

Keywords: aminooxyacetic acid, central nervous system, complement deposition, cystathionine-ß-synthase, early brain injury, endoplasmic reticulum stress, hydrogen sulfide, neuroinflammation, oxidative stress, subarachnoid hemorrhage
Abstract
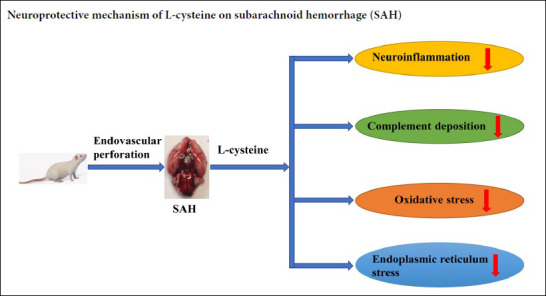
Hydrogen sulfide, which can be generated in the central nervous system from the sulfhydryl-containing amino acid, L-cysteine, by cystathionine-β-synthase, may exert protective effects in experimental subarachnoid hemorrhage; however, the mechanism underlying this effect is unknown. This study explored the mechanism using a subarachnoid hemorrhage rat model induced by an endovascular perforation technique. Rats were treated with an intraperitoneal injection of 100 mM L-cysteine (30 μL) 30 minutes after subarachnoid hemorrhage. At 48 hours after subarachnoid hemorrhage, hematoxylin-eosin staining was used to detect changes in prefrontal cortex cells. L-cysteine significantly reduced cell edema. Neurological function was assessed using a modified Garcia score. Brain water content was measured by the wet-dry method. L-cysteine significantly reduced neurological deficits and cerebral edema after subarachnoid hemorrhage. Immunofluorescence was used to detect the number of activated microglia. Reverse transcription-polymerase chain reaction (RT-PCR) was used to detect the levels of interleukin 1β and CD86 mRNA in the prefrontal cortex. L-cysteine inhibited microglial activation in the prefrontal cortex and reduced the mRNA levels of interleukin 1β and CD86. RT-PCR and western blot analysis of the complement system showed that L-cysteine reduced expression of the complement factors, C1q, C3α and its receptor C3aR1, and the deposition of C1q in the prefrontal cortex. Dihydroethidium staining was applied to detect changes in reactive oxygen species, and immunohistochemistry was used to detect the number of NRF2- and HO-1-positive cells. L-cysteine reduced the level of reactive oxygen species in the prefrontal cortex and the number of NRF2- and HO-1-positive cells. Western blot assays and immunohistochemistry were used to detect the protein levels of CHOP and GRP78 in the prefrontal cortex and the number of CHOP- and GRP78-positive cells. L-cysteine reduced CHOP and GRP78 levels and the number of CHOP- and GRP78-positive cells. The cystathionine-β-synthase inhibitor, aminooxyacetic acid, significantly reversed the above neuroprotective effects of L-cysteine. Taken together, L-cysteine can play a neuroprotective role by regulating neuroinflammation, complement deposition, oxidative stress and endoplasmic reticulum stress. The study was approved by the Animals Ethics Committee of Shandong University, China on February 22, 2016 (approval No. LL-201602022).
Introduction
Subarachnoid hemorrhage is a hemorrhagic stroke subtype that can lead to devastating outcomes (Kanamaru and Suzuki 2019; Liu et al., 2019; Xie et al., 2019). Although advances in early intravascular intervention and intensive care support have successfully increased the survival rate by 17% in recent years, long-lasting cognitive deficits and daily loss of functionality are still prominent (Macdonald and Schweizer, 2017). The failure of the CONSCIOUS-1 and CONSCIOUS-2 clinical trials has shifted research focus from prevention of delayed vasospasm to alleviation of early brain injury, in the time window of the first 72 hours after onset of subarachnoid hemorrhage (SAH) (Macdonald et al., 2008, 2013; Sehba et al., 2012). Multiple factors interact during early brain injury, including but not limited to, neuroinflammation, oxidative stress, and endoplasmic reticulum (ER) stress (Ostrowski et al., 2006).
In sharp contrast to being a highly toxic gas, hydrogen sulfide (H2S) is also a critical neuromodulator in the mammalian central nervous system that is associated with anti-inflammation, anti-oxidation and anti-apoptotic effects (Hu et al., 2007; Kimura, 2013; Wang et al., 2014). H2S is generated mainly from the sulfhydryl-containing amino acid L-cysteine (L-Cys) by cystathionine-β-synthase (CBS) localized in astrocytes (Abe and Kimura, 1996; Kimura, 2013). CBS catalytic activity can be blocked by the drug, aminooxyacetic acid (AOAA) (Asimakopoulou et al., 2013). Accumulating evidence from clinical samples indicates that patients expressing high levels of H2S-producing CBS have a better prognosis after SAH (Grobelny et al., 2011; Hendrix et al., 2018), leading to the hypothesis that exogenous supplementation of H2S might be a treatment for SAH-induced brain injury (Yu et al., 2014; Cui et al., 2016). We previously reported that exogenous L-Cys could successfully elevate the concentration of intracerebral H2S, improving the prognosis of experimental SAH rats (Li et al., 2017); however, the underlying molecular mechanism was not known. The current study investigated whether the protective effect of L-Cys is associated with neuroinflammation, oxidative stress or ER stress.
The complement system contributes greatly to the neuroinflammatory response, in which complement C1q activates the classical complement pathway. This pathway can produce massive up-regulation of proinflammatory mediators, such as C3a and C5a. Meanwhile, the dysregulated deposition of complement fragments can form the membrane attack complex, leading to tissue damage within the central nervous system (Arumugam et al., 2006). In addition, substantial amounts of reactive oxygen species (ROS), including superoxide anions (O2), hydroxyl radicals (OH) and hydrogen peroxide (H2O2) are generated during early brain injury, which disrupt neural cell membranes and the blood-brain barrier (Ostrowski et al., 2006). These components can synergistically interfere with protein synthesis in neural cells, and trigger the unfolded protein response (UPR) to counter ER stress. Excessive expression of the C/EBP homologous protein (CHOP), a downstream protein in the ER stress pathway triggered by dysregulated UPR, can lead to apoptosis (Roussel et al., 2013).
This study aimed to test whether L-Cys administration suppresses neuroinflammation, complement deposition, oxidative stress and ER stress, and whether L-Cys improves outcomes after SAH. We also sought to examine the mechanism of L-Cys action after SAH.
Materials and Methods
Animals and surgical procedure
One hundred and twenty male Wistar rats aged 10 weeks and weighing 280–350 g were purchased from the Laboratory Animal Center of Shandong University, China [license No. SYXK (Lu) 20190005] 1 week before surgery. All rats were kept in a pathogen-free environment at 22 ± 3°C and 40–70% humidity with a 12-hour light/dark cycle and free access to food and water. In accordance with the International Guiding Principles for Animal Research established by the Council for International Organizations of Medical Sciences (CIOMS), all animal surgical procedures were approved (on February 22, 2016) and supervised by the Animal Ethics and Welfare Committee of Shandong University (approval No. LL-201602022). The animal protocols were conducted in accordance with the instructions of the Institutional Animal Care and Use Committee Guidebook (IACUC).
