

Keywords: 3-methyladenine, apoptosis, atorvastatin, autophagy, early brain injury, LC3, neuroprotection, rapamycin, subarachnoid hemorrhage
Abstract
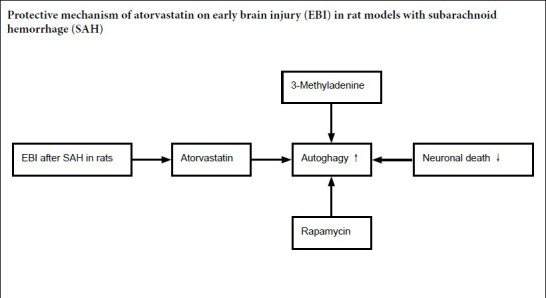
Atorvastatin has been shown to reduce early brain edema and neuronal death after subarachnoid hemorrhage, but its mechanism is not clear. In this study, rat models of subarachnoid hemorrhage were established by autologous blood injection in the cisterna magna. Rat models were intragastrically administered 20 mg/kg atorvastatin 24 hours before subarachnoid hemorrhage, 12 and 36 hours after subarachnoid hemorrhage. Compared with the controls, atorvastatin treatment demonstrated that at 72 hours after subarachnoid hemorrhage, neurological function had clearly improved; brain edema was remarkably relieved; cell apoptosis was markedly reduced in the cerebral cortex of rats; the number of autophagy-related protein Beclin-1-positive cells and the expression levels of Beclin-1 and LC3 were increased compared with subarachnoid hemorrhage only. The ultrastructural damage of neurons in the temporal lobe was also noticeably alleviated. The similarities between the effects of atorvastatin and rapamycin were seen in all the measured outcomes of subarachnoid hemorrhage. However, these were contrary to the results of 3-methyladenine injection, which inhibits the signaling pathway of autophagy. These findings indicate that atorvastatin plays an early neuroprotective role in subarachnoid hemorrhage by activating autophagy. The experimental protocol was approved by the Animal Ethics Committee of Anhui Medical University, China (904 Hospital of Joint Logistic Support Force of PLA; approval No. YXLL-2017-09) on February 22, 2017.
Introduction
Subarachnoid hemorrhage (SAH) occurs frequently in cerebral vascular disease and has high mortality and morbidity with poor outcomes, particularly among hypertension patients. Western countries report an incidence of 6.2–10 per 100,000 (Korja et al., 2016; Mackey et al., 2016; Etminan et al., 2019). Cerebral vasospasm, early brain injury (EBI) and delayed cerebral infarction may lead to neurological dysfunction, especially cerebral vasospasm. However, a recent review showed that vasospasm-targeted medication can remarkably reduce cerebral vasospasm but does not improve outcomes after SAH (Budohoski et al., 2013; Xie et al., 2019). Recent clinical trials also confirmed that medications that ameliorate cerebral vasospasm have no effect on outcomes after SAH (Laskowitz and Kolls, 2010; Macdonald et al., 2011). Our previous study reported that EBI was a very important risk of mortality after SAH, and clinical studies showed that most patient deaths occur within 46 hours (Schievink et al., 1995; Le Roux and Winn, 1998; Sehba et al., 2012; Chen et al., 2019). The possible mechanisms underlying EBI include autophagy, apoptosis and direct neuronal death (Cahill and Zhang, 2009). However, mechanisms to ameliorate EBI remain incompletely understood (Liu et al., 2014).
Apoptosis is a very important mechanism and has been reported after SAH, and our previous study confirmed apoptosis mechanism plays a vital element during EBI (Chen et al., 2016, 2019). However, our previous preliminary experiment found that autophagic changes may also be associated with cell death and apoptosis. Other recent studies confirmed autophagy was linked to neuroprotective effects in several experimental settings of EBI (Liu et al., 2014; Galluzzi et al., 2016; Wu et al., 2016; Cao et al., 2017). Autophagy is an important and well-known mechanism of cell death through lysosomal degradation. In a review, Galluzzi et al. (2016) noted that autophagy played a crucial role in spinal cord injury, SAH and EBI.
Statins, cholesterol-lowering drugs, are inhibitors of HMG-CoA reductase. New clinical and basic research studies have found other pharmacological effects (Tseng et al., 2005; Tousoulis et al., 2006; Luzak et al., 2011; Sehba et al., 2011), especially anti cerebral vasospasm effects (Tseng et al., 2005; Chen et al., 2016). Our previous study also demonstrated that atorvastatin (Ato) can reduce early brain edema and neuronal death in experiment SAH model, but the mechanism of this effect remains incompletely understood (Chen et al., 2016). Han et al. (2018) reported that Ato attenuated the inflammatory response after atherosclerosis by activating autophagy, possibly by regulating the AKT/mTOR molecular pathway. Toepfer et al. (2011) also reported that Ato induced autophagy by activating LC3 transcription through the mTOR autophagic signaling pathway in cancer cells. Further investigations are needed to determine whether Ato regulates EBI by its effect on autophagy activation, EBI inhibition, neuroprotection or signaling pathways.
Therefore, we hope to confirm the neuroprotection by Ato after SAH using a rat SAH model. This study focuses on the role of autophagy by investigating whether the autophagic pathway is activated and to identify the neuroprotective mechanism of Ato in EBI following SAH.
Materials and Methods
Animals
All animal studies were approved by the Animal Ethics Committee of Anhui Medical University, China (904 Hospital of Joint Logistic Support Force of PLA; approval No. YXLL-2017-09) on February 22, 2017. All experiments conformed to the relevant regulations of animal protection. All 90 adult male specific-pathogen-free Sprague-Dawley rats aged 4–6 months and weighing 300–350 g were purchased from Nantong University Laboratories, China (SCXK(Su)2017-0001). All rats were maintained under a natural day and night cycle with free access to food and water at a comfortable temperature. All rats received general anesthesia before any experimental procedures.
