

ABSTRACT
High mobility group box protein 1 (HMGB1) is an evolutionarily conserved non-histone chromatin-binding protein. In a previous study, we showed that treating leukemic cells with chemotherapeutic drugs leads to the translocation of HMGB1, which is involved in autophagy and ultimately promotes chemoresistance in leukemia. However, the underlying translocation mechanism of HMGB1 in chemotherapy-induced autophagy remains unclear. In this study, we showed that knockdown of SIRT6 or PARP1 gene expression significantly inhibited HMGB1 cytoplasmic translocation and autophagy. Meanwhile, we found that SIRT6, an important upstream protein of PARP1, associated with PARP1, leading to the stimulation of polyADP-ribose polymerase activity. We further demonstrated that SIRT6 and PARP1 activation were required for chemotherapy-induced ADP-ribosylation of HMGB1 in leukemic cells and then influenced the acetylation of HMGB1, finally promoting the autophagy of leukemic cells mediated by HMGB1 translocation. These findings provide new insights into the mechanism of chemotherapeutic drug resistance. Targeting the HMGB1 translocation may overcome autophagy-related chemoresistance in leukemia.
KEYWORDS: Autophagy, HMGB1, SIRT6, PARP1, leukemia, chemoresistance
Introduction
Acute lymphoblastic leukemia (ALL) is a common type of cancer in children and is the most curable of cancers. T-cell ALL (T-ALL) is an aggressive disease that accounts for 15% of ALL cases in pediatrics and up to 25% of cases in adults. As a disease of genetic heterogeneity, T-ALL is caused by the accumulation of molecular changes in a multistep pathogenic process.1 Patients with T-ALL have been recognized as a high-risk leukemic group with cure rates of approximately 10% relative to a more favorable response of approximately 40% in B-cell ALL before combination chemotherapy was used in the clinic.2 Despite recent progress in treatment, approximately 25% of children and 50% of adults fail to respond to intensive chemotherapy or die of the disease.3,4 The use of chemotherapeutic drugs to kill fast-growing tumor cells is one of the major methods to treat cancer. However, chemotherapy resistance remains an obstacle to long-term remission of T-ALL and might be the cause of refractory and relapsed cases; therefore, it is a challenge to understand how cancer cells acquire drug resistance.
Autophagy is a form of programmed cell survival and a lysosomal degradation pathway. Macroautophagy (here called autophagy) is the most famous and widely studied type of autophagy. During this process, cells recycle cytoplasm to produce energy under stress conditions, remove redundant and damaged organelles to adapt to changing nutrient conditions and maintain cellular homeostasis.5 Autophagy plays a critical role in cell protection by inhibiting the accumulation of chemotherapy drugs, which is considered an underlying mechanism by which cancer cells resist chemotherapy.6
High mobility group box protein 1 (HMGB1), a member of the HMGB superfamily, has proven to be a critical regulator of autophagy. HMGB1 is abundant in the eukaryotic nucleus, and the translocation of HMGB1 from the nucleus to the cytoplasm is an important molecular event in autophagy.7 Studies have reported that HMGB1 plays an important role as a damage-associated molecular pattern in the pathological processes of several diseases such as sepsis, inflammation, and rheumatic disease.8–10 Furthermore, some studies have demonstrated that changes in HMGB1 expression and subcellular localization are related to tumor occurrence and treatment.11 Our recent study demonstrated that HMGB1 translocation participated in the transformation of the autophagy complex and promoted drug resistance in leukemia.12
Unlike other secretary proteins, HMGB1 triggers atypical lysosome-mediated vesicle transport via lysophosphatidylcholine due to the lack of signaling peptides. HMGB1 is transported from the nucleus to the cytoplasm and then into the lysosome and eventually is released into the extracellular space by exocytosis.13–15 Posttranslational modifications, including methylation, acetylation, phosphorylation and ADP-ribosylation, are crucial for the active secretion of HMGB1.16 Among these modifications, in monocytes and macrophages, acetylation plays the most important role in the secretion of HMGB1, which is induced by certain inflammatory mediators such as LPS or TNF-β.17,18 Therefore, we speculated that acetylation of lysine residues in HMGB1 is the prerequisite for its translocation from the nucleus to the cytoplasm in chemotherapy-induced autophagy. However, more evidence is needed to prove this assumption.
Sirtuin 6 (SIRT6), a chromatin-associated protein belonging to the silent information regulator 2 (Sir2) protein family, performs two enzymatic activities: nicotinamide adenine dinucleotide (NAD)+-dependent deacetylation and monoADP-ribosylation.19 Recently, increasing evidence has indicated that SIRT6 is related to the initiation and development of cancer.20,21 Some studies have demonstrated that SIRT6 is activated by p53 and catalyzes the deacetylation of the FoxO1 protein (Forkhead box O1), which promotes the translocation of FoxO1 from the nucleus to the cytosol.22 FoxO1 in the cytoplasm can combine with autophagy associated protein 7 (Atg7) and participate in the initiation of autophagy. Although the role of deacetylation is widely known, Sir2, the founding member of this family, was initially described as a monoADP-ribosyltransferase.23 The monoADP-ribosylase activity of sirtuins is thought to be important for DNA repair, while deacetylase activity promotes gene silencing.24 The only known substrate of monoADP-ribosylation is poly (ADP-ribose) polymerase (PARP) 1. SIRT6 activates PARP1 through monoADP-ribosylation and then initiates DNA damage repair.25 SIRT6 has been reported to promote autophagy through the FoxO1 or IGF-AKT-mTOR pathway. However, SIRT6 regulates the transposition of HMGB1 through PARP1 and participates in autophagy, this finding that has not been reported at home or abroad.
PARP1, as a nuclear enzyme, is the most abundant member in the PARP superfamily.26 PARP1 catalyzes the transfer of ADP-ribose moieties from NAD+ to itself and other acceptor proteins such as histones, DNA repair proteins, transcription factors and chromatin modulators and plays critical roles in DNA repair, chromatin modulation and transcription.26–28 The nonapoptotic cell death induced by DNA-alkylating agents is dependent on the activation of PARP by ADP-ribosylation modification; meanwhile, DNA damage is considered the most effective inducer of PARP1 activation. In a DNA-alkylation damage model, the activation of PARP1 regulated the nuclear to cytoplasmic translocation of HMGB1.29,30 Recently, Yang, et al reported for the first time that PARP1 can promote the acetylation of HMGB1 through polyADP-ribosylation.31
In a previous study, we demonstrated that the Ulk1-Atg13-FIP200 complex mediates the migration of HMGB1 from the nucleus to the cytoplasm and influences the formation of the downstream HMGB1-Beclin1 complex.12 To further explore the translocation mechanism of HMGB1, we investigated the effect of SIRT6-PARP1 interaction on HMGB1 ADP-ribosylation and acetylation, which could mediate chemotherapy-induced autophagy in leukemic cells by affecting HMGB1 translocation from the nucleus to the cytoplasm.
Result
PARP1 regulated HMGB1 polyADP-ribosylation, which subsequently promoted HMGB1 acetylation
Daunorubicin (DNR), the basic drug for the treatment of leukemia, caused striking damage to Jurkat and RS4:11 cells. Cell viability analysis is not shown here. In our previous research investigating how the ULK-FIP200 complex affects HMGB1 translocation, we found a noteworthy phenomenon in which the acetylation of HMGB1 was significantly enhanced after anticancer therapy, while the acetylation of HMGB1 decreased after chemical treatment in FIP200-silencing cells (Figure 1a). As mention in the introduction, Yang et al. reported that PARP1 could promote the acetylation of HMGB1 through polyADP-ribosylation,31 therefore, in order to elucidate the relationship between PARP1 and HMGB1, we catalyzed the reactions by using the purified recombinant HMGB1 protein and PARP1 enzyme in vitro (The specific method was described in detail in Materials and methods). As shown in Figure 1b, PARP1 could catalyze the PARylation of HMGB1 and then promoted the acetylation of HMGB1 in vitro. As shown in Figure 1c, in leukemic cells, the PARylation and acetylation of HMGB1 increased significantly after DNR treatment. Combining the data in Figure 1, we concluded that PARP1 regulated HMGB1 polyADP-ribosylation, which subsequently promoted HMGB1 acetylation.
