

Abstract
The insulin receptor gene (INSR) undergoes alternative splicing to give rise to two functionally related, but also distinct, isoforms IR-A and IR-B, which dictate proliferative and metabolic regulations, respectively. Previous studies identified the RNA-binding protein CUGBP1 as a key regulator of INSR splicing. In this study, we show that the differential splicing of INSR occurs more frequently in breast cancer than in non-tumor breast tissues. In breast cancer cell lines, the IR-A:IR-B ratio varies in different molecular subtypes, knockdown or overexpression of CUGBP1 gene in breast cancer cells altered IR-A:IR-B ratio through modulation of IR-A expression, thereby reversed or enhanced the insulin-induced oncogenic behavior of breast cancer cells, respectively. Our data revealed the predominant mitogenic role of IR-A isoform in breast cancer and depicted a novel interplay between INSR and CUGBP1, implicating CUGBP1 and IR-A isoform as the potential therapeutic targets and biomarkers for breast cancer.
Introduction
The oncogenic effect of insulin receptor (IR) has been reported in various human carcinomas, such as colon, lung, ovary, thyroid and breast, as arguably a predictor for poor survival (1–4). In cooperation and sometimes dimerization with type I insulin-like growth factor receptor (IGF1R), IR transduces the pro-proliferation signaling cascade upon binding with the cognate ligands, including IGF-I, IGF-II and insulin. Blocking this pro-oncogenic pathway is adopted as a treatment modality in cancers including breast cancer, but it would unavoidably cause serious side effects due to the dual function of IR in both mitogenic and metabolic pathways (5,6).
The IR-encoding gene INSR, containing 22 exons, undergoes alternative splicing generating two distinct isoforms IR-A and IR-B, which differ structurally and functionally in various aspects. IR-A, the product of exon 11 skipping, is mainly expressed in embryonic and less differentiated cells (7). The equally preferential binding of IR-A to insulin and IGF-II, the latter being also linked to fetal development, conforms to its role in growth promotion. The inclusion of the 12 amino acids derived from exon 11 in IR-B confers its binding primarily to insulin and is principally associated with metabolic and differentiation signals after insulin stimulation (8). IR-B isoform is predominantly expressed in liver, muscle and adipose tissue, the salient metabolic targets of insulin. Meanwhile, overexpression of IR-A in many cancer cells is speculated to promote cell survival, mortality and invasiveness when activated by insulin and IGFs (9–11).
Pre-messenger RNA (pre-mRNA) splicing is under the tight control of its enhancers and silencers such as CUG triplet repeat binding protein 1 (CUGBP1), an RNA-binding protein exerting its regulation in RNA splicing, translation and turnover (12). Previous studies identified CUGBP1 as a splicing factor that recognizes sequences in intron 10 and exon 11 of INSR (13,14). Primarily found to be an important regulator in myotonic dystrophy type 1 disease progression (15), the multiple roles of CUGBP1 are extended to liver dysfunction (16) and several types of cancer (17–19). Given the involvement of abnormal insulin signaling in tumor progression, the role of CUGBP1 in INSR splicing may predispose the signaling bifurcation of insulin. On the basis of that, a new perspective of cancer intervention holds promise.
To that purpose, we examined the expression levels of INSR and its alternative splicing products IR-A and IR-B isoforms in the clinical database, tissues and cell lines of breast cancer, representing luminal HER2+ and triple negative breast cancer (TNBC) subtypes. By manipulating CUGBP1 levels with knockdown or overexpression, we investigated the CUGBP1-modulated INSR alternative splicing and the switch of its oncogenic effects in breast cancer cell lines. It is interesting to note that the IR-A isoform-promoted malignant progression of breast cancer and its expression in clinical samples are subtype specific.
Materials and methods
Samples and clinicopathological data
A total of 94 surgically resected breast cancer specimens and adjacent breast tissue were collected from the Second Hospital of Dalian Medical University between January 2008 and January 2014. None of the patients had received chemotherapy or radiotherapy prior to surgery. This study had the inclusion criteria as follows: (i) pathological examination as ER+, PR+ and HER2-, (ii) ≥15 lymph nodes were examined after surgery, (iii) the tumor specimens were intact and incubated uniformly by the IR antibody and (iv) complete medical records were available. The clinical and demographic data of patients were obtained from the medical records. The study protocol was approved by the Ethics Committee in the Second Hospital of Dalian Medical University.
Animals
Mouse mammary carcinoma derived from MMTV-Wnt1 transgenic animals and mammary glands of wild-type mice on FVB/N background were provided by Alison Obr and Teresa L. Wood at the Rutgers New Jersey Medical School.
Cell lines and cell culture
The human breast cancer cell lines MCF7, T47D, SKBR3, HCC1954, MDA-MB-436 and MDA-MB-231 were purchased from American Type Culture Collection (ATCC, Manassas, VA). Cells were cultured in RPMI 1640 medium (Gibco) supplemented with 10% fetal bovine serum (FBS) at 37°C in 5% CO2 incubator. MCF7 and T47D are human estrogen receptor positive breast cancer cell lines. SKBR3 and HCC1954 are human HER2 positive breast cancer cell lines. MDA-MB-436 and MDA-MB-231 are human triple negative breast cancer cell lines.
Small interfering RNA transfection
Lipofectamine 3000 (Invitrogen, CA) was used to transfect CUGBP1 small interfering RNAs (siRNAs) and negative control siRNAs (GenePharma, Suzhou, China). The sequences of the siRNAs used in this study were: CUGBP1 siRNA#1: sense: 5’-GGAUGCAUCACCCUAUACATT-3’, antisense: 5’-UGUAUAGGGUGAUGCAUCCTT-3’; CUGBP1 siRNA#2: sense: 5’- CUCUGUACAACCAGAAUCUTT-3’, antisense: 5’- AGAUUC UGGUUGUACAGAGTT -3’. Cells were collected 24 and 48 h after transfection for PCR and western blot, respectively.
Plasmid construction
CUGBP1 open reading frame was amplified by PCR and the PCR fragment was subcloned into a pcDNA3.1 vector (GenePharma, SuZhou, China) at EcoRI and BamHI sites. The same protocol as the siRNA transfection was applied here.
