

Chlamydia trachomatis is the most reported sexually transmitted infection in the United States and the leading cause of infectious blindness worldwide. Unlike many other intracellular pathogens that have undergone reductive evolution, C. trachomatis is capable of de novo biosynthesis of the essential cofactor tetrahydrofolate using a noncanonical pathway. Here, we identify the biosynthetic precursor to the p-aminobenzoate (pABA) portion of folate in a process that requires the CT610 enzyme from C. trachomatis. We further provide evidence that CT610 is a self-sacrificing or “suicide” enzyme that uses its own amino acid residue(s) as the substrate for pABA synthesis. This work provides the foundation for future investigation of this chlamydial pABA synthase, which could lead to new therapeutic strategies for C. trachomatis infections.
KEYWORDS: Chlamydia, folate biosynthesis, oxygenases, p-aminobenzoate, pABA, suicide enzyme
ABSTRACT
Chlamydia trachomatis lacks the canonical genes required for the biosynthesis of p-aminobenzoate (pABA), a component of essential folate cofactors. Previous studies revealed a single gene from C. trachomatis, the CT610 gene, that rescues Escherichia coli ΔpabA, ΔpabB, and ΔpabC mutants, which are otherwise auxotrophic for pABA. CT610 shares low sequence similarity to nonheme diiron oxygenases, and the previously solved crystal structure revealed a diiron active site. Genetic studies ruled out several potential substrates for CT610-dependent pABA biosynthesis, including chorismate and other shikimate pathway intermediates, leaving the actual precursor(s) unknown. Here, we supplied isotopically labeled potential precursors to E. coli ΔpabA cells expressing CT610 and found that the aromatic portion of tyrosine was highly incorporated into pABA, indicating that tyrosine is a precursor for CT610-dependent pABA biosynthesis. Additionally, in vitro enzymatic experiments revealed that purified CT610 exhibits low pABA synthesis activity under aerobic conditions in the absence of tyrosine or other potential substrates, where only the addition of a reducing agent such as dithiothreitol appears to stimulate pABA production. Furthermore, site-directed mutagenesis studies revealed that two conserved active site tyrosine residues are essential for the pABA synthesis reaction in vitro. Thus, the current data are most consistent with CT610 being a unique self-sacrificing enzyme that utilizes its own active site tyrosine residue(s) for pABA biosynthesis in a reaction that requires O2 and a reduced diiron cofactor.
IMPORTANCE Chlamydia trachomatis is the most reported sexually transmitted infection in the United States and the leading cause of infectious blindness worldwide. Unlike many other intracellular pathogens that have undergone reductive evolution, C. trachomatis is capable of de novo biosynthesis of the essential cofactor tetrahydrofolate using a noncanonical pathway. Here, we identify the biosynthetic precursor to the p-aminobenzoate (pABA) portion of folate in a process that requires the CT610 enzyme from C. trachomatis. We further provide evidence that CT610 is a self-sacrificing or “suicide” enzyme that uses its own amino acid residue(s) as the substrate for pABA synthesis. This work provides the foundation for future investigation of this chlamydial pABA synthase, which could lead to new therapeutic strategies for C. trachomatis infections.
INTRODUCTION
Folates are tripartite molecules consisting of a pteridine ring, p-aminobenzoate (pABA), and glutamate residues. The fully reduced tetrahydrofolate (H4folate) molecule is the active cofactor, in which one-carbon units of various oxidation states are attached to the N-5 and/or N-10 positions for use in central metabolic pathways required for DNA and amino acid biosynthesis (1). Most bacteria and plants synthesize H4folate de novo (2, 3). In the canonical H4folate biosynthetic pathway (Fig. 1), the pteridine portion of the cofactor is derived from GTP in four well-characterized reactions catalyzed by the FolEQBK enzymes, while pABA is synthesized from chorismate through the action of two enzymes, aminodeoxychorismate (ADC) synthase (PabA/PabB) and ADC lyase (PabC) (Fig. 1, red). The pteridine ring and pABA are then combined to generate dihydropteroate, which is subsequently glutamylated and reduced to produce the final H4folate (Fig. 1).
FIG 1.

Shikimate pathway leading to the biosynthesis of the following tetrahydrofolate and aromatic amino acids with corresponding gene names in E. coli: 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase (aroF, aroG, and aroH), dehydroquinate synthase (aroB), 3-dehydroquinate dehydratase (aroD), shikimate dehydrogenase (aroE), shikimate kinase (aroL), 5-enolpyruvylshikimate 3-phosphate synthase (aroA), chorismate synthase (aroC), chorismate mutase/prephenate dehydratase (pheA and tyrA), transaminases (tyrB, aspC, and ilvE), chorismate lyase (ubiC), ADC synthase (pabA and pabB), ADC lyase (pabC), GTP cyclohydrolase (folE), dihydroneopterin triphosphate pyrophosphohydrolase (folQ), dihydroneopterin aldolase (folB), 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase (folK), dihydropteroate synthase (folP). PEP, phosphoenolpyruvate; CoQ, coenzyme Q. Dotted arrows represent multiple steps.
Chlamydia trachomatis is an obligate intracellular bacterial pathogen that is reliant on its host for many essential nutrients, including several metabolic precursors and some cofactors (4). However, C. trachomatis has retained the ability to synthesize H4folate de novo and is sensitive to antibiotics targeting the H4folate biosynthetic pathway (5). Analysis of the C. trachomatis genome revealed a H4folate biosynthetic gene cluster; however, several canonical H4folate biosynthetic genes are not present (6). These missing genes include those encoding the first two enzymes required for the conversion of GTP to the pteridine portion of folate (FolE and FolQ), the enzyme responsible for the addition of γ-linked glutamate residues (FolC), and the enzymes required for pABA biosynthesis from chorismate (PabA/PabB and PabC) (Fig. 1) (6, 7). Genetic and enzymatic experiments have demonstrated that C. trachomatis employs RibA (GTP cyclohydrolase II) and TrpF (N′-5′-phosphoribosylanthranilate isomerase) to bypass FolE and FolQ in order to generate 7,8-dihydroneopterin monophosphate (8), while an archaeal γ-glutamyl ligase was shown to functionally replace FolC (6). Finally, a single gene present in the C. trachomatis H4folate biosynthetic gene cluster, the CT610 gene, was found to complement E. coli mutants with deletions of pabA, pabB, or pabC, indicating that the encoded enzyme is responsible for pABA biosynthesis in C. trachomatis (8). Interestingly, CT610 could still complement E. coli ΔaroA mutants that cannot synthesize chorismate, demonstrating that chorismate is not the substrate for CT610. Further complementation studies showed that the upstream shikimate pathway intermediates shikimate and 3-dehydroquinate (Fig. 1) are also not precursors for CT610-dependent pABA biosynthesis (8).