Figure 1.
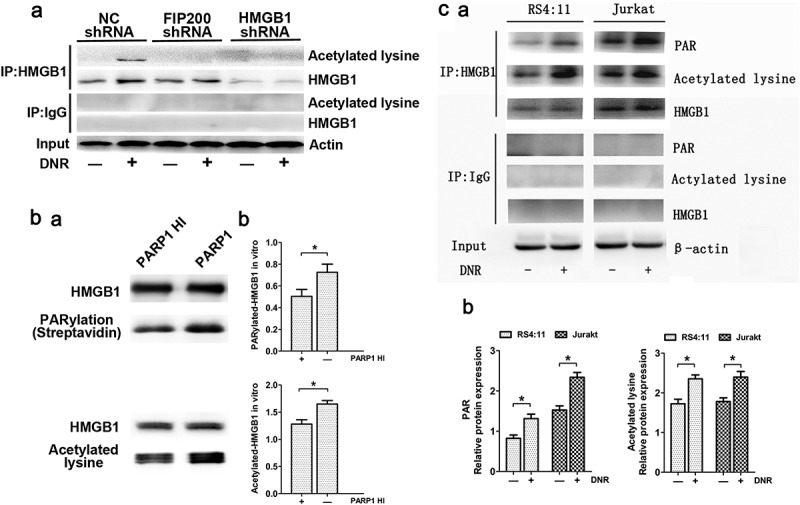
PARP1 regulated HMGB1 polyADP-ribosylation, which subsequently promoted HMGB1 acetylation. (a) Silencing FIP200 reduced chemotherapy-induced HMGB1 acetylation. Jurkat cells were transfected with control or FIP200 shRNA and then treated with or without DNR (0.4 µM) for 24 hours. The cell lysates were pulled down with an HMGB1 antibody and immunoblotted with anti-acetylated lysine and HMGB1 antibodies. β-actin was used as a loading control. (b) PARP1 was required for HMGB1 PARylation and subsequently promoted the acetylation of HMGB1 in vitro. HMGB1 was PARylated and successively acetylated as described in the Materials and Methods. The reactions were analyzed by western blot. Quantified data are presented (PARylation-HMGB1 or Acetylation-HMGB1/HMGB1, n > 3, *p < 0.05). HI, heat-inactivation. (c) Chemotherapy-induced autophagy was accompanied by polyADP-ribosylation and acetylation modification of HMGB1. Jurkat cells and RS4:11 cells were treated with or without DNR (0.4 μM) for 24 hours. The cell lysates were subjected to immunoprecipitation with an HMGB1 antibody followed by western blot analysis, and then the acetylation or PARylation level was measured using specific antibodies against acetylated lysine or PARylated. β-actin was used as a loading control. Quantified data are presented (PARylation-HMGB1 or Acetylation-HMGB1/HMGB1/β-actin, n > 3, *p < 0.05).
SIRT6 served as an upstream signal for the activation of PARP1 through monoADP-ribosylation
To elucidate the possible role of SIRT6-PARP1, SIRT6 or PARP1 shRNA was transfected into the human leukemic Jurkat cells line, resulting in the expression of SIRT6 or PARP1 mRNA, and protein expression decreased significantly (Figure 2a). PARP1 activity in Jurkat cells was significantly increased with the prolongation of chemotherapeutic agents, while PARP1 activity in the SIRT6-silenced cell line did not change (Figure 2b). These results suggested that the activation of SIRT6 served as an upstream signal of PARP1 during autophagy. As mention in the introduction, SIRT6 have two enzymatic activities, deacetylation and monoADP-ribosylation. In our study, we refer to a paper published in science in 2010.25 In that paper, Zhiyong Mao et al developed an in vivo approach to identify the substrate of SIRT6 mono-ADP-ribosylation, including the use of biotin-labeled NAD, a substrate for mono-ADP-ribosyltransferase, as well as the paraquat pretreatment of wild type and SIRT6 knockout cells. They found that SIRT6 physically associates with PARP1 and as the molecular weight of PARP1 immunoprecipitated with SIRT6 is slightly higher than that of the input PARP1 suggesting that PARP1 bound by SIRT6 is mono-ADP-ribosylated. In our study, we carried out relevant experiments in Jurkat cells, as shown in Figure 2c, coimmunoprecipitation indicated that PARP1 could bind to SIRT6, even without chemotherapy stimulation, as demonstrated by the appearance of a band in the second lane. Meanwhile, under DNR stimulation, the molecular weight of PARP1 immunoprecipitated with SIRT6 in lane 4 was slightly higher than that of the input PARP1 in lane 5 and appeared at approximately 120 kDa. Combined with reference, we came up with the conjecture that SIRT6 can form a protein complex with PARP1 and catalyze the monoADP-ribosylation of PARP1.
Figure 2.
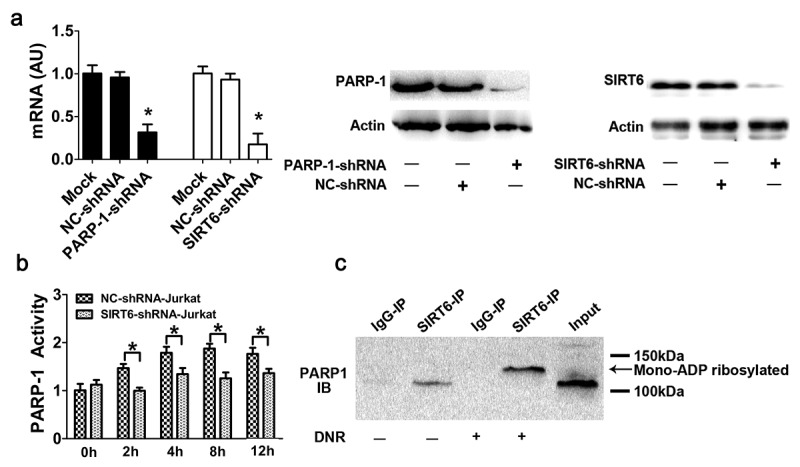
SIRT6 served as an upstream signal for the activation of PARP1 through monoADP-ribosylation. (a) PARP1 or SIRT6 in Jurkat cells was successfully depleted. Jurkat cells were transfected with control, PARP1 or SIRT6 shRNA. The mRNA and protein levels of PARP1 or SIRT6 were assessed via RT-qPCR or western blot analyses, respectively (n > 3, *p < 0.05). AU, arbitrary unit. The mock group was set as 1. (b) The activation of PARP1 induced by DNR was depressed in SIRT6-silencing cells. Cells were exposed to 0.4 μM DNR. The activity of PARP1 was measured by cell ELISA (n > 3, *p < 0.05 compared with the control group without DNR treatment). (c) SIRT6 and PARP1 formed a protein complex, and SIRT6 catalyzed the monoADP-ribosylation of PARP1. Jurkat cells were treated with or without DNR (0.4 μM) for 24 hours. The cell lysates were immunoprecipitated with an anti-SIRT6 antibody and immunoblotted with an anti-PARP1 antibody.
