

ABSTRACT
The recently renewed interest in scientific rigor and reproducibility is of critical importance for both scientists developing new targeted small-molecule inhibitors and those employing these molecule in cellular studies, alike. While off-target effects are commonly considered as limitations for any given small-molecule inhibitor, the ability of a given compound to distinguish between enzyme isoforms is often neglected when employing compounds in cellular studies. To call attention to this issue, we have compared the results of an assay for “direct target engagement”, the Cellular Thermal Shift Assay (CETSA), to the published isoform selectivity of 12 commercially available sphingosine kinase 1 and 2 (SphK 1 and SphK2) inhibitors. Our results suggest that, at the concentrations commonly employed in cellular assay systems, none of the tested SKIs can be considered isoform selective. Thus, caution and complimentary assay strategies must be employed to fully discern isoform selectivity for the SphKs. Moreover, caution must be employed by the scientific community as a whole when designing experiments that aim to discern the effects of one enzyme isoform versus another to ensure that the concentration ranges used are able to distinguish isoform selectivity.
KEYWORDS: Sphingosine kinase, sphingolipids, target engagement, isoform selectivity, CETSA
Introduction
In recent years, a renewed emphasis has been placed on the importance of scientific rigor and reproducibility due to revelations that several high-impact published reports were not able to be independently confirmed1. From the perspective of a drug discovery/development scientist, this renewed emphasis takes the form of ensuring rigor, during lead development, to avoid off-target effects and validate on-target specificity to maximize the utility of a compound as a molecular tool or as a lead for future optimization studies. From the perspective of a cell biologist, the emphasis is placed on the reproducibility of the results obtained with a molecular tool compound across various cell systems. Thus, it is of critical importance for both fields of research to do their due diligence for the benefit of the scientific community and ultimately patients suffering from life threatening diseases such as cancer.
It is not uncommon for small-molecule inhibitors to have off-target effects that can lead to unwanted side-effects and potentially to unwanted toxicities. At the extreme, these off-target effects can lead to the failure of a promising clinical lead compound. Similarly, there is a critical demand for simple methods of demonstrating that a drug candidate binds to its cognate target in cells (i.e. target engagement) as early in the development process as possible.2,3 Indeed, this need became apparent after the Phase III failure of the PARP Inhibitor, Iniparib. Subsequently, it was demonstrated that Iniparib did not inhibit PARP activity, but instead nonselectively modified cysteine residues. Thus, simply demonstrating that Iniparib did, or did not, directly target engaged PARP in intact cells could have saved years and millions of dollars of investment.
Another factor that is frequently encountered in published studies is the use of aberrantly high concentrations of compounds when studying their effects on live cells. This is especially true when attempting to discern which isoform of a particular enzyme is responsible for a given effect. Indeed, an example of this difficulty comes from studies of the Aurora Kinases (AurA and AurB). Early Aurora kinase inhibitors had selectivity for one isoform over the other in the range of 1–2 log fold differences at low nanomolar ranges. For instance, MLN8054 is reportedly ~40 fold more selective for AurA versus AurB (4 nM vs 172 nM).4 However, when employed in cellular studies at 0.5 µM or higher, the observed effects are often attributed to AurA inhibition, when both isoforms are likely inhibited. Indeed, the recent development of a truly selective AurA inhibitor (LY-3295668) has demonstrated that AurA inhibition alone induces mitotic spindle assembly checkpoint arrest, whereas, when AurA and B are co-inhibited, AurB inhibition is dominant and mitotic exit occurs.5,6
Our research interests have focused on the development of small-molecule inhibitors of the Sphingosine kinases (SphK1 and SphK2). Our experiences, both in the development of SphK inhibitors (SKIs) and their use as molecular tools in cell biology experiments has revealed multiple unforeseen factors can influence both the rigorous evaluation of the isoform selectivity of “selective” inhibitors and the implementation of “selective” inhibitors in cellular studies. Early studies suggested that of the two SphK isoforms, SphK1 was the more desirable target for therapeutic targeting. One of the primary reasons for this was the observation that SphK1 activity was stimulated by a host of external and internal stimuli (extensively reviewed in7); whereas SphK2 was thought to have a housekeeping function in the maintenance of physiological levels of S1P. Thus the focus of early inhibitor development programs was on the identification of SphK1-selective small-molecule inhibitors. Concurrent with the development of the SphK1-selective inhibitors, several studies demonstrated that, like SphK1, SphK2 was over-expressed in certain cancer types and that SphK2 also responded to growth factor stimulation.8-10 In fact, rather than having only housekeeping functions, SphK2 was recently shown to have an oncogenic function and to have unique properties such as a nuclear localization that allowed for S1P production in the nucleus and modulation of HDAC activity.11-14 These and many other studies highlighted the need for isoform-selective SKIs to both dissect the physiological roles of the two SphK isoforms, as well as to identify the better pathophysiological target for therapeutic development.
“First-generation” Sphingosine Kinase Inhibitors (SKIs), such as, our small-molecule chemotypes (SKI-I – SKI-IV15), di-methyl sphingosine, etc., were non-isoform selective inhibitors that induced apoptosis, reduced tumor volume in numerous animal studies and provided encouraging results that SKIs would be effective therapeutic agents for multiple cancer types.16 In the intervening years, pharmaceutical industry and academic groups developed “second-generation”, that were reported to be isoform-selective SphK inhibitors. For example, SK1-I, SKI-5 C and PF-543 were reported to be SphK1-selective while ABC294640 and K145 were reported to be SphK2-selective.17-21 Some of these “second generation” inhibitors achieved the goal of nanomolar potency.19,22-28
Simultaneous with the publication of these isoform-selective SKIs, we had begun to optimize our original SKI-I chemotype, due to the fact that SKI-I was the most potently cytotoxic of the four original identified chemotypes (SKI-I – SKI-IV).29 Through a series of Structure-Activity Relationship (SAR) studies we identified a refined SphK1-selective SKI-I analog (SKI-178) that competes for the Sph binding site, is potently cytotoxic, and effectively reduces S1P formation while inducing Cer accumulation against a broad range of cancer cell lines, in vitro.30 The apparent discrepancy between the actions of SKI-178 and these other SphK1 inhibitors led us to suspect that SKI-178 had “off-target” effects that accounted for the ability of SKI-178 to induce apoptosis.
As part of our drug development program, we have extensively examined of the apoptotic mechanism-of-action of a small-molecule inhibitor of sphingosine kinase 1 (SphK1), SKI-178. As part of this examination, we employed the recently developed Cellular Thermal Shift Assay (CETSA31-33) to demonstrate that SKI-178 selectively target engages SphK1 in intact cells.34,35 To our surprise, and contrary to the results of our earlier in-vitro SphK activity assays, we observed that, in addition to SphK1, SKI-178 also target engaged the highly similar isoform, SphK2.30,35 As we considered reasons for the discrepancy between the in-vitro activity assay and CETSA experiment results, it became apparent that one limitation of the in-vitro SphK activity assays, using recombinant proteins, is that they have relied on detergent (e.g. 1% Triton X-100) or high salt (e.g. 1 M KCl) conditions to selectively assay for specific SphK isoforms.36 Clearly, these conditions do not mimic the intracellular environment and, in fact, they put selective pressure on the inhibitor screens by potentially blocking lipophilic or ionic interactions, respectively. Indeed, one of the key advantages of the CETSA experiment is its ability to measure binding in the proper intracellular microenvironment.
A separate issue that we have frequently observed in the literature is that the isoform-selective SKIs, such as PF-543, SK1-I, ABC294640, etc., are often used at very high concentrations in cellular experiments to identify the SphK isoform responsible for a given biological effect. In such cases, it is often assumed that these isoform-selective inhibitors do not inhibit the other SphK isoform at all. While it is very likely that these inhibitors have a greater affinity for one isoform over the other (i.e. selectivity), it is often the case that the inhibitor is employed in a cellular assay system at relatively high concentrations that far exceed the IC50 for the preferred isoform and are, in fact, likely to inhibit both isoforms. This is especially true for the nanomolar potency inhibitors, such as PF-543, when they are employed at micromolar concentrations in cells.
Given the contradictory results we obtained with in-vitro SphK activity assays and CETSA experiments in the development of SKI-178 and the propensity of investigators to use very high concentration of selective SKIs in cellular assays to discern the effects of the different SphK isoforms, we decide to employ the CETSA technique to reevaluate the target selectivity of a cohort of 12 commercially available SphK1-selective, SphK2-selective and nonselective, SphK1/2 inhibitors. Our results confirm that all 12 of the commercially available SphK Inhibitors, are capable of target engaging both human SphK1 and SphK2 at micromolar concentrations typically employed in published studies. Furthermore our results highlight the need to confirm the results obtained using SKIs with molecular tools such as siRNA/shRNA knock-down or Crispr/Cas9 mediated knock-out. Thus, caution should always be employed when using small-molecule inhibitors to attempt to discern the relative contribution of either SphK isoform to an observed biological effect.
Results
One of the major factors complicating the study of the individual SphK isoforms is that they are co-expressed, at relatively low levels, in most cell lines. Because they are co-expressed and because they both utilize Sph to generate S1P, there is no truly reliable method to discern their relative contributions to the intracellular pool of S1P. Various strategies to over-come this limitation have included the use of isoform specific buffer conditions,36 siRNA/shRNA knockdown of individual SphK isoforms or the use of SphK1−/- or SphK2−/- MEF cell lines. Knock-out strategies employing the Crispr/Cas9 technology may overcome these limitations, however, until this technology is validated and extensively utilized in the sphingolipid metabolism field, we are left to rely on a combination of the above technologies and isoform-selective small-molecule inhibitors of SphK1 and SphK2.
