

ABSTRACT
Non-small cell lung cancer (NSCLC) remains recalcitrant to effective treatment due to tumor relapse and acquired resistance. Cancer stem cells (CSCs) are believed to be one mechanism for relapse and resistance and are consequently considered promising drug targets. We report that chetomin, an active component of Chaetomium globosum, blocks heat shock protein 90/hypoxia-inducible factor 1 alpha (Hsp90/HIF1α) pathway activity. Chetomin also attenuated sphere-forming, a stem cell-like characteristic, of NSCLC CSCs (at ~ nM range) and the proliferation of non-CSCs NSCLC cultures and chemoresistant sublines (at ~ μM range). At these concentrations, chetomin exerted a marginal influence on noncancerous cells originating from several organs. Chetomin markedly decreased in vivo tumor formation in a spontaneous KrasLA1 lung cancer model, flank xenograft models, and a tumor propagation flank implanted model at doses that did not produce an observable toxicity to the animals. Chetomin blocked Hsp90/HIF1α pathway activity via inhibiting the Hsp90-HIF1α binding interaction without affecting Hsp90 or Hsp70 protein levels. This study advocates chetomin as a Hsp90/HIF1α pathway inhibitor and a potent, nontoxic NSCLC CSC-targeting molecule.
KEYWORDS: Chetomin, lung cancer, cancer stem cells, heat shock protein
Introduction
Non-small cell lung cancer (NSCLC) is the most prevalent type of lung cancer and is among the most prominent causes of mortality in cancer patients globally1. Great advances have been made in the treatment of NSCLC, particularly with a recent shift from chemotherapy to more targeted treatments, such as tyrosine kinase inhibitors (TKIs)2 and immunotherapy.3 Even with these improved options, survival among patients with NSCLC continues to be dismal.4 Further complicating treatment is cancer progression, particularly for patients that develop tumors with acquired resistance.5 Consequently, research has been directed toward approaches that prevent acquired therapy resistance or cancer relapse.
Cancer stem cells (CSCs) have been suggested as one reason that tumors recur, as they maintain the ability to self-renew and differentiate.6 CSCs have been discovered in numerous cancers, including NSCLC.7 CSCs are believed to be chemoresistant; following treatment, persisting CSCs can continue to divide, giving rise to tumor recurrence and metastatic progression.7 CSC-targeting therapies are therefore regarded as a possible avenue to prevent tumor recurrence.
Hsp90 (heat shock protein 90) is a chaperone protein that helps tumor cells endure and acquire chemoresistance by helping various oncogenic proteins to fold and mature.8,9 Hsp90 is frequently overexpressed in human tumors and is associated with a shorter survival in NSCLC patients.10,11 Hsp90 may also be a promising candidate for targeting CSCs as well as non-CSCs, since CSCs need to endure and preserve their stem cell-like capacity under strenuous environments, including hypoxia and cytotoxic stress.12 Indeed, direct Hsp90 inhibitors have demonstrated efficacy in preclinical models and have progressed to clinical trial assessment.13,14 However, direct Hsp90 inhibitors display several serious limitations, such as inferior solubility, poorly-tolerated formulations, and hepatotoxicity.15,16 Therefore, alternatives to direct Hsp90 inhibition, such as inhibition of Hsp90’s key downstream effectors or Hsp90’s key interaction with its downstream effectors, may be a promising strategy to alleviate the drawbacks apparent in direct Hsp90 inhibition.
Herein, we report that chetomin, purified from Chaetomium globosum,17 functions as an effective agent against NSCLC CSCs and non-CSCs via inhibiting binding between Hsp90 and its key downstream effector hypoxia-inducible factor 1 alpha (HIF1α) without significantly affecting Hsp90 or Hsp70 expression. In vitro, chetomin inhibited sphere-forming by NSCLC CSCs within a nanomolar range, and proliferation of susceptible and chemoresistant NSCLC non-CSCs within a micromolar range. In vivo, chetomin markedly decreased tumor formation in several murine models of NSCLC. At these concentrations and doses, chetomin produced only marginal toxicity to non-cancerous cells and no observable toxicity to animals, demonstrating a promising safety profile. This study advocates chetomin as an inhibitor of NSCLC CSC sphere formation and non-CSC proliferation.
Materials and methods
Ethics statement
All protocols in this study were reviewed and authorized by the Ethics Review Committee at First Affiliated Hospital, Bengbu Medical College. All animals were housed and cared for according to the guidelines outlined in the National Institutes of Health’s (NIH) “Guide for the Care and Use of Laboratory Animals” (8th edition).
Cell lines, antibodies, and general materials
The NSCLC carcinoma lines and other cell lines are described in the Supplementary Information. Primary antibodies were procured from Cell Signaling Technology (Danvers, MA) and are described in the Supplementary Information. Cell culturing media and reagents were obtained from Invitrogen (Beijing, China). Matrigel® was sourced from BD Biosciences. The PrimeScript 1st strand cDNA Synthesis Kit was sourced from Takara (Kyoto, Japan). Propidium iodide (PI), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and additional chemical reagents were obtained from Sigma-Aldrich (St. Louis, MO), except if stated otherwise.
Quantification of proliferation by MTT assay
5 × 103 cells per well were plated onto 96-well culture plates and provided 24 hours to adhere. Methanol extract, chetomin, or vehicle was then added to the cell culture media at specified concentrations and incubated for three days (37°C, 5% CO2). At this stage, MTT was added to the culture media to a final amount of 500 μg/ml and culture plates were returned to the 37°C incubator for 2 to 4 hours. Formazan produced by cells still living was solubilized in DMSO and the colorimetric signal quantified at 570 nm. All experimental results are relative to the control value and expressed as a percent.
Assessment of CSC-like characteristics by sphere-forming assay
Cells were grown in media to sustain sphere formation in ultra-low attachment 96-well culture plates (Corning® Costar®, Corning, NY). The culturing media consisted of DMEM-F12 with B-27™ supplement (Gibco®, Thermo Fisher Scientific), growth factors (bFGF, EGF), and antibiotics. Spheres were incubated with chetomin or vehicle at the specified concentrations in an incubator (37°C, 5% CO2) for two weeks or till spheres were larger than 150 µm3.