The SAH model was induced in rats using an endovascular perforation technique as previously described (Bederson et al., 1995). Briefly, rats were fasted for 12 hours before surgery and anesthetized by inhaling 3% isoflurane (Rayward Life Technology Co., Ltd., Shenzhen, China) in a polymethyl methacrylate box with 5 L of oxygen flow per minute. After anesthetization, rats were mechanically ventilated using a face mask with an air/isoflurane mixture to maintain normal arterial blood gas conditions. Each rat was placed in the supine position, and a midline incision was made on the neck. After identification of appropriate carotid vessels, the left external carotid artery was ligated and then fashioned into a stump. A 3 cm, 3-0 nylon suture with a sharpened tip was advanced into the internal carotid artery from the external carotid artery stump via the common carotid bifurcation. The suture was further inserted until resistance was encountered, and then an additional 3–4 mm was inserted to perforate the blood vessel. The suture was immediately withdrawn after bleeding caused by perforation and the stump was ligated. The skin incision was sutured. The rats were kept in a heated chamber till recovery. A similar procedure was conducted in the sham group except that endovascular perforation was not performed. All efforts were made to reduce the number of animals used and their suffering. Vasospasm and brain injury induced by SAH caused behavioral deficits that manifested as motor and sensory dysfunction, and cognitive, learning and memory impairment. Neurobehavioral evaluation reflected brain injury and was performed using a modified Garcia scoring system (Fujii et al., 2014) to judge the success of model establishment. The best score is 22 and the worst score is 5.
Experimental design and drug administration
L-Cys (Cat#: C1276-10G; Sigma-Aldrich, St. Louis, MO, USA) was dissolved in phosphate buffered saline (PBS, Thermo Fisher Scientific, Waltham, MA, USA) at a working concentration of 100 mM as previously described (Wang et al., 2013a). L-Cys solution (30 μL) was injected into the lateral cerebral ventricle 30 minutes after SAH. Aminooxyacetic acid (AOAA, Cat#: C13408-1G; Sigma-Aldrich), a non-specific inhibitor of CBS, was dissolved in PBS and intraperitoneally administered at 5 mg/kg in parallel with L-Cys injection. The rats were randomly divided into five groups (Figure 1). In the sham group (vehicle; n = 24), rats were administered vehicle (0.01 M PBS, 50 μL) only. In the sham + L-Cys group (n = 24), rats were administered L-Cys only. In the SAH group (vehicle; n = 24), rats were administered vehicle (0.01 M PBS, 50 μL) only at 30 minutes after SAH. In the SAH + L-Cys group (n = 24), rats were administered 30 μL 100 mM L-Cys only. In the SAH + L-Cys + AOAA group (n = 24), rats were administered 30 μL 100 mM L-Cys and 5 mg/kg AOAA.
Figure 1.
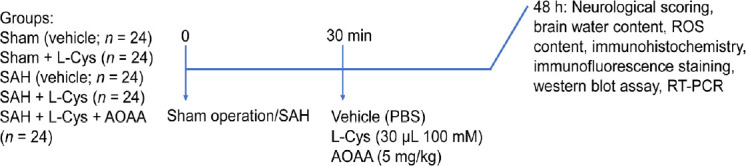
Experimental design and drug administration.
AOAA: Aminooxyacetic acid; L-Cys: L-cysteine; PBS: phosphate buffered saline; ROS: reactive oxygen species; RT-PCR: reverse transcription-polymerase chain reaction; SAH: subarachnoid hemorrhage.
Neurological evaluation
Neurological function was evaluated at 48 hours after SAH by two investigators using a modified Garcia scoring system in a single-blind manner (Garcia et al., 1995; Hasegawa et al., 2011). This rating system consists of the following seven tests: spontaneous activity (0–3 points), reaction to side stroking (1–3 points), reaction to vibrissae touch (1–3 points), limb symmetry (0–3 points), forelimb outstretching (0–3 points), climbing (0–3 points), and beam walking (0–4 points) ability. The combined score reflects neurological function; the higher the Garcia score the better the neurological function (Fujii et al., 2014).
Hematoxylin-eosin staining
Hematoxylin-eosin staining was performed according to our previous method (Li et al., 2017). Briefly, brain tissue was collected 48 hours after SAH and paraffin sections were prepared. The sections were stained with hematoxylin and eosin. Four rats in each group were prepared for hematoxylin-eosin staining. The morphology of the prefrontal cortex (the cerebral cortex that covers the anterior portion of the frontal lobe) was observed under a TE2000U microscope (Nikon, Tokyo, Japan).
Reactive oxygen species contents
To measure ROS production at 48 hours post-SAH, the rat brain was collected and fresh frozen 12 μm coronal sections prepared. These sections were stained with 10 μM dihydroethidium (DHE) (Cat#:810253P-1MG; Sigma-Aldrich) solution for 30 minutes and then rinsed and mounted as previously reported (Wang et al., 2018). Fluorescence images of ROS expression were quantified by Image-pro Plus 6.0 software (Media Cybernetics, Silver Spring, MD, USA). Values are expressed relative to the intensity of fluorescence signals in the sham group.
Western blot assays
The prefrontal cortex was dissected on ice and weighed. Following homogenization in RIPA buffer containing protease/phosphatase inhibitors and phenylmethylsulfonyl fluoride (PMSF) (RIPA:protease/phosphatase inhibitors:PMSF = 100:1:1) for 10 minutes, samples were centrifuged at 13,800 × g for 10 minutes at 4°C. Total protein concentrations were quantified using a bicinchoninic acid protein assay kit (Pierce Biotechnology Inc., Rockford, IL, USA). After mixing with loading buffer (Biotime, Haimen, China), the sample was heated to 100°C for 10 minutes and then left to cool. Afterwards, 30–50 μg of protein was loaded onto each lane of a polyacrylamide gel, electrophoresed, and transferred to a polyvinylidene membrane. The membrane was probed with different ER stress marker antibodies: ATF6α (ER stress marker), CHOP (ER stress maker), eIF2α (ER stress maker), GRP78 (ER stress maker), NRF2 (oxidative stress marker), and p-IRE1α (ER stress maker). β-Actin was used as an internal control. Secondary antibodies were goat-anti-rabbit or goat-anti-mouse IgG. Detailed information of the antibodies used is shown in Table 1. The protein bands were fluorescently detected using an enhanced chemiluminescence detection system (Pierce Biotechnology Inc.) and were analyzed using ImageJ software (National Institutes of Health, Bethesda, MD, USA). The results are expressed as the ratio of the target protein level to the level of β-actin.
Table 1.
