

Abstract
Phage‐displayed synthetic antibody (Ab) repertoires have become a major source of affinity reagents for basic and clinical research. Specific Abs identified from such libraries are often screened as fragments antigen binding (Fabs) produced in bacteria, and those with desired biochemical characteristics are reformatted for production as full‐length immunoglobulin G (IgG) in mammalian cells. The conversion of Fabs to IgGs is a cumbersome and often rate‐limiting step in the development of Abs. Moreover, biochemical properties required for lead IgG development are not always shared by the Fabs, and these issues are not uncovered until a significant effort has been spent on Abs that ultimately will not be useful. Thus, there is a need for simple and rapid techniques to convert phage‐displayed Fabs to IgGs at an early stage of the Ab screening process. We report the generation of a highly diverse phage‐displayed synthetic single‐chain Fab (scFab) library, in which the light and heavy chains were tethered with an optimized linker. Following selection, pools of scFabs were converted to single‐chain IgGs (scIgGs) en masse, enabling facile screening of hundreds of phage‐derived scIgGs. We show that this approach can be used to rapidly screen for and select scIgGs that target cell‐surface receptors, and scIgGs behave the same as conventional IgGs.
Keywords: batch cloning, high‐throughput antibody production, recombinant antibody, scFab, scIgG
Abbreviations
- Ab
antibody
- Abs
antibodies
- CDR
complement determining region
- Fab
fragment antigen binding
- Fc
fragment crystallizable
- IgG
immunoglobulin G
- ORF
open reading frame
- scFab
single‐chain Fab
- scFv
single‐chain variable fragment
- scIgG
single‐chain IgG
- SPR
surface plasmon resonance
1. INTRODUCTION
Phage‐displayed antibody (Ab) libraries have revolutionized the development of affinity reagents and therapeutics by providing a way to rapidly select specific, high‐affinity Abs without the use of animals. While the fragment antigen binding (Fab) format is most often employed, other displayed Ab formats include single‐chain variable fragments (scFvs), 1 autonomous heavy chain variable (VH) domains, 2 , 3 , 4 minibodies, 5 and diabodies, 6 , 7 and each comes with advantages and limitations. Regardless of format, all phage‐displayed libraries provide the crucial advantage of linking an Ab to its encoding gene. The sequence diversity of these libraries can be natural, synthetic, or a mix of the two. 8 , 9 Utilizing the ability to precisely control antigen selection conditions, Abs can be developed against difficult antigens, including RNA, specific protein conformations, protein complexes, and proteins displayed on cells. 9 , 10 Moreover, phage display is very amenable to high‐throughput methods that enable rapid development of Abs for many antigens in parallel. 11 , 12 Because of this versatility, phage‐displayed libraries can generate Abs that can be used as both diagnostic tools and therapeutic leads.
For therapeutic development, the dominant Ab format is the full‐length immunoglobulin G (IgG) comprised of covalently associated heavy and light chains. Compared to monomeric Ab fragments, IgGs typically exhibit higher affinities due to bivalency and long serum half‐lives due to recognition of the crystallizable fragment (Fc) by the recycling neonatal Fc receptor. 13 , 14 However, the functional differences between Fabs and IgGs can be advantageous in certain applications. Fabs are smaller, providing better potential resolution in immunofluorescence and, since they do not interact with the neonatal Fc receptor, they are more quickly cleared from the body, making them excellent in vivo imaging agents. 15 , 16 Thus, an ideal phage‐displayed library should allow for facile reformatting as either Fab or IgG, depending on the intended use.
The standard workflow to produce an IgG from a Fab is to first sequence phage‐displayed Fab clones with desirable binding characteristics, and subsequently, to subclone the DNA sequences encoding for the heavy and light chains separately into mammalian expression vectors designed for production of IgGs. 17 Current methods to facilitate more rapid reformatting from Fab to IgG are often carried out clonally, restrict diversity to a single chain of the Ab, or rely on bicistronic expression using an IRES sequence. 18 , 19 , 20 , 21 , 22 This process is both time‐consuming and expensive.
To enable facile conversion of Fabs to IgGs, the Dübel group developed a single‐chain Fab (scFab) format consisting of light and heavy chains connected by a linker. 23 To enable efficient display on phage, this format required the removal of the intramolecular disulfide bond that connects the C‐termini of the light and heavy chains, but scFabs purified from bacteria exhibited high levels of aggregation, likely due to intermolecular domain swapping in the absence of the covalent disulfide bond. The scFab format was further optimized by the Wells group by reinstalling the disulfide, using longer linkers, and optimizing the secretion signal. 24 Although the scFab format has been adapted to yeast display for the construction of immune libraries from HIV infected patients, 25 naïve scFab libraries capable of generating novel Abs have not been reported in either the yeast or phage display format.
Here, we report further improvement of the phage‐displayed scFab format by optimization of the linker connecting the heavy and light chains to permit high level display, optimization of the elbow region between the light chain variable and constant domains to improve levels of display on phage, and optimization of the protein A binding site on the heavy chain variable domain to improve the efficiency of affinity purification. With this optimized scFab framework, we constructed a highly diverse, naïve synthetic library and we applied an efficient high throughput strategy with an en masse cloning strategy to convert scFabs from selected phage pools into arrays of mammalian expression vectors for production of single‐chain IgGs (scIgGs), without any need for intermediate sequencing or screening of scFab‐phage clones. We demonstrated the utility of the technology by generating scFabs against a panel of human cell‐surface proteins and developing and characterizing high affinity anti‐Her2 scIgGs and IgGs.