Silencing of SIRT6 or PARP1 inhibited the translocation of HMGB1 and suppressed chemotherapy-induced autophagy in leukemic cells
In this study, autophagy characterized by LC3 dots was determined by immunofluorescence staining, and the LC3-II/I ratio and the sequestosome-1 (SQSTM1/p62) level were analyzed by western blot. Additionally, we observed ultrastructural changes via electron microscopy. Based on the western blot analysis, as shown in Figure 3a, high LC3-II/I expression induced by chemotherapy, especially when the cells were treated with the autophagy inhibitor chloroquniene (CQ) in NC shRNA-treated cells. However, chemotherapy-induced LC3-II/I expression was significantly inhibited in SITR6 or PARP1 shRNA-treated cells. What is more, p62, an adaptor between the autophagy machinery and its substrates, was degraded in the control group. In contrast, chemotherapy-induced p62 degradation were significantly inhibited in SITR6 or PARP1 shRNA-treated cells. Transmission electron microscopy analysis showed that compared with the control group, SIRT6 or PARP1 shRNA-treated cells had fewer autophagosomes and autophagolysosomes during chemotherapy (Figure 3b). These results indicated that SIRT6 and PARP1 functions are positive regulators of chemotherapy-induced autophagy. By detecting the expression of HMGB1, the expression of HMGB1 decreased in the nucleus but increased in the cytoplasm after DNR treatment in NC shRNA-treated cells, while this change did not occur with SIRT6 and PARP1 silencing cells (Figure 3a). Immunofluorescence analysis showed that the translocation of HMGB1 in Jurkat cells was consistent with western blot analysis (Figure 3c). As shown in Figure 3, we concluded that activation of the SIRT6-PARP1 complex induces the translocation of HMGB1 from the nucleus to the cytoplasm and chemotherapy-induced autophagy.
Figure 3.
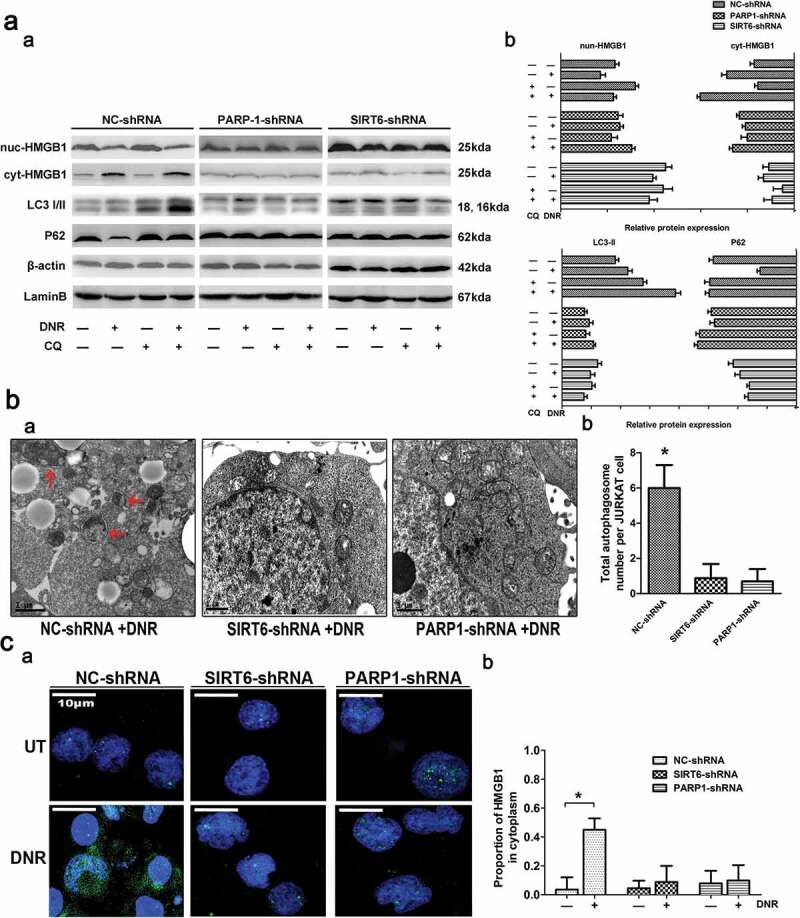
Silencing of SIRT6 or PARP1 inhibited the translocation of HMGB1 and suppressed chemotherapy-induced autophagy in leukemic cells. (a) The chemotherapy-induced autophagy and transition of HMGB1 were depressed in SIRT6 or PARP1 shRNA-treated cells. Jurkat cells were transfected with control, PARP1 or SIRT6 shRNA followed by the presence or the absence of DNR (0.4 μM) or CQ (10μM) for 24 hours. The cell lysates were subjected to western blot to detect the expression of LC3-II/I and p62. Furthermore, the cell lysates were separated into cytosolic and nuclear fractions. Cytosolic and nuclear proteins were subjected to western blot and detected with an HMGB1 antibody. β-actin and lamin B were used as loading controls. Quantified data are presented (p62 or LC3-II/I/β-actin, nuc-HMGB1/lamin B, cyt-HMGB1/β-actin, n > 3, *p < 0.05). CQ: chloroquine, Cyt: cytoplasm, Nuc: nucleus. (b) Autophagosomes and autophagolysosomes in SIRT6 or PARP1 shRNA-treated cells were less numerous than in the NC control group during chemotherapy. Jurkat cells were transfected with control, PARP1 or SIRT6 shRNA followed by the presence or the absence of DNR (0.4 μM) for 24 hours. The cells were subjected to transmission electron microscopy to observe autophagosome-like structures (indicated by the red arrows). (c) The absence of SIRT6 or PARP1 depressed the translocation of HMGB1 from the nucleus to the cytoplasm. Jurkat cells were transfected with control, PARP1 or SIRT6 shRNA followed by the presence or the absence of DNR (0.4 μM) for 24 hours. Intracellular HMGB1 was stained by immunofluorescence and subjected to confocal microscopic analysis to detect the location of HMGB1 (green: HMGB1; blue: nucleus). Quantified data are presented. UT: untreated group.
SIRT6-PARP1 complex was related to the polyADP-ribosylation and acetylation of HMGB1
Considering the significant decrease in HMGB1 translocation in the absence of SIRT6 or PARP1, we next sought to define the mechanisms involved in greater details. Therefore, we immunoprecipitated the cell lysates with an antibody against HMGB1, and these immunoprecipitation proteins were immunoblotted with special anti-polyADP-ribosylation and anti-acetylation antibodies. As shown in Figure 4, we observed a basic level of polyADP-ribosylation and acetylation of HMGB1 without DNR stimulation, while the above modifications of HMGB1 increased after DNR treatment. However, by silencing SIRT6 or PARP1, the above trend disappeared, and the polyADP-ribosylation and acetylation of HMGB1 after DNR treatment were maintained at the basic level. We demonstrated that silencing SIRT6 and PARP1 could inhibit the polyADP-ribosylation and acetylation of HMGB1.
Figure 4.
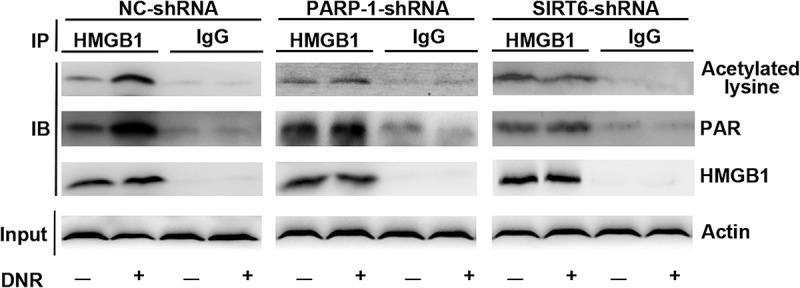
The SIRT6-PARP1 complex was related to the polyADP-ribosylation and acetylation of HMGB1. Jurkat cells were transfected with control, PARP1 or SIRT6 shRNA with or without DNR (0.4 μM) treatment for 24 hours. The cell lysates were pulled down with an HMGB1 antibody and immunoblotted with anti-acetylated lysine, anti-PARylation or HMGB1 antibodies. β-actin was used as a loading control.