Antibodies used in western blot assay and immunohistochemistry in the study
| Description | Company | Catalog number |
|---|---|---|
| Rabbit polyclonal eIF2α antibody, 1:1000 | Proteintech Group, Inc. (Rosemont, IL, USA) | 11233-1-AP |
| Rabbit polyclonal CHOP antibody, 1:1500 | Proteintech Group, Inc. | 15204-1-AP |
| Rabbit polyclonal ATF6α antibody, 1:1000 | Proteintech Group, Inc. | 15794-1-AP |
| Rabbit polyclonal NRF2 antibody, 1:1000 | Proteintech Group, Inc. | 16396-1-AP |
| Rabbit polyclonal HO-1 antibody, 1:100 | Proteintech Group, Inc. | 10701-1-AP |
| Rabbit polyclonal GRP78 antibody, 1:1000/1:100 | Proteintech Group, Inc. | 11587-1-AP |
| Phospho-eIF2α (Ser51) (D9G8) XP® rabbit mAb, 1:1000 | Cell Signaling Technology, Inc. (Boston, MA, USA) | #3398 |
| Rabbit polyclonal IBA1 antibody, 1:100 | Proteintech Group, Inc. | 10904-1-AP |
| Mouse monoclonal beta actin antibody, 1:2000 | Proteintech Group, Inc. | 60008-1-Ig |
| Phosphor-IRE1α antibody, 1:1000 | Abcam (Cambridge, UK) | ab48187 |
| Mouse monoclonal C1q antibody, 1:200 | Abcam | ab71089 |
| IRE1α(14C10) rabbit mAb, 1:1000 | Cell Signaling Technology, Inc. | #3294 |
| Goat-anti-rabbit IgG, 1:10,000 | Boster Biological Technology Co., Ltd. (Pleasanton, CA, USA) | BA1003 |
| Goat-anti-mouse IgG, 1:10,000 | Boster Biological Technology Co., Ltd. | BA1038 |
| Proteintech Group, Inc. | Proteintech Group, Inc. | SA00007-2 |
Reverse transcription-polymerase chain reaction
Tissues were obtained 48 hours post-SAH. Total RNA was extracted using Trizol (01761/20114-1, Haimen, China) and its concentration was measured using a NanoDrop2000 spectrophotometer (Thermo Fisher Scientific). A reverse transcription kit (Fermentas, Vilnius, Lithuania) was used to reverse transcribe 2 µg RNA into cDNA. Specific primers (Table 2) were added to amplify cDNA by reverse transcription-polymerase chain reaction (RT-PCR). A 1.2% agarose/TAE gel containing an ethidium bromide substitute (GoldView™, Solarbio) was used to separate the PCR product and was visualized using a gel-viewing system (Bio-Rad, Hercules, CA, USA). 2–ΔΔCt Values of each group were normalized to β-actin.
Table 2.
Primers in the study
| Gene | Sequence (5′–3′) | Product size (bp) |
|---|---|---|
| IL-1β | Forward: AAG ATG AAG GGC TGC TTC CAA ACC | 106 |
| Reverse: ATA CTG CCT GCC TGA AGC TCT TGT | ||
| CD86 | Forward: TAA GCA AGG TCA CCC GAA AC | 303 |
| Reverse: AGC AGC ATC ACA AGG AGG AG | ||
| C1q | Forward: AAT GAC GCT TGG CAA CGT GGT TAT C | 351 |
| Reverse: ATG AGG AAT CCG CTG AAG ATG CTG | ||
| C3a | Forward: GCC TCT CCT CTG ACC TCT GG | 130 |
| Reverse: AGT TCT TCG CAC TGT TTC TGG | ||
| C3aR1 | Forward: TGT TGG TGG CTC GCA GAT | 141 |
| Reverse: GCA ATG TCT TGG GGT TGA AA | ||
| β-Actin | Forward: CTA TTG GCA ACG AGC GGT | 152 |
| Reverse: GCA ATG TCT TGG GGT TGA AA TCC |
IL-1β: Interleukin-1β.
Brain water content
Brain water content was assessed by the wet/dry method (Li et al., 2017). Briefly, brain samples were removed from the skull 48 hours post-SAH and immediately weighed (wet weight). Samples were then dried for 48 hours in a 100°C chamber and weighed again (dry weight). Brain water content (%) was equal to [(wet weight–dry weight)/wet weight] × 100.
Dihydroethidium staining
Sections were prepared from fresh frozen brains taken 48 hours post-SAH. Each section was incubated with 10 μM superoxide anion fluorescent probe, DHE, for 30 minutes at 37°C. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Images were captured using a TE2000U microscope (Nikon). Six images at 200× magnification (n = 4) were randomly selected for each slice, and the results were quantified and analyzed using the Image-Pro Plus 6.0 image analysis system (Media Cybernetics). The number of positive cells in each section was expressed as the average of six images per section. The calculated value was then expressed as a percentage of positive cells and compared with the sham group.
Immunohistochemistry
Paraffin sections were dewaxed and washed with PBS. After blocking for 30 minutes at room temperature, the sections were incubated with a mouse monoclonal anti-C1q antibody (a complement system maker, 1:200; Abcam, Cambridge, UK), a rabbit monoclonal anti-HO-1 antibody (an oxidative stress marker, 1:100; Proteintech Group, Inc., Chicago, IL, USA), a rabbit monoclonal anti-NRF2 antibody (oxidative stress marker, 1:100; Proteintech Group, Inc.), a rabbit monoclonal anti-GRP78 antibody (ER stress marker, 1:100; Proteintech Group, Inc.), or a rabbit monoclonal anti-CHOP antibody (ER stress marker, 1:100; Proteintech Group, Inc.) at 4°C overnight (Table 1). The slides were then washed and incubated with the appropriate secondary antibody (horseradish peroxidase-labeled goat anti-rabbit IgG or goat anti-mouse, 1:10,000; Proteintech Group, Inc.) for 1 hour at 37°C (Table 1). After incubation with an avidin biotinylated enzyme complex, the probed protein was stained with diaminobenzidine, while the nuclei were counterstained with hematoxylin. Images were captured using a TE2000U microscope (Nikon). Six images at 200× magnification (n = 4) were randomly selected for each section, and the results were analyzed and quantified using the Image-Pro Plus 6.0 image analysis system (Media Cybernetics). The number of positive cells in each section is expressed as the average of six images per section. The calculated value was then expressed as a percentage of positive cells and compared with the sham group.
Immunofluorescence staining
Sections were prepared from fresh frozen brains taken 48 hours post-SAH. Sections were incubated with a rabbit monoclonal anti-IBA1 antibody (microglial marker, 1:100; Proteintech Group, Inc.) for 16 hours at 4°C, followed by a secondary antibody (rhodamine-conjugated goat anti-rabbit IgG, 1:100; Proteintech Group, Inc.) for 30 minutes at 37.4°C and then DAPI for 10 minutes at room temperature (Table 1). Images were captured and analyzed using fluorescence microscopy (BX51; Olympus, Tokyo, Japan) with the Magna Fire system. Six images at 400× magnification (n = 4) were randomly selected for each slice, and the results were analyzed and quantified using the Image-Pro Plus 6.0 image analysis system (Media Cybernetics). The number of positive cells in each section is expressed as the average of six images per section. The results are expressed as a ratio of positive cells and compared with the sham group.
Statistical analysis
Data are presented as the mean ± SD. SPSS 22.0 software (IBM, Armonk, NY, USA) was used for statistical analysis. All data were analyzed by one-way analysis of variance followed by Tukey’s post hoc test. A value of P < 0.05 was considered statistically significant.
Results
Behavioral and morphological changes after L-Cys administration in SAH rats
Blood clots were observed inside the subarachnoid space but mainly around the circle of Willis and cisterna ambiens in the SAH, SAH + L-Cys, and SAH + L-Cys + AOAA groups (Figure 2A). Hematoxylin-eosin staining showed that neuronal cells appeared to be round, pale and edematous in the SAH group while L-Cys treatment alleviated morphological changes. However, AOAA aggravated neuronal loss after SAH (Figure 2B). Consistent with morphological changes between groups, behavioral tests 48 hours after SAH revealed a significant difference between the SAH and the sham groups (P < 0.001), while the SAH + L-Cys group had a better performance than the SAH group (P < 0.05; Figure 2C). In addition, the brain water content was significantly higher in the SAH group compared with the sham group (P < 0.01) at 48 hours after SAH, and L-Cys markedly alleviated the brain edema (P < 0.05; Figure 2D).
Figure 2.

Effects of L-Cys on neurological deficits and brain water content following SAH.
