

Abstract
Because of the technological limitations of de novo DNA synthesis in (i) making constructs containing tandemly repeated DNA sequence units, (ii) making an unbiased DNA library containing DNA fragments with sequence multiplicity in a specific region of target genes, and (iii) replacing DNA fragments, development of efficient and reliable biochemical gene assembly methods is still anticipated. We succeeded in developing a biological standardized genetic parts that are flanked between a common upstream and downstream nucleotide sequences in an appropriate plasmid DNA vector (BioBrick)-based novel assembly method that can be used to assemble genes composed of 25 tandemly repeated BioBricks in the correct format in vitro. We named our new DNA part assembly system: ‘Quick Gene Assembly (QGA)’. The time required for finishing a sequential fusion of five BioBricks is less than 24 h. We believe that the QGA method could be one of the best methods for ‘gene construction based on engineering principles’ at the present time, and is also a method suitable for automation in the near future.
Keywords: BioBrick, engineering principle, gene assembly, gene designing, magnetic beads
1. Introduction
Recent advances in genomics have allowed a great deal of genome and cDNAs information in public databases to be retrieved, catalogued, and accessed by all whom require this data. Such public databases are widely used among molecular and synthetic biologists as fundamental open sources to obtain genetic information. In order to obtain genetic samples until now, researchers requested DNA clones after sending a material transfer agreement, before being sent biological materials from stock centers. However, recent improvements in de novo DNA synthesis have contributed to the removal of these steps, and dramatically reduced the time required to obtain DNA constructs. Nevertheless, current systems for de novo DNA synthesis still have technological limitations. For example, they are not widely applicable for (i) making tandemly repeated DNA sequence units, (ii) for constructing unbiased DNA libraries containing DNA fragments with sequence multiplicity in a specific region of target genes, or (iii) for replacement of DNA fragments with others. To overcome these limitations, new efficient and reliable biochemical gene assembly methods are still required.
Recent common DNA assembly methods can be categorized into two major groups based on the molecular mechanisms employed (1). The first group of common methods relies on the use of a type II restriction enzyme digestion of DNA fragments followed by ligation. This group includes the three-antibiotic (3A) assembly method (2), the BASIC method (3), the Golden Gate assembly method (4,5) employing type IIS restriction enzyme, BsaI, the Golden Braid system (6), the Modular cloning system (7), the VEGAS method (8) that were developed by improving the Golden Gate assembly method, the MASTER ligation method (9) employing type IIM restriction enzyme, MspJI (10–13). The 3A assembly method is one of the typical and rational protocols for gene construction that supports the biological standardized genetic parts that are flanked between a common upstream and downstream nucleotide sequences in an appropriate plasmid DNA vector (BioBrick)-based gene designs. BioBricks, biological standardized genetic parts, were previously proposed by Knight (14). All BioBrick parts are flanked between a common upstream nucleotide sequence (called a ‘prefix’) and a common downstream nucleotide sequence (called a ‘suffix’) in an appropriate plasmid DNA vector (15). Knight also proposed and realized a unified concept of BioBrick, in which each genetic fragment can be fused upstream or downstream by a simple ligation reaction after restriction digestion of BioBricks, using only a limited number of restriction enzymes. This assembly method relies on a combination of positive and negative selection to achieve a high frequency of correctly assembled clones. The greatest advantage of this method is the avoidance of DNA fragment purification using column and agarose gel in all steps. However, in order to perform the 3A assembly method, the destination vector, into which two BioBrick parts will be assembled, must have a different antibiotic resistance marker from the plasmids encoding the two input parts. Therefore, it requires time and effort to construct a large set of BioBricks fused to plasmid DNA vectors with distinct drug resistant markers. Moreover, the number of BioBricks assembled in one cycle of the experiment is limited to two parts. Another typical protocol for gene construction in the first group is the Golden Gate assembly method, which employs the type IIS restriction enzyme, BsaI. This enzyme can cut outside of its recognition site and generates a unique 5’ overhang structure composed of four complementary nucleotides. By using this type of enzyme, digested fragments can be ligated to generate expected products lacking recognition sites of the restriction enzyme between each genetic part. This assembly method can be achieved by adding all DNA fragments and reagents required into one tube. Successful assembly of up to 12 DNA fragments into a destination vector in one reaction was demonstrated (16). However, this method requires manipulations for design and synthesis of various DNA primers containing a BsaI recognition site and different cutting motif at both ends of each DNA fragment. Cutting sequences at the ends of DNA fragments must be sufficiently different to avoid unexpected insertion, deletion, duplication or reversal of genetic parts (5). This method employs a one-pot/one-step protocol. However, it requires much more time and effort to check the fidelity of assembly and to select the correct assembly clone.
The second group of common assembly methods is based on sequence homology at the ends of each DNA fragment and includes techniques such as the in vitro Gibson assembly method (17,18), the overlapping extension polymerase chain reaction (OE-PCR) method (19), the circular polymerase extension cloning method (20,21), the sequence and ligation-independent cloning method (22), the seamless ligation cloning extract method (23), the Urasil-Specific Excision Reagent (USER) method (24) and the in vivo DNA assembler-yeast method (25), and so on (26–30). In the Gibson assembly method, T5 exonuclease chews back 5’-ends to generate single-strand complementary cohesive ends. After specific annealing among DNA fragments, Phusion DNA polymerase fills in the gaps and Taq DNA ligase seals the nicks covalently (17). This assembly method is suitable for making large constructs from long DNA fragments. It was reported that full-length genome (583 kbp) derived from Mycoplasma genitalium was assembled from separate fragments by this method (18). However, this protocol is not suitable for the assembly of short DNA fragments of less than 100 bp in length or tandemly repeated DNA fragments, as T5 exonuclease removes dozens of nucleotides from 5’-ends.