Cell proliferation assay
Cells transfected with siRNA or plasmids were seeded in a 96-well plate (5000 cells/well). Cell proliferation was determined using CCK8 assay (Abbkine, Wuhan, China), according to the manufacturer’s instructions. The CCK8 solution was added (10 μl/well) to each well and incubated at 37°C for 2 h. The optical density of each sample was immediately measured at 450 nm using a microplate reader (Thermo).
Colony formation assay
Cells transfected with siRNA or plasmids were suspended in serum-free medium and seeded in a 6-well plate (1000 per well). Medium with or without 10 nM insulin was added to the wells on the next day. Two weeks were allowed for colonization. The colonies were fixed with formaldehyde, stained with crystal violet and imaged. The number of colonies in each well was quantified using Image J (National Institutes of Health).
Transwell assay
Cell migration assay was performed using Transwell chambers (8 μM pore size; Costar). Cells transfected with siRNA or plasmids were allowed to reach about 75–80% confluence and then serum starved for 24 h before being treated with or without 10 nM insulin for 36 h. After trypsinization, the cells were washed with phosphate-buffered saline (PBS) and suspended in serum-free medium. Next, 100 μl of cell suspension (10 × 10 4cells) were added to the upper chamber of the transwell plates. FBS was added to the bottom chambers. For the assay, the cells that had not migrated after 24 h were removed from the upper surface of the filters using cotton swabs, but the cells that had migrated were fixed with formaldehyde to determine the number of migratory cells. The lower surface of the filters was stained with crystal violet. Images of six different ×10 fields were captured from each membrane, and the number of migratory cells was counted. The corresponding quantitative data were obtained by a spectrophotometer reading of optical density at 590 nm after extraction of the crystal violet staining. The mean of triplicate assays for each experimental condition was calculated.
Wound-healing assay
Cells were plated in six-well plates and grown to 80–90% confluency before being transfected with siRNA or plasmids, then serum starved for 24 h. Wounds were generated using a pipette tip, and the cells were rinsed with PBS prior to the addition with or without 10 nM insulin. Wounded areas were photographed at 0 and 24 h using a phase-contrast microscope (Olympus Japan).
Western blot
Proteins were extracted from total cell lysates using radio-immunoprecipitation assay buffer. The proteins were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis using a 12% polyacrylamide gel and transferred onto nitrocellulose filter membranes. The membranes were probed with monoclonal antibodies against CUGBP1, IR, p-IR, Akt1, p-Akt, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Proteintech, Wuhan, China), C/EBPβ (H-7, Santa Cruz, CA) and β-tubulin (Proteintech, Wuhan, China). SuperLumia ECL HRP Substrate Kit (Abbkine) was used to visualize the protein bands. Quantitative analysis was performed using Quantity One software (Bio-Rad, Hercules, CA).
Isolation of RNA and reverse transcriptase–PCR quantification
Total RNA was extracted from cultured cells using TRIzol total RNA isolation reagent (Takara) according to the manufacturer’s instructions. Complementary DNA was synthesized from total RNA or purified small RNAs using TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (Transgen Biotech) according to the manufacturer’s instructions. PCR was performed using the Taq polymerase (Takara) with specific primers against human CUGBP1 (forward: 5’-ACATCCGAGTCATGTTCTCTTCG-3’ and reverse: 5’-CATTGCCTTGATAGCCGTCTG-3’), INSR (5’- GGAAGAC GTTTGAGGATTACC-3’ and reverse: 5’- TCGAGGAAGTGTTGGGGAAAG-3’), IR-A isoform (forward: 5’- TTTTCGTCCCCAGGCCAT-3’ and reverse: 5’-CCACCGTCACATTCCCAAC-3’), IR-B isoform (forward: 5’-TTTCGT CCCCAGAAAAACCTCT-3’ and reverse: 5’- CCACCGTCACATTCCCAAC-3’), c-jun (forward: 5’-TCCAAGTGCCGAAAAAGGAAG-3’ and reverse: 5’- CGAG TTCTGAGCTTTCAAGGT-3’), junB (forward: 5’- ACGACTCATACACAGCTA CGG-3’ and reverse: 5’- GCTCGGTTTCAGGAGTTTGTAGT-3’), TNFRSF1B (forward: 5’- CGGGCCAACATGCAAAAGTC3’ and reverse: 5’-CAGATGCGGTTCTGTTCCC-3’) and GAPDH (forward: 5’- GGAGCGA GATCCCTCCAAAAT-3’ and reverse: 5’- GGCTGTTGTCATACTTCTCATGG-3’). GAPDH was used for normalization. The PCR conditions were: denaturation at 94°C for 3 min, denaturation at 94°C for 30 s, annealing at 58°C for 45 s, extension at 72°C for 1 min, 35 cycles and final extension step at 72°C for 10 min. The PCR products were loaded on 2% agarose gels. The gels were stained with 10 mg/ml ethidium bromide. The PCR products were visualized with UV light and photographed. Real-time PCR was performed using the TransStart Tip Green qPCR SuperMix (Transgen Biotech) on a ABI 7900HT FAST Real-Time PCR System (Applied Biosystems). For animal mammary glands of breast cancer, transcript levels were normalized to ß-actin and data were analyzed using the Q-Gene software (BioTechniques Software Library) (20). Mouse CUGBP1 primer oligonucleotide pairs were purchased from Qiagen. Primer oligonucleotide pairs for ß-actin, IR-A and IR-B were purchased from IDT. Sequences for ß-actin are: Forward primer: 5’-CTAAGGCCAACCGTGAAAAG-3’, Reverse primer: 5’-ACCAGAGGCATACAGGGACA-3’. Reverse transcriptase–PCR (RT–PCR) conditions and sequences for IR-A and IR-B were described previously (21).
Immunohistochemistry
Immunohistochemistry (IHC) was performed on 94 formalin-fixed, paraffin-embedded primary breast tumors and its adjacent breast tissue that were histopathologically and clinically diagnosed at the Department of Breast Oncology, Second Hospital of Dalian Medical University. Tissue sections were deparaffinized, rehydrated and microwave-treated in a 10 mM citrate buffer (pH 6.0) for 10 min for antigen retrieval. Endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide for 15 min. Sections were incubated with antihuman INSR (1:100 dilution) and antihuman CUGBP1 (1:100 dilution) at 4°C overnight. Next, tissue sections were incubated with horseradish peroxidase immunoglobulin for 30 min and developed using 3’-3’ diaminobenzidine as a chromogen substrate.
