

ABSTRACT
Triple-negative breast cancer (TNBC) displays an aggressive clinical course, heightened metastatic potential, and is linked to poor survival rates. Through its lack of expression of the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), this subtype remains unresponsive to traditional targeted therapies. Undesirable and sometimes life-threatening side effects associated with current chemotherapeutic agents warrant the development of more targeted treatment options. Targeting signal transducer and activator of transcription 3 (STAT3), a transcription factor implicated in breast cancer (BCa) progression, has proven to be an efficient approach to halt cancer growth in vitro and in vivo. Currently, there are no FDA-approved STAT3 inhibitors for TNBC. Although pimozide, a FDA-approved antipsychotic drug, has been attributed a role as a STAT3 inhibitor in several cancers, its role on this pathway remains unexplored in TNBC. As a “one size fits all” approach cannot be applied to TNBC therapies due to the heterogeneous nature of this aggressive cancer, we hypothesized that STAT3 could be a novel biomarker of response to guide pimozide therapy. Using human cell lines representative of four TNBC subtypes (basal-like 1, basal-like 2, mesenchymal-like, mesenchymal stem-like), our current report demonstrates that pimozide significantly reduced their invasion and migration, an effect that was predicted by STAT3 phosphorylation on tyrosine residue 705 (Tyr705). Mechanistically, phosphorylated STAT3 (Tyr705) inhibition resulting from pimozide treatment caused a downregulation of downstream transcriptional targets such as matrix metalloproteinase-9 (MMP-9) and vimentin, both implicated in invasion and migration. The identification of biomarkers of response to TNBC treatments is an active area of research in the field of precision medicine and our results propose phosphorylated STAT3 (Tyr705) as a novel biomarker to guide pimozide treatment as an inhibitor of invasion and migration.
KEYWORDS: STAT3, biomarker, pimozide, triple-negative breast cancer, precision Medicine, drug repurposing
Introduction
Accounting for 15–20% of all breast cancer (BCa) diagnoses, triple-negative breast cancer (TNBC) is the most aggressive subtype of BCa with the poorest short-term prognosis. TNBCs are differentiated from the other BCa subtypes by their lack of expression of the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2).1,2 Due to intrinsic aggressive biology, TNBCs frequently metastasize to distant organs including the lung, brain, liver, and bones.3 These metastases often result in death, as less than 30% of women diagnosed with metastatic TNBC survive beyond 5 years.4 Unfortunately, due to the absence of targetable receptors, women with TNBC are not candidates for anti-hormonal therapies (tamoxifen) or anti-HER2 targeting therapies (trastuzumab), thus making surgery, radiotherapy, and chemotherapy the only treatment options for TNBC patients. Major drawbacks that result from nonspecific chemotherapeutics include cardiotoxicity, neurotoxicity, and neutropenia.5,6 As such, there is an urgent need to develop targeted therapies for the TNBC patient population to reduce the occurrence of systemic toxicities.
Transcription factors, proteins involved in regulating the expression of genes, are commonly dysregulated in cancer and are currently being studied as therapeutic drug targets in TNBC. Signal transducer and activator of transcription 3 (STAT3) is a transcription factor and biomarker indicative of poor survival in TNBC.7–9 Constitutively activated STAT3 expression through mutant constructs has revealed this transcription factor to be an important driver of oncogenic processes including proliferation, cell survival, angiogenesis, invasion, and migration.7,10-14 Published research has demonstrated that constitutive activation of STAT3 led to the transformation of human mammary epithelial cells (HMECs) through upregulation of matrix metalloproteinase-9 (MMP-9) activity.15 Additional reports have shown that STAT3 can directly promote the migration of human breast cancer cells through regulation of the actin-bundling protein, fascin, in response to cytokine stimulation.16 Indeed, STAT3 has emerged as a druggable therapeutic target in BCa. Direct and indirect STAT3 inhibitors are currently being evaluated in clinical trials for the treatment of TNBC.17–20
Translating drug candidates from bench to bedside remains an immense challenge. Drug repurposing, or the repositioning of Food and Drug Administration (FDA)-approved drugs for new indications, has emerged as an effective strategy to reduce the time and cost of bringing a drug to market.21–23 A prime example of drug repurposing is thalidomide. Originally approved by the FDA as a sedative and antiemetic, thalidomide was later withdrawn from the market due to severe teratogenic side effects. Since then, thalidomide has been successfully repurposed by the FDA for the treatment of multiple myeloma and a variant of leprosy referred to as erythema nodosum leprosum.24,25 Numerous drug classes are currently being evaluated as candidates for repurposing in cancer and include antidepressants, cardiovascular agents, antipsychotics, and antibiotics.21,24 However, in order to maximize the efficacy of repurposed drugs, there is an urgent need to identify and validate selection biomarkers that can predict patient response to a specific therapy.
Pimozide is a first-generation typical antipsychotic drug of the diphenylbutylpiperidine class that has been approved by the FDA for the treatment of Tourette syndrome.26 Individuals diagnosed with Tourette syndrome and other chronic psychoses such as schizophrenia exhibit increased dopaminergic activity. Mechanistically, pimozide acts by antagonizing the postsynaptic dopamine receptor D2 in the central nervous system (CNS).27–29 Interestingly, pimozide has been shown to exhibit anti-cancer properties in a variety of solid and hematological cancers including hepatocellular carcinoma,30 pancreatic cancer,31 non-small cell lung cancer (NSCLC),32,33 breast cancer,33–36 and leukemia.37 Multiple reports have demonstrated pimozide to act as a STAT5 inhibitor in leukemia and lymphoma.38–40 Moreover, pimozide treatment seems to be a viable avenue to target STAT3 in multiple malignancies. In fact, published literature shows that pimozide inhibited STAT3 phosphorylation (Tyr705) in multiple myeloma cells, leading to a significant decrease in cell viability as measured in an ATP-dependent luminescence assay.41 In prostate cancer, pimozide reduced the proliferation, migration, and colony/sphere formation in LNCaP and DU145 cells through inhibition of phosphorylated STAT3 (Tyr705).42 In hepatocellular carcinoma, pimozide abrogated interleukin-6 (IL-6)-mediated STAT3 phosphorylation (Tyr705) and reversed cancer stem-like phenotypes in MHCC-97L and Hep 3B cells.43 Lastly, in osteosarcoma, pimozide-induced apoptosis, increased reactive oxygen species (ROS) generation, reduced proliferation, and inhibited colony/sphere formation in U2OS cells via inhibition of phosphorylated STAT3 (Tyr705).44 Collectively, these reports demonstrate pimozide to be a robust anti-cancer agent and a promising inhibitor of STAT3. Although the first report demonstrating the efficacy of pimozide to inhibit TNBC growth was reported almost 30 years ago,34,35 it was only recently that its anti-cancer effects were discovered to regulate the RAN GTPase and AKT signaling pathways in MDA-MB-231 in vitro and in vivo.36 However, due to the innate heterogeneity present in TNBCs,2,45 further investigation is currently warranted to include more TNBC subtypes and to investigate pimozide’s role on the STAT3 pathway, which remains undefined in TNBC. Our current report examines the efficacy of pimozide on cell survival, invasion and migration among different molecular subtypes of TNBC, while using STAT3 phosphorylation (Tyr705) as a biomarker of response.
Herein, our findings show that phosphorylated STAT3 (Tyr705) was constitutively activated in 75% of TNBC subtypes examined, in comparison to non-tumorigenic mammary epithelial cells. TNBC cell lines with constitutively activated phosphorylated STAT3 (Tyr705) expression conferred the most sensitivity to pimozide treatment, as evidenced by a reduction in STAT3 phosphorylation (Tyr705). In contrast, cell lines with low basal expression of phosphorylated STAT3 (Tyr705) were found to be resistant to pimozide. Furthermore, upon treatment with pimozide, inhibition of phosphorylated STAT3 (Tyr705) correlated with the suppression of invasion and migration in TNBC cells, two important processes involved in the development of metastatic disease. Mechanistically, pimozide treatment decreased protein expression of STAT3 downstream targets involved in metastatic progression, including matrix metalloproteinase-9 (MMP-9) and vimentin, in TNBC cell lines sensitive to STAT3 inhibition.
In sum, our research demonstrates that not all molecular subtypes of TNBC are responsive to pimozide treatment. We identified phosphorylated STAT3 (Tyr705) as a novel biomarker of response that is predictive of sensitivity to pimozide treatment in TNBC. We propose that phosphorylated STAT3 (Tyr705) should be validated as an important biomarker using patient-derived TNBC xenografts representative of all molecular subtypes in vivo to effectively predict response to pimozide therapy and further confirm its potential clinical value moving forward.