As described above, each common assembly method is widely used and has significant advantages, but each technique also has its limitations. Here, we will show the development of a novel BioBrick-based DNA assembly method, ‘Quick Gene Assembly (QGA)’, for overcoming the limitations in the techniques described above.
2. Materials and methods
2.1 DNA primers, reagents, magnetic beads, and other tools
Nucleotide sequences of DNA oligomers prepared for this study were as follows, QGA adaptor antisense strand: 5’-Phosphate-CTAGAAGCGGCCGCGAATTC-(dA)18-3’, QGA adaptor sense strand: 5’-TTGAATTCGCGGCCGCTT-3’, 100-bp upstream primer: 5’-AACCTATAAAAATAGGCGTATCAC-3’, and 200-bp downstream primer: 5’-CCCCTGATTCTGTGGATAACCGTATTACCG-3’. Restriction endonucleases, XbaI (20 U/μl), SpeI-HF (20 U/μl), SpeI (10 U/μl), EcoRI (20 U/μl), and solution for digestion (CutSmart) were purchased from New England Biolabs Japan Inc (Tokyo, Japan). DNA polymerase, KOD-Plus-Neo, and the solution for PCR were purchased from TOYOBO Co Ltd (Osaka, Japan). The ligation kit, Mighty Mix, was purchased from TaKaRa BIO INC (Shiga, Japan). Triton X-100 was purchased from Sigma-Aldrich Japan (Tokyo, Japan). Trehalose dehydrate was purchased from Wako Pure chemical Industries Ltd (Osaka, Japan). The magnetic beads, SiMAG-Oligo-dT, were purchased from Chemicell (Berlin, Germany). The neodymium magnet, ND-8R, was purchased from Magna Co Ltd (Tokyo, Japan). The kit for purification of DNA fragments, FastGene Gel/PCR Extraction Kit, was purchased from NIPPON Genetics Co Ltd (Tokyo, Japan).
2.2 Preparation of DNA fragments containing BioBricks
One microliter of template DNA (1 ng/μl) containing BioBrick, 1 μl of 10 μM 100-bp upstream primer (10 pmol), 1 μl of 10 μM 200-bp downstream primer (10 pmol), 11.3 μl of Distilled Water (DW), 2 μl of 10xPCR solution, 2 μl of 2 mM deoxyribonucleotide triphosphate (dNTPs), 1.2 μl of 25 mM MgSO4, and 0.5 μl of 1 U/50 μl DNA polymerase (KOD-Plus-Neo) were mixed for amplification of DNA fragments by PCR. Amplified DNA fragments were purified by using a FastGene PCR Extraction Kit. Amplified DNA fragments were digested by XbaI, separated by agarose gel (1%) electrophoresis, and purified by using a FastGene Gel Extraction Kit. Concentration of each DNA fragment solution was adjusted to 0.1 pmol/μl by adding 10 mM Tris-HCl (pH = 8.0) containing 1 mM EDTA (TE).
2.3 Preparation of adapter DNA solution
Five microliters of 100 μM QGA adaptor antisense strand (500 pmol), 5 μl of 100 μM QGA adaptor sense strand (500 pmol), and 40 μl of TE containing 0.4 M NaCl were mixed and heated at 80 °C for 3 min. The mixture was left for 10 min until temperature went down to 25 °C and diluted 10 times by adding TE containing 0.4 M NaCl to make ‘Adapter Solution’.
2.4 Ligation of adapter to first BioBrick
Two microliters of 0.1 pmol/μl 1st BioBrick solution (0.2 pmol), 0.2 μl of 1 pmol/μl ‘Adapter Solution’ (0.2 pmol), 2.8 μl of deionized water, and 5 μl of Mighty Mix, ligation mixture containing polyethylene glycol for elevation of ligation efficiency (31,32), were mixed and incubated at 16 °C for 15 min followed by mixing by pipetting up and down and additional incubation at 16 °C for 15 min.
2.5 Preparation of magnetic beads
Two microliters of magnetic beads (SiMAG-Oligo-dT) were suspended in TE (50 μl) containing 0.4 M NaCl by agitating the tube by touching it to the head of a sonic toothbrush (OMRON HEALTHCARE Co., Ltd, Kyoto Japan), the beads were collected by attracting them to one side of a 0.2 ml polypropylene tube using a neodymium magnet (4 mm in diameter), and the supernatant was removed.
2.6 Fixation of adapter-conjugated first BioBrick to magnetic beads
Ten microliters of adapter-conjugated first BioBrick solution, 10 μl of TE containing 0.8 M NaCl, and the beads previously rinsed by the above protocol were mixed by agitation. The suspension was incubated at 60 °C for 5 min and left for 35 min until the temperature decreased to 25 °C at a rate of 1 °C/min. The beads were resuspended by agitation and incubated for a further 25 min at 25 °C. The beads were collected using the magnet, and the supernatant containing excess-free DNA fragments was removed.
2.7 Beads rinsing
Twenty microliters of TE containing 0.1% Triton X-100 was added to the beads and the beads were suspended by agitation. The beads were collected using a magnet to remove supernatant by pipetting. Then, the beads were rinsed by repeating the process of suspending them in 20 μl of TE by agitation, and collection by magnet to remove the supernatant.
2.8 Removal of 200-bp downstream DNA fragment
‘Digestion Premix’ containing 1 μl of 10× RE solution, 3 μl of 2 M trehalose, 5.5 μl of DW, and 0.5 μl of restriction endonuclease, SpeI-HF, was added to the beads, and suspended by pipetting up and down. The suspension was incubated at 37 °C for 15 min, followed by mixing by pipetting up and down and further incubation at 37 °C for 15 min. Then, the beads were collected by magnet to remove the supernatant. The beads were rinsed twice, as described in the ‘Beads-rinsing’ section above.