(A) Blood clots at 8 hours after SAH around both the circle of Willis and cisterna ambiens (n = 4). (B) Hematoxylin-eosin staining at 48 hours after SAH at 40× magnification. The number of pale and edematous cells was increased in the SAH group (n = 4). Green arrows: normal cells; red arrows: pale and edematous cells. Scale bars: 50 μm. (C) Evaluation of neurological impairment at 48 hours after SAH. Neurological impairment was markedly improved by 30 μL 100 mM L-Cys treatment (n = 6). (D) Brain water content assessment at 48 hours after SAH. The 100 mM L-Cys (30 μL) treatment significantly reduced brain water content compared with the SAH group, while AOAA (5 mg/kg) blocked this phenomenon (mean ± SD, n = 6, one-way analysis of variance followed by Tukey’s post hoc test). *P < 0.05, **P < 0.01, ***P < 0.001. AOAA: Aminooxyacetic acid; L-Cys: L-cysteine; SAH: subarachnoid hemorrhage.
Effects of L-Cys on microglia and inflammatory gene expression 48 hours post-SAH
Immunohistochemistry identified base-level IBA1+ microglia in the sham group characterized by small cell bodies with long processes. SAH significantly increased the number of microglia, which had a more amoeboid-like appearance, indicating activated microglial cells (P < 0.01). However, both the quantity and morphology of microglia were near base levels in the L-Cys group (P < 0.01; Figure 3A and D). Meanwhile, the transcription of IL-1β and CD86 in brain tissue was significantly upregulated in the SAH group compared with the sham group (P < 0.001). The expression of IL-1β and CD86 was less in the L-Cys group than in the SAH group (P < 0.01). AOAA reversed the effect of L-Cys on microglial activation (P < 0.01; Figure 3B and C).
Figure 3.

Effects of L-Cys on microglia and inflammatory genes in SAH rats.
(A) Immunohistochemistry of IBA1-positive microglia at 48 hours after SAH (n = 4) at 40× magnification. Yellow arrows: IBA1-positive microglia. Scale bars: 50 μm. (B) Reverse transcription-polymerase chain reaction of IL-1β and CD86. (C) Quantitative analysis of IL-1β and CD86 mRNA levels. (D) Quantitative analysis of IBA1-positive cells (ratio to Sham) at 48 hours after SAH (mean ± SD, n = 4, one-way analysis of variance followed by Tukey’s post hoc test). **P < 0.01, ***P < 0.001. AOAA: Aminooxyacetic acid; L-Cys: L-cysteine; IBA1: ionized calcium binding adaptor molecule 1; IL-1β: interleukin 1β; SAH: subarachnoid hemorrhage.
At 48 hours post-SAH, the complement system is down-regulated by L-Cys treatment
Levels of complement mRNAs for C1q, C3α and its receptor C3aR1 were up-regulated 48 hours after SAH compared with the sham group (P < 0.001; Figure 4A–D). Levels of mRNA for C1q, C3α and its receptor C3aR1 were lower in the SAH + L-Cys group compared with the SAH group. Immunohistochemistry showed that C1q was massively deposited in neural cells after SAH, but that L-Cys administration significantly reduced this deposition (P < 0.01; Figure 4E–F). AOAA administration reversed the up-regulation of C1q, C3α and C3aR1 expression of the SAH + L-Cys group (Figure 4).
Figure 4.

Effects of L-Cys on complement proteins C1q, C3a and C3aR1.
(A) Reverse transcription-polymerase chain reaction at 48 hours after SAH (n = 4). (B–D) Quantitative analysis of C1q, C3a and C3aR1 mRNA levels at 48 hours after SAH. (E) Immunohistochemistry images of Cip at 40× magnification at 48 hours after SAH. Red arrows: C1q-positive cells. Scale bar: 50 μm. (F) Quantitative analysis of C1q-immunoreactive (ratio to sham) at 48 hours after SAH (mean ± SD, n = 4, one-way analysis of variance followed by Tukey’s post hoc test). *P < 0.05, **P < 0.01, ***P < 0.001. AOAA: Aminooxyacetic acid; L-Cys: L-cysteine; SAH: subarachnoid hemorrhage.
Effects of L-Cys on ROS and antioxidative stress genes
DHE assays indicated that high levels of ROS were generated after SAH compared with the sham group (P < 0.001). L-Cys treatment markedly reduced ROS content compared with the SAH group (P < 0.01; Figure 5A and C). NRF2 and HO-1 have antioxidative roles in various central nervous system diseases. Immunohistochemistry confirmed increased levels of NRF2 and HO-1 in the SAH group and further up-regulation after L-Cys administration (P < 0.01; Figure 5B and D). Nevertheless, AOAA blocked the effect of L-Cys on ROS production (P < 0.05) and antioxidative gene expression (P < 0.001).
Figure 5.

Effects of L-Cys on ROS and antioxidative stress genes.
(A) DHE-stained images of ROS content at 48 hours after SAH at 20× magnification (n = 4). Scale bar: 50 μm. (B) Immunohistochemistry of NRF2 and HO-1 at 48 hours after SAH at 20× magnification (n = 4). (C) Quantitative analysis of ROS (% sham) at 48 hours after SAH. (D) Quantitative analysis of NRF2/HO-1 expression (ratio to sham) at 48 hours after SAH (mean ± SD, n = 4, one-way analysis of variance followed by Tukey’s post hoc test). Scale bar: 50 μm. *P < 0.05, **P < 0.01, ***P < 0.001. AOAA: Aminooxyacetic acid; DHE: dihydroethidium; HO-1: heme oxygenase-1; L-Cys: L-cysteine; NRF2: nuclear factor erythroid 2 related factor 2; ROS: reactive oxygen species; SAH: subarachnoid hemorrhage.
Effects of L-Cys on ER stress and the UPR after SAH
GRP78 is a marker for ER stress and CHOP leads to cell death at the end stage of ER stress. Western blot assays indicated that both GRP78 and CHOP were increased in the SAH group (P < 0.001 and P < 0.001), while L-Cys treatment reduced their expression (P < 0.01 and P < 0.01), which was reversed by AOAA (P < 0.05 and P < 0.01; Figure 6A). This was also confirmed by immunohistochemistry where levels of both proteins were higher in the SAH group and decreased after L-Cys treatment (P < 0.01; Figure 6B).
Figure 6.

Effects of L-Cys on GRP78 and CHOP expression.
(A) Western blot image and quantitative analysis of GRP78 and CHOP at 48 hours after SAH (n = 4). (B) Immunohistochemistry of CHOP and GRP78 at 20× magnification (n = 4). Scale bars: 50 μm. (C) Quantitative analysis of GRP78 and CHOP in the prefrontal cortex at 48 hours after SAH (n = 4; mean ± SD, one-way analysis of variance followed by Tukey’s post hoc test), *P < 0.05, **P < 0.01, ***P < 0.001. AOAA: Aminooxyacetic acid; CHOP: C/EBP homologous protein; GRP78: 78-kDa glucose-regulated protein; L-Cys: L-cysteine; SAH: subarachnoid hemorrhage.
The UPR pathway is involved in SAH and is affected by L-Cys administration. As shown in Figure 7, western blot assays demonstrate that SAH upregulated the phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) and inositol-requiring enzyme 1α (IRE1α) and increased the cleavage of activating transcription factor 6 (ATF6α) (P < 0.05 and P < 0.01), indicating activation of the three UPR pathways. Treatment with L-Cys decreased eIF2α phosphorylation compared with the SAH group (P < 0.01) but had no influence on IRE1α phosphorylation (P > 0.05). Cleaved ATF6α levels were further up-regulated in the SAH + L-Cys group (P < 0.001). AOAA suppressed the changes induced by L-Cys (P < 0.01).