2. RESULTS
2.1. scFab linker optimization
To develop a scFab display system, we used a phagemid vector that was previously used to construct a highly functional Fab library (library F), 26 modified to use an IPTG‐inducible promoter to drive expression of the fusion protein consisting of the scFab fused to the N‐terminus of the C‐terminal domain of the gene‐3 minor coat protein of M13 bacteriophage, as described previously for a scFv library (library G) 27 (libraries F and G represent earlier manifestations of our Ab library designs and are named alphabetically in the sequence in which they were developed). The Fab framework was identical to that used for library F, except for two substitutions that we have discovered serendipitously over the course of analyzing Fabs isolated from library F. We have found that an Ala to Thr substitution at Position 111 in the elbow region between the light chain variable (VL) domain and light chain constant (CL) domain reduces proteolysis in Escherichia coli, and consequently, improves phage display. In addition, a Gly to Arg substitution at Position 15 in the VH domain enhances affinity for protein A by approximately twofold, and consequently, improves purification of Fab protein from E. coli (data not shown).
It has been shown that a 34‐residue linker connecting the light chain to the heavy chain of a Fab is sufficient to promote assembly of a scFab. 23 Thus, we designed a library in which the C‐terminus of the light chain of the Fab framework was connected to the N‐terminus of the heavy chain by a random 37‐residue linker biased in favor of small amino acids (Gly, Ala, Ser, Thr) that provide for a flexible linkage (see Materials and Methods). After selection for binding to protein A, which enriched clones that displayed scFab efficiently, DNA sequencing of 24 clones revealed a single linker sequence (Figure 1).
FIGURE 1.
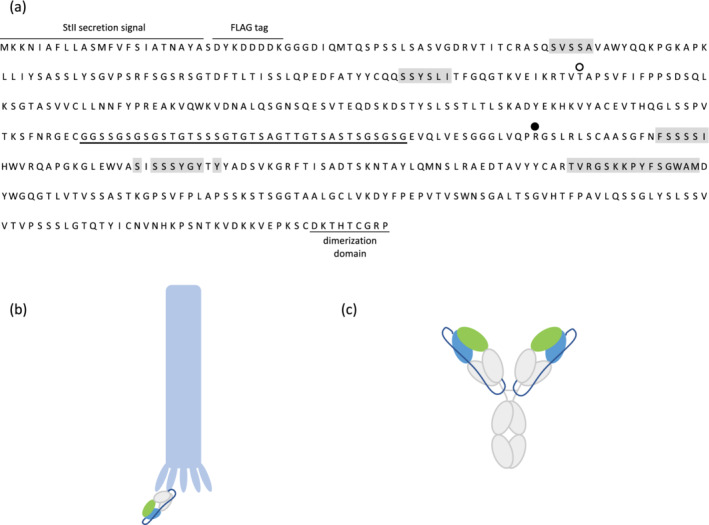
The optimized single‐chain fragment antigen binding (scFab) template used for the construction of phage‐displayed library R. (a) Sequence of the optimized scFab template. For phage display, the open reading frame (ORF) was fused upstream of the ORF for the C‐terminal domain of the M13 gene‐3 minor coat protein. Residues that were diversified in the library are shaded in gray and the linker between the light and heavy chains is underlined. Substitutions in the light chain variable (VL) and heavy chain variable (VH) domains that improve phage display or protein A binding, respectively, are indicated by an open or filled circle, respectively. Cartoon representations of the scFab displayed on M13 phage (b) and in the single‐chain immunoglobulin G (scIgG) format (c). Variable domains are represented as light green (VL) and light blue (VH), constant domains as light grey, and the single‐chain linker in dark blue
It has been noted previously that IgGs in the single‐chain format may exhibit greater aggregation and oligomerization than conventional IgGs. 23 Thus, we applied size‐exclusion chromatography (SEC) to analyze trastuzumab and two Abs derived from library F, purified from mammalian HEK‐293F cells in either the IgG or scIgG format. The major peak for scIgG trastuzumab eluted at the same volume as the IgG monomer peak, but additional peaks indicative of larger aggregates were also present (Figure 2a). A well‐behaved anti‐maltose binding protein Ab eluted almost exclusively as a monomer (98.8%) in the IgG format and exhibited only slightly reduced monomer content (94.0%) in the scIgG format (Figure 2b). A less well‐behaved anti‐Gaussia luciferase Ab eluted predominantly as a monomer (93.7%) but exhibited some evidence of aggregation, and in the scIgG format, the major peak for this Ab (76.1%) eluted with a similar retention volume as the monomer peak for the IgG but there was also a significant fraction that eluted as higher‐order species (Figure 2c). Overall, these results show that the optimized linker enables efficient phage display of scFab and can also be used to produce scIgG proteins that are predominantly monomeric but do show some evidence of aggregation.
FIGURE 2.