2.9 Fusion of second BioBrick to first BioBrick
‘Ligation Premix’ containing 2 μl of second BioBrick solution (0.2 pmol), 3 μl of deionized water, and 5 μl of Mighty Mix was added to the beads and mixed by pipetting up and down. The mixture was incubated at 16 °C for 15 min, followed by mixing by pipetting up and down and further incubation at 16 °C for 15 min. The beads were rinsed twice, as described in ‘Beads-rinsing’. The 200-bp downstream DNA fragment was removed by SpeI restriction enzyme, as described in the ‘Removal of 200-bp downstream DNA fragment’, section, followed by ‘Beads-rinsing’ twice.
2.10 Fusion of more downstream BioBricks
Downstream BioBricks were fused by repeating the protocol described in the ‘Fusion of second BioBrick to first BioBrick’ section. The final round of BioBrick fusion does not contain a step for the removal of the 200-bp downstream DNA fragment, since the downstream fragment is required for amplification of assembled gene by PCR as a primer annealing site.
2.11 Amplification of assembled DNA fragment by PCR
Twenty microliters of TE was added to the beads and the beads were suspended by agitation. A total of 0.2 μl of the suspension, 1 μl (10 pmol) of 10 μM QGA adaptor sense strand, 1 μl (10 pmol) of 10 μM 200-bp downstream primer, 11.3 μl of deionized water, 2 μl of 10xPCR solution, 2 μl of 2 mM dNTPs, 1.2 μl of 25 mM MgSO4, and 0.5 μl of KOD-Plus-Neo (1 U/50 μl) were mixed. At first, the assembled DNA fragments were dissociated from the beads by incubating them at 94 °C for 2 min, then the assembled the DNA fragments were amplified through step-down PCR (5 cycles of incubation at 98 °C for10 s followed by incubation at 74 °C for 1 min, 5 cycles of incubation at 98 °C for 10 s followed by incubation at 72 °C for 1 min, 5 cycles of incubation at 98 °C for 10 s followed by incubation at 70 °C for 1 min, 10 cycles of incubation at 98 °C for 10 s followed by incubation at 68 °C for 1 min). The amplified DNA fragments were purified by using a FastGene PCR Extraction Kit. The DNA fragments were digested by EcoRI and SpeI, digested DNA fragments were separated on 1% agarose gel, then purified by using a FastGene Gel Extraction Kit. A total of 0.1 pmol of EcoRI/SpeI digested linear assembled DNA fragments were ligated with 0.035 pmol of EcoRI/SpeI digested linear plasmid DNA. The ligated DNA constructs were transformed into Escherichia coli DH5α (HIT competent cells, RBC Bioscience), and cells were spread on an Luria-Bertani Broth (LB)-plate containing the appropriate antibiotic for screening transformed clones.
2.12 Notes
(1) Usage of primer set (100-bp upstream primer and 200-bp downstream primer) for gene amplification is recommended to increase the length of the DNA fragment, since recovery of short DNA fragments using affinity columns (for preparation of DNA fragment) is inefficient when fusion of a short BioBrick DNA fragments are required. This process is also helpful to make identification of completely digested DNA fragments easier.
(2) All DNA fragments used in this experiment should be highly purified. Usage of glycogen for DNA purification should be avoided (33).
(3) All steps for beads rinsing should be performed by agitation of the tube by touching to the head of sonic toothbrush to avoid loss of beads by adsorption onto the inner surface of plasticware during pipetting up and down.
(4) All steps for mixing enzyme solutions should be performed by pipetting up and down to avoid reduction of enzyme activity.
(5) The beads should always be fixed on one side of the inner surface of the polypropylene tube by a magnet when a supernatant is removed by pipetting.
(6) We used a very small neodymium magnet (4 mm in diameter) to avoid magnetization of magnetic beads.
3. Results
3.1 Schematic representation of QGA
First, (i) all DNA fragments containing BioBricks were prepared by PCR amplification using a common primer set consisting of 100-bp upstream and 200-bp downstream DNA primers. (ii) Second, all DNA fragments were digested by restriction enzyme (XbaI) to unmask the upstream end of each fragment and make them competent for ligation to their upstream fragment, as shown in Figure 1a. (iii) The upstream end of the first BioBrick should be immobilized by fixing it to the origin of gene assembly, and its downstream end should be kept masked (dephosphorylated) to avoid unexpected fusion with itself when sequentially assembling multiple BioBrick parts. (iv) The downstream 5’-terminus should be removed by restriction enzyme digestion using SpeI to unmask its downstream end for ligation to the second BioBrick. (v) The second BioBrick is ligated downstream of the first BioBrick. The number of BioBricks fused to the downstream end can be increased by repeating this digestion/ligation cycle, as is illustrated in Figure 1b.
Figure 1.