Analysis of gene expression from databases
Gene expression data were downloaded from the ONCOMINE database (Ma Breast 4, Breast Cancer Research, 2009) and the Cancer Genome Atlas (TCGA; Cancer Genome Atlas Network, 2012). Analyses and figures were made using GraphPad Prism. On the dot plot graphs, each dot indicates an individual sample, with results expressed as median with interquartile range.
Statistical analysis
Results were expressed as mean ± standard deviation (n = 3). Data were representative of at least three independent experiments. Two-tailed Student’s t-test and analysis of variance were used for differential comparison between two groups and among three groups, respectively. Correlation analysis was performed using GraphPad Prism.
Results
IR is highly overexpressed in breast cancer
In the examination of the alteration frequencies of INSR gene in four breast cancer cohorts retrieved from the cBioPortal, including TCGA, TCGA_2015, TCGA_pub and METABRIC, the rate of recurrent amplification of INSR gene was approximately 1.2–2.7%, higher than that of mutation and deletion (Figure 1A and B). Consistently, by analyzing gene expression data in the Oncomine database (22), we found that INSR is highly overexpressed in breast cancer (Figure 1C), in accordance with the overexpression of IR protein in breast cancer tissues, as shown by IHC staining (Figure 1D). Notable, however, is the inverse correlation between INSR expression levels and T:tumor, N: node, M: metastasis staging (P = 0.048; Supplementary Table 1, available at Carcinogenesis Online).
Figure 1.

IR is highly overexpressed in breast cancer. (A, B) Amplification of INSR is recurrent in breast carcinoma based on cBioportal database. (C) INSR expression levels are significantly higher in breast carcinoma (n = 11, red circles) than in normal breast tissues (n = 14, yellow circles), data from the Oncomine database (22). Black lines in each group indicate median with interquartile range. P = 9.5 × 10−4 (Student’s t-test). ***P < 0.001. (D) The levels of INSR protein in breast carcinoma and corresponding non-cancerous tissues were measured by IHC.
Alternative splicing levels of INSR vary among molecular subtypes of breast cancer
The splicing isoforms of INSR, IR-A and IR-B have distinct features in their spatiotemporal expressions and functions. The ratio of IR-A and IR-B in breast cancer and normal tissues in TCGA SpliceSeq database was examined. The cross-tumor box plots indicated a lower percent spliced-in (PSI) value of breast cancer than that of normal tissue (Figure 2A; Supplementary Table 2, available at Carcinogenesis Online). Showing in numbers from zero to one, PSI is a common intuitive ratio for quantifying splicing events. A PSI value of 0.8 for an exon skip event would indicate that the exon is included in approximately 80% of the transcripts in the sample (Figure 2B). We found that the exon 11 is included in only 41.8% of the breast cancers, which rendered a higher proportion of IR-A, whereas, in normal tissues, the inclusion rate was 75.6%, indicating an IR-B predominance (Supplementary Table 2, available at Carcinogenesis Online). The differential IR-A and IR-B isoform splicing was also compared between normal and breast cancer tissues from clinical sources (Figure 2C) and between the wild-type and MMTV-Wnt1 transgenic mice on FVB/N background, a widely used metastatic breast cancer model (Figure 2F). In agreement with each other, an IR-A domination and, hence, a higher IR-A:IR-B ratio emanated from both breast cancer samples.
Figure 2.

Exon 11 skip (higher IR-A:IR-B ratio) of IR occurs most frequently in luminal A breast cancer cells. (A) Exon skip PSI in normal tissue and breast cancer. PSI is the ratio of normalized read counts indicating inclusion of a transcript element over the total normalized reads for that event (both inclusion and exclusion reads). (B) Schematic drawing illustrates alternative RNA splicing of exon 11 in the expression of IR-A and IR-B isoform. (C, E) Human clinical samples of breast carcinoma tissues and normal breast tissues were collected, followed by RNA extraction for IR splicing assessment by RT–PCR, and GAPDH served as loading control. The sizes of IR-B and IR-A were 171 and 135 bp, respectively (C). IR-A:IR-B ratio was quantified by scanning densitometry (E). Results are representative of three separate experiments. **P < 0.01. (D) Human clinical samples of breast carcinoma tissues and normal breast tissues were collected, followed by RNA extraction for real-time PCR measurement of relative gene expression of IR-A and IR-B. The IR-A:IR-B ratios are indicated above the bars. Data are means ± SEM. (F) Mammary glands of wild-type mice (n = 5) and mammary tumors of the MMTV-Wnt1 transgenic mice (n = 7) were collected, followed by RNA extraction and relative RNA expression (IR-A and IR-B) measurement by real-time PCR. The IR-A:IR-B ratios are shown above the bars. Data are means ± SEM. (G) Breast cancer cell lines classified as luminal HER2+ and TNBC were cultured in 10% FBS RPMI medium for 48 h and relative RNA expression (IR-A and IR-B) was measured by real-time PCR. The IR-A:IR-B ratios are shown above the bars. Data are means ± SEM. (H) MCF7 and MDA-MB-231 cells were cultured in 10% FBS RPMI medium for 48 h and IR-A and IR-B assessment detected by RT–PCR, GAPDH served as loading control. The size of IR-B and IR-A were 171 and 135 bp, respectively. (I) IR-A:IR-B ratio was quantified by densitometric scan. Results are representative of three separate experiments. *P < 0.05.