In addition to C. trachomatis and other species of Chlamydia, a few other bacteria, including Lactobacillus fermentum IFO 3956 and Nitrosomonas europaea NBRC 14298, are missing pabA, pabB, and pabC, even though they have other genes required for H4folate biosynthesis and are not folate auxotrophs (9–12). The CT610 ortholog from N. europaea, NE1434, has been shown to complement a pABA-auxotrophic E. coli mutant (ΔpabABC) (13). Similarly to studies with CT610, E. coli shikimate pathway knockout mutants were generated in the ΔpabABC background, and all were able to grow in minimal medium when they were complemented with NE1434 (13). These studies demonstrated that none of the intermediates in the shikimate pathway could be precursors in this alternate route for pABA biosynthesis, except for possibly 3-deoxy-d-arabinoheptulosonate-7-phosphate (Fig. 1). Thus, the precursor for this novel route for pABA biosynthesis remains unknown.
CT610 and its orthologs were previously or still are annotated as PqqC or PqqC-like; CT610 shares 18% identity and 26% similarity with PqqC from Klebsiella pneumoniae (14). PqqC is a cofactor-independent oxidase that catalyzes an 8-electron oxidation and ring closure in the final step of coenzyme pyrroloquinoline quinone (PQQ) biosynthesis (15). However, C. trachomatis and other organisms with orthologs to CT610 are not known to use PQQ and do not have the other genes required for PQQ biosynthesis. Interestingly, CT610 was originally identified as a death domain-containing protein based upon sequence similarity and was demonstrated to function in modulating host cell apoptosis during C. trachomatis infection by interacting with the death domains of necrosis factor family receptors (16). Thus, CT610 was named Chlamydia protein associating with death domains (CADD) (16). However, when the crystal structure of CT610 was obtained (17), it did not contain the expected death domain structure and instead revealed overall structural similarity to PqqC (18) and human heme-oxygenase (19), with a diiron active site similar to the hydroxylase component of soluble methane monooxygenase (20) as well as to the β subunit of class Ia ribonucleotide reductase (21, 22).
In this study, we further explore the role of CT610 in the unusual route for pABA biosynthesis by performing isotope feeding studies with CT610 expressed in E. coli ΔpabA cells and through in vitro enzyme assays coupled with site-directed mutagenesis experiments.
RESULTS
Analysis of CT610-dependent pABA production in E. coli ΔpabA.
CT610 from C. trachomatis has been shown to rescue E. coli strains with deletions of pabA, pabB, or pabC (8), indicating that CT610 alone can complete pABA biosynthesis using an unknown precursor. To confirm this activity and serve as a starting point for further studies, we investigated the E. coli ΔpabA mutant (VDC9500) (8) transformed with a pUC19 plasmid encoding CT610 (E. coli ΔpabA-CT610). The E. coli ΔpabA mutant normally requires the addition of pABA when grown on minimal medium, but when CT610 is provided, the cells can grow without supplemental pABA, confirming the involvement of CT610 in pABA biosynthesis (Fig. 2, left). Based upon the established structural similarity of the CT610 active site to enzymes that utilize molecular oxygen as a substrate, such as methane monooxygenase and class Ia ribonucleotide reductase (17), we reasoned that CT610-dependent pABA production would require the presence of O2. Therefore, we cultured E. coli ΔpabA-CT610 anaerobically in minimal medium lacking pABA compared to E. coli ΔpabA with pABA supplemented. The anaerobic culture supplemented with pABA showed growth as expected, while in the anaerobic culture lacking pABA but containing CT610, no growth was observed (Fig. 2, right). This result suggests that CT610 requires O2 for pABA production.
FIG 2.

Complementation studies of E. coli ΔpabA with CT610 in the presence or absence of oxygen. The inability of CT610 to complement the pABA auxotroph under anaerobic conditions indicates that CT610 requires oxygen for pABA production.
To verify the presence of pABA in E. coli ΔpabA-CT610 cells, we originally attempted to identify this compound by liquid chromatography-mass spectrometry (LC-MS) as well as by gas chromatography-mass spectrometry using a trifluoroacetyl-methyl ester derivative. We were not successful at consistently observing pABA in cell extracts, presumably because of its low concentration as a free metabolite. Thus, a more reliable method was desired, especially for future isotope labeling experiments. Since the majority of pABA in cells is present as a component of complete folate cofactors, we isolated and partially purified folates from the E. coli cells. After enzymatic treatment of cell extracts to yield monoglutamylated folate derivatives, the resulting folates were purified on an anion exchange resin, and then reductively cleaved with zinc to yield glutamyl-pABA (Fig. 3A). The zinc cleavage was performed to isolate the pABA-containing moiety from the remainder of the folate cofactor, thus making subsequent isotope incorporation experiments easier to interpret. Glutamyl-pABA was indeed present in the E. coli ΔpabA-CT610 cells based on comparison of the LC retention time (Fig. 3B) and the tandem mass spectrometry (MS-MS) spectrum (Fig. 3C) to authentic glutamyl-pABA and glutamyl-pABA isolated from control E. coli cells that biosynthesize pABA via the canonical route. The ion at 265 m/z corresponds to the [M − H]− molecular ion of glutamyl-pABA.
FIG 3.

Generation of glutamyl-pABA from folates and its analysis by LC-MS. (A) Zinc cleavage of folates extracted from E. coli cells to produce glutamyl-pABA. (B) Total ion current chromatogram of partially purified glutamyl-pABA (9.6 min) from E. coli ΔpabA-CT610 cell extracts; (C) the corresponding MS/MS spectrum of glutamyl-pABA showing the [M − H]− molecular ion and several characteristic fragment ions.
Isotope feeding experiments to determine the biosynthetic precursor of CT610-derived pABA.
It was previously shown that chorismate or shikimate pathway intermediates were not direct precursors to pABA in the CT610-dependent route (8, 13). This was determined by investigating the ability of CT610 to rescue E. coli shikimate pathway deletion strains, which had to be cultured with added aromatic amino acids and p-hydroxybenzoate (pHB) (Fig. 1, green). Therefore, we hypothesized that l-tyrosine (Fig. 1, blue) or pHB could be potential precursors for CT610-dependent pABA biosynthesis. These substrates were previously tested with the purified CT610 ortholog from N. europaea without any observed activity (13); however, we reasoned that these in vitro experiments may have been missing a required reaction component and/or were carried out under suboptimal conditions.
To test whether tyrosine and/or pHB were precursors to pABA in the CT610-dependent pathway, we supplemented the E. coli ΔpabA-CT610 cultures grown in minimal medium with either p-[3,5-2H2]HB or l-[3,5-2H2]tyrosine, where each of the labeled compounds contain two deuteriums on the aromatic ring. Percent incorporation of each isotopically labeled precursor was determined using the isotopic distribution of glutamyl-pABA from cells without any added precursors as a baseline (Fig. 4A). In cultures supplemented with p-[3,5-2H2]HB, about 15% of the isolated glutamyl-pABA contained two deuteriums (Fig. 4B). This is based on the increased intensity of the [M − H + 2]− ion at 267 m/z. When cultured under the same conditions in minimal medium, control E. coli cells with the canonical pABA biosynthetic pathway did not incorporate any deuterium from p-[3,5-2H2]HB into glutamyl-pABA as expected. This indicates that the CT610-dependent route can use pHB as a precursor to pABA.