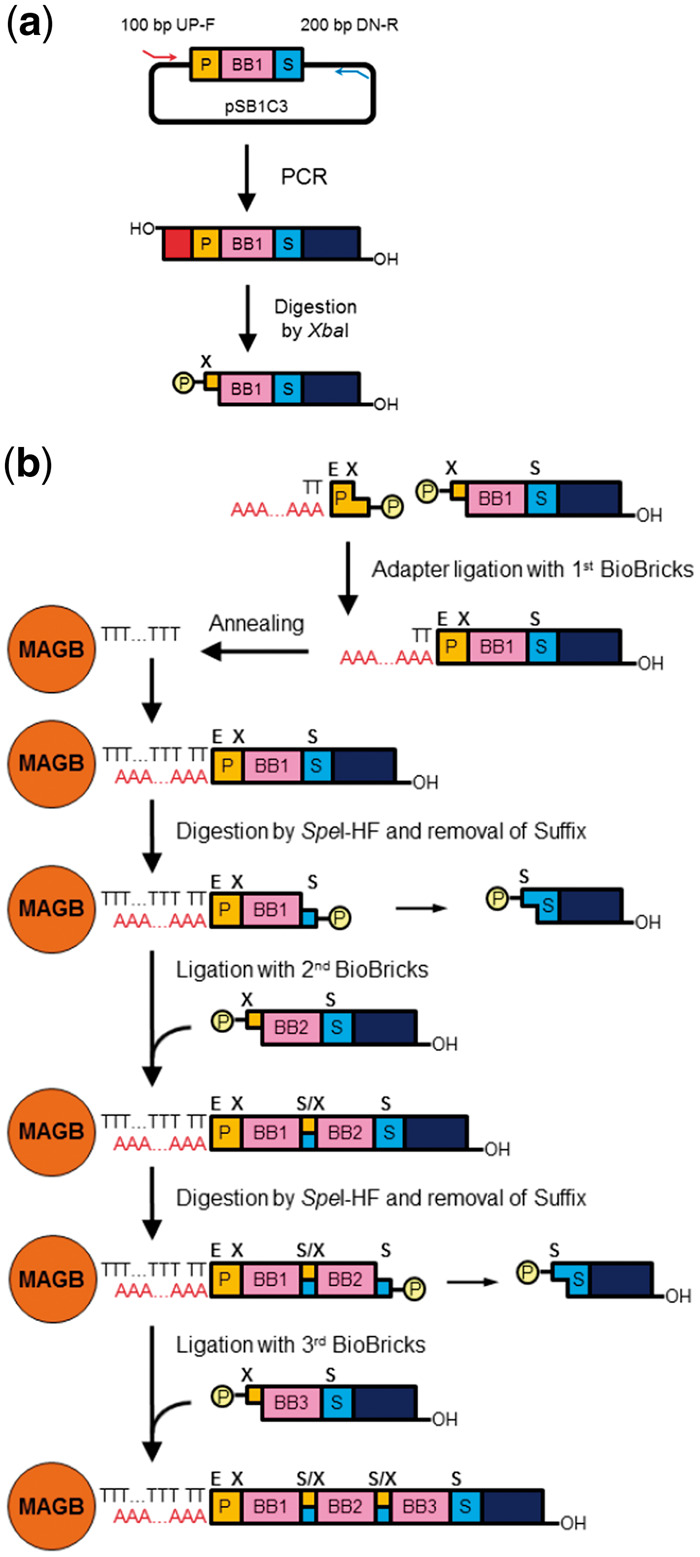
Schematic representation of the QGA protocol. (a) Preparation of a DNA fragment containing a BioBrick part. P: prefix DNA fragment, BB: BioBrick, S: suffix DNA fragment, 100 bp UP-F: 100 base pair upstream forward primer, 200 bp DN-R: 200 base pair downstream reverse primer, circled P: phosphate residue, X: XbaI restriction site. (b) Structural change of extending the DNA molecule in each step of QGA. MAGB: magnetic beads, E: EcoRI restriction site, S: SpeI restriction site, S/X: mixed restriction site (scar), TTT…TTT: oligo-dT conjugated to magnetic beads, AAA…AAA: oligo-dA in DNA adaptor.
The protocol described above requires frequent replacement of reaction solutions for the treatment of both ends of all BioBricks. The most common method for replacement was ethanol precipitation of the DNA fragments. In this case, at least 10 precipitation by ethanol addition steps, 10 rinsing by 70% ethanol steps, 20 centrifugation steps, and 10 drying-up steps are required, just to complete the assembly of 5 BioBricks. These protocols require a long period of time, and DNA sample yield tends to decrease dramatically after sequentially repeated ethanol precipitation and rinsing steps. By taking advantage of the usage of ‘magnetic beads’ conjugated to oligo-dT, SiMAG-Oligo-dT (Chemicell), we succeeded in achieving a dramatic reduction of the time required for the sequential precipitation and rinsing steps. Since oligo-dT conjugated to the beads can hybridize to oligo-dA, we made oligo-dA the central part of the adapter DNA fragment required to fix the first BioBrick to the beads. Utilizing the magnetic properties of the beads allows for much faster isolation and purification of the assembled DNA fragments without the need for multiple sequential centrifugation steps. Replacing the precipitation and rinsing steps by magnetic bead isolation using a magnet largely contributed to the simplification of the whole protocol.
3.2 Possible number of cycles for accurate assembly
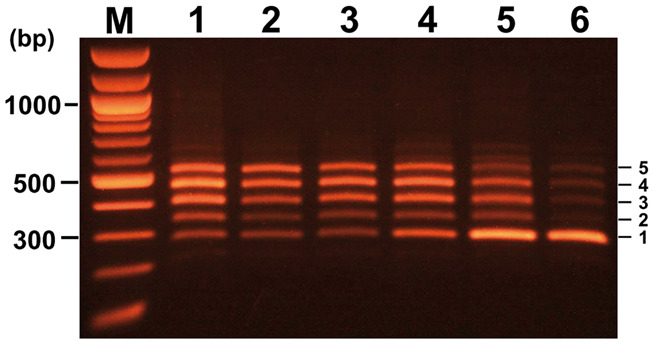
A short DNA fragment containing a promoter-BioBrick, PtetR (BBa_R0040, 54 bp) was used as a building block to test the possible number of cycles for accurate assembly. As seen in Figure 2a, the sizes of PCR products derived from the amplification of templates generated after each cycle of assembly was expected to increase stepwise with an increase in the cycle number of assembly. The PCR product size after each cycle should be 200-bp longer than the DNA fragments composed of only fused BioBricks after each DNA fragment is fused to a common 200-bp downstream fragment. All BioBricks were connected by a 6-bp intervening scar site sequence (ACTAGA). This scar site occurs when the ends resulting from a 5’-overhang restriction digestion by SpeI and XbaI are ligated. The reduction of efficiency of the complete assembly was evaluated by measuring the rate of light intensity associated with the largest PCR products on a gel image using software, ‘Gel Analyzer 2010a (http://www.gelanalyzer.com/download.html)’. Light intensity associated with the largest product in each lane (from lane 1 to lane 4 in Figure 2b) was the highest among products. The efficiency of the complete assembly reaction was calculated by comparing the light intensity of the largest DNA product with other products. The efficiency decreased depending on the number of assembly cycles, where observations of 61.8% (after 2 cycles), 44.3% (after 3 cycles), and 31.1% (after 4 cycles) were made. The efficiency of the complete assembly reaction after 5 cycles was calculated as 22.8%. These observations suggest that the majority of the products are full length and result from up to four cycles of assembly. The potential of this assembly method was tested by repeating more than five BioBrick part assembly cycles. The largest products, corresponding to eight BioBrick parts are still visible, even after eight cycles of assembly (data not shown). The largest product was isolated from agarose for further ligation with a plasmid vector when only perfectly assembled constructs were required.