Figure 7.
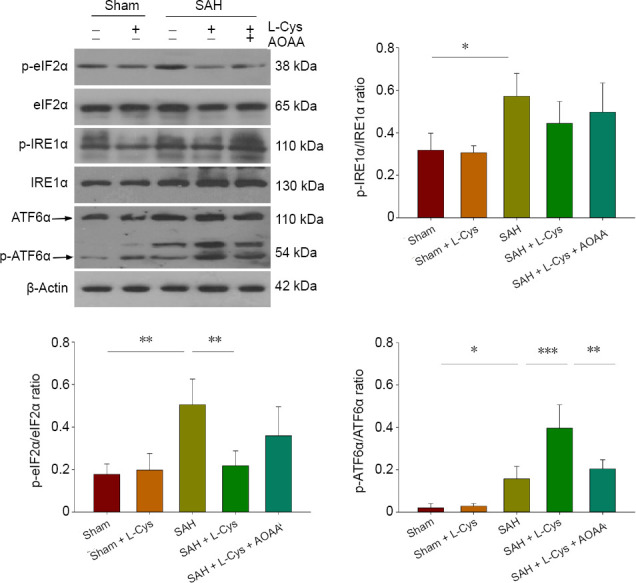
Effects of L-Cys on the UPR pathways.
Western blot image and quantitative analysis of eIF2α, IRE1α and ATF6α phosphorylation at 48 hours after SAH: All three UPR pathways were activated after SAH. The phosphorylation of eIF2α decreased after 100 mM L-Cys (30 μL) treatment, while IRE phosphorylation remained unchanged (mean ± SD, n = 4, one-way analysis of variance followed by Tukey’s post hoc test). *P < 0.05, **P < 0.01, ***P < 0.001. AOAA: Aminooxyacetic acid; ATF6α: activating transcription factor 6α; eIF2α: α-subunit of eukaryotic initiation factor 2; IRE1, inositol-requiring enzyme 1; L-Cys: L-cysteine; SAH: subarachnoid hemorrhage; UPR: unfolded protein response.
Discussion
Reduced generation of H2S has been reported to play important roles in various pathological conditions, including Alzheimer’s disease (Eto et al., 2002), aging (Jin et al., 2015), and hyperglycemia (Coletta et al., 2015). Encouraging clinical results from human samples have identified a close relationship between CBS polymorphisms and clinical sequelae of SAH; increased generation of H2S by certain CBS alleles might be the key reason for better prognosis after SAH (Hendrix et al., 2018). This has led to the hypothesis that H2S might be effective as a treatment for SAH (Yu et al., 2014). Cui et al. (2016) first reported that acute administration of H2S using an NaHS solution as an inorganic H2S donor can ameliorate early brain injury in a SAH rat model. We then demonstrated H2S treatment using L-Cys as an organic donor, which better mimicked physiological conditions (Kimura, 2014; Li et al., 2017). Nonetheless, the underlying molecular mechanism for H2S efficacy remained uninvestigated. Several studies reported potential effects of H2S on cerebral vessels during SAH, such as restoring the blood-brain barrier and reducing vasospasm (Cao et al., 2016; Emmez et al., 2017) but the relationship between H2S and neuroinflammation, oxidative stress, and ER stress was not discussed.
L-Cys contains a sulfhydryl group at the end of its side chain, which provides its reactive capacity. Several clinical trials have investigated its application in human diseases (Clemente Plaza et al., 2018). L-Cys can regulate cellular responses indirectly by generating H2S. For example, L-Cys can relax vascular contraction through H2S, both in vivo and in vitro (Al-Magableh and Hart, 2011; Yetik-Anacak et al., 2016). L-Cys can significantly increase Ca2+ concentration in medullary neurons; however, AOAA, an inhibitor of the H2S-producing enzyme, reversed this effect (Liu et al., 2016). Furthermore, L-Cys, via the CBS/H2S system, can promote the proliferation and differentiation of neural stem cells in vitro (Wang et al., 2013b) and we have also reported that L-Cys exerts neuroprotection on hypoxic-ischemic injury in neonatal mice (Liu et al., 2017; Xin et al., 2018).
Unregulated neuroinflammation plays an important role in early brain injury development (Prunell et al., 2005) and eventually contributes to cell death. Interventions to prevent neuroinflammation and peripheral monocyte infiltration following SAH were predicted to serve protective and beneficial functions. Consistent with a previous study (Schneider et al., 2018), we found that SAH triggers an inflammatory reaction, identified by microglial activation, and striking up-regulation of proinflammatory genes, while L-Cys treatment significantly suppressed microglial activation and the levels of proinflammatory gene expression.
The complement system is part of the inflammatory response and is involved in central nervous system injury (Arumugam et al., 2006). During early brain injury, C1q is activated, and then splits downstream C3 and C5 complement proteins, generating the proinflammatory mediators, C3a and C5a. The increased plasma concentration of C3a positively correlates with delayed morbidity and mortality after aneurysmal rupture (Mack et al., 2007; Kofler et al., 2015). In this study, levels of C1q, C3 and C3aR1 were significantly increased after SAH, indicating activation of the complement system. This activation was, significantly reduced by L-Cys treatment, and was associated with decreased cell death and improved neurological outcomes.
In addition to neuroinflammation, oxidative stress actively participates in the pathogenesis of early brain injury. Antioxidants have beneficial effects on SAH (Ayer and Zhang, 2008; Duan et al., 2016) and H2S can attenuate SAH-increased ROS and MDA levels in vitro and in vivo (Cui et al., 2016). According to our data, ROS and MDA concentrations were significantly increased after SAH, but the administration of L-Cys reversed this phenomenon. Moreover, the protective effect of L-Cys on SAH was blocked by the CBS inhibitor, AOAA, indicating that its antioxidant mechanism was largely dependent on H2S generation. Additionally, a redox-sensitive transcription factor, NRF2, plays a crucial role in regulating redox status. Under normal conditions, NRF2 is bound to an activation inhibitor, Kelch-like ECH-associated protein 1 (KEAP1). However, during oxidative stress. NRF2 dissociates from KEAP1 and then translocates into the nucleus to upregulate expression of the antioxidant gene, HO-1. The present study showed that both NRF2 and HO-1 proteins were upregulated after SAH. Moreover, L-Cys administration further upregulated NRF2/HO-1 expression after SAH, but this was blocked by AOAA. Similar results have been observed in cardiac (Peake et al., 2013) and hepatic (Shimada et al., 2015) ischemia-reperfusion injury and in spinal cord injury (Kesherwani et al., 2013) by activating NRF2/HO-1. These results indicate that oxidative stress may occur via an impaired CBS/H2S pathway following SAH, which leads to neuronal damage and early brain injury. Nevertheless, the antioxidant effects of L-Cys through NRF2/HO-1 activation may be involved in the protective mechanism after SAH.
Another pathophysiological response, ER stress, has been implicated in a wide range of neurological diseases (Roussel et al., 2013). To counter ER stress, UPR is initiated in neural cells by three ER membrane-associated sensors: IRE1, pancreatic endoplasmic reticulum kinase (PERK) and ATF6, which activate three signaling pathways, namely IRE1α/X-box-binding protein 1 (XBP1), PERK/eIF2α/ATF4, and ATF6α pathways (Hammadi et al., 2013). ER stress seems to play dual roles in cell fate; mild ER stress suppresses neuronal death in mouse PD models (Fouillet et al., 2012), whereas severe or long-lasting ER stress causes cell death (Bodalia et al., 2013), possibly because of excessive stimulation of UPR pathways, and the activation of CHOP, which mediates ER stress-induced neural apoptosis by altering the expression of BCL2 family members (Sano and Reed, 2013). Associated with the UPR, GRP78 is an important molecular chaperone in ER stress, and treatments that stimulate the UPR in the ER increase GRP78 expression (Kozutsumi et al., 1988).
