

Summary
Comprehensive understanding of the serological response to SARS-CoV-2 infection is important for both pathophysiologic insight and diagnostic development. Here, we generate a pan-human coronavirus programmable phage display assay to perform proteome-wide profiling of coronavirus antigens enriched by 98 COVID-19 patient sera. Next, we use ReScan, a method to efficiently sequester phage expressing the most immunogenic peptides and print them onto paper-based microarrays using acoustic liquid handling, which isolates and identifies nine candidate antigens, eight of which are derived from the two proteins used for SARS-CoV-2 serologic assays: spike and nucleocapsid proteins. After deployment in a high-throughput assay amenable to clinical lab settings, these antigens show improved specificity over a whole protein panel. This proof-of-concept study demonstrates that ReScan will have broad applicability for other emerging infectious diseases or autoimmune diseases that lack a valid biomarker, enabling a seamless pipeline from antigen discovery to diagnostic using one recombinant protein source.
Keywords: SARS-CoV-2, serology, phage display, COVID-19, diagnostics, assay development
Graphical Abstract
Highlights
ReScan is a whole proteome screen to isolate and identify serologic assay targets
Antibodies to linear peptides in COVID-19 sera bind spike and nucleocapsid proteins
Rapid workflow that seamlessly translates biomarkers into a functional diagnostic
Multiplexing linear S and N SARS-CoV-2 peptides can increase diagnostic specificity
Rapid serologic assay development remains challenging in scenarios in which antigens are unknown and is particularly crucial during a pandemic. Zamecnik et al. use ReScan, which combines paper-based microarrays and programmable phage display, to screen for and isolate the most immunogenic peptides for SARS-CoV-2 antibody diagnostics.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a novel and deadly betacoronavirus that has rapidly spread across the globe.1, 2, 3 Diagnostic assays are still being developed, with reagents often in short supply. Direct detection of the virus in respiratory specimens with SARS-CoV-2 RT-PCR remains the gold standard for identifying acutely infected patients.4 While RT-PCR yields clinically actionable information and can be used to estimate incidence, it does not identify past exposure to virus, information necessary for determining population level disease risk, and the degree of asymptomatic spread. These data are critical as they inform public policy about returning to normal activity at local and state levels.5,6
Targeted, ELISA-based serologies to detect antibodies to the SARS-CoV-2 whole spike (S) glycoprotein, its receptor-binding domain (RBD), or the nucleocapsid (N) protein have been developed and have promising performance characteristics.4,7, 8, 9 However, enhancing the specificity of SARS-CoV-2 serologic assays is particularly important in the context of low seroprevalence, in which even a 1%–2% false positive rate could significantly overestimate population-level viral exposure.10 Comprehensive and agnostic surveys of the antigenic profile across the entire SARS-CoV-2 proteome have the potential to identify highly specific sets of antigens that are less cross-reactive with antibodies elicited by other common human CoV (HuCoV) infections. In addition to their utility as diagnostic tools, multiplexed assays can also provide individualized portraits of the adaptive immune response and help define immunophenotypes that correlate with widely varying coronavirus disease 2019 (COVID-19) clinical outcomes.11,12 These data could inform the design and evaluation of urgently needed SARS-CoV-2 vaccines and therapeutic monoclonal antibodies.
We describe here a programmable phage display HuCoV VirScan13 library with overlapping 38-amino acid (aa) peptides tiled across the genomes of 9 HuCoVs, including both SARS-CoV-1 and SARS-CoV-2. We screened known COVID-19 patient sera (n = 20) against this HuCoV VirScan library and a previously built pan-viral VirScan library14 using phage-immunoprecipitation sequencing (PhIP-seq)15 to identify the most highly enriched viral antigens relative to pre-pandemic controls.
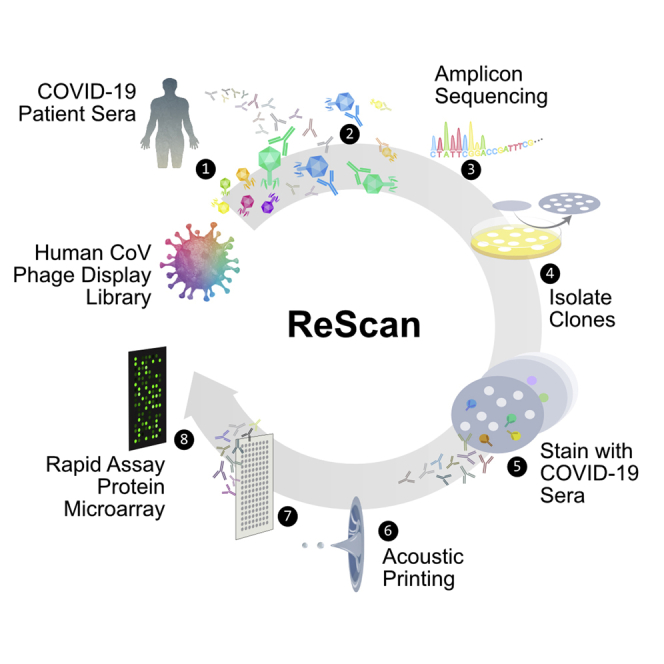
To rapidly progress from broad serological profiling by VirScan to a linear epitope-based serological assay with phage expressing a focused set of highly immunogenic, disease-specific peptides in a microarray format, we used a complementary diagnostic development pipeline, ReScan. With ReScan, we use antibodies from candidate patient sera to physically pan for phage-displaying immunogenic antigens. We then isolate and culture individual phage clones, followed by paper-based microarray production via acoustic liquid handling.
We applied ReScan by immunoprecipitating a focused SARS-CoV-2 T7 phage library containing 534 overlapping 38-aa peptides against COVID-19 patient sera to generate a SARS-CoV-2-specific peptide microarray. A larger cohort of positive and uninfected control patient samples was then screened to identify shared and discriminatory antigens with an automated image-processing algorithm. These microarrays could serve as the basis for a low-cost, disease-specific, multiplex serologic assay whose antigens are generated by an inexhaustible and rapidly scalable reagent source (Figure 1A).
Figure 1.

General Workflow for ReScan and Epitope Mapping SARS-CoV-2 Using PhIP-Seq
(A) VirScan T7 phage display system with SARS-CoV-2 antigens in overlapping 38-aa fragments (1) are incubated with antibodies (2) from patient sera and undergo 2 rounds of amplification (3). Amplified lysates are either analyzed by phage immunoprecipitation sequencing (PhIP-seq), or are lifted, stained, and isolated for ReScan (4 and 5). Microarrays of isolated clonal phage populations are printed via acoustic liquid transfer (6), stained with patient sera, and analyzed for identity (7 and 8) and commonality (9) via dotblotr.
(B and C) Heatmap displaying results from individual patients (B) and (C) cumulative fold enrichment of significant (p < 0.01) SARS-CoV peptides from HuCoV library relative to pre-pandemic controls aligned to the SARS-CoV-2 genome.
(D) Cumulative fold enrichment of significant other seasonal CoV peptides in the HuCoV library relative to pre-pandemic controls aligned to the SARS-CoV-2 genome.
(E) Cumulative enrichment of significant pan-viral library peptides relative to pre-pandemic controls aligned to the SARS-CoV-2 genome. One technical replicate for each library.
Lastly, to demonstrate the translational utility of this approach, we validated the key antigens isolated by ReScan by propagating and conjugating phage directly to spectrally encoded beads in an assay format deployable in a high-throughput clinical laboratory setting. ReScan represents a rapid and scalable approach to antigen discovery and serologic assay development when the predictive antigen set is unknown in a single pipeline.
Results
Patient Cases and Controls
For the VirScan studies, 98 deidentified cases with confirmed SARS-CoV-2 infection as determined by clinically validated qRT-PCR of nasopharyngeal swabs or a 50% neutralizing plasma antibody titer (NT50) >1:350 (Table 1), and 95 deidentified pre-pandemic adult control sera obtained from the New York Blood Center were used. The mean time from symptom onset to sample collection among cases was 30 days (interquartile range [IQR] 23–38 days). Eighteen (19%) of the COVID-19 patients were hospitalized. NT50s were available for 78/98 subjects. An additional 37 qRT-PCR+ COVID-19 cases and 44 deidentified pre-pandemic adult control sera were included in the evaluation of the ReScan Luminex assay.
Table 1.
COVID-19 Patient Demographics
| COVID-19 Cases | |
|---|---|
| n | 98 |
| Age, years, mean (IQR), years | 36 (47–56) |
| Sex, no. (%) | |
| Female | 40 (41) |
| Male | 58 (59) |
| Days from symptom onset, mean (IQR) | 30 (23–38) |
| Immunosuppressed, no. (%) | 4 (22) |
| Hospitalized, no. (%) | 18 (19) |
| Intubated, no. (%) | 7 (7.4) |
| Died, no. (%) | 1 (1.0) |
HuCoV VirScan Library Benchmarking
We designed and built 2 VirScan libraries for SARS-CoV-2 specifically and another for 7 known HuCoVs, including SARS-CoV-1 and SARS-CoV-2, comprising 534 and 3,670 38-mer peptides, respectively (Figure S1). Deep sequencing of each phage library was conducted to assess peptide distribution and representation relative to the original library design. For the SARS-CoV-2 and HuCoV libraries, 83.2% and 92% of sequenced phage were perfect matches for the designed oligonucleotides, respectively. Each library contained 99.8% and 100% of the designed peptide sequences at a sequencing depth of 2–2.5 million reads per library. Benchmarking data on the pan-viral VirScan library were previously described.14
VirScan Detects Coronavirus Antigens in COVID-19 Patient Sera
We identified multiple SARS-CoV-2 peptide sequences that were significantly enriched (p < 0.01, see Method Details) in each of the COVID-19 subjects relative to 95 pre-pandemic healthy controls (HCs) by performing PhIP-seq on both the HuCoV and pan-viral VirScan libraries (Figures 1B–1E). We observed a broad spectrum of enriched peptides covering nearly 88% of the expressed coding space of the virus. Overall, enrichment of antigenic determinants from the S and N proteins dominated. Relative to pre-pandemic control sera in both the HuCoV library and pan-viral VirScan libraries, the SARS-CoV-2 peptides derived from the S and N coding regions accounted for 76% of the total enrichment. In the S protein, both libraries detected two 3′ regions (residues 783–839 and 1,124–1,178). S protein residues 799–836 structurally flank the RBD, while the region where 2 peptides (spanning residues 1,122–1,159 and 1,141–1,178) overlap appear to be on the outer tip of the S protein trimer. Several other antigenic regions in the S protein (residues 188–232 and 681–706) were also enriched in addition to N protein residues 209–265 and residues 142–208. This region of the N protein is a key piece of the N-terminal RNA-binding domain, which aids in viral RNA packaging during virion assembly.16 We also identified a significant antibody response to the ORF3a accessory protein (residues 171–210) with our HuCoV library. Samples separated by geography were qualitatively similar (Figure S1C).