Figure 2.

Size analysis of PCR products after an increase in the number of QGA cycles. (a) Predicted assembled structures of products after increase of cycles of QGA. Cycle numbers of QGA and predicted sizes of assembled DNA fragments after each cycle are indicated at the left of each construct. PtetR: promotor BioBrick of tetracycline resistance gene. (b) Distribution of amplified DNA fragments by PCR when assembled DNA fragments after each cycle were used as a template. QGA cycle numbers are indicated above each lane. Copy numbers of PtetR fragments are indicated at the right side of each band.
3.3 Conditions affect higher yield of correctly assembled product
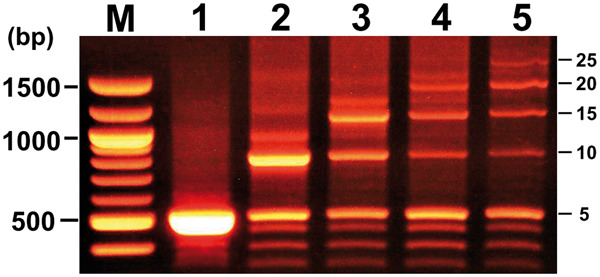
To evaluate each factor affecting the yield of the largest product among all PCR products, only one factor was changed from the optimum condition described in Materials and Methods section. The assembled products generated under optimum condition are shown in lane 1 in Figure 3 as a positive control. In lane 2, the high-fidelity restriction enzyme SpeI (SpeI-HF) in digestion premix was replaced by a standard SpeI enzyme. The yield of the largest product was reduced to 78.6% when the yield obtained in the optimum condition was set as 100%. The yield was further reduced to 72.9% when optimum conditions (30 min at 16 °C) were changed to incubation for 10 min at 25 °C, as shown in lane 3. The yield was reduced to 66.3% when a rinsing by TE containing 0.1% Triton X-100 was replaced by rinsing with TE, as shown in lane 4. Moreover, production of immature product, the smallest product, increased 1.35 times. These observations suggest that the presence of a low concentration of detergent in the rinsing solution is essential for the optimum removal of solution components used in the previous step. The yield of the largest product was reduced to 41.4% when 0.6 M trehalose was removed from ‘Digestion Premix’, as shown in lane 5. Furthermore, production of immature product, the smallest product, increased 2.4 times. Trehalose is a glucose disaccharide synthesized in Saccharomyces cerevisiae and E. coli, and is known to retain the potential to elevate an enzyme reaction in vitro (34,35). Combining all these changes dramatically reduced the yield to 29.2%, as shown in lane 6. These observations shown in Figure 5 suggest that the addition of 0.1% Triton X-100 to TE for rinsing and the addition of 0.6 M trehalose to the digestion premix are essential for successful BioBrick assembly. However, use of the high-fidelity restriction enzyme SpeI-HF and little change of ligation conditions were not essential for higher yield of the largest product.
Figure 3.
Negative effects on the QGA process resulting from the change of each condition away from optimum conditions. Lane 1: assembled products under optimum conditions. Lane 2: replacement of the restriction enzyme from SpeI-HF (NEB) to SpeI (NEB). Lane 3: changing of ligation condition from ‘at 16 °C for 30 min’ to ‘at 25 °C for 10 min’. Lane 4: removal of 0.1% Triton X-100 from the rinsing solution. Lane 5: removal of 0.6 M trehalose from the digestion premix. Lane 6: combination of all changes tested from lanes 2–5. Copy numbers of PtetR fragment are indicated at the right side of each band.
Figure 5.
Effects of the increase of BioBrick length in QGA. Distribution of amplified DNA fragments by PCR (length of each PCR product containing 5, 10, 15, 20, and 25 tandem repeats of PtetR is 514 bp, 814 bp, 1114 bp, 1414 bp, and 1714 bp, respectively) when assembled DNA fragments were used as a template after each cycle. Cycle numbers of QGA correspond to lane numbers. Copy numbers of PtetR fragments are indicated at the right side of each band.
3.4 Optimization of mixing steps
Mixing enzyme solutions by pipetting up and down is generally considered to be one of the best methods to avoid enzyme denaturation. However, a considerable reduction in the number of magnetic beads and a decrease in the yield of the DNA fixed to them were observed through repetition of this manipulation. To address this problem, we tested agitating the test tube to avoid the loss of beads by adsorption onto the inner surface of the plastic pipette tips, and the shear forces associated with repeated pipetting up and down. Complete replacement of the pipetting up and down manipulation by the agitation method considerably reduced the yield of the largest assembled product after assembly, as shown in Figure 4. The efficiency of complete assembly after cycles 1, 2, 3, 4, and 5 were reduced, step by step, from 100% to 57.9%, 38.9%, 21.3%, and 16.2%, respectively. These observations suggest that repeated agitation contributes to avoiding the loss of magnetic beads; however, the manipulation may cause a reduction of enzyme activity. Therefore, the vibration method should be employed for beads rinsing, and the pipetting up and down manipulation should be used for mixing enzyme solutions, such as the ‘Digestion Premix’ and ‘Ligation Premix’ solutions.
Figure 4.