More than two decades of effort in development of small-molecule isoform-selective inhibitors of SphK1 and SphK2 have yielded numerous potential candidates that, even if not clinically useful, have potential value as “tool” compounds to properly evaluate the roles of the individual SphKs in the many biological processes to which they have been linked. Our own recent experience with the development of SKI-178 has led to the discovery that in-vitro kinase assays can mislead investigators into believing that a small-molecule inhibitor is SphK isoform-selective. Indeed, we employed the recently developed Cellular Thermal Shift Assay (CETSA) to demonstrate that SKI-178 directly binds to (e.g. target engages) both SphK1 and SphK2 in intact, growing cells in spite of evidence from in-vitro kinase assays indicating that SKI-178 was SphK1-selective.35 Given our experience, we have decided to evaluate a cohort of commercially available SphK Inhibitors (SKIs; Table 1) and to compare their published isoform selectivity to the results of CETSA target engagement assays.
Table 1.
In vitro Characterization of Sphk1 and SphK2 Inhibitor selectivity.
| Compound # | Name | Structure | Ki (μM) | Ref | |
|---|---|---|---|---|---|
| SphK1 inhibition | SphK2 inhibition | ||||
| 1 | N,N-dimethyl-sphingosine | 16 | 14 | 37 | |
| 2 | SKI-II | 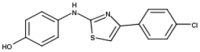 |
16 | 8 | 15,37 |
| 3 | PF-543 | 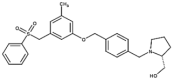 |
0.004 | 0.5 | 19 |
| 4 | SKI-I | 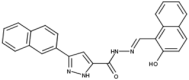 |
IC50 = 1.2 | IC50 = 10 | 15,30 |
| 5 | SK1-I | 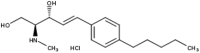 |
10 | ND | 17 |
| 6 | SKI-V |  |
IC50 = 2 | ND | 15 |
| 7 | FTY-720 (Fingolimod) | 2 | Substrate | 38 | |
| 8 | SKI-178 |  |
1.3 | ND | 30 |
| 9 | ABC294640 |  |
ND | 10 | 20 |
| 10 | K145 | ND | 6 | 21 | |
| 11 | SKI-5 C |  |
15 | 46 | 18 |
| 12 | DL-threo-dihydro-sphingosine (Safingol) | 5 | 5 | 39 | |
ND: Not Determined in Published Studies.
Establishment of an over-expression system for SphK1 and SphK2 and validation of isoform specific antibodies
Until recently, the “toolkit” available for studying SphK1 and SphK2 biology has been severely limited by the lack of high quality, well validated, isoform specific antibodies. Two commercial sources of anti-SphK2 antibodies have recently been validated for identification of endogenous levels of human SphK2.40 Conversely, we have had difficulty visualizing endogenous levels of SphK1 protein in most human cell lines. To circumvent these issues, we chose to separately over-express FLAG-His-6XSphK1 (FLAG-SphK1, 384 a.a) and FLAG-His-6XSphK2 (FLAG-SphK2, 618 a.a.), in HEK293 cells. As shown in Figure 1(a), over-expressed FLAG-SphK1 and FLAG-SphK2 are detected by anti-FLAG epitope antibodies. The two SphK isoforms can be easily discerned by western blot analysis due to the large difference in size (FLAG-SphK1 ≈ 47 kDa and FLAG-SphK2 ≈ 75 kDa). FLAG-SphK1 is much more abundant than FLAG-SphK2, assuming equal recognition by the anti-FLAG antibody. This is consistent with previous studies that demonstrate that SphK2 over-expression induces apoptosis due to the fact that SphK2 possesses a BH3-only domain capable of interacting with and sequestering the anti-apoptotic Bcl-2 family member Bcl-XL(REF). Thus, it is likely that high over-expression of SphK2 places selective pro-apoptotic pressure on cells and only those cells expressing tolerable levels of SphK2 survive in culture.
Figure 1.
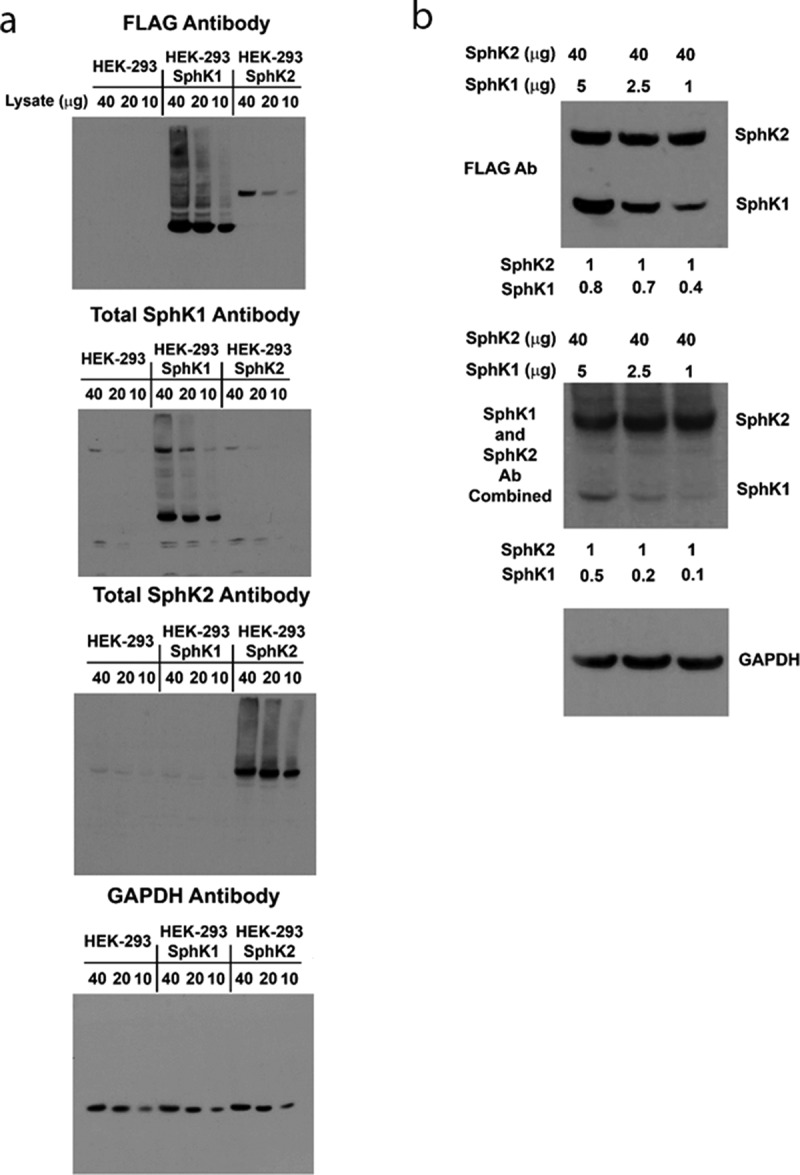
Establishment of SphK1 and SphK2 expressing HEK293 cell lines and validation of the SphK antibodies. (a) Naïve HEK293 cells and separate clones over-expressing FLAG-SphK1 and FLAG-SphK2 were analyzed for expression of SphK1 and SphK2 using anti-FLAG and SphK isoform specific antibodies by western blot analysis, as indicated. GAPDH was included as a loading control. (b) HEK293 cell lysates over-expressing FLAG-SphK1 and FLAG-SphK2 were combined at the indicated ratios and analyzed for expression of SphK1 and SphK2 using anti-FLAG antibodies and simultaneously using both SphK isoform specific antibodies at 1:2000 dilutions by western blot analysis. GAPDH was included as a loading control.
Over-expressed FLAG- SphK1 is detected by a commercially available SphK1 antibody [clone: D1H1 L] from Cell Signaling Technologies (MA, USA; #12071), with no cross-reactivity to either FLAG-SphK2 or endogenous SphK2. Importantly, as stated above, darker exposures did not detect endogenous SphK1 expression, in non-transfected HEK-293 cells, consistent with a very low level of endogenous SphK1 expression. However, because SphK1 lacks a BH3-only domain, high over-expression of SphK1 is well tolerated by cells and, as we have shown previously, provides a pro-survival advantage to HEK-293 cells under serum deprivation conditions (REF). Similarly, over-expressed FLAG-SphK2 was recognized by the SphK2 specific antibody from Proteintech (IL, USA; #17096-1-AP) as previously described40 with no cross-reactivity to either FLAG-SphK1 or endogenous SphK1. Although, this antibody does detect endogenous levels of SphK2 expression in HEK-293 cells and FLAG-SphK1 over-expressing cells.
It seems as though endogenous SphK1 is expressed at extremely low levels (below the limit of detection) in HEK-293 cells, while SphK2 is expressed at detectable levels. It is important to gauge the relative potency of these isoform-specific antibodies if investigators wish to make direct comparisons of SphK1 and SphK2 expression levels in cancer cells, for example. However, it is not possible to make direct comparisons between two antibodies without knowledge of their individual affinities. To circumvent this, we next titrated the FLAG-SphK1 and FLAG-SphK2 HEK-293 cell lysate to approximately equal expression levels based on their equal reactivity to the anti-FLAG antibody. As shown in Figure 1(b), we held FLAG-SphK2 levels steady and titrated the expression of FLAG-SphK1 by mixing lysates on western blots. Total proteins levels of 40 µg from the FLAG-SphK2 lysates and between 10 µg (Figure 1(a)) and 5 µg (Figure 1(b)) of total protein from FLAG-SphK1 lysates gave approximately equal intensities on western blots. This equates to an ~ 8 fold higher expression of SphK1 relative to SphK2 in the HEK-293 over-expression system, based upon the anti-FLAG antibody. We also examined the expression of both FLAG-SphK1 and FLAG-SphK2 on a single blot using a simultaneous exposure to equal 1:2000 dilutions of both the anti-SphK1 and the anti-SphK2 antibodies. Comparing the relative band intensities of SphK1 and SphK2 in the anti-FLAG and anti-SphK1/2 images allows us to state that the anti-SphK2 antibody has a greater affinity for SphK2 than the anti-SphK1 antibody has for SphK1, when employed at 1:2000 dilutions. Nevertheless, it should be feasible for investigators to make direct comparisons of the relative expression levels of SphK1 and SphK2, employing these antibodies in their model system of choice given that the SphK2 antibody has about an ~2–5 fold greater affinity for SphK2 than the SphK1 antibody has for SphK1. Together, these results demonstrate our ability to reliably detect both SphK1 and SphK2 in HEK293 cell lines and that these stably expressing cell lines will be suitable for evaluation of the effects of SKIs on the SphKs both at the expression level and in CETSA experiments, below.
