

Abstract
Scaffolds made from biocompatible polymers provide physical cues to direct the extension of neurites and to encourage repair of damaged nerves. The inclusion of neurotrophic payloads in these scaffolds can substantially enhance regrowth and repair processes. However, many promising neurotrophic candidates are excluded from this approach due to incompatibilities with the polymer or with the polymer processing conditions. This work provides one solution to this problem by incorporating porous silicon nanoparticles (pSiNPs) that are pre-loaded with the therapeutic into a polymer scaffold during fabrication. The nanoparticle-drug-polymer hybrids are prepared in the form of oriented poly(lactic-co-glycolic acid) nanofiber scaffolds. We test three different therapeutic payloads: bpV(HOpic), a small molecule inhibitor of phosphatase and tensin homolog (PTEN); an RNA aptamer specific to tropomyosin-related kinase receptor type B (TrkB); and the protein nerve growth factor (NGF). Each therapeutic is loaded using a loading chemistry that is optimized to slow the rate of release of these water-soluble payloads. The drug-loaded pSiNP-nanofiber hybrids release approximately half of their TrkB aptamer, bpV(HOpic), or NGF payload in 2, 10, and >40 days, respectively. The nanofiber hybrids increase neurite extension relative to drug-free control nanofibers in a dorsal root ganglion explant assay.
Keywords: neuron guidance, tissue engineering, controlled release, PTEN inhibitor, RNAaptamer, tropomyosin-related kinase receptor type B, Nerve Growth Factor
Graphical Abstract
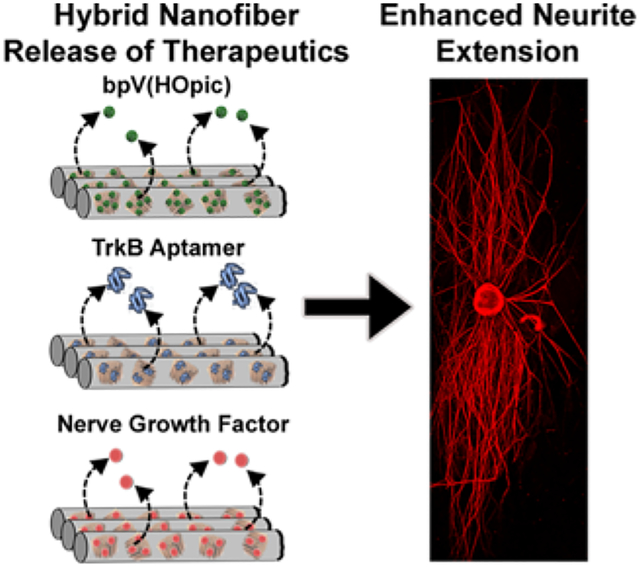
Porous silicon nanoparticles are loaded with bpV(HOpic), a TrkB RNA aptamer, or nerve growth factor using three distinct loading chemistries. They are incorporated into aligned poly(lactic-co-glycolic acid) nanofibers using an airbrush, and the nanofiber hybrids release their payloads over varying timescales. The three released payloads maintain their bioactivity as shown by enhanced neurite extension of dorsal root ganglion explants cultured on the hybrid nanofiber scaffolds.
Polymer nanofibers are used in numerous tissue engineering, regenerative medicine, and in vitro applications due to the ease of their fabrication and their obvious morphological and functional topographical influences on stem cell differentiation,[1] neuron guidance,[2] cardiomyocyte contractility,[3] oligodendrocyte myelination,[4] and many other cellular processes. However, polymer nanofibers have yet to demonstrate utility in the clinic[5–6], and the limitations of polymer nanofiber interventions relying solely on topographical structures for their therapeutic impact have driven a push to imbue nanofibers with additional beneficial properties.[7–9] Of these, tunable drug release is emerging as a necessary component for state-of-the-art tissue engineering scaffolds. While many different fabrication techniques have been devised, these methods still do not allow for the creation of nanofibers that can be readily modified to release distinct types of drugs at tunable rates.
There are now many approaches to incorporate active pharmaceutical ingredients (APIs) into polymer nanofibers. Because organic solvents are used in the preparation of most polymer scaffolds of interest, hydrophobic drugs can be dispersed throughout polymer nanofibers by simply adding them to the organic solvent during fabrication.[10] For hydrophilic drugs or sensitive biologics, which typically are not compatible with organic solvents, other approaches are required. One relatively simple method is to coat or conjugate the drug to the surface of the polymer nanofibers, although this generally leads to a rapid (burst) release of drug from the scaffold over the course of a few hours to a few days.[11] Emulsion electrospinning has been used to disperse biologics throughout the polymer matrix, which results in a relatively quick release profile of the incorporated payload (days).[12–14] Co-axial/tri-axial electrospinning produces core-shell nanofibers and affords more control over the fabrication process and drug release profiles.[15–20] These techniques have been used to generate materials that release sensitive biologics over varying time scales (hours to weeks). However, throughput is low because fabrication parameters must be optimized for each new drug to ensure consistency of the fibers produced.
Hybrid polymer scaffolds that contain drug-loaded nanoparticles dispersed throughout the polymer nanofiber matrix have demonstrated the ability to hold and slowly release hydrophilic drugs in their active forms. For example, chitosan nanoparticles, loaded with nucleic acid therapeutics and incorporated into an electrospinning solution, have been shown to form nanofiber composites that release active RNA over multiple weeks.[21] Similarly, silica nanoparticles loaded with hydrophilic small molecules and incorporated into polymer nanofibers displayed release from hours to multiple days.[22–23] Most relevant to the present study, porous silicon particles have been incorporated into polymer fibers for tissue engineering applications,[24–26] including hybrid nanofibers that incorporated lysozyme-loaded porous silicon nanoparticles (pSiNPs) into polycaprolactone (PCL) nanofibers.[27] Fabricated using a spray nebulization method rather than electrospinning, these latter pSiNP-PCL nanofibers exhibited uniaxial orientation and they released active protein (lysozyme) over 8 weeks. Attractive features of pSiNPs in this context include their low toxicity, degradability,[28–29] intrinsic photoluminescent properties,[30–31] high drug loading capacity,[32–34] and a set of chemistries that tolerate various classes of drugs and biologics.[35–39]
While the prior work demonstrated the potential for tissue engineering applications using a surrogate protein, the ability of hybrid pSiNP-polymer systems to deliver an API that can impact a real biological response (e.g., tissue growth) has not been established. Here we report the effective delivery of three different neurotrophic agents: the small molecule bpV(HOpic) which is an inhibitor of phosphatase and tensin homolog (PTEN); an RNA aptamer that binds to tropomyosin-related kinase receptor type B (TrkB); and the protein nerve growth factor (NGF). We quantify the release rates of each of these agents from polymer/pSiNP nanofiber hybrids, using poly(lactic-co-glycolic acid) (PLGA) as the polymer scaffold. We demonstrate the bioactivity of the released payloads using a dorsal root ganglion (DRG) neurite extension assay. We find that all three API formulations demonstrate guidance of DRG explant neurite outgrowth and enhancement of DRG neurite extension relative to control polymer/pSiNP nanofibers that contain no API. This work represents the first time pSiNP/PLGA nanofiber hybrids have been used to deliver neurotrophic factors to enhance neurite extension—a critically important biological process needed to effect repair of traumatically injured nerves.
The three different payloads were chosen for loading into pSiNPs based on their known ability to enhance neurite extension when administered as free molecules. The small molecule bpV(HOpic) is an inhibitor of PTEN, and PTEN inhibition has previously been shown to increase neurite outgrowth.[40–41] RNA aptamers are single stranded oligonucleotides that fold into defined secondary structures and bind to their targets, such as proteins and cell surface receptors.[42] Activation of the transmembrane receptor, TrkB, leads to the expression of the extracellular signal-regulated kinases (ERK) and mitogen-activated protein kinase kinases (MEK) signaling pathway, which is implicated in sustaining neurite outgrowth.[43] RNA aptamers have been designed that function as ligands for the TrkB receptor, and these aptamers lead to increased neuronal survival in culture and improved functional outcomes in seizure animal models.[44] Nerve growth factor (NGF) is part of the neurotrophin family of molecules, and it binds with high affinity to the transmembrane receptor TrkA, signaling through a similar MEK-ERK pathway as TrkB activation.[45] Many studies have shown the ability of free NGF to increase neurite extension.[46–48] Because of differences in size, shape, and electrostatic charge of the three APIs, three distinct chemistries were used to load them into pSiNPs (Figure 1).
Figure 1.