Negative effects of enzyme solution mixing from pipetting up and down compared to the agitation method. Distribution of amplified DNA fragments by PCR when assembled DNA fragments were used as a template after each cycle. Cycle numbers of QGA are indicated above each lane. Copy numbers of PtetR fragments are indicated at the right side of each band.
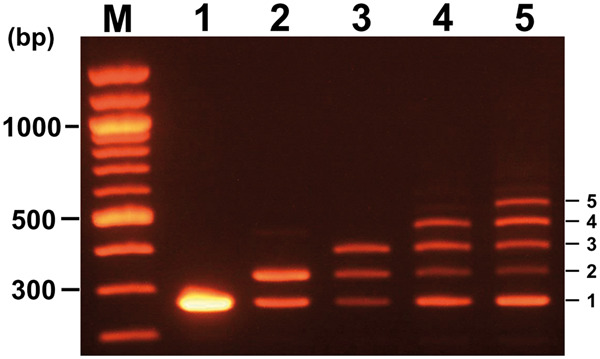
3.5 Effects of increase of BioBrick length in QGA
The results shown in Figures 2–4 indicate that assembly of a longer DNA fragment from five or more short Biobricks by QGA was achieved. To test if this protocol is applicable for the gene assembly of longer BioBricks, a DNA fragment (300 bp) containing five tandem repeats of the short BioBrick part BBa_R0040 was used as a building block for the assembly. The efficiency of complete assembly from longer BioBricks after five cycles was calculated to be 10.4%, shown in Figure 5, lane 5. Although the efficiency of the complete assembly was rather low, the amount of DNA fragment isolated from an agarose gel was enough to be used for an enzyme digestion followed by ligation and transformation of bacterial cells. These observations suggest that genes up to 1500 bp in length can be assembled from 5 BioBricks biochemically in 24 h.
3.6 Assembly of a functional gene through ligation of a promoter, protein coding sequence, and terminator
To test the potential of the assembly system to construction functional genes from distinct BioBricks, Green fluorescent protein (GFP), and Red fluorescent protein (RFP) coding sequences were used as reporter genes. Schematic representations of DNA constructs on the process to finish complete gene assembly are shown in Figure 6a together with their lengths. The efficiency of the assembly of functional genes from three BioBricks was monitored by observing a change in the sizes of PCR products amplified from assembled genes, as shown in Figure 6b. White arrow heads indicate intermediary products and the assembled DNA fragments, as we expected. The sizes of the assembled DNA fragments were successfully increased. The largest PCR products (in lanes 3 and 6 in Figure 6b) containing regions encoding transcription units for expression of GFP or RFP were digested by restriction enzymes, EcoRI and SpeI, ligated with plasmid vector, pSB1A3 which has previously been digested by EcoRI and SpeI, and the ligated samples were subjected for transformation of bacteria strain DH5α.
Figure 6.
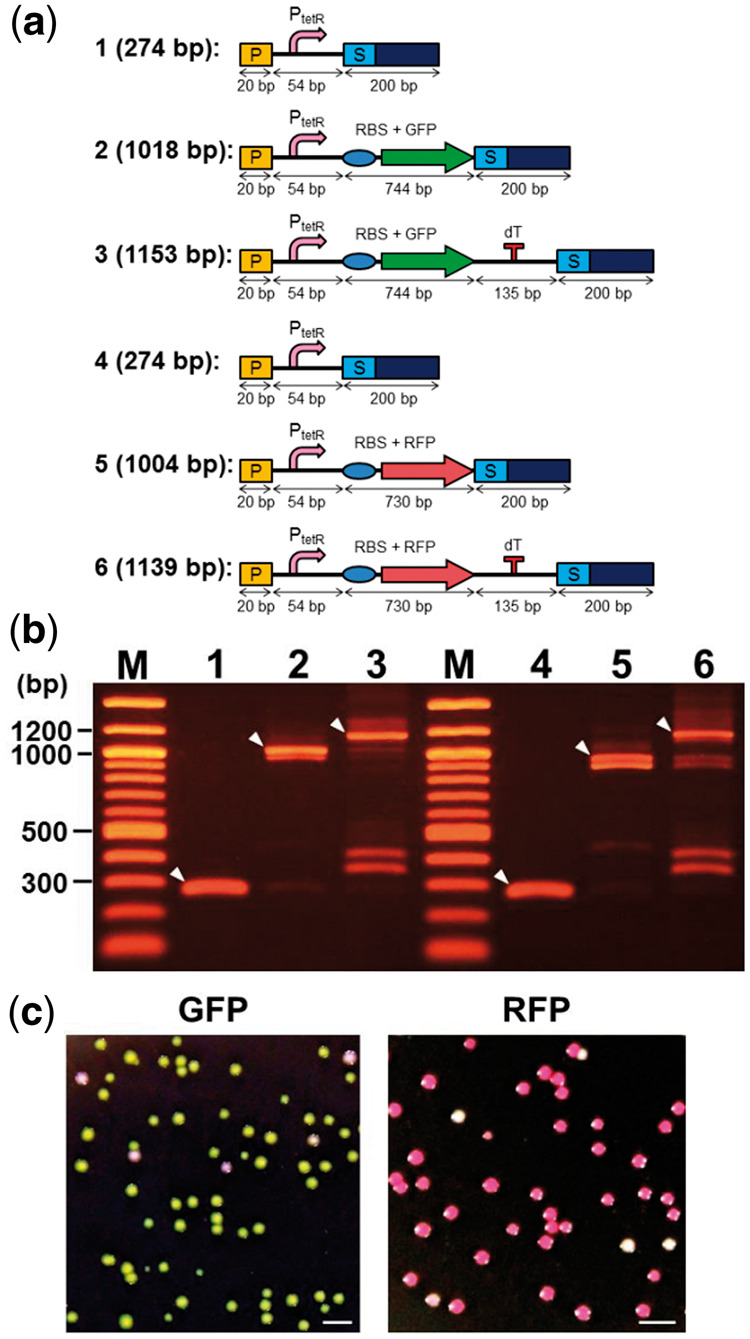
Assembly of functional genes from distinct BioBricks. (a) Schematic representation of the QGA process from a single DNA construct to the finished complete gene assembly is shown together with their lengths. P: prefix DNA fragment, PtetR: TetR promotor, RBS: ribosome binding site, GFP: DNA fragment encoding green fluorescent protein (BBa_I13500), RFP: DNA fragment encoding red fluorescent protein (BBa_K093005), dT: double terminator (BBa_B0015), S: suffix DNA fragment. (b) Sizes of PCR products amplified from assembled genes. White arrowheads indicate the expected DNA fragments. The numbers above each lane are corresponding to the numbers shown in panel A. (c) Images of colonies on agarose LB plates containing 100 μg/mL ampicillin. Expression of GFP and RFP was monitored under white light. Scale bars are 2 mm.