FIG 4.

Incorporation of isotopically labeled precursors into glutamyl-pABA produced in E. coli ΔpabA-CT610. Shown are enhanced-resolution mass spectra of glutamyl-pABA when grown in minimal medium in the absence of isotopically labeled precursors (A), supplemented with p-[3,5-2H2]pHB (B), supplemented with l-[3,5-2H2]tyrosine (C), or supplemented with [15N]tyrosine (D).
When l-[3,5-2H2]tyrosine was supplied, about 40% of the glutamyl-pABA contained two deuteriums (Fig. 4C), while control cells did not incorporate any of the deuterium-labeled tyrosine into pABA. Since an equivalent amount of unlabeled tyrosine was also provided to the cultures in these experiments, the 40% incorporation of l-[3,5-2H2]tyrosine into pABA in the cells containing CT610 is quite significant. This provides strong evidence that tyrosine acts as the precursor to pABA in the CT610-dependent route. To determine if the amino group on pABA was also derived from tyrosine, we cultured the E. coli ΔpabA_CT610 cells in the presence of l-[15N]tyrosine. Based on the increase in the [M − H + 1]− ion at 266, we found that 12% of the glutamyl-pABA derived from these cells contained 15N (Fig. 4D). However, when control E. coli cells containing the canonical pathway for pABA biosynthesis were grown with [15N]tyrosine, about 14% of the glutamyl-pABA contained 15N. Based on these results, the incorporation observed is likely due to transamination reactions occurring in the cells and does not indicate that the amino group on pABA is derived from tyrosine.
In vitro enzymatic activity experiments.
To further investigate the pABA synthesis reaction catalyzed by CT610, the enzyme was overexpressed with an N-terminal His10 tag from pET19b and purified using nickel affinity chromatography (see Fig. S1 in the supplemental material). Enzyme reactions were carried out under various conditions and were analyzed by high-performance liquid chromatography (HPLC) with UV-visible (UV-Vis) detection and/or by LC-MS with a multiple reaction monitoring (MRM) method for sensitive and specific detection of pABA. Interestingly, pABA was observed in reaction mixtures containing CT610 in the absence of any added substrates, where only dithiothreitol (DTT) was necessary to stimulate pABA production (Fig. 5). Other reducing agents such as 2-mercaptoethanol and sodium dithionite with methyl viologen also resulted in some pABA synthesis, but these alternative electron donors resulted in significantly less activity compared to that with DTT. The reducing agent in the CT610 reaction is presumably necessary for the reduction of the diiron cofactor to produce the active diferrous state for subsequent O2 activation (23, 24).
FIG 5.

LC-MS analysis of pABA production by purified CT610. (A) Extracted ion current chromatograms; (B) corresponding calculated concentration of pABA produced in a 0.5-ml reaction mixture. All traces are extracted ion current chromatograms from a multiple reaction monitoring MS method with three pairs—138.1 and 120.4, 138.1 and 94.4, 138.1 and 77.3—to specifically identify pABA, where each trace is the combined intensity of all three pairs. Quantitation was performed using the pABA peak area with the standard curve shown in Fig. S3 in the supplemental material (***, P < 0.001; ****, P < 0.0001; paired t tests versus CT610 only; n = 3). Error bars represent standard deviations. Reactions were carried out in triplicate at 37°C for 14 h in 100 mM Tris, 8 mM MgCl2, and 10% glycerol (pH 8.8). The reaction components were present when indicated at the following concentrations: CT610 (130 μM), dithiothreitol (DTT, 10 mM), l-tyrosine (1 mM), p-hydroxybenzoate (pHB; 1 mM), and p-hydroxyphenylpyruvate (pHPP; 0.5 mM).
The LC-MS peak from the CT610 reactions exhibited the expected [M + H]+ molecular ion at 138.1 m/z, a fragment ion at 120.4 m/z corresponding to a characteristic dehydration product, as well as two additional characteristic fragment ions at 94.4 m/z and 77.3 m/z, all with the expected ratio of intensities observed with a pABA standard. When analyzed by HPLC with UV-Vis detection, the pABA peak from CT610 reactions also displayed the expected absorbance spectrum (see Fig. S2 in the supplemental material). Surprisingly, the addition of l-tyrosine or pHB did not increase pABA production by CT610 (Fig. 5), indicating these are not CT610 substrates. We considered that perhaps p-hydroxyphenylpyruvate (pHPP) (Fig. 1), the transamination product of tyrosine, could instead be the CT610 substrate. But still, no increase in activity was observed when pHPP was included in CT610 reactions (Fig. 5).
The amount of pABA produced by CT610 in in vitro reactions is very low, with only about 0.4 μM resulting from reaction mixtures containing 130 μM CT610 and 10 mM DTT (Fig. 5). Iron quantitation revealed that purified CT610 contained about 0.2 mol of iron per mol of protein. Since each monomer should presumably contain a diiron cofactor, this indicates that at most 10% of the purified protein contained the complete cofactor. Additionally, a major portion of the purified enzyme may only contain a single iron atom. Unfortunately, adding Fe(II) to purified CT610 did not improve activity (see Fig. S4 in the supplemental material) and, surprisingly, it appeared to decrease pABA production. Additionally, protease cleavage of the His tag before activity assays did not result in increased pABA production (see Fig. S5 in the supplemental material). We also tested multiple possible amino group donors and potential cofactors or cosubstrates (see Materials and Methods), but did not observe any significant effect on the amount of pABA generated. Notably, reactions with NADH and NADPH did not produce increased activity compared to that with DTT alone (see Fig. S6 in the supplemental material).
To confirm the requirement for O2 in the CT610 reaction, the purified enzyme was deoxygenated by gentle stirring in an anaerobic chamber for several hours and then assayed in the presence of DTT and l-tyrosine. Only a very minor peak for pABA was detected, corresponding to less than 10% of the pABA present in an aerobic positive-control reaction (see Fig. S7 in the supplemental material), further supporting CT610 requiring O2 for the pABA synthesis reaction. Taken together, the in vitro results are consistent with CT610 being an unusual self-sacrificing enzyme that produces pABA by utilizing an active-site tyrosine residue as a substrate in a reaction that is dependent on a reduced diiron cofactor and O2 as a cosubstrate.
Site-directed mutagenesis studies.
