33.3) | 5 (83.3) | 6 (66.7) | 4 (44.4) | 9 (81.8) | .011 |
| ADAS‐cog 11 score | 7.0 (3.4) | 5 | 6.9 (2.1) | 8.8 (3.4) | 7.2 (3.1) | 7.7 (3.9) | 8.7 (2.7) | 10.2 (3.5) | .172 |
| MMSE score | 29.1 (0.8) | 28 | 27.6 (2.2) | 27.4 (1.4) | 28.2 (2.1) | 28.2 (2.0) | 28.1 (2.1) | 28.2 (2.1) | .169 |
| Progressors b | 2 (7.4) | 0 | 3 (21.4) | 2 (22.2) | 0 | 2 (22.2) | 1 (11.1) | 3 (27.3) | ‐ |
Note: Data were presented as mean (SD) or number (percentage) as appropriate.
Abbreviations: A–, amyloid normal defined by plasma Aβ1‐42/Aβ1‐40 ratio; A+, amyloid abnormal defined by plasma Aβ1‐42/Aβ1‐40 ratio; ADAS‐Cog, Alzheimer Disease Assessment Scale‐Cognitive subscale; APOE, apolipoprotein E; CN, cognitively normal; MCI, mild cognitive impairment; MMSE, Mini‐Mental State Examination; N–, without neurodegeneration defined by plasma NfL; N+, with neurodegeneration defined by plasma NfL; T–, without tau pathology defined by plasma p‐tau181; T+, with tau pathology defined by plasma p‐tau181.
Progressors indicated those who conversed from CN to MCI.
Progressors indicated those who conversed from MCI to AD dementia.
3.3. Prediction of cognitive decline and brain atrophy
Longitudinal changes in cognition and raw brain region volume across the four groups (A–T–N–, A+T–N–, A+T+N–, and A+T+N+) were analyzed, adjusted for age, sex, years of education, and APOE genotype.
Among CN participants, decreases of all three cognitive composite scores (ADNI memory, language, and executive functioning) were observed in the A+T+N+ group (Figure 3A). Comparisons between groups showed that A+T+N+ individuals had faster decline rates than A–T–N– participants (group‐wise difference: –7.16%, 95% CI –11.31% to –3.02%, P = .001). Reduction of the whole brain or hippocampal volumes was observed in all four groups, whereas significant atrophy of the medial temporal region was only found in A+T–N– and A+T+N+ groups (Figure 3B). Differences in annual atrophy rates were not found between groups in pairs (P > .05).
FIGURE 3.
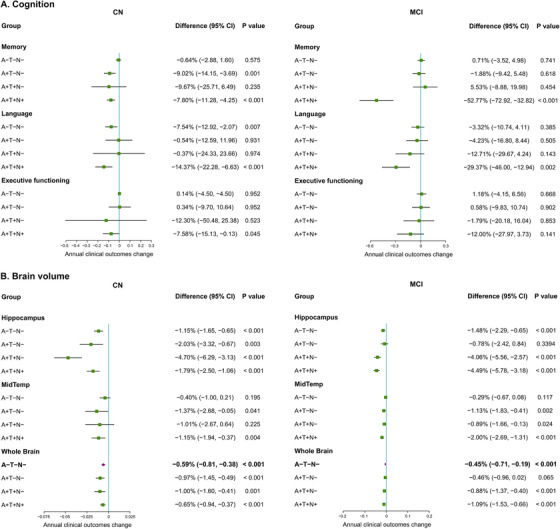
Change in clinical outcomes among four plasma A/T/N groups based on linear mixed‐effects regression models. A. Change in cognition among four plasma A/T/N groups. B. Change in brain volume among four plasma A/T/N groups. Analyses for clinical indicators were adjusted for age, sex, years of education, and APOE ε4 carriage. Change in clinical outcomes expressed as an annual percentage cognitive function scores and volume change, with 95% CI and P value. Abbreviations: A+, amyloid abnormal defined by plasma Aβ1‐42/Aβ1‐40 ratio; APOE, apolipoprotein E; CN, cognitively normal; MCI, mild cognitive impairment; N–, without neurodegeneration defined by plasma NfL; N+, with neurodegeneration defined by plasma NfL; T–, without tau pathology defined by plasma p‐tau181; T+, with tau pathology defined by plasma p‐tau181.