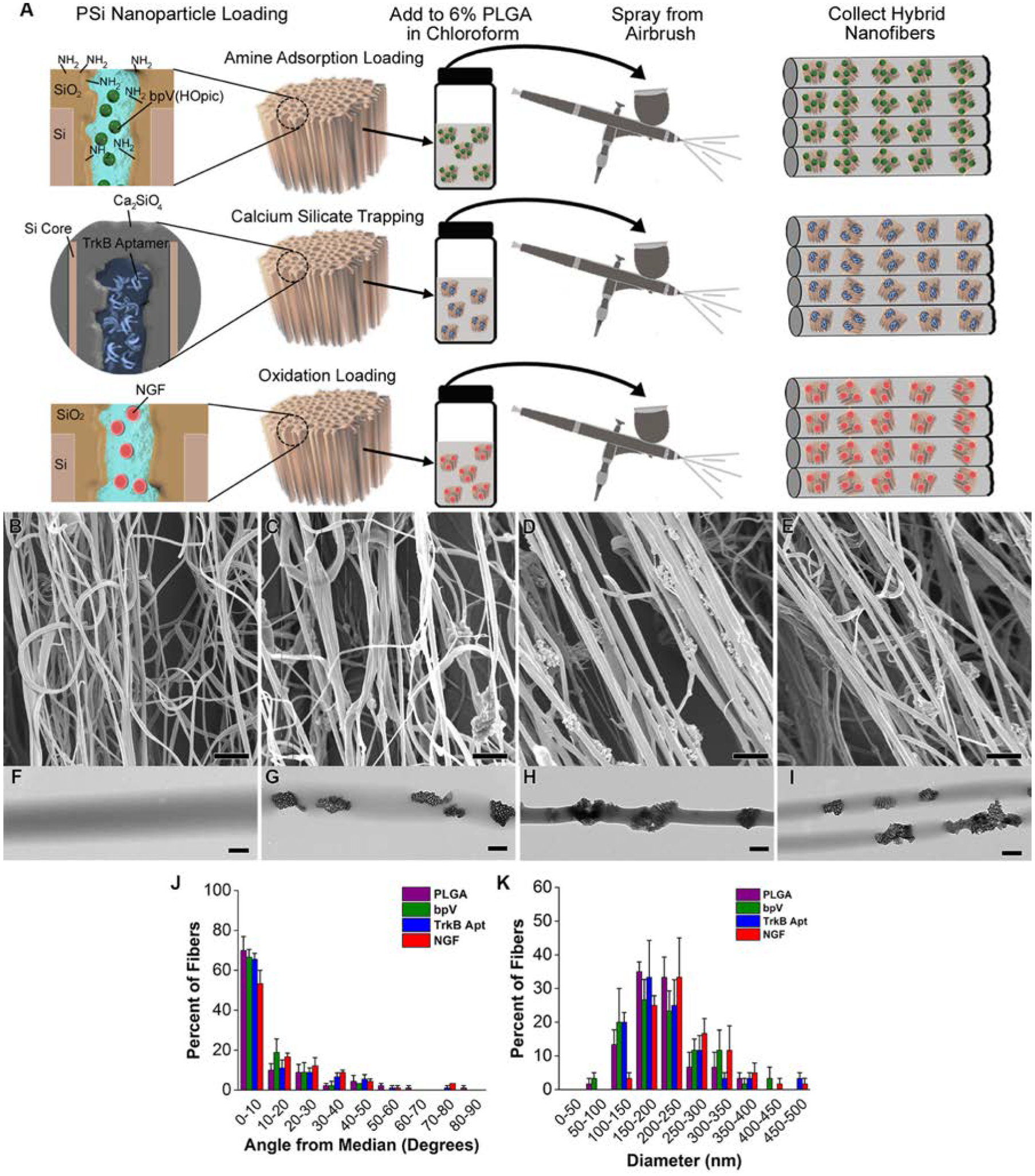
Preparation of drug-loaded pSiNP/PLGA nanofiber scaffolds. (A) Schematic depicting chemistries used to load each drug: “Amine Adsorption Loading” (top) involves pSiNPs whose inner pore walls have been grafted to a primary amine that binds the small molecule drug bpV(HOpic) via electrostatic interactions; “Calcium Silicate Trapping” (middle) involves condensation of the highly negatively charged TrkB aptamer in a calcium silicate matrix contained within the pSiNPs; and “Oxidation Loading” (bottom) involves mild oxidation of the silicon skeleton on the pSiNPs that physically traps the NGF protein within the nanostructure. The relevant particle type is then loaded into a chloroform solution of PLGA and the nanofiber scaffolds are generated by spray nebulization through an airbrush. (B-E) SEM images of (B) PLGA nanofiber controls, (C) bpV(HOpic)-pSiNP/PLGA nanofiber hybrids, (D) TrkB aptamer-pSiNP/PLGA nanofiber hybrids, and (E) NGF-pSiNP/PLGA nanofiber hybrids. (F-I) TEM images of (F) PLGA nanofiber controls, (G) bpV(HOpic)-pSiNP/PLGA nanofiber hybrids, (H) TrkB aptamer-pSiNP/PLGA nanofiber hybrids, and (I) NGF-pSiNP/PLGA nanofiber hybrids. (J) Degree of alignment of nanofibers calculated by measuring the angle of individual fibers relative to the median angle of alignment. All fiber types show similar, uniaxial alignment (n=3). (K) Diameter of nanofibers calculated by measuring the diameter of 30 fibers in each image. Similar diameter ranges are seen between all groups (n=3).
The pSiNP carriers were prepared in a similar fashion for all APIs, following the previously reported “perforation etching” procedure.[49] Briefly, the process involved electrochemical anodization of single crystal silicon to generate a stratified porous silicon layer, removal of the porous layer and creation of the pSiNPs via ultrasonic fracture. As-prepared pSiNPs (prior to drug loading) displayed mean diameters of 187 ± 3 nm, zeta potential of −45 ±1 mV, nominal porosity of 47 ±3%, and pore diameters ranging from 10–20 nm (Figure S1 and Table S1, Supplementary Information). Each API was then loaded into the pSiNPs using chemistries specifically suited to its particular molecular class.
The small molecule drug bpV(HOpic) was loaded via an electrostatic adsorption process. Due to the net negative charge of bpV(HOpic), the inner pore walls of the pSiNPs were first chemically modified to display a positive surface charge. The chemistry involved a ring-opening click-reaction of the cyclic diazasilane 2,2-dimethoxy-1,6-diaza-2-silacyclooctane, which generates a primary amine at the surface of the pSiNPs.[50] Zeta potential measurement (35 ± 5 mV) confirmed the positive surface charge. These amine-functionalized pSiNPs were then loaded with bpV(HOpic) from deionized (DI) water to a mass loading of 16 ± 1%, which resulted in a reduction of the zeta potential to 4 ± 2 mV. The bpV(HOpic)-loaded pSiNPs displayed an average size (by dynamic light scattering, DLS) of 262 ± 1nm.
The TrkB aptamer was loaded into as-etched pSiNPs via a calcium silicate condensation chemistry previously described.[51] Because the process employs high concentrations of Ca (II) ions, this chemistry is particularly well suited for stabilization and trapping of negatively charged, highly water-soluble nucleic acids in porous silicon nanostructures; in the present case the TrkB aptamer was loaded to a mass percentage of 14 ± 0.3%. The loaded particles displayed an average particle size of 257 ± 2 nm and a zeta potential of 3.7 ± 0.4mV.
The NGF protein was loaded using an oxidative trapping technique, where the protein becomes trapped in the pSiNP pores as the pore walls are oxidized in an aqueous buffer.[27, 50] Overnight loading of NGF into pSiNPs from phosphate buffered saline (PBS) resulted in a mass loading of 0.21 ± 0.07%, average particle size of 206 ± 2 nm, and zeta potential of −24 ±4 mV. The mass loading of NGF was substantially lower than what was obtained for the other two techniques due to the low concentration of NGF used in the loading solution. A higher mass loading was not necessary because of the high potency of this molecule. It was estimated that this loading level would result in an in vitro concentration of NGF in excess of the ED50 concentration of 0.3 ng mL−1 (0.01nM) required to induce neurite outgrowth in cultured sensory neurons.[52]
Each of the API-loaded-pSiNPs were then incorporated into uniaxial PLGA nanofibers using an airbrush nebulization technique, following a previously reported method.[27] Scanning electron microscope (SEM) images revealed uniaxial alignment of the collected nanofibers (Figure 1B–E), with average angles from the median angle of alignment of 12.3 ±17.6°, 9.6 ±11.3°, 12.6 ±15.6°, and 15.3 ±16.7° for PLGA, bpV(HOpic)-PLGA, TrkB aptamer-PLGA, and NGF-PLGA nanofibers, respectively (Figure 1J). For all the nanofiber hybrids, more than 70% of the fibers were within 20° of the median angle of alignment, and more than 80% of the fibers were within 30° of the median angle of alignment. SEM measurements (Figure 1F–I) indicated average nanofiber diameters of 209 ± 60 nm, 211 ±78 nm, 213 ±78 nm, and 250 ±79 nm for PLGA, bpV(HOpic)-PLGA, TrkB aptamer-PLGA, and NGF-PLGA nanofibers, respectively (Figure 1K). The diameters of the nanofibers were similar to the diameter of the pSiNPs, and some isolated nanoparticles were observed outside the nanofiber structures in both the SEM (Figure 1C–E) and transmission electron microscope (TEM) (Figure 1G–I) micrographs, although the majority of the nanoparticles appeared to be encapsulated by polymer. In lower magnification TEM images, pSiNPs can be found both dispersed as isolated particles or consolidated into clusters in different regions of the nanofibers (Figure S2, Supplemental Information). Water contact angle values (137 ± 7°, 136 ± 4°, and 133 ± 4° for bpV(HOpic)-PLGA, TrkB aptamer-PLGA, and NGF-PLGA nanofibers, respectively, (Figure S3, Supplemental Information) were similar to the value measured on pure PLGA nanofibers (134 ± 1.8°), indicating that the nanoparticles exerted little impact on the water contact angle of the nanofibers.