Results
Endogenous STAT3 expression levels across four different TNBC subtypes: BT-549 (mesenchymal-like), MDA-MB-231 (mesenchymal stem-like), HCC1806 (basal-like 2), MDA-MB-468 (basal-like 1)
Endogenous protein expression of phosphorylated STAT3 (Tyr705) and total STAT3 was determined in a panel of four human TNBC cell lines (BT-549, MDA-MB-231, HCC1806, MDA-MB-468) as compared to non-tumorigenic human mammary epithelial cells (MCF-10A) (Figure 1(a,b)). The 4 TNBC cell lines selected are representative of different molecular subtypes: BT-549 (mesenchymal-like), MDA-MB-231 (mesenchymal stem-like), HCC1806 (basal-like 2), MDA-MB-468 (basal-like 1).46 Quantitatively, we demonstrate that phosphorylated STAT3 (Tyr705) expression was significantly higher in three of the four TNBC cell lines (BT-549, MDA-MB-231, MDA-MB-468) when compared to non-tumorigenic breast epithelial cells (Figure 1(c)) (BT-549: 5.5-fold, p < .01, n = 3), (MDA-MB-231: 3.4-fold, p < .01, n = 3), (HCC1806: NS, p = .878, n = 3), (MDA-MB-468: 2.5-fold, p < .05, n = 3). Together, these data show that phosphorylated STAT3 (Tyr705) was constitutively activated in the majority of human TNBC cell lines except in the basal-like 2 subtype (HCC1806), compared to non-tumorigenic breast cells.
Figure 1.
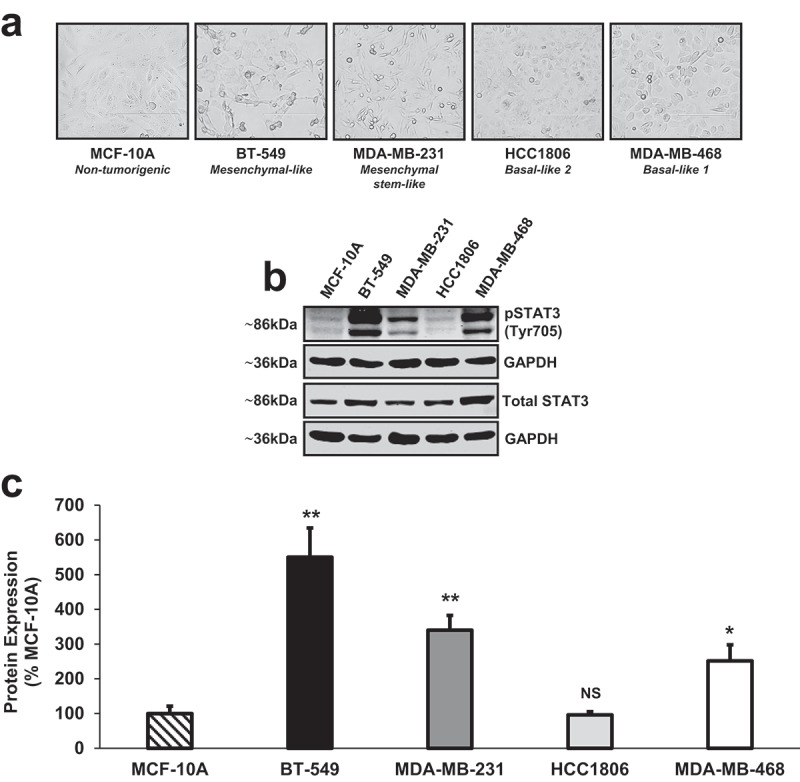
Endogenous STAT3 expression levels across four different TNBC subtypes: BT-549 (mesenchymal-like), MDA-MB-231 (mesenchymal stem-like), HCC1806 (basal-like 2), MDA-MB-468 (basal-like 1).
(a) Phase contrast images captured at 20x objective using the EVOS FL microscope (transmitted setting) depict the morphologies of a non-tumorigenic human mammary epithelial cell line (MCF-10A) and four human TNBC cell lines representative of different molecular subtypes: BT-549 (mesenchymal-like), MDA-MB-231 (mesenchymal stem-like), HCC1806 (basal-like 2), MDA-MB-468 (basal-like 1). (b) Western blot analysis revealed that endogenous phosphorylated STAT3 (Tyr705) protein levels were constitutively activated in three of the four human TNBC cell lines examined (BT-549, MDA-MB-231, MDA-MB-468) as compared to non-tumorigenic breast epithelial cells (MCF-10A). Basal expression of phosphorylated STAT3 (Tyr705) in HCC1806 TNBC cells was not significantly different from phosphorylated STAT3 (Tyr705) expression in MCF-10A cells. GAPDH is shown as a control for equal loading. (c) Densitometry analysis was performed using a LI-COR imager. Quantitatively, phosphorylated STAT3 (Tyr705) and total STAT3 were each normalized to their respective loading controls (GAPDH). Then, a ratio of phosphorylated STAT3 (Tyr705) to total STAT3 was calculated. Data are reported as % MCF-10A. All TNBC cell lines, except HCC1806 cells, showed a significant increase in phosphorylated STAT3 (Tyr705) protein expression when compared to non-tumorigenic mammary epithelial cells (BT-549: 5.5-fold, p < .01, n = 3), (MDA-MB-231: 3.4-fold, p < .01, n = 3), (HCC1806: NS, p = .878, n = 3), (MDA-MB-468: 2.5-fold, p < .05, n = 3).
Inhibition of phosphorylated STAT3 (Tyr705) by pimozide, a FDA-approved antipsychotic
To test the efficacy of pimozide as a STAT3 inhibitor in TNBC, we treated BT-549, MDA-MB-231, HCC1806, and MDA-MB-468 cells with vehicle or increasing concentrations of pimozide. Protein expression of phosphorylated STAT3 (Tyr705) and total STAT3 was then assessed by Western blot (Figure 2(a)). Quantitatively, compared to vehicle control, 10 μM and 20 μM doses of pimozide significantly inhibited the phosphorylation of STAT3 (Tyr705) without affecting total STAT3 protein levels in BT-549, MDA-MB-231, and MDA-MB-468 cells (Figure 2(b)) (BT-549: 5 μM Pimozide, NS, p = .151, n = 3; 10 μM Pimozide, 2.2-fold, p < .01, n = 3; 20 μM Pimozide, 2.3-fold, p < .05, n = 3), (MDA-MB-231: 5 μM Pimozide, NS, p = .281, n = 3; 10 μM Pimozide, 1.9-fold, p < .05, n = 3; 20 μM Pimozide, 3.8-fold, p < .01, n = 3), (MDA-MB-468: 5 μM Pimozide, NS, p = .994, n = 3; 10 μM Pimozide, 2.2-fold, p < .05, n = 3; 20 μM Pimozide, 7.4-fold, p < .01, n = 3). STAT3 phosphorylation (Tyr705) remained unaffected by pimozide treatment in HCC1806 cells (Figure 2(b)) (HCC1806: 5 μM Pimozide, NS, p = .622, n = 3; 10 μM Pimozide, NS, p = .780, n = 3; 20 μM Pimozide, NS, p = .838, n = 3). Qualitatively, as assessed by immunofluorescence, phosphorylated STAT3 (Tyr705) nuclear expression was diminished in BT-549, MDA-MB-231, and MDA-MB-468 cells in response to 10 μM pimozide treatment when compared to vehicle-stimulated counterparts, but was unchanged in HCC1806 cells (Figure 2(c)). These data suggest that TNBC cell lines with constitutively activated phosphorylated STAT3 (Tyr705) expression (BT-549, MDA-MB-231, MDA-MB-468) are more sensitive to pimozide treatment than TNBC cell lines with lower endogenous phosphorylated STAT3 (Tyr705) expression, such as HCC1806, which demonstrated resistance.
Figure 2.
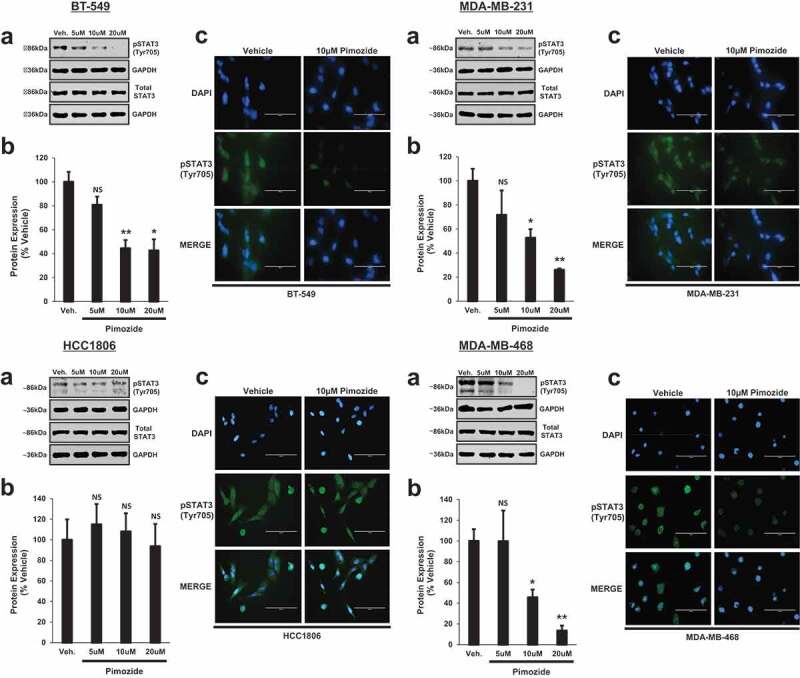
Inhibition of phosphorylated STAT3 (Tyr705) by pimozide, a FDA-approved antipsychotic.