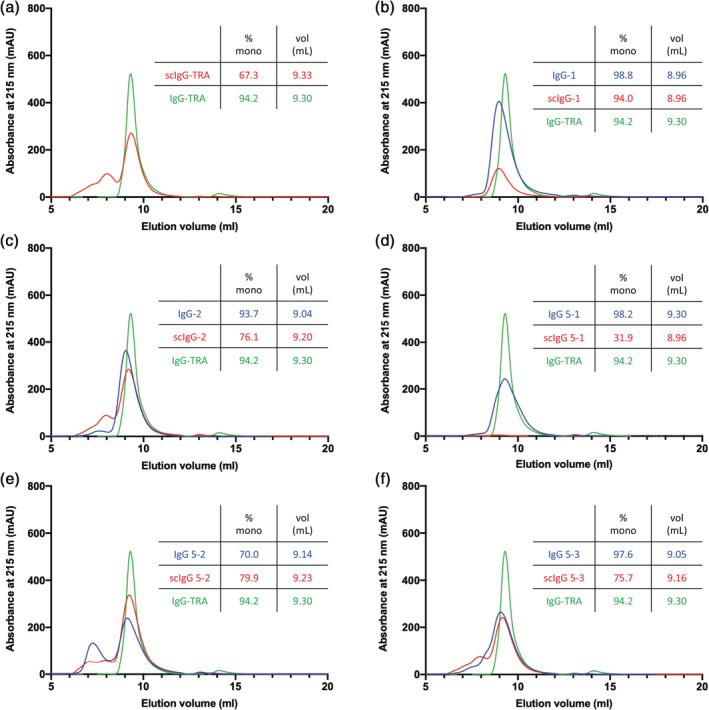
Size‐exclusion chromatography of immunoglobulin G (IgG) and single‐chain IgG (scIgG) proteins. Chromatograms are shown for 5–6.7 μM samples of IgG (blue) and scIgG (red) versions of (a) in‐house produced scIgG‐formatted trastuzumab, (b) anti‐MBP Ab‐1, (c) anti‐Gaussia luciferase Ab‐2, (d) anti‐Her2 clone 5‐1, (e) anti‐Her2 clone 5‐2, and (f) anti‐Her2 clone 5‐3, along with the well‐behaved IgG Trastuzumab (IgG‐TRA, green) as a marker for monomeric IgG. Samples were applied on a Tosoh TSKgel BioAssist G3SWxl column. Percent monomer (% mono) was calculated from the area under the monomer peak (y‐axis) and the elution volume (vol [ml], x‐axis) was calculated at the maximum height of the monomer peak
2.2. Library design and construction
In our final design, the C‐terminus of the CL domain was linked to the N‐terminus of the VH domain by an optimized 37‐residue linker, producing an open reading frame (ORF) encoding for a scFab linked to the C‐terminal domain of the M13 gene‐3 minor coat protein (Figure 1). This optimized scFab framework was used to construct library R, in which five of the six complement determining regions (CDRs) were diversified with a tailored strategy (Figure 3a). The three heavy chain CDRs and CDR‐L3 were diversified as described previously for library F, 26 and solvent‐accessible positions in CDR‐L1 were diversified similarly (Figure 3b). Library R contained 2.3 × 1010 unique clones, and DNA sequencing of 128 naïve clones revealed the incorporation of diversity in approximately 80% of the population within each CDR and the retention of template sequence in the remainder. Overall, 5, 12, and 58% of the library members contained diversity in three, four, or five CDRs, respectively (Figure 3c).
FIGURE 3.
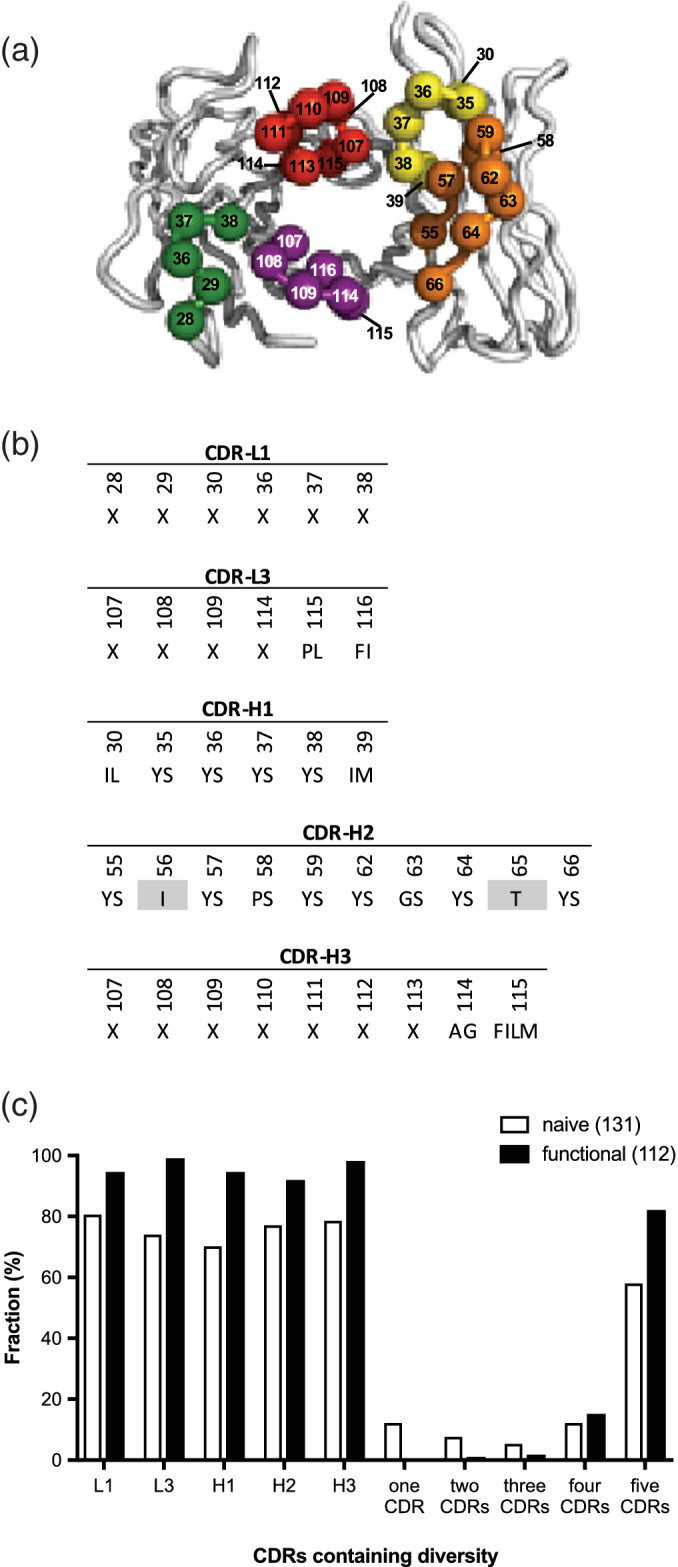
Design and characterization of single‐chain fragment antigen binding (scFab) library R. (a) The backbones of the heavy‐ and light‐chain variable domains are shown as tubes. The framework residues are colored gray and the complement determining region (CDR) loops are colored as follows: CDR‐L1 (green), CDR‐L3 (purple), CDR‐H1 (yellow), CDR‐H2 (orange), and CDR‐H3 (red). Spheres colored according to the CDR coloring scheme represent positions that were diversified. The figure was generated using PyMOL (http://www.pymol.org) with crystal structure coordinates (PDB entry 1FVC). (b) CDR diversity design. Positions shaded in gray were fixed as indicated, and at each diversified position, the allowed amino acids are denoted by the single‐letter code. X denotes a mixture of nine amino acids as follows: Tyr (25%), Ser (20%), Gly (20%), Ala (10%), and Phe, Trp, His, Val, and Pro (5% each). The lengths of CDR‐L1, CDR‐L3, and CDR‐H3 were varied by replacing the positions denoted by X with 5–6, 3–7, or 1–17 degenerate codons, respectively. Residue numbering is according to the IMGT scheme. (c) The fractions of clones containing diversity within a particular CDR or the indicated number of CDRs is shown for 131 naive clones and 112 functional clones that include those shown in Figures 4 and 5