The pSiNPs used in these studies displayed intrinsic photoluminescence (PL) derived from quantum confinement in the silicon nanocrystallites comprising the silicon skeleton.[30] This property has interesting applications in biosensing,[53] self-reporting drug release,[54] two-photon imaging,[55] and time-gated in vivo imaging,[56–57] and in this work it is used to definitively identify the pSiNPs in the polymer fibers. Previously, we showed that photoluminescent pSiNPs incorporated into PCL nanofibers displayed broad PL emission (λem = 600–1000nm), and long emission lifetimes. This latter property allowed separation of the luminescent pSiNPs from polymer autofluorescence and light scattering phenomena using time-gated imaging.[27] All three of the loading chemistries used here generate photoluminescent pSiNPs with emission quantum yields ranging from 8–23%.[27, 50–51, 58]
Using UV excitation (λex = 365nm), bpV(HOpic)-PLGA, TrkB aptamer-PLGA, and NGF-PLGA nanofibers all exhibited the broad photoluminescence emission spectra characteristic of pSiNPs (Figure S4A–E, Supplemental Information). In order to benchmark the ability of time-gated imaging to discriminate these nanoparticles from autofluorescent tissues, images were collected from the pSiNP/PLGA materials and from the excised brain of a mouse. When a 5 μs time gate was used to collect the images, the high level of autofluorescence characteristic of brain tissue was completely removed, while the pSiNP/polymer scaffolds were readily observed (Figure S4F,G, Table S1, Supplemental Information). Measurements of pristine PLGA nanofiber controls (containing no SiNPs) also showed no image contrast in the time-gated images, confirming the presence of pSiNPs in the hybrid scaffolds.
Because of the wide range of indications for which drug-delivering polymer scaffolds might play a role, there is a wide range of temporal release profiles desired. Furthermore, an optimal scaffold chemistry needs to balance potency, duration of action, and side effects/toxicity for a given API. In the present case, we employed three distinct loading chemistries that were previously optimized for specific classes of molecules. For the small molecule drug, bpV(HOpic), a pSi adsorption loading technique was used because this method has previously shown a burst release profile for hydrophilic small molecules over short (minutes-hours) time scales.[59] Calcium-silicate trapping was used for the TrkB aptamer, as this pSi loading chemistry has a release profile in the range of a few hours to 1 day.[60] The oxidation trapping method was used for NGF because it has previously been shown to release proteins over multiple days with minimal loss in activity.[27, 50]
Payload release from each of the pSiNP/PLGA nanofiber systems was determined as a function of time incubated in a PBS buffer at 37 °C. The small molecule API bpV(HOpic) released from the scaffold over a 10 day period (Figure 2), with a pronounced burst release (584 ± 55 pmoles mg−1 nanofibers) during the first 2 days (Fig. S5A, Supplemental Information). The release then slowed to a steady, relatively linear profile for the remaining 8 days, and after 10 days of incubation a total of 744 ± 16 pmoles of API per mg of nanofibers was released. Of the incorporated bpV(HOpic), 38 ± 4% was released over the first 2 days, with 49 ± 6% released over the 10 day study period (Figure 2B). The nanofibers containing TrkB aptamer-loaded pSiNPs released 77 ±5% of the aptamer payload over a 20 day period (Fig. 2C). A relatively large quantity was released in the first 2 days (326 ± 5 pmoles mg−1 nanofibers), which slowed between days 2–4 (67 ± 5 pmoles mg−1 nanofibers), and then a slower, linear release was observed for the remaining 20 days (average of 9 ± 3 pmoles mg−1 nanofibers 48h−1), for a total of 464 ± 17 pmoles mg−1 nanofibers released over 20 days (Fig. 2C and Fig. S5B, Supplemental Information). Nanofibers containing NGF-loaded pSiNPs released the protein growth factor payload over a 6 week period (Fig. 2D and Fig. S5C, Supplemental Information). The temporal release profile for this chemistry showed a delayed burst: 0.30 ± 0.05 ng NGF mg−1 nanofibers was released in the first 2 days, 3 ± 2 ng mg−1 nanofibers released between days 2–4, and 10 ± 5 ng mg−1 nanofibers released between days 4–6. Release slowed again at later times, with an average of 4 ± 1 ng mg−1 nanofibers released between days 6 to 12 and 1.5 ± 0.9 ng mg−1 nanofibers 48h−1 for the remaining time of the release study (cumulative release of 48 ± 16 ng mg−1 nanofibers, or 32 ±11% of the incorporated NGF was released over the 6 week study period).
Figure 2.
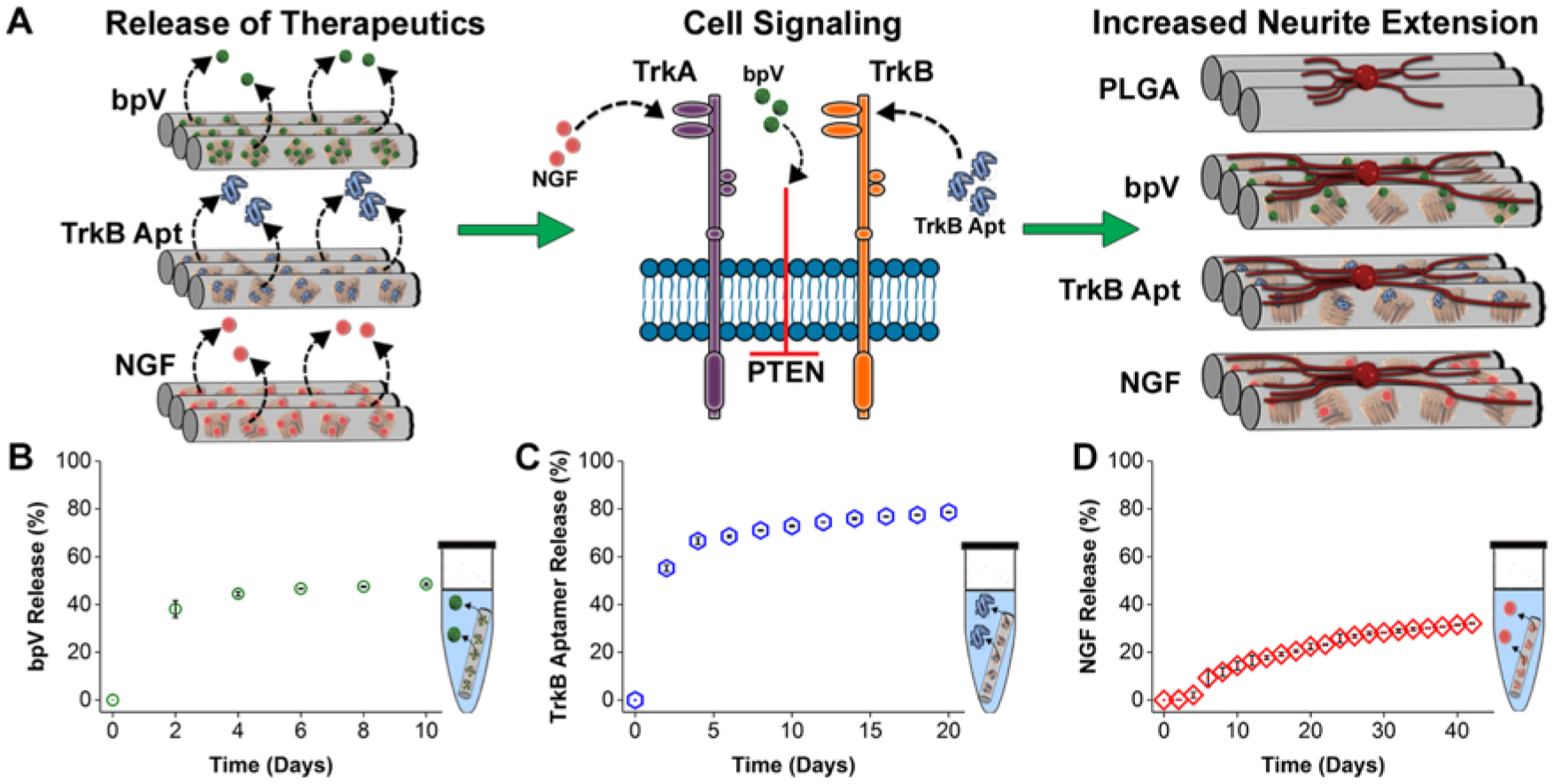
Mode of action of the three neurotrophic agents studied in this work and their temporal release characteristics from hybrid pSiNP/PLGA nanofibers. (A) Schematic depicting the different modes of action of each of the three payloads released from the hybrid nanofiber scaffolds. Once released from the relevant scaffold (left), bpV(HOpic) blocks PTEN intracellularly, TrkB aptamer acts as a ligand for TrkB, and NGF binds to its receptor (TrkA) (middle). Growth of neurites from DRG explants placed on these nanofibers is hypothesized to be enhanced compared to a PLGA nanofiber control (right). (B-D) Percent of API payload released from hybrid PLGA nanofibers as a function of time in PBS buffer at 37 °C. (A) 49 ± 5.6% of incorporated bpV(HOpic) was released over the 10-day release period. (B) 77 ±4.9% of incorporated TrkB aptamer was released over the 20 day release period. (C) 32 ±11% of incorporated NGF was released over the 42 day release period.