Images of colonies appeared on agarose LB plates containing ampicillin (100 µg/mL), which are shown in Figure 6c. GFP and RFP expression was monitored under white light. Ratios of green colonies to all colonies and red colonies to all colonies were calculated by counting the total number of colonies. Colonies of 92% or 90% successfully produced GFP or RFP, respectively. These results suggest that the majority of the largest products after the assembly and gene amplification have a potential for successful expression.
4. Discussion
We have often encountered a requirement to construct hundreds of recombinant genes and genetic circuits to investigate the function of various biological systems at the molecular level, or to generate novel biological systems in the field of synthetic biology. This process always requires continuous effort, especially given the increasing demand for new constructs in research. Therefore, the reduction of the time required for each design and construction cycle for gene assembly, is a critical issue facing the field as a whole.
First, the existence of a common engineering principle based gene-design-concept is important to reduce the time required for the design cycle of genes, since the sharing of ideas and frequent discussions among researchers and students is crucial to achieve the best designs. The most well-known gene-design-concept based on engineering principles is the ‘standardized genetic part (BioBrick)-based gene design protocol’ proposed by Knight (14). To take full advantage of the gene-design-concept, making good use of a highly reliable BioBrick collection is essential. Continuous effort to evaluate the quality of each BioBrick part, accumulation of measurement and characterization information of each BioBrick part, and the sharing of these data are critical to improve the quality and quantity of the collection. Furthermore, BioBrick parts should be widely used under different conditions and in different organisms to evaluate the performance of each part. The performance of each BioBrick part should be measured under a variety of conditions, and the information should be shared widely and publically. With this accumulated body of knowledge, BioBricks can be used appropriately under various conditions for researchers to achieve the best designs possible.
Second, the development of an efficient gene assembly method is also important for the construction of hundreds of recombinant genes, and this development could lead to a reduction in the time required for the ‘construction cycle’ of genes. Furthermore, the method should be based on engineering principles so that the technique can be applied to automated genetic part assembly. In the introduction section, we described several other gene assembly protocols as well as their advantages and limitations. In this study, we succeeded in making a novel BioBrick-based DNA assembly method, and named it ‘Quick Gene Assembly (QGA)’. This method enables us to assemble at least five BioBricks in vitro in 12 h. All steps (including amplification of DNA fragments by PCR, and assembly of the plasmid DNA vector) can be finished in 24 h.
The advantages of QGA are listed as follows: (i) Use of only one universal DNA primer set is enough to prepare any kind of BioBrick or other building block for the assembly of genetic parts. (ii) Genetic parts can be fused correctly in as short a time as 12 h. (iii) We can successfully assemble repetitive DNA sequences or short DNA fragments (less than 100 bp). (iv) Filling gaps among building blocks and plasmid DNA vector by DNA polymerase is not required. (v) More than 95% of DNA products in the largest product after PCR were accurately assembled as expected. (vi) Quick and easy replacement of each solution after each step dramatically reduced the time required to complete the whole assembly. (vii) Entire steps in QGA can be achieved in vitro, without the need for an in vivo clone screening process. Consequently, we believe the QGA system is amenable to automation because of its interactive workflow and its reliance on magnetic beads for purification of fragments. The system could also be used for the construction of unbiased focused libraries.
Conflict of interest statement. None declared.
References
- 1. Chao R., Yuan Y., Zhao H. (2014) Recent advances in DNA assembly technologies. FEMS Yeast Res., 15, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shetty R., Lizarazo M., Rettberg R., Knight T.F. (2011) Assembly of BioBrick standard biological parts using three antibiotic assembly. Methods Enzymol., 498, 311–326. [DOI] [PubMed] [Google Scholar]
- 3. Storch M., Casini A., Mackrow B., Fleming T., Trewhitt H., Ellis T., Baldwin G.S. (2015) BASIC: a new biopart assembly standard for idempotent cloning provides accurate, single-tier DNA assembly for synthetic biology. ACS Synth. Biol., 4, 781–787. [DOI] [PubMed] [Google Scholar]
- 4. Engler C., Kandzia R., Marillonnet S. (2008) A one pot, one step, precision cloning method with high throughput capability. PLoS One, 3, e3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Engler C., Gruetzner R., Kandzia R., Marillonnet S. (2009) Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS One, 4, e5553.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sarrion-Perdigones A., Falconi E.E., Zandalinas S.I., Juárez P., Fernández-del-Carmen A., Granell A., Orzaez D. (2011) GoldenBraid: An iterative cloning system for standardized assembly of reusable genetic modules. PLoS One, 6, e21622.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weber E., Engler C., Gruetzner R., Werner S., Marillonnet S. (2011) A modular cloning system for standardized assembly of multigene constructs. PLoS One, 6, e16765.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mitchell L.A., Chuang J., Agmon Ne., Khunsriraksakul C., Phillips N.A., Cai Y., Truong D.M., Veerakumar A., Wang Y., Mayorga M.. et al. (2015) Versatile genetic assembly system (VEGAS) to assemble pathways for expression in S. cerevisiae. Nucleic Acids Res., 43, 6620–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen W.H., Qin Z.J., Wang J., Zhao G.P. (2013) The MASTER (methylation-assisted tailorable ends rational) ligation method for seamless DNA assembly. Nucleic Acids Res., 41, e93.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sleight S.C., Bartley B.A., Lieviant J.A., Sauro H.M. (2010) In-Fusion BioBrick assembly and re-engineering. Nucleic Acids Res., 38, 2624–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhu B., Cai G., Hall E.O., Freeman G.J. (2007) In-Fusion assembly: seamless engineering of multidomain fusion proteins, modular vectors, and mutations. Biotechniques, 43, 354–359. [DOI] [PubMed] [Google Scholar]
- 12. Wang R.Y., Shi Z.Y., Guo Y.Y., Chen J.C., Chen G. Q. (2013) DNA fragments assembly based on nicking enzyme system. PLoS One, 8, e57943.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blake W.J., Chapman B.A., Zindal A., Lee M.E., Lippow S.M., Baynes B.M. (2010) Pairwise selection assembly for sequence-independent construction of long-length DNA. Nucleic Acids Res., 38, 2594–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Knight T. (2006) Idempotent Vector Design for Standard Assembly of Biobricks. MIT Synthetic Biology Working Group. [Google Scholar]
- 15. Registry of standard biological parts. http://partsregistry.org (27th December 2016, date last accessed).