(a) Western blot analysis shows phosphorylated (Tyr705) and total STAT3 protein expression in BT-549, MDA-MB-231, HCC1806, MDA-MB-468 TNBC cells upon treatment with vehicle (DMSO), 5 μM, 10 μM, or 20 μM pimozide for 24 h. Final DMSO concentration was kept at less than 0.2%. GAPDH is shown as a loading control. (b) Using a LI-COR imager, densitometry was used to quantitate the inhibition of STAT3 by pimozide. The ratio of phosphorylated STAT3 (Tyr705) to total STAT3 was calculated upon normalizing to respective loading controls. Data are reported as % vehicle. Significant inhibition of STAT3 phosphorylation (Tyr705) in BT-549, MDA-MB-231, and MDA-MB-468 cell lines was observed at 10 μM and 20 μM doses of pimozide as compared to vehicle-treated counterparts. No change in STAT3 phosphorylation (Tyr705) was observed in HCC1806 cells with pimozide treatment (BT-549: 5 μM Pimozide, NS, p = .151, n = 3; 10 μM Pimozide, 2.2-fold, p < .01, n = 3; 20 μM Pimozide, 2.3-fold, p < .05, n = 3), (MDA-MB-231: 5 μM Pimozide, NS, p = .281, n = 3; 10 μM Pimozide, 1.9-fold, p < .05, n = 3; 20 μM Pimozide, 3.8-fold, p < .01, n = 3), (HCC1806: 5 μM Pimozide, NS, p = .622, n = 3; 10 μM Pimozide, NS, p = .780, n = 3; 20 μM Pimozide, NS, p = .838, n = 3), (MDA-MB-468: 5 μM Pimozide, NS, p = .994, n = 3; 10 μM Pimozide, 2.2-fold, p < .05, n = 3; 20 μM Pimozide, 7.4-fold, p < .01, n = 3). (c) Qualitatively, immunofluorescence analysis demonstrated a reduction in phosphorylated STAT3 (Tyr705) nuclear expression in response to 10 μM pimozide treatment compared to vehicle treatment in BT-549, MDA-MB-231, and MDA-MB-468 cells. Nuclear expression of phosphorylated STAT3 (Tyr705) was unchanged in HCC1806 cells upon treatment with pimozide. Images were captured at 40x objective using the EVOS FL microscope (blue: DAPI immunostaining; green: phosphorylated STAT3 (Tyr705) immunostaining). DAPI was used as a nuclear counterstain. Immunofluorescence experiments were performed in triplicate on cells derived from three independent passages.
Dose-dependent cytotoxicity of pimozide in TNBC cells: a 10-μM dose of pimozide is non-cytotoxic to non-tumorigenic mammary epithelial cells
Next, we utilized a caspase-3/7 cleavage assay to determine the presence of apoptosis following pimozide treatment in all 4 TNBC molecular subtypes and in non-tumorigenic breast epithelial cells (MCF-10A). As depicted in Figure 3(a), an apoptotic profile was generated upon treatment of cells with vehicle or pimozide. The percentage of live, (early) apoptotic, (late) apoptotic/dead, and dead cells are displayed in each plot. Quantitatively, as depicted in Figure 3(b), the percentage of live cells and total apoptotic cells (early apoptotic + late apoptotic) are shown for each treatment condition. Interestingly, a 10 μM dose of pimozide was found to be non-cytotoxic to BT-549, MDA-MB-231, and HCC1806 TNBC cells, with only minor cytotoxicity observed in MDA-MB-468 TNBC cells (BT-549: 10 μM Pimozide, live cells, NS, p = .298, n = 3; total apoptotic cells, NS, p = .803, n = 3), (MDA-MB-231: 10 μM Pimozide, live cells, NS, p = .089, n = 3; total apoptotic cells, NS, p = .154, n = 3), (HCC1806: 10 μM Pimozide, live cells, NS, p = .121, n = 3; total apoptotic cells, NS, p = .078, n = 3), (MDA-MB-468: 10 μM Pimozide, live cells, 1.2-fold, p < .05, n = 3; total apoptotic cells, 1.7-fold, p < .05, n = 3). Importantly, treatment with 10 μM pimozide was non-cytotoxic to MCF-10A cells, suggesting a lack of off-target toxicity to non-tumorigenic breast cells at this dose of pimozide (MCF-10A: 10 μM Pimozide, live cells, NS, p = .297, n = 3; total apoptotic cells, NS, p = .313, n = 3). Furthermore, a higher 20 μM dose of pimozide led to a significant decrease in live cells and a significant increase in total apoptotic cells in the BT-549, MDA-MB-231, and MDA-MB-468 cell lines and, to a lesser extent, HCC1806 and MCF-10A cells (BT-549: 20 μM Pimozide, live cells, 1.4-fold, p < .001, n = 3; total apoptotic cells, 2.1-fold, p < .001, n = 3), (MDA-MB-231: 20 μM Pimozide, live cells, 1.2-fold, p < .001, n = 3; total apoptotic cells, 7.5-fold, p < .001, n = 3), (HCC1806: 20 μM Pimozide, live cells, 1.1-fold, p < .05, n = 3; total apoptotic cells, 1.6-fold, p < .05, n = 3), (MDA-MB-468: 20 μM Pimozide, live cells, 5.1-fold, p < .001, n = 3; total apoptotic cells, 5.2-fold, p < .001, n = 3), (MCF-10A: 20 μM Pimozide, live cells, 1.1-fold, p < .001, n = 3; total apoptotic cells, 2.4-fold, p < .001, n = 3).
Figure 3.
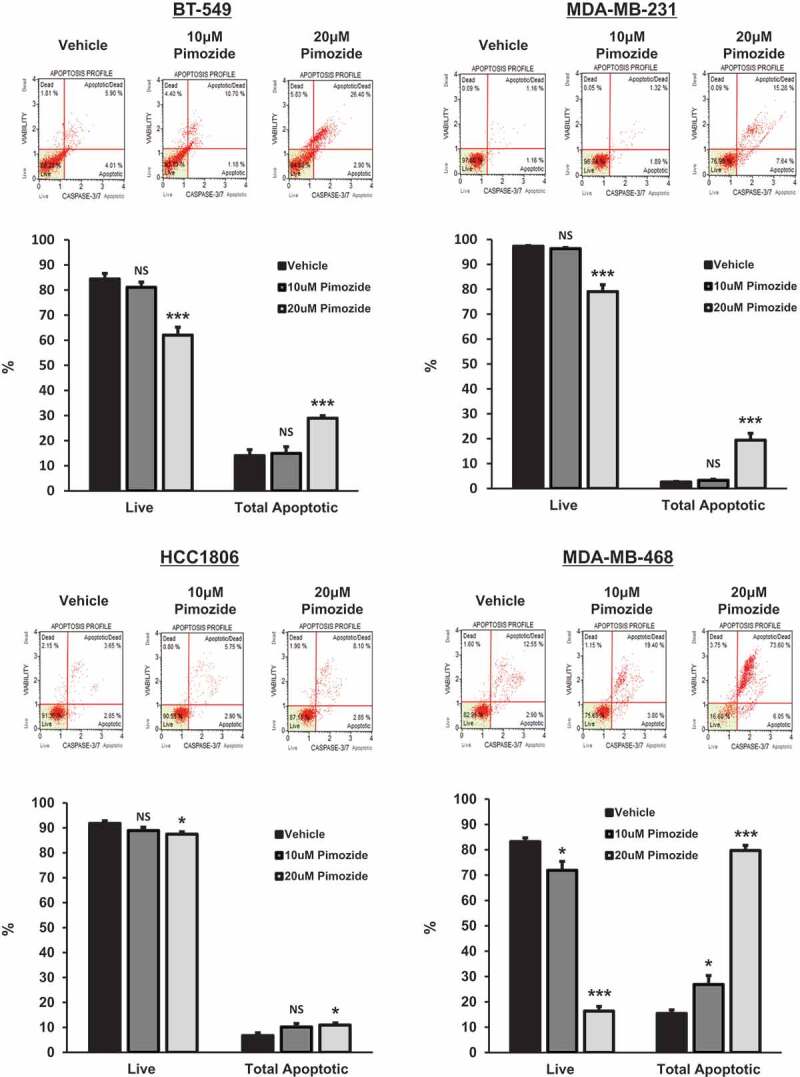
Dose-dependent cytotoxicity of pimozide in TNBC cells: a 10 μM dose of pimozide is non-cytotoxic to non-tumorigenic mammary epithelial cells.
Top panels show caspase-3/7 activation profiles for BT-549, MDA-MB-231, HCC1806, and MDA-MB-468 TNBC cells and MCF-10A non-tumorigenic mammary epithelial cells treated with vehicle (DMSO), 10 μM, or 20 μM pimozide for 48 h. The raw percentage of live, (early) apoptotic, (late) apoptotic/dead, and dead cells in each sample are displayed. Bottom graphs represent quantitatively live and apoptotic populations. Cell lines dosed with 20 μM pimozide for 48 h showed elevated levels of caspase-3/7 activation compared to vehicle-treated counterparts, as evidenced by decreases in live cells and increases in total apoptotic cell populations. A lower 10 μM dose of pimozide was non-cytotoxic to normal MCF-10A cells and all TNBC cell lines when compared to vehicle, with the exception of MDA-MB-468 cells, which showed only minor cytotoxicity. Total apoptotic cell populations refer to the sum of (early) apoptotic and (late) apoptotic/dead cells (BT-549: 10 μM Pimozide, live cells, NS, p = .298, n = 3; total apoptotic cells, NS, p = .803, n = 3; 20 μM Pimozide, live cells, 1.4-fold, p < .001, n = 3; total apoptotic cells, 2.1-fold, p < .001, n = 3), (MDA-MB-231: 10 μM Pimozide, live cells, NS, p = .089, n = 3; total apoptotic cells, NS, p = .154, n = 3; 20 μM Pimozide, live cells, 1.2-fold, p < .001, n = 3; total apoptotic cells, 7.5-fold, p < .001, n = 3), (HCC1806: 10 μM Pimozide, live cells, NS, p = .121, n = 3; total apoptotic cells, NS, p = .078, n = 3; 20 μM Pimozide, live cells, 1.1-fold, p < .05, n = 3; total apoptotic cells, 1.6-fold, p < .05, n = 3), (MDA-MB-468: 10 μM Pimozide, live cells, 1.2-fold, p < .05, n = 3; total apoptotic cells, 1.7-fold, p < .05, n = 3; 20 μM Pimozide, live cells, 5.1-fold, p < .001, n = 3; total apoptotic cells, 5.2-fold, p < .001, n = 3), (MCF-10A: 10 μM Pimozide, live cells, NS, p = .297, n = 3; total apoptotic cells, NS, p = .313, n = 3; 20 μM Pimozide, live cells, 1.1-fold, p < .001, n = 3; total apoptotic cells, 2.4-fold, p < .001, n = 3).
