

Abstract
Biosensors are valuable and versatile tools in synthetic biology that are used to modulate gene expression in response to a wide range of stimuli. Ligand responsive transcription factors are a class of biosensor that can be used to couple intracellular metabolite concentration with gene expression to enable dynamic regulation and high-throughput metabolite producer screening. We have established the Saccharomyces cerevisiae WAR1 transcriptional regulator and PDR12 promoter as an organic acid biosensor that can be used to detect varying levels of para-hydroxybenzoic acid (PHBA) production from the shikimate pathway and output green fluorescent protein (GFP) expression in response. The dynamic range of GFP expression in response to PHBA was dramatically increased by engineering positive-feedback expression of the WAR1 transcriptional regulator from its target PDR12 promoter. In addition, the noise in GFP expression at the population-level was controlled by normalising GFP fluorescence to constitutively expressed mCherry fluorescence within each cell. These biosensor modifications increased the high-throughput screening efficiency of yeast cells engineered to produce PHBA by 5,000-fold, enabling accurate fluorescence activated cell sorting isolation of producer cells that were mixed at a ratio of 1 in 10,000 with non-producers. Positive-feedback, ratiometric transcriptional regulator expression is likely applicable to many other transcription-factor/promoter pairs used in synthetic biology and metabolic engineering for both dynamic regulation and high-throughput screening applications.
Keywords: biosensor, adaptive laboratory evolution, synthetic biology, metabolic engineering, yeast
1. Introduction
Advances in synthetic biology and metabolic engineering are beginning to enable alternative routes for chemical, fuel, and pharmaceutical manufacturing. Microorganisms can now be engineered to convert renewable agricultural resources into a wide array of metabolites that can serve as either replacements or alternatives to existing industrial products. However, there are significant challenges involved in engineering microbial cells to produce desired products at commercially viable titres, rates, and yields. Traditional approaches involve balancing the expression levels of metabolic pathway enzymes leading to the desired product, the elimination of enzymes that compete for carbon flux, and the balancing of cellular redox and energy states (Nielsen and Keasling 2016). These biological components often interact synergistically to control complex metabolic regulation, and their rearrangement/optimisation therefore entails a combinatorial explosion of complex traits that require building and testing for performance. Although in silico modelling approaches have proven invaluable for understanding metabolism and improving metabolite production (Wiechert 2002; Patil et al. 2004), metabolic engineering ‘design principles’ are yet to be fully elucidated due to our incomplete knowledge of living systems. Subsequently, it can take many iterations of the classical design-build-test cycle to achieve engineering objectives, some of which may even be impossible to meet using available biological knowledge.
An elegant way to overcome the challenges associated with engineering in biology is to apply a selective pressure to a genetically diverse population so that cells with the desired phenotype can be isolated. Using selective pressure in this way means that biological knowledge and the limited capacity to design-build-test no longer limit the solutions available to biological engineering problems. While methods such as adaptive laboratory evolution can be used to generate populations with superior tolerance to growth inhibiting chemicals (Goodarzi et al. 2010; Kildegaard et al. 2014; Almario et al. 2013; Brennan et al. 2015), altered substrate specificity (Wisselink et al. 2009; Garcia Sanchez et al. 2010; Quan et al. 2012; Zhou et al. 2012), and increased temperature tolerance (Riehle et al. 2003; Caspeta et al. 2014), this approach cannot be readily applied to the selection of high metabolite yields because phenotypes such as metabolic productivity are usually not naturally coupled to cell survival. One of the most promising ways to make this connection and select for cells with higher metabolic productivity is to use a biosensor that detects the intracellular concentration of a metabolite of interest and outputs a survival function in response (Williams et al. 2016). The output is typically green fluorescent protein (GFP) expression so that cells that are more productive within an evolving or mutated population can be isolated using fluorescence activated cell sorting (FACS) (Williams et al. 2016). The advantage of using FACS is that thousands of cells can be screened per second, as opposed to per week or month using traditional analytical methods for metabolites.
Biosensors are gaining prominence both for the dynamic control of metabolic pathways in response to metabolites, and as conduits between metabolite productivity and cell survival for high-throughput screening (Liu et al. 2015; Zhang et al. 2015; Williams et al. 2016). Allosterically regulated transcription factors are abundant in nature, with a large number characterised in terms of their activating ligands and target promoters (Tropel et al. 2004; Ramos et al. 2005; Gallegos et al. 1997; Taylor et al. 2016). These proteins have therefore typically been the first option for those seeking metabolite biosensors for metabolic engineering applications and high-throughput screening (Taylor et al. 2016; Williams et al. 2016). However, many naturally occurring transcription factors have activation dynamics that are not ideal for high-throughput metabolite-producer cell screening. Features such as high basal expression levels, low dynamic range of expression, and stochastic variation in gene expression across a population (noise) can all reduce the effective screening throughput by making it difficult to distinguish ‘producer’ cells from non-producers within a genetically diverse population.
With the aim of establishing a generic synthetic biology approach to select for enhanced metabolite producers we have developed an amplified ligand-responsive transcription-factor biosensor with a built-in ratiometric noise suppressor to enhance the efficacy of high-throughput screening. As a proof of concept, we focused on transcriptional regulators that respond to organic acids, as there is great interest in producing these compounds biologically (Chen and Nielsen 2016). Organic acids are used to make a variety of products such as plastics, solvents, polymers and chemical ‘building blocks’, animal feed, nylons, flavours and fragrances, as well as food and beverage ingredients (Sauer et al. 2008). Most organic acids are currently produced from petrochemical feed-stocks, and there is therefore significant interest in implementing renewable and more environmentally friendly production processes using microbial hosts (Sauer et al. 2008). The yeast Saccharomyces cerevisiae is a well characterised eukaryotic model organism, industrial work-horse, and synthetic biology ‘chassis’ cell (Pretorius 2016). S. cerevisiae is also preferred industrial producer of organic acids due to its ability to grow at a low pH, enabling low purification costs and reducing microbial contamination from non-sterile substrates (Sauer et al 2008; Abbott et al. 2009). Consequently, there have been intensive efforts in metabolic engineering of yeast for the production of valuable organic acids such as lactic (Ishida et al. 2006; Baek et al. 2015), succinic (Raab et al. 2010; Agren et al. 2013; Ito et al. 2014), malic (Zelle et al. 2008), 3-hydroxypropionic (Borodina et al. 2015; Kildegaard et al. 2015), 4-hydroxybenzoic and para-aminobenzoic (Krömer et al. 2012; Averesch and Krömer 2014; Williams et al. 2015), itaconic (Blazeck et al. 2014), and muconic (Curran et al. 2013) acids.
2. Materials and methods
2.1 Growth media
All S. cerevisiae strains were grown in synthetic dropout (SD) media containing Yeast Nitrogen Base Without Amino Acids mix (Sigma-Aldrich Y0626) supplemented with 10 g/l glucose, and amino acids at 100 mg/l to complement auxotrophies as appropriate. pH was adjusted to either 6.5 or 3.5 as indicated in individual experiments. Escherichiacoli DH5α strains were grown in LB medium with ampicillin.
2.2. Growth conditions
For all dose–response GFP measurement and cell-sorting experiments, culturing methods and pre-culture methods were as follows. Glycerol stocked strains were inoculated into 5 ml of SD medium in a 50-ml sterile Falcon tube and grown for 8–10 hours at 30 °C, 200 rpm in a shaking incubator. These cultures were then used to inoculate 10 ml of SD medium at an OD600nm of 0.02–0.04 and grown overnight for approximately 18 hours. The next morning, exponentially growing cultures (OD600nm 0.1–1.5) were used to inoculate experimental cultures in triplicate, at an OD600nm of 0.1. Populations were grown in 24-well micro-plates in a total volume of 1.5 ml containing any organic acids involved in promoter screening or dose–response experiments at the indicated concentrations. Weak acid response experiments were carried out in SD medium at pH 3.5 such that acid molecules outside of cells would freely diffuse inside, potentially being available for promoter/GFP activation (Holyoak et al. 1999). Cell sorting and GFP measurement experiments with para-hydroxybenzoic acid (PHBA) producer populations were carried out in SD medium at pH 6.5 such that any organic acids produced and secreted by individual cells would not enter other cells (Holyoak et al. 1999) and interfere with the GFP readout from their own PHBA production capacity (or lack of). For cell sorting experiments, pre-cultured producer and non-producer populations were inoculated at exactly OD600nm 0.05 in separate tubes prior to being mixed together at the indicated ratios within the same 50-ml Falcon tube. To reach the dilutions ranging from 1:10 to 1:105 producers to non-producers, serial 10-fold dilutions were made starting from the 1:10 population. Both multi-well plate and Falcon-tube cultures were grown at 30 °C with 200 rpm shaking in an InFors incubator for 3 hours prior to GFP measurement.