Multiple SKIs induce proteasomal degradation of SphK1 in HEK293 cells
The reported Kis for SphK inhibition of the SKIs, listed in Table 1, range from low-nanomolar (PF-543 (3)19 for SphK1) to mid micromolar ranges (ABC294640 (9)20 for SphK2). Similarly, there is a wide range of cytotoxic IC50s for these compounds, including the report that PF-543 is non-cytotoxic;19 although it has subsequently been shown to induce necrosis at mid micromolar concentration in colorectal cancer cells.41 Regardless of the reported IC50s, most investigations employ these compounds at 10–20 μM for approximately 24 hours to evaluate their effects on sphingolipid signaling. Using this as a starting point for our evaluations, we first determined whether any of the SKIs were overtly cytotoxic to naïve HEK-293 cells. HEK-293 cells were treated with the various SKIs for 72 hours and the relative increase in cell density relative to time 0 was determined by sulforhodamine B assay.42 As shown in Figure 2(a), none of the SKIs were overtly cytotoxic and, with the exception of SKI-I (4),15,30 none of the agents, including the PD98059 control, blocked the growth of cells over the 72 hour treatment period. Thus, these concentration were employed below to determine the target engagement of SphK proteins in live cells.
Figure 2.
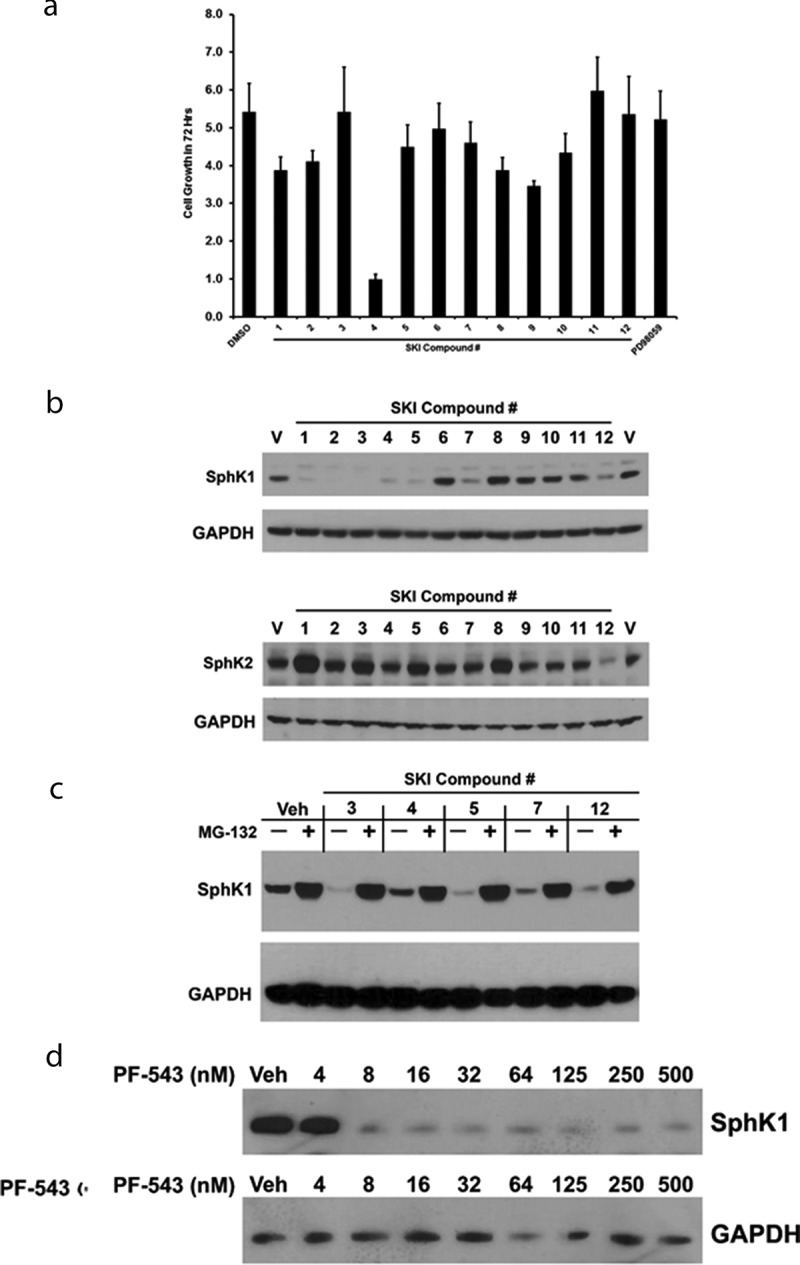
Sphingosine Kinase Inhibitor (SKI)-induced degradation of SphK1. (a) Naïve HEK293 cells were incubated with the indicated SKIs at the following concentrations: 1: DMS (20 µM), 2: SKI-II (10 µM), 3: PF-543 (10 µM), 4: SKI-I (10 µM), 5: SK1-I (10 µM), 6: SKI-V (10 µM), 7: FTY720 (10 µM), 8: SKI-178 (10 µM), 9: ABC294640 (20 µM), 10: K145 (10 µM), 11: SKI-5 c (20 µM), 12: Safingol (20 µM), for 72 hours. Sulforhodamine B staining was performed at time 0 and time 72 hours to determine relative increase in cell number. n = 3. (b) A representative western blot of HEK293 cells over-expressing FLAG-SphK1 or FLAG-SphK2 were incubated with the indicated SKIs at the indicated concentrations for 24 hrs. Whole cell lysates were prepared and analyzed by western blot detection of SphK1, SphK2 and GAPDH using the appropriate primary antibodies. (c) A representative western blot of HEK293 cells over-expressing FLAG-SphK1 were treated with the indicated SKIs in the presence or absence of MG-132 (10 µM) for 24 hrs. Whole cell lysates were prepared and analyzed by western blot detection of SphK1 and GAPDH using the appropriate primary antibodies. (d) A representative western blot of HEK293 cells over-expressing FLAG-SphK1 were treated with the indicated concentrations of PF-543 for 24 hrs. Whole cell lysates were prepared and analyzed by western blot detection of SphK1 and GAPDH using the appropriate primary antibodies.
There are numerous reports in the literature that some SKIs will induce either lysosomal or proteasomal degradation of SphK1.43-45 This may be the result of target engagement of SphK1, by the SKIs, leading to the destabilization, ubiquitination and proteasomal degradation of SphK1 protein. However, it is also possible is that the degradation of SphK1 occurs, indirectly, as a result of apoptotic events/caspase activation, thus, inhibitors of the proteasome, lysosomal cathepsins and caspases should be employed to clarify the mechanism of degradation. To date, there has been no detailed examination of the ability of multiple SKIs to induce degradation of either SphK1 or SphK2.
To this end, HEK-293 cells over-expressing either FLAG-SphK1 or FLAG-SphK2 were treated with the 12 SKIs (Table 1) and the effects of the SKIs on both SphK1 and SphK2 expression were determined 24 hrs later by western blot analysis. Consistent with previous reports,43-45 DMS (1),37 SKI-II (2),15,37 PF-543 (3) and FTY720 (7),38 all induced degradation of SphK1 relative to vehicle treated controls (Figure 2(b)). Similarly, Figure 2(b) represents the first observation that SKI-I (4), SK1-I (5),17 and Safingol (12)39 also induce SphK1 degradation. SKI induced degradation of SphK1 provides clear evidence of target engagement of SphK1 by these compounds, thus we did not further evaluate their ability to target engage SphK1 by CETSA experiments. Notably, ABC294640 (9) has been reported to induce proteasomal degradation of SphK1 in prostate cancer cells;46 however, we did not observe any effects of ABC294640 (9) on SphK1 levels in our transformed HEK293 over-expression model system. It is possible that induction of apoptosis/activation of caspases may have accounted for some of the degradation of SphK1 in LNCAP cells as there was a dose dependent decrease of SphK1 in both untreated and MG132 treated conditions, in response to ABC294640 (9).46 Nevertheless, our SKI-V (6)15 compound, SKI-5 C (11),18 K145 (10),21 ABC294640 (9) and our SKI-178 (8)30 compound did not affect levels of SphK1 (Figure 2(b)). Conversely, with the exception of Safingol (12), none of the SKIs induced degradation of SphK2 (Figure 2(b)).
To further examine whether SKI-I (4), SK1-I (5) and Safingol (12) induce proteasomal degradation of SphK1, we treated FLAG-SphK1 HEK-293 cells with the SKIs in the presence or absence of the proteasomal inhibitor, MG-132. As shown in Figure 2(c), both SK1-I (5) and Safingol (12), and to a lesser extent SKI-I (4), induced the degradation of SphK1 through the proteasomal pathway as MG-132 abrogated the effects of these SKIs, as well as, the PF-543 (3) and FTY-720 (7) treated controls. Presuming that the degradation of SphK1 occurs due to binding of these SKIs to SphK1, this phenomenon provides evidence of target engagement, in cells, by these degradation-inducing SKIs. Thus, it should be possible to determine an IC50 for SKI induced degradation for any given SKI that induces proteasomal degradation.