PARylation of HMGB1 facilitated its acetylation and promoted HMGB1 translocation-related autophagy
Referring to the study of Bonaldi T et al., which found that the migration of HMGB1 depended on the acetylation of some special lysine residues, we mutated the lysine residues at 28,29,30,180,182,183,184 and 185 in HMGB1 into alanine as mutant type1 cells (HMGB1MT1).17 Ditsworth D et al. and Davis K et al. found that HMGB1 is PARylated by PARP1 and released from the nucleus; therefore, we mutated the glutamate residues at 40,47 and 179 into alanine as mutant type2 cells (HMGB1MT2), as the above glutamate residues were found to be frequently modified.26,27 The expression of lentivirus was detected by western blot with an anti-Flag antibody. As shown in Figure 5a, HMGB1MT1, HMGB1MT2 and wild-type Jurkat cells (HMGB1WT) were successfully constructed. Compared with high PARylation and acetylation of HMGB1 in HMGB1WT and normal control Jurkat cells (HMGB1NC) after DNR treatment, the expression of PARylated HMGB1 was not affected in HMGB1MT1, while the acetylation of HMGB1 was significantly reduced. In HMGB1MT2, not only the expression of PARylated HMGB1 but also the acetylation level of HMGB1 significantly decreased after DNR treatment, which indicated that the PARylation of HMGB1 might facilitate its acetylation (Figure 5b). By using immunoblotting, we found that the expression of HMGB1 in HMGB1WT and HMGB1NC cytoplasm increased after DNR treatment, but these changes were not observed in HMGB1MT1 and HMGB1MT2 (Figure 5c). Moreover, through the indirect immunofluorescence labeling of HMGB1, the green fluorescence relocated to the cytoplasm in HMGB1WT and HMGB1NCafter DNR treatment, while HMGB1MT1 and HMGB1MT2 did not respond to DNR treatment, and the green fluorescence remained nuclear (Figure 5e). According to western blot analysis, LC3-II showed high expression and p62 showed low expression after DNR treatment in HMGB1WT and HMGB1NC, which indicated an increased level of autophagy; however, the changes in LC3-II and p62 were not observed in HMGB1MT1 and HMGB1MT2(Figure 5c). Based on transmission electron microscopy analysis that compared HMGB1WT and HMGB1NC, HMGB1MT1 and HMGB1MT2 had fewer autophagosomes and autophagolysosomes during chemotherapy, which was consistent with the results of the western blot analysis (Figure 5d). Blocking the acetylation and PARylation of HMGB1 significantly reduced its chemotherapy-induced migration from the nucleus to the cytoplasm and inhibited the generation of autophagy during chemotherapy in Jurkat cells. These data indicated that the PARylation of HMGB1 might influence its acetylation, which could promote HMGB1 translocation and ultimately promote chemotherapy-induced autophagy of leukemic cells.
Figure 5.
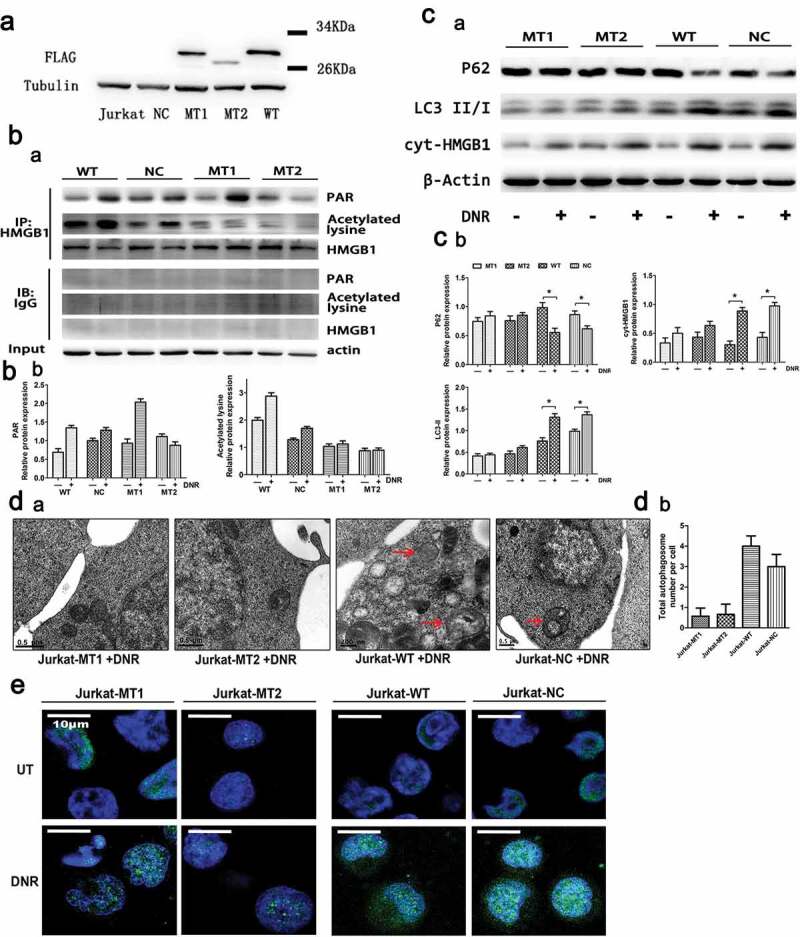
PARylation of HMGB1 facilitated its acetylation and promoted HMGB1 translocation-related autophagy. (a) HMGB1MT1, HMGB1MT2, HMGB1WT were successfully constructed. HMGB1MT1, HMGB1MT2, HMGB1WT, HMGB1NC and Jurkat cells were subjected to western blot analysis to detect the expression of lentivirus with a specific anti-Flag antibody. Tubulin was used as a loading control. (b) Jurkat cells were transfected with lentivirus, and then HMGB1NC, HMGB1MT1, HMGB1MT2 and HMGB1WT were treated with or without DNR (0.4 μM) for 24 hours. The cell lysates were pulled down with an HMGB1 antibody and immunoblotted with anti-acetylated lysine, anti-PARylation and HMGB1 antibodies. β-actin was used as a loading control. Quantified data are presented (PARylation-HMGB1 or Acetylation-HMGB1/HMGB1/β-actin, n > 3, *p < 0.05). (c) Jurkat cells were transfected with lentivirus, and then HMGB1NC, HMGB1MT1, HMGB1MT2 and HMGB1WT were treated with or without DNR (0.4 μM) for 24 hours. The cell lysates were subjected to western blot to detect the expression of LC3-II/I and p62. Furthermore, the cell lysates were separated into cytosolic and nuclear fractions. Cytoplasmic proteins were subjected to western blot and detected with an HMGB1 antibody. β-actin and lamin B were used as loading controls. Quantified data are presented.
Discussion
HMGB1 is a non-histone chromatin-binding nuclear protein that was found in calf thymus 40 years ago. Now, we have found that HMGB1 also exists in the cytoplasm and can be secreted into the extracellular space.13,28 The complex functions of HMGB1 in tumorigenesis and treatment have been reported to depend on its subcellular location: (1) nuclear HMGB1 induces the apoptosis of tumor cells by participating in the transcription of heat shock protein β1 (Hsp-β1); (2) cytosolic HMGB1 is bound to Beclin1 following Bcl-2 phosphorylation during autophagy and facilitates the formation of the PI3KC3-Beclin1 complex, which plays an essential role in the formation of the autophagosome; and (3) extracellular HMGB1 promotes tumor cells growth, invasion and metastasis by maintaining autophagy and inhibiting apoptosis by binding to receptor for advanced glycation end products (RAGE), which is always overexpressed on tumor cells.11,16,29 Our previous study showed that the function of Ulk1-Atg13-FIP 200 complexes upstream of HMGB1-Beclin1 and PI3KC3-Beclin1 complexes is related to HMGB1 translocation.
However, the specific mechanism of HMGB1 transposition is still unclear. Previous studies have shown that translocation and secretion of HMGB1 may involve but not be limited to the following mechanisms: posttranslational modifications, the calcium signaling pathway, reactive oxygen species (ROS) signaling pathway, JAK-STAT signaling pathway, p53 pathway, and the inflammasome pathway.32,33 In our study, we observed that silencing the ULK1-FIP200 complex inhibited the translocation of HMGB1 from the nucleus to the cytoplasm and that the acetylation level of HMGB1 decreased during this process (Figure 1a). This interesting phenomenon attracted our attention to the posttranscriptional modification of HMGB1. Posttranscriptional modification includes methylation, acetylation, phosphorylation and ADP-ribosylation. Acetylation is considered a prerequisite for the migration of HMGB1 to the cytoplasm.34 Sterner R et al. found that HMGB1 could be acetylated on lysines 2 and 11, and Bonaldi T et al. identified 17 acetylated lysine residues and indicated that two sets of lysine residues in nuclear localization sequence (NLS)1 and NLS2 appeared to be acetylated concomitantly in HMGB1.17,35 Lu B et al. identified that JAK/STAT1 played a key role in HMGB1 hyperacetylation and cytoplasmic accumulation.33 In our study, we mutated eight lysines into alanines (which cannot be modified) in HMGB1MT1; surprisingly, the translocation of HMGB1 was affected. In HMGB1MT1, the expression of HMGB1 in the cytoplasm was lower than that in HMGB1WT and HMGB1NC. At the same time, autophagy was also inhibited in HMGB1MT1 (Figure 5). Therefore, we concluded that the alternative subcellular location of HMGB1 depended on acetylation.