Our analysis of the previously reported CT610 crystal structure (17) revealed five tyrosine residues that could potentially act as the sacrificial substrate for pABA production (Fig. 6). Additionally, positioned near Y27 and Y43 is a lysine residue, K152, which we hypothesized could be the necessary amino group donor in the pABA synthesis reaction (Fig. 6). Importantly, all three of these residues are conserved in the CT610 ortholog from N. europaea (see Fig. S8 in the supplemental material). To determine the involvement of the active site tyrosine residues and K152 in pABA synthesis, we utilized site-directed mutagenesis followed by in vitro assays in the presence of DTT, l-tyrosine, and O2. Intriguingly, pABA was not detected in reactions with the Y27F and Y43F variants, while Y47F, Y141F, and Y170F exhibited activity comparable that of to the wild-type enzyme (Fig. 7). Additionally, converting K152 to an arginine nearly completely abolished pABA production, where the K152R variant exhibited only about 2% of the activity of the wild type (Fig. 7). Thus, Y27 and Y43 are essential for pABA synthesis by CT610, and one or both of these residues may be the precursor to pABA in the proposed self-sacrificing reaction. The data also indicate that K152 is critical for catalysis, which is consistent with its possible function as an internal amino group donor.
FIG 6.

Active site of CT610 with residues coordinating the diiron cofactor and potential residues required for pABA production highlighted. Iron atoms are shown as orange spheres. Image generated in PyMOL Molecular Graphics System (version 2.0) using PDB accession number 1RCW (17).
FIG 7.
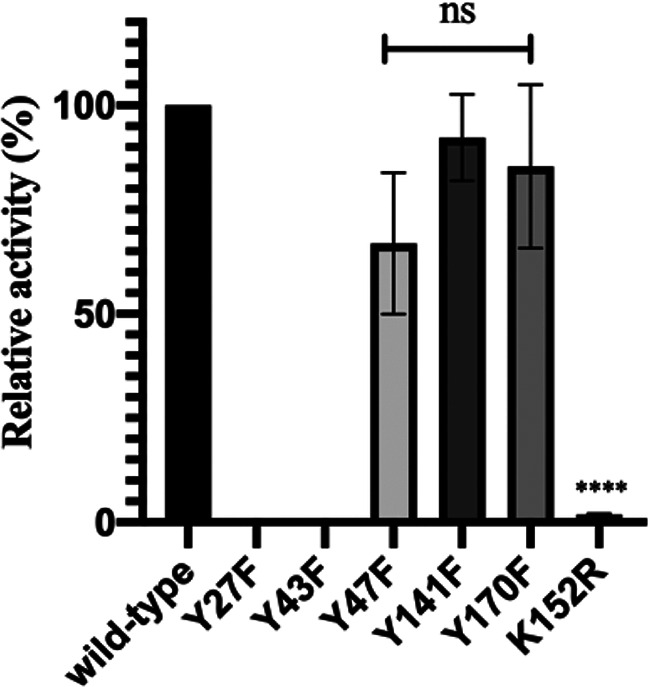
Relative activity of CT610 variants compared to that of the wild type. The activity was determined by the same in vitro enzymatic assay analyzed by LC-MS described and depicted in Fig. 5 (ns, no significant difference compared to wild type; ****, P < 0.0001; n = 3). Error bars represent standard deviations.
DISCUSSION
In the established route for folate biosynthesis, pABA is synthesized from chorismate in two enzymatic steps (Fig. 1, red). However, C. trachomatis is missing the pabABC genes required for pABA biosynthesis via this route. Previous work demonstrated that a single gene from C. trachomatis, the CT610 gene, can replace the functions of these three genes normally involved in pABA biosynthesis, but the precursor is not chorismate or other shikimate pathway intermediates (8). In this study, we further explored this CT610-dependent route for pABA biosynthesis with isotope feeding studies in E. coli and with purified CT610.
When shikimate pathway genes are deleted in E. coli, the mutants require supplementation with aromatic amino acids as well as pHB for coenzyme Q (CoQ) synthesis (Fig. 1). Since CT610-dependent pABA synthesis was demonstrated to still occur in shikimate pathway deletion strains, it seemed plausible that the precursor for pABA in the CT610-dependent route was one of these supplemented aromatic compounds. When p-[3,5-2H2]HB was provided to cultures of E. coli ΔpabA-CT610, we saw low but detectable incorporation of the label into the aromatic portion of pABA, and when l-[3,5-2H2]tyrosine was provided, we observed substantial incorporation of the label into pABA (Fig. 4). We therefore inferred that l-tyrosine was the substrate for CT610 with pHB as a possible intermediate in the reaction. Growth studies with E. coli ΔpabA-CT610 and subsequent in vitro enzyme assays showed that the CT610-dependent route for pABA biosynthesis requires O2 (Fig. 2; also see Fig. S7 in the supplemental material). Further, purified CT610 was shown to generate pABA in a reaction that requires a reducing agent but does not require added l-tyrosine or other potential substrates (Fig. 5). This led us to the proposal that a tyrosine residue in the CT610 active site serves as the precursor to pABA in a reaction that requires O2 and a reduced diiron cofactor. Although unusual, self-sacrificing or “suicide” mechanisms have been described in eukaryotic thiamine biosynthesis, in which a cysteine residue is used as a sulfur source to produce the thiazole ring in the reaction catalyzed by THI4 (25), and a histidine residue is a precursor to the pyrimidine portion of thiamine in the THI5-catalyzed reaction (26). Additionally, nickel pincer cofactor biosynthesis involves a cysteine residue as a sulfur donor (27). Most recently, a suicide enzyme was characterized in biotin biosynthesis that utilizes a lysine residue as a sacrificial amino donor (28).
Despite the sequence and overall structural similarity of CT610 to PqqC, the active sites of the two proteins are not conserved (17), consistent with the fact that Chlamydia does not synthesize PQQ and that the two enzymes must catalyze different reactions. Instead, the CT610 active site resembles the diiron active sites present in the β (R2) subunits of class Ia ribonucleotide reductase (RNR) as well as in methane monooxygenase hydroxylase (MMOH). In RNR catalysis, the β subunit generates a diferric-tyrosyl radical cofactor in the presence of O2 and reducing equivalents (29, 30). This radical is transferred via proton coupled electron transfer through several amino acid residues to produce a tyrosyl radical that is eventually transferred to a cysteine residue in the α (R1) subunit, generating the catalytic thiyl radical (30, 31). The thiyl radical then initiates catalysis for the eventual generation of a deoxynucleotide product catalyzed by RNR (29–31). On the other hand, MMOH uses the diiron center to activate O2 in order to hydroxylate methane and a variety of other hydrocarbon substrates using a complex radical recombination process. Both iron ions begin in the ferrous state before reacting with oxygen to generate the reactive intermediate, bis-μ-oxo-iron(IV), known as compound Q, which abstracts a hydrogen atom to initiate the radical sequence that results in a hydroxylated product (32, 33). More recently described diiron monooxygenases involved in natural product biosynthesis utilize a diiron active site and O2 to carry out comparable but distinct catalytic cycles (34). We propose that related chemistry is occurring in the CT610 active site to generate a pHB intermediate from a tyrosine residue. In our proposed reaction scheme (Fig. 8), a monooxygenase reaction first activates a tyrosine residue by hydroxylating the Cβ position. The side chain of the residue is subsequently released from the protein backbone via cleavage of the Cα-Cβ bond initiated by base catalysis to form p-hydroxybenzaldehyde (Fig. 8b), leaving a glycine in place of the original tyrosine residue. Another monooxygenase hydroxylation then generates pHB (Fig. 8c). Amination could occur on the quinone form, which is then dehydrated and finally rearranged to produce the final pABA (Fig. 8e to g).