Figure 3.
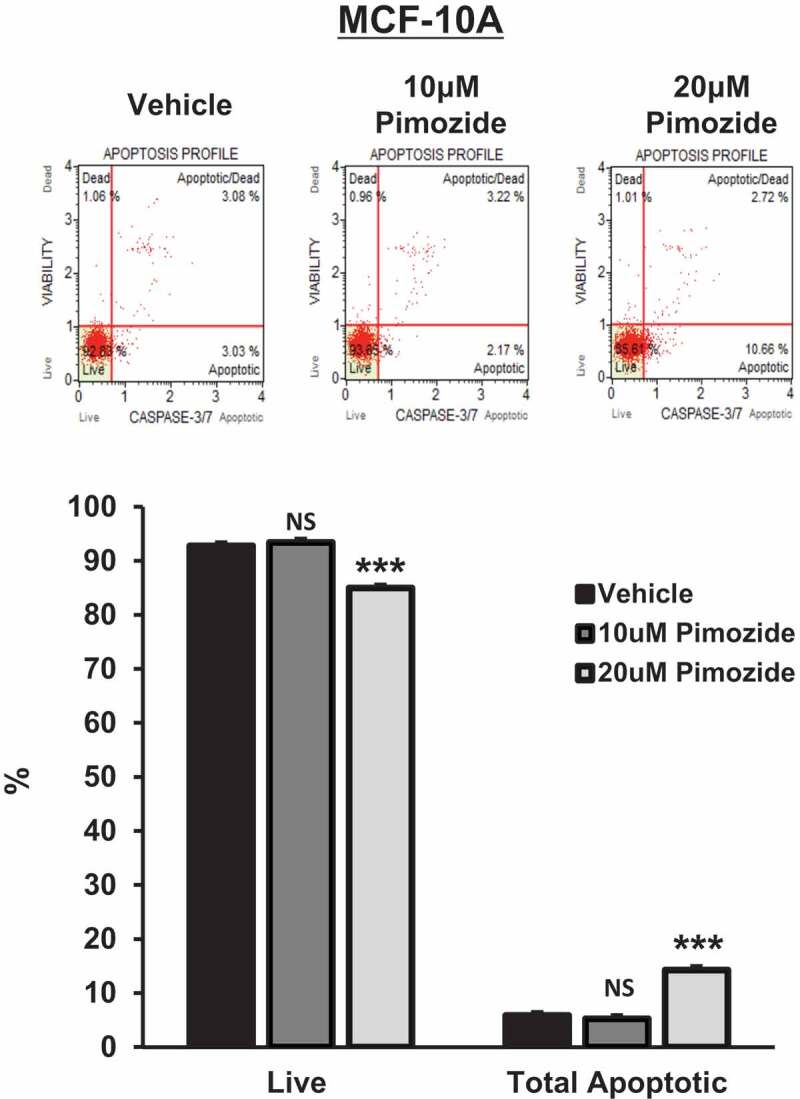
(Continued).
Pimozide inhibits the invasive potential of BT-549, MDA-MB-231, and MDA-MB-468 TNBC cells in vitro
As previously discussed, TNBCs are among the most aggressive breast cancers that exhibit increased metastatic potential.1,3 STAT3 is an important regulator of such oncogenic processes, including invasion and migration.10,13,14 As pimozide inhibited phosphorylated STAT3 (Tyr705) in 75% of TNBC cell lines examined, we set out to determine if this biomarker could predict its efficacy at inhibiting invasion. We selected a 10 μM dose of pimozide to carry out the current and remaining experiments since 10 μM pimozide was the lowest effective dose in inhibiting STAT3 phosphorylation (Tyr705) and was non-cytotoxic to normal breast cells and most TNBC subtypes. Qualitatively, images of the invaded cells after treatment with vehicle or pimozide are displayed in Figure 4(a). BT-549, MDA-MB-231, and MDA-MB-468 cells exposed to 10 μM pimozide showed a significant decrease in invasion compared to vehicle-treated counterparts, whereas the invasive capacity of HCC1806 cells was unaffected by pimozide treatment (Figure 4(b)) (BT-549: 2.4-fold, p < .001, n = 3), (MDA-MB-231: 4.1-fold, p < .001, n = 3), (HCC1806: NS, p = .446, n = 3), (MDA-MB-468: 5.4-fold, p < .001, n = 3). These data demonstrate that pimozide suppresses invasion only in TNBC cell lines sensitive to phosphorylated STAT3 (Tyr705) inhibition.
Figure 4.
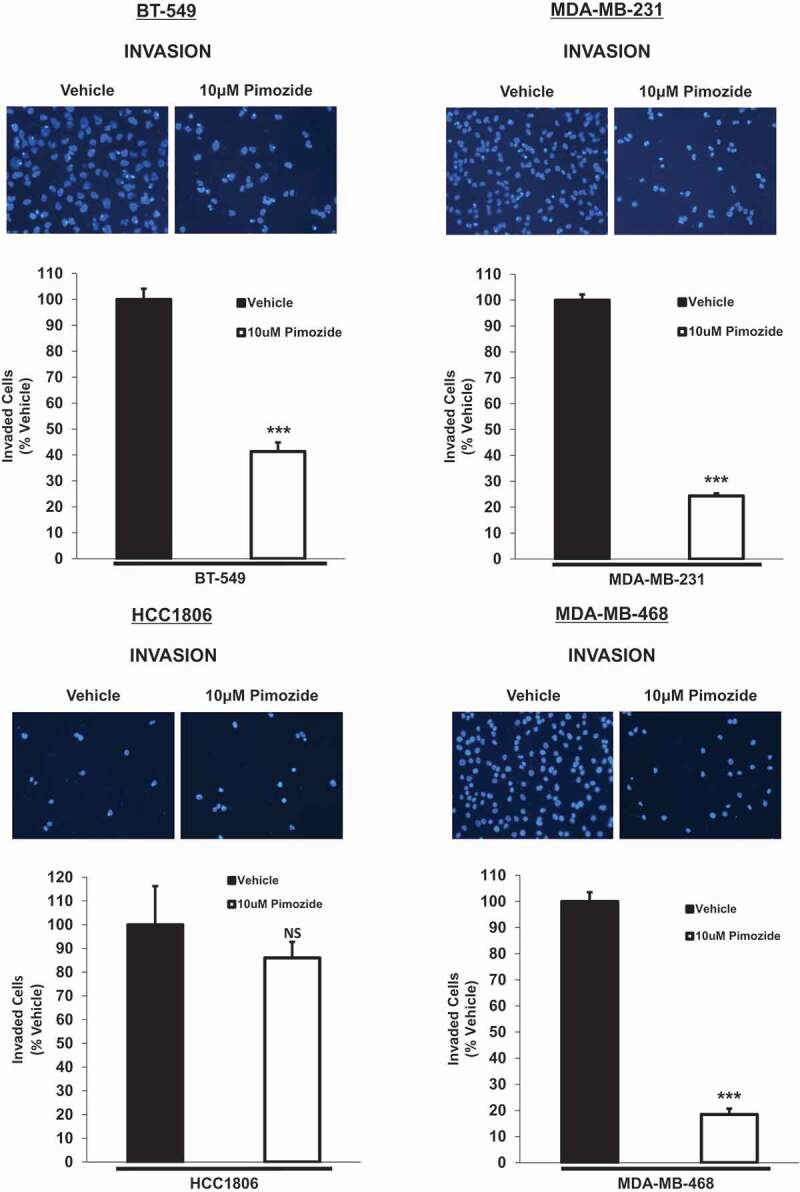
Pimozide inhibits the invasive potential of BT-549, MDA-MB-231, and MDA-MB-468 TNBC cells in vitro, an effect predicted by phosphorylated STAT3 (Tyr705) inhibition.
Qualitatively, transwell invasion assay results depict DAPI-stained invaded cells after 24 h treatment with vehicle (DMSO) or 10 μM pimozide (top images). Images of the invaded cells were captured using the DAPI channel on the EVOS FL microscope at 20x objective. Quantitatively (bottom graphs), the number of invaded cells in five representative fields of view were averaged for each membrane. Data are reported as % vehicle. All TNBC cell lines except HCC1806 cells showed a significant decrease in invasion when treated with 10 μM pimozide for 24 h in comparison to vehicle-stimulated cells (BT-549: 2.4-fold, p < .001, n = 3), (MDA-MB-231: 4.1-fold, p < .001, n = 3), (HCC1806: NS, p = .446, n = 3), (MDA-MB-468: 5.4-fold, p < .001, n = 3).