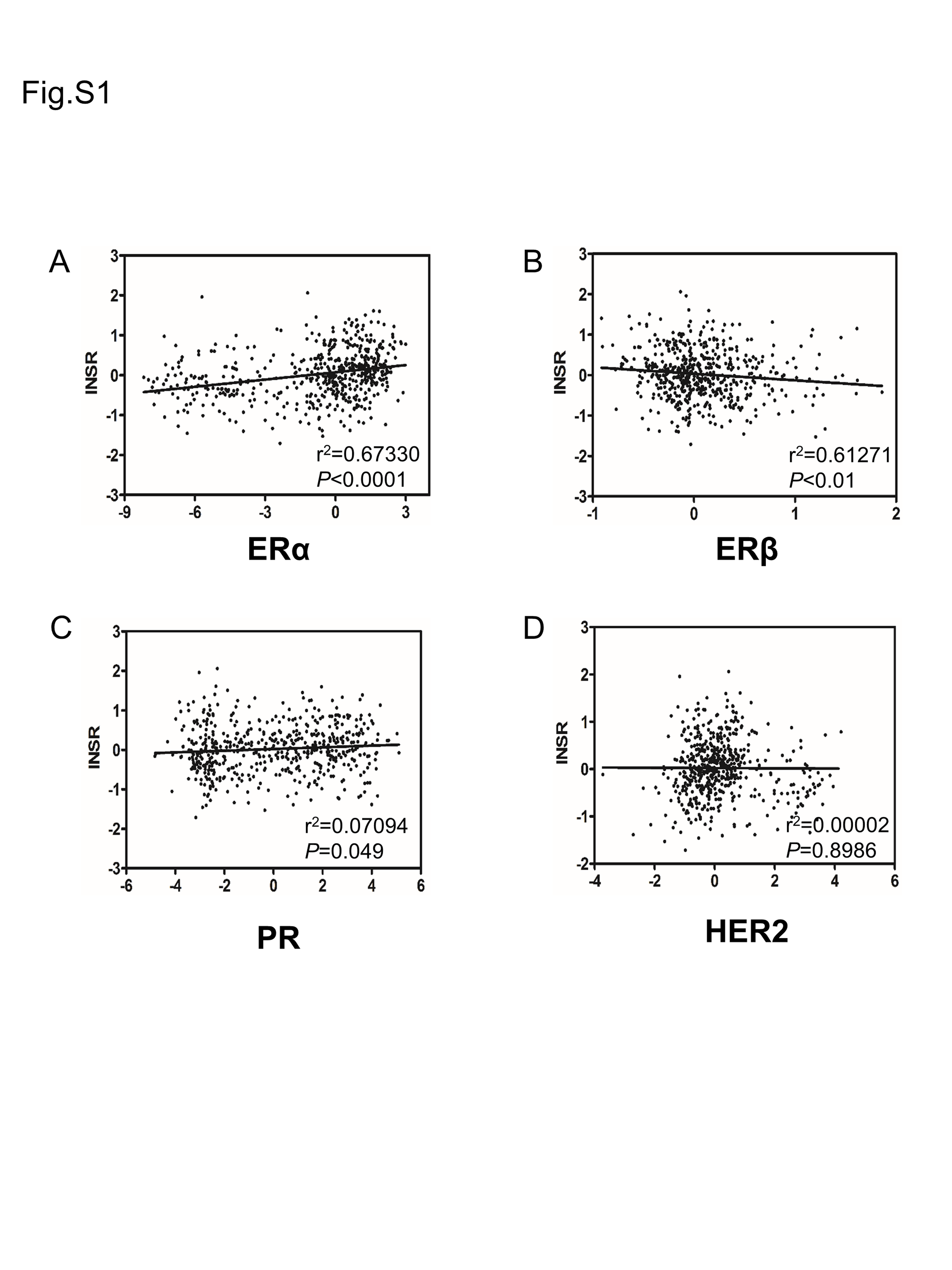
IR-A and IR-B mRNA levels in six breast cancer cell lines, representing different molecular subtypes of luminal HER2+ and TNBC, respectively, were compared by real-time PCR (Figure 2G). We found that the luminal cells had the highest and the TNBC cells the lowest IR-A level. The relative abundance of the two isoforms in MCF7 and MDA-MB-231 cells was also examined by RT–PCR, followed by a calculation of IR-A:IR-B ratio. As expected, the IR-A:IR-B ratio was higher in the luminal cell line MCF7 than the TNBC cell line MDA-MB-231 (Figure 2H and I). Consistent with the overexpression of IR-A in the luminal breast cancer cell lines, we found that INSR expression had a positive correlation with the expression of luminal markers estrogen receptor α (ERα) and progesterone receptors but negative with ERβ (P <0.05) and was not related to HER2 (Supplementary Figure S1A–D, available at Carcinogenesis Online). The data suggested that the INSR gene is alternatively spliced in breast cancer cells in a molecular subtype-dependent manner.
CUGBP1 promotes exon 11 exclusion and favors IR-A expression in breast cancer cells
The exclusion of exon 11 of INSR pre-mRNA promoted by CUGBP1 results in more IR-A isoform production (14). We next examined whether the expression of CUGBP1 correlates with the abundance and distribution of IR-A isoform in breast cancer. IHC, quantitative real-time PCR and data extracted from TCGA (Cancer Genome Atlas Network, 2012) (23) revealed that CUGBP1 was highly expressed in breast carcinomas in both animal and human samples (Figure 3A–C). In keeping with the results of IR-A:IR-B ratio (Figure 2G), the luminal MCF7 and T47D cells had the highest mRNA expression of CUGBP1 among the six breast cancer cell lines representing luminal HER2+ and TNBC (Figure 3D). A similar trend of protein levels was observed with western blot (P < 0.05) (Figure 3E and G). Using the TCGA database, a significant correlation was also displayed between CUGPB1 and INSR expression (P < 0.01) (Figure 3F).
Figure 3.

CUGBP1 expressions among molecular subtypes of breast cancer. (A) CUGBP1 expression levels are significantly higher in breast carcinoma (n = 525, red circles) than in normal breast tissues (n = 22, yellow circles) in the TCGA breast cancer cohort (Cancer Genome Atlas Network, 2012). Black lines in each group indicate the median with interquartile range. ***P < 0.001. (B) The levels of CUGBP1 protein in clinical samples of breast carcinoma and normal breast tissues were measured by IHC. (C) Mammary glands of wild-type mice (n = 5) and mammary tumors of the MMTV-Wnt1 transgenic mice (n = 7) were collected, followed by RNA extraction and real-time PCR measurement of the relative RNA expression of CUGBP1. Data are means ± SEM. (D) Breast cancer cell lines classified as luminal HER2+ and TNBC were cultured in 10% FBS RPMI medium for 48 h, followed by RNA extraction and real-time PCR measurement of the relative RNA expression of CUGBP1. Data are means ± SEM. (E, G) Immunoblotting analysis of CUGBP1 protein in breast cancer cell lines of luminal HER2 and TNBC subtypes. GAPDH served as a loading control (E). Quantitative protein levels were obtained by densitometric analyses normalized to GAPDH (G). Data are means ± SEM. ns denotes not significant. *P < 0.05. (F) CUGBP1 and INSR expression are positively correlated. Each circle represents an individual sample of human breast carcinoma (n = 547 from TCGA data set). Correlation analysis was performed by GraphPad Prism. P < 0.01.