2.3 Flow cytometry
A BD Influx flow cytometer was used for all fluorescent protein measurements and cell sorting. For GFP measurement a 200 mW 488-nm laser was used for excitation with emission filters at 540 ± 30 nm. A GFP negative control strain (415.C, Table 3) was used to measure auto-fluorescence in parallel to GFP positive strains, and the 540 ± 30 nm PMT voltage was set such that the mean auto-fluorescence value was either 2 or 4, as indicated in the supplementary data (fluorescence of no-GFP control strain 415.C, Table 3). Mean GFP values from experimental populations were divided by auto-fluorescence, and all conditions were measured in triplicate cultures, with mean and SDs reported. Mixed producer/non-producer populations were sorted according to GFP or GFP:mCherry fluorescence whereby gates that excluded any cells below a GFP fluorescence level that occurred in a yeast population containing the pPDR12-GFP or pPDR12-GFP-pPDA1-mCherry biosensor (measured in parallel), but not containing a production pathway (strains GFP.415, +FB.GFP.415, +FB.GFP.mCherry.415, Table 3). Alternatively, in the case of the GFP-only sorting experiment (Fig. 2d), a gate encompassing the top 3.4% of the mixed population was used for sorting. mCherry fluorescence was measured simultaneously to GFP fluorescence in individual cells using the 488-nm laser. A 692/40-emission filter was used to measure mCherry fluorescence with PMT gain set to 75 and the 540/30 PMT gain set to 35. These gain settings were imposed with the goal of enhancing mCherry signal while reducing the affect of GFP emission leakage into the mCherry channel. Likewise the 692/40-emission filter was used in place of the 610/20 filter (closer to the mCherry emission optima) in order to reduce GFP leakage into the mCherry channel. mCherry expression was verified by comparing the mCherry.415 strain 692/40 values to those of the 415.C control strain. Overlay histograms of GFP and GFP:mCherry distributions were made using GraphPad Prism 7 software. 10,000 events were recorded and FCS files were converted to csv prior to being imported into GraphPad Prism, where GFP fluorescence values were grouped into 100 arbitrary units (au) bins, and GFP:mCherry values into 0.05 au bins. Fluorescence values/ratios were plotted on the x-axes with event numbers on the y-axes.
Table 3.
Yeast strains used in this study.
| Name | Genotype, plasmids | Notes | Origin |
|---|---|---|---|
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Haploid auxotrophic laboratory strain, mating type ‘a’ | Euroscarf |
| BY4742 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Haploid auxotrophic laboratory strain, mating type ‘α’ | Euroscarf |
| PDR12 | BY4742, pPDR12-GFP-415 | PDR12 promoter regulated GFP expression | This study |
| YGP1 | BY4742, pYGP1-GFP-415 | YGP1 promoter regulated GFP expression | This study |
| TPO2 | BY4742, pTPO2-GFP-415 | TPO2 promoter regulated GFP expression | This study |
| 415.C | BY4741, pRS415, pRS416 | No GFP control strain | This study |
| 425.C | BY4741, pRS425, pRS416 | No GFP control strain | This study |
| GFP.415 | BY4741, pPDR12-GFP-415, pRS416 | Centromeric plasmid biosensor strain | This study |
| GFP.415.U | BY4741, pPDR12-GFP-415, U-416 | Centromeric plasmid biosensor strain with UBiC expression | This study |
| GFP.415.UA | BY4741, pPDR12-GFP-415, UA-416 | Centromeric plasmid biosensor strain with UBiC and ARO4 expression | This study |
| GFP.415.UA.KanMX | BY4741, pPDR12-GFP-415, UA-KanMX-416 | Centromeric plasmid biosensor strain with UBiC, ARO4, and KanMX expression | This study |
| GFP.425 | BY4741, pPDR12-GFP-425, pRS416 | Episomal plasmid biosensor strain | This study |
| +FB.GFP.415 | BY4741, pWAR1::pPDR12, pPDR12-GFP-415, pRS416 | Positive feedback, centromeric plasmid biosensor strain | This study |
| +FB.GFP.415.U | BY4741, pWAR1::pPDR12, pPDR12-GFP-415, U-416 | Positive feedback, centromeric plasmid biosensor strain with UBiC expression | This study |
| +FB.GFP.415.UA | BY4741, pWAR1::pPDR12, pPDR12-GFP-415, UA-416 | Positive feedback, centromeric plasmid biosensor strain with UBiC and ARO4 expression | This study |
| +FB.GFP.mCherry.415 | BY4741, pWAR1::pPDR12, pPDR12-GFP-pPDA1-mCherry-415, pRS416 | Positive feedback, ratiometric centromeric plasmid biosensor strain | This study |
| +FB.GFP.mCherry.415. UA | BY4741, pWAR1::pPDR12, pPDR12-GFP-pPDA1-mCherry-415, UA-416 | Positive feedback, ratiometric centromeric plasmid biosensor strain with UBiC and ARO4 expression | This study |
Figure 2.

Detection of intracellular PHBA and shikimate pathway flux variants. (a) The expression of chorismate pyruvate lyase and 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase (UBiC and ARO4 genes) from strong TEF1 promoters in yeast enables the production of PHBA (Williams et al. 2016). The pPDR12-GFP biosensor was tested for responsiveness to intracellular PHBA production via co-expression in a PHBA producing strain. (b) The GFP fluorescence of pPDR12-GFP biosensor containing strains increases with increasing PHBA production. Mean GFP values from triplicate cultures are shown with error bars representing ± 1 SD. (c) As a proof of principle, equal amounts of biosensor containing PHBA producers and non-producers were mixed in the same culture and cells were sorted onto agar plates according to GFP fluorescence. (d and e) Flow cytometry density plots are shown with GFP fluorescence on the y-axis and forward scatter on the x-axis. (d) A non-PHBA producing strain (GFP.415, Table 3). (e) The fluorescence level of a strain with TEF1 promoter-mediated expression of UBiC, ARO4Q166K, and KanMX (strain GFP.415.UA.KanMX, Table 3) for PHBA production.
2.4 Strain and plasmid construction
Primers, plasmids, and strains used in this study are shown in Tables 1, 2, and 3, respectively. Strains were constructed by transforming plasmids into the relevant yeast strain using the lithium acetate method (Gietz et al. 2007) and selecting for growth on appropriate auxotrophic dropout agar plates. All in silico cloning, Gibson assembly, and primer design was carried out using Geneious Pro software (Kearse et al. 2012), version 9.1.5. E. coli DH5α was used for cloning using standard techniques (Sambrook and Russell 2001) unless otherwise mentioned. For the construction of pRS415 biosensor plasmids, PDR12 (primers 1/2), YGP1 (primers 3/4), and TPO2 (primers 5/6) promoter regions were amplified from S. cerevisiae BY4741 DNA and Gibson Assembled (Gibson et al. 2009) 5′ of the yEGFP-ADH1t sequence amplified from the p413-TR-SSRE-GFP plasmid (Chen and Weiss 2005) (primers 7/8, 9/10, 11/12 for amplification of yEGFP-ADH1t and assembly with pPDR12, pYGP1, and pTPO2 respectively). For pPDR12-yEGFP assembly the pRS415 plasmid was linearized via PCR amplification with primers pRS-F and pRS-R, while for pYGP1-yEGFP and pTPO2-yEGFP, pRS415 was linearized via digestion with SmaI and Eco53KI enzymes respectively. The high copy-number pPDR12-yEGFP-pRS425 plasmid was made using the same approach as for pPDR12-yEGFP-pRS415, except with pRS425.
Table 1.
Primers used in this study.