Experimental design
All rats were assigned randomly to five groups: sham (n = 18), SAH (n = 18), SAH + Ato (n = 18), SAH + 3-methyladenine (3-MA) (n = 18) and SAH + rapamycin (Rap) (n = 18). Ato (Pfizer, Wuxi, China) 20 mg/kg/day was orally administered for 3 days (24 hours before SAH, 12 hours after SAH and 36 hours after SAH). 3-MA (Sigma-Aldrich, St. Louis, MO, USA) and Rap (Tocris Biosciences, Bristol, UK) were administered via intracerebral ventricular injection 30 minutes before SAH onset in their respective groups. All rats were sacrificed 72 hours after SAH. Shortly before the rats were sacrificed, each rat had their final assessment of their behavioral scores and intracranial pressure (ICP). Six rats from each group were used for terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining. Six rats were evaluated for brain edema. The remaining six rats were used for electron microscopy to evaluate autophagosomes, double immunohistochemistry and western blot assay.
SAH models
SAH models were established by rat autologous-blood injection as our previous studies (Chen et al., 2016, 2018). Briefly, rats were intraperitoneally anesthetized with 10% chloral hydrate (0.25 mg/kg) and then positioned in a stereotaxic frame (Stoelting, Wood Dale, IL, USA) under an animal respirator (Harvard Apparatus, Holliston, MA, USA). A trocar was stereoscopically inserted into the cisterna magna. Then 0.3 mL of nonheparinized fresh autologous artery blood from the femoral artery was injected into the cisterna magna for approximately 20 seconds. In the sham group 0.3 mL of saline was injected instead. To avoid cerebrospinal fluid leakage and bleeding from the trocar, bone wax was used as a plug before inserting the trocar. All rats were maintained in a 45° head-down body posture for approximately 30 minutes. After the operation, all rats received 5 mL of 0.9% saline, to make up for lost fluids, and each returned to a single, comfortable cage. The vital signs were monitored; ICP and behavior scores were recorded throughout the experiment.
Intracerebral ventricular injection
An autophagy inhibitor, 3-MA, was used to block the autophagy pathway and Rap was chosen to activate the autophagy pathway (He et al., 2008; Toepfer et al., 2011; Han et al., 2018). After anesthesia, a 10-μL syringe was used to insert the 3-MA or Rap into the left lateral ventricle guided by the rat stereotaxic frame. The puncture point was located 1.5 mm posterior to, 3.5 mm below the horizontal plane of bregma and 0.8 mm lateral to bregma. 3-MA (400 nmol) or Rap (20 pmol) was injected by a microinfusion pump (1 μL/min) 30 minutes before SAH onset. The drug management was based on previous studies (He et al., 2008; Jing et al., 2012).
Intracranial pressure monitoring
ICP was measured using a Codman microsensor basic kit (Probe diameter, 1.2 mm, Codman, New Brunswick, NJ, USA) that was implanted at the intraventricular injection point 30 minutes before model establishment. After 3-MA or Rap injection, the dura was opened, and the Codman ICP probe was inserted into the subdural space. Bone wax (Codman) was used to prevent leakage of cerebrospinal fluid.
Neurological function assessment
The severity of early brain injury was evaluated by neurological function at 72 hours after SAH using our previously described neurological grading system (Cao et al., 2017; Chen et al., 2019). The scoring system consisted of six tests and specific standards are shown in Additional Table 1, and the neurological score ranged from 3 to 18. All rats from every group received behavioral assessment (n = 18), and a higher score represented a better neurological function.
Additional Table 1.
Neurological score
| Test | Score | |||
|---|---|---|---|---|
| 0 | 1 | 2 | 3 | |
| Spontaneous activity (in cage for 5 min) | No movement | Barely moves | Moves but does not approach at least three sides of cage | Moves and approaches at least three sides of cage |
| Symmetry of movements (four limbs) | Left side: no movement | Left side: slight movement | Left side: moves slowly | Both sides: move symmetrically |
| Symmetry of forelimbs (outstretching while held by tail) | Left side: no movement, no outreaching | Left side: slight movement to outreach | Left side: moves and outreaches less than right side | Symmetrical outreach |
| Climbing wall of wire cage | Left side: no movement, no outreaching | Fails to climb | Left side is weak | Normal climbing |
| Reaction to touch on either side of trunk | Left side: no movement, no outreaching | No response on left side | Weak response on left side | Symmetrical response |
| Response to vibrissae touch | Left side: no movement, no outreaching | No response on left side | Weak response on left side | Symmetrical response |
Brain water content
The severity of brain edema was evaluated by brain water content, which was determined by the standard wet-dry method as in our previous studies (Chen et al., 2016, 2018). The rats were sacrificed 72 hours after SAH, and the entire brain was harvested and weighed directly (wet weight). The brains of six rats from each group were dehydrated at 105°C for 24 hours to obtain the dry weight. The percentage of brain water content was equal to (wet weight – dry weight)/wet weight × 100.
TUNEL staining
The severity of cell apoptosis in the cerebral cortex was evaluated by TUNEL assay according to the manufacturer’s instructions and our previous standard methods (Chen et al., 2016, 2019). Rats were sacrificed with an overdose of sodium pentobarbital (100 mg/kg). The sample of the cerebral cortex on each slide was treated with TUNEL staining mixture (Roche Inc., Basel, Switzerland) and incubated in a humidified dark chamber at 37°C for 60 minutes. DAPI substrate was added in the dark for 5 minutes at 22°C. All processes were monitored by fluorescence microscopy (Olympus, Tokyo, Japan). The procedure was performed according to the manufacturer’s instructions with the TUNEL staining kit. A negative control (without the TUNEL reaction mixture) was used. The apoptotic index (%) was the ratio of the number of TUNEL-positive cells/total number of cells × 100. The cell count was confirmed in four randomly high-power fields, and the data were averaged.