ReScan Pans COVID-19 Sera to Generate a Focused SARS-CoV-2 Antigen Microarray
To isolate the phage clones displaying candidate antigens from the parent SARS-CoV-2 T7 phage library, we immunostained and picked plaque colonies from phage immunoprecipitated by COVID-19 patient sera. We sequenced phage grown from each plaque pick and found that a mean sequence purity of 98% (51%–100%) and a total of 31 unique peptides were represented across 364 positively staining plaques. Nine of these antigens were identified in >4 plaques and had >80% sequence clonality, with a mean of 98.8% of reads mapping to the top sequence. The 9 antigens were restricted to specific regions of the N and S proteins, with the exception of a single ORF3a peptide (residues 172–209). The peptide sequences are listed in Table S1.
We repurposed an acoustic liquid handler (Labcyte Echo) to form 1 × 2 cm 384 spot microarrays of printed phage on nitrocellulose. For the analysis, we used dotblotr (https://github.com/czbiohub/dotblotr), a customized microarray image processing software built specifically for ReScan. After staining microarrays with COVID-19 patient (n = 20) and a subset of pre-pandemic control (n = 37) sera, dotblotr successfully detected 99.84% of all printed dots on the 57 arrays analyzed. No significant difference in spot detection was seen between the positive and healthy groups (Figure S2).
We found that 19/20 (95%) COVID-19 subjects had a significant number of positively stained dots on at least 1 of the 9 candidate antigens on the array compared to HCs (p < 0.05, Fisher’s exact test) (Figure 2). One subject (subject 16) was positive on all 9 peptides. A single patient (subject 19) did not have any significant hits by ReScan but was subsequently noted to be 1 of 2 in the cohort of positive patients on chronic immunosuppression for a solid organ transplant.
Figure 2.

ReScan Analysis Using SARS-CoV-2 Phage Display Library
(A) Schematic of microarray design.
(B) ReScan assay configuration layout (left) and map of each clonal phage population on plate (right).
(C) Examples of staining patterns on patients positive for nucleocapsid and spike peptides; scale bar, 5 mm. Anti-T7 tag signal is magenta, anti-IgG signal is green.
(D) SARS-CoV-2 peptide analysis from ReScan microarrays; dot size is proportional to the fraction of dots for a given peptide that stained positive by a given patient sample. One technical replicate per serum sample.
Peptides spanning residues 799–836 and 1,141–1,178 on the S protein were the most broadly reactive antigen targets, called significant on 12/20 (60%) and 14/20 (80%) of subjects, respectively. Staining on peptides spanning residues 134–171 and 153–190 of the N protein was detected across the 8/12 (66.7%) of the same patients, indicating a likely shared motif in the overlapping region between these consecutive tiled peptides. The single peptide from ORF3a (residues 171–210) was positive on 6/20 patients (30%). We performed multiple sequence alignments of a total of 9,950 spike protein sequences and 9,866 nucleoprotein sequences available from NCBI to the reference Wuhan genome used in the generation of the T7 library. Using the sequence alignment data, we calculated Shannon entropies for each aa position and overlaid the peptides found by ReScan. We found that they are located in areas of low relative mutational diversity, indicating the potential for use in a diagnostic setting (Figure S3).
Similarity Analysis of SARS-CoV-2 Antigens Identified by ReScan
Of the 4 S peptides isolated by ReScan, fragment 30 (residues 552–589) had 0% aa similarity, fragment 43 (residues 799–836) had 15.8% aa similarity, fragment 44 (residues 818–855) had 26.3% aa similarity, and fragment 61 (residues 1,141–1,178) had 5.3% aa similarity with other HuCoVs (Figure 3). In contrast, residues 1,141–1,178 of the S protein were identical between SARS-CoV-2 and SARS-CoV-1, and the first 16 aas of this region of the S protein (residues 1,141–1,156) shared 56% similarity with 9 other human and non-human CoVs from which peptides spanning this region of the S protein were also significantly enriched in our patient samples relative to pre-pandemic healthy sera.
Figure 3.

Similarity of Enriched ReScan and VirScan SARS-CoV-2 Peptides to Other CoVs
(A and B) Comparing underlined SARS-CoV-2 spike (A) and nucleocapsid (B) regions found by ReScan with other CoV sequences that were enriched over healthy controls in the HuCoV library (S552−589 = red, S799−836 = light green, S818−855 = dark green, S1,141−1,178 = purple; N134−171 = blue, N153−190 = teal, N210−247 = orange, N362−399 = pink). Homology maps with SARS-CoV-1 and seasonal coronaviruses in the HuCoV libraries are seen below.
(C and D) Diverse CoV peptide sequences with partial similarity to the regions spanning (1,141−1,178) and (153−190) of the S and N proteins that were enriched in both the HuCoV and pan-viral libraries.
(E and F) CryoEM structures of prefusion S glycoprotein (6VSB) and RNA-binding domain of N protein (6WKP) with color-coded ReScan peptides overlaid in colors corresponding to those in the coverage maps, and the S glycoprotein receptor-binding domain (RBD) highlighted in black.
Of the 4 N peptides isolated, fragments 8 (residues 134–171), 9 (153–190), 12 (210–247), and 20 (362–399) had 5.3%, 2.6%, 0%, and 0% aa similarity with other human CoVs. These fragments had aa similarities of 94.7%, 97.4%, 86.8%, and 89.5% with SARS-CoV-1. The multiple sequence alignment of fragment 9 (residues 153–190), the most conserved of the 4 N protein peptides we identified, demonstrated an N-terminal conserved motif found in peptides from 2 other human and 3 non-human CoV species (NXXAIV, 66.6% aa similarity) as well as a C-terminal motif (YAEGSRGXSXXSSRXSSRX, 73.7% aa similarity) found only in SARS-CoV-2 and 3 bat CoV species.
Validation on High-Throughput Bead-Based Serologic Assay
Phage expressing the 9 antigens identified by ReScan, along with 2 negative controls, were each propagated, purified, and conjugated to a different spectrally encoded set of beads. Phage-conjugated beads were then pooled and incubated with 100 of the pre-pandemic controls, of which 80 were used to establish a negative cutoff value using a Luminex-based anti-immunoglobulin G (IgG) secondary fluorescence assay of 3 standard deviations from the mean. Signal was calculated as the ratio of the mean fluorescence intensity of a specific phage-conjugated bead to 2 negative control conjugations in each well. Using these cutoff values, 87/98 confirmed COVID-19+ patients were positive for at least a single ReScan peptide fragment (false negative rate [FNR] = 0.112) (Figure 4). We compared the performance of our set of peptide antigens to a whole-protein bead-based panel consisting of whole SARS-CoV-2 N, S ectodomain, and the S RBD, which together achieved an FNR of 3/98, or 0.031. For the remaining 20 pre-pandemic controls used in the test set, the ReScan peptides were positive on 2/20 (false positive rate [FPR] = 0.1), while the whole-protein panel identified 6/20 (FPR = 0.3) as positive. As with the peptide conjugated beads, a single positive cell among the 3 whole-protein conjugations was considered a positive result.
Figure 4.

ReScan Peptides as High-Throughput Serology Assay for COVID-19
(A) Test set of patients who were analyzed by either VirScan and ReScan, with each column representing either a ReScan-identified peptide or a whole-protein antigen. The outline around a result indicates a significant (i.e., exceeds Z score of 3) signal established by the background population of pre-pandemic controls. Adjudication is made from either a single peptide or a single protein determined to be positive within their respective panels by exceeding 3 standard deviations from the mean of pre-pandemic control distribution. The prior disease status columns represent patients either with a historically positive qPCR swab or neutralizing antibodies against SARS-CoV-2.
(B) Validation set of new patient sera from blinded plate.
(C) Second blinded validation set of new patient samples with an accompanying clinical serology IgG result measuring anti-Spike antibodies. All of the data are represented as the log of secondary fluorescence signal on each antigen normalized to 2 separate negative controls in each well.
In all of the panels, each row represents a unique patient. n = 2 technical replicates averaged for each data point.