To investigate if and how CUGBP1 dictates INSR splicing in breast cancer, we used siRNA and pcDNA3.1-CUGBP1 plasmid to knockdown or overexpress CUGBP1, respectively. The impact of CUGBP1 manipulation on IR-A and IR-B isoform expression was examined (Figure 4). The inhibition of CUGBP1 gene expression by siRNA in both MCF7 and T47D cell lines, monitored by western blot (P < 0.01; Figure 4A and F), resulted in a significant decrease of the IR-A:IR-B ratio (Figure 4B and G). Conversely, transfection of the pcDNA3.1-CUGBP1 plasmid into MDA-MB-231 cells led to an overexpression of CUGBP1 (P < 0.05; Figure 4K) and increased the IR-A:IR-B ratio (Figure 4L) as analyzed by RT–PCR (P < 0.05). It is worth noting that the changes of IR-A:IR-B ratio following CUGBP1 manipulation were mainly attributed to the altered IR-A proportion, whereas IR-B was left intact, as indicated by agarose separation (Figure 4B, G and L). These data suggested that CUGBP1 affects INSR splicing by promoting exon11 exclusion to favor IR-A expression in breast cancer.
Figure 4.

CUGBP1 regulates IR-A:IR-B ratio in breast cancer cells. (A, D, F, I) Western blotting demonstrated that CUGBP1 protein level was significantly decreased by siRNA-mediated CUGBP1 silencing in MCF7 (A) and T47D cells (F). GAPDH was used as a loading control. Relative protein levels were measured by densitometric analyses normalized to GAPDH (D, I). Data are means ± SEM. **P < 0.01. (B, E, G, J) MCF7 (B) and T47D cells (G) were transfected with control or CUGBP1 siRNA for 24 h, followed by RNA extraction and IR-A and IR-B assessment using RT–PCR. GAPDH served as the loading control. The sizes of IR-B and IR-A were 171 and 135 bp, respectively. IR-A:IR-B ratio was quantified by scanning densitometry (E, J). Results are representative of three separate experiments. *P < 0.05. (C, H) Relative RNA expressions of IR-A and IR-B in MCF7 (C) and T47D cells (H) were measured by real-time PCR. The IR-A:IR-B ratios are shown above the bars. Data are means ± SEM. (K, N) Western blotting demonstrated that CUGBP1 protein level was significantly increased 24 h after pcDNA3.1-CUGBP1 plasmid transfection in MDA-MB-231 cells (K). GAPDH was used as the loading control. Relative protein expressions were measured by densitometric analyses normalized to GAPDH (N). Data are means ± SEM. *P < 0.05. (L, O) MDA-MB-231 cells were transfected with pcDNA3.1 vector or pcDNA3.1-CUGBP1 plasmid for 24 h and IR-A and IR-B isoforms were detected by RT–PCR. GAPDH served as the loading control. The sizes of IR-B and IR-A were 171 and 135 bp, respectively (L). IR-A:IR-B ratio was quantified by scanning densitometry (O). Results are representative of three separate experiments. *P < 0.05. (M) Relative RNA expressions of IR-A and IR-B in MDA-MB-231 cells were measured by real-time PCR. The IR-A:IR-B ratios are shown above the bars. Data are means ± SEM.
Changes in CUGBP1 expression affect the oncogenic behavior of the breast cancer cells
The pro-tumorigenic nature of IR-A isoform has been confirmed by its link with tumor cell survival, mortality and invasiveness in several cancers (9,10). Based on the finding that CUGBP1 regulates INSR splicing and favors IR-A isoform expression, the proliferative and metastatic capacity of MCF7, T47D and MDA-MB-231 cells upon CUGBP1 modification were examined. CCK8 assay showed that the cell viability was decreased by 33–41% when CUGBP1 was suppressed in luminal MCF7 and T47D cells but increased by 30% in TNBC cell line MDA-MB-231 with CUGBP1 overexpression (P < 0.05; Figure 5A and B). A more than 50% decrease of colony formation was detected in CUGBP1-silenced cells, whereas it was enhanced by almost 40% in CUGBP1 overexpression cells (Figure 5C–F). Next, the effect of CUGBP1 on breast cancer cell invasion and migration was tested using transwell and wound-healing assays. The results showed that silencing CUGBP1 inhibited the cell invasion in MCF7 and T47D cells (Figure 5G and H) and migration in MCF7 cells (Figure 5K and M). In the meantime, both the migration and wound-healing capability of MDA-MB-231 cells were significantly elevated by CUGBP1 overexpression (Figure 5I, J, L and N). These data suggested that CUGBP1 promotes breast cancer aggressiveness.
Figure 5.

CUGBP1 regulates cell growth, colony formation, migration and invasion in breast cancer cells. (A, B) CUGBP1 was silenced by siRNA in MCF7 and T47D cells (A), and overexpressed by plasmid transfection in MDA-MB-231 cells (B). Cell viability was assessed using CCK8 assay. ns denotes not significant. *P < 0.05. **P < 0.01. ***P < 0.001. (C–F) Representative images and the quantitative analysis (D, F) of colony formation after CUGBP1 knockdown in MCF7 and T47D cells (C) and overexpression in MDA-MB-231 cells (E). Cells were seeded at a density of 1000 cells/well in six-well plates and allowed for colonization for 14 days. *P < 0.05. **P < 0.01. (G–J) Representative images of the transwell assay after CUGBP1 knockdown in MCF7 cells and T47D cells (G) and overexpression in MDA-MB-231 cells (I). The corresponding quantitative data were obtained by a spectrophotometer reading of optical density at 590 nm after extraction of the crystal violet staining (H, J). *P < 0.05. **P < 0.01. (K–N) Representative images and quantification of the wound-healing assay after CUGBP1 knockdown in MCF7 cells (K, M) and overexpression in MDA-MB-231 cells (L, N). *P < 0.05. **P < 0.01.