Quantification of aldehyde dehydrogenase (ALDH) activity
The subpopulation of H1299 cells with high ALDH activity (ALDHhigh) was identified using an AldeRed™ ALDH Detection Assay (Merck Millipore), according to the manufacturer’s instructions. Briefly, one million H1299 cells were suspended in AldeRed™ Assay Buffer and a cell-permeable, fluorescent, nontoxic ALDH substrate, AldeRed™ 588-A, was added and cells were incubated at 37°C for 40 minutes. A sample prepared in parallel served as the control and additionally contained the ALDH inhibitor diethylaminobenzaldehyde (DEAB). The readout was performed by flow cytometry; the control sample (containing DEAB) was used for gating, and ALDH activity was measured as the fluorescence intensity.
Assessment of colony-forming capacity by an anchorage-dependent assay
300 cells per well were plated to 6-well culture plates and chetomin or vehicle with/without caspase-3 inhibitor was added at the specified concentrations and incubated for two weeks (37°C, 5% CO2). Fresh culture media was replenished 1 or 2 times per week. At the end of the incubation period, fixation of colonies was performed with methanol (100%), followed by staining with crystal violet (0.002% in water), and by multiple rinse steps using deionized water. Colonies were visualized and then quantified by ImageJ (NIH, Bethesda MA).
Assessment of colony-forming capacity by an anchorage-independent (soft agar) assay
Cells were suspended in sterile agar (1%), resulting in a 0.4% agar solution, which was added into the wells of a 24-well culture plate that had been pre-coated with agar (1%). Culture media with chetomin or vehicle with/without caspase-3 inhibitor at the specified concentrations was overlaid over the solidified agar and incubated for two weeks (37°C, 5% CO2). Fresh culture media was replenished 1 or 2 times per week. At the end of the incubation period, staining of live colonies was performed by MTT solution, which were then visualized, and then quantified by ImageJ.
Evaluation of apoptosis by cell-cycle analysis
Cells were cultured with varying levels of chetomin or vehicle for 24 hours. Both suspended and adherent cells were harvested, rinsed with phosphate buffered saline (PBS), and fixation was performed with methanol (100%). Staining was achieved with PI supplemented with RNase A (both at 50 μg/ml) for 30 minutes, room temperature (RT). Readout was performed by flow cytometry on a BD FACSCalibur® (BD Biosciences); a control sample was used for gating and fluorescence intensity was measured. BD CellQuest™ (also BD Biosciences) was employed to analyze cell-cycle progression.
Quantification of endothelial cell tube formation
Preparation of conditioned media (CM): H1299 cells were cultured with chetomin or vehicle for 24 hours, followed by an additional incubation period of 4 hours in a hypoxic (1% O2) or normoxic (room air, 21% O2) environment. At this point, the media containing chetomin or vehicle was replaced by new, serum-deficient culture media and incubated an additional 24 hours. The resultant CM was collected for subsequent experiments.
Tube formation assay: The procedure adopted for assessing tube-forming capacity was executed according to published reports.18 In brief, HUVECs were plated into the wells of CellBIND® 96-well culture plates (Corning®) and given time to adhere. They were then treated with regular media or CM (normoxic or hypoxic) from chetomin- or vehicle-treated H1299 cultures. HUVECs were visualized, and alterations in morphology were scored.
Analysis of NSCLC culture protein and mRNA content
Cell cultures were incubated with specified concentrations of chetomin or vehicle for 24 hours. For protein analyses, cell lysates were subjected to Western blotting as described in the Supplementary Information. For mRNA analyses, a RNA extraction kit (BioTeke, Beijing, China) was used to purify total RNA from cells. Reverse transcription PCR (RT-PCR) was conducted according to published methods19 using primer pairs outlined in Supplementary Table 3. The amplified DNA was resolved by agarose gel (1.5%) electrophoresis and imaged with a Gel Doc™ EZ System (Bio-Rad Laboratories).
Reporter gene assay
The reporter assay of HIF-response element (HRE) activity was conducted according to published reports18 as described in the Supplementary Information.
Generation of Hsp90-expressing H1299 cells
H1299 cultures were transiently transfected with either a plasmid overexpressing Hsp90 or the corresponding empty plasmid control with jetPRIME® transfection reagent. H1299 cultures were stably transfected with either lentiviruses bearing a short hairpin RNA (shRNA) against Hsp90 (Hsp90-shRNA) or a scrambled control (scr-shRNA) within a pLKO.1 vector (Sigma-Aldrich).
Pull-down, immunoprecipitation (IP), and competitive ATP-binding assays
Pull-down, IP assays, and purification of Hsp90 with chetomin or vehicle were conducted according to published reports.18
Mouse models
Animals were accommodated in housing that was maintained at a constant temperature and humidity level under alternating 12-hour light-dark cycles. They received water and standard chow ad libitum. The mouse models are fully described in the Supplementary Information.
Tumor-propagation from dissociated tumor xenografts
Following the 8-week regimen of chetomin or vehicle, mice were sacrificed, tumors excised, and dissociated using a Tumor Dissociation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Viable individual cancer tells were counted with trypan blue and 500 to 50,000 cells were implanted in NOD/SCID animals by subcutaneous injection in their right flank. Mice were monitored, and tumor burden assessed.
Fluorescence immunohistochemistry (IHC)
IHC was performed on lung tumor sections from KrasLA1 transgenic animals and H1299 xenograft sections embedded in paraffin according to published reports.18 Primary antibodies against cleaved caspase-3 (Cl-Cas3), HIF1α, Oct4, and CD34 were employed to assess levels of those proteins.
Interaction between Hsp90 and chetomin by fluorescence titration
Fluorescence titration experiments were conducted in order to evaluate the interaction between Hsp90 and chetomin, and to identify the possible binding location(s) of chetomin to Hsp90, as described in the Supplementary Information.
Statistical tests and analyses
All results are expressed as means ± standard errors of the mean (SEMs). All data were entered into Microsoft Excel (Microsoft Corporation, Redmond, WA), and statistical tests were implemented with GraphPad Prism 6 (La Jolla, CA), including IC50 determination by non-linear regression analysis and two-tailed Student’s t-test to calculate p-values. Comparisons were taken to be significant when p < .05.
Results
The C. globosum methanol extract attenuates NSCLC CSC sphere-forming and non-CSC proliferation
We sought to identify Hsp90 inhibitors due to the prevalence of elevated Hsp90 protein levels in tumors16,20 and the possibility that Hsp90 may help CSCs endure adverse conditions.12 We conducted a literature screen of natural compounds known to possess antitumor properties,21–23 centering on molecules isolated from Chaetomium globosum.17 Natural products were extracted from C. globosum with methanol and hexanes,24 and the bioactivities of those isolates were tested against Hsp90 activity (Supplementary Figure 1A). Binding between Hsp90 and HIF1α was substantially attenuated in H1299 cultures incubated with the C. globosum methanol extract, but not the hexanes extract (Supplementary Figure 1B). Furthermore, the methanol extract also lowered HIF1α protein levels in H1299 cells (Supplementary Figure 1C).