Double immunofluorescence staining
Double immunofluorescence staining for Beclin-1 or LC-3 (autophagy related proteins) with NeuN (neuronal staining specific marker) was performed to count cells and neurons undergoing autophagy (Toepfer et al., 2011; Han et al., 2018). The specific method has been described in previous studies (Liu et al., 2014; Cao et al., 2017). Briefly, the rat brain samples were fixed in 4% paraformaldehyde for 24 hours at 4°C, then dehydrated in a 30% sucrose solution. Brain samples were sectioned at a thickness of 10 µm. Double immunofluorescence staining for Beclin-1 with NeuN was similar among the groups. The primary antibodies were rabbit anti NeuN polyclonal antibody (1:200, rabbit polyclonal, ab128886; Abcam, Cambridge, UK), rabbit anti LC-3 polyclonal antibody (1:300, ab48394; Abcam) and rabbit anti Beclin-1 polyclonal antibody (1:500, ab62557; Abcam), and were diluted in PBS overnight at 4°C. The sections were washed with PBS and incubated with goat anti-rabbit IgG secondary monoclonal antibody (1:500; Beyotime, Shanghai, China) at room temperature for 1 hour. The sections were incubated and covered with DAPI, rinsed again and covered with glycerol. The results were observed and analyzed by fluorescence microscopy (Leica Microsystems, Wetzlar, Germany).
Western blot assay
Cerebral cortex samples were collected, dissolved and proteins separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis in 10% polyacrylamide gels. Proteins were transferred onto Immobilon nitrocellulose membranes. The membranes were incubated with primary antibodies for 1 hour at room temperature and washed with Tris-buffered saline + 5% Tween-20 (TBST). The following primary antibodies were used: a rabbit polyclonal antibody for anti-LC-3 (1:2000, ab48394; Abcam), a rabbit polyclonal antibody for anti caspase-3 (cell apoptosis marker; 1:300, ab4051; Abcam), and a rabbit polyclonal antibody for anti Beclin-1 (1:3000, ab62557; Abcam). After washing in TBST again, the blots were incubated with horseradish peroxidase-conjugated goat anti-rabbit secondary antibodies (1:1000; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) for 1.5 hours at room temperature. Following two rinses and four washes with TBST, the optical density results were quantified by Quantity 4.5 Software (Bio-Rad Laboratories, Hercules, CA, USA) and ImageJ (NIH, Bethesda, MD, USA). GAPDH (1:1000, ab8245; Abcam) was used as internal standards.
Transmission electron microscopy
After rats were sacrificed, the 1 mm3 temporal lobe samples were collected and fixed in 2.5% glutaraldehyde and 1% osmium tetroxide at 4°C. After four or five washes in distilled water, the samples were dehydrated through a graded ethanol series at a high concentration, and then embedded in an epoxy resin. Ultrathin sections were made and stained with uranyl acetate and Reynold’s lead citrate. The autophagosome and organelles were observed under a transmission electron microscope (JSM-IT300LV; JEOL, Tokyo, Japan).
Statistical analysis
The data are presented as the mean ± SD. SPSS 14.0 (SPSS, Chicago, IL, USA) and GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA) were used for the statistical analysis. The mortality between the experimental groups was analyzed by Fisher’s exact test. Multiple-group comparisons were tested by one-way analysis of variance. P < 0.05 was considred statistically significant.
Results
General health and mortality of SAH rats
Blood gas levels, mean arterial blood pressure and blood glucose (90–120 mg/dL) were all within normal ranges before the operation. Mortality was 38.9% (7/18) in the SAH group, 27.8% (5/18) in the SAH + Ato group, 33.3% (6/18) in the SAH + Rap group, and 38.9% (7/18) in SAH + 3-MA group. No rats died in the sham group (0/18).
Ato improves neurological function of SAH rats
The neurological scores were recorded 72 hours after SAH (Figure 1). The neurological scores were significantly lower in the SAH group compared with the sham group (n = 18, P < 0.05). Ato significantly improved the neurological scores of rats in the SAH + Ato group compared with the SAH group (n = 18, P < 0.05). Similarly, the scores were higher in the SAH + Rap group than in the SAH group (n = 18, P < 0.05). However, the scores were lower in the SAH + 3-MA group than in the SAH group (n = 18, P < 0.05). The scores were not significantly different between the SAH + Rap group and the SAH + Ato group (n = 18, P > 0.05; Figure 1).
Figure 1.
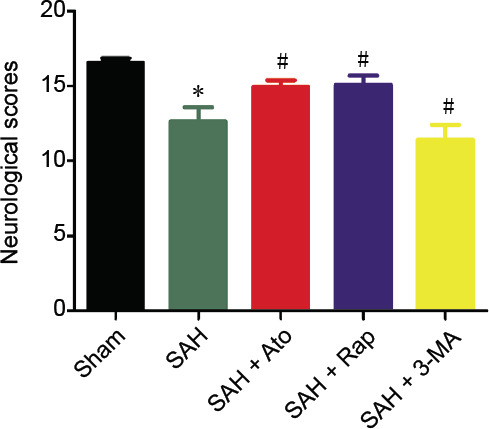
Effect of atorvastatin on neurological function of subarachnoid hemorrhaged rats.
The neurological function was assessed using the neurological grading system (Cao et al., 2017; Chen et al., 2019). Data are expressed as the mean ± SD (n = 18). *P < 0.01, vs. sham group; #P < 0.05, vs. SAH group (one-way analysis of variance). 3-MA: 3-Methyladenine; Ato: atorvastatin; Rap: rapamycin; SAH: subarachnoid hemorrhage.
Ato decreases brain edema of SAH rats
Brain edema was evaluated by brain water content after SAH. There was highly significant brain edema after SAH, but this was significantly alleviated in the SAH + Ato and SAH + Rap groups compared with the SAH group (n = 6, P < 0.01). On the other hand, pretreatment with 3-MA further aggravated the effects of SAH in the SAH + 3-MA group compared with the SAH group (n = 6, P < 0.01; Figure 2).
Figure 2.
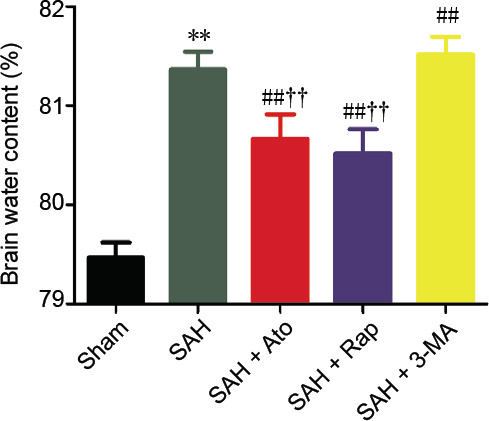
Effect of atorvastatin on brain edema of subarachnoid hemorrhage rats.