In patients with MCI, the A+T+N+ group showed obvious decreases in ADNI memory and language composite scores (Figure 3A), which were faster compared to A–T–N− patients (memory domain: −53.49%, 95% CI −74.04% to −33.01%, P < .001; and language domain: −26.05%, 95% CI −44.08% to −8.00%, P = .006). Annual decreases in the hippocampal and whole brain volumes were detected in all groups except for the A+T–N− group (Figure 3B). Medial temporal atrophy was observed in A+T–N−, A+T+N−, and A+T+N+ groups, but not in A–T–N− patients (Figure 3B). Furthermore, A+T+N+ patients had faster change rates in hippocampus, medial temporal regions, and the whole brain, compared to A–T–N− individuals (hippocampus: −3.01%, 95% CI −4.54% to −1.32%, P = .001; medial temporal region: −1.70%, 95% CI −2.48% to −0.93%, P < .001; whole brain: −0.63%, 95% CI −1.15% to −0.12%, P = .017). In addition,, the A+T+N– group showed faster rates of hippocampal atrophy than the A–T–N− group (−2.58%, 95% CI −4.29% to −0.85%, P = .002).
3.4. Prediction of clinical progression
Figure 4 exhibits the results of the Kaplan‐Meier analyses with log‐rank tests across four groups (A–T–N−, A+T–N−, A+T+N−, and A+T+N+ groups). Survival curves showed differences across four groups in both CN and MCI participants (log‐rank P < .0001 and P = .0002, respectively). Compared to A–T–N− individuals, A+T+N− and A+T+N+ participants presented faster decline of the non‐progression proportion to CDR‐SB > or = 0.5 (P = .005 and P < .0001, respectively; Figure 4A). Among MCI patients, A+T+N− and A+T+N+ participants had higher risk of having progressive cognitive deterioration than A–T–N− patients during the 8‐year follow‐up period (P = .001 and P < .0001, respectively; Figure 4B). The A+T‐N– group did not show any difference with the A–T–N− group. In addition, we did not observe difference between A+T+N− and A+T+N+ groups. In both unadjusted and adjusted Cox proportional‐hazards models with A–T–N− groups as reference, A+T+N− and A+T+N+ individuals presented increased progression risks (Table 2).
FIGURE 4.
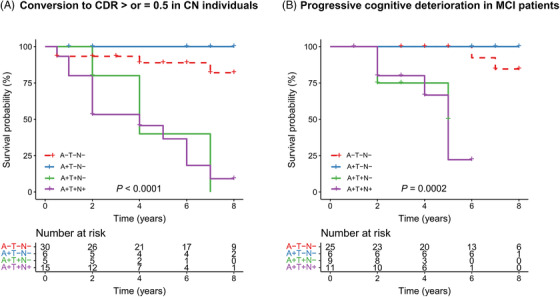
Kaplan‐Meier curves showing cumulative probability of disease progression. A. Kaplan‐Meier curves showing cumulative probability of progressing to CDR > or = 0.5 among cognitively normal participants. B. Kaplan‐Meier curves showing cumulative probability of progressive cognitive deterioration in patients with MCI. Abbreviations: A−, amyloid normal defined by plasma Aβ1‐42/Aβ1‐40 ratio; A+, amyloid abnormal defined by plasma Aβ1‐42/Aβ1‐40 ratio; MCI, mild cognitive impairment; N−, without neurodegeneration defined by plasma NfL; N+, with neurodegeneration defined by plasma NfL; T−, without tau pathology defined by plasma p‐tau181; T+, with tau pathology defined by plasma p‐tau181
TABLE 2.