Previously, it was demonstrated that the 75:25 (L:G) composition of PLGA nanofibers used in this work show very small changes in mass over an 8-week time period when in PBS solutions at 37℃.[61–62] We used SEM and AFM measurements to probe how inclusion of pSiNPs alters the nanofiber surface after 7 days in PBS at 37℃. Control PLGA nanofibers showed minimal surface roughness at both 0 and 7 days in PBS at 37℃ (Figure S6A, B,E,F Supplemental Information). By contrast, the pSiNP/PLGA hybrid nanofibers displayed obvious evidence of degradation (Figure S6C,D, Supplemental Information). The porous nanoparticle structure is evident in the SEM images prior to degradation (Figure S6C, Supplemental Information). After 7 days in PBS the pSiNPs have visibly degraded (Figure S6D, Supplemental Information) and the polymer fibers displayed a marked increase in surface roughness, with surface indentations (measured by AFM) of 10–40 nm in depth (Figure S6G, H, Supplemental Information). Prior to degradation, PLGA and pSiNP/PLGA hybrid nanofibers demonstrated similar Young’s moduli of 122 ± 33 kPa and 90 ± 4 kPa, respectively (Figure S6I, Supplemental Information). These values lie in the range of PLGA moduli previously reported using similar AFM nanoindentation methods.[63] Interestingly, while the PLGA nanofibers actually displayed a lower Young’s modulus after 7 days (32 ± 8 kPa), pSiNP/PLGA hybrid nanofibers showed an increase in Young’s modulus (to 198 ± 41 kPa). The degradation of the pSiNPs at the surface of the nanofibers observed following the 1 week incubation period is consistent with the rapid release of bpV(HOpic) and the TrkB aptamer observed in the first week of PBS exposure.
We next evaluated the impact of the released therapeutics on neurite extension using whole DRG explants from P2 C57BL/6J mouse pups. DRG seeded onto the pSiNP/PLGA nanofiber hybrids were immersed in neurobasal media supplemented with B27 and N2, but no additional neurotrophic factors, so that the media itself had a limited capacity to encourage the outgrowth of neurites from the DRG. In addition, the PLGA scaffolds were coated with laminin (5μg mL−1) to improve the adhesion of DRG explants to the hydrophobic scaffolds. Following 7 days in culture, the DRG were fixed and stained with NF200 (neurites) and Hoechst (nuclei) for analysis.
The alignment of the neurites extending on the hybrid nanofibers was analyzed and the results are presented in Figure 3. Degree of alignment is an important parameter because neurite growth alignment has been shown to improve nerve regeneration following injury.[64–66] Neurites growing from the DRG were uniaxially oriented due to the uniaxial alignment of the hybrid nanofiber scaffolds (Fig. 3A–E), with 54 ± 8%, 64 ± 13%, 69 ± 3%, and 59 ± 2% of the extending neurites aligned within 15° of the median angle of neurite alignment for PLGA, bpV(HOpic)-PLGA, TrkB aptamer-PLGA, and NGF-PLGA nanofibers, respectively (Fig. 3F). When DRG explants were cultured on flat PLGA films, a significantly lower percentage (16 ± 6.5%) of the extending neurites were within 15° of the median angle of neurite alignment compared to all the nanofiber groups. There were also significantly fewer neurites extending between 75–90° from the median angle of alignment (Fig. 3F). These results demonstrate the ability of the topography created by the hybrid nanofibers to uniaxially orient the outgrowth of extending neurites from cultured DRG explants.
Figure 3.
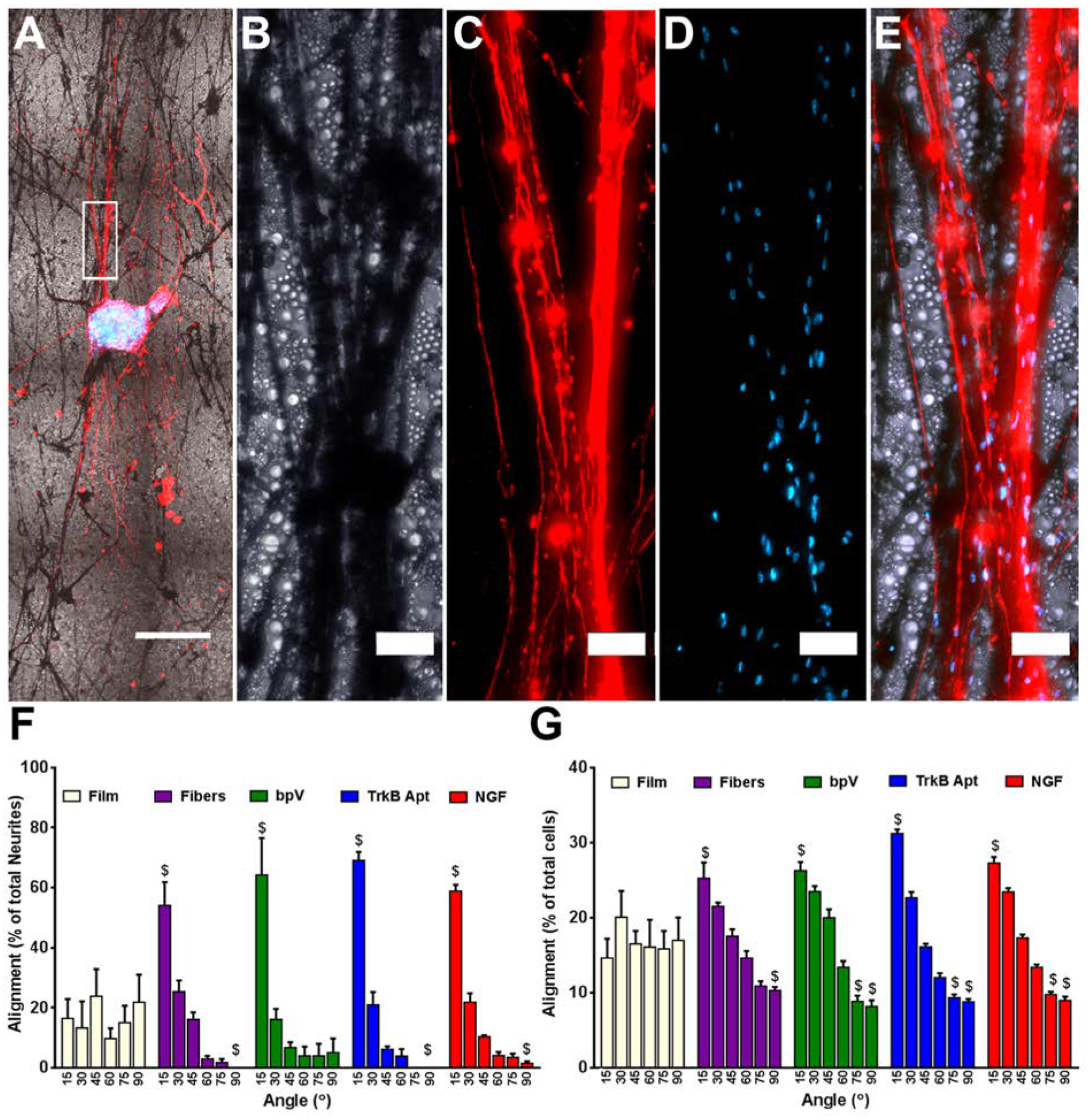
Alignment of extending neurites and migrating cells cultured on the different nanofiber scaffolds. (A) Low magnification (5x) image of NF200 stained DRG explant growing along pure PLGA nanofibers. (B) High magnification bright field image of nanofibers, (C) neurites, (D) Hoechst stained nuclei, and (E) the merged image of all three. (F) Alignment of extending neurites from DRG explants. When compared to DRG explants grown on flat PLGA films, significantly more neurites extended within 15° and significantly fewer grew between 75–90° of the median angle of neurite alignment on all four fiber groups. No differences were seen between any of the fiber groups. (G) Angle of the long axis of cells migrating away from the DRG explants. When compared to DRG explants grown on films, significantly more long axes of cells were within 15° and significantly fewer grew between 75–90° of the median angle of cellular alignment on all four fiber groups. For bpV(HOpic), TrkB aptamer, and NGF pSiNP/PLGA hybrid nanofibers, significantly fewer cellular long axes were with 60–75° of the median angle of cellular alignment compared to explants on PLGA films. Scale Bar=500 μm (A), 50 μm (B-E); n=5; $= p < 0.05 compared to PLGA films.