Our present study shows that both GRP78 and CHOP were upregulated after SAH, which is consistent with the study of Yan et al. (2014). Nonetheless, this expression was remarkably decreased following L-Cys treatment and this decrease was blocked by the CBS inhibitor, AOAA. This evidence indicates that ER stress is involved in the pathogenesis of early brain injury and that L-Cys can ameliorate ER stress after SAH by generating H2S. To our surprise, three of the UPR pathways changed in different ways after L-Cys administration. The increased eIF2α phosphorylation indicated activation of the PERK pathway, which was consistent with a previous study (Yan et al., 2017), and L-Cys administration reduced this increase. The IRE1α pathway was similarly activated but L-Cys administration did not change its activation. The most interesting phenomenon appeared in the ATF6α pathway where ATF6α expression is upregulated after L-Cys injection into SAH rats. Upon its release from the ER by GRP78 in response to ER stress, ATF6α also translocates to the Golgi complex, where it is processed to its active form to regulate numerous genes. ATF6α has antioxidant activity by inducing antioxidant proteins that reside inside and outside the ER (Jin et al., 2017). We speculate that increased levels of ATF6α following L-Cys treatment most likely exert its antioxidant effects after SAH, but this requires further investigation.
Our study has several limitations. First, the specific target cells of L-Cys have not been studied. Second, the mechanism of activation of the NRF2/HO-1 system by L-Cys has not been fully elucidated. Third, the interaction of the complement system with endoplasmic reticulum stress after SAH and L-Cys administration was not investigated. Further research of these aspects is highly warranted.
In summary, L-Cys treatment has a therapeutic effect against early brain injury. L-Cys treatment can successfully supply H2S to remarkably ameliorate early brain injury following SAH, attenuate neuroinflammation and complement deposition, relieve oxidative stress and ER stress, and alter the UPR response.
Additional file: Open peer review reports 1 (91.2KB, pdf) , 2 (96.9KB, pdf) and 3 (92.1KB, pdf) .
Footnotes
Conflicts of interest: None declared.
Financial support: The study was supported by the National Natural Science Foundation of China, Nos. 81873768 and 81671213 (to ZW), 81571284 and 81874083 (to GL); the Key Research and Development Foundation of Shandong Province of China, No. 2017GSF218091 (to ZW); the Natural Science Foundation of Shandong Province of China, No. ZR2016HM33 (to DXL); the Shandong Medical and Health Science and Technology Development Plan Project of China, No. 2017WS068 (to QH); the Taishan Scholars of Shandong Province of China, No. ts201511093 (to GL). The funding sources had no role in study conception and design, data analysis or interpretation, paper writing or deciding to submit this paper for publication.
Institutional review board statement: The study was approved by the Animals Ethics Committee of Shandong University, China on February 22, 2016 (approval No. LL-201602022). The experimental procedure followed the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996).
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: Peng Luo, Fourth Military Medical University, China; Guilherme R. França, Universidade Federal Fluminense Laboratory of Neurochemistry, Brazil; Susan Goulding, Cork Institute of Technology Biological Sciences, Ireland.
Funding: The study was supported by the National Natural Science Foundation of China, Nos. 81873768 and 81671213 (to ZW), 81571284 and 81874083 (to GL); the Key Research and Development Foundation of Shandong Province of China, No. 2017GSF218091 (to ZW); the Natural Science Foundation of Shandong Province of China, No. ZR2016HM33 (to DXL); the Shandong Medical and Health Science and Technology Development Plan Project of China, No. 2017WS068 (to QH); the Taishan Scholars of Shandong Province of China, No. ts201511093 (to GL).
Chinese Library Classification No. R453; R363; R364
P-Reviewers: Luo P, França GR, Goulding S; C-Editor: Zhao M; S-Editors: Wang J, Li CH; L-Editors: Jeremy Allen, de Souza M, Qiu Y, Song LP; T-Editor: Jia Y
References
- 1.Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci. 1996;16:1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-Magableh MR, Hart JL. Mechanism of vasorelaxation and role of endogenous hydrogen sulfide production in mouse aorta. Naunyn Schmiedebergs Arch Pharmacol. 2011;383:403–413. doi: 10.1007/s00210-011-0608-z. [DOI] [PubMed] [Google Scholar]
- 3.Arumugam TV, Magnus T, Woodruff TM, Proctor LM, Shiels IA, Taylor SM. Complement mediators in ischemia-reperfusion injury. Clin Chim Acta. 2006;374:33–45. doi: 10.1016/j.cca.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 4.Asimakopoulou A, Panopoulos P, Chasapis CT, Coletta C, Zhou ZM, Cirino G, Giannis A, Szabo C, Spyroulias GA, Papapetropoulos A. Selectivity of commonly used pharmacological inhibitors for cystathionine synthase (CBS) and cystathionine lyase (CSE) Brit J Pharmacol. 2013;169:922–932. doi: 10.1111/bph.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ayer RE, Zhang JH. Oxidative stress in subarachnoid haemorrhage: significance in acute brain injury and vasospasm. Acta Neurochir Suppl. 2008;104:33–41. doi: 10.1007/978-3-211-75718-5_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bederson JB, Germano IM, Guarino L. Cortical blood flow and cerebral perfusion pressure in a new noncraniotomy model of subarachnoid hemorrhage in the rat. Stroke. 1995;26:1086–1091. doi: 10.1161/01.str.26.6.1086. [DOI] [PubMed] [Google Scholar]
- 7.Bodalia A, Li H, Jackson MF. Loss of endoplasmic reticulum Ca2+ homeostasis: contribution to neuronal cell death during cerebral ischemia. Acta Pharmacol Sin. 2013;34:49–59. doi: 10.1038/aps.2012.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao S, Zhu P, Yu X, Chen J, Li J, Yan F, Wang L, Yu J, Chen G. Hydrogen sulfide attenuates brain edema in early brain injury after subarachnoid hemorrhage in rats: Possible involvement of MMP-9 induced blood-brain barrier disruption and AQP4 expression. Neurosci Lett. 2016;621:88–97. doi: 10.1016/j.neulet.2016.04.018. [DOI] [PubMed] [Google Scholar]