2.3. Selection of functional Abs
To assess performance, we used library R to generate Abs against six human cell‐surface receptors, including the natural killer cell marker CD161 and the receptor tyrosine kinases EphA3, EphB2, EphB4, Her2, and Her3. For CD161 and the Eph receptors, we followed a standard phage display workflow to identify and characterize positive Abs. After four rounds of selections, 48–96 individual clones were assessed by phage ELISAs and positive clones that bound to cognate antigen but not to negative control proteins were subjected to DNA sequence analysis. This process identified 47 unique Abs with diverse CDR sequences (Figure 4). For Her2 and Her3, we took advantage of the modular nature of the library to directly convert selected pools en masse from the phage‐displayed format to arrays of mammalian expression vectors for production of individual scIgG proteins. This was achieved by using the PCR with round four phage pools as template to amplify scFab ORFs and cloning these amplified, pooled ORFs into a mammalian expression vector, between an ORF for a secretion signal and an ORF for the Fc of human IgG1, which generated ORFs for scIgG versions of the scFabs. Following this conversion process, we purified 96 individual mammalian expression plasmids for each antigen, and for those containing inserts of the correct size, we used the plasmid directly for transient transfection of HEK‐293T cells in 24‐well plates. The supernatant from these cell cultures was used in ELISAs to assess scIgG binding to antigen, and plasmids representing positive supernatants were subjected to DNA sequence analysis. This process yielded six and five unique scIgGs that bound to Her2 or Her3, respectively (Figure 5). Taken together, these results showed that library R is a rich source of Abs for diverse antigens, and a streamlined strategy can be used to generate and screen panels of scIgGs derived from scFab‐phage pools by en masse cloning in a high‐throughput manner.
FIGURE 4.
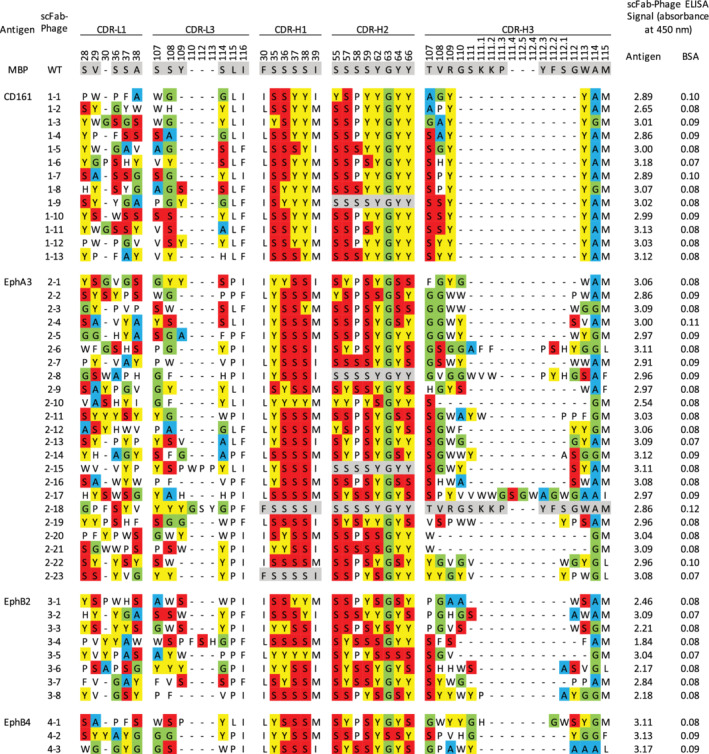
Sequences and ELISAs for single‐chain fragment antigen binding (scFab)‐phage clones selected for binding to antigens. Complement determining region (CDRs) shaded in gray match the template sequence and dashes indicate gaps in the alignment. Amino acids are shaded as follows: Tyr (yellow), Ser (red), Gly (green), and Ala (blue)
FIGURE 5.