Data are expressed as the mean ± SD (n = 6). **P < 0.01, vs. sham group; ##P < 0.01, vs. SAH group; ††P < 0.01, vs. SAH + 3-MA group (one-way analysis of variance). 3-MA: 3-Methyladenine; Ato: atorvastatin; Rap: rapamycin; SAH: subarachnoid hemorrhage.
Ato decreases ICP of SAH rats
All rats received regular monitoring of ICP. After all operations, the ICP of all rats immediately increased from 8 to 80 mmHg and fell back to 38 mmHg within 5 minutes (Figure 3). After SAH induction, rats had a higher ICP at 15 minutes than the sham group, which had returned to normal, and the ICPs were relatively stable for 1 day. However, ICPs were higher in the SAH groups and progressively increased compared with the sham group at 24, 48 and 72 hours (n = 18, P < 0.01). More importantly, after Ato and Rap intervention, ICP did not increase as much compared with the SAH groups (n = 6, P < 0.01; Figure 3).
Figure 3.
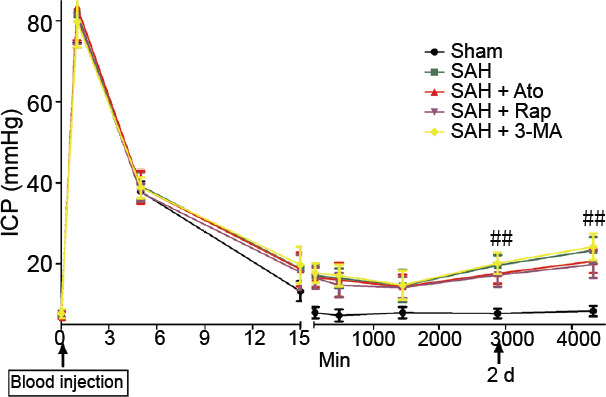
Intracranial pressure over a 3-day period after subarachnoid hemorrhage.
Data are expressed as the mean ± SD (n = 18). ##P < 0.01, vs. SAH group (one-way analysis of variance). 3-MA: 3-Methyladenine; Ato: atorvastatin; ICP: intracranial pressure; Rap: rapamycin; SAH: subarachnoid hemorrhage.
Ato decreases cell apoptosis in the cerebral cortex of SAH rats
TUNEL staining showed that almost no cell apoptosis was detected in sham animals. There was significantly more cell apoptosis in the cerebral cortex of rats 72 hours after SAH (Figure 4A and B). Treatment with Ato and Rap dramatically reduced the amount of cell apoptosis compared with the SAH group (n = 6, P < 0.01), and the neuroprotective effects were similar in the SAH + Ato and SAH + Rap groups. However, 3-MA pretreatment significantly increased cell apoptosis compared with the SAH group (n = 6, P < 0.01).
Figure 4.

TUNEL staining of the cerebral cortex after subarachnoid hemorrhage.
(A) TUNEL staining: cell apoptosis (arrows) reduced in the SAH + Ato and SAH + Rap groups. Cell apoptosis aggravated in the SAH + 3-MA group. Original magnification, 400×. (B) Results of quantitative analysis. The data are presented as the mean ± SD (n = 6). **P < 0.01, vs. sham group; #P < 0.05, ##P < 0.01, vs. SAH group (one-way analysis of variance). 3-MA: 3-Methyladenine; Ato: atorvastatin; Rap: rapamycin; SAH: subarachnoid hemorrhage; TUNEL: terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling.
Ato decreases Beclin-1-positive cells in the hippocampus of SAH rats
Double-labeled immunofluorescent staining showed that Beclin-1-positive cells were widespread among the hippocampus after SAH induction. Additionally, more Beclin-1 positive cells were observed after Ato and autophagy activator-Rap treatment, but there were fewer NeuN-positive cells after 3-MA administration (Figure 5).
Figure 5.
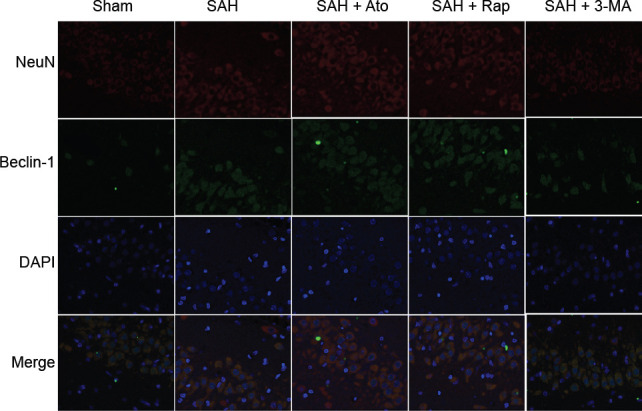
Effect of Ato on Beclin-1 immunoreactivity in the hippocampus of subarachnoid hemorrhage rats (double immunofluorescent staining).
Immunofluorescent staining of Beclin-1/NeuN/DAPI in the hippocampus exhibited enhanced Beclin-1 immunoreactivity in the SAH + Ato treated group and the effect was very similar to that in the SAH + Rap group; Beclin-1 immunoreactivity decreased after pretreatment with 3-MA. Fluorescence colors: NeuN (red marked by Alexa Fluor 647), Beclin-1 (green marked by Alexa Fluor 488), and DAPI (blue). 3-MA: 3-Methyladenine; Ato: atorvastatin; DAPI: 4′,6-diamidino-2-phenylindole; Rap: rapamycin; SAH: subarachnoid hemorrhage.
Effect of Ato on cerebral cortex autophagy-related proteins and caspase-3 expression of SAH rats
Western blot (Figure 6A and B) assays showed that the expression levels of LC3 and Beclin-1 were significantly increased after SAH induction compared with the sham group (n = 6, P < 0.05). The expression levels of Beclin-1 and LC3 were increased more after Ato and Rap treatments compared with the SAH only group (n = 6, P < 0.05). There was no difference in expression levels of Beclin-1 and LC3 between SAH + Rap and SAH + Ato groups (n = 6, P > 0.05). However, there were contrary effects after pretreatment with 3-MA compared with the SAH group (n = 6, P < 0.05) Figure 6C and D).