Pimozide prevents scratch gap closure of BT-549, MDA-MB-231, and MDA-MB-468 TNBC cells in an in vitro wound-healing migration assay
To further characterize the functional role of pimozide in TNBC, we performed a wound healing assay to assess migration. As observed in Figure 5(a), images of the scratch gap before and after treatment with vehicle or pimozide are shown for each cell line. A larger scratch gap area is indicative of less migration, whereas a smaller scratch gap area is suggestive of more migration. Quantitatively, the median scratch gap area measured over time (T = 48 h relative to T = 0) for pimozide treated cells was significantly greater than the median scratch gap area measured over time (T = 48 h relative to T = 0) for vehicle-treated counterparts in BT-549, MDA-MB-231, and MDA-MB-468 cells (Figure 5(b)) (BT-549: Vehicle (T = 48/T = 0), 0.21 median, 0.05 IQR; Pimozide (T = 48/T = 0), 0.50 median, 0.02 IQR; p < .01, n = 3), (MDA-MB-231: Vehicle (T = 48/T = 0), 0.10 median, 0.02 IQR; Pimozide (T = 48/T = 0), 0.48 median, 0.05 IQR; p < .01, n = 3), (MDA-MB-468: Vehicle (T = 48/T = 0), 0.46 median, 0.10 IQR; Pimozide (T = 48/T = 0), 0.79 median, 0.05 IQR; p < .01, n = 3). As predicted, no significant difference in median scratch gap area over time (T = 48 h relative to T = 0) was observed between vehicle and pimozide treated groups in HCC1806 cells (Figure 5(b)) (HCC1806: Vehicle (T = 48/T = 0), 0.42 median, 0.07 IQR; Pimozide (T = 48/T = 0), 0.42 median, 0.07 IQR; NS, p = .936, n = 3). These results are in concordance with the transwell invasion data shown above: pimozide can inhibit migration only in TNBC cells that are sensitive to suppression of STAT3 phosphorylation (Tyr705).
Figure 5.
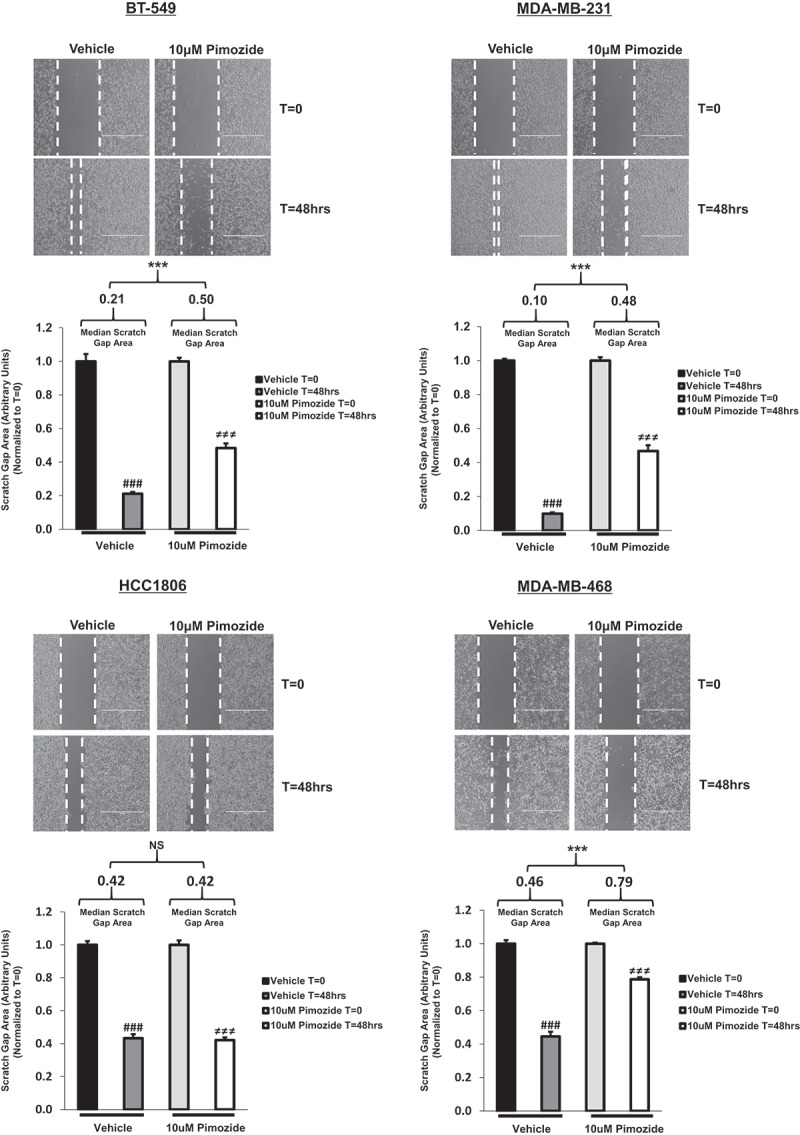
Pimozide prevents scratch gap closure of BT-549, MDA-MB-231, and MDA-MB-468 TNBC cells in an in vitro wound-healing migration assay.
Using the transmitted setting on the EVOS FL microscope, phase contrast images captured at 4x objective depict wound-healing migration assay results (top images). Upon inducing a scratch on a monolayer of confluent cells, TNBC cells were treated with vehicle (DMSO) or 10 μM pimozide for 48 h. Closure of the scratch gap for each treatment condition was monitored over time. The area contained within the scratch gap was measured using the “polygon selections” tool in Image J software. Data are displayed as scratch gap area in arbitrary units, normalized to T = 0 measurements (bottom graphs). Quantitatively, the scratch gap area at T = 48 h relative to T = 0 was calculated independently for each treatment group and reported as median scratch gap area (displayed in graph) with an interquartile range (IQR) (BT-549: Vehicle (T = 48/T = 0), 0.21 median, 0.05 IQR, p < .001, n = 3; Pimozide (T = 48/T = 0), 0.50 median, 0.02 IQR, p < .001, n = 3), (MDA-MB-231: Vehicle (T = 48/T = 0), 0.10 median, 0.02 IQR, p < .001, n = 3; Pimozide (T = 48/T = 0), 0.48 median, 0.05 IQR, p < .001, n = 3), (HCC1806: Vehicle (T = 48/T = 0), 0.42 median, 0.07 IQR, p < .001, n = 3; Pimozide (T = 48/T = 0), 0.42 median, 0.07 IQR, p < .001, n = 3), (MDA-MB-468: Vehicle (T = 48/T = 0), 0.46 median, 0.10 IQR, p < .001, n = 3; Pimozide (T = 48/T = 0), 0.79 median, 0.05 IQR, p < .001, n = 3). In BT-549, MDA-MB-231, and MDA-MB-468 cells, a significant difference in median scratch gap area at T = 48 h relative to T = 0 was found between vehicle and pimozide treated groups (BT-549: p < .01, n = 3), (MDA-MB-231: p < .01, n = 3), (MDA-MB-468: p < .01, n = 3). In HCC1806 cells, no significant difference in median scratch gap area at T = 48 h relative to T = 0 was observed between vehicle- and pimozide-treated groups (HCC1806: p = .936, n = 3). Furthermore, upon observing each treatment condition independently over time, vehicle-treated cells showed a significant difference in scratch gap area from T = 0 to T = 48 h (###p < .001, n = 3) and pimozide-treated cells showed a significant difference in scratch gap area from T = 0 to T = 48 h (≠≠≠p < .001, n = 3).
Pimozide suppresses the migratory potential of BT-549, MDA-MB-231, and MDA-MB-468 TNBC cells in vitro
To expand on our findings generated from the wound healing assay, we treated cells with vehicle or pimozide in transwell migration chambers. Images of the migrated cells for each treatment condition are displayed in Figure 6(a). Quantitative analysis revealed that BT-549, MDA-MB-231, and MDA-MB-468 cells exposed to 10 μM pimozide showed a significant decrease in migration compared to vehicle-treated counterparts (Figure 6(b)) (BT-549: 1.4-fold, p < .001, n = 3), (MDA-MB-231: 1.7-fold, p < .001, n = 3), (MDA-MB-468: 2.2-fold, p < .001, n = 3). At this same timepoint, very few HCC1806 cells migrated upon stimulation with vehicle alone, and it could not be determined if treatment of HCC1806 cells with pimozide altered their ability to migrate due to lack of events (data not shown). The decreased basal migratory and invasive potential of HCC1806 cells observed in our transwell assays coincide with published literature demonstrating that HCC1806 cells invade to a lesser extent and are less aggressive compared to other TNBC cell lines, including MDA-MB-231 and MDA-MB-468 cells.47 However, upon extending the assay timepoint from 16 h to 48 h, treatment of HCC1806 cells with 10 μM pimozide did not alter their ability to migrate (Figure 6(b)) (HCC1806: NS, p = .707, n = 3). These data indicate a positive correlation between suppression of migration upon pimozide treatment and inhibition of phosphorylated STAT3 (Tyr705).
Figure 6.