Progression risk of participants in the study
| Group | Crude | Adjusted | ||
|---|---|---|---|---|
| Hazard ratio (95% CI) a | P | Hazard ratio (95% CI) b | P | |
| Risk of progressing to CDR > or = 0.5 in CN subjects | ||||
| A–T–N− | Reference | — | — | — |
| A+T‐N− | 0 (0 ‐ Inf.) | .998 | 0 (0 ‐ Inf.) | .998 |
| A+T+N− | 7.32 (1.60–33.53) | .010 | 9.68 (1.50–62.58) | .017 |
| A+T+N+ | 10.20 (3.21–32.42) | < .001 | 16.08 (3.09–83.67) | .001 |
| Risk of progressive cognitive deterioration in MCI patients | ||||
| A–T–N− | Reference | — | — | — |
| A+T–N− | 0 (0−Inf.) | .998 | 0 (0−Inf.) | .999 |
| A+T+N− | 20.65 (1.90–224.20) | .013 | 16.87 (1.05–269.81) | .046 |
| A+T+N+ | 25.70 (2.83–233.50) | .004 | 18.07 (1.19–273.62) | .037 |
Abbreviations: A−, amyloid normal defined by plasma Aβ1‐42/Aβ1‐40 ratio; A+, amyloid abnormal defined by plasma Aβ1‐42/Aβ1‐40 ratio; CI, confidence interval; CN, cognitively normal; MCI, mild cognitive impairment; N−, without neurodegeneration defined by plasma NfL; N+, with neurodegeneration defined by plasma NfL; T−, without tau pathology defined by plasma p‐tau181; T+, with tau pathology defined by plasma p‐tau181. P values which indicated statistical significance were displayed in bold.
Hazard ratio (95% CI) calculated using Cox regression analyses.
Hazard ratio (95% CI) calculated using Cox regression analyses and corrected for baseline age, sex, APOE ε4 status, and years of education.
4. DISCUSSION
There is an urgent need for blood‐based biomarkers in defining early preclinical or prodromal stages of AD. The present study showed that the AD pathology could be defined by plasma amyloid, tau, and neurodegeneration biomarker profiles, and these profiles exhibited promising accuracy for predicting clinical progression in older adults without dementia. Our data proved that plasma Aβ1‐42/Aβ1‐40 concentration ratio, p‐tau181, and NfL level had relatively good diagnostic capacities in predicting cerebral amyloid, tau pathologies, and neurodegeneration, respectively. Defined by these plasma profiles, individuals who had amyloid and tau pathologies, with or without neurodegeneration, presented a significantly higher risk of clinical progression compared to those without. Plasma biomarker profiles provided modest but reliable complementary information beyond clinical and genetic data, and possess great potential in the population screening of clinical trials.
It is critical to classify the plasma candidate marker into abnormal or normal. We used the Youden index in ROC analyses to define eligible plasma cut‐offs, and further tested them in the logistic regression model. 33 , 34 Adapting amyloid PET as the reliable standard of truth, plasma Aβ1‐42/Aβ1‐40 ratio provided relatively good diagnostic performance with balanced sensitivity and specificity. The resultant value was slightly lower than that previously reported using the same measurements (ratio < 0.1243 as abnormal), 11 which may be due to different samples. The optimal cut‐off point for plasma p‐tau181 provided favorable negative predictive value (91.1%, see supporting information), which supported its promising role for target population screening in clinical practice. In this study, plasma NfL presented moderately good performance in predicting cerebral neurodegeneration defined by FDG PET or hippocampal volume. The cut‐off for plasma NfL level was higher than that generated in a dataset combined diagnosis and CSF biomarkers (concentration > 25.7 pg/mL as abnormal). 35 Because our cut‐offs set stricter entry criteria for abnormality of plasma biomarker profiles, it tended to be more conservative and led to lower proportions of A+, T+, or N+.
Our study showed a relative concordance of plasma biomarker definition with the CSF/PET classification. Restricted by the small sample size, our cohort did not show difference in A+/A− proportions between CN and MCI. But participants with A+ had significantly higher proportions of APOE ɛ4 carriers than those with A−, whether in CN (44.7% vs 23.7%) or MCI individuals (68.6% vs 27.5%). This was in line with investigations using CSF/PET definitions on cognitively normal subjects and MCI patients. 32 , 36 , 37 The proportion of the preclinical AD defined by plasma profiles was 39.2%, similarly with that in populations classified by CSF Aβ1‐42, t‐tau, and p‐tau. 33 Further, in our cohort, older adults without dementia displayed a high proportion (50.3%) of plasma abnormal neurodegeneration biomarkers. Though the proportion ratio was slightly higher than previous studies on CSF/PET, 38 , 39 our findings contributed to the extension of the assumption that neurodegenerative pathology was harbored among a large proportion of cognitively normal older adults, without abnormal amyloid burden. 38 In addition, participants with N+ had more clinical progressors (converse from CN to MCI, or from MCI to AD dementia) compared to those with N−. Using plasma data from ADNI, Mattsson et al. have found that the faster increment of plasma NfL was correlated with faster changes of CSF biomarkers presenting neuronal injury, faster rates of brain atrophy and hypometabolism, and faster rates of cognitive decline. 18 , 19 It is reasonable to believe that plasma NfL, concordant with CSF or imaging biomarkers, was potentially applicable in supplementing the biological definition of cognitive disorders. These findings support the validity of plasma biomarker‐based classification of AD pathology.