We also tested the extracts’ ability to inhibit CSC sphere-forming and non-CSC proliferation by MTT assay in NSCLC cell lines. The methanol extract markedly decreased proliferation (Supplementary Figure 1D) and sphere-forming (Supplementary Figure 1E) in H1299 cultures compared to the hexanes extract. To verify the results in vivo, we employed a transgenic KrasLA1 murine model that develops lung tumors spontaneously.25 Animals were administered methanol extract or vehicle daily for two months, and in vivo bioluminescence was used to monitor tumor formation. Methanol extract substantially inhibited tumor development, resulting in fewer and smaller lesions (Supplementary figure 1f and 1g).
Chetomin is an inhibitor of Hsp90/HIF1α activity
Chetomin is found in the methanol extract of C. globosum24 and has been previously demonstrated to inhibit HIF1α binding to other key protein moieties.26,27 Therefore, its ability to block Hsp90/HIF1α pathway activity was evaluated in the NSCLC cell lines H1299 and H460. In the following experiments, micromolar levels of chetomin were used for monolayer experiments, while nanomolar levels of chetomin were employed for spheroid cultures of NSCLC cell lines due to differences in IC50 levels (Supplementary Tables 1 and 2).
Consistent with previous research on Hsp90 inhibition,28 hypoxic H1299 and H460 monolayers incubated with increasing concentrations of chetomin for 24 hours showed progressively lower expression of several survival-promoting proteins promoted by Hsp90/HIF1α activity, including insulin-like growth factor 1 (IGF1 R), epidermal growth factor receptor (EGFR), Src, mitogen-activated protein kinase kinase 1/2 (MEK1/2), activation of protein kinase B (Akt), and mammalian target of rapamycin (mTOR).9 Consistent with these findings, the angiogenesis-promoting factors platelet-derived growth factor (PDGF) and basic fibroblast growth factor (bFGF), which are both regulated by IGF1R,29 were also downregulated by chetomin exposure in hypoxic H460 and H1299 monolayers (Figure 1A). Consistent with the monolayer findings, hypoxic H1299 spheres treated with two weeks of chetomin exposure also showed decreased expression of IGF1R, EGFR, Src, MEK1/2, and Akt (Figure 1B). Chetomin exposure for 24 hours in hypoxic H1299 monolayers also dose-dependently decreased Hsp90-HIF1α binding and HIF1α protein expression as assessed by a pull-down assay (Figure 1C). Follow-up experiments in hypoxic H1299 and H460 monolayers revealed dose-dependent downregulation of HIF1α’s target vascular endothelial growth factor (VEGF) in response to 24 hours of chetomin exposure (Figure 1D). Consistent with previous research,18 hypoxia had a profound stimulatory effect on HRE activity in hypoxic A549 and H1299 monolayers transfected with a reporter plasmid (Figure 1E). Twenty-four hours of chetomin exposure attenuated HRE activity in these cell lines (Figure 1E). Consistent with previous research,18 hypoxia-derived conditioned media (CM) had no discernible effect on HUVEC angiogenesis-like tube formation compared to normoxia-derived CM (Figure 1F). Notably, CM from chetomin-treated NSCLC cultures induced a lesser extent of angiogenesis-like tube formation in HUVECs (Figure 1F).
Figure 1.
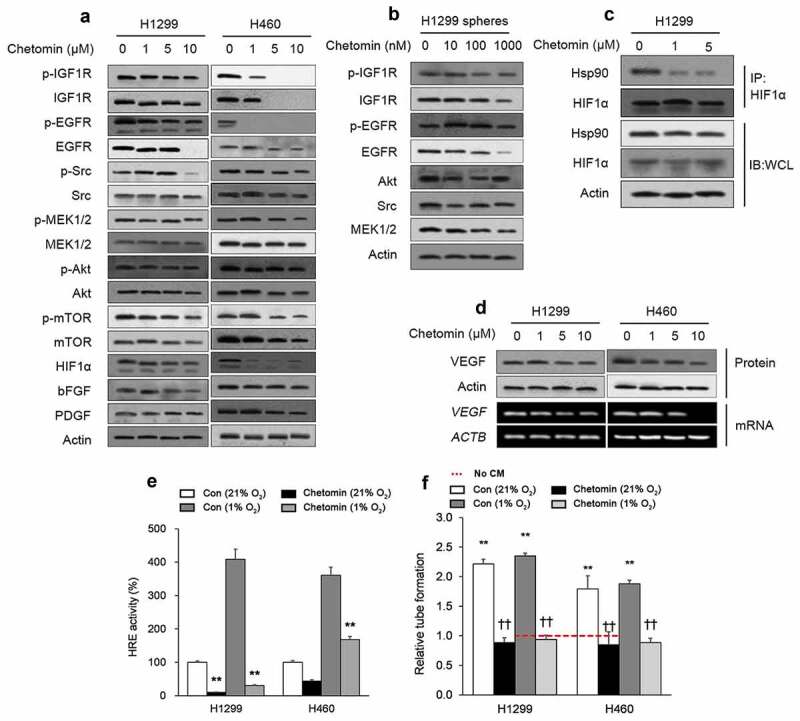
Chetomin is an inhibitor of Hsp90/HIF1α pathway activity.
Chetomin exerts its inhibitory action on NSCLC CSCs by eliciting apoptosis
We had a particular interest in targeting CSCs as a strategy for overcoming tumor recurrence; therefore, we next tested whether chetomin could target CSCs, initially testing chetomin on adhered monolayer cultures. Chetomin exposure for 24 hours dose-dependently lowered expression of stem cell biomarkers, SRY (sex determining region Y)-box 2 (Sox2), homeobox protein Nanog, and octamer-binding transcription factor 4 (Oct4), in H1299 monolayer cultures (Figure 2A). Additionally, chetomin pretreatment of monolayered H460 and H1299 cells for 24 hours attenuated their sphere formation capacity and number of cells per sphere (Figure 2B).
Figure 2.