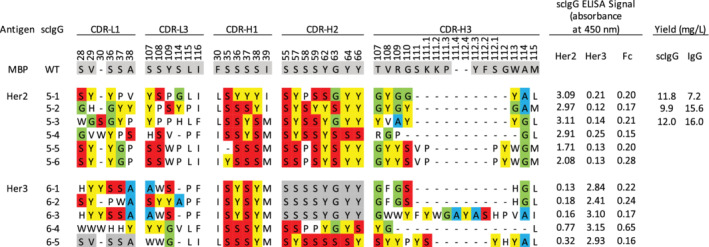
Sequences and ELISAs for single‐chain immunoglobulin G (scIgG) binding to Her2 or Her3. Complement determining regions (CDRs) shaded in gray match the template sequence and dashes indicate gaps in the alignment. Amino acids are shaded as follows: Tyr (yellow), Ser (red), Gly (green), and Ala (blue). ELISAs were performed directly with cell culture supernatants from HEK‐293T cells secreting the indicated scIgG. Yields of scIgG or IgG are shown for proteins purified from HEK‐293F cells
2.4. Characterization of anti‐Her2 Abs
To compare scIgGs with IgGs, we chose three anti‐Her2 scIgGs that exhibited strong binding by ELISA and produced them in both formats. When expressed in HEK‐293T cells, Abs 5‐1, 5‐2, and 5‐3 could be purified in similar yields in the scIgG or IgG format (Figure 5). SDS‐PAGE analysis under nonreducing conditions showed single bands with molecular weights of ~150 kDa for both formats, as expected for intact tetramers of two light‐chains and two heavy chains. In contrast, under reducing conditions that result in breaking of disulfide bonds, the IgG samples migrated as two bands with expected molecular weights for heavy‐chains (~50 kDa) and light chains (~25 kDa), whereas the scIgG samples exhibited a single band of ~75 kDa, consistent with a single chain consisting of a light‐chain linked to a heavy‐chain (Figure 6). When analyzed by SEC, we observed largely monomeric species for the IgG and scIgG formats for Abs 5‐2 and 5‐3; however, Ab 5‐1 in scIgG format did not elute in discrete fractions, indicating either aggregation or nonspecific interactions with the column resin (Figure 2d–f). Kinetic binding measurements by surface plasmon resonance (SPR) showed very similar affinities for both formats and showed that the three Abs bound specifically to Her2 with apparent affinities in the low nanomolar range (Table 1). Notably, Ab 5‐1 displayed evidence of aggregation and nonspecific interactions on the SEC column, and it also displayed a lower off‐rate and apparent K D in the scIgG format compared with the IgG format, and thus, we hypothesize that increased affinity resulted from aggregation‐driven avidity. Taken together, these results show that library R yielded tight and specific anti‐Her2 scFabs that could be converted directly to scIgGs, which behaved the same as conventional IgGs in terms of protein yield and specificity and affinity for antigen.
FIGURE 6.
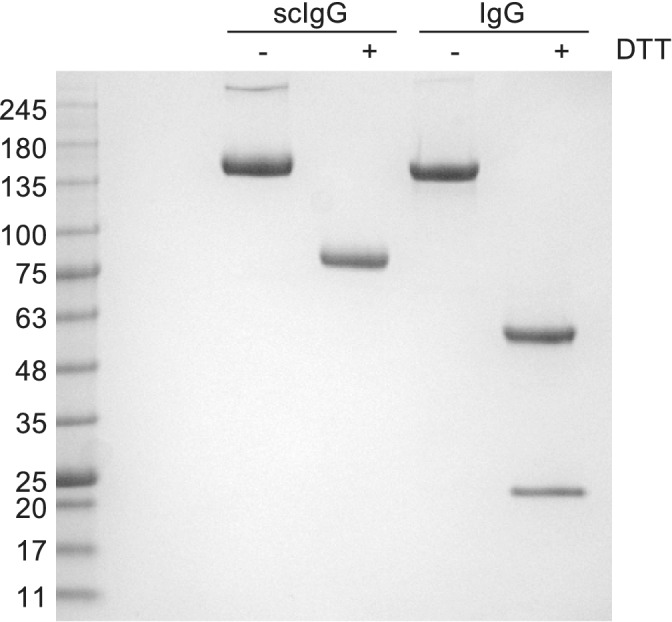
SDS‐PAGE analysis of single‐chain immunoglobulin G (scIgG) and IgG proteins. Samples of purified anti‐Her2 scIgG or IgG 5‐3 were run under nonreducing (−DDT) or reducing (+DDT) conditions. Markers with the indicated MW (kDa) are shown on the left
TABLE 1.