FIG 8.

Proposed reaction scheme catalyzed by CT610. Dotted arrows represent multiple chemical steps analogous to the reaction catalyzed by soluble methane monooxygenase.
The identity of the specific tyrosine residue potentially serving as a precursor to pABA in the proposed reaction scheme was probed in this work using site-directed mutagenesis followed by in vitro assays. Interestingly, the Y27F and Y43F variants were inactive, while converting the other three tyrosine residues in the active site to phenylalanines did not significantly affect pABA production (Fig. 7). Notably, the result that Y170 is not required for activity (Fig. 7) does not support the hypothesis by Schwarzenbacher et al. that Y170 forms a catalytic tyrosyl radical (17), unless Y170 is involved in an additional activity outside pABA synthesis. The residues corresponding to Y27 and Y43 are conserved in the CT610 ortholog from N. europaea (Fig. S8); thus, taken together with the mutagenesis data, one or both of these tyrosine residues may be the precursor to pABA. Since the distance between the diiron site and Y27/Y43 is measured to be about 13 Å based on the crystal structure (17), a radical transfer chain may be required similar to that of class I RNRs. Interestingly, upon further examination of the crystal structure, a probable covalent link is observed between the OH of Y43 and an aromatic carbon (CE1) of Y47, which supports the suggestion of radical transfer occurring in the CT610 active site. Along with the mutagenesis data, this further suggests that Y43 may be the direct radical source for hydrogen abstraction from the β carbon of Y27 for subsequent hydroxylation and eventual conversion to pABA (Fig. 8). If this were the case, the hydroxylation chemistry would not directly involve the oxo-iron cofactor.
The proposed suicide mechanism will inherently result in low enzyme activity, since the protein is a cosubstrate rather than a true enzyme and is left in an inactive state after a single turnover. However, the CT610 activity in our in vitro reactions is exceedingly low; the amount of pABA produced is only about 0.3% of the enzyme concentration. This indicates that the majority of isolated enzyme is in an inactive form and/or a required activator or cosubstrate is missing. The possible covalent link observed in the crystal structure between Y43 and Y47 described above may partially explain the low in vitro activity of CT610, in which this species may be an inactive side product. Additionally, the amount of bound iron to purified CT610 indicates that no more than ∼10% of the protein contained a complete diiron cofactor and, unfortunately, adding Fe(II) to CT610 enzyme reactions did not increase pABA production (Fig. S4). Schwarzenbacher et al. also reported that the diiron active site was not fully occupied during their analysis of crystal structure data (17). Thus, future work should focus on improving iron incorporation and retention, which may entail altering expression and purification conditions, expressing in a different host or coexpression with potential cofactor assembly proteins, using different purification tags, and/or developing in vitro reconstitution methods. Another complication with the CT610 reaction that is expected to be a major contributor to the low in vitro activity is that a separate reductase enzyme is likely required to supply the reducing equivalents for the reaction. The enzyme reactions reported here contained DTT as a reducing agent, which was previously reported to be capable of activating RNR to produce the tyrosyl radical (24). This use of DTT as a chemical reductant to produce the reduced diiron cluster is expected to be inefficient; thus, future work will be required to identify and characterize the putative reductase likely required to activate CT610. Finally, a possible alternative to our proposed suicide mechanism that could also partially explain the results reported here as well as the low in vitro activity is that the enzyme copurifies with a minor amount of its requisite small-molecule substrate(s) that can be converted to pABA in the presence of O2 and DTT.
The second part of the proposed CT610 reaction involves the conversion of a phenol to an aniline (Fig. 8d to g). This reaction has been demonstrated to occur in methanopterin (folate analog in methanogenic archaea) biosynthesis, where aspartate is the amino group donor in a proposed reaction sequence requiring activation by ATP and the action of two enzymes (35). The CT610 reaction does not appear to be stimulated by the addition of ATP and/or aspartate in vitro; however, it is possible that another enzyme present in C. trachomatis and E. coli assists in the reaction in vivo. The mutagenesis studies described here indicate that K152 is important for catalysis since the K152R variant resulted in dramatically decreased pABA production (Fig. 7). Since the addition of several potential external amino group donors did not increase pABA production in vitro (see Materials and Methods), K152 may act as an internal amino group donor, similarly to the role of an active site lysine in the BioU suicide reaction required for biotin biosynthesis in cyanobacteria (28). Alternatively, K152 may instead or additionally have a more traditional role as a general acid/base during CT610 catalysis.
CT610-catalyzed pABA production can finally be compared to the biogenesis of tyrosine-derived quinone cofactors, where enzymes such as copper amine oxidases utilize Cu(II) and O2 to autocatalytically synthesize their requisite cofactor from an active site tyrosine residue (36). Although the individual proposed reaction steps catalyzed by this novel pABA synthase have biochemical precedent, the entirety of the reaction that must occur to go from a tyrosine residue to pABA is quite remarkable. This work provides the foundation for future mechanistic studies of this interesting and important suicide enzyme required for folate biosynthesis in Chlamydia.
MATERIALS AND METHODS
Chemicals.
l-Tyrosine (15N, 98%) was obtained from Cambridge Isotope Laboratories. l-[3,5-2H2]tyrosine and p-[3,5-2H2]hydroxybenzoate were prepared by exchange in the presence of 2H2O and 2H2SO4 as previously described (37). All other chemicals and reagents were purchased from typical suppliers.
Culture conditions for feeding and complementation experiments.
A single colony of E. coli BW25113 ΔpabA::kan (VDC9500) (8) transformed with pAJM95 (pUC19::CT610) (8) from an LB agar plate was used to inoculate 5 ml of LB broth supplemented with 50 μg/ml kanamycin and 100 μg/ml ampicillin and grown overnight at 37°C with shaking at 300 rpm. A volume (1 ml) of the culture was pelleted by centrifugation, cells were washed with M9 medium and centrifuged again, and the remaining pellet was used to inoculate 100 ml M9 medium containing 2% glucose, 50 μM FeCl3, 50 μg/ml kanamycin, 100 μg/ml ampicillin, and 1% arabinose. When p-[3,5-2H2]hydroxybenzoate was provided, it was added at a concentration of 0.6 mg/ml. When the cells were grown in the presence of l-[3,5-2H2]-tyrosine or l-[15N]tyrosine, they were added at a concentration of 0.6 mg/ml in addition to 0.6 mg/ml of unlabeled tyrosine, phenylalanine, and tryptophan. The aromatic amino acids were added to minimize aromatic amino acid biosynthesis and increase potential incorporation of the labeled tyrosine. Since unlabeled tyrosine was also added, the highest incorporation of l-[3,5-2H2]tyrosine or l-[15N]tyrosine expected was 50%. The cells were grown at 37°C with shaking for 18 h, harvested by centrifugation, and stored at −20°C. For anaerobic growth, minimal medium was supplemented with sodium nitrate (20 mM) as an electron acceptor, and tubes containing 5 ml medium were sparged with anaerobic nitrogen gas for 1 h. After inoculation, these anaerobic cultures were incubated at 37°C with shaking (200 rpm) for 48 h.