- 9.Clemente Plaza N, Reig Garcia-Galbis M, Martinez-Espinosa RM. Effects of the usage of L-cysteine (l-Cys) on human health. Molecules. 2018;23:E575. doi: 10.3390/molecules23030575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coletta C, Modis K, Szczesny B, Brunyanszki A, Olah G, Rios EC, Yanagi K, Ahmad A, Papapetropoulos A, Szabo C. Regulation of vascular tone, angiogenesis and cellular bioenergetics by the 3-mercaptopyruvate sulfurtransferase/h2s pathway: functional impairment by hyperglycemia and restoration by DL-alpha-lipoic acid. Mol Med. 2015;21:1–14. doi: 10.2119/molmed.2015.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui Y, Duan X, Li H, Dang B, Yin J, Wang Y, Gao A, Yu Z, Chen G. Hydrogen sulfide ameliorates early brain injury following subarachnoid hemorrhage in rats. Mol Neurobiol. 2016;53:3646–3657. doi: 10.1007/s12035-015-9304-1. [DOI] [PubMed] [Google Scholar]
- 12.Duan X, Wen Z, Shen H, Shen M, Chen G. Intracerebral hemorrhage, oxidative stress and antioxidant therapy. Oxid Med Cell Longev. 2016;2016:1203285. doi: 10.1155/2016/1203285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emmez H, Borcek AO, Gonul, Belen HB, Solaroglu I, Baykaner MK. The effect of hydrogen sulphide on experimental cerebral vasospasm. Turk Neurosurg. 2017;27:374–379. doi: 10.5137/1019-5149.JTN.16101-15.1. [DOI] [PubMed] [Google Scholar]
- 14.Eto K, Asada T, Arima K, Makifuchi T, Kimura H. Brain hydrogen sulfide is severely decreased in Alzheimer’s disease. Biochem Bioph Res Commun. 2002;293:1485–1488. doi: 10.1016/S0006-291X(02)00422-9. [DOI] [PubMed] [Google Scholar]
- 15.Fouillet A, Levet C, Virgone A, Robin M, Dourlen P, Rieusset J, Belaidi E, Ovize M, Touret M, Nataf S, Mollereau B. ER stress inhibits neuronal death by promoting autophagy. Autophagy. 2012;8:915–926. doi: 10.4161/auto.19716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fujii M, Sherchan P, Krafft PR, Rolland WB, Soejima Y, Zhang JH. Cannabinoid type 2 receptor stimulation attenuates brain edema by reducing cerebral leukocyte infiltration following subarachnoid hemorrhage in rats. J Neurol Sci. 2014;342:101–106. doi: 10.1016/j.jns.2014.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26:627–635. doi: 10.1161/01.str.26.4.627. [DOI] [PubMed] [Google Scholar]
- 18.Grobelny BT, Ducruet AF, DeRosa PA, Kotchetkov IS, Zacharia BE, Hickman ZL, Fernandez L, Narula R, Claassen J, Lee K, Badjatia N, Mayer SA, Connolly ES. Gain-of-function polymorphisms of cystathionine beta-synthase and delayed cerebral ischemia following aneurysmal subarachnoid hemorrhage. J Neurosurg. 2011;115:101–107. doi: 10.3171/2011.2.JNS101414. [DOI] [PubMed] [Google Scholar]
- 19.Hammadi M, Oulidi A, Gackiere F, Katsogiannou M, Slomianny C, Roudbaraki M, Dewailly E, Delcourt P, Lepage G, Lotteau S, Ducreux S, Prevarskaya N, Van Coppenolle F. Modulation of ER stress and apoptosis by endoplasmic reticulum calcium leak via translocon during unfolded protein response: involvement of GRP78. FASEB J. 2013;27:1600–1609. doi: 10.1096/fj.12-218875. [DOI] [PubMed] [Google Scholar]
- 20.Hasegawa Y, Suzuki H, Altay O, Zhang JH. Preservation of tropomyosin-related kinase B (TrkB) signaling by sodium orthovanadate attenuates early brain injury after subarachnoid hemorrhage in rats. Stroke. 2011;42:477–483. doi: 10.1161/STROKEAHA.110.597344. [DOI] [PubMed] [Google Scholar]
- 21.Hendrix P, Foreman PM, Harrigan MR, Fisher WS, Vyas NA, Lipsky RH, Lin M, Walters BC, Tubbs RS, Shoja MM, Pittet JF, Mathru M, Griessenauer CJ. Association of cystathionine beta-synthase polymorphisms and aneurysmal subarachnoid hemorrhage. J Neurosurg. 2018;128:1771–1777. doi: 10.3171/2017.2.JNS162933. [DOI] [PubMed] [Google Scholar]
- 22.Hu LF, Wong PT, Moore PK, Bian JS. Hydrogen sulfide attenuates lipopolysaccharide-induced inflammation by inhibition of p38 mitogen-activated protein kinase in microglia. J Neurochem. 2007;100:1121–1128. doi: 10.1111/j.1471-4159.2006.04283.x. [DOI] [PubMed] [Google Scholar]
- 23.Jin JK, Blackwood EA, Azizi K, Thuerauf DJ, Fahem AG, Hofmann C, Kaufman RJ, Doroudgar S, Glembotski CC. ATF6 decreases myocardial ischemia/reperfusion damage and links ER stress and oxidative stress signaling pathways in the heart. Circ Res. 2017;120:862–875. doi: 10.1161/CIRCRESAHA.116.310266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin S, Pu SX, Hou CL, Ma FF, Li N, Li XH, Tan B, Tao BB, Wang MJ, Zhu YC. Cardiac H2S generation is reduced in ageing diabetic mice. Oxid Med Cell Longev. 2015;2015:758358. doi: 10.1155/2015/758358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanamaru H, Suzuki H. Potential therapeutic molecular targets for blood-brain barrier disruption after subarachnoid hemorrhage. Neural Regen Res. 2019;14:1138–1143. doi: 10.4103/1673-5374.251190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kesherwani V, Nelson KS, Agrawal SK. Effect of sodium hydrosulphide after acute compression injury of spinal cord. Brain Res. 2013;1527:222–229. doi: 10.1016/j.brainres.2013.06.023. [DOI] [PubMed] [Google Scholar]
- 27.Kimura H. Physiological role of hydrogen sulfide and polysulfide in the central nervous system. Neurochem Int. 2013;63:492–497. doi: 10.1016/j.neuint.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 28.Kimura H. Production and physiological effects of hydrogen sulfide. Antioxid Redox Signal. 2014;20:783–793. doi: 10.1089/ars.2013.5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kofler M, Schiefecker A, Ferger B, Beer R, Sohm F, Broessner G, Hackl W, Rhomberg P, Lackner P, Pfausler B, Thome C, Schmutzhard E, Helbok R. Cerebral taurine levels are associated with brain edema and delayed cerebral infarction in patients with aneurysmal subarachnoid hemorrhage. Neurocrit Care. 2015;23:321–329. doi: 10.1007/s12028-015-0140-y. [DOI] [PubMed] [Google Scholar]
- 30.Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462–464. doi: 10.1038/332462a0. [DOI] [PubMed] [Google Scholar]
- 31.Li T, Wang L, Hu Q, Liu S, Bai X, Xie Y, Zhang T, Bo S, Gao X, Wu S, Li G, Wang Z. Neuroprotective Roles of l-cysteine in attenuating early brain injury and improving synaptic density via the CBS/H2S pathway following subarachnoid hemorrhage in rats. Front Neurol. 2017;8:176. doi: 10.3389/fneur.2017.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu S, Xin D, Wang L, Zhang T, Bai X, Li T, Xie Y, Xue H, Bo S, Liu D, Wang Z. Therapeutic effects of L-Cysteine in newborn mice subjected to hypoxia-ischemia brain injury via the CBS/H2S system: Role of oxidative stress and endoplasmic reticulum stress. Redox Biol. 2017;13:528–540. doi: 10.1016/j.redox.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu X, Zhang N, Ding Y, Cao D, Huang Y, Chen X, Wang R, Lu N. Hydrogen sulfide regulates the [Ca(2+)]i level in the primary medullary neurons. Oxid Med Cell Longev. 2016;2016:2735347. doi: 10.1155/2016/2735347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu ZW, Zhao JJ, Pang HG, Song JN. Vascular endothelial growth factor A promotes platelet adhesion to collagen IV and causes early brain injury after subarachnoid hemorrhage. Neural Regen Res. 2019;14:1726–1733. doi: 10.4103/1673-5374.257530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macdonald RL, Schweizer TA. Spontaneous subarachnoid haemorrhage. Lancet. 2017;389:655–666. doi: 10.1016/S0140-6736(16)30668-7. [DOI] [PubMed] [Google Scholar]