Binding kinetic constants and associated errors for IgGs and scIgGs binding to Her2
| Antibody | k a (M/s × 105) | k d (s−1 × 10−3) | Apparent K D (nM) |
|---|---|---|---|
| 5‐1 | |||
| scIgG | 12.3 ± 0.07 | 0.0454 ± 0.007 | 0.037 |
| IgG | 10.5 ± 0.08 | 0.119 ± 0.008 | 0.11 |
| 5‐2 | |||
| scIgG | 6.99 ± 0.04 | 0.807 ± 0.01 | 1.2 |
| IgG | 7.25 ± 0.05 | 0.983 ± 0.01 | 1.4 |
| 5‐3 | |||
| scIgG | 4.84 ± 0.03 | 2.12 ± 0.01 | 4.4 |
| IgG | 5.10 ± 0.03 | 1.97 ± 0.01 | 3.9 |
Abbreviations: IgG, immunoglobulin G; scIgG, single‐chain immunoglobulin G.
3. DISCUSSION
This report represents the first example of a synthetic, naïve scFab library. By physically linking the light and heavy chains of a Fab, we were able to produce a highly diverse phage‐displayed scFab library from which pools of binding clones could be converted en masse into a scIgG format optimized for expression in mammalian cells. Following selection, the library design permitted facile screening of output clones either as scFab‐phage or as scIgG protein. Screening of scFab‐phage is a rapid means for determining the success of a selection. Subsequently, pools from successful selections can be efficiently converted en masse into mammalian expression vectors to enable direct screening of scIgG protein for affinity and specificity, either with purified antigen or on cells. Thus, the time‐consuming process of converting individual Fab‐phage clones into IgG expression vectors is avoided, enabling research efforts to focus on functional Abs.
The scFab library is analogous to established scFv libraries, in which the displayed Ab fragment consists of the two variable domains tethered by a flexible linker. While scFv‐phage clones can be converted to a scFv‐Fc format, 28 , 29 , 30 scFvs are much more prone to aggregation than Fabs and the scFv‐Fc format is not an accurate mimic of natural IgGs. In contrast, scIgGs are identical to natural IgGs in terms of the structure and orientation of the binding sites and the hinge region connecting Fabs to the Fc. However, the scIgG format does exhibit somewhat greater aggregation than the IgG format (Figure 2). Thus, while the scIgG format is highly suitable for rapid screening and most affinity reagent applications, the IgG format may still be preferred for antagonist and agonist functions, which are often strongly influenced by valency. In this regard, we have demonstrated that the scIgG linker can be genetically removed from lead candidates through conventional cloning techniques without altering the affinity or specificity of the resulting IgG (Figure 5, Table 1). Thus, the scIgG format is a very close mimic of the natural IgG format, but with the significant advantage of being rapidly convertible with the scFab format that is well suited for phage display. Taken together, these advantages establish scFab libraries and the complementary scIgG proteins as a powerful means for more rapidly and efficiently accessing the full diversity of phage‐displayed synthetic Ab libraries.
4. MATERIALS AND METHODS
4.1. scFab linker optimization
An IPTG‐inducible phagemid previously used for phage display of a scFv library 27 was modified by replacing the DNA fragment encoding for the scFv with a DNA fragment encoding for a Fab, derived from a previously described Fab, 26 with an Ala to Thr substitution at Position 111 of the light chain and a Gly to Arg substitution at Position 15 of the heavy chain. Standard molecular biology techniques were used to create an ORF consisting of the following: a secretion signal, a FLAG epitope tag, the Fab light chain, a short linker (sequence: GGSSGSGGSGSG), the Fab heavy chain, a dimerization domain (sequence: DKTHTCGRP), the C‐terminal domain of the gene‐3 minor coat protein. The resulting phagemid was used as the template for large‐scale oligonucleotide‐directed mutagenesis with established methods 31 to insert 25 [A/G][C/G]T degenerate codons within the linker sequence between the Fab light and heavy chains, thus generating a combinatorial linker library (~1010 unique members) with the following amino acid sequence: GGSSGSG‐X25‐GSGSG (X = A/G/S/T). Phage pools representing the scFab linker library were cycled through five rounds of binding selections with immobilized protein A and individual clones were subjected to DNA sequence analysis, as described. 32
4.2. Size‐exclusion chromatography
Anti‐maltose binding protein and anti‐Gaussia luciferase scIgGs and IgGs were expressed and purified as described. 33 Each Ab (37.5–50 μg) was analyzed using a BioRAD NGC chromatography system with a C‐96 autosampler equipped with a Tosoh TSKgel BioAssist G3SWxl SEC column (catalogue number 20026) with guard column (catalogue number 18008).
4.3. Library construction and Ab selections
The scFab library R was constructed using the IPTG‐inducible phagemid designed to display the scFab with an optimized linker (Figure 1). Library R was constructed using previously described methods and mutagenic oligonucleotides 26 with the addition of mutagenic oligonucleotides designed to diversify CDR‐L1 (Figure 3). Binding selections and scFab‐phage ELISAs to select binding clones for particular antigens were carried out as described. 26 The following protein antigens were obtained from R&D Systems (catalogue numbers in parentheses): CD161 (7448‐CD); EphA3, (6444‐A3); EphB2 (5189‐B2); EphB4 (3038‐B4); Her2 (1129‐ER); and Her3 (348‐RB).