| Primer number/name | 5′ to 3′ sequence |
|---|---|
| 1/pPDR12F | CGAGGTCGACGGTATCGATCTAAACCAAAGATGGATTGTTTACCA |
| 2/pPDR12R | ACCTTTAGACATTTTTTTATTAATAAGAACAATAACA |
| 3/pYGP1F | ATCGAATTCCTGCAGCCCAGCGTGCTATTTTTTAAAAAGGGC |
| 4/pYGP1R | CCTTTAGACATTTTCTATTACTGTATTACTTAACTGACGA |
| 5/pTPO2F | GCCGCCACCGCGGTGGAGCCTATGCAAAAACCCTTCCCC |
| 6/pTPO2R | CCTTTAGACATATTTGTTTTGTGTATTATTTTTGTGA |
| 7/pPDR12-yEGFPF | ATTAATAAAAAAATGTCTAAAGGTGAAGAATTATTCACTGG |
| 8/pPDR12-yEGFPR | GGAATTCGATATCAAGCTTAATATTACCCTGTTATCCCTAGCGG |
| 9/pYGP1-yEGFPF | AGTAATAGAAAATGTCTAAAGGTGAAGAATTATTCACTGG |
| 10/pYGP1-yEGFPR | AGAACTAGTGGATCCCCCATATTACCCTGTTATCCCTAGCGG |
| 11/pTPO2-yEGFPF | ACAAAACAAATATGTCTAAAGGTGAAGAATTATTCACTGG |
| 12/pTPO2-yEGFPR | AGGGAACAAAAGCTGGAGATATTACCCTGTTATCCCTAGCGG |
| 13/crRNAF | CGAGGTCGACGGTATCGAGCTTCTTTGAAAAGATAATGT |
| 14/crRNAR | GGAATTCGATATCAAGCTTAGGCCGCAAATTAAAGCCTTC |
| 15/pRSF | TAAGCTTGATATCGAATTCC |
| 16/pRSR | TCGATACCGTCGACCTCG |
| 17/Cas9F | ATCGAATTCCTGCAGCCCCATAGCTTCAAAATGTTTCTACTCCT |
| 18/Cas9R | AGAACTAGTGGATCCCCCGGCCGCAAATTAAAGCCTTC |
| 19/pWAR1-crRNAF | TAGTGTGTATTGACTGTGATGTTTTAGAGCTAGAAATAGCAAGTTA |
| 20/pWAR1-crRNAR | ATCACAGTCAATACACACTAGATCATTTATCTTTCACTGCG |
| 21/pPDR12F | TTACAAGTTCGTGCATATATAGAAAGAATTCTGTTGTTGTAATTGTCATAACTATTGAGCTCTAAACCAA AGATGGATTGTTTACCA |
| 22/pPDR12R | ATTATCATTATTGATTTCTTTCCCGACGGCAACGCCAGTTATTGCAATCTGCGTGTCCATTTTTTTATTAA TAAGAACAATAACA |
| 23/pWAR1checkF | AACCTGCTGAACCAACAAAACC |
| 24/pWAR1checkR | AACTTTTTGGTCGGTCTTTG |
| 25/4HB-KanMX F | GCCGCCACCGCGGTGGAGTAGGTCTAGAGATCTGTTTAGCTTGC |
| 26/4HB-KanMX R | AGGGAACAAAAGCTGGAGATTAAGGGTTCTCGAGAGCTCG |
| 27/pPDA1 F | GTATTCTGATAAATCTAAAGAGA |
| 28/pPDA1 R | TGGCACAAATGTGGTTTCCT |
| 29/mCherry F | ATGGTTTCTAAGGGTGAAGAAGACA |
| 30/mCherry R | TTACTTGTACAATTCGTCCATACCAC |
| 31/CYC1t F | AAGGCCCCTTTTCCTTTGTC |
| 32/CYC1t R | CGACGATGAGAGTGTAAACTGC |
| 33/UBiC-pTEF1 F | TATTATGTCGACCTCGAGGCACACACCATAG |
| 34/UBiC-CYC1t R | TATTATGCGGCCGCACGATGAGAGTGTAAACTGC |
Table 2.
Plasmids used in this study.
| Name | Details | Origin |
|---|---|---|
| pRS415 | Yeast centromeric plasmid, LEU2 marker | Euroscarf 52 |
| pRS416 | Yeast centromeric plasmid, URA3 marker | Euroscarf (Sikorski and Hieter 1989) |
| pRS425 | Yeast 2 micron plasmid, LEU2 marker | Euroscarf (Christianson et al. 1992) |
| pUG6 | Contains G418 resistance marker KanMX | Euroscarf (Güldener et al. 1996) |
| TR-SSRE | yEGFP containing plasmid | Chen and Weiss (2005) |
| pPDR12-GFP-415 | pPDR12-yEGFP-ADH1t-pRS415 | This study |
| pYGP1-GFP-415 | pYGP1-yEGFP-ADH1t-pRS415 | This study |
| pTPO2-GFP-415 | pTPO2-yEGFP-ADH1t-pRS415 | This study |
| pPDR12-GFP-425 | pPDR12-yEGFP-ADH1t-pRS425 | This study |
| U-416 | pTEF1-UBiC-CYC1t-pRS416 | This study |
| UA-416 | pTEF1-ARO4-CYC1t-pTEF1-UBiC-CYC1t-pRS416 | This study |
| UA-KanMX-416 | pTEF1-ARO4-CYC1t-pTEF1-UBiC-CYC1t-KanMX-pRS416 | This study |
| pPDR12-GFP- mCherry-415 | pPDR12-yEGFP-ADH1t-pPDA1-mCherry-CYC1t-pRS415 | This study |
| mCherry-415 | pPDA1-mCherry-CYC1t-pRS415 | This study |
The native WAR1 promoter was replaced with the DNA binding target of the WAR1p transcriptional regulator (PDR12 promoter) to create a positive feedback loop using CRISPR-Cas9-mediated (Clustered Regularly Interspersed Short Palindromic Repeat) targeting and homologous recombination. The CRISPR guide RNA expression cassette and Streptococcus pyogenes Cas9 gene from the work reported by DiCarlo et al. (2013) were first combined on the same high copy number plasmid with the HIS3 marker (pRS423). The guide RNA expression cassette comprising the SNR52 promoter, CAN1.Y targeting guide RNA, structural crRNA, and CYC1 terminator from the p426-gRNA.CAN1.Y plasmid (DiCarlo et al. 2013) were PCR amplified (primers 13/14) and Gibson assembled into PCR linearised pRS423 (primers 15/16) to create the ‘gRNA-423’ plasmid. The TEF1 promoter, Cas9 gene, and CYC1 terminator from pTEF1-Cas9-pRS414 (DiCarlo et al. 2013) were PCR amplified with primers 17/18 to create 40-bp homologous overlaps to the SmaI digested gRNA-423 plasmid, and the two were Gibson Assembled to make Cas9-gRNA-423. This plasmid still contained the guide RNA sequence designed to target the CAN1.Y locus from the original CRISPR study (DiCarlo et al. 2013). To generate a WAR1 promoter-targeting guide RNA for CRISPR-mediated knock-in of the PDR12 promoter, the entire Cas9-gRNA-423 plasmid was PCR amplified using primers that bind either side of the existing CAN1.Y guide RNA sequence. These primers (19/20) contained 20 nt 5′ extensions that encode a new CRISPR guide RNA specific for the WAR1 promoter region. The new CRISPR guide RNA encoding extensions on the forward and reverse primers for the Cas9-gRNA-423 plasmid were also 100% homologous to one another so that the linearized PCR product could be circularised to create Cas9-pWAR1-crRNA-423 using Gibson Assembly (Gibson et al. 2009) or Yeast Assembly (Gibson et al. 2008; Shao et al. 2009). In this case, Yeast Assembly was used where PCR linearized and DpnI-treated plasmid (to remove template DNA) was co-transformed with the PDR12 promoter DNA. The PDR12 promoter was PCR amplified from S. cerevisiae BY4741 genomic DNA using primers (21/22) with 5′ extensions that encode 60 bp of homology with the WAR1 promoter region such that the 221 bp 5′ of the WAR1 start codon would be replaced with the 780 bp PDR12 promoter. CRISPR-mediated promoter replacement was achieved by co-transforming approximately 200 ng of Cas9-pWAR1-crRNA-423 and 5 μg of PDR12 promoter PCR product. Cells were plated on SD –His medium to select for the CRISPR plasmid and screened using primers that anneal outside of the WAR1 promoter region (23/24). Only 3 out of 28 colonies screened had the WAR1 promoter replaced with the PDR12 promoter using this method.
The PHBA production pathway comprised a feedback-resistant version (Q166K) (Hartmann et al. 2003; Fukuda et al. 1992) of the 3-deoxy-D-arabino-heptulosonate-7-phosphate (DAHP) synthase enzyme (ARO4 gene) and a chorismate pyruvate lyase enzyme encoded by a codon optimised version of the E. coli UBiC gene, which was previously shown to function in S. cerevisiae using a synthetic quorum sensing circuit (Williams et al. 2013, 2015). Here, UBiC and ARO4 were expressed constitutively using the TEF1 promoter, from the pRS416 plasmid. A previously described plasmid (pTEF1-UBiC-CYC1t-pTEF1-ARO4-CYC1t-pRS406) (Williams et al. 2016) was digested with SalI and NotI to release the UBiC-ARO4 expression cassette, which was gel purified prior to cloning into SalI/NotI digested pRS416 to create pTEF1-UBiC-CYC1t-pTEF1-ARO4-CYC1t-pRS416 (named UA-416). A pRS416 plasmid containing only the pTEF1-UBiC-CYCt expression cassette (U-416) was created by digesting this cassette from the UA-416 plasmid using SalI/EcoRI enzymes and cloning the gel purified product into pRS416. To facilitate easy identification of sorted-cells that contain the PHBA production genes, the G418 antibiotic resistance marker (KanMX) was PCR amplified from pUG6 DNA using primer pair 25/26 and Gibson assembled onto the Eco53KI linearised UA-416 plasmid to make UA-KanMX-416.
A ratiometric biosensor was made by expressing a red fluorescent protein (mCherry) from the constitutive PDA1 promoter (Peng et al. 2015) on the same pRS415 plasmid that the pPDR12-yEGFP biosensor is encoded on. The PDA1 promoter and CYC1 terminator were PCR amplified from BY4741 genomic DNA using primer pairs 27/28 and 31/32, while the mCherry open reading frame was synthesised by IDT as a gBlock and PCR amplified using primer pair 29/30 for Gibson assembly with pPDA1 and CYC1t into SmaI linearised pPDR12-GFP-415 plasmid to make pPDR12-GFP-mCherry-415. The same assembly reaction was carried out with empty, SmaI linearised pRS415 vector to make mCherry-415.