Figure 6.
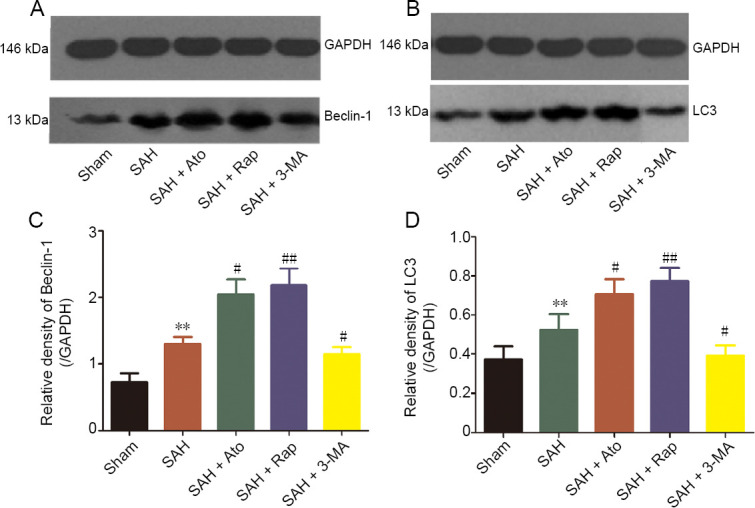
Autophagy-related protein expression levels in the cerebral cortex.
(A, B) Levels of Beclin-1 (A) and LC3 (B). (C, D) Results of quantitative analysis. The densities of the Beclin-1 (C) and LC3 (D) protein bands were analyzed and normalized to GAPDH. Data are expressed as the mean ± SD (n = 6; one-way analysis of variance). **P < 0.01, vs. sham group; #P < 0.05, ##P < 0.01, vs. SAH group. 3-MA: 3-Methyladenine; Ato: atorvastatin; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; Rap: rapamycin; SAH: subarachnoid hemorrhage.
The levels of caspase-3 (Figure 7) were upregulated significantly after SAH compared with the sham group (n = 6, P < 0.01). Ato and Rap treatment significantly downregulated the levels of caspase-3 compared with the SAH group (n = 6, P < 0.01). However, caspase-3 was enhanced after pretreatment with the autophagy inhibitor 3-MA compared with SAH group (n = 6, P < 0.05) (Figure 7B).
Figure 7.
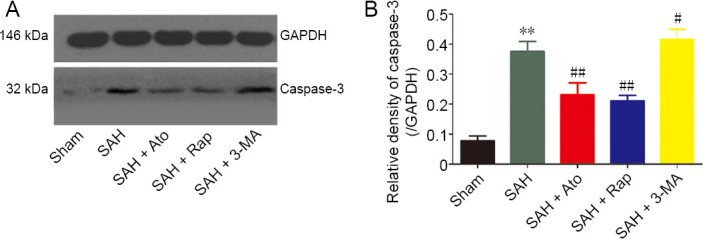
Caspase-3 protein levels in the cerebral cortex.
(A) Caspase-3 levels. (B) Relative density of caspase-3: The densities of the protein bands were analyzed and normalized to GAPDH. Data are expressed as the mean ± SD (n = 6; one-way analysis of variance). **P < 0.01, vs. sham group; #P < 0.05, ##P < 0.01, vs. SAH group. 3-MA: 3-Methyladenine; Ato: atorvastatin; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; Rap: rapamycin; SAH: subarachnoid hemorrhage.
Effect of Ato on the ultrastructure of autophagosome and organelles in the temporal lobe of SAH rats
Transmission electron microscopy showed that autophagosome and organelles in neurons, all mitochondria, endoplasmic reticulum, lysosomes and nuclei, were normal and not swollen in the sham group (Figure 8A). However, diverse morphological changes, such as swollen mitochondria, were observed in the SAH group (Figure 8B–E). Some neurons displayed severe damage, as indicated by multiple vacuole-related structures containing autophagosomes (red arrow; Figure 8B–E) and electron-dense materials (blue arrow; Figure 8B–E). Ato treatment alleviated these morphological changes in mitochondria, as did Rap, however, 3-MA aggravated the SAH damage.
Figure 8.

Effect of Ato on the ultrastructure of autophagosome and organelles in the temporal lobe of subarachnoid hemorrhage rats (transmission electron microscopy).
(A) The structure of neurons was normal. All mitochondria, endoplasmic reticulum, lysosomes, and nuclei showed no pathological changes in the sham group. (B) However, diverse morphological changes were found after SAH, such as swollen mitochondria (triangle) and broken endoplasmic reticula (five-pointed star). Multiple double-membrane autophagosomes (red arrow) and electron-dense materials (blue arrow) were seen in the SAH-damaged neurons. (C) Atorvastatin treatment lessened the morphological changes in the mitochondria. (D) Rap treatment presented similar changes. (E) 3-MA aggravated the effects of SAH. Original magnification, 15,000×. 3-MA: 3-Methyladenine; Ato: atorvastatin; Rap: rapamycin; SAH: subarachnoid hemorrhage.
Discussion
Our study found that autophagy was activated after SAH in neurons, as revealed by autophagy-related proteins levels, double immunohistochemistry, indicating elevated LC3 and NeuN expression levels in injured brains, and our estimation of the number of autophagosomes by transmission electron microscopy. The SAH induced deterioration in neurological function, neuronal apoptosis and brain edema were further worsened in rats treated with 3-MA. However, neurological function and SAH-induced EBI improved after autophagy was activated by Rap. Notably, Ato treatment gave the same results after SAH. This suggests that the neuroprotective role of Ato in SAH is similar to that of Rap, acting as a promoter of autophagy in neurons. However, the largest advantage that Ato has a treatment for EBI is that it can be administered orally whereas Rap is given intraventricularly. This study also clarified that autophagy was the key to the neuroprotective effect of Ato treatment in EBI after SAH.