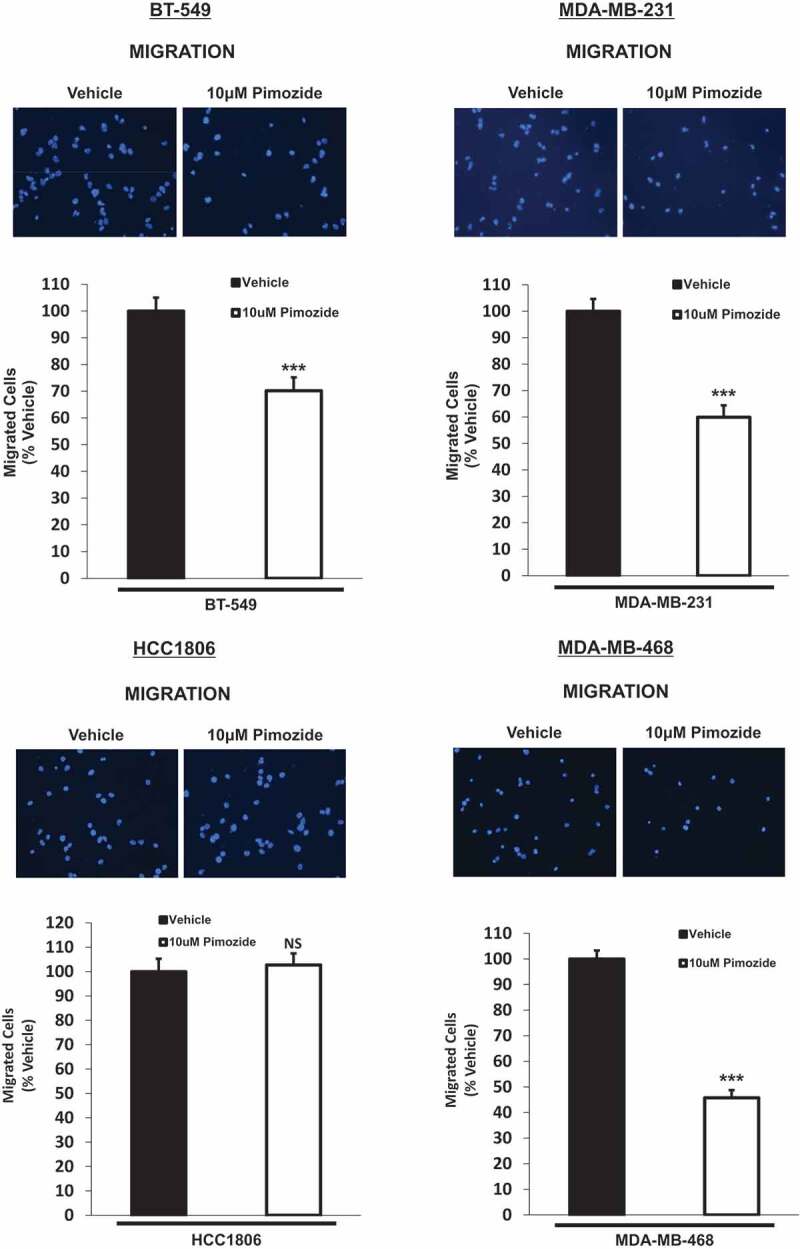
Pimozide suppresses the migratory potential of BT-549, MDA-MB-231, and MDA-MB-468 TNBC cellsin vitro.
Qualitatively, transwell migration assay results depict DAPI-stained migrated cells after 16-h treatment with vehicle (DMSO) or 10 μM pimozide (top images). Images of the migrated cells were captured using the DAPI channel on the EVOS FL microscope at 20x objective. Note: HCC1806 cells (5.0 × 104) were treated with vehicle or 10 μM pimozide for an extended timepoint of 48 h to ensure a reasonable number of migrated cells for quantitative analysis. Quantitatively, the number of migrated cells in five representative fields of view were averaged for each membrane (bottom graphs). Data are reported as % vehicle. All TNBC cell lines except HCC1806 cells showed a significant decrease in migration when treated with 10 μM pimozide in comparison to vehicle-stimulated cells (BT-549: 1.4-fold, p < .001, n = 3), (MDA-MB-231: 1.7-fold, p < .001, n = 3), (HCC1806: NS, p = .707, n = 3), (MDA-MB-468: 2.2-fold, p < .001, n = 3).
Pimozide inhibits downstream STAT3 transcriptional targets involved in metastatic progression in BT-549, MDA-MB-231, and MDA-MB-468 TNBC cells
To further delineate the mechanistic role of pimozide in suppressing the invasive and migratory potential of TNBC cells, we assessed the expression of markers known to be transcriptionally regulated by STAT3. Matrix metalloproteinase-9 (MMP-9) and vimentin are two proteins commonly associated with metastatic processes including migration, invasion, and epithelial-mesenchymal transition (EMT).48–53 As depicted in Figure 7(a), protein expression of MMP-9 and vimentin was assessed by Western blot upon treatment of cells with vehicle or 10 μM pimozide. Quantitatively, MMP-9 expression was significantly reduced in BT-549, MDA-MB-231, and MDA-MB-468 cells exposed to pimozide in comparison to vehicle-stimulated counterparts, whereas no change in MMP-9 expression was observed in HCC1806 cells upon treatment with pimozide (Figure 7(b)) (BT-549: MMP-9, 3.5-fold, p < .01, n = 3), (MDA-MB-231: MMP-9, 2.6-fold, p < .01, n = 3), (HCC1806: MMP-9, NS, p = .204, n = 3), (MDA-MB-468: MMP-9, 8.2-fold, p < .01, n = 3). We were surprised to observe the highest endogenous expression of MMP-9 in the least aggressive TNBC cell line (HCC1806). However, as other published work has demonstrated, it is possible for less aggressive cells to initially undergo EMT, but lose attributes of the EMT-derived phenotype over time.54 Nonetheless, MMP-9 expression remained unaffected by pimozide treatment in HCC1806 cells. Similarly, in BT-549, MDA-MB-231, and MDA-MB-468 cells, protein expression of vimentin was also significantly downregulated in response to pimozide treatment when compared to vehicle-stimulated cells. Endogenous vimentin expression was not detectable in HCC1806 cells. 293T cells were used as a positive control for vimentin expression due to undetectable expression in HCC1806 cells (Figure 7(b)) (BT-549: Vimentin, 2.0-fold, p < .05, n = 3), (MDA-MB-231: Vimentin, 1.4-fold, p < .05, n = 3), (HCC1806: Vimentin, not detected, n = 3), (MDA-MB-468: Vimentin, 13.5-fold, p < .001, n = 3). Perhaps HCC1806 cells do not rely on as many pro-metastatic proteins since these cells invade and migrate to a lesser extent compared to the other TNBC cell lines examined and are overall less aggressive.47 Taken together, these data demonstrate that pimozide treatment suppresses the invasion and migration of TNBC cells, a process that can also be predicted by endogenous phosphorylated STAT3 (Tyr705) expression levels.
Figure 7.
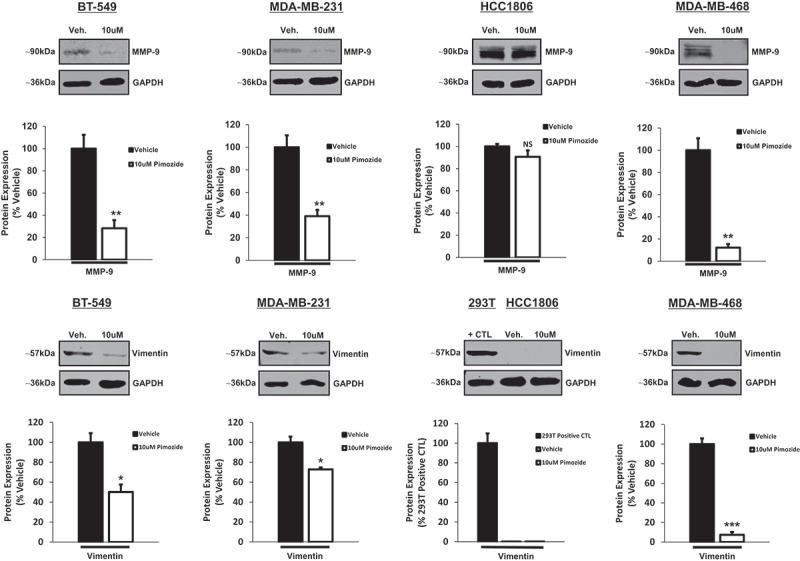
Pimozide inhibits downstream STAT3 transcriptional targets involved in metastatic progression in BT-549, MDA-MB-231, and MDA-MB-468 TNBC cells.