4.4. Conversion of scFab‐phage pools to scIgG expression constructs for screening
Following selections for antigen binding, pooled phage were diluted in PBS and used as the template for a PCR to amplify DNA fragments encoding the scFab. The DNA fragments were digested with EcoRV and AgeI (New England Biolabs) and ligated into a similarly digested version of the pFuse‐hIgG1 expression vector (InvivoGen) designed for the expression of scIgG. Individual scIgG expression vectors were purified from E. coli and the plasmid DNA was used to transfect HEK‐293T cells grown in 24‐well tissue culture plates (Falcon) using Xtremegene 9 (Roche), following the manufacturer's instructions. Transfected cells were grown at 37°C in a humidified incubator supplemented with 5% CO2 for 48 hr and culture supernatant was harvested and used directly for antigen‐binding ELISAs. For supernatants that exhibited specific, positive signals by ELISA, the gene encoding the scIgG was subjected to DNA sequencing.
4.5. Production and characterization of scIgG and IgG proteins
Plasmids designed for the expression of scIgG or IgG protein were transfected into HEK‐293F cells using 293fectin according to the manufacturer's instructions (Invitrogen). For each scIgG, a single plasmid was used, as described above. For each IgG, a dual plasmid system was used in which the light and heavy chains were encoded on separate plasmids, pFusess‐CHIg‐hG1 (human IgG1 heavy chain, InvivoGen) and pFuse2ss‐CLIg‐hK (human kappa light chain, InvivoGen). Then, 48 hr after transfection, cell cultures were supplemented with 20% tryptone to a final concentration of 0.5%. After 5 days, scIgG or IgG protein was purified from the cell culture supernatant using Protein A‐sepharose (GE Healthcare) affinity chromatography. Purity was assessed by SDS‐PAGE analysis using 4–15% acrylamide tris‐glycine SDS‐PAGE (Bio‐Rad) and coomassie blue staining.
4.6. Surface plasmon resonance
Binding kinetics were measured by SPR using a ProteOn XPR36 (Bio‐Rad). Fc‐tagged Her2 (110 RUs) was immobilized to a GLC chip via amine coupling. Four twofold serial dilutions of Abs (beginning at 50 nM) were injected over antigen and blank channels at a rate of 100 μl/min for 60 s, followed by 10 min of buffer injection to monitor disassociation. Association and dissociation buffer was composed of PBS supplemented with 0.05% Tween 20. The chip was regenerated with 0.85% H3PO4 between analyte runs. Kinetic parameters were calculated by globally fitting a reference cell‐subtracted data series to a 1:1 (Langmuir) binding model.
AUTHOR CONTRIBUTIONS
Rachel Hanna: Conceptualization; formal analysis; investigation; methodology; writing‐original draft. Lia Cardarelli: Formal analysis; visualization; writing‐original draft. Nish Patel: Conceptualization; methodology. Levi L. Blazer: Formal analysis; investigation; writing‐review and editing. Jarrett J. Adams: Conceptualization; methodology; writing‐review and editing. Sachdev S. Sidhu: Conceptualization; formal analysis; funding acquisition; methodology; supervision; writing‐original draft.
ACKNOWLEDGMENTS
The authors thank N. Jarvik, K. Nagal, A. Pavlenco, and S. Suvarna for their technical assistance. The authors also thank A. Gakhal, I. Leung, and P. Garg for helpful discussions. This work was supported by two operating grants from the Canadian Institutes of Health Research (MOP‐93725 and MOP‐136944).
Hanna R, Cardarelli L, Patel N, Blazer LL, Adams JJ, Sidhu SS. A phage‐displayed single‐chain Fab library optimized for rapid production of single‐chain IgGs . Protein Science. 2020;29:2075–2084. 10.1002/pro.3931
Funding information Canadian Institutes of Health Research, Grant/Award Numbers: MOP‐93725, MOP‐136944
REFERENCES
- 1. Skerra A, Pluckthun A. Assembly of a functional immunoglobulin Fv fragment in Escherichia coli . Science. 1988;240:1038–1041. [DOI] [PubMed] [Google Scholar]
- 2. Ward ES, Gussow D, Griffiths AD, Jones PT, Winter G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli . Nature. 1989;341:544–546. [DOI] [PubMed] [Google Scholar]
- 3. Barthelemy PA, Raab H, Appleton BA, et al. Comprehensive analysis of the factors contributing to the stability and solubility of autonomous human VH domains. J Biol Chem. 2008;283:3639–3654. [DOI] [PubMed] [Google Scholar]
- 4. Nilvebrant J, Sidhu SS. Construction of synthetic antibody phage‐display libraries. Methods Mol Biol. 2018;1701:45–60. [DOI] [PubMed] [Google Scholar]
- 5. Hu S, Shively L, Raubitschek A, et al. Minibody: A novel engineered anti‐carcinoembryonic antigen antibody fragment (single‐chain Fv‐CH3) which exhibits rapid, high‐level targeting of xenografts. Cancer Res. 1996;56:3055–3061. [PubMed] [Google Scholar]
- 6. Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23:1126–1136. [DOI] [PubMed] [Google Scholar]
- 7. Holliger P, Prospero T, Winter G. "Diabodies": Small bivalent and bispecific antibody fragments. Proc Natl Acad Sci U S A. 1993;90:6444–6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hoet RM, Cohen EH, Kent RB, et al. Generation of high‐affinity human antibodies by combining donor‐derived and synthetic complementarity‐determining‐region diversity. Nat Biotechnol. 2005;23:344–348. [DOI] [PubMed] [Google Scholar]