In 2 independent validation sample sets of 34 patients who were previously positive by a clinically validated qRT-PCR for SARS-CoV-2 as well as 45 pre-pandemic negative patient sera, we found that the ReScan peptide panel correctly identified 31/34 positive patients (FNR = 0.088) and 42/45 negative patients (FPR = 0.067). The combined whole-protein panel again showed slightly higher sensitivity correctly identifying 32/34 positive patients (FNR = 0.059) with reduced specificity (5/45 or FPR = 0.111). In a subset of the validation cohort in which clinical spike antibody testing data were available, the ReScan peptide panel correctly identified 2 patients as negative who were incorrectly classified as positive by a clinical serology assay. Notably, the panel correctly identified 4 patients as positive who were below threshold for positivity on clinical assay. There was 1 concordant false positive between the whole-protein panel and clinical serology testing. All of the data are summarized in Table S2. ReScan peptide assay performance was 100% sensitive in sera with the highest neutralizing titers across 78 patients for which NT50 data were available and 75% sensitive in patients with low titers (top and bottom half of distribution respectively, see Figure S4).
Discussion
Diagnostic testing is a critical component of an effective public health response to a rapidly spreading pandemic. While deploying these assays is time sensitive, prematurely releasing tests that are not sufficiently sensitive or specific can stymie timely interventions and undermine confidence in government and public health institutions. Optimizing specificity is particularly important for serologic assays that are typically not being used to diagnose acutely infected patients but instead are used to make epidemiologic measurements such as infection and case fatality rates or as 1 potential surrogate marker for protective immunity. Rather than a priori selecting 1 or 2 antigens upon which to base a serologic assay, we used a proteome-wide custom HuCoV VirScan library and COVID-19 patient sera to agnostically identify the antigens that were the most enriched relative to pre-pandemic control sera. In addition to using next-generation sequencing (NGS) and statistical analysis (PhIP-seq), we developed ReScan to rapidly select and propagate the most immunogenic peptide-bearing phage clones by immunofluorescence without the hurdles of expressing recombinant proteins or chemically synthesizing peptides. With this approach, the most critical antigens are identified, and the phages expressing them are simultaneously isolated and cultured, rendering an immediate, renewable protein source that is amendable to a wide range of ELISA platforms, including microarrays, beads, and plates. Here, ReScan identified 9 antigens derived from 3 SARS-CoV-2 proteins as candidates for more specific, multiplexed SARS-CoV-2 serologic assays.
ReScan has several key advantages over traditional peptide arrays for antigen screening. Diversity of the parent library is not restricted by physical space on the array, and the cost of peptide synthesis is decreased by at least 2 orders of magnitude as the input library is generated via an oligonucleotide pool cloned en masse into a phage backbone. Through IP and phage amplification, IgG antibodies from patients with a disease of interest (e.g., COVID-19) select a specific subset of peptides for use in downstream diagnostic assays. This approach is an improvement over standalone phage IPs, which can errantly amplify clones due to nonspecific binding of phage to beads and protein-protein interactions outside of antibody-antigen events. These nonspecific binding events can pose significant challenges for the interpretation of PhIP-seq datasets.17 ReScan circumvents these problems by using plaque lift immunoblotting,18 a well-characterized technique that verifies that a particular phage colony was generated based on a true antibody-mediated interaction, to select for antigens that also perform well in the context of an immunostaining assay.
Typically, peptides can be challenging to spot or print on microarrays due to their widely variable physical properties and solubility, necessitating bespoke solvent systems and resuspension procedures.19 By hosting these peptides on a stable T7 bacteriophage carrier, the treatment of every clone is standardized, with each species undergoing high-throughput, plate-based amplification simultaneously. In addition, carrier proteins provide better physical access for antibody binding and inter-antigen assay reproducibility.20 This standardization, combined with acoustic liquid handling, permits the generation of protein microarrays through a flexible and rapid printing process with low variability between arrays.
We found that COVID-19 serum IgG antibodies were predominantly directed against epitopes in the S and N proteins consistent with the rich literature on the sensitivity and specificity of ELISAs using these proteins for SARS-CoV-1.21, 22, 23, 24, 25 While the majority of the focus thus far on serology development for SARS-CoV-2 has been on the S protein,4,7,26,27 our data suggest that N protein-based ELISAs should be pursued in parallel.8 Because of its high degree of glycosylation, peptides from the S protein displayed on phage cannot capture all possible epitope diversity, although 2 of the S protein peptides we detected with VirScan and ReScan (residues 783–839 and residues 1,124–1,178) do contain N-linked glycosylated sites in the native S protein.28 We identified 3 critical regions that were concordant between both ReScan and VirScan. All 3 S protein fragments are on the surface of the protein and appear to be solvent accessible based on the 6VSB cryoEM structure.29
In addition to our identification of the most enriched N protein peptides in the region of the N-terminal RNA binding domain, previous work with SARS-CoV-1 peptides has shown C-terminal dimerization regions to be particularly immunogenic.30 This region, spanning peptides 362–399, was also a significant target in both ReScan and HuCoV VirScan methods, although less frequent (seen in 15% and 30% of patients, respectively).
The only additional epitope identified by ReScan outside the N and S proteins was a single region mapping to the ORF3a protein between positions 172 and 209. ORF3a plays several important roles in viral trafficking, release, and fitness,31 and this particular region spanning the di-acidic central domain facilitates export from the host cell endoplasmic reticulum to the Golgi apparatus.32 It was also identified as an antibody target in SARS-CoV-1-infected patients.33,34 While only 6/20 patients had significant hits to this region by ReScan, it is a unique accessory protein to the SARS-like family, and therefore may represent an attractive candidate to enhance the specificity of SARS-CoV-2 diagnostic assays.
ReScan allows for rapid and agnostic antigen validation across multiple patients, but serves an important secondary purpose—once peptide-bearing phage are sequenced for identity and the breadth of their antigenicity is determined, it is straightforward to return a source colony to propagate and extend their use in alternative, high-throughput formats. From ReScan source wells, we produced high-concentration, sequence-verified stocks of each of the 9 identified antigens in the test cohort of patients and conjugated them to spectrally encoded beads to perform secondary immunofluorescence assays in a format that was amenable to a clinical diagnostics setting at <US$2 reagent cost per sample.
Without knowing the antigens in advance, ReScan identified a panel of linear peptides that have comparable sensitivity to a panel of known immunogenic whole proteins. We observed improved specificity with this peptide-based approach, with only 5 as opposed to 11 total false positives out of 65 patients in both test and validation cohorts. Residues mapping to S protein residues 552–589 and ORF3a accessory protein residues 172–209 produced the least hits specific to SARS-CoV-2 patients, and their removal from the panel serve only to improve the FDR to 2.2% (compared to the whole-protein FDR of 11.1%) in our validation cohort without a reduction in sensitivity (Table S2). This particular S protein region was recently reported to be a target of neutralizing antibodies, although in a very small patient cohort.35 Our data suggest that antibodies to this antigen are not particularly prevalent among patients with otherwise strong SARS-CoV-2 antibody responses, as it was significant in only 10 of the total 137 COVID-19 patients who were assessed by this high-throughput assay.
Limitations of Study
To fully characterize the test performance characteristics of these 9 candidate antigens as the basis for a more specific, high-throughput clinical diagnostic assay, additional patient samples need to be analyzed. However, both the statistically significant enrichment of these antigens above pre-pandemic HCs by PhIP-seq, and the rigorous comparison between staining on HCs and COVID-19 subjects to determine a positive signal on the ReScan microarrays are both supportive of these antigens being highly specific for SARS-CoV-2 exposure. The current IP strategy focuses only on IgG, but future work may center on IgM targets, given their role in the early humoral immune response. In addition, post-translational modifications and tertiary or quaternary conformational epitopes are not represented in the T7 phage display system used here.15,36 As a result, antibodies to spatial motifs within the S protein and its RBD are not captured by peptide display systems, which may account for the higher FNR seen with the ReScan peptides alone as compared to the whole-protein panel, which may justify a complementary panel using a combination of linear and whole-protein antigens in the future.
While these observed test characteristics were lower than expected compared to other published whole-protein data, it is crucial to note that the patient population used in this study reflects a cross-section of the real-world patient population that has contracted COVID-19, as we have included patients with broadly variable disease status (both inpatient and >70 convalescent outpatient samples), 2 different geographic centers, gender, and time from symptom onset. In a head-to-head comparison using this sample set, we see similar or better diagnostic performance of this multiplexed peptide assay compared to whole-protein-based diagnostics, both within the same assay and benchmarked against a clinical serology ELISA test.