HMGB1 has previously been characterized as a receptor protein for poly (ADP)-ribosylation.31,36 PARP1 can catalyze the transfer of ADP-ribose moieties from NAD+ to the glutamic acid residues of acceptor proteins, thereby resulting in O-linked PAR, and PARP1 is the master regulator of PARylation.37 There have been reports that LPS induces PARP1 activation in macrophage cells and ultimately releases HMGB1 from the nucleus as HMGB1 via PARylation by PARP1. As showed in Figure 1c, chemotherapy-induced polyADP-ribosylation and acetylation of HMGB1 increased which showed the same trend in leukemic cell lines. Therefore, it is reasonable to believe that the polyADP-ribosylation of HMGB1 may affect its acetylation modification. In order to eliminate the influence of other intracellular factors on acetylation modification, we established an enzyme reaction system in vitro according to previous research.26 In this system, polyADP-ribosylation of HMGB1 in the PARP1 group was significantly upregulated, and the acetylation level of HMGB1 was also significantly higher than that in the control group (Figure 1b). In the vitro enzyme reaction system, the only reason for the difference in acetylation level is the different levels of polyADP-ribosylation of HMGB1. Therefore, according to Figure 1c and 1b, we concluded that the polyADP-ribosylation of HMGB1 promoted its acetylation.
However, the mechanism underlying the acetylation modification of HMGB1 is unclear, although a few studies have obtained preliminary results. Bonaldi T et al. showed that the mutation of some special lysine residues into alanines, which cannot be acetylated, in the HMGB1 NLS site rendered HMGB1 unable to be transposed. Ditsworth D et al. demonstrated that the C-terminal of HMGB1 was a potential target for polyADP-ribosylation by PARP1, as glutamic acid residues were abundant in the C-terminus, but he also observed that both full-length and truncated HMGB1 can be modified by PARP1.26 Although the glutamate-rich C-tail is not the only substrate for PARP1, it plays an important role in this process. The A-box may also be involved in the process, but the specific modification sites need to be further studied. Another study demonstrated that PARP1 decreased HDAC activities, whereas CREB binding protein (CBP)/p300 and PCAF were activated, which resulted in the hyperacetylation of HMGB1.34,38 Therefore, we believed that the posttranslational modifications in the NLS of HMGB1 played an important role in regulating the subcellular localization of intracellular protein, directing its localization.30,34 However, previous studies failed to mention the relationships among PARP1, PARylation and acetylation of HMGB1. Consistently, our study showed that in leukemic cells, PARP1 regulated the translation of HMGB1 to the cytoplasm by upregulating the acetylation of HMGB1 (Figures 3a,c, and 4). Unlike the mechanisms mentioned above, we hypothesized that PARylation of the glutamic acid residues would expose the acetylation site, promote the acetylation of lysine residues of HMGB1, and then affect the nuclear localization of HMGB1.
As mentioned previously, the most effective inducer of PARP1 activation is DNA damage, and the only known substrate for monoADP-ribosylation of SIRT6 is PARP1.39,40 In our study, we found that knockdown of SIRT6 or PARP1 gene expression significantly inhibited HMGB1 cytoplasmic translocation and autophagy (Figure 3). In previous studies, Lombard DB, Toiber D, et al. and McCord RA et al. indicated that SIRT6 executed its antiaging function by participating in DNA double strand break (DBS) repair.41–43 Xu Z et al. demonstrated that SIRT6 contributed to the maintenance of genomic stability in a PARP1-dependent manner.44 Van Meter M et al. suggested that SIRT6 physically associated with PARP1, leading to the stimulation of PARP1 polyADP-ribose polymerase activity and enhancing DSB repair under oxidative stress.39 The activity of SIRT6 deacetylase is weak, but point mutants with monoADP-ribosylation activity could strongly stimulate PARP1 at lysine 521.21,25 In our study, we also demonstrated that SIRT6 not only acted upstream of PARP1 but also formed a protein complex with PARP1. SIRT6 associated with PARP1 directly and catalyzed PARP1 monoADP-ribosylation, which was necessary for PARP1 activity (Figure 2). Furthermore, by combining the results shown in Figures 3–5, these data demonstrated that the SIRT6-PARP1 complex was associated with HMGB1 via polyADP-ribosylation and acetylation modification. Meanwhile, the polyADP-ribosylation of HMGB1 promoted its acetylation, which led to the translocation of HMGB1.
As cancer is becoming one of the biggest problems worldwide, chemotherapy is playing an important role in anticancer therapy, and the pivotal cellular target of chemotherapy is genomic DNA. The induction of autophagy is a common response to multiple forms of chemotherapy, including DNR, and the role of autophagy is complex.5 Autophagy, a lysosome-dependent degradation process, has been demonstrated to be involved in DNA damage by Czarny P et al. and Qiang L et al.45,46 Shao J et al. and Takasaka et al. reported that SIRT6 induced autophagy by targeting the AKT/mTOR signaling pathway under oxidative stress damage.47,48 Lee et al. found that SIRT6 was associated with HMGB1 release after cerebral ischemia.49 Rodriguez V et al. and Munoz G et al. reported that PARP1 activation induced by DNA damage was involved in amplifying cytoprotective autophagy in cancer cells.50 Furthermore, Yang, M et al. demonstrated that PARP1-mediated HMGB1 PARylation was required for TNFSF10-induced HMGB1 cytoplasmic translocation and autophagy.31 Kang R et al. observed that upregulated HMGB1 expression or HMGB1 release contributed to drug resistance in several cancer cells, such as leukemia, colon cancer and liver cancer cells.32 Meanwhile, in our previous study, the complex function of HMGB1 in tumor development and therapy was reported to depend on its subcellular location. Unfortunately, the functions of SIRT6, PARP1 and HMGB1 in chemotherapy-induced autophagy in ALL remains underestimated, and the interplay among them has garnered little attention.
By combining previous data with our present results, we have elucidated the process of chemotherapy-induced autophagy in leukemic cells, showing that the translocation of HMGB1 was the most critical step during this process and demonstrating that the SIRT6-PARP1-HMGB1 pathway mediated chemotherapy-induced autophagy in leukemic cells. In conclusion, SIRT6 activated PARP1 by monoADP-ribosylation, thus promoting PARP1 to modify HMGB1 by polyADP-ribosylation, which subsequently enhanced the acetylation of HMGB1 and finally promoted the autophagy of leukemic cells, mediated by the translocation of the HMGB1(Figure 6). By considering the results shown in Figure 4, we found a notable phenomenon in which the levels of polyADP-ribosylation and acetylation of HMGB1 did not decrease below the baseline level in SIRT6 or PARP1 silenced cells after DNR treatment. We speculated that the SIRT6-PARP1 pathway may not be the only HMGB1 translocation-related modification channel; thus, further research is needed.
Figure 6.