Western blot analysis shows MMP-9 and vimentin protein expression in BT-549, MDA-MB-231, HCC1806, MDA-MB-468 TNBC cells treated with vehicle (DMSO) or 10 μM pimozide for 24 h. 293T cells were utilized as a positive control for vimentin expression in HCC1806 cells. GAPDH is shown as a control for equal loading. Using a LI-COR imager, densitometry analysis was used to quantitate Western bands. Each marker was normalized to its respective loading control. Data are displayed as % vehicle or % positive control (for vimentin expression in HCC1806 cells). Both MMP-9 and vimentin protein expression were significantly suppressed in BT-549, MDA-MB-231, and MDA-MB-468 cell lines after 24-h treatment with a 10 μM dose of pimozide as compared to vehicle-treated counterparts. No change in MMP-9 protein expression was observed in HCC1806 cells treated with vehicle or pimozide. Basal protein expression of vimentin was undetectable in HCC1806 cells (293T cells were used as a positive control for vimentin expression) (BT-549: MMP-9, 3.5-fold, p < .01, n = 3; Vimentin, 2.0-fold, p < .05, n = 3), (MDA-MB-231: MMP-9, 2.6-fold, p < .01, n = 3; Vimentin, 1.4-fold, p < .05, n = 3), (HCC1806: MMP-9, NS, p = .204, n = 3; Vimentin, undetectable expression, n = 3), (MDA-MB-468: MMP-9, 8.2-fold, p < .01, n = 3; Vimentin, 13.5-fold, p < .001, n = 3).
Discussion
The first reports of pimozide displaying anti-cancer properties date back to the early 1990s, where it was demonstrated that pimozide inhibited the growth of MDA-MB-231 and MDA-MB-453 TNBC cells in vitro.34,35 However, a deeper understanding of the involvement of pimozide as an anti-cancer agent in TNBC was most recently explored. It was discovered that pimozide inhibited proliferation, promoted apoptosis, caused cell cycle arrest, induced double-strand DNA breaks, suppressed migration, and inhibited epithelial–mesenchymal transition in MDA-MB-231 TNBC cells in vitro.36 Although intriguing, the effects of pimozide were focused on a specific TNBC subtype (mesenchymal stem-like).46 The molecular heterogeneity in TNBCs is a prominent reason underlying the failure of targeted therapies for this complex disease.55 It is critical to study this heterogeneous disease by testing potential therapies on different molecular subtypes to better align TNBC patients to the most appropriate therapy later on.45,46,55
As such, there is an urgent and unmet need to discover biomarkers that are predictive of response to targeted therapies in TNBC.56,57 Since inhibition of STAT3 phosphorylation (Tyr705) by pimozide was demonstrated in leukemia,41 prostate cancer,42 hepatocellular carcinoma43 and osteosarcoma,44 we hypothesized if STAT3 could be an effective biomarker of response to this treatment in TNBC. We thus incorporated TNBC cell lines representative of four different molecular subtypes in our current report: BT-549 (mesenchymal), MDA-MB-231 (mesenchymal stem-like), HCC1806 (basal-like 2), and MDA-MB-468 (basal-like 1).46 Herein, our data show that TNBC cell lines exhibiting constitutive activation of phosphorylated STAT3 (Tyr705) (BT-549, MDA-MB-231, MDA-MB-468) were most sensitive to pimozide compared to lower expressors (HCC1806) which exhibited resistance to its inhibitory effects on invasion and migration.
These findings could be clinically relevant for the treatment of TNBC patients, as recently, STAT3 was identified as a potential biomarker of response to immunotherapy in non-small cell lung cancer (NSCLC).58 By inducing the upregulation of programmed death-ligand 1 (PD-L1) on the surface of tumor cells and by recruiting immunosuppressive cells of the tumor microenvironment such as myeloid-derived suppressor cells (MDSCs), STAT3 facilitates NSCLC cells to escape immune surveillance and promotes resistance toward immune checkpoint inhibitors.58 As such, STAT3 has emerged as both a biomarker and a target in oncogene-addicted NSCLC. Biomarker guided therapy has value for TNBC treatment management. A prime example of a successful biomarker in TNBC is the use of breast cancer gene (BRCA) mutation selection, a gene involved in homologous recombination to repair DNA double-stranded breaks. TNBC patients harboring germline mutations in BRCA were recently shown to benefit from improved sensitivity to the FDA-approved poly-ADP-ribose polymerase (PARP) inhibitor Olaparib, by rendering DNA repair mechanisms dysfunctional during therapy.59,60 Furthermore, biomarkers predictive of response to epidermal growth factor receptor (EGFR) targeted therapy are under investigation for the treatment of TNBC. EGFR methylation is being evaluated as a biomarker indicative of resistance to anti-EGFR monoclonal antibody therapeutics for TNBC patients.61 Collectively, these studies demonstrate the importance of validating biomarkers that could better stratify TNBC patients most likely to benefit from a given targeted therapy.
Similar to pimozide, metformin, a first-line anti-diabetic drug for the treatment of type 2 diabetes, has shown promise as a repurposed anti-cancer drug for TNBC, through the inhibition of the STAT3 pathway. Published work has revealed, in this case, that lowest STAT3 (Tyr705) expression was linked to highest TNBC growth inhibition by metformin. In fact, overexpression of constitutively active STAT3 in TNBC cell lines (representative of basal-like 1, basal-like 2, mesenchymal stem-like, and unclassified molecular subtypes), reduced the efficacy of metformin as a STAT3 inhibitor whereas lentiviral-mediated stable knockdown of STAT3 enhanced its inhibitory effects on growth inhibition and apoptosis induction.62 These results are very intriguing, and are in contrast to our results with pimozide showing that higher expression levels of phosphorylated STAT3 (Tyr705) predicted an improved efficacy of pimozide treatment in TNBC cells. Although metformin and pimozide both inhibit STAT3 phosphorylation (Tyr705) in TNBC, these divergences could be attributed to different mechanisms of action by which metformin and pimozide inhibit STAT3 phosphorylation (Tyr705), which currently remain undefined.41–44,62 Additional avenues for investigation will involve delineating the molecular mechanism(s) by which pimozide inhibits STAT3 phosphorylation (Tyr705) compared to metformin. While very little work has focused on the exact mechanism of action of pimozide as a STAT3 inhibitor, extensive research efforts have been directed at uncovering its role as an inhibitor of STAT5 phosphorylation (Tyr694). Thus far, published reports indicate that pimozide does not inhibit the BCR/ABL kinase, an upstream activator of STAT5 in CML.38 While progress in identifying the mechanisms underlying phosphorylated STAT3 (Tyr705) inhibition by pimozide in solid and hematological malignancies has been much slower, further studies should examine its role on phosphatases, inhibition of upstream tyrosine kinases, direct targeting of the SH2 domain of STAT3, and inhibition of micro RNAs (miRNAs).17–20 Our results presented here seem to emphasize that although pimozide and metformin both inhibit STAT3 phosphorylation on tyrosine 705, their efficacies seem to differ on the phosphorylation levels of this transcription factor. This should be taken into consideration for future pre-clinical studies that might compare the effects of both drugs together or separate on the progression of TNBC. Our findings further emphasize the need for selection biomarkers for the treatment of TNBC. In fact, the Biotechnology Innovation Organization (BIO) conducted a study that tracked the clinical development success rates of investigational drugs in terms of Likelihood of Approval (LOA) by the FDA. They found that the use of selection biomarkers for patient stratification in clinical trial programs resulted in a three-fold higher LOA from Phase 1 compared to clinical trials that did not utilize selection biomarkers.63 This discovery underscores the importance of clinically validating biomarkers for TNBC to better predict patient response to investigational therapies.
Interestingly, pimozide does not seem to be the only antipsychotic with anti-cancer properties in TNBC. Another first-generation antipsychotic drug of the diphenylbutylpiperidine class recently demonstrated efficacy in TNBC. Approved by the FDA for the treatment of schizophrenia, penfluridol was shown to suppress the growth, invasion, and migration of MDA-MB-231, HCC1806, and 4T1 TNBC cells in vitro.64 Mechanistically, penfluridol treatment reduced the proliferation of TNBC cells by suppressing the integrin α6β4 signaling axis. Orthotopic and intracranial mouse models revealed that penfluridol inhibited TNBC tumor growth in the breast and brain, respectively, through suppression of integrin signaling.64 Considering the structural similarities of penfluridol and pimozide, it would be interesting to examine if phosphorylated STAT3 (Tyr705) could also be a biomarker of response to penfluridol treatment in the different molecular subtypes of TNBC.
The role of STAT3 in driving tumorigenesis is well established.7,10-14 In addition to promoting cancer cell proliferation and survival, STAT3 has been robustly shown to function as a key regulator of metastatic progression, accelerating the invasion and migration of breast cancer cells.13–16 STAT3 was recently shown to behave as a direct modulator of gene signatures enriched for pathways related to invasion.65 Being regarded as an important therapeutic target in TNBC,66 our current report emphasizes endogenous phosphorylated STAT3 (Tyr705) as a predictive biomarker of sensitivity to pimozide treatment with regards to suppression of invasion and migration in vitro. Future directions will involve generating patient-derived xenograft (PDX) mouse models to capture different TNBC subtypes as well as high vs low STAT3 expressors, to further characterize the heterogeneity of this aggressive BCa and response to this antipsychotic and other related analogues. According to our current results, future experiments should be designed using in vivo models to evaluate the anti-cancer efficacy of pimozide in preventing metastatic spread to distant organs and reducing the incidence of tumor metastases that are already established using STAT3 (Tyr705) as a guided biomarker of response. Testing pimozide treatment in already established metastatic TNBC is especially relevant because there are no effective therapies for patients with Stage IV metastatic TNBC, characterized by the presence of life-threatening brain, liver, lung, and/or bone metastases.3 Also of particular interest and as shown in our results, MMP-9, an endopeptidase that degrades the extracellular matrix to facilitate invasion and metastasis, should also be followed in response to pimozide treatment, as its levels correlated with STAT3 inhibition in our system.48,49 This is novel and could be clinically relevant, as recent failures of nonselective MMP inhibitors in clinical trials warrant the need to uncover additional molecules that could inhibit this pathway in TNBC.67 Finally, the role of pimozide in combination therapy with traditional chemotherapy should also be considered and tested using clinically relevant models. For example, the use of pimozide as a targeted STAT3 inhibitor could sensitize specific TNBC subtypes to chemotherapies, as STAT3 is linked to chemoresistance in TNBC.66 Combination therapy is particularly advantageous5,6 because the dose of chemotherapy as well as the dose of pimozide could be reduced, resulting in less systemic and antipsychotic off-target effects, respectively.