Longitudinally, plasma biomarker profiles showed good predictive value in clinical progression. Brain atrophy emerged over time in older adults without dementia. In our study, all groups illustrated annual changes of brain atrophy during the 8‐year follow‐up period. Though groups in CN individuals did not prove any difference in decline rates, MCI patients with A+T+N+ presented significantly faster rates of atrophy in hippocampal, medial temporal regions, and the whole brain volume, compared to A–T–N− patients. MCI A+T+N− patients possessed comparable atrophy rates of the hippocampus to A+T+N+, and obviously faster than A–T–N−. In addition, A+T+N+ participants presented significant cognition decline in memory and language domains. Furthermore, MCI patients with A+T+N+ proved to worsen much faster than A–T–N− patients in memory and language performance, whereas CN subjects with A+T+N+ showed a faster rate of decline in the memory aspect. These outcomes were consistent with our previous findings using CSF/PET biomarkers for the definition of AD continuum profiles, 6 which supported the implementation of plasma biomarkers in clinical trials. Other research, which focused only on “A/N” profiles defined by CSF/PET biomarkers, reported similar but not identical findings. 33 , 40 , 41 , 42 A study on MCI found significantly greater changes of MMSE and hippocampus volume in A+N+ individuals compared to A–N−. 32 Soldan et al. reported a significantly greater rate of cognitive decline in stage 2 (with both amyloid pathology and tau‐related neurodegeneration) of preclinical AD compared to other groups. 40 Differences between brain atrophy and cognitive decline could be partially explained by the fact that cognitive symptoms emerged later than AD‐related pathological features. 43 , 44 More longitudinal case‐control studies are required to evaluate whether cortical or hippocampal atrophy were earlier and more sensitive indicators for progression of dementia than cognition tests.
Our data indicated that plasma amyloid, tau, and neurodegeneration biomarker profiles were associated with the high risk of clinical progression. A+T+N+ participants had the largest proportions of diagnostic conversion (from CN to MCI, or from MCI to AD dementia), and were at greater risk of clinical progression in comparison to those with A–T–N−. These were in accord with other CSF/PET studies, 33 , 45 including the previous “A/T/N” findings from our team. 6 As we used the combination of plasma amyloid, tau, and the plasma NfL as neurodegenerative biomarkers, these findings would be more precise in assessing the predictive capacity of clinical progression compared to other research that adapted one or two of these profiles. 32 , 40 , 45
Limitations are acknowledged in the present study. One is that the study lacked follow‐up data of plasma markers, which restricted us from investigating whether these plasma markers parallel their CSF or PET equivalent, which would be useful in efficacy monitoring. Also, the relatively small sample size reduced the power of the tests. This was due to the fact that only a small proportion of ADNI participants had the plasma amyloid data analyzed by the immunoprecipitation mass spectrometry technique. It could be difficult to add this measuring method into clinical routine, thus accurate, alternative detection methods are needed in future clinical practice. Also, the population of this ADNI cohort was predominantly White, which limited generalizability to other racial/ethnic groups. As plasma NfL is associated with age, it would cause individuals at the lower end of ages in this cohort to possibly have been under‐categorized as N+ and those at the upper end over‐categorized as N+. Replications in other, larger cohorts with different population are required to ascertain robustness thresholds of these plasma biomarker profiles.
In conclusion, the present study provided evidence that plasma amyloid, tau pathology, and neurodegeneration biomarker profiles could predict AD pathology. We further demonstrated the predictive capacity of these profiles in clinical progression, supporting the potential use of plasma biomarkers in clinical trials. Future investigations are needed on longitudinal changes of plasma biomarkers over time and robust plasma biomarker framework.
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.