- 16. Nielsen A.A.K., Der B.S., Shin J., Vaidyanathan P., Paralanov V., Strychalski E.A., Ross D., Densmore D., Voigt A.. et al. (2016) Genetic circuit design automation. Science, 352, aac7341.. [DOI] [PubMed] [Google Scholar]
- 17. Gibson D.G. (2011) Enzymatic assembly of overlapping DNA fragments. Methods Enzymol., 498, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A. 3rd, Smith H.O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods, 6, 343–345. [DOI] [PubMed] [Google Scholar]
- 19. Horton R.M., Hunt H.D., Ho S.N., Pullen J.K., Pease L.R. (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene, 77, 61–68. [DOI] [PubMed] [Google Scholar]
- 20. Quan J., Tian J. (2009) Circular polymerase extension cloning of complex gene libraries and pathways. PLoS One, 4, e6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Quan J., Tian J. (2011) Circular polymerase extension cloning for high-throughput cloning of complex and combinatorial DNA libraries. Nat. Protoc., 6, 242–251. [DOI] [PubMed] [Google Scholar]
- 22. Li M.Z., Elledge S.J. (2007) Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods, 4, 251–256. [DOI] [PubMed] [Google Scholar]
- 23. Zhang Y., Werling U., Edelmann W. (2012) SLiCE: A novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res., 40, e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stein V., Hollfelder F. (2009) An efficient method to assemble linear DNA templates for in vitro screening and selection systems. Nucleic Acids Res., 37, e122.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shao Z., Zhao H., Zhao H. (2009) DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acids Res., 37, e16.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gibson D.G., Benders G.A., Axelrod K.C., Zaveri J., Algire M.A., Moodie M., Montague M.G., Venter J.C., Smith H.O., Hutchison C.A. 3rd. (2008) One-step assembly in yeast of 25 overlapping DNA fragments to form a complete synthetic Mycoplasma genitalium genome. Proc. Natl. Acad. Sci. U.S.A., 105, 20404–20409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Itaya M., Fujita K., Kuroki A., Tsuge K. (2008) Bottom-up genome assembly using the Bacillus subtilis genome vector. Nat. Methods, 5, 41–43. [DOI] [PubMed] [Google Scholar]
- 28. Yonemura I., Nakada K., Sato A., Hayashi J., Fujita K., Kaneko S., Itaya M. (2007) Direct cloning of full-length mouse mitochondrial DNA using a Bacillus subtilis genome vector. Gene, 391, 171–177. [DOI] [PubMed] [Google Scholar]
- 29. Zhang Y., Muyrers J.P.P., Testa G., Stewart A.F. (2000) DNA cloning by homologous recombination in Escherichia coli. Nat. Biotechnol., 18, 1314–1317. [DOI] [PubMed] [Google Scholar]
- 30. Fu J., Bian X., Hu S., Wang H., Huang F., Seibert P.M., Plaza A., Xia L., Müller R., Stewart A.F.. et al. (2012) Full-length RecE enhances linear-linear homologous recombination and facilitates direct cloning for bioprospecting. Nat. Biotechnol., 30, 440–446. [DOI] [PubMed] [Google Scholar]
- 31. Hayashi K., Nakazawa M., Ishizaki Y., Hiraoka N., Obayashi A. (1986) Regulation of inter- and intramolecular ligation with T4 DNA ligase in the presence of polyethylene glycol. Nucleic Acids Res., 14, 7617–7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pheiffer B.H., Zimmerman S.B. (1983) Polymer-stimulated ligation: enhanced blunt- or cohesive-end ligation of DNA or deoxyribooligonucleotides by T4 DNA ligase in polymer solutions. Nucleic Acids Res., 11, 7853–7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gaillard C., Strauss F.O. (1990) Ethanol precipitation of DNA with linear polyacrylamide as carrier. Nucleic Acids Res., 18, 378.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carninci P., Nishiyama Y., Westover A., Itoh M., Nagaoka S., Sasaki N., Okazaki Y., Muramatsu M., Hayashizaki Y. (1998) Thermostabilization and thermoactivation of thermolabile enzymes by trehalose and its application for the synthesis of full length cDNA. Proc. Natl. Acad. Sci. U. S. A., 95, 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Spiess A.N., Ivell R. (2002) A highly efficient method for long-chain cDNA synthesis using trehalose and betaine. Anal. Biochem., 301, 168–174. [DOI] [PubMed] [Google Scholar]