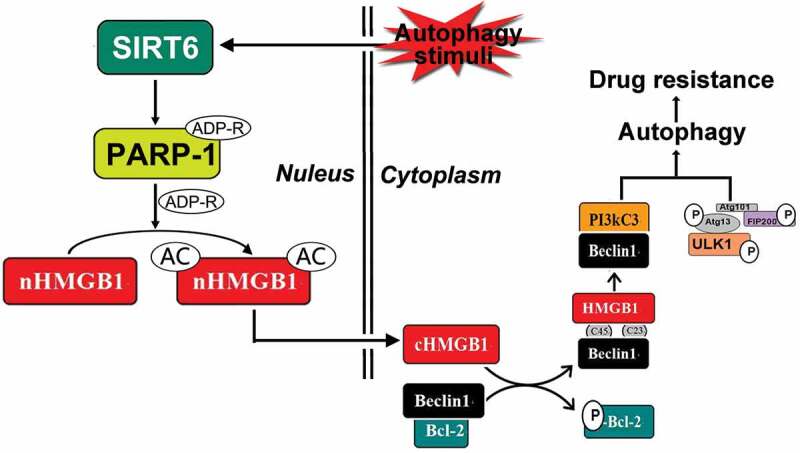
Schematic representation of the potential HMGB1-translocation related chemotherapy-induced autophagy in leukemia. Under stress condition, SIRT6 was activated, SIRT6 not only served as an upstream signal of PARP1, but also formed the complex with PARP1. SIRT6 activated PARP1 through monoADP-ribosylation and the activated PARP1 modified HMGB1 by polyADP-ribosylation, which subsequently enhanced the acetylation of HMGB1 and promoted the translocation of the HMGB1 from nucleus to cytoplasm. The translocation of HMGB1 was the most critical step during this process and mediated chemotherapy-induced autophagy in leukemic cells.
This study mainly focuses on the mechanism research of HMGB1 translocation-related chemotherapy-induced autophagy. In the next step, we will continue to explore whether SIRT6-PARP1-HMGB1 contributes to chemotherapy resistance. What is more, it is of great significance to further explore the drug resistance mechanism of various commonly used chemotherapy drugs and verify it with various leukemia cell lines. By further understanding these mechanisms and signaling pathways, we are likely to open up new avenues for the development of targeted therapies for drug-resistant leukemia.
Materials and methods
Antibodies and reagents The antibody to streptavidin was obtained from Santa Cruz (sc-363872). The antibody to PAR (4336-BPC-100) was obtained from Trevigen. The antibodies to acetylated lysine (9441S), rabbit mAb IgG XPTM isotype control (3900S), PARP1 (5625T), and SIRT6 (12486S) were obtained from Cell Signaling Technology. The antibodies to HMGB1 (H9539), p62 (P0067), and LC3 (L8918) were obtained from Sigma-Aldrich. The antibodies to β-actin (7D2C10), lamin B (12987-1-AP), and tubulin (11224-1-AP) were obtained from Proteintech. The secondary antibodies, including sheep anti-mouse IgG-HRP (RM3001), sheep anti-rabbit IgG-HRP (RM3002) were obtained from Beijing Ray Antibody Biotech. FITC-conjugated secondary antibody (ZF-0134) was obtained from Zsbio Commerce Store. IPKine HRP AffiniPure Goat Anti-Rabbit IgG Light Chain (Abbkine A25012) was used as a secondary antibody at a dilution of 1:10000 for detecting target proteins without interference from denatured IgG in the western blot. Daunorubicin (DNR) was purchased from MedChemExpress and chloroquniene (CQ) was purchased from Sigma-Aldrich.
Cell culture The human acute leukemic cell line, Jurkat cells and RS4:11 cells were purchased from the Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China. Jurkat cells and RS4:11 cells were grown in RPMI-1640 (Gibco) with 10% fetal bovine serum (FBS; HyClone), 2 mM L-glutamine, and 1% penicillin/streptomycin (HyClone) at 37°C in 5% CO2 air.
Western blot analysis Cells with different treatments were lysed in RIPA buffer (Beyotime Institute of Biotechnology) with protease inhibitor mixture. After boiling for 10 min with Laemmli buffer, the samples were resolved by denaturing 10% or 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride membrane (PVDF; Millipore Billerica). After blocking with 5% nonfat dried milk in phosphate-buffered saline (PBS; Sigma-Aldrich) for 1 hour at room temperature and washing with TBS containing 1% Tween 20 (TBST; Sigma) 3 times, the membranes were incubated with primary antibodies at a dilution of 1:1000 overnight at 4°C. After washing thrice with TBST, secondary antibodies were applied at a 1:5000 dilution for 1 hour at room temperature and then washed in TBST thrice for 10 min. The blots were detected with enhanced chemiluminescence reagent (Millipore) by G:BOX XT4 (Syngene).
PARylation and acetylation of proteins in vitro PARylation was done using biotin-NAD instead of (32P)NAD.26 The reaction was performed in 30 μl of mix buffer containing 50 mM Tris-HCl (pH 8.0), 25 mM MgCl2, 5 μg of PARP-activated DNA (R&D Systems), 20 μM 6-biotin-17-NAD (R&D Systems), and 3 μg of purified recombinant HMGB1 protein (R&D Systems). One microliter of PARP1 enzyme (Trevigen, highly specific activity enzyme) was added to the reaction and then incubated at room temperature for 30 min. The acetylation reaction was performed in 30 μl of HAT buffer including 50 mM Tris-HCl (pH 8.0), 10% glycerol, 1 mM DTT, 1 mM PMSF, 10 mM sodium butyrate, 1 mM acetyl CoA (Sigma), and 3 μg of purified recombinant HMGB1 protein. One microliter of CBP (Merck-Millipore) catalytic domain was added at room temperature for 30 min. For successive acetylation, 30 μl mixtures of products from the control group and the experimental group after the PARylation reaction in the last step were precipitated with a 20% final concentration of trichloroacetic acid (TCA; Sigma) at −20°C overnight. After rotation at 12000 g for 15 min, the solutions were washed with cold acetone three times and dissolved in 5 μl of TE buffer (pH 8.0). Heat-inactivated enzyme was used as a control treatment. Loading buffer was added to stop the reaction, and samples were boiled for 10 min. The samples were analyzed by SDS-PAGE.
Lentivirus infection Experiments were performed in 6-well plates at a density of 5 × 106 cells/well. Lentiviruses were purchased from Genepharma (Shanghai, China) and Obio Technology (Shanghai, China). Transfection was conducted with lentivirus using 5 μg/ml polybrene and selected with 1 μg/ml puromycin, following the protocol. The efficiency and specificity of shRNA gene knockdown of target proteins were detected by RT-qPCR and western blotting, and the cells were used in subsequent experiments. The optimal target sequence for SIRT6 was 5ʹ-GGGACAAACTGGCAGAGCT-3ʹ, for PARP1 was 5ʹ- CAGAACGACCTGATCTGGAACATCA-3ʹ and for control was 5ʹ-TTCTCCGAACGTGTCACGTAA-3ʹ. In this paper, the lysine residues were mutated at amino acids 28, 29, 30, 180, 182, 183, 184 and 185 in HMGB1 to alanine in order to generate mutant type-1 cells (HMGB1MT1), and the glutamate residues were mutated at amino acids 40, 47 and 179 to alanine to generate mutant type-2 cells (HMGB1MT2).
PARP activity assay The activity of PARP was measured by cell ELISA. A total of 5 × 104 cells was plated in 96-well plates. After incubation at 37°C for 30 min in 100 μl of PARP buffer with 0.01% digitonin and 10 μM biotin-NAD+, cells were treated with 200 μl of 95% ethanol at −20°C for 10 min. Then, cells were washed once with PBS and blocked in 1% BSA for 30 min. After that, cells were incubated in 50 μl of streptavidin-HRP (1:500) at 37°C for 30 min. Cells were washed three times with PBS, followed by incubation in 100 μl of TACS-Sapphire for 15 min at room temperature. The reaction was stopped by 1M H2SO4. The absorbance was detected at 450 nm with a microplate reader.
Immunoprecipitation analysis Whole-cell lysates (1000 μg) harvested in RIPA buffer were precleared with Protein G Magnetic Beads (Bimake) for 1 hour at room temperature. The precleared samples were then incubated with normal, nonspecific IgG (control group) or specific antibodies (5 μg/ml) overnight at 4°C to form the immune complexes. The next day, the samples were applied in a Protein G Magnetic Bead reaction. After washing 3 times with sample wash buffer, the samples were eluted under denaturing conditions in 50μl of 2 × SDS sample buffer and analyzed by western blot as previously described.