As an example of this, we chose to examine the dose-dependence of PF-543 (3) induced degradation of SphK1. As shown in Figure 2(d), PF-543 (3) induces degradation of SphK1, within 24 hrs, at concentrations as low as 8 nM, which is consistent with the published in-vitro IC50 for inhibition of SphK1 catalytic activity. Together, these data demonstrate that one way to classify, or sub-divide SKIs, is by whether or not they induce degradation of SphK1. To our knowledge, there are no reports of SKI induced degradation of SphK2 and further examination of the effects of Safingol (12) on SphK2 indicated that it did not induce either proteasomal or lysosomal degradation of SphK2 (data not shown). If an SKI induces proteasomal degradation of SphK1, evidence of target engagement can be assumed. However, for a number of SphK1 inhibitors including our SKI-V (6) and SKI-178 (8) and SKI-5 C (11), and other SKIs that do not induce degradation, other methods of demonstrating target engagement must be employed.
Demonstration of target engagement of SphK1 using CETSA
CETSA measures the ability of a compound to directly bind to and stabilize/destabilize its target protein(s) when exposed to a thermal denaturation gradient. In a CETSA experiment, cells are treated with the compound of interest, harvested and subjected to a thermal denaturation gradient, lysed, and the soluble proteins, after thermal treatment, are analyzed by western blot analysis. If a test compound directly binds to its target, we may see a thermal stability shift in the presence of compound relative to vehicle treated cells. We have previously identified thermal denaturation gradients suitable for evaluation of SphK1 and SphK2 target engagement and employed them to demonstrate that SKI-178 target engages SphK1 and SphK2 and induces their stabilization, relative to vehicle.35 Thus, we next employed CETSA to confirm that the SphK1-selective inhibitors SKI-V (6) and SKI-5 C (11) target engage SphK1 and to determine whether the SphK2-selective inhibitors ABC294640 (9) and K145 (10), are capable of target engagement of SphK1. As shown in Figure 3(a), the specificity of the CETSA target engagement assay is demonstrated by the fact that the MEK 1/2 inhibitor, PD98059, does not target engage SphK1 using the FLAG-SphK1 over-expressing HEK-293 cell system. Similarly, we demonstrate that actin is relatively heat stable protein across the thermal denaturation gradient between vehicle and SKI-178 treated lysates. Conversely, SKI-178 (8) does target engage SphK1 and stabilize the protein over the temperature gradient relative to vehicle treated cells, as previously demonstrated.35 This demonstrates the ability to quantitatively analyze the stability of SphK1 and SphK2 without inadvertent error arising from inefficient loading/transfer of samples (i.e. pipette error) during the course of the CETSA protocol. Similarly, as shown in Figure 3(b), we observed target engagement of SphK1 by the SphK1-selective inhibitors SKI-V (6) and SKI-5 C (11). Interestingly, both ABC294640 (9) and K145 (10), which are reported in the literature to be SphK2-selective, were observed to target engage and stabilize SphK1 protein.
Figure 3.
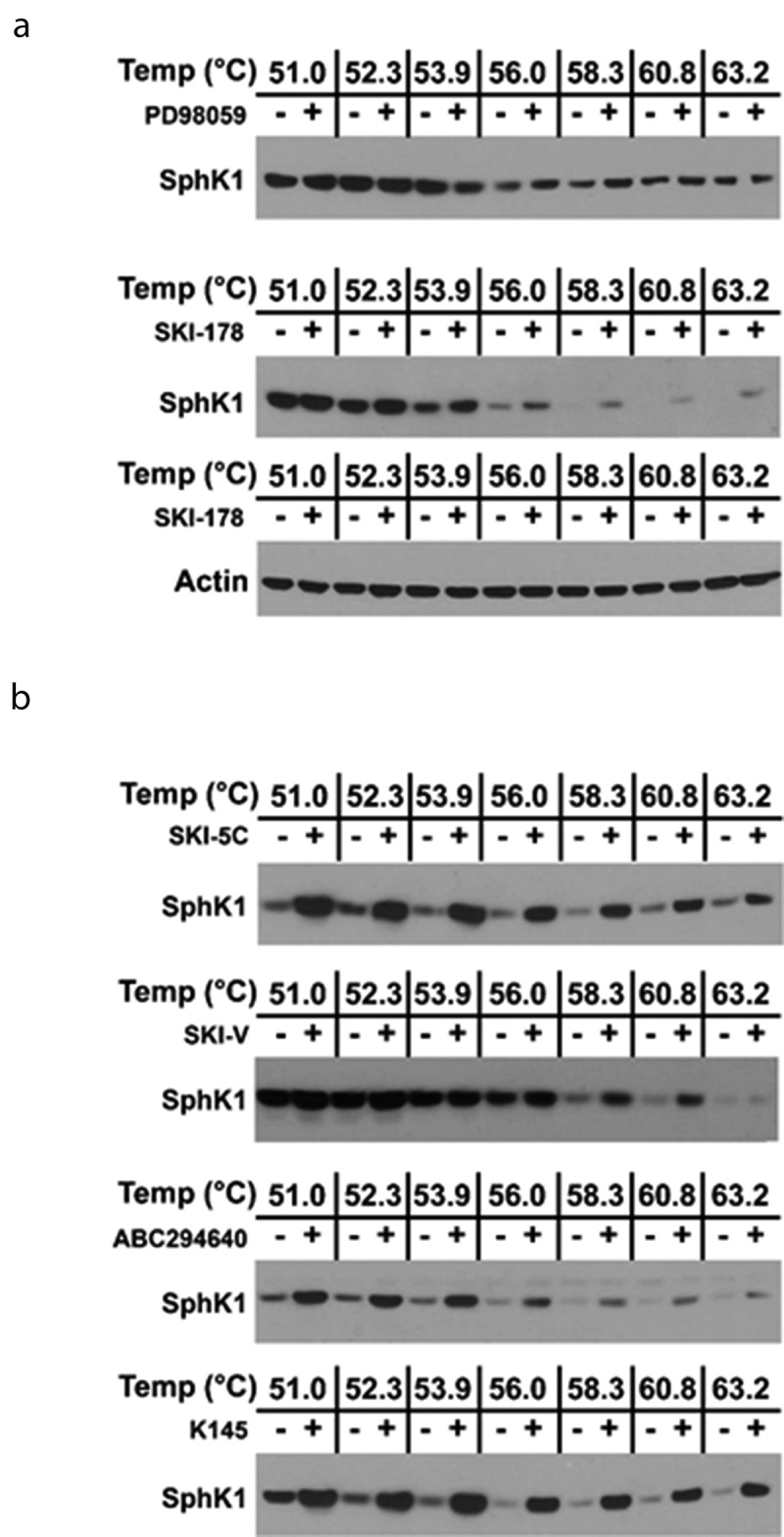
Demonstration of Target Engagement of SphK1 in HEK293 cells by SKIs using CETSA. (a) A representative western blot of HEK293 cells over-expressing FLAG-SphK1 where cells were incubated with PD98059 (10 μM) or 8: SKI-178 (10 µM) and DMSO, for 24 hrs. CETSA experiments were performed as detailed in the Materials and Methods and analyzed by western blot detection of SphK1 and Actin using the appropriate primary antibodies. (b) Representative western blots of HEK293 cells over-expressing FLAG-SphK1 where cells were incubated with the indicated SKIs at the indicated concentrations [6: SKI-V (10 µM), 9: ABC294640 (20 µM), 10: K145 (10 µM), and 11: SKI-5 c (20 µM)] and DMSO, for 24 hrs. CETSA experiments were performed as detailed in the Materials and Methods and analyzed by western blot detection of SphK1 using the appropriate primary antibody.
Demonstration of target engagement of SphK2 using CETSA
Between SKI-induced proteasomal degradation and CETSA confirmed target engagement, we confirmed that 12 of the 12 SKIs target engaged SphK1 including the 2 SphK2-selective inhibitors ABC294640 (9) and K145 (10). In contrast to SphK1, none of the SKIs tested induced proteasomal degradation of SphK2. Thus, we employed CETSA to determine whether the SKIs target engaged SphK2 and also to determine whether any of the reported SphK1-selective inhibitors were, indeed, specific for SphK1. We chose to focus our studies on the more recently described SKIs and as such, have excluded SKI-I (4), SKI-V (6), DMS (1) and Safingol (12) from our CETSA analysis. Similarly, it is well know that FTY720 (7) is a substrate of SphK2 and therefore we eliminated it from our CETSA analysis of SphK2 target engagement, as well.47
The remaining SKIs were evaluated for their ability to target engage SphK2, using the FLAG-SphK2 over-expressing HEK-293 system, by CETSA experiments. As shown in Figure 4(a), SKI-178 (8) target engaged SphK2 as previously demonstrated.35 Again, actin controls were consistent across the thermal denaturation gradient indicating that there was no inadvertent error arising from inefficient loading/transfer of samples. As expected, we observed, in Figure 4(b), target engagement of SphK2 with both of the reported SphK2-selective SKIs, ABC294640 (9) and K145 (10). Also, we observed target engagement by our nonselective inhibitor, SKI-II (2). Surprisingly, we once again observed target engagement of SphK2 with the reportedly SphK1-selective SKIs, SKI-5 C (11), SK1-I (5) and very strong target engagement by PF-543 (3).
Figure 4.