DRG explants are not composed entirely of DRG neurons, and the other cells present in the explants are known to be mainly Schwann cells and fibroblasts.[67] We also examined the ability of the nanofibers to alter the alignment of the longest axis of cells that migrated out of the DRG explant using a previously published image analysis technique.[68] We found that the nanofibers induced orientation of the longest axis of migrating cell nuclei, with 25 ± 2%, 26 ± 2%, 31 ± 0.6%, and 27 ± 0.8% of the longest axes within 15° of the median angle of nuclei alignment for PLGA, bpV(HOpic)-PLGA, TrkB aptamer-PLGA, and NGF-PLGA nanofibers, respectively (Fig. 3G). When DRG explants were cultured on PLGA films, a significantly lower percentage (14 ± 3%) of the longest axes on the migrating cell nuclei were within 15° of the median angle of nuclei alignment. Again, there were fewer migrating cell nuclei with their longest axes between 75–90° of the median angle of alignment compared to DRG explants on films (Fig. 3G). Taken together, these results demonstrate that the orientation of both neurite outgrowth and migrating cell nuclei are uni-axially aligned due to the presentation of the nanofiber topography for all the different hybrid nanofiber scaffolds tested.
We next evaluated the ability of the neurotrophic APIs to enhance neurite extension (Figure 4). It should first be pointed out that nanofibers can induce neurite extension even in the absence of a neurotrophic factor. This has been attributed to the topological features of nanofibers, which offer adhesion points and direct the process of neurite elongation along the nanofiber scaffolds.[69] Such elongation was also observed in the present system, and DRG explants cultured on pure PLGA nanofiber scaffolds (Fig. 4B) showed significantly longer neurite extension compared to those cultured on pure PLGA films (Fig. 4A). Both the range of neurite extension (Fig. 4F), and the average of the 10 longest neurites on each scaffold (Fig. 4H) were larger, consistent with previous reports.[69]
Figure 4.
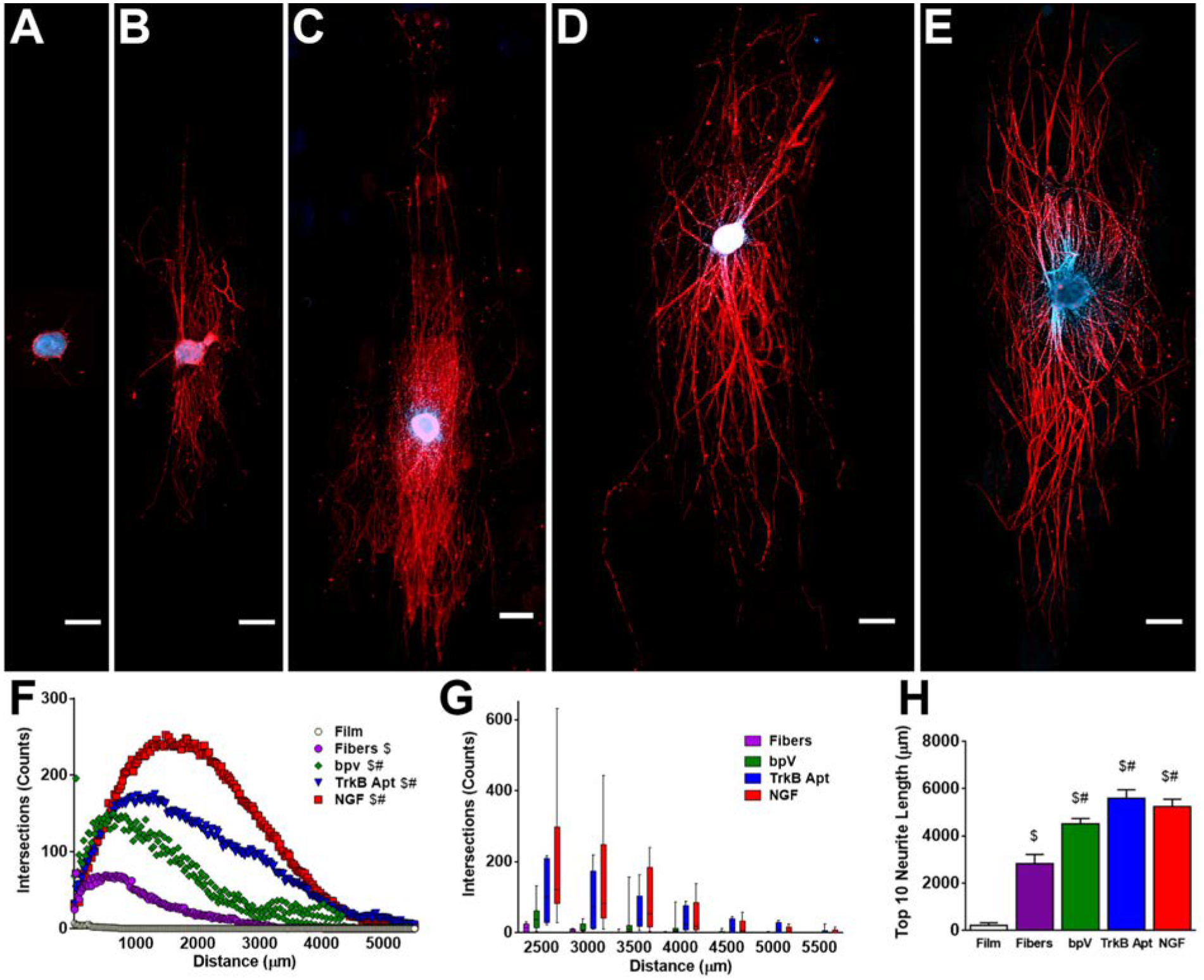
Enhancement of neurite extension induced by release of therapeutics from hybrid PLGA nanofibers. (A-E) DRG explants cultured on (A) PLGA film control, (B) PLGA fiber control, (C) bpV(HOpic)-pSiNP/PLGA nanofiber hybrids, (D) TrkB aptamer-pSiNP/PLGA nanofibers, and (E) NGF-PSiNP/PLGA nanofiber hybrids. (F) Range of neurite extension on each scaffold type. Pure PLGA fibers caused more robust growth of neurite extension from DRG explants compared to PLGA films. The addition of bpV(HOpic)-pSiNPs, TrkB aptamer-pSiNPS, or NGF-pSiNPs further increased the number of neurite intersections over the range of neurite extension. (G) Box and whisker plot of select ranges beyond 2500 μm of neurite extension, showing that few neurite intersections were counted from any PLGA nanofiber scaffold controls at these distances from the DRG explants. (H) Average of the ten longest extending neurites on each scaffold. PLGA fibers alone induced longer neurite extension from DRG explants compared to PLGA films. The addition of bpV(HOpic)-pSiNPs, TrkB aptamer-pSiNPS, or NGF-pSiNPs further increased the length of neurite intersection. Scale Bar = 500 μm; n = 5; $ = p < 0.05 compared to PLGA film; # = p < 0.05 compared to PLGA fibers.
In order to test if the incorporation of a neurotrophic payload in the nanofibers would further enhance neurite extension, the nanofibers and their nanoparticle delivery vehicles were engineered such that each API would reach a steady state concentration in vitro that would be effective in enhancing neuronal growth. The inputs to these designs were: culture conditions (2 mg of nanofiber scaffolds, 0.5 mL neurobasal media); the measured release rate of the API (Figure S5, Supporting Information); and the published minimum concentration needed to elicit a therapeutic impact on DRG extension,[40, 44, 52] which allowed us to calculate estimates of the concentration of API in the DRG cultures for the duration of the culture period (Figure S7, Supporting Information). Importantly, because these concentrations were an estimate using the entire volume of the culture media, DRG explants were potentially exposed to higher local concentrations of the therapeutics due to their proximity to the scaffolds.
A series of control experiments designed to eliminate the possibility that surface chemistry, surface charge, or other factors played a role in enhancement of neurite extension were then performed. We tested PLGA nanofibers containing empty pSiNPs grafted with the same amine surface chemistry used to load bpV(HOpic) but with no small molecule payload (Figure S8, Supporting Information), PLGA nanofibers containing pSiNPs loaded with a control DNA sequence using the same calcium silicate chemistry used to load the aptamer (Figure S9, Supporting Information), and PLGA nanofibers containing pSiNPs that had been loaded with bovine serum albumin (BSA) using the same oxidative trapping procedure used to load the NGF protein (Figure S10, Supporting Information). All of these controls showed no significant difference in the average of the 10 longest neurites or in overall DRG extension as measured by the number of neurites that intersected with radiating outlines of the DRG body at 50 μm intervals from each scaffold. These results showed that the pSiNPs and all of the loading chemistries used in the formulations did not alter neurite extension from DRG explants on nanofiber scaffolds.