2.5 PCR-mediated identification of producers
Cells with GFP:mCherry fluorescence values deemed to indicate the presence of the UBiC and ARO4Q166K genes were sorted directly onto SD minus leucine and uracil agar plates. After 3–4 days DNA was extracted from single colonies by resuspending them in 50 μl of MilliQ water with 16 units of zymolyase enzyme (Zymo Research catalogue number E1005), incubating at 37 °C for 30–60 minutes then at 95 °C for 10 minutes. Two microlitres of crude DNA extract was used as template for PCR using primers 33/34 that anneal to the TEF1 promoter and CYC1 terminator of the UBiC gene. Touch-down PCR (Korbie and Mattick 2008) was used with the annealing temperature decreasing from 65 to 50 °C at 1 °C per cycle, then staying constant at 50 °C for an additional 20 cycles. Denaturation at 95 °C for 30 seconds and extension at 72 °C for 1 minute 20 seconds was used for each cycle. GoTaq 2x master mix (Promega) was used with a total reaction volume of 20 μl in 96-well plates. Known non-producer and producer colonies were used as negative and positive controls, respectively. All PCR products were visualised on 1% agarose gels stained with SYBR safe (LifeTechnologies) and ran at 100 V for 30 minutes.
3. Results and discussion
3.1 Identification and testing of organic acid responsive promoters
A literature search identified two native yeast promoters that respond to organic acid bio-products. The first operates via the War1p transcriptional regulator protein, which up-regulates transcription of the membrane acid efflux protein encoding PDR12 gene (Piper et al. 1998). Although the exact mechanism of War1p activation is incompletely understood, it appears to involve a combination of direct interaction between intracellular dissociated acid molecules and War1p, and phosphorylation of the protein via an unknown mechanism (Kren et al. 2003; Schüller et al. 2004; Gregori et al. 2008). The second weak acid response module results in the up-regulation of expression of the membrane protein encoding TPO2 and YGP1 genes from the Haa1p transcriptional regulator as part of a rapid response to acid stress (Fernandes et al. 2005). We therefore tested the capacity of the PDR12, YGP1, and TPO2 promoters to regulate the expression of GFP in response to two industrially relevant organic acids, PHBA and propionic acid (PA) (Fig. 1a). The YGP1 promoter showed no difference in GFP expression levels when treated with either PHBA or PA, while the TPO2 promoter displayed slightly up-regulated GFP expression in the presence of PA. In contrast, the PDR12 promoter up-regulated GFP expression approximately 3.4- and 6.2-fold above the level of the control in the presence of PHBA and PA, respectively. This positive response indicated that the War1p-mediated PDR12 promoter (Fig. 1b) has potential to act as a biosensor of exogenously added weak organic acids such as PHBA and PA.
Figure 1.

PHBA and PA responsive promoter screening. (a) Three different promoters that were previously reported to be up-regulated in the presence of organic acids (PDR12, YGP1, and TPO1) were used to control GFP expression in cells treated with water (control), 50 mM PHBA, or 50 mM PA. Mean GFP values from triplicate cultures are shown with error bars representing ± 1 SD. Flow cytometry was used to quantify GFP expression after 3 hours of cultivation. (b) The weak acid response system in yeast maintains neutral cytosolic pH via the exportation of organic acid anions and hydrogen ions form the cell. Organic acids can freely diffuse into the cell in their non-dissociated form (R-COOH) when the pH is below their pKa. Once inside the cell they encounter a neutral pH and exist in a dissociated anionic form (R-COO-) (Holyoak et al. 1999). The hydrogen ions accumulated after acid dissociation are exported from the cell via the Pma1p membrane protein, while acid anions bind to the War1p transcription factor which up-regulates expression of the PDR12 gene encoding a membrane transporter. Pdr12p then transports organic acid anions outside of the cell (Piper et al. 1998).
3.2 Discrimination of intracellular production levels and genetic variants
Critical characteristics for a metabolite-reporter are to: detect metabolites originating from within the cell; distinguish between a wide range of different levels of cellular production from within a genetically diverse population of rapidly growing cells; and facilitate rapid selection by high-throughput screening methods. The pPDR12-GFP biosensor was tested against these performance criteria using PHBA production from the shikimate pathway in yeast (Fig. 2a). PHBA is an aromatic molecule used in liquid crystal polymers, with applications in the electronics and fibre industries and an estimated market of $150 million (USD) per annum (Krömer et al. 2012). It has previously been established that expression of an E.coli chorismate pyruvate lyase (UBiC gene), and feedback resistant S. cerevisiae DAHP synthase results in PHBA production in yeast (Williams et al. 2015, 2016). As a proof-of-concept, the UBiC and ARO4Q166K genes were expressed from strong-constitutive TEF1 promoters in a pPDR12-GFP biosensor-containing strain (Fig. 2a). Expression of only the UBiC gene resulted in a slight but significant (P = 0.009) increase in average GFP expression (Fig. 2b) from 28 to 35 arbitrary units (au). This is highly consistent with previous results that demonstrated a small increase in PHBA production from yeast cultures expressing UBiC (26–46 μM) (Williams et al. 2015). The ARO4Q166K enzyme resists feedback inhibition by downstream metabolites in the shikimate pathway (Fukuda et al. 1992; Hartmann et al. 2003), and over-expression of feedback resistant versions of this enzyme is known to result in an approximately 4- to 5-fold increase in shikimate pathway flux (Luttik et al. 2008), and a 6-fold increase in PHBA production (from 46 to 297 μM)35. In concordance with these results, we observed a significant (P = 1.7 × 10−6) 3-fold increase in pPDR12-GFP expression from cells expressing both UBiC and ARO4Q166K genes (Fig. 2b). These results indicate that War1p-mediated pPDR12-GFP expression can be modulated by intracellular PHBA levels and is sensitive to variations in metabolic flux though a production pathway.
The second performance objective for a metabolite production biosensor is the capacity to enable selection of productive cells from a mixed population of producers and non-producers (Fig. 2c). The pPDR12-GFP biosensor was tested for this characteristic using a simple experiment where cells that contain the genes required for PHBA production (UBiC and ARO4Q166K) also express the geneticin resistance gene KanMX. This strain (GFP.415.UA.KanMX, Table 3) was mixed in a 1:1 ratio with an equivalent biosensor containing strain without PHBA production genes (GFP.415, Table 3). After 3 hours of growth cells within the top 3.4% of GFP fluorescence values were sorted from the population directly onto YPD agar plates. This gate was drawn to visually encompass the top fraction of the population where more events from the producer-only population were likely to occur. To determine if sorted cells actually contain the PHBA production genes, these colonies were replica-plated onto YPD agar containing geneticin (G418), which only producer cells have the capacity to grow on. The majority of sorted colonies (23/35) tested using this method were found to have the G418 resistance and were therefore correctly isolated from the mixed producer/non-producer population based on GFP fluorescence. Conversely, when cells in the bottom 10% of GFP fluorescence values were sorted, as expected none were found to be producers (0/35 able to grow on YPD-G418 plates). This experiment was carried out in growth medium at a pH of 6.5 so that any PHBA molecules (pKa = 4.54) excreted by producer cells exist in their dissociated form outside of the cell, and are unable to diffuse into other cells that are non-producers (Holyoak et al. 1999), thereby preventing GFP expression that would result in the sorting of false positives. These results validate the use of the War1p-mediated pPDR12-GFP system as a biosensor for War1p interacting organic acid metabolites produced from engineered metabolic pathways in yeast.
An interesting feature of the pPDR12-GFP biosensor populations was the high noise level of the non-producer control strain, as assessed by flow cytometry (Fig. 2d). This strain was cultured without PHBA production pathway genes, and without the addition of PHBA to the growth medium, yet 49% of the cells had the same high level of GFP expression seen in the producer population (Fig. 2d). Furthermore there were two sub-populations, with most cells (51%) residing in a low-GFP state that is equivalent to yeast auto-fluorescence. When biosensor-containing cells also had UBiC and ARO4Q166K genes for PHBA production the majority of cells (85%) existed in the high-GFP state (Fig. 2d). By only sorting cells in the top 3.4% of fluorescence levels, it was possible to isolate producers based on GFP expression with 66% accuracy (23/35 colonies able to grow on YPD-G418 plates). This relatively low level of sorting accuracy probably arose because the gate used to isolate producers overlapped significantly with the non-producer population. A greater separation of producer and non-producer fluorescence levels would enable the use of a gate that excluded all GFP fluorescence levels observed in the non-producer population. With these strains this is not possible (Fig. 2d–e), making the system impractical for further high-throughput screening applications. Ideally, a much higher separation of producer-GFP expression from non-producer-GFP expression would exist so that producers could be more easily isolated. This led us to consider ways of improving the dynamic range of GFP expression, and reducing the noise levels of the pPDR12-GFP biosensor.