Supporting information
Supplementary information
ACKNOWLEDGMENTS
This study was supported by grants from the National Natural Science Foundation of China (91849126, 81571245, and 81771148), the National Key R&D Program of China (2018YFC1314700), Shanghai Municipal Science and Technology Major Project (No.2018SHZDZX01) and ZHANGJIANG LAB, Tianqiao and Chrissy Chen Institute, and the State Key Laboratory of Neurobiology and Frontiers Center for Brain Science of Ministry of Education, Fudan University. Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI; National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). Data used in the preparation for this article were derived from the ADNI database (http://adni.loni.usc.edu/). The authors express appreciation to contributors of Alzheimer's Disease Neuroimaging Initiative (ADNI) database. ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie; Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Shen X‐N, Li J‐Q, Wang H‐F, et al. Plasma amyloid, tau, and neurodegeneration biomarker profiles predict Alzheimer's disease pathology and clinical progression in older adults without dementia. Alzheimer's Dement. 2020;12:e12104 10.1002/dad2.12104
REFERENCES
- 1. Scheltens P, Blennow K, Breteler MM, et al. Alzheimer's disease. Lancet. 2016;388:505‐517. [DOI] [PubMed] [Google Scholar]
- 2. Qian J, Wolters FJ, Beiser A, et al. APOE‐related risk of mild cognitive impairment and dementia for prevention trials: an analysis of four cohorts. PLoS Med. 2017;14:e1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hansson O, Seibyl J, Stomrud E, et al. CSF biomarkers of Alzheimer's disease concord with amyloid‐beta PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 2018;14:1470‐1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jack CR, Jr , Bennett DA, Blennow K, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. 2016;87:539‐547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yu JT, Li JQ, Suckling J, et al. Frequency and longitudinal clinical outcomes of Alzheimer's AT(N) biomarker profiles: a longitudinal study. Alzheimers Dement. 2019;15:1208‐1217. [DOI] [PubMed] [Google Scholar]
- 7. Soldan A, Pettigrew C, Fagan AM, et al. ATN profiles among cognitively normal individuals and longitudinal cognitive outcomes. Neurology. 2019;92:e1567‐e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jack CR, Jr , Bennett DA, Blennow K, et al. NIA‐AA Research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Janelidze S, Stomrud E, Palmqvist S, et al. Plasma β‐amyloid in Alzheimer's disease and vascular disease. Sci Rep. 2016;6:26801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vergallo A, Megret L, Lista S, et al. Plasma amyloid beta 40/42 ratio predicts cerebral amyloidosis in cognitively normal individuals at risk for Alzheimer's disease. Alzheimers Dement. 2019;15:764‐775. [DOI] [PubMed] [Google Scholar]
- 11. Ovod V, Ramsey KN, Mawuenyega KG, et al. Amyloid beta concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement. 2017;13:841‐849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vogelgsang J, Shahpasand‐Kroner H, Vogelgsang R, Streit F, Vukovich R, Wiltfang J. Multiplex immunoassay measurement of amyloid‐beta42 to amyloid‐beta40 ratio in plasma discriminates between dementia due to Alzheimer's disease and dementia not due to Alzheimer's disease. Exp Brain Res. 2018;236:1241‐1250. [DOI] [PubMed] [Google Scholar]
- 13. Nakamura A, Kaneko N, Villemagne VL, et al. High performance plasma amyloid‐beta biomarkers for Alzheimer's disease. Nature. 2018;554:249‐254. [DOI] [PubMed] [Google Scholar]
- 14. Schindler SE, Bollinger JG, Ovod V, et al. High‐precision plasma β‐amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019;93:e1647‐e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fossati S, Ramos Cejudo J, Debure L, et al. Plasma tau complements CSF tau and P‐tau in the diagnosis of Alzheimer's disease. Alzheimer's & dementia (Amsterdam, Netherlands: ). 2019;11:483‐492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Palmqvist S, Insel PS, Stomrud E, et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer's disease. EMBO Mol Med. 2019;11:e11170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Park JC, Han SH, Yi D, et al. Plasma tau/amyloid‐beta1‐42 ratio predicts brain tau deposition and neurodegeneration in Alzheimer's disease. Brain. 2019;142:771‐786. [DOI] [PubMed] [Google Scholar]