The immediate focus of this study was to develop a blueprint for candidate antigens in a scenario such as a global outbreak where discriminatory biomarkers are unknown. We used this systematic and antigen-agnostic pipeline to develop confirmatory SARS-CoV-2 serologic assays and identify a panel of linear antigens with significant discriminatory power in regions of the virus that have low mutational diversity. Beyond their use for diagnostic purposes, the next and critical question is to what degree a patient’s antibody repertoire confers protective immunity. Because our assay characterizes patients’ antibodies to multiple SARS-CoV-2 antigens, it enables deeper phenotyping of the adaptive immune response that, in future studies, can be correlated with acute and long-term clinical outcomes.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-T7 tag | Invitrogen | Cat# PA1-32386; RRID:AB_1960902 |
| Anti-Human IgG | Li-Cor | Cat# 926-32232, RRID:AB_10806644 |
| Anti-Rabbit IgG | Li-Cor | Cat# 926-68071, RRID:AB_10956166 |
| Anti-Glial Fibrillary Associated Protein | Agilent | Cat# Z033429-2 |
| Anti-IgG conjugated to Phycoerythrin | Thermo Scientific | Cat# 12-4998-82, RRID:AB_465926 |
| Bacterial and Virus Strains | ||
| BL5403 | Novagen, T7 Select Kit | Cat# 70550-3 |
| T7 Bacteriophage | Novagen, T7 Select Kit | Cat# 70550-3 |
| Biological Samples | ||
| COVID-19 Patient Sera | This paper | N/A |
| Pre-Pandemic Control Sera | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Blocking Powder | BioRad | Cat# 1706404 |
| Protein A conjugated magnetic beads | Invitrogen | Cat# 10008D |
| Protein G conjugated magnetic beads | Invitrogen | Cat# 10009D |
| T4 ligase | New England Bio | Cat# M0202S |
| Phusion DNA Polymerase | New England Bio | Cat# M0530L |
| MagPlex Magnetic Beads | Luminex | Cat# MC10012 |
| Coupling Kit | Luminex | Cat# 40-50016 |
| Critical Commercial Assays | ||
| T7 Select 10-3b Cloning kit | EMD Millipore | Cat# 70550-3 |
| Ampure XP Beads | Beckman Coulter | Cat# A63881 |
| Deposited Data | ||
| VirScan tabulated peptide counts | This paper | https://github.com/UCSF-Wilson-Lab/sars-cov-2_ReScan_VirScan_complete_analysis |
| ReScan image analysis and hit tables | This paper | https://github.com/UCSF-Wilson-Lab/sars-cov-2_ReScan_VirScan_complete_analysis |
| Recombinant DNA | ||
| Human Coronavirus Synthetic DNA | Twist Bioscience | https://www.twistbioscience.com/ |
| Software and Algorithms | ||
| dotblotr | This paper | https://github.com/czbiohub/dotblotr |
| cd-hit | Li and Godzik, 200637 | http://weizhongli-lab.org/cd-hit/ |
| Bowtie2 | Langmead and Salzberg38 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 200939 | http://samtools.sourceforge.net/ |
| Flash | Magoč and Salzberg, 201140 | https://ccb.jhu.edu/software/FLASH/ |
| R | Open Source | https://ccb.jhu.edu/software/FLASH/ |
| Bioconductor | Open Source | https://www.bioconductor.org |
| numpy | Open Source | https://dx.doi.org/10.1109/MCSE.2011.37 |
| skimage | Open Source | https://peerj.com/articles/453 |
| scipy | Open Source | https://www.nature.com/articles/s41592-019-0686-2 |
| pandas | Open Source | https://zenodo.org/record3509134 |
| Matplotlib | Open Source | https://ieeeexplore.ieee.org/document/4160265 |
| openCV | Open Source | https://github.com/skvark/opencv-python |
| Other | ||
| 0.2μm filter plates | Arctic White | Cat# AWFP-F20080 |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Wilson (Michael.Wilson@ucsf.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
VirScan, ReScan and clinical data analyzed in the manuscript have been made available in the supplementary material. Dotblotr package and source code are available on the Chan Zuckerberg Biohub GitHub repository.
Raw data and analysis code for this study are publicly available on the Wilson Lab GitHub Repository (https://github.com/UCSF-Wilson-Lab/sars-cov-2_ReScan_VirScan_complete_analysis).
Experimental Model and Subject Details
Patient Enrollment and Data Collection
Inclusion criteria for patients with a confirmed diagnosis of COVID-19 were 1) age > 18 years and 2) a positive SARS-CoV-2 RT-PCR from a nasopharyngeal swab performed in a clinical laboratory. Patient 1 was enrolled in a research study at UCSF (IRB number 13-12236). Patient 2 was enrolled in a research study at UCSF (IRB number 10-00476). Patients 3-6 and 9-20 were enrolled in a research study at UCSF (IRB number 18-25287). Patients 7 and 8 were enrolled in a research study at the Vitalant Research Institute. Because patient samples were received de-identified, the UCSF Institutional Review Board did not require further institutional review. Electronic and paper medical records of patients 2 and 3 from UCSF were reviewed for demographic details, clinical data and laboratory results, and outcome at last follow-up. Patients 3-6, 7, 8, and 9-20 were de-identified with only limited clinical and demographic data available. Their ages were rounded to the nearest mid-decade (e.g., 45 years-old, 55 years-old, etc). Patients 21-114 were enrolled as part of a recently published study.41 Patients 115-156 were enrolled in a research study at the Vitalant Research Institute. Patients 157-166 were enrolled in a research study at UCSF (IRB number 18-25287). Patients 167-193 were enrolled in a research study at UCSF (IRB 10-02598). The 95 de-identified healthy control sera were obtained from adults who donated blood to the New York Blood Bank before the SARS-CoV-2 pandemic. Sera were transported on dry ice to our laboratory at UCSF and stored immediately upon receipt at −80°C. Upon thawing, aliquots were diluted 1:1 into a glycerol containing storage buffer as described below. There is incomplete information on the handling of all specimens analyzed between the time they were obtained from the patient and ultimately sent to UCSF as these samples were collected by clinical labs. Since the COVID-19 cases and uninfected control samples were all collected from hospital clinical labs it is unlikely there was differential handling procedures between the cases and controls in this study.
Method Details
PhIP-Seq/VirScan Protocols
Coronavirus (CoV) Library Design and Cloning
RefSeq sequences for SARS-CoV-2 (NC_045512), SARS-CoV-1 (NC_004718) and 7 other CoVs known to infect humans, including betaCoV England 1 (NC_038294), HuCoV 229E (NC_002645), HuCoV HKU1 (NC_006577), HuCoV NL63 (NC_005831), HuCoV OC43 (NC_006213), Infectious Bronchitis virus (NC_001451), and MERS CoV (NC_019843) were downloaded from the National Center for Biotechnology Information (NCBI). All open reading frames for each virus were divided into sequences of 38-mer peptides, with consecutive peptides sharing a 19 amino acid overlap. All peptide sequences were collapsed on 99% amino acid sequence identity using cd-hit. After collapsing on sequence identity, peptide amino acid sequences were converted to DNA sequences using an R language script, randomizing codon selection. Twenty-one (21) nucleotide 5′ linker sequences as well as a nucleic acid sequence encoding a FLAG tag (DYKDDDDK) at the 3′ end of each oligonucleotide sequence were added. The following 5′ linker sequences were: GCTGTTGCAGTTGTTGGCGCT (SARS-CoV-1 and SARS-CoV-2), GTAGCTGGTGTTGTAGCTGCC (SARS-CoV-1 only), GCAGGAGTAGCTGGTGTTGTG (SARS-CoV-2 only), and AGCCATCCGCAGTTCGAGAAA (betaCoV England 1, 229E, HKU1, NL63, OC43, Infectious Bronchitis virus, MERS) for a combination SARS-CoV-1/SARS-CoV-2 library, an exclusive SARS1-CoV-1 library, an exclusive SARS-CoV-2 library, and a library spanning other CoVs known to cause human infections. The final oligonucleotide sequences were 159 nucleotides in length, output to a FASTA file and sent to Twist Biosciences for synthesis.
A single vial of 10pmoles of lyophilized DNA was received from Twist Bioscience. The lyophilized oligonucleotides were resuspended in TE to a final concentration of 0.2nM and PCR amplified for cloning into T7 phage vector arms (Novagen/EMD Millipore, Inc). The following amplification primers and cycling conditions were used:
Forward:
TAGTTAAGCGGAATTCAGCTGTTGCAGTTGTTGGCGCT (SARS-Cov-1/SARS-CoV-2)
TAGTTAAGCGGAATTCAGTAGCTGGTGTTGTAGCTGCC (SARS-CoV-1)
TAGTTAAGCGGAATTCAGCAGGAGTAGCTGGTGTTGTG (SARS-CoV-2)
TAGTTAAGCGGAATTCAAGCCATCCGCAGTTCGAGAAA (Other CoVs)
Reverse:
ATCCTGAGCTAAGCTTTTACTTATCATCATCATCCTTGTAATCACC
Cycling conditions:
| STEP | TEMP | TIME |
|---|---|---|
| Initial Denaturation | 98°C | 30 s |
| 98°C | 5 s | |
| 70°C | 15 s | |
| 20 Cycles | 72°C | 10 s |
| Final Extension | 72°C | 2 minutes |
| Hold | 10°C | - |
After amplification, PCR reactions were bead cleaned using Ampure XP beads at 1:1 ratio per the manufacturer’s instructions. Bead cleaned PCR products were quantified by using Qubit reagent (Life Technologies, Inc) and 1 μg of PCR product was digested with 1 μL of EcoRI-HF (NEB) and 1 μL of HindIII-HF (NEB) with 1X CutSmart buffer (NEB) in a total volume of 50 μL for 1h at 37°C and heat inactivated at 65°C for 10 minutes. Digested DNA was cleaned again using Ampure XP beads and cutting efficiency confirmed by running both the uncut and restriction digested PCR products on a high sensitivity dsDNA Bioanalyzer chip (Applied Biosystems, Inc.).
Cut PCR amplified oligonucleotide pools were ligated into pre-cut T7 vector arms in a 5 μL volume at a ratio of 3:1 (0.06 pmol of insert and 0.03 pmol of T7 vector arms) with T4 DNA ligase (NEB) at 16°C for 12h, after which ligations were heat inactivated for 15 minutes at 65°C. A total of 2 μL of the ligation reaction was mixed with 12 μL of T7 packaging extract, stirring with a the pipet tip and incubating at room temperature for 2h. At the end of two hours, 150 μL of cold LB + Carbenicillin was added to the packaging reaction and it was stored at 4°C. Ten-fold serial dilutions of the raw packaging reaction were done, followed by plaque assays to determine the titer the number of infectious phage in the packaging reaction.