3.3 Fine-tuning sensor dynamic range using positive feedback
An ideal biosensor has a large dynamic-range of output levels (e.g. GFP expression), low non-induced expression, and is effective at differentiating between a wide-range of input concentrations. The pPDR12-GFP biosensor was tested for dose-dependent response to externally applied PHBA with pPDR12-GFP expression from low- or high-copy vectors (pRS415 or pRS425) (Fig. 3a). In each case, there was a dose-dependent increase in GFP expression, with a 2.1-fold dynamic range from the high-copy vector and a 2.9-fold dynamic range with the low-copy vector, and a response range between 10 and 75 mM PHBA. Increasing GFP copy-number via expression from the pRS425 vector had little effect on these parameters and served primarily to increase the uninduced GFP expression level (Fig. 3a). The fold-change observed in this native system was not ideal for use as a high-throughput screening biosensor, we therefore sought to implement genetic modifications in the yeast weak acid response module that would increase the dynamic range and sensitivity of the biosensor output.
Figure 3.

Tuning biosensor output using positive feedback. (a) PHBA dose response curves for strains with pPDR12-GFP expression form low- or high-copy vectors (pRS415 or pRS425). (b) Circuit configuration of a positive feedback biosensor. The native WAR1 promoter was replaced with the PDR12 promoter such that the organic acid anion responsive War1p transcription factor regulates its own expression as part of a positive feedback loop, in addition to pPDR12-GFP expression. pPDR12-GFP expression levels in strains with the native WAR1 promoter or positive-feedback pPDR12-WAR1 expression in response to PHBA (c) or propionic acid (d). Mean GFP values and SDs from triplicate cultures are shown.
Positive feedback loops are commonly used synthetic biology devices that can reduce non-specific expression while increasing dynamic range and sensitivity (Ingolia et al. 2007; Williams et al. 2013). In order to implement a positive feedback loop in the weak acid response system in yeast, we replaced the native promoter that regulates the expression of the War1p transcription factor with its target PDR12 promoter (Fig. 3b) using CRISPR-Cas9-mediated homologous recombination (see Methods section for details). With this re-configuration the War1p transcription factor should in theory only be expressed at low-levels from ‘leaky’ non-induced PDR12 promoter expression in a cell without weak acid production/exposure. Upon response to weak acid molecules, the amount of War1p available for GFP expression induction from the PDR12 promoter should sharply increase as War1p starts to induce its own expression as part of a positive-feedback loop (Fig. 3b). When PHBA and PA dose–response experiments (Fig. 3c and d) were carried out on cells with positive feedback War1p expression, there was a significant increase in the dynamic range of GFP expression. The dynamic range increased from to 2.9- to 4.2-fold in the presence of PHBA and from 3.6- to 10-fold with PA (Fig. 3c and d). The basal non-induced GFP-expression levels were also reduced by between 7 and 19% due to positive-feedback War1p expression, as expected from the positive-feedback model (zero acid GFP values not shown in Fig. 3a–d due to the log scale on the x-axis).
In theory, positive feedback expression of War1p results in a lower basal-concentration of GFP in the absence of inducer, and a higher maximum expression-level upon induction, resulting in a higher dynamic range. While our findings are consistent with this model, it is also possible for positive-feedback loops to alter the timing and rate of gene expression (Ingolia et al. 2007; Williams et al. 2013). Therefore, another possible explanation for our observations could be that the dynamic ranges of the native and positive feedback War1p expression systems are actually the same, but that the positive-feedback system is simply induced faster. We therefore tested the pPDR12-GFP expression levels from both systems after PA induction over 7 hours (Fig. 4). Although the initial rate of increase in GFP expression was much higher in the positive-feedback strain (0 to 2.5 hours, Fig. 4), a constant difference of ∼130 au was reached after 2.5 hours and the high-GFP state of the positive-feedback system was not attained by the native one within the time-frame of this experiment. These observations further demonstrate the utility of the positive feedback system for reducing basal expression levels and increasing dynamic range, and validate the use of the 3-hour sampling point that we employed for other experiments in this study.
Figure 4.
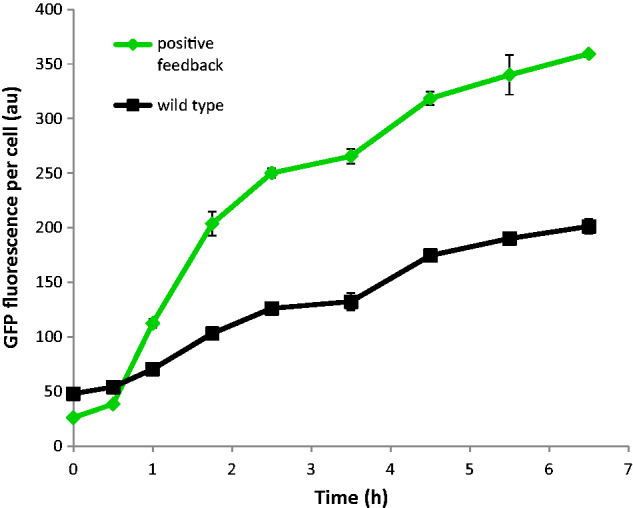
Time-course biosensor induction. Average GFP expression-levels of native (wild-type, black squares) and positive-feedback (green diamonds) War1p expression systems. Each strain was induced with 1.5 mM of propionic acid at time zero. Mean GFP values and standard deviations from triplicate cultures are shown.
3.4 Controlling noise using a ratiometric biosensor
High noise-levels in gene expression across a population are a universal feature of biological systems (Elowitz et al. 2002), and a constant bane to synthetic biologists. Gene expression noise can be attributed to extrinsic factors such as differences in cell size and cycle, substrate uptake rates and metabolic fluxes, as well as variations in the concentrations of other biomolecules that affect a cell’s capacity to express genes such as; plasmid copy number, transcription factors, RNA polymerase, ribosomes, and ATP (Elowitz et al. 2002). Although variations in plasmid copy-number are known to contribute significantly to noise levels (Zhang et al. 2016), we chose to use the low-copy yeast vector pRS415 for biosensor expression instead of genomic integration to facilitate rapid prototyping of different biosensors. Even with genomic integration of reporter systems such as GFP, there are still significant levels of noise due to stochastic variations in the concentrations of other biomolecules between individual cells in a population (Elowitz et al. 2002). In the context of biosensor-mediated cell sorting, noisiness in gene expression can mean that the throughput of screening is greatly reduced. Although a responsive population may have a much higher average GFP level, there can be significant overlap between ligand-responsive and control populations. Populations can be gated such that only cells with GFP levels higher than a non-productive control population are sorted, but if there is too much noise then the number of cells that are available for selection is dramatically reduced. Mitigating the effects of highly variable gene expression is therefore of critical importance to biosensor-mediated high-throughput screening. One potential way to achieve this is to ‘normalise’ the level of biosensor output (GFP) to another constitutively expressed fluorescent protein. Because the second fluorescent protein is constitutively regulated, its expression level should serve as a proxy for aspects of cellular physiology that contribute to gene expression noise. In theory, cells can then be selected based on the ratio of biosensor-mediated GFP expression to constitutive fluorescent protein expression within a single cell.
In order to reduce extrinsic noise in biosensor output and increase screening throughput, we converted our positive-feedback biosensor into a ratiometric sensor. This was achieved by expressing a red fluorescent protein (mCherry) from the weak-constitutive PDA1 promoter (Peng e al. 2015) alongside the weak-acid responsive pPDR12-GFP biosensor on the same plasmid (pPDR12-GFP- mCherry-415, Table 2), in the same strain (+FB.GFP.mCherry.415, Table 3) (Fig. 5a). By plotting mCherry fluorescence on the x-axis and GFP fluorescence on the y-axis, the ratio of GFP to mCherry in individual cells can be measured using flow-cytometry (Fig. 5b). In theory, the level of mCherry reflects a cell’s general capacity for gene expression, and enables the inclusion of low GFP expressing, but organic acid responding/producing cells in high-throughput screening. The fact that there is a positive linear relationship between mCherry fluorescence and GFP fluorescence (Fig. 5b) supports the idea that a constitutively expressed fluorescent protein can be used as a kind of ‘internal standard’ for a cell’s gene expression capacity. When a ratiometric biosensor strain was treated with and without a saturating concentration of PA (1.5 mM) and analysed using only GFP fluorescence, there was a large overlap between the groups with approximately 34% of the cells in the treated population within the range of GFP fluorescence values observed in the non-treated population (Fig. 5c). When the same comparison was made using the ratio of GFP to mCherry within each cell, the overlap between the two populations was approximately 1% (Fig. 5d). This demonstrated the efficiency of the ratiometric approach for controlling noise in the biosensor population. The same trend was observed with PHBA treatment, although there was a less pronounced effect (Figure S1 in the Supplementary Data). This is consistent with the previously observed weaker biosensor induction using PHBA relative to PA (Fig. 3c and d).
Figure 5.