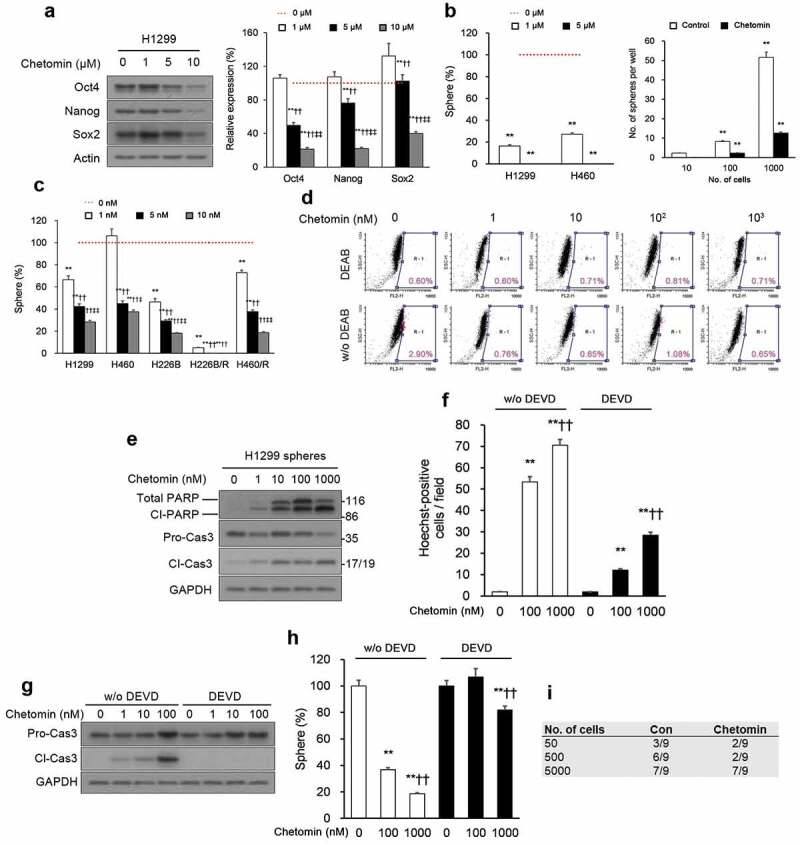
Chetomin exerts its inhibitory action on NSCLC CSCs by eliciting apoptosis.
We tested the action of chetomin on spheroid cultures of NSCLC cell lines, including two lines with paclitaxel-acquired resistance H460/R and H226B/R, where “R” denotes resistance in comparison to parental lines,30 since chemoresistance is a key property of CSCs.31 Nanomolar levels of chetomin for two weeks curbed sphere formation by both susceptible NSCLC cell lines and paclitaxel-resistant sub-lines (Figure 2C; Supplementary Table 1). Aldehyde dehydrogenases (ALDH) exhibit high levels of activity in CSCs;32 chetomin markedly decreased the proportion of cells with elevated ALDH activity (ALDHhigh) in H1299 cultures (Figure 2D). It also exerted enhanced cleaved caspase-3 (Cl-Cas3) and poly (ADP ribose) polymerase (PARP) (Figure 2E) and Hoechst-stained condensed chromatin (Figure 2F), indicating a greater fraction of apoptotic H1299 spheres in comparison to vehicle-treated spheres. Apoptosis was suppressed in H1299 spheres when Z-DEVD-FMK, a caspase-3 inhibitor, was included with chetomin, partially rescuing Cl-Cas3 expression (Figure 2G) and chromatin condensation (Figure 2F, right). The caspase-3 inhibitor also attenuated sphere-forming by H1299 cells (Figure 2H), underlying the relevance of caspase 3-induced apoptosis on chetomin’s cytotoxicity.
The in vitro findings were corroborated by in vivo experiments of tumorigenesis by CSCs. H1299 cultures were incubated with chetomin or vehicle, and flow cytometry was used to isolate the subset of ALDHhigh cells from each condition. Serial dilutions of chetomin- or vehicle-exposed ALDHhigh cells subjected to three days of exposure were then subcutaneously injected into NOD/SCID animals. Tumorigenesis was substantially lower in mice injected with chetomin-exposed ALDHhigh cells (Figure 2I).
Chetomin exerts its inhibitory action on NSCLC non-CSCs by eliciting apoptosis
The majority of tumor cells are non-CSCs, so eliminating them constitutes an important aspect of cancer treatment. Therefore, we next assessed chetomin’s effects on NSCLC non-CSCs. Activating KRAS and inactivating TP53 mutations are frequently recurring features of NSCLC lung cancer cells33 and CSCs.34,35 Hence, we compared the efficacy of chetomin on immortalized human bronchial epithelial (hBE) cells against hBE cells with: (i) a constitutively active KRASG12V mutation (KRASG12V-hBE), (ii) TP53 deficient via a short interfering RNA (siRNA) (siTP53-hBE), or (iii) both mutant KRASG12V and TP53 deficient (KRAS;G12V siTP53-hBE) (Supplementary Figure 2).36 Chetomin exposure for three days did not significantly inhibit proliferation of hBE cells but did adversely affect the proliferation of KRASG12V-hBE, siTP53-hBE, and KRAS;G12V siTP53-hBE cells (Figure 3A; Supplementary Table 2) and of the diverse NSCLC cell lines and the two paclitaxel-resistant sublines (H460/R and H226B/R; Figure 3B; Supplementary Table 2). The IC50s against the NSCLCs were in the micromolar range, almost a hundred times less potent than the inhibitory action of chetomin on sphere formation, which was in the nanomolar range. Therefore, while chetomin demonstrates some efficacy against non-CSCs, it is more potent against CSCs.
Figure 3.
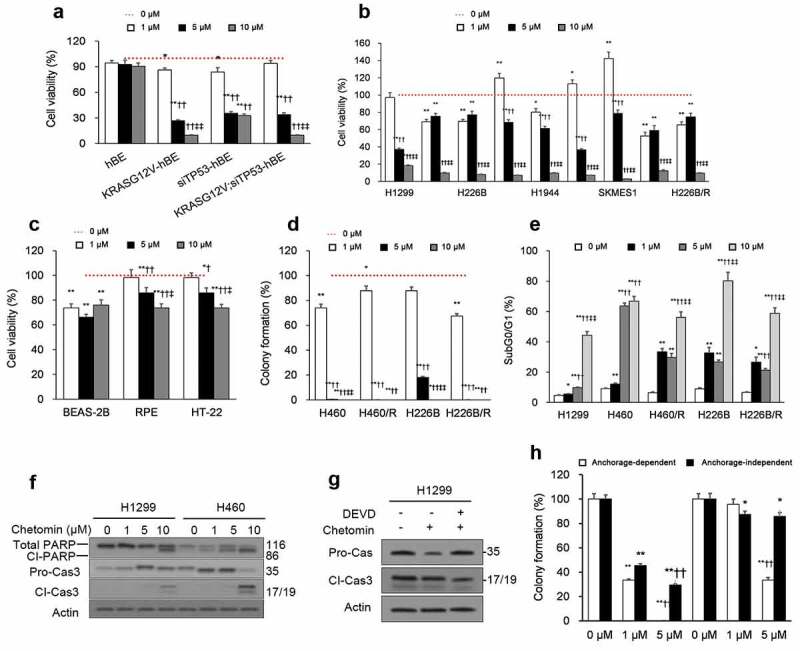
Chetomin exerts its inhibitory action on NSCLC non-CSCs by eliciting apoptosis.