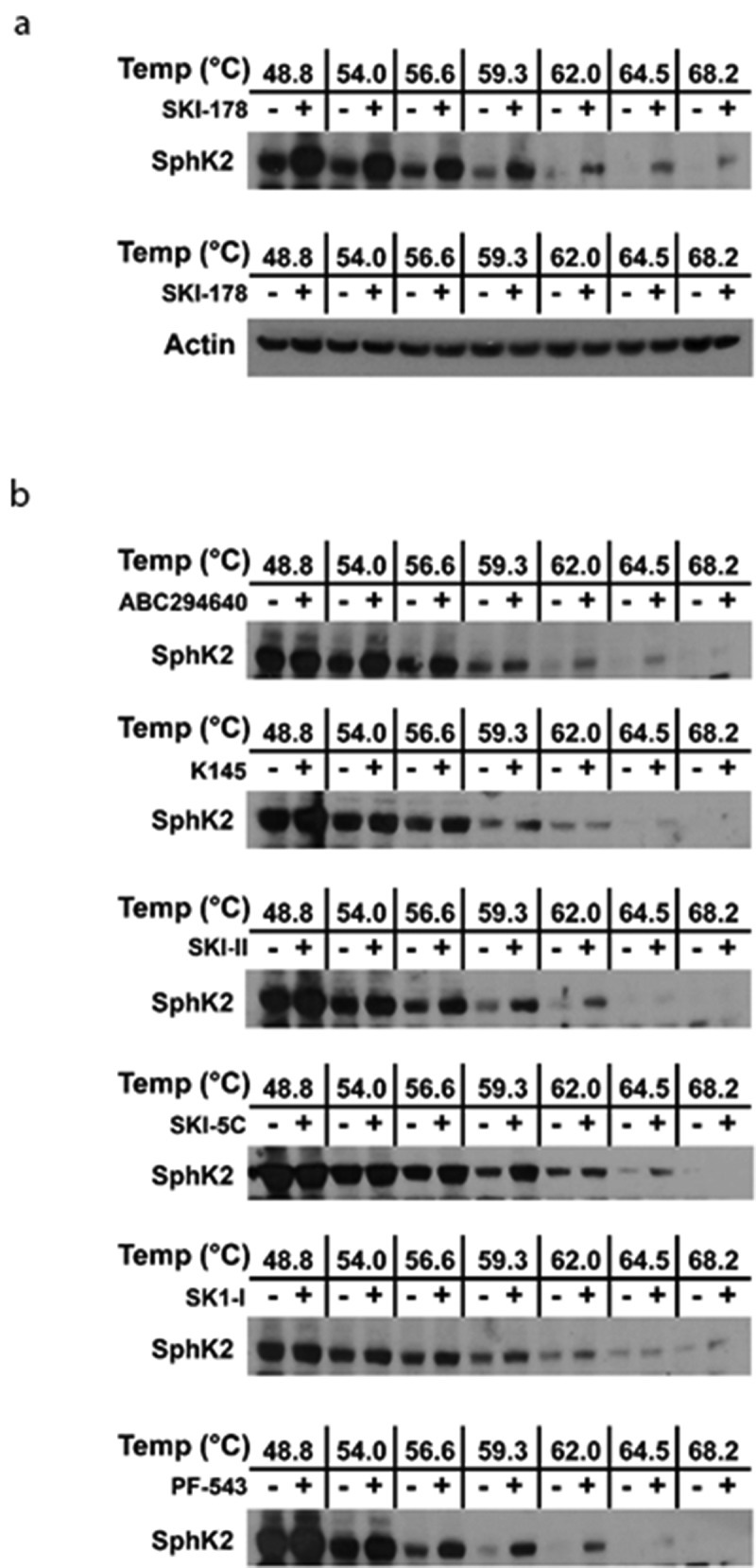
Demonstration of Target Engagement of SphK2 in HEK293 cells by SKIs using CETSA. (a) A representative western blot of HEK293 cells over-expressing FLAG-SphK2 where cells were incubated with 8: SKI-178 (10 µM) and DMSO, for 24 hrs. CETSA experiments were performed as detailed in the Materials and Methods and analyzed by western blot detection of SphK1 and Actin using the appropriate primary antibodies. (b) Representative western blots of HEK293 cells over-expressing FLAG-SphK2 where cells were incubated with the indicated SKIs at the indicated concentrations [2: SKI-II (10 µM), 3: PF-543 (10 µM), 5: SK1-I (10 µM), 8: SKI-178 (10 µM), 9: ABC294640 (20 µM), 10: K145 (10 µM), 11: SKI-5 c (20 µM)] and DMSO, for 24 hours. CETSA experiments were performed as detailed in the Materials and Methods and analyzed by western blot detection of SphK2 using the appropriate primary antibody.
Determination of the IC50 of target engagement of PF-543 for SphK2
The standard CETSA experiment can be modified to generate an IsoThermal Dose-Response Fingerprint (ITDRFCETSA) curve for the protein of interest at a temperature where the test compound is known to stabilize/destabilize its targets.31 We have employed the ITDRFCETSA method to determine the IC50 for target engagement of PF-543 (3) for SphK2. As shown in Figure 5, PF-543 (3) target engages SphK2 with an approximate IC50 of 2.5 μM. Given these results and the fact that the IC50 of PF-543 (3) induced SphK1 degradation was approximately 10 nM (Figure 2(d)), treatment of cells with approximately 5 μM PF-543 (3) should be sufficient to completely block S1P production.
Figure 5.
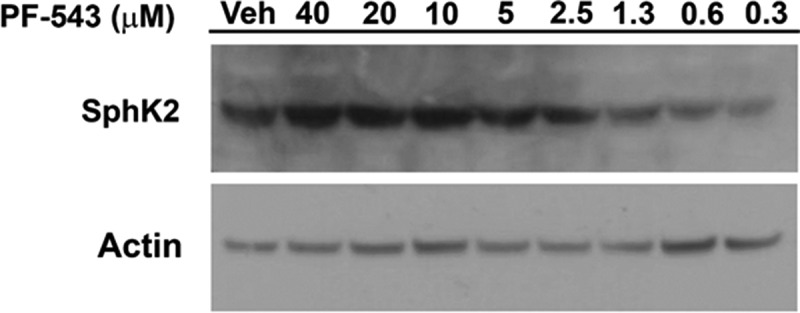
Determination of the IC50 of Target Engagement of SphK2 by PF-543 using the IsoThermal Dose Response Fingerprint (ITDRF) Assay. A representative western blot of HEK293 cells over-expressing FLAG-SphK2 where cells were incubated with the indicated concentrations of (3: PF-543) and DMSO for 24 hrs. ITDRF assays were performed at 63.0°C as detailed in the Materials and Methods and analyzed by western blot detection of SphK2 using the appropriate primary antibody. Actin was included as a loading control.
Sphingolipid analysis in intact cells
To further confirm that PF-543 (3) inhibited the activities of both SphK1 and SphK2 in cells and to confirm the results of the CETSA experiments, we generated SphK1 and SphK2 knock-out clones of DLD-1 cells using Crispr/Cas9 technology. As shown in Figure 6(a) we isolated single knock-out clones of SphK1 (clone #1) and SphK2 (clone #3). To determine the ability of PF-543 (3) to block the generation of S1P at higher concentrations, we treated DLD-1 colorectal cancer (CRC) cells and the DLD-1 SphK1 and SphK2 knock-out cell lines (SphK1 KO and SphK2 KO) with either vehicle or PF-543 (3; 5 μM) for 48 hours and conducted sphingolipidomic analysis (Lipidomic Shared Resource Facility; Hollings Cancer Center, Medical University of South Carolina, Charleston SC). As shown in Figure 6(b), dhS1P and S1P production were reduced in DLD-1 cells treated with PF-543 (3) relative to vehicle treatment. PF-543 (3) treatment of DLD-1 cells devoid of SphK1 protein (SphK1 KO) reduced dhS1P and S1P production similarly to naïve DLD-1 cells indicating that PF-543 (3) does indeed target and inhibit SphK2 in intact cells. Similarly, SphK2 KO dramatically reduced both dhS1P and S1P levels in vehicle treated cells indicating that SphK2 is the predominant isoform expressed in DLD-1 cells. Concomitantly, we observed an increase in Sph and dhSph levels in response to PF-543 in naïve DLD-1 cells, as well as the SphK1 KO and SphK2 KO cells (Figure 6(c)). Consistent with the idea that SphK2 is the predominant isoform in DLD-1 cells, the levels of dhSph and Sph were elevated in vehicle treated SphK2 KO cells relative to naïve and SphK1 KO vehicle controls. dhSph and Sph levels were further elevated upon treatment of the SphK2 KO cells with PF-543 indicating that PF-543 inhibited SphK1 in the SphK2 KO cells.
Figure 6.