Insulin stimulation accentuates the oncogenic regulation of CUGBP1 in breast cancer cells
Dysregulations of insulin-signaling pathway play a major role in the development of breast cancer. The cellular response to insulin stimulation was next assessed with opposing modulations of CUGBP1. The optimal concentration of insulin at 10 nM was first determined by a dose-response experiment using CCK8 assay. Both MCF7 and MDA-MB-231 cells achieved peak proliferation after 24 h of treatment (Supplementary Figure S2A, available at Carcinogenesis Online). The insulin administration significantly stimulated cell proliferation in the scrambled control but not in the cells transfected with CUGBP1 siRNA (Supplementary Figure S2B, available at Carcinogenesis Online). In contrast, insulin-induced cell proliferation in MDA-MB-231 cells was 30% higher in CUGBP1 overexpression cells than in the control (Supplementary Figure S2C, available at Carcinogenesis Online). In parallel, the insulin-enabled colony formation was significantly impeded by CUGBP1 siRNA silencing in MCF7 and T47D cells (P < 0.001; Supplementary Figure S2D and E, available at Carcinogenesis Online) but enhanced by CUGBP1 overexpression in MDA-MB-231 cells (Supplementary Figure S2F, G, available at Carcinogenesis Online). Transwell and wound-healing assays showed that the effectiveness of insulin to activate cell invasion and migration was reduced in CUGBP1 siRNA-treated cells (Supplementary Figure S2H, I, L and N, available at Carcinogenesis Online) but strengthened when CUGBP1 was upregulated (Supplementary Figure S2J, K, M and O, available at Carcinogenesis Online).
CUGBP1 regulates the phosphorylation events in insulin-signaling pathway
The IR-mediated mitogenic signaling plays a pivotal role in breast cancer progression and poses the main cause for the failure of therapeutic targeting IGF1R (24). The phosphorylation events following IR activation by insulin treatment was investigated to determine whether the IR pathway is mediating the altered tumorigenic characteristics arisen from CUGBP1 manipulation in breast cancer cells. The total and phosphorylated IR were significantly diminished when CUGBP1 was knocked down in MCF7 cells (Figure 6A–C) but increased when CUGBP1 was overexpressed in MDA-MB-231 cells (Figure 6E–G). Insulin-induced downstream Akt phosphorylation at S473 was inhibited by CUGBP1 suppression (Figure 6D). In MDA-MB-231 cells, CUGBP1 overexpression induced higher downstream Akt phosphorylation at S473, which was further remarkably increased by insulin (Figure 6H). These results implied that, by regulating IR activity, CUGBP1 has a direct influence on insulin-signaling cascade.
Figure 6.

CUGBP1 regulates the expression of the IR and the insulin-signaling pathway. (A) Cell lysate were collected from MCF7 cells transfected with control or CUGBP1 siRNA, with or without 10 nM insulin pretreatment for 20 min. Protein levels associated with insulin-signaling pathway were determined by western blotting. GAPDH was used as an internal control. (B–D) Relative protein expressions of p-IR (B), IR (C) and p-Akt (D) in MCF7 cells were measured by densitometric analyses normalized to GAPDH. Data are means ± SEM. ns denotes not significant. *P < 0.05, **P < 0.01. (E) Cell lysate were collected from MDA-MB-231 cells transfected with pcDNA3.1 vector control or pcDNA3.1-CUGBP1 plasmid, with or without 10 nM insulin pretreatment for 20 min. Protein levels associated with insulin-signaling pathway were determined by western blotting. GAPDH was used as an internal control. (F–H) Relative protein expression of p-IR (F), IR (G) and p-Akt (H) in MDA-MB-231 cells were measured by densitometric analyses normalized to GAPDH. Data are means ± SEM. ns denotes not significant. *P < 0.05, **P < 0.01, ***P < 0.001.
As schematically described in Supplementary Figure S3, available at Carcinogenesis Online, a causal relationship between CUGBP1 expression, INSR splicing and breast cancer phenotype is proposed. Inhibition of CUGBP1 expression may serve to reverse the insulin-induced oncogenic progression of breast cancer cells through the switch of IR-A isoform dominance.
Discussion
In accordance with the literature showing an approximately 80% higher INSR expression in breast tumors (3), we confirmed the overexpression of INSR gene and IR protein in breast carcinomas (Figure 1C and D). A positive correlation between INSR and the RNA-binding protein CUGBP1 was detected in the clinical database and tissue samples.
Previous studies identified CUGBP1 as the splicing factor governing the alternative splicing of INSR (13,14). By examining breast cancer tissues and mining TCGA database, we found that CUGBP1 was overexpressed at mRNA and protein levels in breast carcinoma samples from both mice and humans. In conjunction with the differentially expressed CUGBP1, IR-A was the predominant isoform in breast carcinoma tissues, and IR isoforms displayed a molecular subtype-specific distribution among the breast cancer cell lines, where IR-A was the highest in the luminal cancer cells and the lowest in TNBC. Manipulation of the expression of CUGBP1 in breast cancer cell lines changed IR-A levels accordingly, which is consistent with the reports showing that CUGBP1 in different cells determined the degree of exon inclusion (14,25). We also found that the increased IR-A:IR-B ratio was largely attributable to an increase in IR-A regulated by CUGBP1, whereas, in a previous study, it was caused by the decrease of IR-B (26). The discrepancy may reflect differences in the experimental approaches (tumor samples versus molecular subtypes of cancer cell lines).