The effect of chetomin was also examined on non-cancerous cell lines originating from diverse tissues. Across the board, chetomin exposure for three days was only marginally toxic to these non-cancerous cell lines (Figure 3C). When NSCLC cultures were grown under monolayer conditions, chetomin exposure for two weeks curbed colony formation in both anchorage-dependent (Figure 3D) and -independent (Supplementary Figure 3) assays. Chetomin (24 hours) elicited apoptosis of non-CSCs, arresting cells in the sub-G0/G1 cell-cycle phase (Figure 3e) and enhancing caspase-3 and PARP cleavage (Figure 3F). When the caspase-3 inhibitor Z-DEVD-FMK was included with chetomin, caspase-3 cleavage (Figure 3G) and colony formation (Figure 3H) were partially rescued in NSCLC cultures. As for CSCs, the partial rescue by a caspase-3 inhibitor underlies the relevance of caspase 3-induced apoptosis on chetomin’s cytotoxicity to NSCLC non-CSCs.
Chetomin inhibits lung tumorigenesis in NSCLC mouse models
To corroborate our in vitro findings, we next tested chetomin in vivo in several mouse models. In the KrasLA1 mouse model of spontaneous lung tumorigenesis,25 administration of chetomin for eight weeks strikingly lowered the number of lesions compared to vehicle (Figure 4A). Microexamination of lung tissue revealed dramatically lower lesion counts, size, and total burden in chetomin- versus vehicle-administered groups (Figure 4B). Fluorescence immunohistochemistry (IHC) of lung sections revealed dysregulation of Cl-Cas3, HIF1α, CD34, and Oct4 expression in chetomin- versus vehicle-administered groups (Figure 4C, D). Chetomin exposure did not produce any observable variation in body weight in KrasLA1 mice (Supplementary Figure 4a).
Figure 4.
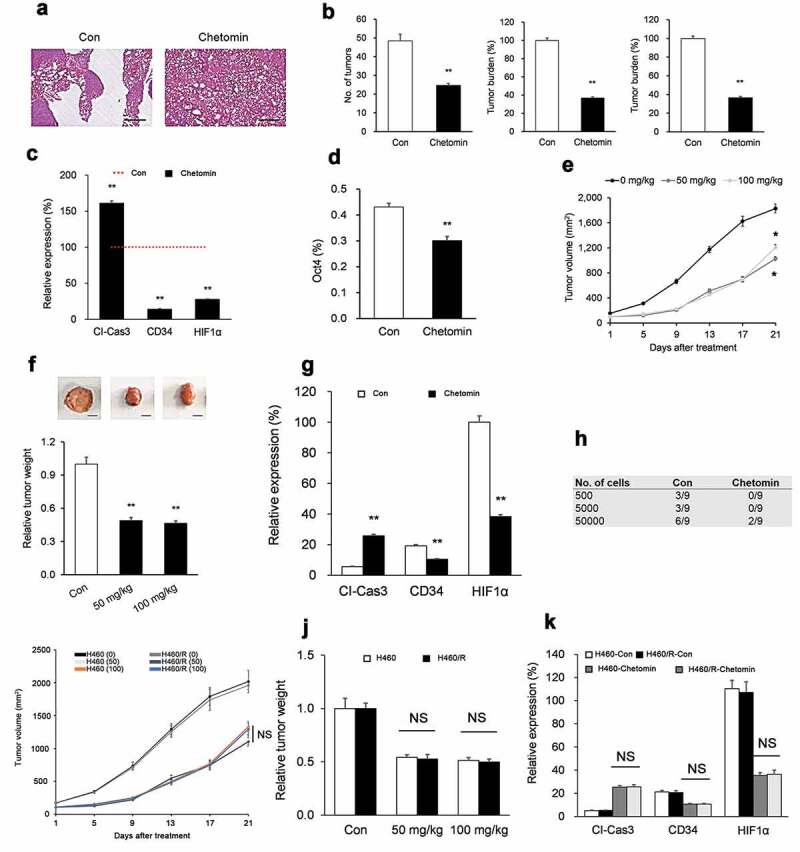
Chetomin inhibits lung tumorigenesis in NSCLC mouse models.
The second model we tested chetomin in vivo on was an H1299 flank xenograft model. When xenografts attained a size ranging from 50 to 150 mm3, mice were randomized to chetomin (50 or 100 mg/kg, p.o.) or vehicle for three weeks. At both doses, chetomin markedly lowered xenograft tumor size (Figure 4E) and mass (Figure 4F). Fluorescence IHC of lung sections revealed a higher intensity of Cl-Cas3 (more apoptosis) and lower intensities of HIF1α and CD34 (less angiogenesis) in chetomin- versus vehicle-administered groups (Figure 4G). Chetomin exposure did not produce any observable variation in body weight in H1299 xenograft mice (Supplementary Figure 4b).
Due to their ability to self-renew, CSCs can propagate tumors when inoculated into mice;37 therefore, we also tested the impact of chetomin on tumor propagation of CSCs. First, H1299 flank xenografts were established and mice received chetomin or vehicle as described above. The tumors were excised, dissociated, and serially-diluted cells were implanted into recipient NOD/SCID animals. As observed in vitro (Figure 2E), recipients inoculated with chetomin-exposed cells exhibited drastically lower tumorigenesis compared to recipients of vehicle-exposed cells (Figure 4H).