Preparation of Phage Libraries
Phage libraries were prepared and amplified fresh from packaging reactions. To prepare phage libraries, a 300 mL culture of E. coli BLT5403 was incubated at 37oC with shaking until mid-log phase, defined as OD600 = 0.4. The culture was then inoculated at a multiplicity of infection (MOI) of 0.001 and lysed at 37°C for 2.5 hours. 5M NaCl was added to the lysate on ice for 5 minutes at a 1:10 volume ratio to complete lysis. Lysate was then spun at 4000 g for 15 minutes to remove cellular debris. Phage-containing supernatant was then precipitated with PEG/NaCl (PEG-8000 20%, NaCl 2.5 mM) overnight at 4°C. Precipitated phage were then pelleted for 15 minutes at 3220 g at 4°C and resuspended in storage media (20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 6 mM MgCl2) before 0.22 μM filtration. Resulting phage libraries were titered by plaque assay and adjusted to a working concentration of 1010-1011 pfu/mL before incubation with patient sera.
Immunoprecipitation of Phage-Bound Patient Antibodies
96-well, 2mL deep well plates (Genesee Scientific) were incubated with blocking buffer of 3% BSA in 1xTBST overnight to prevent nonspecific binding to plate walls. Blocking buffer was then replaced with 500 μL of either library type (HuCoV or pan-viral) and 1 μL of human sera in 1:1 storage buffer (0.04% NaN3, 40% Glycerol, 40mM HEPES to preserve antibody integrity), or 1 μg of commercial antibody for a positive immunoprecipitation control. To facilitate antibody phage binding, the deep well plates with library and sample were incubated overnight at 4°C on a rocker platform in secondary containment. Pierce Protein A/G Beads (ThermoFisher Scientific) were aliquoted 20 μL of A and 20 μL of G per reaction and were washed 3 times in TNP-40 (140mM NaCl, 10mM Tris-HCL, 0.1% NP-40). After the final wash, beads were resuspended in TNP-40 in half the volume (20uL) and added to the phage-patient antibody mixture and incubated on the rocker at 4°C for 1 hour. Phage bound to antibody bound to bead was then washed five times with RIPA (140mM NaCl, 10mM Tris-HCl, 1% Triton X, 0.1% SDS) using a P1000 multichannel pipet to remove supernatant and add RIPA each time, with each wash incubating the bound A/G beads for 5 minutes with gentle rocking at 4°C. After the fifth wash, immunoprecipitated solution was resuspended in 150 μL of LB-Carb and then added to 500 μL of log phase E. coli for amplification (OD600 = 0.4). After amplification, 5M NaCl was added to lysed E. coli to a final concentration of 0.5M NaCl to ensure complete lysis. The lysed solution was spun at 3220 rcf. for 20 minutes and the top 500 μL was filtered through a 0.2 μM PVDF filter plates (Arctic White, #AWFP-F20080) to remove cell debris. Filtered solution was transferred to a new deep well pre-blocked plate and patient sera was added and subjected to another round of immunoprecipitation and amplification. The final lysate was spun, filtered and stored at 4°C for NGS library prep and subsequent ReScan plaque lifts and downstream colony selection.
NGS Library Prep
All liquid handling steps were performed using the Labcyte Echo 525 and the Integra Via Flow 3520. Immunoprecipitated phage lysate was diluted 1:10 and heated to 70°C for 15 minutes to expose DNA. DNA was then thermocycled in two subsequent reactions, the first of which amplified the unique, programmed peptide insert of the phage genome. The second served to the add on custom 8-12nt TruSeq sample barcode primers as well as adaptor sequences for Illumina flow cell. Libraries were then pooled at equal volumes, followed by cleaning and size selection using Ampure XP magnetic beads at 0.9X ratio according to manufacturer instructions. Eluted libraries were quantified by High Sensitivity DNA Qubit and qualified by High Sensitivity DNA Bioanalyzer after dilution to approximately 1ng/μL. Sequencing was then performed on a NovaSeq S1 (300 cycle kit with 1.3 billion clusters) averaging 5 million reads per sample for HuCoV and pan-viral libraries, and 500,000 reads for SARS-CoV-2 specific libraries.
PCR #1 Master Mix
| Reagent | Volume (uL) per reaction | Stock concentration |
|---|---|---|
| Nuclease-free water | 4.125 | – |
| Phusion HF Buffer (NEB) | 2.5 | 5x |
| 10 mM dNTPs | 0.25 | 10mM |
| 10 uM Forward Primer | 0.25 | 10uM |
| 10 uM Reverse Primer | 0.25 | 10uM |
| Polymerase | 0.125 | 1U/uL |
| Phage Lysis | 5 | Diluted 1:10 |
PCR #1 Cycle Protocol
| STEP | TEMP | TIME |
|---|---|---|
| Initial Denaturation | 98°C | 30 s |
| 98°C | 5 s | |
| 70°C | 20 s | |
| 16 Cycles | 72°C | 15 s |
| Final Extension | 72°C | 2 minutes |
| Hold | 10°C | - |
PCR #2 Master Mix
| Reagent | Volume (μL) per reaction | Stock concentration |
|---|---|---|
| Nuclease-free water | 8.125 | – |
| Phusion HF Buffer (NEB) | 2.5 | 5x |
| 10 mM dNTPs | 0.25 | 10mM |
| Barcode Forward i5/Reverse i7 mixed Primers∗ | 1 | 5mM |
| Polymerase | 0.125 | 1U/uL |
| PCR 1 | 0.5 | 1x |
PCR #2 Cycle Protocol
| STEP | TEMP | TIME |
|---|---|---|
| Initial Denaturation | 98°C | 30 s |
| 5 Cycles | 98°C | 5 s |
| 70°C | 20 s | |
| 72°C | 15 s | |
| Final Extension | 72°C | 2 minutes |
| Hold | 10°C | - |
Primers for DNA Amplification:
| SARS1/2 Forward_0N | ACACTCTTTCCCTACACGACGCTCTTCCGATCTGCTGTTGCAGTTGTTGGCGCT |
| SARS1/2_Fwd_1N | ACACTCTTTCCCTACACGACGCTCTTCCGATCTNGCTGTTGCAGTTGTTGGCGCT |
| SARS1/2_Fwd_2N | ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNGCTGTTGCAGTTGTTGGCGCT |
| SARS1/2_Fwd_3N | ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNGCTGTTGCAGTTGTTGGCGCT |
| SARS2_Fwd_0N | ACACTCTTTCCCTACACGACGCTCTTCCGATCTGCAGGAGTAGCTGGTGTTGTG |
| SARS2_Fwd_1N | ACACTCTTTCCCTACACGACGCTCTTCCGATCTNGCAGGAGTAGCTGGTGTTGTG |
| SARS2_Fwd_2N | ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNGCAGGAGTAGCTGGTGTTGTG |
| SARS2_Fwd_3N | ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNGCAGGAGTAGCTGGTGTTGTG |
| sCoV_Fwd_0N | ACACTCTTTCCCTACACGACGCTCTTCCGATCTAGCCATCCGCAGTTCGAGAAA |
| sCov_Fwd_1N | ACACTCTTTCCCTACACGACGCTCTTCCGATCTNAGCCATCCGCAGTTCGAGAAA |
| sCov_Fwd_2N | ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNAGCCATCCGCAGTTCGAGAAA |
| sCov_Fwd_3N | ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNAGCCATCCGCAGTTCGAGAAA |
| FLAG_Rev_0N | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTATCATCATCATCCTTGTAGTCACC |
| FLAG_Rev_1N | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNATCATCATCATCCTTGTAGTCACC |
| FLAG_Rev_2N | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNATCATCATCATCCTTGTAGTCACC |
| FLAG_Rev_3N | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNATCATCATCATCCTTGTAGTCACC |
Bioinformatic Analysis of PhIP-Seq Data
Sequencing reads were aligned to a reference database of the full viral peptide library using the Bowtie2 aligner, outputting a SAM file. All SAM format files were converted to BAM using the samtools view command. For each aligned sequence, the CIGAR string was examined, and all alignments where the CIGAR string did not indicate a perfect match were removed. The filtered set of aligned sequences was then translated and the translated peptides that were perfect matches to the input library were identified. Any peptides that were not perfect matches were excluded from further analysis. The final set of peptides was tabulated to generate counts for each peptide in each individual sample. All of this analysis was automated using an R language script in RStudio.
Peptides counts were normalized for read depth by dividing them by the total number of peptides in the sample and multiplying by 100,000, resulting in a reads/100,000 reads for each peptide.40,42, 43, 44 For all VirScan libraries, the null distribution of each peptide’s log10(rpK) was modeled using a set of 95 healthy control sera. All counts were augmented by 1 to avoid zero counts in the healthy control sera samples. Multiple distribution fits were examined for these data, including Poisson, Negative Binomial and Normal distributions. Quantile-quantile plots for each distribution demonstrated that the Normal distribution had the best fit across all peptides, as measured by its median linear correlation coefficient. These null distributions were used to calculate p values for the observed log10(rpK) of each peptide within a given sample. The calculated p values were corrected for multiple hypothesis using the Benjamini-Hochberg method. Any peptide with a corrected p value of < 0.01 was considered significantly enriched over the healthy background. For peptides where the number of unique values was less than 10 across the healthy controls, and for which it was thus impossible to estimate a Normal null distribution because of sparsity of data, they were combined into a single distribution and this common distribution was used to calculate p values. As before, these p values were corrected for multiple hypothesis testing and a corrected p value of < 0.01 was used as a significance threshold.
To identify regions targeted by host antibodies, all library peptides were aligned to the SARS-CoV-2 reference genome, concatenating all of its open reading frames (ORFs) into a single sequence. This reference sequence was used to generate a blastp database, and all peptides in the library were aligned to it using NCBI blastp. Using these data, the summation of enrichment relative to the healthy background was calculated at each position across SARS-CoV-2 for all significant peptides for each experimental sample. Finally, the results were summed at each position across all experimental samples and the summed enrichment plotted by position using the R ggplot2 library. Only alignments that were at least 38 amino acids in length (the length of the peptides in the CoV VirScan libraries) were included in this analysis.