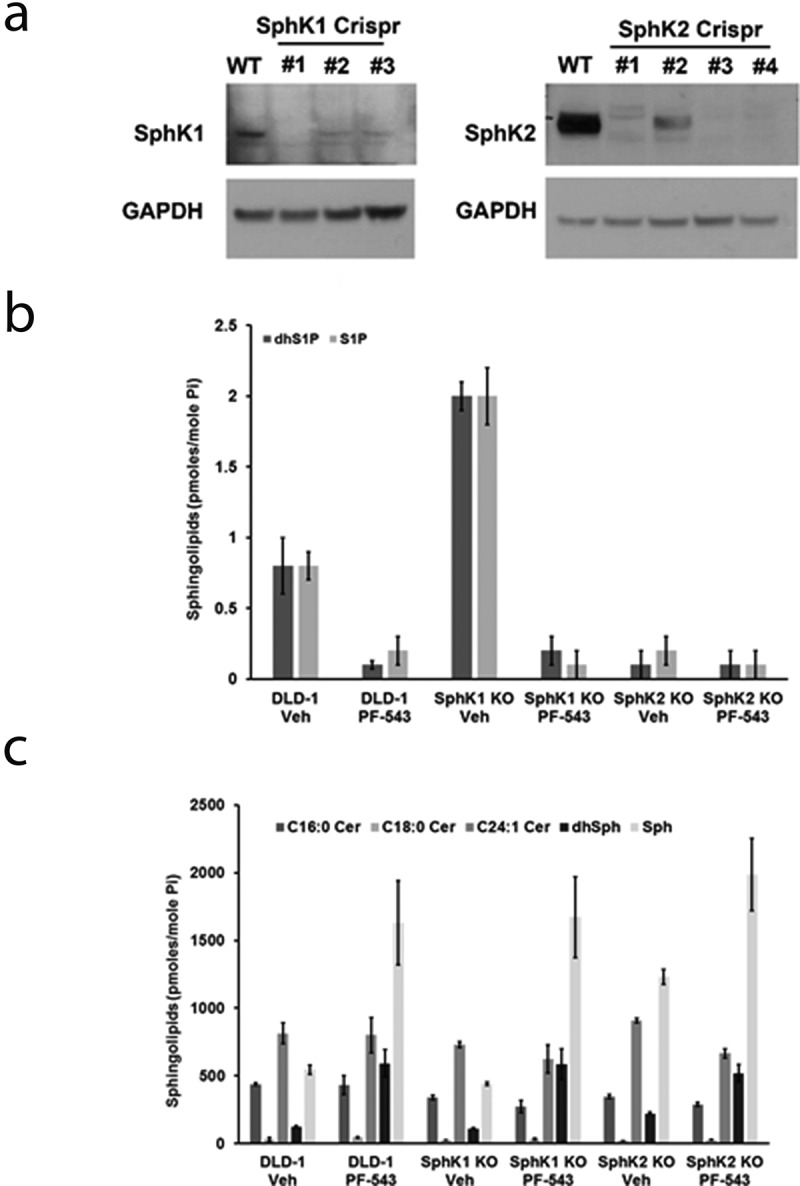
Crispr/Cas9 knock-out clones of SphK1 and SphK2 in DLD1 cells. (a) Representative western blots of DLD1 cells transfected with gRNAs directed to either SphK1 or SphK2. Detection of SphK1 and SphK2 was performed using the appropriate primary antibodies. GAPDH was included as a loading control. (b) Sphingolipid analysis of dhS1P and S1P levels in naïve DLD-1 cells and Crispr/Cas9 knock-out DLD-1 clones of SphK1 (SphK1 KO; clone 1) and SphK2 (SphK2 KO; clone 3) treated with DMSO or PF-543 (3: 5 µM) for 48 h. Sphingolipid levels are expressed as pmoles/mole of inorganic phosphate (Pi) for normalization purposes. (c) Sphingolipid analysis of C16:0 Cer, C18:0 Cer, C24:1 Cer, dhSph and Sph levels in naïve DLD-1 cells and Crispr/Cas9 DLD-1 SphK1 KO and SphK2 KO clones treated with DMSO or PF-543 (3: 5 µM) for 48 h. Sphingolipid levels are expressed as pmoles/mole of inorganic phosphate (Pi) for normalization purposes.
Discussion
Taken together, our results demonstrate that all 12 of the commercially available SphK Inhibitors, tested herein, are capable of entering intact cells and target engaging both human SphK1 and SphK2 at micromolar concentrations. Such concentrations are typically employed in published studies and often a biological effect is attributed to one SphK isoform or the other based upon the use of a single “selective” SphK inhibitor. The major take home message of this study is that caution should always be employed when using small-molecule inhibitors to attempt to discern the relative contribution of either SphK isoform to an observed biological effect. Furthermore we stress the need for investigators to confirm the results obtained with SKIs by the use of molecular tools such as siRNA/shRNA knock-down or, more ideally Crispr/Cas9 mediated knock-out.
Rather than point fingers at any particular study that might have employed an aberrantly high concentration of a particular SKI, we would draw attention to several studies that employed multiple strategies to diligently determine whether a particular SphK isoform was involved in the observed phenomenon.48-51 These, and many more, studies employed multiple strategies to confirm the results obtained with SKIs. Such strategies include the use of multiple SKIs, the use of molecular tools such as siRNA/shRNA knock-down and/or the use of knock-out mouse embryonic fibroblasts (MEFs). Given the need for increased rigor and reproducibility, it is incumbent upon investigators to employ these strategies and to make these tools available to the scientific community.
While our primary goal was to raise awareness of the need for caution in studies employing SKIs, this study also provides/validates several other research tools for the SphK “toolkit”. Indeed, as Neubauer et al.40 has previously done for SphK2, we have identified and validated a SphK1 antibody that recognizes endogenous levels of SphK1 (Figure 6). Moreover, we identified Crispr/Cas9 guide RNAs (gRNAs) that are capable of inducing knock-out of both human Sphk1 and SphK2. Finally, we have described assay conditions for the use of CETSA techniques to demonstrate the target engagement of both SphK1 and SphK2 in intact cells. This simple technique should facilitate the discovery and development of new, more isoform-selective SKIs.
The CETSA protocol can also be adapted to demonstrate target engagement in whole animal tissues, by isolating cells from organs of interest and conducting ex-vivo CETSA experiments. This use of CETSA will be extremely valuable in the demonstration of target engagement of the SphKs in the tumor cells of orthotopic tumor models, for instance. Similarly, CETSA has also been adapted to be employed as a discovery technique in LC/MS/MS-based proteomic studies to identify unknown proteins to which a particular compound binds.52 We believe that this use of CETSA could help clarify the controversy regarding whether cytotoxic SKIs induce apoptosis through “on-target” inhibition of the SphKs or through “off-target” effects such as inhibition of Dihydroceramide Desaturase (DES1) as has been reported for SKI-II and ABC294640.46,53 Indeed, CETSA experiment conditions can be developed for DES1 and all current and future SKIs can be easily tested for target engagement of DES1 as part of the drug development process. In this manner, it should be relatively easy to optimize an SKI away from DES1 binding.
We must acknowledge that one caveat to our findings is that they demonstrate that the tested SKIs bind to both SphK1 and SphK2, somewhere. Stabilization of SphK protein does not automatically indicate that the SKIs bind to the active site and inhibit SphK activity. It is possible that they could bind to allosteric sites on the proteins and stabilize/destabilize them in this manner. However, given that these SKIs all share, in common, the fact that they are Sph competitive inhibitors, it is much more likely that they are target engaging the Sph binding sites of both SphK isoforms.
In conclusion, we strongly encourage investigators to be cognizant of the concentrations of SKIs used in their studies. We also encourage the use of the CETSA experiment to evaluate the IC50s for target engagement using the ITDRF technique when selecting concentrations of SKIs to use in studies. Furthermore, the complimentary use of molecular tools to supplement the use of small-molecule inhibitors is warranted. Together, a more cautious approach will enhance the rigor and reproducibility of future SphK studies and potentially aid in the transition of SKIs form “bench to bedside”.
Materials and methods
Reagents
Sphingosine Kinase Inhibitors (SKIs) were purchased as follows: N,N-dimethyl-sphingosine, SKI-II and SK1-I were from Enzo Life Sciences (Farmingdale NY, USA), PF-543 was from Selleck Chemicals (Houston TX, USA), SKI-I was from Abcam (Cambridge MA, USA), FTY720, SKI-178 and ABC294640 were from Cayman Chemicals (Ann Arbor MI, USA), SKI-V, K145, SKI-5 c, Safingol and MG-132 were from Sigma-Aldrich (St. Louis, MO). Antibodies against SphK1 (clone D1H1 L, Cell Signaling Technology, Danvers, MA), SphK2 (Proteintech, Rosemont IL), FLAG epitope (Sigma-Aldrich, St. Louis, MO), Actin (Cell Signaling Technology, Danvers, MA) and GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA).
Cell lines, constructs, and culture conditions
The human colorectal cancer (CRC) derived cell line, DLD-1 (CCL-221), was obtained from the American Type Culture Collection (ATCC, Manassas, Virginia, USA) and were maintained in DMEM (Thermo Fisher Scientific, Waltham, Massachusetts, USA) supplemented with 10% FBS (Denville Scientific, South Plainfield, NJ, USA). FLAG-His6X-SphK1 and FLAG-His6X-SphK2 constructs were created by site-directed mutagenesis of the Xpress epitope of pcDNA3.1/HisB to the FLAG epitope. HEK293 cells stably over-expressing FLAG-SphK1 and FLAG-SphK2 were maintained in DMEM (Thermo Fisher Scientific, Waltham, MA) supplemented with 10% FBS and G418 as described in.35 All cell lines were maintained at 37°C with 5% CO2 in a humidified incubator.
Western blot analysis
Briefly, whole cell lysates were harvested from cells treated with various agents in 1X RIPA buffer (20 mM Tris-HCL (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, 1 mM sodium orthovandate, 30 mM sodium fluoride, and complete protease inhibitor cocktail tablet (Roche Diagnostics, Mannheim, Germany)). Lysates were centrifuged (20,000 x g) for 15 min at 4°C to remove cell debris. Total protein concentrations were quantified using the BCA assay from Pierce (Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of denatured total protein were resolved by NuPAGE 4–12% Bis-Tris gel electrophoresis (Life technologies Carlsbad, CA, USA) and transferred to PVDF membranes (Life technologies Carlsbad, CA, USA). Membranes were blocked for 1 h at room temperature in 5% milk/TBS-T, incubated overnight at 4°C with primary antibodies (1:2000), and immunodetection was done with corresponding secondary IgG HRP-linked antibodies (1:5000) using the ECL chemiluminescence reagents (Thermo Fisher Scientific, Waltham, Massachusetts, USA).