Finally, we comparatively evaluated the effects of chetomin in vivo on a H460 flank xenograft model versus a H460/R flank xenograft model in order to test whether chetomin has a differential effect on chemoresistant H460/R xenografts. When xenografts attained a size ranging from 50 to 150 mm3, mice were randomized to chetomin (50 or 100 mg/kg, p.o.) or vehicle for three weeks. At both doses, H460/R xenograft tumor size (Figure 4I) and mass (Figure 4J) were not significantly different than those of H460 xenografts. Moreover, fluorescence IHC of lung sections revealed no significant differences in Cl-Cas3 (apoptosis) or HIF1α and CD34 (angiogenesis) between chetomin-treated H460/R xenografts versus chetomin-treated H460 xenografts (Figure 4K). Chetomin exposure did not produce any observable variation in body weight in either H460 xenograft mice or H460/R xenograft mice (Supplementary Figure 4C, D).
Chetomin blocks Hsp90’s binding to HIF1α’s N-terminus
We explored the molecular details of the inhibitory action of chetomin on Hsp90/HIF1α binding. We confirmed that chetomin did not affect Hsp90 or Hsp70 protein levels in 24 hour-exposed monolayer NSCLC cultures (Figure 5A). Since chetomin did not impact Hsp90 protein levels, we next sought whether it directly inhibits the interaction between Hsp90 and HIF1α. It has been well-established that the Hsp90 protein binds to HIF1α’s basic helix–loop–helix–PER–ARNT–SIM (bHLH–PAS) domain, which is located at HIF1α’s N-terminus.38,39 Chetomin was therefore titrated into full-length (“FL”), N-terminus domain (“N”), and C-terminus domain (“C”) variants of HIF1α in the presence of Hsp90. Addition of chetomin inhibited the interaction of Hsp90 to the “FL” and “N” variants but not to the “C” variant (Figure 5B), suggesting chetomin’s specifically inhibits the Hsp90-HIF1α binding interaction in HIF1α’s N-terminus.
Figure 5.
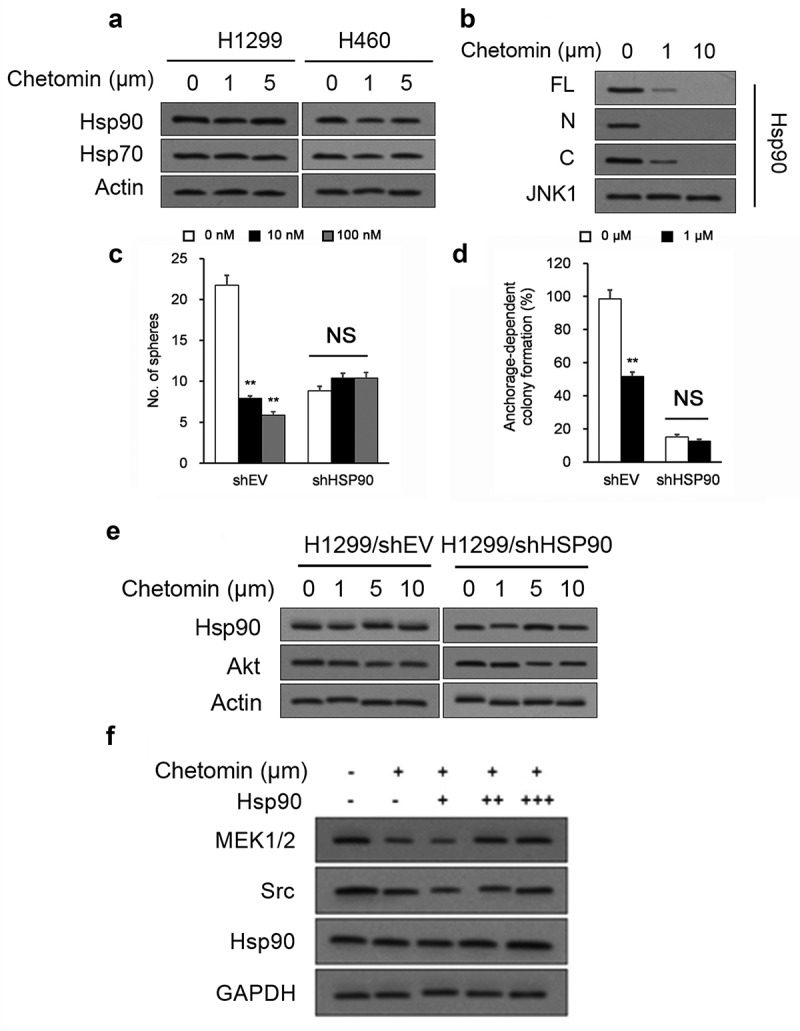
Chetomin blocks Hsp90’s binding to HIF1α’s N-terminus.
We generated stable lines of H1299 cells with short hairpin RNA (shRNA)-mediated knockdown of Hsp90 (Hsp90-shRNA-H1299) and scrambled control (scr-shRNA-H1299) to determine whether chetomin’s anti-cancer properties are associated with its inhibition of Hsp90’s binding to HIF1α. As expected, chetomin exposure for two weeks lowered colony- and sphere-forming capacity in scr-shRNA-H1299 cells (Figure 5C, D). However, chetomin exposure did not exert any discernible impact on these characteristics in Hsp90-shRNA-H1299 cultures (Figure 5C, D). Furthermore, chetomin did not affect levels of Akt, an Hsp90 substrate, in Hsp90-shRNA-H1299 cultures (Figure 5E). On the other hand, H1299 cultures overexpressing Hsp90 reestablished levels of Hsp90 substrates MEK1/2 and Src (Figure 5F). These results suggest that chetomin’s anti-cancer properties are dependent upon the presence of Hsp90.
Discussion
CSCs are believed to be one mechanism for tumor recurrence, metastasis, and acquired therapy resistance in NSCLC.40 Discovering the molecular mechanisms that help CSCs survive could pinpoint possible treatment avenues. Hsp90 aids protein folding, an essential process for CSCs enduring cytotoxic pressure, and several genes associated with Hsp90 are upregulated in CSCs.9,12 Numerous Hsp90 proteins are involved in the proliferative and metastatic potential of tumor cells and elevated Hsp90 protein levels are associated with worse prognosis in NSCLC patients.10,11 These features suggest that Hsp90 inhibitors may serve as potential anti-cancer drugs.13,14 As drugs to prevent tumor relapse would need to be administered over months to years, long-term direct Hsp90 inhibition displays several drawbacks, such as inferior solubility, poorly-tolerated formulations, and hepatotoxicity15,16_ENREF_20 in addition to induced resistance via Hsp70.41 Therefore, an indirect Hsp90 inhibition strategy, such as inhibition of Hsp90’s key downstream effectors or Hsp90’s key interaction with its downstream effectors, may be a promising approach in NSCLC patients. That being said, an ideal indirect Hsp90 inhibitor would need a particularly favorable safety profile and exert no significant effects on either Hsp90 or Hsp70 expression.