Cell extraction and partial purification of glutamyl-pABA.
Pelleted cells were resuspended in 2 ml 50% methanol and incubated at 100°C for 10 min. After centrifugation to remove insoluble debris, the supernatant was evaporated down to ∼300 μl with a stream of nitrogen gas. This solution was adjusted to a pH of ∼5 with 15 μl 0.5 M HCl, and 200 μl of rat plasma (Pel-Freez Biologicals) was added to deglutamate the folylpolyglutamates to folate (38). After incubation at 37°C overnight, the sample was applied to a DEAE–Sephadex A-25 column (2 by 10 mm) equilibrated with water. The column was washed with water and the folates were eluted with 2 M ammonium bicarbonate. The ammonium bicarbonate was removed by evaporation under a stream of nitrogen gas, and the sample was dissolved in 1.2 ml 1 M HCl. Zinc dust (5 mg) was added, and the sample was shaken at room temperature for 1 h. After centrifugation to remove excess zinc dust, the supernatant was evaporated to dryness and resuspended in 150 μl of water for LC-MS analysis of glutamyl-pABA.
LC-MS analysis for in vivo isotope incorporation.
An AB Sciex 3200 QTrap mass spectrometer attached to an Agilent 1200 series liquid chromatograph with a Kromasil 100-5-C18 column (4.6 by 250 mm) was used for the identification and isotopic analysis of glutamyl-pABA. Solvent A was 25 mM ammonium acetate, and solvent B was methanol. The flow rate was set at 0.7 ml/min, and the elution profile consisted of a 3-min wash at 100% A, followed by a 15-min linear gradient to 65% B. The injection volume was 15 μl. Under these conditions, glutamyl-pABA eluted at 9.6 min. MS and MS-MS data were acquired in negative enhanced resolution (ER) and enhanced product ion (EPI) mode with electrospray ionization set at −4,500 V at a temperature of 400°C. The curtain gas was set at 35, and ion source gas 1 and ion source gas 2 were 60 and 50, respectively. Analyst software (Applied Biosystems/MDS Sciex) was used for system operation and data processing.
Overexpression and purification of CT610.
The CT610 gene in pET19b was originally obtained from Anthony Maurelli (University of Florida). Two constructs were obtained, one containing CT610 with an ATG start site from the STDgen prediction and the other containing CT610 with an ATG start site based on proteomics/mass spectrometry data that is 15 nucleotides downstream (8). Upon sequencing, both constructs contained errors (an insertion and a point mutation) that we repaired using the Phusion site-directed mutagenesis kit (Thermo Fisher Scientific) according to the manufacturer’s protocols. In our hands, the construct with the proteomics predicted start codon expressed better and was more stable during purification. Thus, the reported in vitro data herein are from CT610 expressed with the downstream proteomics predicted start codon. Both versions of CT610 with either the proteomics- or the genomics-predicted start codon were previously shown to rescue E. coli pABA auxotrophs (8).
For overexpression and purification of CT610 containing an N-terminal histidine tag, a single colony of E. coli BL21 cells transformed with CT610_pET19b from an LB agar plate was used to inoculate 10 ml of LB broth supplemented with 100 μg/ml ampicillin and grown overnight at 37°C with shaking at 250 rpm. The entire overnight culture was used to inoculate 1 liter LB broth with 100 μg/ml ampicillin and was incubated at 37°C with shaking at 250 rpm until the optical density at 600 nm (OD600) reached ∼0.7. Expression of CT610 was induced upon addition of 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG), and the cells were cultured for an additional 4 h at 37°C before being harvested by centrifugation and stored at −20°C.
A routine purification was from 2 liters of culture resulting in a cell pellet of ∼6 g. The pellet was thawed and resuspended in 15 ml 50 mM sodium phosphate, 300 mM sodium chloride, and 20 mM imidazole (pH 7.4) (buffer A). The cells were sonicated on ice and then centrifuged (15,000 rpm for 30 min) to remove insoluble cell debris. The lysate was applied to an equilibrated gravity flow column (1 by 5 cm, ∼2 ml of resin) of Ni-nitrilotriacetic acid (Ni-NTA) metal affinity resin (Prometheus), washed with 10 ml buffer A followed by 6 ml of buffer A with 100 mM imidazole. The desired protein was eluted with 6 ml of buffer A containing 250 mM imidazole and collected in three 2-ml fractions. After concentrating to 2.5 ml using an Amicon centrifuge concentrator (10-kDa cutoff, 15 ml; EMD Millipore), CT610 was exchanged into 100 mM Tris, 8 mM MgCl2, and 10% glycerol (pH 8.8) using a PD-10 desalting column (GE Healthcare). The final purified protein (3.5 ml, ∼200 μM) was flash frozen in liquid nitrogen and stored at −80°C. Protein concentrations were determined by the Bradford method (39) with bovine serum albumin as a standard. The amount of iron bound to purified CT610 was determined using previously described methods (40).
In vitro enzyme assays.
The purified protein from above was thawed at room temperature and enzyme assays were performed using similar conditions reported in early work on ribonucleotide reductase (24). A typical reaction (500 μl) was carried out in 100 mM Tris, 8 mM MgCl2, and 10% glycerol (pH 8.8) and contained one or more of the following components: 130 μM CT610, 10 mM DTT, 1 mM l-tyrosine, 1 mM pHB, and/or 0.5 mM pHPP. Additional potential reaction components added in attempts to increase activity included ferrous ammonium sulfate (100 μM and 1 mM), ammonium chloride (1 mM), glutamate (1 mM), glutamine (1 mM), aspartate (1 mM), ATP (1 mM), pyridoxal phosphate (100 μM), NADH (1 mM), NADPH (1 mM), and flavin adenine dinucleotide (FAD) (1 mM). Enzyme reaction mixtures were incubated at 37°C for ∼14 h and then precipitated with 50% CH3CN. The sample was centrifuged at high speed, and the resulting supernatant was concentrated under vacuum to 100 μl before analysis by HPLC and/or LC-MS.
LC-MS analysis of in vitro CT610 reactions.