The presence of a neurotrophic payload in the nanofiber scaffold substantially enhanced neurite extension relative to pure PLGA nanofibers, for all three of the APIs studied. Neurites extending from DRG cultured on bpV(HOpic)-loaded pSiNP/nanofiber scaffolds (Fig. 4C), TrkB aptamer-loaded pSiNP/nanofiber scaffolds (Fig. 4D), and NGF-loaded pSiNP/nanofiber scaffolds (Fig. 4E) all showed significantly more NF200 stained intersections across the range of distances when compared to pure PLGA nanofiber controls (Fig. 4B,F) and their respective nanoparticle scaffold controls (Supplemental Figures S8, S9, S10, Supplemental Information). Using a box and whisker plot, distances over 2500 μm were further depicted in 500 μm intervals to demonstrate NF200 counts far from the body of the DRG explant. Few neurite intersections from DRG explants on PLGA nanofibers were seen beyond 3000 μm, whereas neurites extending on all 3 API-releasing nanofibers had intersections out to 4500 μm (Fig. 4G). TrkB aptamer and NGF releasing scaffolds showed neurite intersections even out to 5500 μm.
In order to analyze changes in the extension of the longest neurites, the average length of the 10 longest neurites in each scaffold was determined. The longest neurites extending on bpV(HOpic)-loaded pSiNP/nanofibers averaged 4511 ± 241 μm, on TrkB aptamer-loaded pSiNP/nanofibers averaged 5592 ± 360 μm, and on NGF-loaded pSiNP/nanofibers averaged 5248 ± 323 μm, all of which were significantly longer than the longest neurites extending on control PLGA nanofibers (2835 ± 389 μm) (Figure 4H). Neurites from DRG explants extending on NH2 pSiNP control nanofibers (2653 ± 605 μm) (Figure S8, Supplemental Information), DNA control pSiNP nanofibers (3732 ± 394 μm) (Figure S9, Supplemental Information), and BSA pSiNP control nanofibers (3262 ± 511 μm) (Figure S10, Supplemental Information), were all significantly shorter than their corresponding API-releasing nanofiber scaffolds, and they showed no difference compared to neurites extending on PLGA nanofiber control scaffolds. These data demonstrate that all three API-releasing nanofibers promote more robust neurite growth as determined by an increase in the number of neurite intersections (Figure 4F), and the length of neurite extension (Figure 4G,H). Importantly, these results verify the retention of activity of all three payloads, showing that each of these drug-loading chemistries is suitable for payload incorporation into polymer nanofibers.
Cellular migration out of the DRG explants was quantified in order to assess how the various hybrid scaffolds changed the growth of Schwann cells and fibroblasts (Figure 5). Cells migrating from DRG cultured on bpV(HOpic)-loaded pSiNP/nanofibers (Fig. 5C), TrkB aptamer-loaded pSiNP/nanofibers (Fig. 5D), and NGF-loaded pSiNP/nanofibers (Fig. 5E) all showed significantly more Hoechst stained intersections across the range of distances when compared to PLGA nanofiber and film controls (Figure 5B,F), as well as their respective nanoparticle scaffold controls (Supplemental Figure S11, S12, S13, Supplemental Information). This signifies that there were generally more cells migrating out of the DRG explants in the presence of the three APIs. We then looked at the average length of the 10 cells that migrated farthest from each DRG explant. The farthest cells migrated 2794 ± 174 μm on bpV(HOpic)-loaded pSiNP/nanofibers, 4290 ± 764 μm on TrkB aptamer-loaded pSiNP/nanofibers, and 3847 ± 347 μm on NGF-loaded pSiNP/nanofibers (Fig. 5G). Cells migrated significantly farther on TrkB aptamer and NGF-loaded pSiNP/nanofiber scaffolds compared to PLGA nanofiber controls (1867 ± 195 μm) (Fig. 5B,G), the DNA-pSiNP control nanofibers (1968 ± 172 μm) (Figure S12, Supplemental Information), and BSA-pSiNP control nanofibers (2668 ±563 μm) (Figure S13, Supplemental Information). However, no statistically significant differences were seen between bpV(HOpic)-loaded pSiNP/nanofibers and PLGA controls or NH2-pSiNP control nanofibers (1967 ± 22 μm) (Figure S11, Supplemental Information). Previously, inhibition of PTEN was shown to increase fibroblast cellular migration.[70] While the bpV(HOpic) results here show that cells are not migrating significantly farther than DRG explants on control scaffolds, more cells are migrating out of the DRG explants on bpV(HOpic) scaffolds demonstrating mobilization of cells from the DRG explants (Figure 5F,G).
Figure 5.
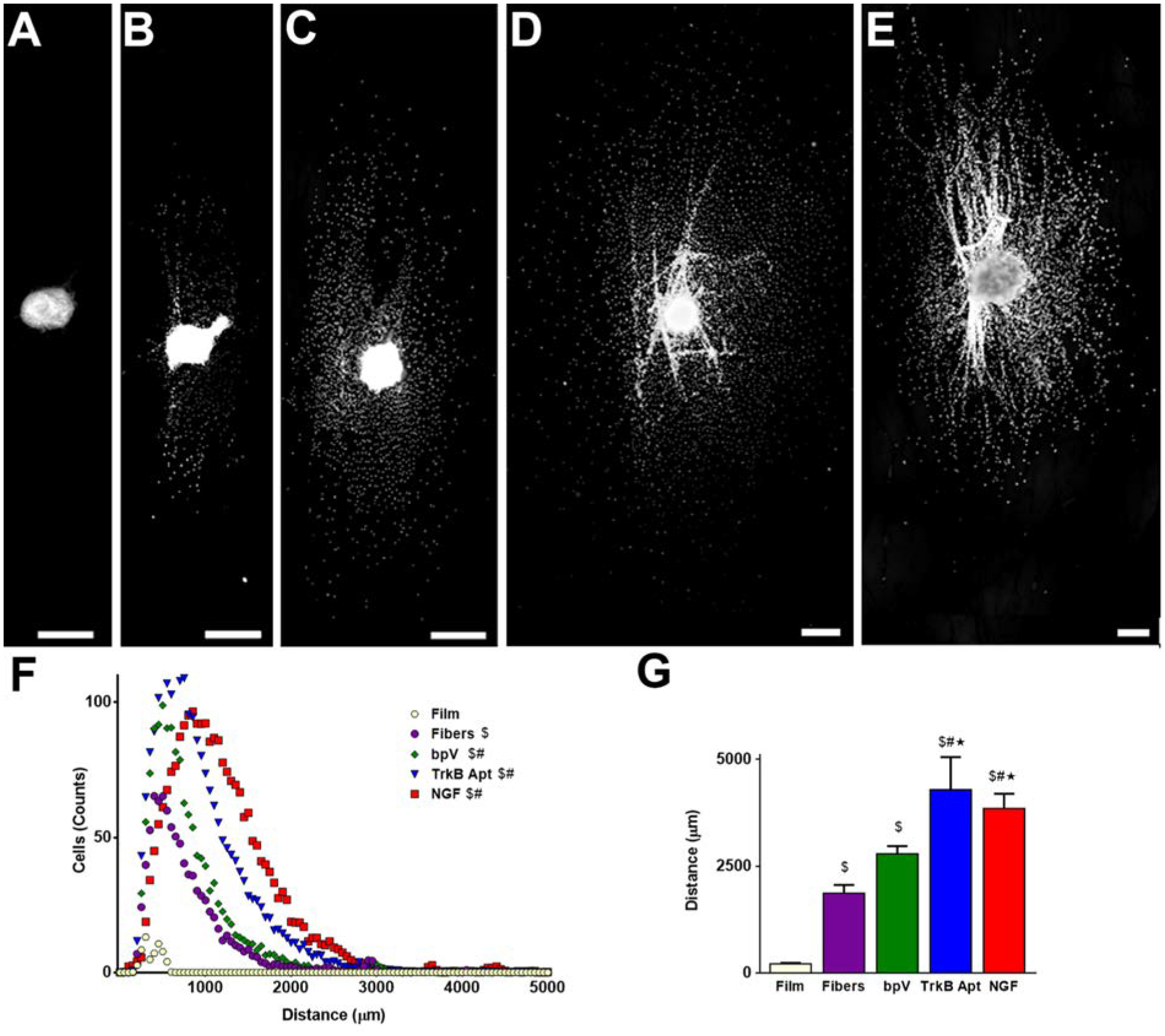
Increase in neurite migration induced on hybrid PLGA nanofibers. (A-E) Hoechst stained DRG explants cultured on (A) pure PLGA film control, (B) pure PLGA fiber control, (C) bpV(HOpic)-pSiNP/PLGA nanofiber hybrids, (D) TrkB Aptamer-pSiNP/PLGA nanofibers, and (E) NGF-PSiNP/PLGA nanofiber hybrids. (F) Range of cellular migration from DRG explants on each scaffold type. Pure PLGA fibers showed more counts of cellular migration from DRG explants compared to pure PLGA films. Scaffolds containing bpV(HOpic)-pSiNPs, TrkB aptamer-pSiNPS, or NGF-pSiNPs showed further increase in the number of nuclei intersections over the range of cellular migration. (G) Average of the ten farthest migrating nuclei on each scaffold. PLGA fibers alone induced more extensive cellular migration from DRG explants compared to PLGA films. Scaffolds containing TrkB aptamer-pSiNPS or NGF-pSiNPs, but not bpV(HOpic)-pSiNPs, further increased the distance of cellular migration compared to pure PLGA fibers. Scale Bar = 500μm; n = 5; $ = p < 0.05 compared to PLGA film; # = p < 0.05 compared to PLGA fibers; * = p < 0.05 compared to bpV(HOpic)-pSiNP/PLGA nanofiber hybrids.