Cellular Thermal Shift Assay (CETSA)
Target engagement assays of SphK1, and SphK2 were performed as described in.35 Briefly, nearly confluent 10 cm culture dishes of HEK293 cells, overexpressing either FLAG-SphK1 or FLAG-SphK2, were treated with either vehicle control or the indicated SKIs for 24 h. Cells were collected by trypsinization, neutralized by the addition of DMEM containing 10% FBS, pelleted at 300 x g for 5 min, and washed with 1X PBS. After washing, cells were pelleted again at 300 x g for 5 min and resuspended in 1 mL of 20 mM Tris pH 7.4 containing a complete protease inhibitor cocktail tablet (Roche Diagnostics, Mannheim, Germany). 100 μL aliquots of the cell suspension were heated to designated temperatures for 3 min. After cooling for 3 min at room temperature, cells were freeze-thawed three times with liquid nitrogen and the soluble fraction (lysate) was separated from cell debris as well as the precipitated and aggregated proteins by centrifugation at 20,000 x g for 20 min at 4°C. The supernatant was transferred to a new tube and quantification of the remaining soluble protein was achieved by Western blot analysis of equal volumes of the remaining soluble protein.
Isothermal Dose Response Fingerprint (ITDRF)
ITDRF assays were performed as described in Jafari et al. with minor modifications.31 Briefly, nearly confluent 10 cm culture dishes of HEK293 cells, overexpressing FLAG-SphK2, were treated with either vehicle control or the indicated concentration of PF-543 for 24 h. Cells were collected by trypsinization, neutralized by the addition of DMEM containing 10% FBS, pelleted at 300 x g for 5 min, and washed with 1X PBS. After washing, cells were pelleted again at 300 x g for 5 min and resuspended in 1 mL of 20 mM Tris pH 7.4 containing a complete protease inhibitor cocktail tablet (Roche Diagnostics, Mannheim, Germany). 100 μL aliquots of the cell suspension were heated to 63.0°C for 3 min. After cooling for 3 min at room temperature, cells were freeze-thawed three times with liquid nitrogen and the soluble fraction (lysate) was separated from cell debris as well as the precipitated and aggregated proteins by centrifugation at 20,000 x g for 20 min at 4°C. The supernatant was transferred to a new tube and quantification of the remaining soluble SphK2 protein was achieved by Western blot analysis of equal volumes of the remaining soluble protein.
Crispr/Cas9 knock-out of SphK1 and SphK2 in DLD-1 cells
SphK1 and SphK2 knock-out clones of DLD-1 cells were generated using the LentiCRISPR v2.0 system with guide RNAs (gRNAs; SphK1; 5ʹ-GACGCTCTGGTGGTCATGTC-3ʹ and SphK2; 5ʹ-CTACGAAGAGAACGTGCCG-3ʹ) purchased from GenScript (Piscataway, NJ USA). DLD-1 cells were transiently transfected using Lipofectamine 2000. After 48 h, cell were selected in puromycin (10 ug/mL) for 24 h and maintained in 1 ug/mL puromycin until single clones were isolated. Screening for SphK expression knock-out was conducted by western blot analysis using the appropriate SphK antibodies.
Acknowledgments
This work was supported by the National Institutes of Health (P01 CA171983 to J.K.Y), (P01 CA203628 to C.D.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was also supported by the Four Diamonds (to C.D.S.) and The Jake Gittlen Memorial Golf Tournament (to J.K.Y.)
Funding Statement
This work was supported by the National Cancer Institute [CA203628]; National Cancer Institute [CA171983].
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Begley CG, Ellis LM.. Drug development: raise standards for preclinical cancer research. Nature. 2012;483:531–533. [DOI] [PubMed] [Google Scholar]
- 2.Liu X, Shi Y, Maag DX, Palma JP, Patterson MJ, Ellis PA, Surber BW, Ready DB, Soni NB, Ladror US, et al. Iniparib nonselectively modifies cysteine-containing proteins in tumor cells and is not a bona fide PARP inhibitor. Clin Cancer Res. 2012;18:510–523. doi: 10.1158/1078-0432.CCR-11-1973. [DOI] [PubMed] [Google Scholar]
- 3.Patel AG, De Lorenzo SB, Flatten KS, Poirier GG, Kaufmann SH.. Failure of iniparib to inhibit poly(ADP-Ribose) polymerase in vitro. Clin Cancer Res. 2012;18:1655–1662. doi: 10.1158/1078-0432.CCR-11-2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, Galvin KM, Hoar KM, Huck JJ, LeRoy PJ, et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci USA. 2007;104:4106–4111. doi: 10.1073/pnas.0608798104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du J, Yan L, Torres R, Gong X, Bian H, Marugan C, Boehnke K, Baquero C, Hui Y-H, Chapman SC, et al. Aurora A selective inhibitor LY3295668 leads to dominant mitotic arrest, apoptosis in cancer cells and shows potent preclinical antitumor efficacy. Mol Cancer Ther. 2019;18:2207–2219. doi: 10.1158/1535-7163.MCT-18-0529. [DOI] [PubMed] [Google Scholar]
- 6.Gong X, Du J, Parsons SH, Merzoug FF, Webster Y, Iversen PW, Chio L-C, Van Horn RD, Lin X, Blosser W, et al. Aurora A kinase inhibition is synthetic lethal with loss of the RB1 tumor suppressor gene. Cancer Discov. 2019;9:248–263. doi: 10.1158/2159-8290.CD-18-0469. [DOI] [PubMed] [Google Scholar]
- 7.Pulkoski-Gross MJ, Obeid LM.. Molecular mechanisms of regulation of sphingosine kinase 1. Biochim Biophys Acta Mol Cell Biol Lipids. 2018;1863:1413–1422. doi: 10.1016/j.bbalip.2018.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song DD, Zhou JH, Sheng R. Regulation and function of sphingosine kinase 2 in diseases. Histol Histopathol. 2018;33:433–445. doi: 10.14670/HH-11-939. [DOI] [PubMed] [Google Scholar]
- 9.Neubauer HA, Pitson SM. Roles, regulation and inhibitors of sphingosine kinase 2. Febs J. 2013;280:5317–5336. doi: 10.1111/febs.12314. [DOI] [PubMed] [Google Scholar]
- 10.Hait NC, Bellamy A, Milstien S, Kordula T, Spiegel S. Sphingosine kinase type 2 activation by ERK-mediated phosphorylation. J Biol Chem. 2007;282:12058–12065. doi: 10.1074/jbc.M609559200. [DOI] [PubMed] [Google Scholar]
- 11.Neubauer HA, Pham DH, Zebol JR, Moretti PA, Peterson AL, Leclercq TM, Chan H, Powell JA, Pitman MR, Samuel MS, et al. An oncogenic role for sphingosine kinase 2. Oncotarget. 2016;7:64886–64899. doi: 10.18632/oncotarget.11714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Igarashi N, Okada T, Hayashi S, Fujita T, Jahangeer S, Nakamura S. Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J Biol Chem. 2003;278:46832–46839. doi: 10.1074/jbc.M306577200. [DOI] [PubMed] [Google Scholar]
- 13.Ding G, Sonoda H, Yu H, Kajimoto T, Goparaju SK, Jahangeer S, Okada T, Nakamura S-I. Protein kinase D-mediated phosphorylation and nuclear export of sphingosine kinase 2. J Biol Chem. 2007;282:27493–27502. doi: 10.1074/jbc.M701641200. [DOI] [PubMed] [Google Scholar]
- 14.Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S, et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325:1254–1257. doi: 10.1126/science.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, Yun JK, Smith CD. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63:5962–5969. [PubMed] [Google Scholar]
- 16.Plano D, Amin S, Sharma AK. Importance of sphingosine kinase (SphK) as a target in developing cancer therapeutics and recent developments in the synthesis of novel SphK inhibitors. J Med Chem. 2014;57:5509–5524. doi: 10.1021/jm4011687. [DOI] [PubMed] [Google Scholar]
- 17.Paugh SW, Paugh BS, Rahmani M, Kapitonov D, Almenara JA, Kordula T, Milstien S, Adams JK, Zipkin RE, Grant S, et al. A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood. 2008;112:1382–1391. doi: 10.1182/blood-2008-02-138958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong L, Tan SS, Lam Y, Melendez AJ. Synthesis and evaluation of sphingosine analogues as inhibitors of sphingosine kinases. J Med Chem. 2009;52:3618–3626. doi: 10.1021/jm900121d. [DOI] [PubMed] [Google Scholar]
- 19.Schnute ME, McReynolds MD, Kasten T, Yates M, Jerome G, Rains JW, Hall T, Chrencik J, Kraus M, Cronin C, et al. Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem J. 2012;444:79–88. doi: 10.1042/BJ20111929. [DOI] [PubMed] [Google Scholar]
- 20.French KJ, Zhuang Y, Maines LW, Gao P, Wang W, Beljanski V, Upson JJ, Green CL, Keller SN, Smith CD, et al. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J Pharmacol Exp Ther. 2010;333:129–139. doi: 10.1124/jpet.109.163444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu K, Guo TL, Hait NC, Allegood J, Parikh HI, Xu W, Kellogg GE, Grant S, Spiegel S, Zhang S, et al. Biological characterization of 3-(2-amino-ethyl)-5-[3-(4-butoxyl-phenyl)-propylidene]-thiazolidine-2,4-dione (K145) as a selective sphingosine kinase-2 inhibitor and anticancer agent. PloS One. 2013;8:e56471. doi: 10.1371/journal.pone.0056471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gustin DJ, Li Y, Brown ML, Min X, Schmitt MJ, Wanska M, Wang X, Connors R, Johnstone S, Cardozo M, et al. Structure guided design of a series of sphingosine kinase (SphK) inhibitors. Bioorg Med Chem Lett. 2013;23:4608–4616. doi: 10.1016/j.bmcl.2013.06.030. [DOI] [PubMed] [Google Scholar]