All protein sequences for SARS-CoV-2 were downloaded from the NCBI GenBank database on 7/22/20 and sequences for the spike and nucleoprotein identified by their annotated names. Multiple sequence alignment was performed in R using the DECIPHER package and the Shannon entropy (-∑ Pi∗Ln(Pi), with i ∈ {1,20} for each of 20 acids), calculated for each position using multiple sequence alignment data to provide a measure of sequence diversity at each position in the S and N proteins. Data were plotted using the R ggplot2 package.
ReScan Protocol
Plaque Lift Immunoblots
Immunoprecipitations were performed as previously described with a T7 phage display library containing the full-length genome of SAR-CoV-2 with 38 amino acid insert length and 19 amino acid tiling. Standard top-agar plaque assays were performed on freshly filtered second round lysate from a candidate patient IP at 106 to 107 dilution in LB-Carbenicillin. Plaques were left to grow for 5-6 hours at 37°C or until approximately 2mm in diameter, then placed for 10 min at 4°C to fully set top agar. Pre-cut nitrocellulose (Protran, #10401116) was placed onto the agar plates for 1 minute to adhere phage colonies then carefully lifted and left to dry. Nitrocellulose blots were then washed with TBST for 3 minutes, blocked for 30 minutes at room temperature in TBST with 5% non-fat milk (BioRad, #1706404). The primary buffer consisted of the serum from the patient IP that the plaque assay was cast from diluted to 1:10,000 in blocking buffer, with serum being previously diluted 1:1 in storage buffer. Blots were then incubated overnight with gentle agitation at 4°C. Immunoblots were then washed 3x for 5 minutes with TBST at room temperature, then stained with anti-human IgG antibody (LICOR, #926-32232) at 1:20,000 concentration for 1h and then washed 4x in TBST for 8 minutes each. Immunoblots were then given a final wash in PBS to remove detergent for 2 minutes at room temperature, dried on Whatman 3MM paper, and immediately imaged on a LICOR Odyssey CLX imager.
Plaque Picking and Sub-Library Generation
Plaques were picked with clean 200 μL pipette tips from corresponding agar plate using immunoblot anti-IgG staining as a guide for which phage colonies represented antibody-specific events. Approximately 40 plaques were each picked from 7 corresponding different patient plaque lifts into 2mL deep well plates into storage media (1xTBS supplemented with 5mM MgCl2) and pipetted up and down to free phage from tip. 0.5mL of BL5403 E. coli at an OD600 = 0.4 were then added to each plate and left to lyse for 2.5h on an INFORS shaker at 37°C and 720rpm with hydrophobic breathable seal. 100 μL of lysis were added to 700 μL of a second propagation of BL5403 E. coli at OD600 = 0.4 to improve titer and minimize variation between wells. This was similarly lysed for 1.5h or until visually complete, then 80 μL of 5M NaCl was added to complete lysis for 5 minutes on ice. Plates were spun at 3200 rcf. for 15 minutes at 4°C to pellet debris, then filtered through 0.2 μm PVDF filter membranes for storage at 4°C. Lysis was used within two weeks.
Acoustic Microarray Printing and Staining
60 μL of each monoclonal phage stock was added to a unique well on an ECHO qualified source plate (Labycte, #PPT-0200) and spun at 3200 rcf. for 20 min to remove bubbles. Nitrocellulose (0.2 μm, Whatman) was cut and adhered to a standard Framestar 384 skirted plate (4titude, #0384) as a backing frame and used as a destination plate. Arrays were printed using a Labcyte ECHO 525 instrument on the 384_PP_PLUS_BP setting at a 6,144 well pattern with 2 row spacing between arrays, with 9 arrays per 384 well plate. 25nL from each source well was printed onto each microarray spot. Arrays were thoroughly dried for 30 minutes, then removed from backing plate, diced and placed into black 7.4cm x 2.4cm western-style boxes at 4°C until staining.
Individual arrays were wetted with 1xTBST for 5 minutes, then blocked with 1X TBST with 5% non-fat milk as previously described for 30 minutes. Primary buffer master mix consisted of cold blocking buffer with 0.02% sodium azide as a preservative, and 1:2000 concentration of anti-T7 Tag (Invitrogen, # PA132386). Each microarray was incubated with patient serum previously diluted 1:1 in storage buffer at 1:5000 dilution for a final dilution of 1:10,000. Arrays were allowed to incubate overnight at 4°C. Blots were then washed 3x for 5 minutes each in 1x TBST and immediately stained in secondary buffer consisting of anti-human IgG 800 and anti-rabbit IgG 680 (LICOR, 926-68071) at 1:20,000 dilution each for 1h under gentle agitation at room temperature. Arrays were then washed 4x in 1X TBST for 8 minutes each, with a final wash in PBS to remove detergent, then dried fully and imaged on a LICOR Odyssey CLX imager.
Miniaturized Next Generation Sequencing of Monoclonal Microarray Library
All liquid transfer steps were done with a Labcyte ECHO 525 acoustic handler. 0.25 μL of phage stock was transferred from library source plate into 2.25 μL of nuclease free water in a 384 well PCR plate. Phage were lysed as previously described at 70°C for 15 minutes, then the following concentrated master mix was dispensed into each well for a 6.25 μL total reaction volume:
SARS CoV-2 ReScan PCR#1 Master Mix
| Reagents | μL/reaction |
|---|---|
| 5x HF buffer | 1.25 |
| 10 mM dNTPs | 0.125 |
| 10mM Forward Primer | 0.125 |
| 10mM Reverse Primer | 0.125 |
| Phusion HF Polymerase | 0.0625 |
| Water | 2.0625 |
| DNA template | 2.5 |
SARS-CoV-2 Library Prep Sequencing Primers same as described above.
PCR thermocycler sequence was identical to that used above for library preparation for IP sequencing described above with 13 cycles. 0.5 μL of product from PCR#1 was used as input for PCR#2, with 2.75 μL of the following concentrated master mix dispensed into each well:
SARS CoV-2 ReScan PCR#2 Master Mix
| Reagents | μL/reaction |
|---|---|
| 5x HF buffer | 1.25 |
| 10 mM dNTPs | 0.125 |
| Phusion HF Polymerase | 0.0625 |
| Water | 1.3125 |
| DNA template | 0.5 |
8-12 base pair TruSeq barcoding primers were kindly provided by the Chan Zuckerberg BioHub at 5mM concentration in water. These were diluted to 1:5 in nuclease free water and pooled into a 384 well ECHO qualified source plate. 3 μL of each diluted primer was dispensed into the corresponding well for PCR#2 giving a unique barcode to each reaction after thermocycling protocol identical to what was used above for IP library preparation.
Once complete, the entire 6.25 μL reaction volume from each well was pooled and purified with Ampure XP bead clean up at 0.9x ratio per manufacturer instructions, then eluted into 600uL total volume. Pooled and cleaned DNA was quantified by Qubit and Bioanalyzer and sequenced on an Illumina iSeq for an average read depth of 4000 reads per well.
dotblotr Image Processing
Image Processing of ReScan blots
Image processing and visualization of the rescan blot data was done using dotblor, a python package for analyzing dotblot data.
First, LI-COR Image Studio was used to crop and rotate the images of the blot such that each image contained a single strip with the rows and columns of the dot array aligned with the rows and columns of pixels in the image. The images were then loaded into numpy arrays using skimage and the image split into the reference channel (anti-t7 tag antibody) and the probe channel (anti-human IgG antibody).
Each dot was detected using the dot signal in the reference image. Pixels corresponding to a dot were segmented using a threshold selected using Otsu’s algorithm and connected components labeling. The detected spots were filtered based on circularity and size. Each detected dot that passed the quality control filters was assigned a coordinate (letters from rows, numbers for columns) by clustering. Finally, the label image where each dot was given a unique label.
Next, the dot locations were determined from the reference channels to segment the dots in the probe channel. For each dot, the normalized intensity (probe channel / reference channel) was calculated. The calculated data were then output as a results_table in a pandas DataFrame. The resulting plots were generated using matplotlib and openCV.
Analysis code python notebook, hitcalling.ipynb, is available on the data repository for this manuscript, which takes the results_table using dotblotr (specifically, using the multifile example available on the dotblotr github repo) from both healthy and positive control cohorts.
Array Identification with Clustering
The row and column corresponding to each dot were determined by clustering the x and y coordinates of the dot centroids, respectively. To determine the row of each dot, only the y coordinate of the centroid of each dot were considered along with the number of rows in the array based on the assay configuration. The y coordinates were then clustered with k-means where k (the number of clusters) was set to the number of rows. Each cluster then corresponds with a row and the identity of the row was determined by the order. The same process was applied for the columns by applying clustering in the same way to the x coordinates of the dot centroids. Thus, the row and column were assigned for each detected dot.
Image Processing and Hit Calling
To determine which dots were positive, the magnitude of the dots’ normalized signal (human IgG signal intensity / anti-T7 tag signal intensity) was compared to the distribution of normalized signal from the negative controls (phage expressing GFAP or Tubulin 1a1). We calculated the mean and standard deviation of the negative control dots on the same strip and called dots ‘positive’ with a normalized signal at least 3 standard deviations above the mean of the negative control dots.
Since a given peptide was represented by multiple dots on each strip, all dots corresponding to the same peptide were grouped for hit calling. To determine if a given strip, s, was positive for a given peptide, p, the counts of positive and negative dots for peptide p in strip s were compared to the counts of positive and negative dots for peptide p in all healthy control strips using Fisher’s exact test. If peptide p was more likely to yield positive dots in in strip s than in the healthy controls with p < 0.05, strip s was called positive for peptide p.