Enhanced population separation using a ratiometric biosensor. (a) Positive-feedback, ratiometric biosensor configuration (+FB.GFP.mCherry.415 strain, Table 3) whereby War1p-mediated biosensor output (GFP expression) is normalised to constitutively expressed mCherry in each individual cell. (b) GFP:mCherry fluorescence distribution of the positive-feedback ratiometric biosensor strain (+FB.GFP.mCherry.415, Table 3). (c) The same strain treated with (black bars) and without (grey bars) 1.5 mM propionic acid is analysed using only GFP fluorescence or (d) using the ratio of GFP to mCherry fluorescence within each cell. Data from single cultures which are representative of repeated experiments are shown here.
The concept of using ratiometric fluorescence normalisation to increase the signal to noise ratios of sensors has existed for some time (Demchenko et al. 2010) and has most commonly been exploited in the form of Forster Resonance Energy Transfer (FRET) systems where a different emission spectrum results from close fluorophore proximity. FRET systems have previously been used as in vivo metabolite biosensors (Michener et al. 2012), but are yet to be used for high-throughput screening of producer-cells. Similarly, ratiometric fluorescent protein normalisation was recently employed by Zhang et al (2016) to improve the signal-to-noise ratio of an in vivo NADP+/NADPH reporter with great success. However, the ratiometric approach was not used for high-throughput metabolite producer screening, and as far as we are aware, ratiometric biosensor expression has not previously been applied to high-throughput metabolite-producer screening. Given the efficiency of ratiometric fluorescent protein normalisation at reducing the signal to noise ratio of biosensor output, we sought to explore how this mode of biosensor expression affects the efficiency of high-throughput screening.
The decrease in overlap that we observed between treated and non-treated biosensor populations (Fig. 5c and d) should also result in increased high-throughput screening power, as a greater proportion of a given population is available for selection using gates that exclude control population fluorescence values. The producer-screening efficiency of the positive-feedback-ratiometric biosensor was therefore tested by attempting to sort producer cells mixed with non-producers at a variety of ratios. The success of fluorescence based PHBA-producer sorting was determined using PCR with a forward primer specific to the TEF1 promoter and a reverse primer specific to the CYC1 terminator region of the UBiC gene expression cassette, on DNA extracted from single colonies arising from sorted cells on agar plates.
Mixed populations with producer to non-producer ratios varying from 1:1 through to 1:1×105 were used to test the limits of our biosensor’s sorting accuracy. When pure producer and non-producer populations from the initial non-positive-feedback, and positive-feedback biosensor strains were compared it was not possible to make a sorting gate in the producer population that did not overlap with the non-producer population, as previously observed (Fig. 2d and e). In contrast, when the GFP:mCherry fluorescence levels of producer and non-producer strains containing the positive-feedback ratiometric biosensor (strains +FB.GFP.mCherry.415.UA and +FB.GFP. mCherry.415, Table 3) were compared there was a clear separation of fluorescence levels that could be used to define a sorting gate (Fig. 6a). It should be noted that we used a high-power blue laser (200 mW, 488 nm) to excite mCherry (excitation maximum 587 nm) so it is likely that using a green laser, which is common option on many flow sorters, may significantly improve the separation of producers from non-producers. Furthermore, when mixed producer/non-producer populations were visualised using density plots after 3 hours of co-culturing, the two populations were still clearly distinguishable (Fig. 6a). This again indicates that the technique of culturing mixed populations at a pH above the pKa of the secreted organic acid product (pH 6.5 and PHBA pKa 4.54 in this case) prevents the PHBA molecules produced by one cell entering another cell in the population and potentially activating the biosensor of a non-producer. This would lead to an averaging-out of the fluorescence levels observed in the two pure populations, and was not observed here (Fig. 6a). When cells were sorted from mixed populations ranging from 1:1 to 1:105 producer to non-producers, the positive-feedback ratiometric biosensor was highly efficient at enabling the correct identification of producers based on GFP:mCherry ratios (Fig. 6b). 100% of isolates from the 1:1 through to 1:102 producer:non-producer populations were confirmed as producers via PCR. Sorting accuracy dropped to ∼88% at 1:103 and to ∼66% at 1:104, while no producers could be correctly identified at 1:105 using this strategy (Fig. 6b). This a dramatic increase in high-throughput screening power when compared with the original non-positive feedback non-ratiometric version of the biosensor (strain GFP.415, Table 3), which only enabled 66% sorting accuracy with equal amounts of producers and non-produces in a mixed population (Fig. 2c–e). The 66% sorting accuracy we observed with the positive-feedback, ratiometric biosensor therefore represents a 5,000-fold improvement in high-throughput screening power. The magnitude of this improvement can partly be attributed to the poor-performance of the original War1p-pPDR12 system as a biosensor. It is therefore likely that positive-feedback, ratiometric biosensor expression would result in less dramatic fold-improvements of biosensors that have inherently lower noise-levels and higher dynamic ranges.
Figure 6.

Positive-feedback, ratiometric biosensor mixed population cell sorting. Strains with positive-feedback WAR1 expression, pPDR12-GFP expression and constitutive PDA1 promoter-mediated mCherry expression, containing either the control pRS416 plasmid or the UBiC-ARO4 expression plasmid were mixed together in different ratios to test the efficiency of biosensor mediated high-throughput screening. a) Flow cytometry density plot of PHBA producers mixed with non-producers at a ratio of 1 in 10. One hundred thousand events were recorded with increasing density indicated by the transition through blue, green, yellow, and red with mCherry (x-axis) and GFP expression (y-axis) measured simultaneously for each cell. The red coloured ‘sorting gate’ indicates the range of fluorescence values used to sort producers from non-producers. b) Producers were diluted with non-producers by the indicated factors and cultured together for 3 hours prior to sorting cells through the gate depicted in (a) onto agar plates. DNA extracted from colonies arising from individual sorted cells was used to test for the presence of the UBiC gene using PCR. Colonies giving rise to amplification were identified as being correctly sorted cells based on biosensor fluorescence. Between three and eight colonies were analysed for each dilution level with the proportion of producers identified expressed as a percentage (y-axis). The experiment was done in biological duplicate on different days, with raw data plotted (replicate 1 in black diamonds, replicate 2 in grey triangles).
These results demonstrate the utility of the positive-feedback ratiometric biosensor approach for improving high-throughput screening efficiency via controlling for noise, reducing basal biosensor output, and increasing dynamic range. When applying this sensor to isolate producer cells from randomly mutated or evolving populations, the frequency of cells containing mutations that encode for increased metabolite production are likely to be much lower than 1 in 104, the limit of accuracy for our biosensor. However it has recently been demonstrated that repeated cycles of enrichment for cells with high biosensor output using FACS can be used to concentrate high producers over time and identify rare mutational events that lead to higher productivity (Mahr et al. 2015). We envisage that a similar process of iterative growth and FACS enrichment could be successfully employed to evolve PHBA or PA-producing yeast using our biosensor. The highest previously recorded titre of PHBA in yeast is ∼ 1 mM, and this level was achieved by simultaneously expressing three pathway enzymes (UBiC, ARO4, TKL1) and dynamically repressing two enzymes (ARO7, CDC19) that compete for carbon flux (Williams et al. 2015). Although the dynamic range of our biosensor is nearly saturated via the expression of UBiC and ARO4 genes (Fig. 2b), it should be possible to discover novel mutations that affect shikimate pathway flux by carrying out biosensor-mediated directed evolution experiments beginning with only UBiC gene expression. In theory this approach should enable the exploration of evolutionary trajectories that are independent of the traditional feedback resistant ARO4 enzyme (Luttik et al. 2008). It is also possible that this biosensor could be used to screen PHBA production (or other organic acids) from other cells in microtiter plates as part of a co-culture system. PHBA in particular has been produced at high levels in E. coli (12 g/L) (Barker and Frost 2001), Klebsiella pneumonia, and Pseudomonas putida (317 mg/L) (Verhoef et al. 2010), and this biosensor has the potential to be used to screen modified or mutated strains of these producers, albeit at a much lower throughput compared with FACS.
4. Summary and Conclusions
Ligand-responsive transcriptional regulators and their cognate promoters are widely used tools in synthetic biology (Taylor et al. 2016), and are becoming increasingly valuable as metabolite biosensors for high-throughput strain screening and dynamic pathway regulation in metabolic engineering (Zhang and Keasling 2011; Liu et al. 2015; Mahr et al. 2016; Rogers et al. 2016; Williams et al. 2016). We have demonstrated that positive-feedback biosensor expression significantly reduces the basal expression level and increases the dynamic range. Furthermore ratiometric fluorescent protein ‘normalisation’ within single cells provides a control system for extrinsic noise, increasing the efficiency and accuracy of high-throughout strain screening. By combining positive-feedback biosensor expression with ratiometric fluorescence normalisation, high-throughput screening efficiency was improved 5,000-fold. These phenomena were demonstrated using the WAR1 organic acid responsive transcriptional regulator in yeast, which holds great promise for use as a biosensor for isolating highly productive organic acid production strains from randomly mutated or evolving populations. Due to the fact that gene expression noise is a universal feature of biological systems (Elowitz et al. 2002; Keren et al. 2015) the design principles of positive-feedback, ratiometric biosensor expression are likely to be relevant to many other transcription-factor promoter pairs used in synthetic biology.