Quantitative real-time PCR(RT-qPCR) Cells were harvested in 1 ml TRIzolreagen (Takara Bio Inc, Otsu, Shiga, Japan). The final RNA pellet was dissolved in 50 ul of nuclease-free water and then reverse-transcribed into cDNA. The sequences of primers used were as follows: GAPDH: forward, 5ʹ-GGTCGGAGTCAACGGATTTGGTCG-3ʹ and reverse, 5ʹ-CCTCCGACGCCTGCTTCACCAC-3ʹ, for SIRT6: forward 5ʹ-GCAGTCTTCCAGTGTGGTGT-3ʹ and reverse 5ʹ-CCATGGTCCAGACTCCGT-3ʹ, for PARP1: forward 5ʹ-CGGAGTCTTCGGATAAGCTCT-3ʹ and reverse 5ʹ-TTTCCATCAAACATGGGCGAC-3ʹ. Reactions were carried out in a Roche Light Cycler 480 system (Roche Diagnostics GmbH, Mannheim, Germany) with a SYBR Premix ExTaq kit (Takara Bio Inc., Otsu, Shiga, Japan). With the 2−ΔΔCt method. Data were normalized to GAPDH expression. The control group was set as 1.
Preparation of cytoplasmic and nuclear extracts Cytoplasmic and nuclear extracts were prepared using the Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime Institute of Biotechnology) according to the manufacturer’s instructions. First, cells were washed in ice-cold PBS and then resuspended in 200 μl of ice-cold cytoplasmic extraction buffer A with 1% protease inhibitor cocktail (Bimake) for 10 min. After incubation with cytoplasmic extraction buffer B for 1 min in an ice bath and vortexing for 5 s, cell lysates were centrifuged at 12000 g for 5 min at 4°C. Supernatants were aliquoted and stored at −80°C. Nuclear pellets were washed with PBS three times and then resuspended in 50 μl of nuclear extraction buffer. After vortexing four times for 15 s every 7 min, lysates were centrifuged at 12000 g for 5 min at 4°C. Nuclear extracts were aliquoted and stored at −80°C.
Immunofluorescence analysis Different cells were fixed with 4% formaldehyde for 15 min and washed three times with PBS. After permeabilizing in 0.3% Triton X-100 in PBS for 10 min, cells were incubated in blocking buffer for 1 hour at room temperature. Then, the cells were incubated with primary antibody in 1% BSA overnight at 4°C. Cells were washed three times with 1% Tween in PBS followed by incubation with FITC-conjugated secondary antibody for 1 hour at room temperature in the dark and then with DAPI for 5 min. After another three washes, the cells were resuspended and added to a laser confocal Petri dish. Images were acquired with a confocal microscope (Carl Zeiss Inc).
Transmission electron microscopic analysis Cells were fixed with 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 mol/l phosphate buffer (pH 7.4) at 4°C for 2 hours, followed by 1% OsO4. After dehydration, thin sections (50–80 nm) were stained with uranyl acetate and lead citrate respectively at 4°C for 15 min, and observed by Tecnai G2 Spirit Twin election microscope (FEI, Hillsboro, OR, USA).
Statistical analysis All data are expressed as the mean ± standard deviation (SD). Statistical analysis was performed using Student’s t-test. A value of P < 0.05 was regarded as statistically significant. All statistical analyses were conducted by SPSS version 18.0 software (SPSS, Inc).
Funding Statement
This work was supported by the National Natural Science Foundation of China under [Grant no.81570140]; and the Doctoral Start-up Fund of Natural Science Foundation of Guangdong Province under Grant [no.2016A030310161].
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Competing interests
The authors declare that they have no competing interests.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Vadillo E, Dorantes-Acosta E, Pelayo R, Schnoor M.. 2018. T cell acute lymphoblastic leukemia (T-ALL): new insights into the cellular origins and infiltration mechanisms common and unique among hematologic malignancies. Blood Rev. 32:36–51. doi: 10.1016/j.blre.2017.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Greaves MF, Janossy G, Peto J, Kay H. 1981. Immunologically defined subclasses of acute lymphoblastic leukaemia in children: their relationship to presentation features and prognosis. Br J Haematol. 48:179–197. doi: 10.1111/j.1365-2141.1981. [DOI] [PubMed] [Google Scholar]
- 3.Richter-Pechanska P, Kunz JB, Hof J, Zimmermann M, Rausch T, Bandapalli OR, Orlova E, Scapinello G, Sagi JC, Stanulla M, et al. 2017. Identification of a genetically defined ultra-high-risk group in relapsed pediatric T-lymphoblastic leukemia. Blood Cancer J. 7:e523. doi: 10.1038/bcj.2017.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belver L, Ferrando A. 2016. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer. 16:494–507. doi: 10.1038/nrc.2016.63. [DOI] [PubMed] [Google Scholar]
- 5.Feng Y, He D, Yao Z, Klionsky DJ. 2014. The machinery of macroautophagy. Cell Res. 24:24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parzych KR, Klionsky DJ. 2014. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 20:460–473. doi: 10.1089/ars.2013.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bustin M. 1999. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol Cell Biol. 19:5237–5246. doi: 10.1128/mcb.19.8.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stevens NE, Chapman MJ, Fraser CK, Kuchel TR, Hayball JD, Diener KR. 2017. Therapeutic targeting of HMGB1 during experimental sepsis modulates the inflammatory cytokine profile to one associated with improved clinical outcomes. Sci Rep. 7:5850. doi: 10.1038/s41598-017-06205-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee W, Kwon OK, Han MS, Lee YM, Kim SW, Kim KM, Lee T, Lee S, Bae JS. 2015. Role of moesin in HMGB1-stimulated severe inflammatory responses. Thromb Haemost. 114:350–363. doi: 10.1160/th14-11-0969. [DOI] [PubMed] [Google Scholar]
- 10.Harris HE, Andersson U, Pisetsky DS. 2012. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nature Rev Rheumatol. 8:195–202. doi: 10.1038/nrrheum.2011.222. [DOI] [PubMed] [Google Scholar]
- 11.Tang D, Kang R, Zeh HJ 3rd, Lotze MT. 2010. High-mobility group box 1 and cancer. Biochim Biophys Acta. 1799:131–140. doi: 10.1016/j.bbagrm.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kong Q, Xu LH, Xu W, Fang JP, Xu HG. 2015. HMGB1 translocation is involved in the transformation of autophagy complexes and promotes chemoresistance in leukaemia. Int J Oncol. 47:161–170. doi: 10.3892/ijo.2015.2985. [DOI] [PubMed] [Google Scholar]
- 13.Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, Rubartelli A. 2002. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 3:995–1001. doi: 10.1093/embo-reports/kvf198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oh YJ, Youn JH, Ji Y, Lee SE, Lim KJ, Choi JE, Shin JS. 2009. HMGB1 is phosphorylated by classical protein kinase C and is secreted by a calcium-dependent mechanism. J Immunol (Baltimore, Md: 1950). 182:5800–5809. doi: 10.4049/jimmunol.0801873. [DOI] [PubMed] [Google Scholar]
- 15.Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, Nakasato M, Lu Y, Hangai S, Koshiba R, et al. 2009. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. 462:99–103. doi: 10.1038/nature08512. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Q, Wang Y. 2010. HMG modifications and nuclear function. Biochim Biophys Acta. 1799:28–36. doi: 10.1016/j.bbagrm.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME. 2003. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. Embo J. 22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Y, Wang J, Yang Q, Wu S, Yang Z, Zhu H, Zheng M, Liu W, Wu W, He J, et al. 2014. Shikonin inhibits the lipopolysaccharide-induced release of HMGB1 in RAW264.7 cells via IFN and NF-kappaB signaling pathways. Int Immunopharmacol. 19:81–87. doi: 10.1016/j.intimp.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 19.Gil R, Barth S, Kanfi Y, Cohen HY. 2013. SIRT6 exhibits nucleosome-dependent deacetylase activity. Nucleic Acids Res. 41:8537–8545. doi: 10.1093/nar/gkt642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Azuma Y, Yokobori T, Mogi A, Altan B, Yajima T, Kosaka T, Onozato R, Yamaki E, Asao T, Nishiyama M, et al. 2015. SIRT6 expression is associated with poor prognosis and chemosensitivity in patients with non-small cell lung cancer. J Surg Oncol. 112:231–237. doi: 10.1002/jso.23975. [DOI] [PubMed] [Google Scholar]