This work provides one solution to the problem of formulating and delivering therapeutic agents in conventional biocompatible/biodegradable polymers by incorporating porous silicon nanoparticles (pSiNPs) that are pre-loaded with the therapeutic into the polymer scaffold during fabrication. The pSiNPs were loaded with three classes of compounds: a highly water-soluble small molecule, a nucleic acid construct (RNA aptamer), and a protein. The three molecules allowed the evaluation of three distinct pSiNP loading chemistries, and the ability of these chemistries to control payload release profiles from PLGA nanofibers. We found that the nanofiber topography directs the growth of cells and extending neurites in a DRG explant culture system, and that the small molecule drug, the RNA aptamer, and the protein growth factor all retained their function and were able to enhance DRG explant neurite extension when formulated with the pSiNP/PLGA nanofiber system. Additionally, this work represents the first example of release of an RNA aptamer from polymer nanofibers to enhance neurite extension. The ultimate goal of these studies was to develop implantable scaffolds that incorporate slow release of therapeutic agents to enhance nerve repair after traumatic injury. This study demonstrates a versatile, modular approach to deliver a variety of therapeutic payloads from polymer scaffolds for such an application; the approach may also be applicable to other tissue engineering problems.
Supplementary Material
Acknowledgements
JMZ and CMD contributed equally to this work. The authors thank Dr. Kendell Pawelec and Prof. Jeff Sakamoto for helpful discussions regarding this research. This study was supported by the National Science Foundation under Grant No. CBET-1603177 (MJS), and in vitro DRG studies were supported in part by the National Institutes of Health under Grant No. NIBIB-RO1-EB005678 (LDS). Transmission electron micrographs were obtained in the Cellular and Molecular Medicine Electron microscopy core facility, which is supported in part by National Institutes of Health Award number S10OD023527. This work was performed in part at the San Diego Nanotechnology Infrastructure (SDNI) of UCSD, a member of the National Nanotechnology Coordinated Infrastructure, which is supported by the National Science Foundation (Grant ECCS-1542148). This project received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 704120 (“MIRNANO”). Dr. AB is a Global Marie Sklodowska-Curie Fellow. Dr. JK acknowledges financial support from the UCSD Frontiers of Innovation Scholars Program (FISP) fellowship. JW thanks the NIH for the predoctoral training grant 2T32CA1539106A1 (CRIN).
Conflict Disclosure
MJS holds an appointment as a “High-Level Talent” at the Key Laboratory of Organosilicon Chemistry and Material Technology of Hangzhou Normal University, China, he is a Guest Professor at Zhejiang University, China, and he is a scientific founder of Spinnaker Biosciences, Inc., a member of the Board of Directors, and has an equity interest in the company. Although one or more of the grants that supported this research has been identified for conflict of interest management based on the overall scope of the project and its potential benefit to Spinnaker Biosciences, Inc., the research findings included in this particular publication may not necessarily relate to the interests of Spinnaker Biosciences, Inc. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Jonathan M. Zuidema, Department of Chemistry and Biochemistry, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA
Courtney M. Dumont, Department of Biomedical Engineering, University of Miami, 1251 Memorial Drive, Coral Gables, FL 33146, USA John T. Macdonald Foundation Biomedical Nanotechnology Institute, University of Miami, 1951 NW 7th Avenue, Miami, FL 33136, USA.
Joanna Wang, Materials Science and Engineering, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA.
Wyndham M. Batchelor, Department of Biomedical Engineering, University of Miami, 1251 Memorial Drive
Yi-Sheng Lu, Materials Science and Engineering, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA.
Jinyoung Kang, Department of Nanoengineering, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, CA.
Alessandro Bertucci, Department of Chemistry and Biochemistry, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA; Department of Chemical Sciences and Technologies, University of Rome Tor Vergata, Via della Ricerca Scientifica 1, 00133 Rome, Italy.
Noel M. Ziebarth, Department of Biomedical Engineering, University of Miami, 1251 Memorial Drive
Lonnie D. Shea, Department of Biomedical Engineering, University of Michigan, 2200 Bonisteel Boulevard, Ann Arbor, MI 48109, USA Department of Chemical Engineering, University of Michigan, 2200 Bonisteel Boulevard, Ann Arbor, MI 48109, USA.
Michael J. Sailor, Department of Chemistry and Biochemistry, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA
References
- [1].Xie JW, Willerth SM, Li XR, Macewan MR, Rader A, Sakiyama-Elbert SE, Xia YN, Biomaterials 2009, 30, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yang F, Murugan R, Wang S, Ramakrishna S, Biomaterials 2005, 26, 2603. [DOI] [PubMed] [Google Scholar]
- [3].Zong XH, Bien H, Chung CY, Yin LH, Fang DF, Hsiao BS, Chu B, Entcheva E, Biomaterials 2005, 26, 5330. [DOI] [PubMed] [Google Scholar]
- [4].Lee S, Leach MK, Redmond SA, Chong SYC, Mellon SH, Tuck SJ, Feng ZQ, Corey JM, Chan JR, Nat. Methods 2012, 9, 917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ingavle GC, Leach JK, Tissue Eng. Part B Rev 2014, 20, 277. [DOI] [PubMed] [Google Scholar]
- [6].Sperling LE, Reis KP, Pranke P, Wendorff JH, Drug Discov. Today 2016, 21, 1243. [DOI] [PubMed] [Google Scholar]
- [7].Kai D, Tan MJ, Prabhakaran MP, Chan BQY, Liow SS, Ramakrishna S, Loh XJ, Colloid Surf. B 2016, 148, 557. [DOI] [PubMed] [Google Scholar]
- [8].Smith MK, Singh V, Kalaitzidou K, Cola BA, ACS Appl. Mater. Interfaces 2016, 8, 14788. [DOI] [PubMed] [Google Scholar]
- [9].Chen ML, Li YF, Besenbacher F, Adv. Healthc. Mater 2014, 3, 1721. [DOI] [PubMed] [Google Scholar]
- [10].Lin XL, Tang DY, Du HF, J. Pharm. Pharmacol 2013, 65, 673. [DOI] [PubMed] [Google Scholar]
- [11].Lee MS, Ahmad T, Lee J, Awada HK, Wang YD, Kim K, Shin H, Yang HS, Biomaterials 2017, 124, 65. [DOI] [PubMed] [Google Scholar]
- [12].Briggs T, Arinzeh TL, J. Biomed. Mater. Res. A 2014, 102, 674. [DOI] [PubMed] [Google Scholar]
- [13].Li XQ, Su Y, Liu SP, Tan LJ, Mo XM, Ramakrishna S, Colloid Surf. B 2010, 75, 418. [DOI] [PubMed] [Google Scholar]
- [14].Yazgan G, Popa AM, Rossi RM, Maniura-Weber K, Puigmarti-Luis J, Crespy D, Fortunato G, Polymer 2015, 66, 268. [Google Scholar]
- [15].Jiang HL, Wang LQ, Zhu KJ, Control J. Release 2014, 193, 296. [DOI] [PubMed] [Google Scholar]
- [16].Su Y, Su QQ, Liu W, Lim M, Venugopal JR, Mo XM, Ramakrishna S, Al-Deyab SS, El-Newehy M, Acta Biomater. 2012, 8, 763. [DOI] [PubMed] [Google Scholar]
- [17].Yang C, Yu DG, Pan D, Liu XK, Wang X, Bligh SWA, Williams GR, Acta Biomater. 2016, 35, 77. [DOI] [PubMed] [Google Scholar]
- [18].Han D, Sherman S, Filocamo S, Steckl AJ, Acta Biomater. 2017, 53, 242. [DOI] [PubMed] [Google Scholar]
- [19].Yang Y, Xia T, Zhi W, Wei L, Weng J, Zhang C, Li XH, Biomaterials 2011, 32, 4243. [DOI] [PubMed] [Google Scholar]