Herein, we report that chetomin, an indole heteropentacyclic compound isolated from C. globosum,17 functions as an Hsp90/HIF1α pathway inhibitor that decreases the viability and elicits apoptosis of NSCLC CSCs spheres and non-CSCs monolayer cultures in vitro within nanomolar and micromolar range, respectively. Moreover, we found that chetomin inhibits tumorigenesis in a spontaneous KrasLA1 lung cancer model, a H1299 flank xenograft model, and a tumor propagation flank implanted model. Notably, chetomin does not significantly affect Hsp90 or Hsp70 expression in vitro, is only marginally toxic to non-cancerous cell lines in vitro, and produces no discernible toxicity to mice in vivo. These properties make chetomin particularly attractive as a potential anti-cancer drug in NSCLC patients.
On a molecular level, chetomin has been shown to target the CH1 domain of the transcriptional co-activator p300, thereby blocking the p300-HIF-1α interaction, which attenuates HIF-1α’s hypoxia-inducible transcription.27 As we found that chetomin blocks Hsp90’s binding to HIF1α, these findings suggest that chetomin indirectly produces this effect through p300. On a cellular level, chetomin inhibits human triple-negative breast cancer cell proliferation via apoptosis induction42 and displays anti-myelomatous activity without toxicity to normal hematopoietic cells.43 This evidence supports our contention that chetomin displays potent anti-cancer properties with a favorable safety profile. In conclusion, our study advocates chetomin as a Hsp90/HIF1α pathway inhibitor and a potent, nontoxic NSCLC CSC-targeting molecule.
Supplementary Material
Funding Statement
This work was supported by the National Natural Science Foundation of China [81772493]; Science and Technology Program of Anhui Province [2017070503B037, YDZX20183400002554].
Author Contributions
Conceived and designed the study: TW, NW, and XJW.
Performed the experimental procedures: SPM, QYD, HYG, XCL, YBS, YY, WL, YC, and CLZ.
Analyzed the data: TW, NW, YBS, and XXW.
Drafted the manuscript: XJW and YQC.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A.. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Schrank Z, Chhabra G, Lin L, Iderzorig T, Osude C, Khan N, Kuckovic A, Singh S, Miller R, Puri N, et al. Current molecular-targeted therapies in NSCLC and their mechanism of resistance. Cancers. 2018;10(7):224. doi: 10.3390/cancers10070224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El Rassy E, Botticella A, Kattan J, Le Péchoux C, Besse B, Hendriks L.. Non-small cell lung cancer brain metastases and the immune system: from brain metastases development to treatment. Cancer Treat Rev. 2018;68:69–79. doi: 10.1016/j.ctrv.2018.05.015. [DOI] [PubMed] [Google Scholar]
- 4.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 5.Tan C-S, Gilligan D, Pacey S. Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer. Lancet Oncol. 2015;16(9):e447–e59. doi: 10.1016/S1470-2045(15)00246-6. [DOI] [PubMed] [Google Scholar]
- 6.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 7.Medema JP. Cancer stem cells: the challenges ahead. Nat Cell Biol. 2013;15(4):338. doi: 10.1038/ncb2717. [DOI] [PubMed] [Google Scholar]
- 8.Jego G, Hazoumé A, Seigneuric R, Garrido C. Targeting heat shock proteins in cancer. Cancer Lett. 2013;332(2):275–285. doi: 10.1016/j.canlet.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 9.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11(7):515. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 10.Wu Y, Huang B, Liu Q, Liu Y. Heat shock protein 90-β over-expression is associated with poor survival in stage I lung adenocarcinoma patients. Int J Clin Exp Pathol. 2015;8:8252. [PMC free article] [PubMed] [Google Scholar]
- 11.Ruiz MIG, Floor K, Roepman P, Rodriguez JA, Meijer GA, Mooi WJ, Jassem E, Niklinski J, Muley T, van Zandwijk N, et al. Integration of gene dosage and gene expression in non-small cell lung cancer, identification of HSP90 as potential target. PLoS One. 2008;3(3):e0001722. doi: 10.1371/journal.pone.0001722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan G-C. 2012. Role of heat shock proteins in stem cell behavior. In: Progress in molecular biology and translational science. Elsevier; 2012. p. 305–322. doi: 10.1016/B978-0-12-398459-3.00014-9. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isaacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell. 2003;3(3):213–217. doi: 10.1016/S1535-6108(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 14.Modi S, Stopeck AT, Linden HM, Solit DB, Chandarlapaty S, Rosen N, D’Andrea G, Dickler M, Moynahan ME, Sugarman S, et al. HSP90 inhibition is effective in breast cancer: a phase 2 trial of tanespimycin (17AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin Cancer Res. 2011;17(15):5132–5139. clincanres. 0072.2011. doi: 10.1158/1078-0432.CCR-11-0072 [DOI] [PubMed] [Google Scholar]
- 15.Akram A, Khalil S, Halim SA, Younas H, Iqbal S, Mehar S. Therapeutic uses of HSP90 inhibitors in non-small cell lung carcinoma (NSCLC). Curr Drug Metab. 2018;19(4):335–341. doi: 10.2174/1389200219666180307122441. [DOI] [PubMed] [Google Scholar]
- 16.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18(1):64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu G-B, He G, Bai -H-H, Yang T, Zhang G-L, Wu L-W, Li G-Y. Indole Alkaloids from Chaetomium globosum. J Nat Prod. 2015;78(7):1479–1485. doi: 10.1021/np5007235. [DOI] [PubMed] [Google Scholar]
- 18.Oh SH, Woo JK, Jin Q, Kang HJ, Jeong JW, Kim KW, Hong WK, Lee H-Y. Identification of novel antiangiogenic anticancer activities of deguelin targeting hypoxia-inducible factor-1 alpha. Int J Cancer. 2008;122(1):5–14. doi: 10.1002/ijc.23075. [DOI] [PubMed] [Google Scholar]