Several studies reported that CUGBP1 is overexpressed in glioma, oral squamous cell carcinomas and hepatocellular carcinoma (17,19,27), acting as an oncogenic target involved in cell proliferation, growth and cell cycle. According to David et al. (28), the expression of CUPBP1 mRNA is not correlated with breast cancer invasiveness, where TCGA panel was also used. The authors concluded that its prognostic value in breast cancer resides only in combination with ELAVL1 mRNA levels. In the present study, CUGBP1 overexpression activated the insulin-signaling pathway through phosphorylation of Akt on the site S473 and, subsequently, promoted cell viability, colony formation and invasion, whereas the downregulation of CUGBP1 had opposite impact. Overactivation of Akt by phosphorylation on either or both residue T308 and S473 is of great pathological and therapeutic interests in multiple malignancies, including breast carcinomas, as it confers cancer cell survival, proliferation and chemo-resistance (29). Although phosphorylation on both sites is essential for a full Akt activity and they concomitantly occur in some breast cancers, Akt T308 phosphorylation has a much higher frequency in breast malignancies than on S473 (30). While T308 is phosphorylated by PDK1, the main kinase for Akt S473 is the mechanistic target of rapamycin complex 2 (mTORC2), which is PI3K-dependently stimulated by growth-promoting factors such as insulin (31). Whether and how the different IR conformations fine-tune the Akt phosphorylation and the subsequent network are still elusive. However, CUGBP1 certainly plays a key role at least by controlling IR isoform splicing (Figure 6). Insulin exhibits higher affinity to IR-A than to IR-B (32,33); on the other hand, the hybrid receptor IR-A/IGF1R (HR-A) has a higher affinity for insulin, IGF-1 and IGF-2 than IR-B/IGF1R (HR-B) hybrid (34).
In addition to its regulation in RNA splicing and decay, CUGBP1 also exhibits pleiotropic roles in post-transcriptional events that are closely linked to the etiology and progression of many tumors, including breast cancer. The ratio of the LIP and LAP isoform translations of the CCAAT/enhancer-binding protein beta (C/EBP β), a leucine-zipper transcription factor associated with breast cancer aggressiveness, was found to be increased along with CUGBP1 overexpression (Supplementary Figure S4A and B, available at Carcinogenesis Online). Interestingly, as part of a large nuclear complex, C/EBP β regulates and is regulated by insulin signaling, where differential IR transcription is involved (35).
However, there are some important outstanding issues requiring clarification by further research. First, it is not clear if other regulations of CUGBP1 work in tandem with or counteract IR isoform splicing to affect cancer aggressiveness. For instance, it is known that CUGBP1 mediates mRNA degradation of cancer-promoting genes such as c-jun and junB (36,37), which is demonstrated by the modulation of CUGBP1 with siRNA or plasmid transfection into breast cancer cell lines (Supplementary Figure S4C and D, available at Carcinogenesis Online). Second, a more compelling chain of causation threading CUGBP1 regulation, IR-A expression and cancerous behavior would necessitate a manipulation of IR-A level to reverse the effects of CUGBP1, as a very wide range of factors including EMT and the apoptosis machinery is under control of CUGBP1 in cancer. Third, apart from the elevated response to insulin, the upregulated IR-A might potentiate higher cellular responsiveness to other mitogens and growth factors too, which might contribute a considerable part to the tumor aggressiveness in breast cancer. In particular, the distinction between IR-A and IR-B is the high affinity of IR-A to IGF-II. It still awaits clarification whether the upregulated IR-A by CUGBP1 alters its affinity to IGFs and the functions of the hybrid receptor IR/IGF1R. Another deduction from the results is that the higher IR-A ratio in luminal breast cancer patients would potentially amplify the detrimental complications in patients with concurrent hyperinsulinaemia.
The IR-A:IR-B ratio derived from the alternative exon 11 splicing of INSR presents as an adjustable switch between mitogenic- and metabolic-dominant effects of the IR. To date, antibodies have not been developed that can distinguish IR-A and IR-B isoforms; thus, only the mRNA levels of each isoform can be decided by PCR. Across the cell lines covering luminal HER2+ and TNBC molecular subtypes, the highest IR-A level was detected in the ERα+ breast cancer cell lines (Figure 2G–I), which is consistent with the previous evidence that IGF system cross talks with ERα and contributes to the regulation of ERα-positive breast cancer: the IGF pathway stimulates malignant behavior of breast cancer cells by activating (mTOR)/S6 kinase 1 (S6K1) axis and affects ERα-regulated genes, whereas ERα can increase the expression of both IGF1R and IRS-1 in a feed-forward way (38–40). Although higher IR-A confers more aggressiveness of the cancer (LumA/B), some breast cancer types (Her+, TNBC) might escape the growth dependency on IR/ERa regulation loop that probably makes the splicing ratio irrelevant.
Despite more questions raised than answered, we conclude that CUGBP1 regulates INSR splicing to favor IR-A expression and impact intrinsic oncogenic signaling through the insulin-signaling pathway in breast cancer. Co-targeting CUGBP1 and IR-A receptors might be clinically beneficial. Besides, understanding the coordination between varying IR-A:IR-B ratios and other signal receptors such as IGF1R and ERα is the key to unveiling the breast cancer evolution.
Supplementary material
Supplementary data are available at Carcinogenesis online.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgement
We are grateful to all patients for enrolling in this study.
Funding: This study was funded by the Nature Science Foundation of China (81471000, 31871163 to Y.W., 81872156 to M.L.) and the Ministry of Science and Technology (2014DFA32120 to Y.W. and the National Cancer Institute (NCI) grant (R01CA128799 to D.LeR.).
Conflict of Interest Statement: The authors declare no conflict of interest.