Results Table Schema
The results_table contains the following columns. Each row contains the results for a single dot. The list is formatted: column_name [data type]: < description > .
assay_id [str]: the name of the assay configuration file
strip_id [str]: the unique identifier for the strip containing the dot
dot_name [str]: the name of the well in the source plate the dot came from
source_plate_id [str]: the name of the source plate the dot came from
source_plate_row [str]: the name of the source plate row the dot came from
source_plate_column [str]: the name of the source plate column the dot came from
exp_group [str]: the name of the experimental group
zscore_threshold [float]: the value of the z score threshold used to call a positive hit
top_hit [str]: name of the most common peptide sequence in the phage source
top_hit_pct [str]: fraction of total reads in the phage source corresponding to the top_hit.
row [str]: row name in the strip the dot is in
col [str]: column name in the strip the dot is in
x [float]: x coordinate of the dot centroid in the image (in pixels)
y [float]: y coordinate of the dot centroid in the image (in pixels)
mean_intensity_control [float]: mean intensity of the dot in the reference channel
area [float]: area of the dot in pixels
mean_intensity_probe [float]: mean intensity of the dot in the probe channel
norm_probe_intensity [float]: mean_intensity_probe / mean_intensity_control
positive_threshold [float]: threshold for norm_probe_intensity value required for the dot to be positive (set by zscore_threshold).
pos_hit [bool]: True of the dot was called positive (based on positive_threshold)
Luminex Assay
High concentration phage stocks were propagated from ReScan wells and grown to high (> 1011 PFU/mL) titer using the protocol described above, resuspended in PBS, then each was conjugated to unique bead IDs according to manufacturer’s Antibody Coupling Kit instructions (Luminex). Whole N protein (RayBiotech) beads were conjugated similarly using manufacturer instructions with 5μg of protein per 1 million beads. For other whole protein Luminex-based beads, MagPlex-Avidin Microspheres (Luminex) were coated with either the S protein RBD (residues 328-533) or the trimeric S protein ectodomain (residues 1-1213). Expression plasmids for both proteins were transfected into Expi293 cells co-expressing BirA using Expifectamine Expression System Kits (Thermo Fisher Scientific) in accordance with the manufacturer’s recommended protocol. During expression, cultures were maintained in biotin-supplemented media to permit biotinylation of a C-terminal Avi-tag. Proteins were then purified using Ni-NTA chromatography and purity assessed using SDS-PAGE. MagPlex Microspheres were coated following the manufacturer’s suggested protocol. Briefly, beads were incubated with 0.2 μg protein/million beads at a bead concentration of 2,000,000 beads/mL in PBS + 0.05% Tween 20 (PBST) for 30 minutes at room temperature. The beads were then washed twice with PBST using 30 s magnetic separation before resuspension in PBST and subsequent use in the Luminex-based serology panel.
All beads were blocked overnight before use and pooled on day of use. 2000-2500 beads per ID were pooled per incubation with patient serum at a final dilution of 1:500 for 1 hour, washed, then stained with an anti-IgG pre-conjugated to phycoerythrin (Thermo Scientific, #12-4998-82) for 30 minutes at 1:2000. Primary incubations were done in PBST supplemented with 2% nonfat milk and secondary incubations were done in PBST. Beads were processed in 96 well format and analyzed on a Luminex LX 200 cytometer.
Median Fluorescence Intensity from each set of beads within each bead ID were retrieved directly from the LX200 and log transformed after normalizing to the mean signal across two negative controls (GFAP and Tubulin phage peptide conjugated beads). The negative threshold was generated for each peptide-bead combination by randomly selecting 80 of the 100 pre-pandemic controls from the New York Blood Center to serve as a background distribution, where a cutoff of 3 standard deviations from the mean of this distribution was then set to define a positive result for that specific bead ID.
Quantification and Statistical Analysis
For statistical analyses of VirScan data, null distributions of each peptide from 95 pre-pandemic controls were generated and p values were calculated for the observed peptide rpKs, and multiple hypothesis corrected using the Benjamini-Hochberg method. All peptides with a corrected p value of < 0.01 were considered significantly enriched over the healthy background for that specific peptide. For ReScan data, anti-IgG fluorescent signal was considered significant if exceeding 3 standard deviations from negative intra-assay controls. Fisher’s exact test was used to assess significance of antigens between pre-pandemic and COVID-19 positive sera with a threshold of p < 0.05. Other statistical details can be found in figure legends. No data were excluded.
Acknowledgments
The authors would like to thank Dr. Nicole Repina for graphic design assistance with Figure 1, as well as the entire Chan Zuckerberg Biohub Sequencing Team for sequencing support. We thank Dr. John Pak (Chan Zuckerberg Biohub) and Dr. Florian Krammer (Mt. Sinai Icahn School of Medicine) for plasmids containing the S protein ectodomain, as well as Dr. Shion Lim for cloning the RBD construct. The authors would also like to acknowledge Dr. Michael Busch and Vitalant Research Institute for additional serum samples and controls, as well as the New York Blood Center for pre-pandemic control sera. We thank Andrew Kung and Ravi Dandekar for their help using Chimera. We thank Dr. Stephen Hauser (University of California, San Francisco Weill Institute for Neurosciences) for support. We thank Kanishka Koshal for help with consenting patients. We thank Irene Lui, Kevin Leung, and Xin Zhou for their help making purified proteins. C.R.Z. and J.V.R. wish to acknowledge Dr. Caleigh Mandel-Brehm for immunoblotting protocols and extensive discussion on PhIP-seq analysis. We thank the patients and their families for participating in this study. This work is supported by David Friedberg (C.R.Z. and M.R.W.); NIH grant K08NS096117, UCSF Weill Institute for Neurosciences, and an endowment from the Rachleff family (M.R.W.); NIH grant P01-AI138398-S1 and 2U19AI111825 (M.C.N.); NIH grant R01AI152161 (J.V.R.); George Mason University Fast Grant and the European ATAC Consortium EC 101003650 (D.F.R.); the Robert S. Wennett Post-Doctoral Fellowship, in part by the National Center for Advancing Translational Sciences (National Institutes of Health Clinical and Translational Science Award program, grant UL1 TR001866), and by the Shapiro-Silverberg Fund for the Advancement of Translational Research (C.G.); the Howard Hughes Medical Institute (M.C.N.); the Chan Zuckerberg Biohub (J.L.D., J.A.W., and K.A.Y.); the Sandler Foundation, and the William K. Bowes, Jr. Foundation (M.R.W., K.C.Z., C.Y.C., S.A.M., and J.L.D.); the UCSF Dean’s Office Medical Student Research Program (G.M.S.); National Cancer Institute 5F32CA239417 (J.R.B.); and the Damon Runyon Cancer Research Foundation (X.X.Z.). The findings and conclusions in this report are those of the author(s) and do not necessarily represent the official position of the NIH.
Author Contributions
J.V.R. computationally designed and cloned the VirScan peptide libraries. S.A.M., B.D.A., and C.R.Z. performed the VirScan experiments. C.R.Z., G.M.S., and B.D.A. performed the ReScan experiments. C.R.Z. and S.A.M. performed the library preparation for the sequencing of the phage libraries. K.A.Y. wrote the code for dotblotr and with C.R.Z. performed the assay development and analysis for ReScan. K.C.Z., W.G., C.Y.C., M.S., C.G., M.C., D.F.R., M.C.N., D.N., and P.J.N. identified patients, performed clinical phenotyping, and provided patient samples. J.V.R., C.R.Z., M.R.W., and J.L.D. analyzed the VirScan and ReScan data. R.P.L. performed the Luminex assay, and C.R.Z. performed the bead conjugations. J.R.B., R.P.L., X.X.Z., and C.R.Z. developed the Luminex assay. C.R.Z., J.V.R., J.L.D., and M.R.W. conceived of and wrote the manuscript. All of the authors discussed the results and critically reviewed the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: September 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrm.2020.100123.