- 23.Xiang Y, Asmussen G, Booker M, Hirth B, Kane JL Jr., Liao J, Noson KD, Yee C. Discovery of novel sphingosine kinase 1 inhibitors. Bioorg Med Chem Lett. 2009;19:6119–6121. doi: 10.1016/j.bmcl.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 24.Xiang Y, Hirth B, Kane JL Jr., Liao J, Noson KD, Yee C, Asmussen G, Fitzgerald M, Klaus C, Booker M, et al. Discovery of novel sphingosine kinase-1 inhibitors. Part 2. Bioorg Med Chem Lett. 2010;20:4550–4554. doi: 10.1016/j.bmcl.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 25.Mathews TP, Kennedy AJ, Kharel Y, Kennedy PC, Nicoara O, Sunkara M, Morris AJ, Wamhoff BR, Lynch KR, Macdonald TL, et al. Discovery, biological evaluation, and structure-activity relationship of amidine based sphingosine kinase inhibitors. J Med Chem. 2010;53:2766–2778. doi: 10.1021/jm901860h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raje MR, Knott K, Kharel Y, Bissel P, Lynch KR, Santos WL. Design, synthesis and biological activity of sphingosine kinase 2 selective inhibitors. Bioorg Med Chem. 2012;20:183–194. doi: 10.1016/j.bmc.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patwardhan NN, Morris EA, Kharel Y, Raje MR, Gao M, Tomsig JL, Lynch KR, Santos WL. Structure−Activity relationship studies and in vivo activity of guanidine-based sphingosine kinase inhibitors: discovery of SphK1- and SphK2-selective inhibitors. J Med Chem. 2015;58:1879–1899. doi: 10.1021/jm501760d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Childress ES, Kharel Y, Brown AM, Bevan DR, Lynch KR, Santos WL. Transforming sphingosine kinase 1 inhibitors into dual and sphingosine kinase 2 selective inhibitors: design, synthesis, and in vivo activity. J Med Chem. 2017;60:3933–3957. doi: 10.1021/acs.jmedchem.7b00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma AK, Sk UH, Gimbor MA, Hengst JA, Wang X, Yun J, Amin S. Synthesis and bioactivity of sphingosine kinase inhibitors and their novel aspirinyl conjugated analogs. Eur J Med Chem. 2010;45:4149–4156. doi: 10.1016/j.ejmech.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Hengst JA, Wang X, Sk UH, Sharma AK, Amin S, Yun JK. Development of a sphingosine kinase 1 specific small-molecule inhibitor. Bioorg Med Chem Lett. 2010;20:7498–7502. doi: 10.1016/j.bmcl.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 31.Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundback T, Nordlund P, Molina DM. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc. 2014;9:2100–2122. doi: 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- 32.Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 33.Martinez Molina D, Nordlund P. The cellular thermal shift assay: a Novel biophysical assay for in situ drug target engagement and mechanistic biomarker studies. Annu Rev Pharmacol Toxicol. 2016;56:141–161. doi: 10.1146/annurev-pharmtox-010715-103715. [DOI] [PubMed] [Google Scholar]
- 34.Dick TE, Hengst JA, Fox TE, Colledge AL, Kale VP, Sung SS, Sharma A, Amin S, Loughran TP, Kester M, et al. The apoptotic mechanism of action of the sphingosine kinase 1 selective inhibitor SKI-178 in human acute myeloid leukemia cell lines. J Pharmacol Exp Ther. 2015;352:494–508. doi: 10.1124/jpet.114.219659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hengst JA, Dick TE, Sharma A, Doi K, Hegde S, Tan SF, Geffert L, Fox T, Sharma A, Desai D, et al. SKI-178: a multitargeted inhibitor of sphingosine kinase and microtubule dynamics demonstrating therapeutic efficacy in acute myeloid leukemia models. Cancer Transl Med. 2017;3:109–121. doi: 10.4103/ctm.ctm_7_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu H, Sugiura M, Nava VE, Edsall LC, Kono K, Poulton S, Milstien S, Kohama T, Spiegel S. Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J Biol Chem. 2000;275:19513–19520. doi: 10.1074/jbc.M002759200. [DOI] [PubMed] [Google Scholar]
- 37.Gao P, Peterson YK, Smith RA, Smith CD. Characterization of isoenzyme-selective inhibitors of human sphingosine kinases. PloS One. 2012;7:e44543. doi: 10.1371/journal.pone.0044543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lim KG, Tonelli F, Li Z, Lu X, Bittman R, Pyne S, Pyne NJ. FTY720 analogues as sphingosine kinase 1 inhibitors: enzyme inhibition kinetics, allosterism, proteasomal degradation, and actin rearrangement in MCF-7 breast cancer cells. J Biol Chem. 2011;286:18633–18640. doi: 10.1074/jbc.M111.220756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olivera A, Kohama T, Tu Z, Milstien S, Spiegel S. Purification and characterization of rat kidney sphingosine kinase. J Biol Chem. 1998;273:12576–12583. doi: 10.1074/jbc.273.20.12576. [DOI] [PubMed] [Google Scholar]
- 40.Neubauer HA, Pitson SM. Validation of commercially available sphingosine kinase 2 antibodies for use in immunoblotting, immunoprecipitation and immunofluorescence. F1000Res. 2016;5:2825. doi: 10.12688/f1000research.10336.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ju T, Gao D, Fang ZY. Targeting colorectal cancer cells by a novel sphingosine kinase 1 inhibitor PF-543. Biochem Biophys Res Commun. 2016;470:728–734. doi: 10.1016/j.bbrc.2016.01.053. [DOI] [PubMed] [Google Scholar]
- 42.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 43.Byun HS, Pyne S, Macritchie N, Pyne NJ, Bittman R. Novel sphingosine-containing analogues selectively inhibit sphingosine kinase (SK) isozymes, induce SK1 proteasomal degradation and reduce DNA synthesis in human pulmonary arterial smooth muscle cells. Medchemcomm. 2013;4:1394. doi: 10.1039/c3md00201b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tonelli F, Lim KG, Loveridge C, Long J, Pitson SM, Tigyi G, Bittman R, Pyne S, Pyne NJ. FTY720 and (S)-FTY720 vinylphosphonate inhibit sphingosine kinase 1 and promote its proteasomal degradation in human pulmonary artery smooth muscle, breast cancer and androgen-independent prostate cancer cells. Cell Signal. 2010;22:1536–1542. doi: 10.1016/j.cellsig.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loveridge C, Tonelli F, Leclercq T, Lim KG, Long JS, Berdyshev E, Tate RJ, Natarajan V, Pitson SM, Pyne NJ, et al. The sphingosine kinase 1 inhibitor 2-(p-hydroxyanilino)-4-(p-chlorophenyl)thiazole induces proteasomal degradation of sphingosine kinase 1 in mammalian cells. J Biol Chem. 2010;285:38841–38852. doi: 10.1074/jbc.M110.127993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McNaughton M, Pitman M, Pitson SM, Pyne NJ, Pyne S. Proteasomal degradation of sphingosine kinase 1 and inhibition of dihydroceramide desaturase by the sphingosine kinase inhibitors, SKi or ABC294640, induces growth arrest in androgen-independent LNCaP-AI prostate cancer cells. Oncotarget. 2016;7:16663–16675. doi: 10.18632/oncotarget.7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paugh SW, Payne SG, Barbour SE, Milstien S, Spiegel S. The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2. FEBS Lett. 2003;554:189–193. doi: 10.1016/S0014-5793(03)01168-2. [DOI] [PubMed] [Google Scholar]
- 48.Lima S, Takabe K, Newton J, Saurabh K, Young MM, Leopoldino AM, Hait NC, Roberts JL, Wang H-G, Dent P, et al. TP53 is required for BECN1- and ATG5-dependent cell death induced by sphingosine kinase 1 inhibition. Autophagy. 2018;14:942–957. doi: 10.1080/15548627.2018.1429875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Young MM, Takahashi Y, Fox TE, Yun JK, Kester M, Wang HG. Sphingosine kinase 1 cooperates with autophagy to maintain endocytic membrane trafficking. Cell Rep. 2016;17:1532–1545. doi: 10.1016/j.celrep.2016.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qin Z, Dai L, Trillo-Tinoco J, Senkal C, Wang W, Reske T, Bonstaff K, Del Valle L, Rodriguez P, Flemington E, et al. Targeting sphingosine kinase induces apoptosis and tumor regression for KSHV-associated primary effusion lymphoma. Mol Cancer Ther. 2014;13:154–164. doi: 10.1158/1535-7163.MCT-13-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wallington-Beddoe CT, Powell JA, Tong D, Pitson SM, Bradstock KF, Bendall LJ. Sphingosine kinase 2 promotes acute lymphoblastic leukemia by enhancing MYC expression. Cancer Res. 2014;74:2803–2815. doi: 10.1158/0008-5472.CAN-13-2732. [DOI] [PubMed] [Google Scholar]
- 52.Savitski MM, Reinhard FB, Franken H, Werner T, Savitski MF, Eberhard D, Molina DM, Jafari R, Dovega RB, Klaeger S, et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014;346:1255784. doi: 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- 53.Cingolani F, Casasampere M, Sanllehi P, Casas J, Bujons J, Fabrias G. Inhibition of dihydroceramide desaturase activity by the sphingosine kinase inhibitor SKI II. J Lipid Res. 2014;55:1711–1720. doi: 10.1194/jlr.M049759. [DOI] [PMC free article] [PubMed] [Google Scholar]