Author Contributions
Conceived of the project (T.C.W.), designed the experiments (T.C.W., I.T.P.), performed the experiments (T.C.W., X.X.), analysed the data (T.C.W.), trained T.C.W. in flow cytometry techniques and assisted with experimental design (M.O.), wrote the manuscript (T.C.W., I.T.P.), edited and reviewed the manuscript (T.C.W., X.X., M.O., I.S.P., I.T.P.), project support for T.C.W. and X.X. (I.S.P., I.T.P.).
Supplementary Material
Acknowledgements
The Synthetic Biology initiative at Macquarie University is financially supported by an internal grant from the University, and external grants from Bioplatforms Australia, the New South Wales (NSW) Chief Scientist and Engineer, and the NSW Government’s Department of Primary Industries. Ian Paulsen is supported by an Australian Research Council Laureate Fellowship.
Data availability
All raw data are available with the online version of this article.
Supplementary data
Supplementary data are available at Synthetic Biology online.
Conflict of interest: None declared.
References
- Abbott D. A., Zelle R. M., Pronk J. T., van Maris A. J. A. (2009) ‘Metabolic Engineering of Saccharomyces cerevisiae for Production of Carboxylic Acids: Current Status and Challenges’, FEMS Yeast Research, 9: 1123–36. [DOI] [PubMed] [Google Scholar]
- Agren R., Otero J. M., Nielsen J. (2013) ‘Genome-Scale Modeling Enables Metabolic Engineering of Saccharomyces cerevisiae for Succinic Acid Production’, Journal of Industrial Microbiology and Biotechnology, 40: 735–47. [DOI] [PubMed] [Google Scholar]
- Almario M. P., Reyes L. H.,, Kao K. C. (2013) ‘Evolutionary Engineering of Saccharomyces cerevisiae for Enhanced Tolerance to Hydrolysates of Lignocellulosic Biomass’, Biotechnology Bioengineering 110: 2616–23. [DOI] [PubMed] [Google Scholar]
- Averesch N. J. H., Krömer J. O. (2014) ‘Tailoring Strain Construction Strategies for Muconic Acid Production in S. cerevisiae and E. coli’, Metabolic Engineering Communications, 1: 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek S. H., Kwon E. Y., Kim Y. H., Hahn J. S. (2015) ‘Metabolic Engineering and Adaptive Evolution for Efficient Production of D-Lactic Acid in Saccharomyces cerevisiae’, Applied Microbiology and Biotechnology 100: 2737–48 [DOI] [PubMed] [Google Scholar]
- Barker J. L., Frost J. W. (2001) ‘Microbial Synthesis of p-Hydroxybenzoic Acid From Glucose’, Biotechnology Bioengineering, 76: 376–90. [DOI] [PubMed] [Google Scholar]
- Blazeck J. et al. (2014) ‘Metabolic Engineering of Saccharomyces cerevisiae for Itaconic Acid Production’, Applied Microbiology and Biotechnology, 98: 8155–64. [DOI] [PubMed] [Google Scholar]
- Borodina I. et al. (2015) ‘Establishing a Synthetic Pathway for High-Level Production of 3-Hydroxypropionic Acid in Saccharomyces cerevisiae via β-Alanine’, Metabolic Engineering, 27: 57–64. [DOI] [PubMed] [Google Scholar]
- Brennan T. C. et al. (2015) ‘Evolutionary Engineering Improves Tolerance for Replacement Jet Fuels in Saccharomyces cerevisiae’, Applied and Environmental Microbiology, 81: 3316–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspeta L. et al. (2014) ‘Altered Sterol Composition Renders Yeast Thermotolerant’, Science, 346: 75–8. [DOI] [PubMed] [Google Scholar]
- Chen Y., Nielsen J. (2016) ‘Biobased Organic Acids Production by Metabolically Engineered Microorganisms’, Current Opinion in Biotechnology, 37: 165–72. [DOI] [PubMed] [Google Scholar]
- Chen M.-T.,, Weiss R. (2005) ‘Artificial Cell-Cell Communication in Yeast Saccharomyces cerevisiae Using Signaling Elements From Arabidopsis thaliana’, Nature Biotechnology, 23: 1551–5. [DOI] [PubMed] [Google Scholar]
- Christianson T. W. et al. (1992) ‘Multifunctional Yeast High-Copy-Number Shuttle Vectors, Gene 110: 119–22 [DOI] [PubMed] [Google Scholar]
- Curran K. A., Leavitt J. M., Karim A. S.,, Alper H. S. (2013) ‘Metabolic Engineering of Muconic Acid Production in Saccharomyces cerevisiae’, Metabolic Engineering, 15: 55–66. [DOI] [PubMed] [Google Scholar]
- Demchenko A. P. (2010) ‘The Concept of λ-Ratiometry in Fluorescence Sensing and Imaging’, Journal of Fluorescence, 20: 1099–128. [DOI] [PubMed] [Google Scholar]
- DiCarlo J. E. et al. (2013) ‘Genome Engineering in Saccharomyces cerevisiae Using CRISPR-Cas Systems’, Nucleic Acids Research 41: 4336–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz M. B., Levine A. J., Siggia E. D.,, Swain P. S. (2002) ‘Stochastic Gene Expression in a Single Cell’, Science, 297: 1183–6. [DOI] [PubMed] [Google Scholar]
- Fernandes A. R. et al. (2005) ‘Saccharomyces cerevisiae Adaptation to Weak Acids Involves the Transcription Factor Haa1p and Haa1p-Regulated Genes’, Biochemical and Biophysical Research Communications, 337: 95–103. [DOI] [PubMed] [Google Scholar]
- Fukuda K., Asano K., Ouchi K.,, Takasawa S. (1992) ‘Feedback-Insensitive Mutation of 3-Deoxy-d-Arabino-Hepturosonate-7-Phosphate Synthase Caused by a Single Nucleotide Substitution of ARO4 Structural Gene in Saccharomyces cerevisiae’, Journal of Fermentation and Bioengineering, 74: 117–9. [Google Scholar]
- Garcia Sanchez R. et al. (2010) ‘Improved Xylose and Arabinose Utilization by an Industrial Recombinant Saccharomyces cerevisiae Strain Using Evolutionary Engineering’, Biotechnology for Biofuels 3: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos M. T. et al. (1997) ‘Arac/XylS Family of Transcriptional Regulators’, Microbiology and Molecular Biology Reviews, 61: 393–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson D. G. et al. (2008) ‘One-Step Assembly in Yeast of 25 Overlapping DNA Fragments to Form a Complete Synthetic Mycoplasma genitalium Genome’, Proceedings of the National Academy of Sciences 105: 20404–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson D. G. et al. (2009) ‘Enzymatic Assembly of DNA Molecules Up To Several Hundred Kilobases’, Nature Methods, 6: 343–5. [DOI] [PubMed] [Google Scholar]
- Gietz R. D.,, Schiestl R. H. (2007) ‘Quick and Easy Yeast Transformation Using the LiAc/SS Carrier DNA/PEG Method’, Nature Protocols, 2: 35–7 [DOI] [PubMed] [Google Scholar]
- Goodarzi H. et al. (2010) ‘Regulatory and Metabolic Rewiring During Laboratory Evolution of Ethanol Tolerance in E. coli’, 6: 378. [DOI] [PMC free article] [PubMed]
- Gregori C. et al. (2008) ‘Weak Organic Acids Trigger Conformational Changes of the Yeast Transcription Factor War1 in Vivo to Elicit Stress Adaptation, Journal of Biological Chemistry 283: 25752–64. [DOI] [PubMed] [Google Scholar]
- Güldener U. et al. (1996) ‘A New Efficient Gene Disruption Cassette for Repeated Use in Budding Yeast’, Nucleic Acids Research, 24: 2519–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann M. et al. (2003) ‘Evolution of Feedback-Inhibited β/α Barrel Isoenzymes by Gene Duplication and a Single Mutation,’ Proceedings of the National Academy of Sciences 100: 862–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holyoak C. D. et al. (1999) ‘The Saccharomyces cerevisiae Weak-Acid-Inducible ABC Transporter Pdr12 Transports Fluorescein and Preservative Anions from the Cytosol by an Energy-Dependent Mechanism’, Journal of Bacteriology, 181: 4644–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N. T.,, Murray A. W. (2007) ‘Positive-Feedback Loops as a Flexible Biological Module’ Current Biology 17: 668–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida N. et al. (2006) ‘Metabolic Engineering of Saccharomyces cerevisiae for Efficient Production of Pure L-(+)-Lactic Acid’, Applied Biochemistry and Biotechnology, 129–132: 795–807. [PubMed] [Google Scholar]
- Ito Y., Hirasawa T.,, Shimizu H. (2014) ‘Metabolic Engineering of Saccharomyces cerevisiae to Improve Succinic Acid Production Based on Metabolic Profiling’, Bioscience, Biotechnology, and Biochemistry, 78: 151–9. [DOI] [PubMed] [Google Scholar]