- 18. Mattsson N, Andreasson U, Zetterberg H, Blennow K. Association of plasma neurofilament light with eurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2017;74:557‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mattsson N, Cullen NC, Andreasson U, Zetterberg H, Blennow K. Association between longitudinal plasma neurofilament light and neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2019;76:791‐799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Benedet AL, Ashton NJ, Pascoal TA, et al. Plasma neurofilament light associates with Alzheimer's disease metabolic decline in amyloid‐positive individuals. Alzheimers Dement (Amst) 2019;11:679‐689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Verberk IMW, Slot RE, Verfaillie SCJ, et al. Plasma amyloid as prescreener for the earliest Alzheimer pathological changes. Ann Neurol. 2018;84:648‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Palmqvist S, Janelidze S, Stomrud E, et al. Performance of fully automated plasma assays as screening tests for Alzheimer disease‐related β‐amyloid status. JAMA Neurol. 2019;76:1060‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Landau SM, Harvey D, Madison CM, et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology. 2010;75:230‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weiner MW, Veitch DP, Aisen PS, et al. 2014 Update of the Alzheimer's Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement. 2015;11:e1‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kolibas E, Korinkova V, Novotny V, Vajdickova K, Hunakova D. ADAS‐cog (Alzheimer's Disease Assessment Scale‐cognitive subscale)–validation of the Slovak version. Bratisl Lek Listy. 2000;101:598‐602. [PubMed] [Google Scholar]
- 27. Karikari TK, Pascoal TA, Ashton NJ, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer's disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020;19:422‐433. [DOI] [PubMed] [Google Scholar]
- 28. Gisslén M, Price RW, Andreasson U, et al. Plasma concentration of the neurofilament light protein (NFL) is a biomarker of CNS injury in HIV infection: a cross‐sectional study. EBioMedicine. 2016;3:135‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jack CR, Jr , Wiste HJ, Therneau TM, et al. Associations of amyloid, tau, and neurodegeneration biomarker profiles with rates of memory decline among individuals without dementia. JAMA. 2019;321:2316‐2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Landau SM, Harvey D, Madison CM, et al. Associations between cognitive, functional, and FDG‐PET measures of decline in AD and MCI. Neurobiol Aging. 2011;32:1207‐1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mormino EC, Betensky RA, Hedden T, et al. Synergistic effect of β‐amyloid and neurodegeneration on cognitive decline in clinically normal individuals. JAMA Neurol. 2014;71:1379‐1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Caroli A, Prestia A, Galluzzi S, et al. Mild cognitive impairment with suspected nonamyloid pathology (SNAP): prediction of progression. Neurology. 2015;84:508‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vos SJ, Xiong C, Visser PJ, et al. Preclinical Alzheimer's disease and its outcome: a longitudinal cohort study. Lancet Neurol. 2013;12:957‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shaw LM, Vanderstichele H, Knapik‐Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lewczuk P, Ermann N, Andreasson U, et al. Plasma neurofilament light as a potential biomarker of neurodegeneration in Alzheimer's disease. Alzheimers Res Ther. 2018;10:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Knopman DS, Jack CR, Jr , Wiste HJ, et al. Brain injury biomarkers are not dependent on beta‐amyloid in normal elderly. Ann Neurol. 2013;73:472‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Petersen RC, Aisen P, Boeve BF, et al. Mild cognitive impairment due to Alzheimer disease in the community. Ann Neurol. 2013;74:199‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wirth M, Villeneuve S, Haase CM, et al. Associations between Alzheimer disease biomarkers, neurodegeneration, and cognition in cognitively normal older people. JAMA Neurol. 2013;70:1512‐1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jack CR, Jr , Knopman DS, Weigand SD, et al. An operational approach to National Institute on Aging‐Alzheimer's Association criteria for preclinical Alzheimer disease. Ann Neurol. 2012;71:765‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Soldan A, Pettigrew C, Cai Q, et al. Hypothetical preclinical Alzheimer disease groups and longitudinal cognitive change. JAMA Neurol. 2016;73:698‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mormino EC, Betensky RA, Hedden T, et al. Synergistic effect of beta‐amyloid and neurodegeneration on cognitive decline in clinically normal individuals. JAMA Neurol. 2014;71:1379‐1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wirth M, Oh H, Mormino EC, Markley C, Landau SM, Jagust WJ. The effect of amyloid beta on cognitive decline is modulated by neural integrity in cognitively normal elderly. Alzheimers Dement. 2013;9:687‐698.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jack CR, Jr , Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gordon BA, Blazey TM, Su Y, et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer's disease: a longitudinal study. Lancet Neurol. 2018;17:241‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Knopman DS, Jack CR, Jr , Wiste HJ, et al. Short‐term clinical outcomes for stages of NIA‐AA preclinical Alzheimer disease. Neurology. 2012;78:1576‐1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information