Autophagy regulates the turnover of cellular constituents to ensure the removal and recycling of toxins and is very important in cell homeostasis. The role of autophagy has been confirmed in many central nervous system diseases, including acute brain injury, intracerebral hemorrhage, SAH and Huntington’s disease (Nixon, 2006; He et al., 2008; Lee et al., 2009; Smith et al., 2011; Chen et al., 2019). Lee et al. (2009) and Zhao et al. (2013) reported that the autophagy mechanism occurred and had an important role in controlling cell death in EBI caused by experimental SAH. Rap can induce autophagy and decrease expression levels of Bax and caspase-3, thereby inhibiting neuronal cell death and improving EBI (Jing et al., 2012). However, 3-MA inhibits the autophagy pathway, thereby increasing neuronal cell death and aggravating EBI (Jing et al., 2012; Zhao et al., 2013). Therefore, autophagy activation can decrease neuronal apoptosis and ameliorate EBI and may present a potentially effective target for reducing EBI. Autophagy and apoptosis are activated after SAH (Jing et al., 2012; Zhao et al., 2013; Wang et al., 2015; Chen et al., 2016). Altay et al. (2012) reported that the neuronal apoptosis signaling pathway was a very important and vital pathologic mechanism in SAH-induced EBI. Bcl-2, Bax and their proapoptotic family members are widely expressed (Chen et al., 2014; Shao et al., 2016). Both of these proteins are necessary for the activation of caspase-9, which can activate caspase-3 and thereby contribute to neuronal apoptosis (Reed, 2002; Cheng et al., 2009; Chen et al., 2016).
Our previous studies showed that Ato can protect the autoregulation of cerebral vessels and ameliorate brain edema and EBI after SAH in animal experiments (Chen et al., 2016, 2018). However, the specific mechanism of the neuroprotective effects of Ato remained unclear. Numerous experiments have confirmed that statins have many anti-inflammatory and antioxidative effects (Tseng et al., 2005; Tousoulis et al., 2006; Luzak et al., 2011). Han et al. (2018) verified that Ato exerts an anti-inflammatory effect in atherosclerosis via the autophagy pathway, possibly by acting on the AKT/mTOR signaling pathway to enhance autophagy. Wang et al. (2015) found that statin treatment of spontaneous hypertensive rat cardiac cells regulated the Akt/mTOR pathway and inhibited apoptotic and autophagic cell death. Yang et al. (2017) believed that fluvastatin-induced anti-bone metastatic properties were based on the mechanism of autophagy in lung adenocarcinoma cells, as it can increase nuclear p53 expression. Qi et al. (2018) concluded that Ato improved EBI by reducing apoptosis and endoplasmic reticulum stress. Nevertheless, there are few studies on the possibility of activation of autophagy by Ato as its neuroprotective mechanism.
The mechanism by which Ato induces autophagy activation remains unclear. Previous studies showed that the Akt/mTOR pathway was one of the mechanisms involved in autophagy that may be activated by Ato (Wang et al., 2015). Ato can enhance LC3 transcription, promote LC3 expression and lead to the activation of autophagy (Han et al., 2018). Toepfer et al. (2006) reported other pathways and mechanisms involved in atorvastatin-induced autophagy, including the non-mTOR signaling pathway, PI-3 kinase activity, and JNK and Erk signaling pathways. Whether these pathways and mechanisms that are involved in statin-induced autophagy activation exert similar effects after SAH is still unclear. In our future research we will focus on exploring the underlying pathways and mechanisms by which Ato regulates autophagy. In particular, mTOR-independent autophagic signaling through LC3 activation may represent an important pathway that regulates statin-induced autophagy and deserves further investigation. In this study, we clarified that autophagy was the key to the neuroprotective effect of Ato treatment in EBI after SAH. However, the mechanism by which autophagy reduces neuronal apoptosis is unclear, and whether autophagy helps in the overall neuronal survival or death remains unclear (Shintani and Klionsky, 2004; Toepfer et al., 2011). We suspect that the activation of Ato-related autophagy can result in apoptosis-related proteins, such as caspase-3, being broken down or engulfed and thus vastly decreasing their effects or even removing them completely. After Ato or Rap intervention, the expression levels of caspase-3 were downregulated and neuronal apoptosis was also significantly decreased. However, the specific molecular mechanism underlying these effects, or the involvement of other mechanisms requires further study.
In summary, Ato could be a beneficial treatment for SAH-induced EBI in a SAH model. Ato treatment enhanced the expression of autophagy-related proteins, possibly resulting in autophagy. Caspase-dependent apoptosis was alleviated by Ato and Rap treatment after SAH. Ato can be a potential neuroprotective drug that reduces SAH-induced EBI and improves clinical outcomes via the autophagy pathway. However, the precise mechanisms underlying Ato activities, how autophagy reduces neuronal apoptosis and deeper signaling pathways, need to be studied further.
Additional files:
Additional Table 1: Neurological score.
Additional file 1: Open peer review report 1 (83.1KB, pdf) .
Footnotes
Conflicts of interest: The authors declare that they have no conflicts of interest.
Financial support: This study was supported by the Wuxi Foundation for Development of Science and Technology of China, No. WX18IIAN041 (to JHC); the Major Project of Nanjing Military Area Research Fund of China, No. 15DX003 (to JHC); the Wuxi Youth Medical Fund of China, No. QNRC046 (to TW). The funding bodies played no role in the study design, in the collection, analysis and interpretation of data, in the writing of the paper, or in the decision to submit the paper for publication.
Institutional review board statement: The experimental protocol was approved by the Animal Ethics Committee of Anhui Medical University, China (904 Hospital of Joint Logistic Support Force of PLA; approval No. YXLL-2017-09) on February 22, 2017.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Jérôme Dormoi, Institut de Recherche Biomédicale des Armées, France.
Chinese Library Classification No. R453; R741; Q255
Funding: This study was supported by the Wuxi Foundation for Development of Science and Technology of China, No. WX18IIAN041 (to JHC); the Major Project of Nanjing Military Area Research Fund of China, No. 15DX003 (to JHC); the Wuxi Youth Medical Fund of China, No. QNRC046 (to TW).