- 19.Min H-Y, Yun HJ, Lee J-S, Lee H-J, Cho J, Jang H-J, Park S-H, Liu D, Oh S-H, Lee JJ, et al. Targeting the insulin-like growth factor receptor and Src signaling network for the treatment of non-small cell lung cancer. Mol Cancer. 2015;14(1):113. doi: 10.1186/s12943-015-0392-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciocca DR, Arrigo AP, Calderwood SK. Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: an update. Arch Toxicol. 2013;87:19–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fridlender M, Kapulnik Y, Koltai H. Plant derived substances with anti-cancer activity: from folklore to practice. Front Plant Sci. 2015;6:799. doi: 10.3389/fpls.2015.00799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sznarkowska A, Kostecka A, Meller K, Bielawski KP. Inhibition of cancer antioxidant defense by natural compounds. Oncotarget. 2017;8(9):15996. doi: 10.18632/oncotarget.13723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leite ML, da Cunha NB, Costa FF. Antimicrobial peptides, nanotechnology, and natural metabolites as novel approaches for cancer treatment. Pharmacol Ther. 2018. Mar;183:160–176. doi: 10.1016/j.pharmthera.2017.10.010. Epub 2017 Oct 10. Review. [DOI] [PubMed] [Google Scholar]
- 24.Marmouzi I, Faouzi MEA, Saidi N, Cherrah Y, Rehberg N, Ebada SS, Ebrahim W, Kalscheuer R, Proksch P. Bioactive secondary metabolites from chaetomium globosum, an endophyte from the moroccan plant avena sativa. Chem Nat Compd. 2017;53(6):1208–1211. doi: 10.1007/s10600-017-2242-6. [DOI] [Google Scholar]
- 25.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410(6832):1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 26.Staab A, Loeffler J, Said HM, Diehlmann D, Katzer A, Beyer M, Fleischer M, Schwab F, Baier K, Einsele H, et al. Effects of HIF-1 inhibition by chetomin on hypoxia-related transcription and radiosensitivity in HT 1080 human fibrosarcoma cells. BMC Cancer. 2007;7(1):213. doi: 10.1186/1471-2407-7-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, Cornell-Kennon S, Lee J, Wang B, Wang J, et al. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell. 2004;6(1):33–43. doi: 10.1016/j.ccr.2004.06.009 [DOI] [PubMed] [Google Scholar]
- 28.Le HT, Nguyen HT, Min H-Y, Hyun SY, Kwon S, Lee Y, Le THV, Lee J, Park JH, Lee H-Y, et al. Panaxynol, a natural Hsp90 inhibitor, effectively targets both lung cancer stem and non-stem cells. Cancer Lett. 2018;412:297–307. doi: 10.1016/j.canlet.2017.10.013. [DOI] [PubMed] [Google Scholar]
- 29.Kim J-H, Choi DS, Lee O-H, Oh S-H, Lippman SM, Lee H-Y. Antiangiogenic antitumor activities of IGFBP-3 are mediated by IGF-independent suppression of Erk1/2 activation and Egr-1-mediated transcriptional events. Blood. 2011. Sep 1; 118(9):2622–31. doi: 10.1182/blood-2010-08-299784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee S-C, Min H-Y, Choi H, Bae SY, Park KH, Hyun SY, Lee HJ, Moon J, Park S-H, Kim JY, et al. Deguelin analogue SH-1242 inhibits Hsp90 activity and exerts potent anticancer efficacy with limited neurotoxicity. Cancer Res. 2016;76(3):686–699. doi: 10.1158/0008-5472.CAN-15-1492. [DOI] [PubMed] [Google Scholar]
- 31.Abdullah LN, Chow EK-H. Mechanisms of chemoresistance in cancer stem cells. Clin Transl Med. 2013;2(1):3. doi: 10.1186/2001-1326-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu X, Chai S, Wang P, Zhang C, Yang Y, Yang Y, Wang K. Aldehyde dehydrogenases and cancer stem cells. Cancer Lett. 2015;369(1):50–57. doi: 10.1016/j.canlet.2015.08.018. [DOI] [PubMed] [Google Scholar]
- 33.Pikor LA, Ramnarine VR, Lam S, Lam WL. Genetic alterations defining NSCLC subtypes and their therapeutic implications. Lung Cancer. 2013;82(2):179–189. doi: 10.1016/j.lungcan.2013.07.025. [DOI] [PubMed] [Google Scholar]
- 34.Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A, Sebolt-Leopold J, Shannon KM, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40(5):600. doi: 10.1038/ng.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao Z, Zuber J, Diaz-Flores E, Lintault L, Kogan SC, Shannon K, Lowe SW. p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes Dev. 2010;24(13):1389–1402. doi: 10.1101/gad.1940710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boo H-J, Min H-Y, Jang H-J, Yun HJ, Smith JK, Jin Q, Lee H-J, Liu D, Kweon H-S, Behrens C, et al. The tobacco-specific carcinogen-operated calcium channel promotes lung tumorigenesis via IGF2 exocytosis in lung epithelial cells. Nat Commun. 2016;7(1):12961. doi: 10.1038/ncomms12961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 38.Antonsson C, Whitelaw ML, McGuire J, J-A G, Poellinger L. Distinct roles of the molecular chaperone hsp90 in modulating dioxin receptor function via the basic helix-loop-helix and PAS domains. Mol Cell Biol. 1995;15(2):756–765. doi: 10.1128/MCB.15.2.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minet E, Mottet D, Michel G, Roland I, Raes M, Remacle J, Michiels C. Hypoxia‐induced activation of HIF‐1: role of HIF‐1α‐Hsp90 interaction. FEBS Lett. 1999;460(2):251–256. doi: 10.1016/S0014-5793(99)01359-9. [DOI] [PubMed] [Google Scholar]
- 40.Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13(10):727. doi: 10.1038/nrc3597. [DOI] [PubMed] [Google Scholar]
- 41.Ramanathan RK. Phase I pharmacokinetic-pharmacodynamic study of 17-(allylamino)-17-demethoxygeldanamycin (17AAG, NSC 330507), a novel inhibitor of heat shock protein 90, in patients with refractory advanced cancers. Clin Cancer Res. 2005;11:3385–3391. [DOI] [PubMed] [Google Scholar]
- 42.Dewangan J, Srivastava S, Mishra S, Pandey PK, Divakar A, Rath SK. Chetomin induces apoptosis in human triple-negative breast cancer cells by promoting calcium overload and mitochondrial dysfunction. Biochem Biophys Res Commun. 2018;495(2):1915–1921. doi: 10.1016/j.bbrc.2017.11.199. [DOI] [PubMed] [Google Scholar]
- 43.Viziteu E, Grandmougin C, Goldschmidt H, Seckinger A, Hose D, Klein B, Moreaux J. Chetomin, targeting HIF-1α/p300 complex, exhibits antitumour activity in multiple myeloma. Br J Cancer. 2016;114(5):519. doi: 10.1038/bjc.2016.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.