- Kearse M. et al. (2012) ‘Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data’, Bioinformatics, 28: 1647–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren L. et al. (2015) ‘Noise in Gene Expression Is Coupled to Growth Rate, Genome Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kildegaard K. R., Wang Z., Chen Y., Nielsen J., Borodina I. (2015) ‘Production of 3-Hydroxypropionic Acid From Glucose and Xylose by Metabolically Engineered Saccharomyces cerevisiae’, Metabolic Engineering Communications, 2: 132–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kildegaard K. R. et al. (2014) ‘Evolution Reveals a Glutathione-Dependent Mechanism of 3-Hydroxypropionic Acid Tolerance’, Metabolic Engineering, 26: 57–66. [DOI] [PubMed] [Google Scholar]
- Korbie D. J.,, Mattick J. S. (2008) ‘Touchdown PCR for Increased Specificity and Sensitivity in PCR Amplification’, Nature Protocols, 3: 1452–6. [DOI] [PubMed] [Google Scholar]
- Kren A. et al. (2003) ‘War1p, a Novel Transcription Factor Controlling Weak Acid Stress Response in Yeast’, Molecular Cell Biology, 23: 1775–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krömer J. O. et al. (2012) ‘Production of Aromatics in Saccharomyces cerevisiae—A Feasibility Study’, Journal of Biotechnology 163: 184–93. [DOI] [PubMed] [Google Scholar]
- Nielsen J.,, Keasling J. D. (2016) ‘Engineering Cellular Metabolism’, Cell, 164; 1185–97. [DOI] [PubMed] [Google Scholar]
- Liu D., Evans T.,, Zhang F. (2015) ‘Applications and Advances of Metabolite Biosensors for Metabolic Engineering’, Metabolic Engineering, 31; 35–43. [DOI] [PubMed] [Google Scholar]
- Luttik M. A. H. et al. (2008) ‘Alleviation of Feedback Inhibition in Saccharomyces cerevisiae Aromatic Amino Acid Biosynthesis: Quantification of Metabolic Impact’, ‘Metabolic Engineering, 10: 141–53. [DOI] [PubMed] [Google Scholar]
- Mahr R. et al. (2015) ‘Biosensor-Driven Adaptive Laboratory Evolution of l-Valine Production in Corynebacterium glutamicum’, Metabolic Engineering, 32: 184–94. [DOI] [PubMed] [Google Scholar]
- Mahr R.,, Frunzke J. (2016) ‘Transcription Factor-Based Biosensors in Biotechnology: Current State and Future Prospects’, Applied Microbiology and Biotechnology, 100: 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michener J. K., Thodey K., Liang J. C.,, Smolke C. D. (2012) ‘Applications of Genetically-Encoded Biosensors for the Construction and Control of Biosynthetic Pathways’, Metabolic Engineering, 14: 212–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil K. R., Åkesson M.,, Nielsen J. (2004) ‘Use of Genome-Scale Microbial Models for Metabolic Engineering’, Current Opinion in Biotechnology, 15: 64–9. [DOI] [PubMed] [Google Scholar]
- Peng B. et al. (2015) ‘Controlling Heterologous Gene Expression in Yeast Cell Factories on Different Carbon Substrates and Across the Diauxic Shift: A Comparison of Yeast Promoter Activities’, Microbial Cell Factories, 14: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper P. et al. (1998) ‘The pdr12 ABC Transporter Is Required for the Development of Weak Organic Acid Resistance in Yeast’, The EMBO Journal, 17: 4257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pretorius I. S. (2016) ‘Synthetic Genome Engineering Forging New Frontiers for Wine Yeast’, Critical Reviews in Biotechnology 37: 112–36. [DOI] [PubMed] [Google Scholar]
- Quan S. et al. (2012) ‘Adaptive Evolution of the Lactose Utilization Network in Experimentally Evolved Populations of Escherichia coli’, PLoS Genet. 8: e1002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab A. M. et al. (2010) ‘Metabolic Engineering of Saccharomyces cerevisiae for the Biotechnological Production of SUCCINIC Acid’, Metabolic Engineering, 12: 518–25. [DOI] [PubMed] [Google Scholar]
- Ramos J. L. et al. (2005) ‘The TetR Family of Transcriptional Repressors’, Microbiology and Molecular Biology Reviews, 69: 326–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riehle M. M., Bennett A. F., Lenski R. E.,, Long A. D. (2003) ‘Evolutionary Changes in Heat-Inducible Gene Expression in Lines of Escherichia coli Adapted to High Temperature’, Physiological Genomics, 14: 47–58. [DOI] [PubMed] [Google Scholar]
- Rogers J. K., Taylor N. D.,, Church G. M. (2016) ‘Biosensor-Based Engineering of Biosynthetic Pathways’, Current Opinion in Biotechnology, 42: 84–91. [DOI] [PubMed] [Google Scholar]
- Sambrook J.,, Russell D. W. (2001) Molecular Cloning: A Laboratory Manual, 1, pp. 11–258. New York, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Sauer M., Porro D., Mattanovich D., Branduardi P. (2008) ‘Microbial Production of Organic Acids: Expanding the Markets’, Trends in Biotechnology, 26: 100–8. [DOI] [PubMed] [Google Scholar]
- Schüller C. et al. (2004) ‘Global Phenotypic Analysis and Transcriptional Profiling Defines the Weak Acid Stress Response Regulon in Saccharomyces cerevisiae’, Molecular Biology of the Cell, 15: 706–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski R. S.,, Hieter P. (1989) ‘A System of Shuttle Vectors and Yeast Host Strains Designed for Efficient Manipulation of DNA in Saccharomyces cerevisiae’, Genetics, 122: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z., Zhao H.,, Zhao H. (2009) ‘DNA Assembler, an In Vivo Genetic Method for Rapid Construction of Biochemical Pathways, Nucleic Acids Research, 37: e16–e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor N. D. et al. (2016) ‘Engineering an Allosteric Transcription Factor to Respond to New Ligands’, Nature Methods 13: 177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropel D.,, van der Meer J. R. (2004) ‘Bacterial Transcriptional Regulators for Degradation Pathways of Aromatic Compounds’, Microbiology and Molecular Biology Reviews, 68: 474–500, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoef S. et al. (2010) ‘Comparative Transcriptomics and Proteomics of p-Hydroxybenzoate Producing Pseudomonas putida S12: Novel Responses and Implications for Strain Improvement’, Applied Microbiology and Biotechnology, 87: 679–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiechert W. (2002) ‘Modeling and Simulation: Tools for Metabolic Engineering’, Journal of Biotechnology 94: 37–63. [DOI] [PubMed] [Google Scholar]
- Williams T. C. et al. (2015) ‘Quorum-Sensing Linked RNA Interference for Dynamic Metabolic Pathway Control in Saccharomyces cerevisiae’, Metabolic Engineering, 29: 124–34. [DOI] [PubMed] [Google Scholar]
- Williams T. C., Nielsen L. K., Vickers C. E. (2013) ‘Engineered Quorum Sensing Using Pheromone-Mediated Cell-to-Cell Communication in Saccharomyces cerevisiae. ACS Synthetic Biology, 2: 136–49. [DOI] [PubMed] [Google Scholar]
- Williams T. C., Peng B., Vickers C. E.,, Nielsen L. K. (2016) ‘The Saccharomyces cerevisiae Pheromone-Response Is a Metabolically Active Stationary Phase for Bio-Production’, Metabolic Engineering Communications 3: 142–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T. C., Pretorius I. S.,, Paulsen I. T. (2016) ‘Synthetic Evolution of Metabolic Productivity Using Biosensors’, Trends Biotechnology, 34: 371–81. [DOI] [PubMed] [Google Scholar]
- Wisselink H. W. et al. (2009) ‘Novel Evolutionary Engineering Approach for Accelerated Utilization of Glucose, Xylose, and Arabinose Mixtures by Engineered Saccharomyces cerevisiae Strains’, Applied and Environmental Microbiology, 75: 907–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelle R. M. et al. (2008) ‘Malic Acid Production by Saccharomyces cerevisiae: Engineering of Pyruvate Carboxylation, Oxaloacetate Reduction, and Malate Export’, Applied and Environmental Microbiology, 74: 2766–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. et al. (2016) ‘Engineering an NADPH/NADP+ Redox Biosensor in Yeast’ ACS Synthetic Biology 5: 1546–56. [DOI] [PubMed] [Google Scholar]
- Zhang F.,, Keasling J. (2011) ‘Biosensors and Their Applications in Microbial Metabolic Engineering’, Trends in Microbiology, 19: 323–9. [DOI] [PubMed] [Google Scholar]
- Zhang J., Jensen M. K.,, Keasling J. D. (2015) ‘Development of Biosensors and Their Application in Metabolic Engineering’, Current Opinion in Chemical Biology, 28: 1–8. [DOI] [PubMed] [Google Scholar]
- Zhou H. et al. (2012) ‘Xylose Isomerase Overexpression Along With Engineering of the Pentose Phosphate Pathway and Evolutionary Engineering Enable Rapid Xylose Utilization and Ethanol Production by Saccharomyces cerevisiae’, Metabolic Engineering, 14: 611–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data are available with the online version of this article.