Supplemental Information
References
- 1.Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., China Novel Coronavirus Investigating and Research Team A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolfel R., Corman V.M., Guggemos W., Seilmaier M., Zange S., Muller M.A., Niemeyer D., Jones T.C., Vollmar P., Rothe C. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020;581:465–469. doi: 10.1038/s41586-020-2196-x. [DOI] [PubMed] [Google Scholar]
- 5.Petherick A. Developing antibody tests for SARS-CoV-2. Lancet. 2020;395:1101–1102. doi: 10.1016/S0140-6736(20)30788-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lipsitch M., Swerdlow D.L., Finelli L. Defining the Epidemiology of Covid-19 - Studies Needed. N. Engl. J. Med. 2020;382:1194–1196. doi: 10.1056/NEJMp2002125. [DOI] [PubMed] [Google Scholar]
- 7.Stadlbauer D., Amanat F., Chromikova V., Jiang K., Strohmeier S., Arunkumar G.A., Tan J., Bhavsar D., Capuano C., Kirkpatrick E. SARS-CoV-2 Seroconversion in Humans: A Detailed Protocol for a Serological Assay, Antigen Production, and Test Setup. Curr. Protoc. Microbiol. 2020;57:e100. doi: 10.1002/cpmc.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qu J., Wu C., Li X., Zhang G., Jiang Z., Li X., Zhu Q., Liu L. Profile of IgG and IgM antibodies against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Clin. Infect. Dis. 2020 doi: 10.1093/cid/ciaa489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu W., Liu L., Kou G., Zheng Y., Ding Y., Ni W., Wang Q., Tan L., Wu W., Tang S. Evaluation of nucleocapsid and spike protein-based ELISAs for detecting antibodies against SARS-CoV-2. J. Clin. Microbiol. 2020 doi: 10.1128/JCM.00461-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson B.R. The dangers of false-positive and false-negative test results: false-positive results as a function of pretest probability. Clin. Lab. Med. 2008;28:305–319. doi: 10.1016/j.cll.2007.12.009. vii. [DOI] [PubMed] [Google Scholar]
- 11.Guan W.J., Ni Z.Y., Hu Y., Liang W.H., Ou C.Q., He J.X., Liu L., Shan H., Lei C.L., Hui D.S.C. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl.J. Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhatraju P.K., Ghassemieh B.J., Nichols M., Kim R., Jerome K.R., Nalla A.K., Greninger A.L., Pipavath S., Wurfel M.M., Evans L. Covid-19 in Critically Ill Patients in the Seattle Region - Case Series. N. Engl. J. Med. 2020;382:2012–2022. doi: 10.1056/NEJMoa2004500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu G.J., Kula T., Xu Q., Li M.Z., Vernon S.D., Ndung’u T., Ruxrungtham K., Sanchez J., Brander C., Chung R.T. Viral immunology. Comprehensive serological profiling of human populations using a synthetic human virome. Science. 2015;348:aaa0698. doi: 10.1126/science.aaa0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schubert R.D., Hawes I.A., Ramachandran P.S., Ramesh A., Crawford E.D., Pak J.E., Wu W., Cheung C.K., O’Donovan B.D., Tato C.M. Pan-viral serology implicates enteroviruses in acute flaccid myelitis. Nat. Med. 2019;25:1748–1752. doi: 10.1038/s41591-019-0613-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larman H.B., Zhao Z., Laserson U., Li M.Z., Ciccia A., Gakidis M.A., Church G.M., Kesari S., Leproust E.M., Solimini N.L., Elledge S.J. Autoantigen discovery with a synthetic human peptidome. Nat. Biotechnol. 2011;29:535–541. doi: 10.1038/nbt.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang C.K., Hou M.H., Chang C.F., Hsiao C.D., Huang T.H. The SARS coronavirus nucleocapsid protein--forms and functions. Antiviral Res. 2014;103:39–50. doi: 10.1016/j.antiviral.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan T., Mohan D., Laserson U., Ruczinski I., Baer A.N., Larman H.B. Improved Analysis of Phage ImmunoPrecipitation Sequencing (PhIP-Seq) Data Using a Z-score Algorithm. bioRxiv. 2018 doi: 10.1101/285916. [DOI] [Google Scholar]
- 18.Tsui P., Sweet R.W., Sathe G., Rosenberg M. An efficient phage plaque screen for the random mutational analysis of the interaction of HIV-1 gp120 with human CD4. J. Biol. Chem. 1992;267:9361–9367. [PubMed] [Google Scholar]
- 19.Szymczak L.C., Kuo H.Y., Mrksich M. Peptide Arrays: Development and Application. Anal. Chem. 2018;90:266–282. doi: 10.1021/acs.analchem.7b04380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dubois M.E., Hammarlund E., Slifka M.K. Optimization of peptide-based ELISA for serological diagnostics: a retrospective study of human monkeypox infection. Vector Borne Zoonotic Dis. 2012;12:400–409. doi: 10.1089/vbz.2011.0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer B., Drosten C., Müller M.A. Serological assays for emerging coronaviruses: challenges and pitfalls. Virus Res. 2014;194:175–183. doi: 10.1016/j.virusres.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan P.K., To W.K., Liu E.Y., Ng T.K., Tam J.S., Sung J.J., Lacroix J.M., Houde M. Evaluation of a peptide-based enzyme immunoassay for anti-SARS coronavirus IgG antibody. J. Med. Virol. 2004;74:517–520. doi: 10.1002/jmv.20207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woo P.C., Lau S.K., Wong B.H., Tsoi H.W., Fung A.M., Kao R.Y., Chan K.H., Peiris J.S., Yuen K.Y. Differential sensitivities of severe acute respiratory syndrome (SARS) coronavirus spike polypeptide enzyme-linked immunosorbent assay (ELISA) and SARS coronavirus nucleocapsid protein ELISA for serodiagnosis of SARS coronavirus pneumonia. J. Clin. Microbiol. 2005;43:3054–3058. doi: 10.1128/JCM.43.7.3054-3058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao J., Wang W., Wang W., Zhao Z., Zhang Y., Lv P., Ren F., Gao X.M. Comparison of immunoglobulin G responses to the spike and nucleocapsid proteins of severe acute respiratory syndrome (SARS) coronavirus in patients with SARS. Clin. Vaccine Immunol. 2007;14:839–846. doi: 10.1128/CVI.00432-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhong X., Yang H., Guo Z.F., Sin W.Y., Chen W., Xu J., Fu L., Wu J., Mak C.K., Cheng C.S. B-cell responses in patients who have recovered from severe acute respiratory syndrome target a dominant site in the S2 domain of the surface spike glycoprotein. J. Virol. 2005;79:3401–3408. doi: 10.1128/JVI.79.6.3401-3408.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okba N.M.A., Muller M.A., Li W., Wang C., GeurtsvanKessel C.H., Corman V.M., Lamers M.M., Sikkema R.S., de Bruin E., Chandler F.D. Severe Acute Respiratory Syndrome Coronavirus 2-Specific Antibody Responses in Coronavirus Disease 2019 Patients. Emerg. Infect. Dis. 2020;26:1478–1488. doi: 10.3201/eid2607.200841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haveri A., Smura T., Kuivanen S., Österlund P., Hepojoki J., Ikonen N., Pitkäpaasi M., Blomqvist S., Rönkkö E., Kantele A. Serological and molecular findings during SARS-CoV-2 infection: the first case study in Finland, January to February 2020. Euro Surveill. 2020;25:2000266. doi: 10.2807/1560-7917.ES.2020.25.11.2000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watanabe Y., Allen J.D., Wrapp D., McLellan J.S., Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science. 2020;369:330–333. doi: 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He Q., Chong K.H., Chng H.H., Leung B., Ling A.E., Wei T., Chan S.W., Ooi E.E., Kwang J. Development of a Western blot assay for detection of antibodies against coronavirus causing severe acute respiratory syndrome. Clin. Diagn. Lab. Immunol. 2004;11:417–422. doi: 10.1128/CDLI.11.2.417-422.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Haan C.A., Masters P.S., Shen X., Weiss S., Rottier P.J. The group-specific murine coronavirus genes are not essential, but their deletion, by reverse genetics, is attenuating in the natural host. Virology. 2002;296:177–189. doi: 10.1006/viro.2002.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McBride R., Fielding B.C. The role of severe acute respiratory syndrome (SARS)-coronavirus accessory proteins in virus pathogenesis. Viruses. 2012;4:2902–2923. doi: 10.3390/v4112902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiu M., Shi Y., Guo Z., Chen Z., He R., Chen R., Zhou D., Dai E., Wang X., Si B. Antibody responses to individual proteins of SARS coronavirus and their neutralization activities. Microbes Infect. 2005;7:882–889. doi: 10.1016/j.micinf.2005.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan Y.J., Goh P.Y., Fielding B.C., Shen S., Chou C.F., Fu J.L., Leong H.N., Leo Y.S., Ooi E.E., Ling A.E. Profiles of antibody responses against severe acute respiratory syndrome coronavirus recombinant proteins and their potential use as diagnostic markers. Clin. Diagn. Lab. Immunol. 2004;11:362–371. doi: 10.1128/CDLI.11.2.362-371.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poh C.M., Carissimo G., Wang B., Amrun S.N., Lee C.Y., Chee R.S., Fong S.W., Yeo N.K., Lee W.H., Torres-Ruesta A. Two linear epitopes on the SARS-CoV-2 spike protein that elicit neutralising antibodies in COVID-19 patients. Nat. Commun. 2020;11:2806. doi: 10.1038/s41467-020-16638-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu J., Larman H.B., Gao G., Somwar R., Zhang Z., Laserson U., Ciccia A., Pavlova N., Church G., Zhang W. Protein interaction discovery using parallel analysis of translated ORFs (PLATO) Nat. Biotechnol. 2013;31:331–334. doi: 10.1038/nbt.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li W., Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 38.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G. Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Magoč T., Salzberg S.L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robbiani D.F., Gaebler C., Muecksch F., Lorenzi J.C.C., Wang Z., Cho A., Agudelo M., Barnes C.O., Gazumyan A., Finkin S. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature. 2020;584:437–442. doi: 10.1038/s41586-020-2456-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J., Kobert K., Flouri T., Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2014;30:614–620. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mandel-Brehm C., Dubey D., Kryzer T.J., O’Donovan B.D., Tran B., Vazquez S.E., Sample H.A., Zorn K.C., Khan L.M., Bledsoe I.O. Kelch-like Protein 11 Antibodies in Seminoma-Associated Paraneoplastic Encephalitis. N. Engl. J. Med. 2019;381:47–54. doi: 10.1056/NEJMoa1816721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Donovan B., Mandel-Brehm C., Vazquez S.E., Liu J., Parent A.V., Anderson M.S., Kassimatis T., Zekeridou A., Hauser S.L., Pittock S.J. High-resolution epitope mapping of anti-Hu and anti-Yo autoimmunity by programmable phage display. Brain Commun. 2020;2:a059. doi: 10.1093/braincomms/fcaa059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
VirScan, ReScan and clinical data analyzed in the manuscript have been made available in the supplementary material. Dotblotr package and source code are available on the Chan Zuckerberg Biohub GitHub repository.
Raw data and analysis code for this study are publicly available on the Wilson Lab GitHub Repository (https://github.com/UCSF-Wilson-Lab/sars-cov-2_ReScan_VirScan_complete_analysis).