- 21.Cagnetta A, Soncini D, Orecchioni S, Talarico G, Minetto P, Guolo F, Retali V, Colombo N, Carminati E, Clavio M, et al. 2018. Depletion of SIRT6 enzymatic activity increases acute myeloid leukemia cells’ vulnerability to DNA-damaging agents. Haematologica. 103:80–90. doi: 10.3324/haematol.2017.176248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livesey KM, Kang R, Vernon P, Buchser W, Loughran P, Watkins SC, Zhang L, Manfredi JJ, Zeh HJ 3rd, Li L, et al. 2012. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 72:1996–2005. doi: 10.1158/0008-5472.Can-11-2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanny JC, Dowd GJ, Huang J, Hilz H, Moazed D. An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell. 1999;99:735–745. doi: 10.1016/S0092-8674(00)81671-2. [DOI] [PubMed] [Google Scholar]
- 24.Imai S, Armstrong CM, Kaeberlein M, Guarente L. 2000. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 25.Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V. 2011. SIRT6 promotes DNA repair under stress by activating PARP1. Sci (New York, NY). 332:1443–1446. doi: 10.1126/science.1202723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ditsworth D, Zong WX, Thompson CB. 2007. Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J Biol Chem. 282:17845–17854. doi: 10.1074/jbc.M701465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis K, Banerjee S, Friggeri A, Bell C, Abraham E, Zerfaoui M. 2012. Poly(ADP-ribosyl)ation of high mobility group box 1 (HMGB1) protein enhances inhibition of efferocytosis. Mol Med (Cambridge, Mass). 18:359–369. doi: 10.2119/molmed.2011.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu B, Wang H, Andersson U, Tracey KJ. 2013. Regulation of HMGB1 release by inflammasomes. Protein Cell. 4:163–167. doi: 10.1007/s13238-012-2118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boone BA, Orlichenko L, Schapiro NE, Loughran P, Gianfrate GC, Ellis JT, Singhi AD, Kang R, Tang D, Lotze MT, et al. 2015. The receptor for advanced glycation end products (RAGE) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther. 22:326–334. doi: 10.1038/cgt.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito I, Fukazawa J, Yoshida M. 2007. Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J Biol Chem. 282:16336–16344. doi: 10.1074/jbc.M608467200. [DOI] [PubMed] [Google Scholar]
- 31.Yang M, Liu L, Xie M, Sun X, Yu Y, Kang R, Yang L, Zhu S, Cao L, Tang D. 2015. Poly-ADP-ribosylation of HMGB1 regulates TNFSF10/TRAIL resistance through autophagy. Autophagy. 11:214–224. doi: 10.4161/15548627.2014.994400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, Huang J, Yu Y, Fan XG, Yan Z, et al. 2014. HMGB1 in health and disease. Mol Aspects Med. 40:1–116. doi: 10.1016/j.mam.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu B, Antoine DJ, Kwan K, Lundback P, Wahamaa H, Schierbeck H, Robinson M, Van Zoelen MA, Yang H, Li J, et al. 2014. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc Natl Acad Sci U S A. 111:3068–3073. doi: 10.1073/pnas.1316925111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.JY Z, Crews FT. 2014. Release of neuronal HMGB1 by ethanol through decreased HDAC activity activates brain neuroimmune signaling. PLoS One. 9:e87915. doi: 10.1371/journal.pone.0087915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sterner R, Vidali G, Allfrey VG. Studies of acetylation and deacetylation in high mobility group proteins identification of the Sites of acetylation in HMG-1. The J Biol Che. 1979;254:11577–11583. [PubMed] [Google Scholar]
- 36.Yang Z, Li L, Chen L, Yuan W, Dong L, Zhang Y, Wu H, Wang C. 2014. PARP-1 mediates LPS-induced HMGB1 release by macrophages through regulation of HMGB1 acetylation. J Immunol (Baltimore, Md: 1950). 193:6114–6123. doi: 10.4049/jimmunol.1400359. [DOI] [PubMed] [Google Scholar]
- 37.Luo X, Kraus WL. 2012. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 26:417–432. doi: 10.1101/gad.183509.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evankovich J, Cho SW, Zhang R, Cardinal J, Dhupar R, Zhang L, Klune JR, Zlotnicki J, Billiar T, Tsung A. 2010. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J Biol Chem. 285:39888–39897. doi: 10.1074/jbc.M110.128348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Meter M, Mao Z, Gorbunova V, Seluanov A. 2011. Repairing split ends: SIRT6, mono-ADP ribosylation and DNA repair. Aging. 3:829–835. doi: 10.18632/aging.100389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woodhouse BC, Dianov GL. 2008. Poly ADP-ribose polymerase-1: an international molecule of mystery. DNA Repair (Amst). 7:1077–1086. doi: 10.1016/j.dnarep.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 41.Lombard DB. 2009. Sirtuins at the breaking point: SIRT6 in DNA repair. Aging. 1:12–16. doi: 10.18632/aging.100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Toiber D, Erdel F, Bouazoune K, Silberman DM, Zhong L, Mulligan P, Sebastian C, Cosentino C, Martinez-Pastor B, Giacosa S, et al. 2013. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol Cell. 51:454–468. doi: 10.1016/j.molcel.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCord RA, Michishita E, Hong T, Berber E, Boxer LD, Kusumoto R, Guan S, Shi X, Gozani O, Burlingame AL, et al. 2009. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging. 1:109–121. doi: 10.18632/aging.100011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu Z, Zhang L, Zhang W, Meng D, Zhang H, Jiang Y, Xu X, Van Meter M, Seluanov A, Gorbunova V, et al. 2015. SIRT6 rescues the age related decline in base excision repair in a PARP1-dependent manner. Cell Cycle (Georgetown, Tex). 14:269–276. doi: 10.4161/15384101.2014.980641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Czarny P, Pawlowska E, Bialkowska-Warzecha J, Kaarniranta K, Blasiak J. 2015. Autophagy in DNA damage response. Int J Mol Sci. 16:2641–2662. doi: 10.3390/ijms16022641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qiang L, Zhao B, Shah P, Sample A, Yang S, He YY. 2016. Autophagy positively regulates DNA damage recognition by nucleotide excision repair. Autophagy. 12:357–368. doi: 10.1080/15548627.2015.1110667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shao J, Yang X, Liu T, Zhang T, Xie QR, Xia W. 2016. Autophagy induction by SIRT6 is involved in oxidative stress-induced neuronal damage. Protein Cell. 7:281–290. doi: 10.1007/s13238-016-0257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takasaka N, Araya J, Hara H, Ito S, Kobayashi K, Kurita Y, Wakui H, Yoshii Y, Yumino Y, Fujii S, et al. 2014. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J Immunol (Baltimore, Md: 1950). 192:958–968. doi: 10.4049/jimmunol.1302341. [DOI] [PubMed] [Google Scholar]
- 49.Lee OH, Kim J, Kim JM, Lee H, Kim EH, Bae SK, Choi Y, Nam HS, Heo JH. 2013. Decreased expression of sirtuin 6 is associated with release of high mobility group box-1 after cerebral ischemia. Biochem Biophys Res Commun. 438:388–394. doi: 10.1016/j.bbrc.2013.07.085. [DOI] [PubMed] [Google Scholar]
- 50.Rodriguez-Vargas JM, Ruiz-Magana MJ, Ruiz-Ruiz C, Majuelos-Melguizo J, Peralta-Leal A, Rodriguez MI, Munoz-Gamez JA, de Almodovar MR, Siles E, Rivas AL, et al. 2012. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res. 22:1181–1198. doi: 10.1038/cr.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.