- 9. Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat Biotechnol. 2005;23:1105–1116. [DOI] [PubMed] [Google Scholar]
- 10. Bradbury AR, Sidhu S, Dübel S, McCafferty J. Beyond natural antibodies: The power of in vitro display technologies. Nat Biotechnol. 2011;29:245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mersmann M, Meier D, Mersmann J, et al. Towards proteome scale antibody selections using phage display. N Biotechnol. 2010;27:118–128. [DOI] [PubMed] [Google Scholar]
- 12. Schofield DJ, Pope AR, Clementel V, et al. Application of phage display to high throughput antibody generation and characterization. Genome Biol. 2007;8:R254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roopenian DC, Akilesh S. FcRn: The neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–725. [DOI] [PubMed] [Google Scholar]
- 14. Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol. 2010;10:345–352. [DOI] [PubMed] [Google Scholar]
- 15. Leelawattanachai J, Kwon KW, Michael P, Ting R, Kim JY, Jin MM. Side‐by‐side comparison of commonly used biomolecules that differ in size and affinity on tumor uptake and internalization. PLoS One. 2015;10:e0124440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. El‐Sayed A, Bernhard W, Barreto K, et al. Evaluation of antibody fragment properties for near‐infrared fluorescence imaging of HER3‐positive cancer xenografts. Theranostics. 2018;8:4856–4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li J, Menzel C, Meier D, Zhang C, Dubel S, Jostock T. A comparative study of different vector designs for the mammalian expression of recombinant IgG antibodies. J Immunol Methods. 2007;318:113–124. [DOI] [PubMed] [Google Scholar]
- 18. Jostock T, Vanhove M, Brepoels E, et al. Rapid generation of functional human IgG antibodies derived from Fab‐on‐phage display libraries. J Immunol Methods. 2004;289:65–80. [DOI] [PubMed] [Google Scholar]
- 19. Jones ML, Seldon T, Smede M, et al. A method for rapid, ligation‐independent reformatting of recombinant monoclonal antibodies. J Immunol Methods. 2010;354:85–90. [DOI] [PubMed] [Google Scholar]
- 20. Chen CG, Fabri LJ, Wilson MJ, Panousis C. One‐step zero‐background IgG reformatting of phage‐displayed antibody fragments enabling rapid and high‐throughput lead identification. Nucleic Acids Res. 2014;42:e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Quinlan BD, Gardner MR, Joshi VR, Chiang JJ, Farzan M. Direct expression and validation of phage‐selected peptide variants in mammalian cells. J Biol Chem. 2013;288:18803–18810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen CG, Sansome G, Wilson MJ, Panousis C. High‐throughput IgG reformatting and expression. Methods Mol Biol. 2018;1701:447–461. [DOI] [PubMed] [Google Scholar]
- 23. Hust M, Jostock T, Menzel C, et al. Single chain Fab (scFab) fragment. BMC Biotechnol. 2007;7:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koerber JT, Hornsby MJ, Wells JA. An improved single‐chain Fab platform for efficient display and recombinant expression. J Mol Biol. 2015;427:576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Walker LM, Bowley DR, Burton DR. Efficient recovery of high‐affinity antibodies from a single‐chain Fab yeast display library. J Mol Biol. 2009;389:365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Persson H, Ye W, Wernimont A, et al. CDR‐H3 diversity is not required for antigen recognition by synthetic antibodies. J Mol Biol. 2013;425:803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van Deventer JA, Kelly RL, Rajan S, Wittrup KD, Sidhu SS. A switchable yeast display/secretion system. Protein Eng Des Sel. 2015;28:317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang H, Wilson IA, Lerner RA. Selection of antibodies that regulate phenotype from intracellular combinatorial antibody libraries. Proc Natl Acad Sci U S A. 2012;109:15728–15733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ho M, Pastan I. Mammalian cell display for antibody engineering. Methods Mol Biol. 2009;525:337–352. xiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boder ET, Raeeszadeh‐Sarmazdeh M, Price JV. Engineering antibodies by yeast display. Arch Biochem Biophys. 2012;526:99–106. [DOI] [PubMed] [Google Scholar]
- 31. Fellouse FA, Sidhu SS. Making antibodies in bacteria In: Howard GC, Kaser MS, editors. Making and using antibodies. Boca Raton, FL: CRC Press, 2007; p. 157–180. [Google Scholar]
- 32. Tonikian R, Zhang Y, Boone C, Sidhu SS. Identifying specificity profiles for peptide recognition modules from phage‐displayed peptide libraries. Nat Protoc. 2007;2:1368–1386. [DOI] [PubMed] [Google Scholar]
- 33. Newsted D, Banerjee S, Watt K, et al. Blockade of TGF‐β signaling with novel synthetic antibodies limits immune exclusion and improves chemotherapy response in metastatic ovarian cancer models. Onco Targets Ther. 2019;8:e1539613. [DOI] [PMC free article] [PubMed] [Google Scholar]