P-Reviewer: Dormoi J; C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Dawes EA, Raye W, Qiu Y, Song LP; T-Editor: Jia Y
References
- 1.Altay O, Hasegawa Y, Sherchan P, Suzuki H, Khatibi NH, Tang J, Zhang JH. Isoflurane delays the development of early brain injury after subarachnoid hemorrhage through sphingosine-related pathway activation in mice. Crit Care Med. 2012;40:1908–1913. doi: 10.1097/CCM.0b013e3182474bc1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Budohoski KP, Czosnyka M, Kirkpatrick PJ, Smielewski P, Steiner LA, Pickard JD. Clinical relevance of cerebral autoregulation following subarachnoid haemorrhage. Nat Rev Neurol. 2013;9:152–163. doi: 10.1038/nrneurol.2013.11. [DOI] [PubMed] [Google Scholar]
- 3.Cahill J, Zhang JH. Subarachnoid hemorrhage: is it time for a new direction. Stroke. 2009;40:S86–87. doi: 10.1161/STROKEAHA.108.533315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao S, Shrestha S, Li J, Yu X, Chen J, Yan F, Ying G, Gu C, Wang L, Chen G. Melatonin-mediated mitophagy protects against early brain injury after subarachnoid hemorrhage through inhibition of NLRP3 inflammasome activation. Sci Rep. 2017;7:2417. doi: 10.1038/s41598-017-02679-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen JH, Yang LK, Chen L, Wang YH, Wu Y, Jiang BJ, Zhu J, Li PP. Atorvastatin ameliorates early brain injury after subarachnoid hemorrhage via inhibition of AQP4 expression in rabbits. Int J Mol Med. 2016;37:1059–1066. doi: 10.3892/ijmm.2016.2506. [DOI] [PubMed] [Google Scholar]
- 6.Chen JH, Wu T, Yang LK, Chen L, Zhu J, Li PP, Hu X, Wang YH. Protective effects of atorvastatin on cerebral vessel autoregulation in an experimental rabbit model of subarachnoid hemorrhage. Mol Med Rep. 2018;17:1651–1659. doi: 10.3892/mmr.2017.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, Wang L, Wu C, Hu Q, Gu C, Yan F, Li J, Yan W, Chen G. Melatonin-enhanced autophagy protects against neural apoptosis via a mitochondrial pathway in early brain injury following a subarachnoid hemorrhage. J Pineal Res. 2014;56:12–19. doi: 10.1111/jpi.12086. [DOI] [PubMed] [Google Scholar]
- 8.Chen J, Xuan Y, Chen Y, Wu T, Chen L, Guan H, Yang S, He J, Shi D, Wang Y. Netrin-1 alleviates subarachnoid haemorrhage-induced brain injury via the PPARγ/NF-KB signalling pathway. J Cell Mol Med. 2019;23:2256–2262. doi: 10.1111/jcmm.14105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng G, Wei L, Zhi-Dan S, Shi-Guang Z, Xiang-Zhen L. Atorvastatin ameliorates cerebral vasospasm and early brain injury after subarachnoid hemorrhage and inhibits caspase-dependent apoptosis pathway. BMC Neurosci. 2009;10:7. doi: 10.1186/1471-2202-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Etminan N, Chang HS, Hackenberg K, de Rooij NK, Vergouwen MDI, Rinkel GJE, Algra A. Worldwide incidence of aneurysmal subarachnoid hemorrhage according to region, time period, blood pressure and smoking prevalence in the population: a systematic review and meta-analysis. JAMA Neurol. 2019;76:588–597. doi: 10.1001/jamaneurol.2019.0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galluzzi L, Bravo-San Pedro JM, Blomgren K, Kroemer G. Autophagy in acute brain injury. Nat Rev Neurosci. 2016;17:467–484. doi: 10.1038/nrn.2016.51. [DOI] [PubMed] [Google Scholar]
- 12.Han F, Xiao QQ, Peng S, Che XY, Jiang LS, Shao Q, He B. Atorvastatin ameliorates LPS-induced inflammatory response by autophagy via AKT/mTOR signaling pathway. J Cell Biochem. 2018;119:1604–1615. doi: 10.1002/jcb.26320. [DOI] [PubMed] [Google Scholar]
- 13.He Y, Wan S, Hua Y, Keep RF, Xi G. Autophagy after experimental intracerebral hemorrhage. J Cereb Blood Flow Metab. 2008;28:897–905. doi: 10.1038/sj.jcbfm.9600578. [DOI] [PubMed] [Google Scholar]
- 14.Jing CH, Wang L, Liu PP, Wu C, Ruan D, Chen G. Autophagy activation is associated with neuroprotection against apoptosis via a mitochondrial pathway in a rat model of subarachnoid hemorrhage. Neuroscience. 2012;213:144–153. doi: 10.1016/j.neuroscience.2012.03.055. [DOI] [PubMed] [Google Scholar]
- 15.Korja M, Lehto H, Juvela S, Kaprio J. Incidence of subarachnoid hemorrhage is decreasing together with decreasing smoking rates. Neurology. 2016;87:1118–1123. doi: 10.1212/WNL.0000000000003091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laskowitz DT, Kolls BJ. Neuroprotection in subarachnoid hemorrhage. Stroke. 2010;41:S79–84. doi: 10.1161/STROKEAHA.110.595090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le Roux PD, Winn HR. Management of the ruptured aneurysm. Neurosurg Clin N Am. 1998;9:525–540. [PubMed] [Google Scholar]
- 18.Lee J-Y, He Y, Sagher O, Keep R, Hua Y, Xi G. Activated autophagy pathway in experimental subarachnoid hemorrhage. Brain Res. 2009;1287:126–135. doi: 10.1016/j.brainres.2009.06.028. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Li J, Wang Z, Yu Z, Chen G. Attenuation of early brain injury and learning deficits following experimental subarachnoid hemorrhage secondary to Cystatin C: possible involvement of the autophagy pathway. Mol Neurobiol. 2014;49:1043–1054. doi: 10.1007/s12035-013-8579-3. [DOI] [PubMed] [Google Scholar]