An AB Sciex 3200 QTrap mass spectrometer attached to an Agilent 1200 series liquid chromatograph with a Phenomenex Kinetex Polar C18 column (4.6 by 150 mm) was used with solvent A as 0.1% formic acid in water and solvent B as 100% methanol. The LC program consisted of 2 min at 100% A and an 18-min linear gradient to 50% B at a flow rate of 0.6 ml/min. The injection volume was 15 μl. The MS method was a multiple reaction-monitoring method scanning three pairs: 138.1 and 120.4, 138.1 and 94.4, and 138.1 and 77.3. Electrospray ionization was set at 4,500 V with a temperature of 400°C. The curtain gas was 35, ion source gas 1 was 50, ion source gas 2 was 40, and the collision energy was 30 V. Analyst software (Applied Biosystems/MDS Sciex) was used for system operation and data processing.
UV-Vis analysis of pABA produced in CT610 reactions.
CT610 reactions were also analyzed with an Agilent 1100 series high-performance liquid chromatograph equipped with a diode array detector and a Kromasil 100-5-C18 column (4.6 by 250 mm; Sigma). Solvent A was 0.1% formic acid in water, and solvent B was 100% methanol. The column was equilibrated with 95% A and the program consisted of 2 min at 95% A followed by a 10 min linear gradient to 50% B. The injection volume was 50 μl. Under these conditions, pABA eluted at 10.2 min. The low sensitivity of UV-Vis compared to MS detection made this method less useful, but it was employed to obtain the UV-Vis spectrum in Fig. S2 in the supplemental material, which, along with the MS data, confirms the identity of pABA produced in CT610 reactions.
Site-directed mutagenesis.
The Phusion site-directed mutagenesis kit (Thermo Fisher Scientific) was used to generate all CT610 mutants according to the manufacturer’s instructions. The primers are listed in Table S1 in the supplemental material. Sequences were verified by Sanger sequencing at the Virginia Tech Genomics Sequencing Center. The resulting CT610 variants were expressed and purified in the same manner as described for the wild-type enzyme. The in vitro assays were carried out in triplicate (from the same protein purification) with 130 μM protein, 10 mM DTT, and 1 mM l-tyrosine in 100 mM Tris, 8 mM MgCl2, and 10% glycerol (pH 8.8).
Supplementary Material
ACKNOWLEDGMENTS
We thank Megan Zimbelman (Gonzaga University) and Monica Labine (Virginia Tech) for assistance with preliminary work on CT610 and Anthony Maurelli (University of Florida) for providing the CT610-pUC19 and CT610-pET19b plasmids.
This work was supported by USDA National Institute of Food and Agriculture Hatch project VA-160115 (K.D.A.), National Science Foundation grant MCB-1120346 (R.H.W), and National Science Foundation grant MCB-1153413 (V.D.C.-L).
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Matthews RG. 1996. One-carbon metabolism, 2nd ed ASM Press, Washington, DC. [Google Scholar]
- 2.Green JM, Matthews RG. 21 March 2007, posting date Folate biosynthesis, reduction, and polyglutamylation and the interconversion of folate derivatives. EcoSal Plus 2007. doi: 10.1128/ecosalplus.3.6.3.6. [DOI] [PubMed] [Google Scholar]
- 3.Hanson AD, Gregory JF III. 2011. Folate biosynthesis, turnover, and transport in plants. Annu Rev Plant Biol 62:105–125. doi: 10.1146/annurev-arplant-042110-103819. [DOI] [PubMed] [Google Scholar]
- 4.Zomorodipour A, Andersson SG. 1999. Obligate intracellular parasites: Rickettsia prowazekii and Chlamydia trachomatis. FEBS Lett 452:11–15. doi: 10.1016/s0014-5793(99)00563-3. [DOI] [PubMed] [Google Scholar]
- 5.Fan H, Brunham RC, McClarty G. 1992. Acquisition and synthesis of folates by obligate intracellular bacteria of the genus Chlamydia. J Clin Invest 90:1803–1811. doi: 10.1172/JCI116055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Crecy-Lagard V, El Yacoubi B, de la Garza RD, Noiriel A, Hanson AD. 2007. Comparative genomics of bacterial and plant folate synthesis and salvage: predictions and validations. BMC Genomics 8:245. doi: 10.1186/1471-2164-8-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Crecy-Lagard V. 2014. Variations in metabolic pathways create challenges for automated metabolic reconstructions: examples from the tetrahydrofolate synthesis pathway. Comput Struct Biotechnol J 10:41–50. doi: 10.1016/j.csbj.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adams NE, Thiaville JJ, Proestos J, Juárez-Vázquez AL, McCoy AJ, Barona-Gómez F, Iwata-Reuyl D, de Crécy-Lagard V, Maurelli AT. 2014. Promiscuous and adaptable enzymes fill “holes” in the tetrahydrofolate pathway in Chlamydia species. mBio 5:e01378-14. doi: 10.1128/mBio.01378-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuratsu M, Hamano Y, Dairi T. 2010. Analysis of the Lactobacillus metabolic pathway. Appl Environ Microbiol 76:7299–7301. doi: 10.1128/AEM.01514-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, Koonin EV, Davis RW. 1998. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282:754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- 11.Morita H, Toh H, Fukuda S, Horikawa H, Oshima K, Suzuki T, Murakami M, Hisamatsu S, Kato Y, Takizawa T, Fukuoka H, Yoshimura T, Itoh K, O'Sullivan DJ, McKay LL, Ohno H, Kikuchi J, Masaoka T, Hattori M. 2008. Comparative genome analysis of Lactobacillus reuteri and Lactobacillus fermentum reveal a genomic island for reuterin and cobalamin production. DNA Res 15:151–161. doi: 10.1093/dnares/dsn009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chain P, Lamerdin J, Larimer F, Regala W, Lao V, Land M, Hauser L, Hooper A, Klotz M, Norton J, Sayavedra-Soto L, Arciero D, Hommes N, Whittaker M, Arp D. 2003. Complete genome sequence of the ammonia-oxidizing bacterium and obligate chemolithoautotroph Nitrosomonas europaea. J Bacteriol 185:2759–2773. doi: 10.1128/jb.185.9.2759-2773.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Satoh Y, Kuratsu M, Kobayashi D, Dairi T. 2014. New gene responsible for para-aminobenzoate biosynthesis. J Biosci Bioeng 117:178–183. doi: 10.1016/j.jbiosc.2013.07.013. [DOI] [PubMed] [Google Scholar]
- 14.Meulenberg JJ, Sellink E, Riegman NH, Postma PW. 1992. Nucleotide sequence and structure of the Klebsiella pneumoniae pqq operon. Mol Gen Genet 232:284–294. doi: 10.1007/BF00280008. [DOI] [PubMed] [Google Scholar]