Glossary
Abbreviations
- cDNA
complementary DNA
- ER
estrogen receptor
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- IGF1R
type I insulin-like growth factor receptor
- INSR
insulin receptor gene
- IR
insulin receptor
- mRNA
messenger RNA
- PBS
phosphate-buffered saline
- PSI
percent spliced-in
- RT–PCR
reverse transcriptase–PCR
- siRNA
small interfering RNA
- TCGA
the Cancer Genome Atlas
- TNBC
triple negative breast cancer
REFERENCE
- 1. Belfiore A., et al. (2009) Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev., 30, 586–623. [DOI] [PubMed] [Google Scholar]
- 2. Vella V., et al. (2001) The IGF system in thyroid cancer: new concepts. Mol. Pathol., 54, 121–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vigneri R., et al. (2016) Insulin, insulin receptors, and cancer. J Endocrinol Invest., 39, 1365–1376. [DOI] [PubMed] [Google Scholar]
- 4. Papa V., et al. (1990) Elevated insulin receptor content in human breast cancer. J. Clin. Invest., 86, 1503–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yee D. (2012) Insulin-like growth factor receptor inhibitors: baby or the bathwater? J. Natl. Cancer Inst., 104, 975–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang Y. et al. (2012) Targeting insulin and insulin-like growth factor signaling in breast cancer. J. Mammary Gland Biol. Neoplasia, 17, 251–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Frasca F., et al. (1999) Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell. Biol., 19, 3278–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sciacca L., et al. (2003) Signaling differences from the A and B isoforms of the insulin receptor (IR) in 32D cells in the presence or absence of IR substrate-1. Endocrinology, 144, 2650–2658. [DOI] [PubMed] [Google Scholar]
- 9. Sciacca L., et al. (2002) In IGF-I receptor-deficient leiomyosarcoma cells autocrine IGF-II induces cell invasion and protection from apoptosis via the insulin receptor isoform A. Oncogene, 21, 8240–8250. [DOI] [PubMed] [Google Scholar]
- 10. Vella V., et al. (2002) A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. J. Clin. Endocrinol. Metab., 87, 245–254. [DOI] [PubMed] [Google Scholar]
- 11. Kalli K.R. et al. (2002) Functional insulin receptors on human epithelial ovarian carcinoma cells: implications for IGF-II mitogenic signaling. Endocrinology, 143, 3259–3267. [DOI] [PubMed] [Google Scholar]
- 12. Dasgupta T. et al. (2012) The importance of CELF control: molecular and biological roles of the CUG-BP, Elav-like family of RNA-binding proteins. Wiley Interdiscip. Rev. RNA, 3, 104–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paul S., et al. (2006) Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J., 25, 4271–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sen S., et al. (2009) SRp20 and CUG-BP1 modulate insulin receptor exon 11 alternative splicing. Mol. Cell Biol., 29, 871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuyumcu-Martinez N.M., et al. (2007) Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell, 28, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jones K., et al. (2012) The role of CUGBP1 in age-dependent changes of liver functions. Ageing Res. Rev., 11, 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xia L., et al. (2015) CELF1 is up-regulated in glioma and promotes glioma cell proliferation by suppression of CDKN1B. Int. J. Biol. Sci., 11, 1314–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang X., et al. (2016) CUG-binding protein 1 (CUGBP1) expression and prognosis of brain metastases from non-small cell lung cancer. Thorac. Cancer, 7, 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Talwar S. et al. (2013) Overexpression of RNA-binding protein CELF1 prevents apoptosis and destabilizes pro-apoptotic mRNAs in oral cancer cells. RNA Biol., 10, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Muller P.Y. et al. (2002) Processing of gene expression data generated by quantitative real-time RT-PCR. Biotechniques, 32, 1372–1374, 1376,, 1378–1379. [PubMed] [Google Scholar]
- 21. Rowzee A.M. et al. (2009) Insulin-like growth factor type 1 receptor and insulin receptor isoform expression and signaling in mammary epithelial cells. Endocrinology, 150, 3611–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ma X.J. et al. (2009) Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res., 11, R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cancer Genome Atlas Network. (2012) Comprehensive molecular portraits of human breast tumours. Nature, 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rostoker R., et al. (2015) Highly specific role of the insulin receptor in breast cancer progression. Endocr. Relat. Cancer, 22, 145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sen S., et al. (2010) Muscleblind-like 1 (Mbnl1) promotes insulin receptor exon 11 inclusion via binding to a downstream evolutionarily conserved intronic enhancer. J. Biol. Chem., 285, 25426–25437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang J., et al. (2011) Altered expression of insulin receptor isoforms in breast cancer. PLoS One, 6, e26177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim J.H., et al. (2017) Inhibition of CUG-binding protein 1 and activation of caspases are critically involved in piperazine derivative BK10007S induced apoptosis in hepatocellular carcinoma cells. PLoS One, 12, e0186490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Géraldine D. et al. (2018) The RNA-binding proteins CELF1 and ELAVL1 cooperatively control alternative splicing. bioRxiv. doi:10.1101/373704. [DOI] [PubMed]
- 29. Yang Z.Y. et al. (2015) The prognostic value of phosphorylated Akt in breast cancer: a systematic review. Sci. Rep., 5, 7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lin H.J. et al. (2005) Elevated phosphorylation and activation of PDK-1/AKT pathway in human breast cancer. Br. J. Cancer, 93, 1372–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Manning B.D. et al. (2017) AKT/PKB signaling: navigating the network. Cell, 169, 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yamaguchi Y., et al. (1991) Functional properties of two naturally occurring isoforms of the human insulin receptor in Chinese hamster ovary cells. Endocrinology, 129, 2058–2066. [DOI] [PubMed] [Google Scholar]
- 33. Yamaguchi Y., et al. (1993) Ligand-binding properties of the two isoforms of the human insulin receptor. Endocrinology, 132, 1132–1138. [DOI] [PubMed] [Google Scholar]
- 34. Pandini G., et al. (2002) Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J. Biol. Chem., 277, 39684–39695. [DOI] [PubMed] [Google Scholar]
- 35. Zahnow C.A. (2009) CCAAT/enhancer-binding protein beta: its role in breast cancer and associations with receptor tyrosine kinases. Expert Rev. Mol. Med., 11, e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vlasova I.A., et al. (2008) Conserved GU-rich elements mediate mRNA decay by binding to CUG-binding protein 1. Mol. Cell, 29, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arnal-Estapé A., et al. (2010) HER2 silences tumor suppression in breast cancer cells by switching expression of C/EBPß isoforms. Cancer Res., 70, 9927–9936. [DOI] [PubMed] [Google Scholar]
- 38. Fagan D.H. et al. (2008) Crosstalk between IGF1R and estrogen receptor signaling in breast cancer. J. Mammary Gland Biol. Neoplasia, 13, 423–429. [DOI] [PubMed] [Google Scholar]
- 39. Becker M.A., et al. (2011). The IGF pathway regulates ERalpha through a S6K1-dependent mechanism in breast cancer cells. Mol. Endocrinol., 25, 516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Harrington S.C., et al. (2012) Quantifying insulin receptor isoform expression in FFPE breast tumors. Growth Horm. IGF Res., 22, 108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.