- 20.Luzak B, Boncler M, Rywaniak J, Wilk R, Stanczyk L, Czyz M, Rysz J, Watala C. The effect of a platelet cholesterol modulation on the acetylsalicylic acid-mediated blood platelet inhibition in hypercholesterolemic patients. Eur J Pharmacol. 2011;658:91–97. doi: 10.1016/j.ejphar.2011.02.026. [DOI] [PubMed] [Google Scholar]
- 21.Macdonald RL, Higashida RT, Keller E, Mayer SA, Molyneux A, Raabe A, Vajkoczy P, Wanke I, Bach D, Frey A, Marr A, Roux S, Kassell N. Clazosentan, an endothelin receptor, antagonist in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: a randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2) Lancet Neurol. 2011;10:618–625. doi: 10.1016/S1474-4422(11)70108-9. [DOI] [PubMed] [Google Scholar]
- 22.Mackey J, Khoury JC, Alwell K, Moomaw CJ, Kissela BM, Flaherty ML, Adeoye O, Woo D, Ferioli S, De Los Rios La Rosa F, Martini S, Khatri P, Broderick JP, Zuccarello M, Kleindorfer D. Stable incidence but declining case-fatality rates of subarachnoid hemorrhage in a population. Neurology. 2016;87:2192–2197. doi: 10.1212/WNL.0000000000003353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat. Trends Neurosci. 2006;29:528–535. doi: 10.1016/j.tins.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 24.Qi W, Cao D, Li Y, Peng A, Wang Y, Gao K, Tao C, Wu Y. Atorvastatin ameliorates early brain injury through inhibition of apoptosis and ER stress in a rat model of subarachnoid hemorrhage. Biosci Rep. 2018;38:BSR20171035. doi: 10.1042/BSR20171035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reed JC. Apoptosis-based therapies. Nat Rev Drug Discov. 2002;1:111–121. doi: 10.1038/nrd726. [DOI] [PubMed] [Google Scholar]
- 26.Schievink WI, Wijdicks EF, Parisi JE, Piepgras DG, Whisnant JP. Sudden death from aneurysmal subarachnoid hemorrhage. Neurology. 1995;45:871–874. doi: 10.1212/wnl.45.5.871. [DOI] [PubMed] [Google Scholar]
- 27.Sehba FA, Pluta RM, Zhang JH. Metamorphosis of subarachnoid hemorrhage research: from delayed vasospasm to early brain injury. Mol Neurobiol. 2011;43:27–40. doi: 10.1007/s12035-010-8155-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sehba FA, Hou J, Pluta RM, Zhang JH. The importance of early brain injury after subarachnoid hemorrhage. Prog Neurobiol. 2012;97:14–37. doi: 10.1016/j.pneurobio.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shao A, Wang Z, Wu H, Dong X, Li Y, Tu S, Tang J, Zhao M, Zhang J, Hong Y. Enhancement of autophagy by histone deacetylase inhibitor trichostatin A ameliorates neuronal apoptosis after subarachnoid hemorrhage in rats. Mol Neurobiol. 2016;53:18–27. doi: 10.1007/s12035-014-8986-0. [DOI] [PubMed] [Google Scholar]
- 30.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith CM, Chen Y, Sullivan ML, Kochanek PM, Clark RSB. Autophagy in acute brain injury: feast, famine or folly. Neurobiol Dis. 2011;43:52–59. doi: 10.1016/j.nbd.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Toepfer N, Childress C, Parikh A, Rukstalis D, Yang W. Atorvastatin induces autophagy in prostate cancer PC3 cells through activation of LC3 transcription. Cancer Biol Ther. 2011;12:691–699. doi: 10.4161/cbt.12.8.15978. [DOI] [PubMed] [Google Scholar]
- 33.Tousoulis D, Antoniades C, Katsi V, Bosinakou E, Kotsopoulou M, Tsioufis C, Stefanadis C. The impact of early administration of low-dose atorvastatin treatment on inflammatory process, in patients with unstable angina and low cholesterol level. Int J Cardiol. 2006;109:48–52. doi: 10.1016/j.ijcard.2005.05.055. [DOI] [PubMed] [Google Scholar]
- 34.Tseng MY, Czosnyka M, Richards H, Pickard JD, Kirkpatrick PJ. Effects of acute treatment with pravastatin on cerebral vasospasm, autoregulation and delayed ischemic deficits after aneurysmal subarachnoid hemorrhage: a phase II randomized placebo-controlled trial. Stroke. 2005;36:1627–1632. doi: 10.1161/01.STR.0000176743.67564.5d. [DOI] [PubMed] [Google Scholar]
- 35.Wang W, Wang H, Geng QX, Wang HT, Miao W, Cheng B, Zhao D, Song GM, Leanne G, Zhao Z. Augmentation of autophagy by atorvastatin via Akt/mTOR pathway in spontaneously hypertensive rats. Hypertens Res. 2015;38:813–820. doi: 10.1038/hr.2015.85. [DOI] [PubMed] [Google Scholar]
- 36.Wu H, Niu H, Wu C, Li Y, Wang K, Zhang J, Wang Y, Yang S. The autophagy-lysosomal system in subarachnoid haemorrhage. J Cell Mol Med. 2016;20:1770–1778. doi: 10.1111/jcmm.12855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie YK, Zhou X, Yuan HT, Qiu J, Xin DQ, Chu XL, Wang DC, Wang Z. Resveratrol reduces brain injury after subarachnoid hemorrhage by inhibiting oxidative stress and endoplasmic reticulum stress. Neural Regen Res. 2019;14:1734–1742. doi: 10.4103/1673-5374.257529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Z, Su Z, DeWitt JP, Xie L, Chen Y, Li X, Han L, Li D, Xia J, Zhang Y, Yang Y, Jin C, Zhang J, Li S, Li K, Zhang Z, Qu X, He Z, Chen Y, Shen Y, et al. Fluvastatin prevents lung adenocarcinoma bone metastasis by triggering autophagy. EBioMedicine. 2017;19:49–59. doi: 10.1016/j.ebiom.2017.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao H, Ji Z, Tang D, Yan C, Zhao W, Gao C. Role of autophagy in early brain injury after subarachnoid hemorrhage in rats. Mol Biol Rep. 2013;40:819–827. doi: 10.1007/s11033-012-2120-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.