- 15.Bonnot F, Iavarone AT, Klinman JP. 2013. Multistep, eight-electron oxidation catalyzed by the cofactorless oxidase, PqqC: identification of chemical intermediates and their dependence on molecular oxygen. Biochemistry 52:4667–4675. doi: 10.1021/bi4003315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stenner-Liewen F, Liewen H, Zapata JM, Pawlowski K, Godzik A, Reed JC. 2002. CADD, a Chlamydia protein that interacts with death receptors. J Biol Chem 277:9633–9636. doi: 10.1074/jbc.C100693200. [DOI] [PubMed] [Google Scholar]
- 17.Schwarzenbacher R, Stenner-Liewen F, Liewen H, Robinson H, Yuan H, Bossy-Wetzel E, Reed JC, Liddington RC. 2004. Structure of the Chlamydia protein CADD reveals a redox enzyme that modulates host cell apoptosis. J Biol Chem 279:29320–29324. doi: 10.1074/jbc.M401268200. [DOI] [PubMed] [Google Scholar]
- 18.Magnusson OT, Toyama H, Saeki M, Rojas A, Reed JC, Liddington RC, Klinman JP, Schwarzenbacher R. 2004. Quinone biogenesis: structure and mechanism of PqqC, the final catalyst in the production of pyrroloquinoline quinone. Proc Natl Acad Sci U S A 101:7913–7918. doi: 10.1073/pnas.0402640101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schuller DJ, Wilks A, Ortiz de Montellano PR, Poulos TL. 1999. Crystal structure of human heme oxygenase-1. Nat Struct Biol 6:860–867. doi: 10.1038/12319. [DOI] [PubMed] [Google Scholar]
- 20.Rosenzweig AC, Brandstetter H, Whittington DA, Nordlund P, Lippard SJ, Frederick CA. 1997. Crystal structures of the methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath): implications for substrate gating and component interactions. Proteins 29:141–152. doi: 10.1002/(SICI)1097-0134(199710)29:2<141::AID-PROT2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 21.Eriksson M, Jordan A, Eklund H. 1998. Structure of Salmonella typhimurium nrdF ribonucleotide reductase in its oxidized and reduced forms. Biochemistry 37:13359–13369. doi: 10.1021/bi981380s. [DOI] [PubMed] [Google Scholar]
- 22.Brignole EJ, Ando N, Zimanyi CM, Drennan CL. 2012. The prototypic class Ia ribonucleotide reductase from Escherichia coli: still surprising after all these years. Biochem Soc Trans 40:523–530. doi: 10.1042/BST20120081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wallar BJ, Lipscomb JD. 1996. Dioxygen activation by enzymes containing binuclear non-heme iron clusters. Chem Rev 96:2625–2658. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 24.Fontecave M, Gerez C, Mansuy D, Reichard P. 1990. Reduction of the Fe(III)-tyrosyl radical center of Escherichia coli ribonucleotide reductase by dithiothreitol. J Biol Chem 265:10919–10924. [PubMed] [Google Scholar]
- 25.Chatterjee A, Abeydeera ND, Bale S, Pai PJ, Dorrestein PC, Russell DH, Ealick SE, Begley TP. 2011. Saccharomyces cerevisiae THI4p is a suicide thiamine thiazole synthase. Nature 478:542–546. doi: 10.1038/nature10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai RY, Huang S, Fenwick MK, Hazra A, Zhang Y, Rajashankar K, Philmus B, Kinsland C, Sanders JM, Ealick SE, Begley TP. 2012. Thiamin pyrimidine biosynthesis in Candida albicans: a remarkable reaction between histidine and pyridoxal phosphate. J Am Chem Soc 134:9157–9159. doi: 10.1021/ja302474a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Desguin B, Soumillion P, Hols P, Hausinger RP. 2016. Nickel-pincer cofactor biosynthesis involves LarB-catalyzed pyridinium carboxylation and LarE-dependent sacrificial sulfur insertion. Proc Natl Acad Sci U S A 113:5598–5603. doi: 10.1073/pnas.1600486113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakaki K, Ohishi K, Shimizu T, Kobayashi I, Mori N, Matsuda K, Tomita T, Watanabe H, Tanaka K, Kuzuyama T, Nishiyama M. 2020. A suicide enzyme catalyzes multiple reactions for biotin biosynthesis in cyanobacteria. Nat Chem Biol 16:415–422. doi: 10.1038/s41589-019-0461-9. [DOI] [PubMed] [Google Scholar]
- 29.Kolberg M, Strand KR, Graff P, Andersson KK. 2004. Structure, function, and mechanism of ribonucleotide reductases. Biochim Biophys Acta 1699:1–34. doi: 10.1016/j.bbapap.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 30.Huang M, Parker MJ, Stubbe J. 2014. Choosing the right metal: case studies of class I ribonucleotide reductases. J Biol Chem 289:28104–28111. doi: 10.1074/jbc.R114.596684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minnihan EC, Nocera DG, Stubbe J. 2013. Reversible, long-range radical transfer in E. coli class Ia ribonucleotide reductase. Acc Chem Res 46:2524–2535. doi: 10.1021/ar4000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang X, Groves JT. 2017. Beyond ferryl-mediated hydroxylation: 40 years of the rebound mechanism and C-H activation. J Biol Inorg Chem 22:185–207. doi: 10.1007/s00775-016-1414-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ross MO, Rosenzweig AC. 2017. A tale of two methane monooxygenases. J Biol Inorg Chem 22:307–319. doi: 10.1007/s00775-016-1419-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Komor AJ, Jasniewski AJ, Que L, Lipscomb JD. 2018. Diiron monooxygenases in natural product biosynthesis. Nat Prod Rep 35:646–659. doi: 10.1039/C7NP00061H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.White RH. 2011. The conversion of a phenol to an aniline occurs in the biochemical formation of the 1-(4-aminophenyl)-1-deoxy-d-ribitol moiety in methanopterin. Biochemistry 50:6041–6052. doi: 10.1021/bi200362w. [DOI] [PubMed] [Google Scholar]
- 36.Brazeau BJ, Johnson BJ, Wilmot CM. 2004. Copper-containing amine oxidases. Biogenesis and catalysis; a structural perspective. Arch Biochem Biophys 428:22–31. doi: 10.1016/j.abb.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 37.Matthews HR, Matthews KS, Opella SJ. 1977. Selectively deuterated amino acid analogues. Synthesis, incorporation into proteins and NMR properties. Biochim Biophys Acta 497:1–13. doi: 10.1016/0304-4165(77)90134-9. [DOI] [PubMed] [Google Scholar]
- 38.Pribat A, Blaby IK, Lara-Núñez A, Gregory JF III, de Crécy-Lagard V, Hanson AD. 2010. FolX and FolM are essential for tetrahydromonapterin synthesis in Escherichia coli and Pseudomonas aeruginosa. J Bacteriol 192:475–482. doi: 10.1128/JB.01198-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 40.Beinert H. 1978. Micro methods for the quantitative determination of iron and copper in biological material. Methods Enzymol 54:435–445. doi: 10.1016/s0076-6879(78)54027-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.