- [20].Han D, Steckl AJ, ACS Appl. Mater. Inter 2013, 5, 8241. [DOI] [PubMed] [Google Scholar]
- [21].Chen ML, Gao S, Dong MD, Song J, Yang CX, Howard KA, Kjems J, Besenbacher F, ACS Nano 2012, 6, 4835. [DOI] [PubMed] [Google Scholar]
- [22].Ke M, Wahab JA, Hyunsik B, Song KH, Lee JS, Gopiraman M, Kim IS, RSC Adv. 2016, 6, 4593. [Google Scholar]
- [23].Qiu KX, He CL, Feng W, Wang WZ, Zhou XJ, Yin ZQ, Chen L, Wang HS, Mo XM, J Mater Chem B 2013, 1, 4601. [DOI] [PubMed] [Google Scholar]
- [24].Kashanian S, Harding F, Irani Y, Klebe S, Marshall K, Loni A, Canham L, Fan D, Williams KA, Voelcker NH, Coffer JL, Acta Biomater. 2010, 6, 3566. [DOI] [PubMed] [Google Scholar]
- [25].Fan D, Akkaraju GR, Couch EF, Canham LT, Coffer JL, Nanoscale 2011, 3, 354. [DOI] [PubMed] [Google Scholar]
- [26].McInnes SJP, Macdonald TJ, Parkin IP, Nann T, Voelcker NH, Nanomaterials 2018, 8, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zuidema JM, Kumeria T, Kim D, Kang JY, Wang JAN, Hollett G, Zhang X, Roberts DS, Chan N, Dowling C, Blanco-Suarez E, Allen NJ, Tuszynski MH, Sailor MJ, Adv. Mater 2018, 30, 1706785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Park J-H, Gu L, Maltzahn G. v., Ruoslahti, Bhatia SN, Sailor MJ, Nat. Mater 2009, 8, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Low SP, Voelcker NH, Canham LT, Williams KA, Biomaterials 2009, 30, 2873. [DOI] [PubMed] [Google Scholar]
- [30].Cullis AG, Canham LT, Nature 1991, 353, 335. [Google Scholar]
- [31].Bley RA, Kauzlarich SM, Davis JE, Lee HWH, Chem. Mat 1996, 8, 1881. [Google Scholar]
- [32].Wang CF, Sarparanta MP, Makila EM, Hyvonen MLK, Laakkonen PM, Salonen JJ, Hirvonen JT, Airaksinen AJ, Santos HA, Biomaterials 2015, 48, 108. [DOI] [PubMed] [Google Scholar]
- [33].Secret E, Smith K, Dubljevic V, Moore E, Macardle P, Delalat B, Rogers ML, Johns TG, Durand JO, Cunin F, Voelcker NH, Adv. Healthc. Mater 2013, 2, 718. [DOI] [PubMed] [Google Scholar]
- [34].Tasciotti E, Liu XW, Bhavane R, Plant K, Leonard AD, Price BK, Cheng MMC, Decuzzi P, Tour JM, Robertson F, Ferrari M, Nat. Nanotechnol 2008, 3, 151. [DOI] [PubMed] [Google Scholar]
- [35].Beavers KR, Werfel TA, Shen TW, Kavanaugh TE, Kilchrist KV, Mares JW, Fain JS, Wiese CB, Vickers KC, Weiss SM, Duvall CL, Adv. Mater 2016, 28, 7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Anglin EJ, Schwartz MP, Ng VP, Perelman LA, Sailor MJ, Langmuir 2004, 20, 11264. [DOI] [PubMed] [Google Scholar]
- [37].Cifuentes-Rius A, Ivask A, Sporleder E, Kaur I, Assan Y, Rao S, Warther D, Prestidge CA, Durand JO, Voelcker NH, Small 2017, 13, 1701201. [DOI] [PubMed] [Google Scholar]
- [38].Shahbazi MA, Almeida PV, Correia A, Herranz-Blanco B, Shrestha N, Makila E, Salonen J, Hirvonen J, Santos HA, Control J. Release 2017, 249, 111. [DOI] [PubMed] [Google Scholar]
- [39].Kwon EJ, Skalak M, Bertucci A, Braun G, Ricci F, Ruoslahti E, Sailor MJ, Bhatia SN, Adv. Mater 2017, 29, 1701527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Christie KJ, Webber CA, Martinez JA, Singh B, Zochodne DW, J. Neurosci 2010, 30, 9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kim MS, El-Fiqi A, Kim JW, Ahn HS, Kim H, Son YJ, Kim HW, Hyun JK, ACS Appl. Mater. Inter 2016, 8, 18741. [DOI] [PubMed] [Google Scholar]
- [42].Keefe AD, Pai S, Ellington A, Nat. Rev. Drug Discov 2010, 9, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nagahara AH, Tuszynski MH, Nat. Rev. Drug Discov 2011, 10, 209. [DOI] [PubMed] [Google Scholar]
- [44].Huang YZ, Hernandez FJ, Gu B, Stockdale KR, Nanapaneni K, Scheetz TE, Behlke MA, Peek AS, Bair T, Giangrande PH, McNamara JO, Mol. Pharm 2012, 82, 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sofroniew MV, Howe CL, Mobley WC, Annu. Rev. Neurosci 2001, 24, 1217. [DOI] [PubMed] [Google Scholar]
- [46].Gundersen RW, Barrett JN, Science 1979, 206, 1079. [DOI] [PubMed] [Google Scholar]
- [47].Baron-Van Evercooren A, Kleinman HK, Ohno S, Marangos P, Schwartz JP, Dubois-Dalcq ME, J. Neurosci. Res 1982, 8, 179. [DOI] [PubMed] [Google Scholar]
- [48].Yu XJ, Dillon GP, Bellamkonda RV, Tissue Eng. 1999, 5, 291. [DOI] [PubMed] [Google Scholar]
- [49].Qin Z, Joo J, Gu L, Sailor MJ, Part. Part. Syst. Charact 2014, 31, 252. [Google Scholar]
- [50].Kim D, Zuidema JM, Kang J, Pan Y, Wu L, Warther D, Arkles B, Sailor MJ, J. Am. Chem. Soc 2016, 138, 15106. [DOI] [PubMed] [Google Scholar]
- [51].Kang J, Joo J, Kwon EJ, Skalak M, Hussain S, She ZG, Ruoslahti E, Bhatia SN, Sailor MJ, Adv. Mater 2016, 28, 7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Mohiuddin L, Fernandez K, Tomlinson DR, Fernyhough P, Neurosci. Lett 1995, 185, 20. [DOI] [PubMed] [Google Scholar]
- [53].Sailor MJ, Wu EC, Adv. Funct. Mater 2009, 19, 3195. [Google Scholar]
- [54].Wang J, Kumeria T, Bezem MT, Wang J, Sailor MJ, ACS Appl. Mater. Inter 2018, 10, 3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kim D, Kang J, Wang T, Ryu HG, Zuidema JM, Joo J, Kim M, Huh Y, Jung J, Ahn KH, Kim KH, Sailor MJ, Adv. Mater 2017, 29, 1703309. [DOI] [PubMed] [Google Scholar]
- [56].Joo J, Liu X, Kotamraju VR, Ruoslahti E, Nam Y, Sailor MJ, ACS Nano 2015, 9, 6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Gu L, Hall DJ, Qin Z, Anglin E, Joo J, Mooney DJ, Howell SB, Sailor MJ, Nat. Commun 2013, 4, 2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Joo J, Cruz JF, Vijayakumar S, Grondek J, Sailor MJ, Adv. Funct. Mater 2014, 24, 5688. [Google Scholar]
- [59].Salonen J, Laitinen L, Kaukonen AM, Tuura J, Bjorkqvist M, Heikkila T, Vaha-Heikkila K, Hirvonen J, Lehto VP, Control J. Release 2005, 108, 362. [DOI] [PubMed] [Google Scholar]
- [60].Hussain S, Joo J, Kang J, Kim B, Braun GB, She ZG, Kim D, Mann AP, Molder T, Teesalu T, Carnazza S, Guglielmino S, Sailor MJ, Ruoslahti E, Nat. Biomed. Eng 2018, 2, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].von Burkersroda F, Schedl L, Gopferich A, Biomaterials 2002, 23, 4221. [DOI] [PubMed] [Google Scholar]
- [62].Shin HJ, Lee CH, Cho IH, Kim YJ, Lee YJ, Kim IA, Park KD, Yui N, Shin JW, Biomat J. Sci-Polym. E 2006, 17, 103. [DOI] [PubMed] [Google Scholar]
- [63].Xin XJ, Hussain M, Mao JJ, Biomaterials 2007, 28, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Dumont CM, Carlson MA, Munsell MK, Ciciriello AJ, Strnadova K, Park J, Cummings BJ, Anderson AJ, Shea LD, Acta Biomater. 2019, 86, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Koffler J, Zhu W, Qu X, Platoshyn O, Dulin JN, Brock J, Graham L, Lu P, Sakamoto J, Marsala M, Chen SC, Tuszynski MH, Nat. Med 2019, 25, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Pawelec KM, Koffler J, Shahriari D, Galvan A, Tuszynski MH, Sakamoto J, Biomed. Mater 2018, 13, 044104. [DOI] [PubMed] [Google Scholar]
- [67].Almqvist P, Pschera H, Samuelsson EB, Lunell NO, Seiger A, Exp. Neurol 1994, 125, 1. [DOI] [PubMed] [Google Scholar]
- [68].Zuidema JM, Hyzinski-Garcia MC, Van Vlasselaer K, Zaccor NW, Plopper GE, Mongin AA, Gilbert RJ, Biomaterials 2014, 35, 1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Hurtado A, Cregg JM, Wang HB, Wendell DF, Oudega M, Gilbert RJ, McDonald JW, Biomaterials 2011, 32, 6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Tamura M, Gu JG, Matsumoto K, Aota S, Parsons R, Yamada KM, Science 1998, 280, 1614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.