- 36.Macdonald RL, Kassell NF, Mayer S, Ruefenacht D, Schmiedek P, Weidauer S, Frey A, Roux S, Pasqualin A, Investigators C. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1): randomized, double-blind, placebo-controlled phase 2 dose-finding trial. Stroke. 2008;39:3015–3021. doi: 10.1161/STROKEAHA.108.519942. [DOI] [PubMed] [Google Scholar]
- 37.Macdonald RL, Higashida RT, Keller E, Mayer SA, Molyneux A, Raabe A, Vajkoczy P, Wanke I, Bach D, Frey A, Marr A, Roux S, Kassell N. Randomised trial of clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid hemorrhage undergoing surgical clipping (CONSCIOUS-2) Acta Neurochir Suppl. 2013;115:27–31. doi: 10.1007/978-3-7091-1192-5_7. [DOI] [PubMed] [Google Scholar]
- 38.Mack WJ, Ducruet AF, Hickman ZL, Garrett MC, Albert EJ, Kellner CP, Mocco J, Connolly ES., Jr Early plasma complement C3a levels correlate with functional outcome after aneurysmal subarachnoid hemorrhage. Neurosurgery. 2007;61:255–260. doi: 10.1227/01.NEU.0000255518.96837.8E. [DOI] [PubMed] [Google Scholar]
- 39.Ostrowski RP, Colohan AR, Zhang JH. Molecular mechanisms of early brain injury after subarachnoid hemorrhage. Neurol Res. 2006;28:399–414. doi: 10.1179/016164106X115008. [DOI] [PubMed] [Google Scholar]
- 40.Peake BF, Nicholson CK, Lambert JP, Hood RL, Amin H, Amin S, Calvert JW. Hydrogen sulfide preconditions the db/db diabetic mouse heart against ischemia-reperfusion injury by activating Nrf2 signaling in an Erk-dependent manner. Am J Physiol Heart Circ Physiol. 2013;304:H1215–1224. doi: 10.1152/ajpheart.00796.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prunell GF, Svendgaard NA, Alkass K, Mathiesen T. Inflammation in the brain after experimental subarachnoid hemorrhage. Neurosurgery. 2005;56:1082–1092. [PubMed] [Google Scholar]
- 42.Roussel BD, Kruppa AJ, Miranda E, Crowther DC, Lomas DA, Marciniak SJ. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2013;12:105–118. doi: 10.1016/S1474-4422(12)70238-7. [DOI] [PubMed] [Google Scholar]
- 43.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460–3470. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schneider UC, Xu R, Vajkoczy P. Inflammatory events following subarachnoid hemorrhage (SAH) Curr Neuropharmacol. 2018;16:1385–1395. doi: 10.2174/1570159X16666180412110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sehba FA, Hou J, Pluta RM, Zhang JH. The importance of early brain injury after subarachnoid hemorrhage. Prog Neurobiol. 2012;97:14–37. doi: 10.1016/j.pneurobio.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shimada S, Fukai M, Wakayama K, Ishikawa T, Kobayashi N, Kimura T, Yamashita K, Kamiyama T, Shimamura T, Taketomi A, Todo S. Hydrogen sulfide augments survival signals in warm ischemia and reperfusion of the mouse liver. Surg Today. 2015;45:892–903. doi: 10.1007/s00595-014-1064-4. [DOI] [PubMed] [Google Scholar]
- 47.Wang JF, Li Y, Song JN, Pang HG. Role of hydrogen sulfide in secondary neuronal injury. Neurochem Int. 2014;64:37–47. doi: 10.1016/j.neuint.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Huang Y, Xu Y, Ruan W, Wang H, Zhang Y, Saavedra JM, Zhang L, Huang Z, Pang T. A dual AMPK/Nrf2 activator reduces brain inflammation after stroke by enhancing microglia M2 polarization. Antioxid Redox Signal. 2018;28:141–163. doi: 10.1089/ars.2017.7003. [DOI] [PubMed] [Google Scholar]
- 49.Wang Z, Liu DX, Wang FW, Zhang Q, Du ZX, Zhan JM, Yuan QH, Ling EA, Hao AJ. (L)-cysteine promotes the proliferation and differentiation of neural stem cells via the Cbs/H2s pathway. Neuroscience. 2013a;237:106–117. doi: 10.1016/j.neuroscience.2012.12.057. [DOI] [PubMed] [Google Scholar]
- 50.Wang Z, Liu DX, Wang FW, Zhang Q, Du ZX, Zhan JM, Yuan QH, Ling EA, Hao AJ. L-Cysteine promotes the proliferation and differentiation of neural stem cells via the CBS/H(2)S pathway. Neuroscience. 2013b;237:106–117. doi: 10.1016/j.neuroscience.2012.12.057. [DOI] [PubMed] [Google Scholar]
- 51.Xie YK, Zhou X, Yuan HT, Qiu J, Xin DQ, Chu XL, Wang DC, Wang Z. Resveratrol reduces brain injury after subarachnoid hemorrhage by inhibiting oxidative stress and endoplasmic reticulum stress. Neural Regen Res. 2019;14:1734–1742. doi: 10.4103/1673-5374.257529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xin D, Chu X, Bai X, Ma W, Yuan H, Qiu J, Liu C, Li T, Zhou X, Chen W, Liu D, Wang Z. l-Cysteine suppresses hypoxia-ischemia injury in neonatal mice by reducing glial activation, promoting autophagic flux and mediating synaptic modification via H2S formation. Brain Behav Immun. 2018;73:222–234. doi: 10.1016/j.bbi.2018.05.007. [DOI] [PubMed] [Google Scholar]
- 53.Yan F, Li JR, Chen JY, Hu Q, Gu C, Lin W, Chen G. Endoplasmic reticulum stress is associated with neuroprotection against apoptosis via autophagy activation in a rat model of subarachnoid hemorrhage. Neurosci Lett. 2014;563:160–165. doi: 10.1016/j.neulet.2014.01.058. [DOI] [PubMed] [Google Scholar]
- 54.Yan F, Cao SL, Li JR, Dixon B, Yu XB, Chen JY, Gu C, Lin W, Chen G. Pharmacological inhibition of PERK attenuates early brain injury after subarachnoid hemorrhage in rats through the activation of akt. Mol Neurobiol. 2017;54:1808–1817. doi: 10.1007/s12035-016-9790-9. [DOI] [PubMed] [Google Scholar]
- 55.Yetik-Anacak G, Sevin G, Ozzayim O, Dereli MV, Ahmed A. Hydrogen sulfide: a novel mechanism for the vascular protection by resveratrol under oxidative stress in mouse aorta. Vascul Pharmacol. 2016;87:76–82. doi: 10.1016/j.vph.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 56.Xie YK, Zhou X, Yuan HT, Qiu J, Xin DQ, Chu XL, Wang DC, Wang Z. Resveratrol reduces brain injury after subarachnoid hemorrhage by inhibiting oxidative stress and endoplasmic reticulum stress. Neural Regen Res. 2019;14:1734–1742. doi: 10.4103/1673-5374.257529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu YP, Chi XL, Liu LJ. A Hypothesis: hydrogen sulfide might be neuroprotective against subarachnoid hemorrhage induced brain injury. ScientificWorldJournal. 2014;2014:432318. doi: 10.1155/2014/432318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.