In conclusion, we have characterized phospho-STAT3 (Tyr705) as a potential biomarker of response to pimozide treatment in metastatic TNBC cell lines undergoing rapid invasion and migration in vitro. Our study demonstrates that while most TNBC cells responded to pimozide treatment, those with the lowest amount of phospho-STAT3 (Tyr705) are resistant to the inhibitory effects of pimozide on invasion and migration. Our current study highlights the need to further investigate the value of pre-screening TNBCs for their levels of phospho-STAT3 (Tyr705) before pimozide treatment and following its level as well as the levels of MMP-9 or vimentin as markers of pimozide efficacy. Further validation using patient-derived TNBC models will be required to capture the tumor heterogeneity and metastatic progression and to accurately predict if phospho-STAT3 (Tyr705) could be a reliable predictor of pimozide treatment for TNBC.
Materials and methods
Cell lines
BT-549 cells (Cat#HTB-122, ATCC) were cultured in RPMI-1640 growth medium (Cat#30-2001, ATCC) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Cat#16140071, ThermoFisher Scientific), 0.023IU/mL bovine insulin (Cat#I0516, Sigma-Aldrich), and 1% penicillin/streptomycin (Cat#15140163, ThermoFisher Scientific). MDA-MB-231 cells (Cat#HTB-26, ATCC) were cultured in Dulbecco’s modified eagle medium (DMEM) (Cat#11965118) supplemented with 10% heat-inactivated FBS (Cat#16140071), 1% sodium pyruvate (Cat#11360070), and 1% penicillin/streptomycin (Cat#15140163) (ThermoFisher Scientific). HCC1806 cells (Cat#CRL-2335, ATCC) were cultured in RPMI-1640 growth medium (Cat#30-2001, ATCC) supplemented with 10% heat-inactivated FBS (Cat#16140071) and 1% penicillin/streptomycin (Cat#15140163) (ThermoFisher Scientific). MDA-MB-468 cells (Cat#HTB-132, ATCC) were cultured in Leibovitz’s L-15 growth medium (Cat#30-2008, ATCC) supplemented with 10% heat-inactivated FBS (Cat#16140071) and 1% penicillin/streptomycin (Cat#15140163) (ThermoFisher Scientific). MCF-10A cells (Cat#CRL-10317, ATCC) were cultured in mammary epithelial basal medium (MEBM) (Cat#CC-3151, Lonza) supplemented with 5% horse serum (Cat#16050-122, Invitrogen), 20 ng/mL epidermal growth factor (EGF) (Cat#CC-4136, Lonza), 0.5 mg/mL hydrocortisone (Cat#CC-4136, Lonza), 10 μg/mL insulin (Cat#CC-4136, Lonza), 100 ng/mL cholera toxin (Cat#C8052, Sigma-Aldrich), and 1% penicillin/streptomycin (Cat#15140122, ThermoFisher Scientific). 293T cells (Cat#CRL-3216, ATCC) were cultured in DMEM medium (Cat#11965118) supplemented with 10% FBS (Cat#16140071) and 1% penicillin/streptomycin (Cat#15140163) (ThermoFisher Scientific). All cell lines were incubated at 37°C in a 5% CO2 incubator, except for MDA-MB-468 cells, which were incubated at 37°C in atmospheric air.
Antibodies
Anti-mouse monoclonal antibodies (mAbs) against phosphorylated STAT3 (Tyr705) (Cat#4113S) and total STAT3 (Cat#9139S) were purchased from Cell Signaling Technology. An anti-mouse mAb against MMP-9 (Cat#sc-393859) was purchased from Santa Cruz Biotechnology. An anti-mouse mAb against vimentin (Cat#ab8978) was purchased from abcam. An anti-mouse mAb against glyceradehyde-3-phosphate dehydrogenase (GAPDH) (Cat#10R-2932) was purchased from Fitzgerald Industries International. An IRDye 680RD goat anti-mouse IgG secondary antibody (Cat#926-68070) was purchased from LI-COR.
Western blot
Samples were homogenized in RIPA lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS) supplemented with complete mini protease inhibitor cocktail (Cat#NC0969110, Roche Diagnostics) and phosphatase inhibitor cocktail (Cat#78428, ThermoFisher Scientific). After homogenization, samples were sonicated and centrifuged at 10,000 x rpm for 10 min at 4°C. Supernatant was then collected for measurement of protein concentration using a bicinchoninic acid (BCA) kit (Cat#23225, ThermoFisher Scientific). Samples (50-100 μg) were separated by SDS-PAGE (12% acrylamide) and transferred to a nitrocellulose membrane (Cat#45-004-001, GE Healthcare) for probing. Subsequent wash buffers consisted of 10 mM Tris pH 8.0, 150 mM NaCl, 0.05% Tween 20 (TBS-T). Membranes were blocked in TBS-T supplemented with 5% bovine serum albumin (BSA) (Cat#BP1600-100, ThermoFisher Scientific) or 5% nonfat dry milk (Cat#50-447-778, Quality Biological Inc.) for 1 h at room temperature. The membranes were subsequently incubated with a given primary antibody (1:100 to 1:500 dilution) overnight at 4°C. IRDye 680RD secondary antibodies (1:15,000 dilution) were used to visualize bound primary antibodies (LI-COR). The Odyssey CLx Imaging System was utilized for near-infrared (NIR) fluorescent detection of proteins (LI-COR). Image Studio software version 5 on the Odyssey CLx was used to quantify Western bands (LI-COR).
Immunofluorescence
Briefly, 1.5 × 105 cells were seeded on Poly-L-Lysine coated coverslips (Cat#P4707, Sigma-Aldrich) in 6 well plates (Cat#3516, Corning). Subsequently, cells were treated with vehicle (dimethyl sulfoxide (DMSO)) (Cat#D128-500, ThermoFisher Scientific) or 10 μM pimozide (Cat#P1793, Sigma-Aldrich) for 24 h. Cells were then fixed in 4% paraformaldehyde (Cat#159-SP, Electron Microscopy Sciences) for 15 min at room temperature. Fixed cells were washed three times with Dulbecco’s phosphate-buffered saline (DPBS) (1x) and then permeabilized in 100% methanol for 10 min at −20°C. Following another DPBS (1x) wash, cells were blocked in blocking buffer (DPBS 1x, 5% normal serum, 0.3% Triton X-100) for 60 min at room temperature. Cells were then incubated with a given primary antibody (diluted 1:100 in DPBS 1x, 1% BSA, 0.3% Triton X-100) for 1 h at 37°C. After three DPBS (1x) washes, cells were incubated with an Alexa Fluor 647 conjugated goat anti-mouse secondary antibody (diluted 1:500 in DPBS 1x, 1% BSA, 0.3% Triton X-100) for 1 h at 37°C (Cat#A-21236, ThermoFisher Scientific). Cells were washed three times with DPBS (1x) before being mounted with ProLong Gold Antifade with 4ʹ6-diamidino-2-phenylindole (DAPI) (Cat#P36931, ThermoFisher Scientific). Using the EVOS FL microscope, images were acquired at 40x objective using the Cy5.5 light cube (ThermoFisher Scientific).
Caspase-3/7 assay
Briefly, 1.5 × 106 cells were seeded in 10 cm cell culture dishes. Cells were treated with vehicle (DMSO) (Cat#D128-500, ThermoFisher Scientific), 10 μM pimozide, or 20 μM pimozide (Cat#P1793, Sigma-Aldrich) for 48 h. After 48 h of drug treatment, adherent and non-adherent cells were collected, counted using a Cell Count and Viability solution (Cat#MCH600103), and analyzed using a Caspase-3/7 assay kit (Cat#MCH100108) on the MUSE Cell Analyzer (Sigma-Aldrich). More specifically, 5 μL of the Caspase-3/7 working solution was added to 50 μL of cell suspension at a concentration of 1 × 106 cells/mL and incubated for 30 min at 37°C. After 30 min, 150 μL of the Caspase 7-AAD working solution was added to the cell suspension and incubated for 5 min in the dark at room temperature before being analyzed on the MUSE Cell Analyzer. An apoptotic profile was generated for each sample, displaying the percentage of live, apoptotic, apoptotic/dead, and dead cells.
Transwell invasion assay
Briefly, 7.5 × 104 cells were resuspended in 500 μL of complete growth medium containing a reduced serum concentration of 0.5% FBS. Culture medium was supplemented with vehicle (DMSO) (Cat#D128-500, ThermoFisher Scientific) or 10 μM pimozide (Cat#P1793, Sigma-Aldrich). Cells were then seeded in transwell invasion chambers with membranes (8 μm pore size) pre-coated with growth factor reduced matrigel (Cat#354483, Corning). Invasion chambers were placed in 24 well plates (Cat#353047, Corning), with each well containing 750 μL of complete growth medium at a normal serum concentration of 10% FBS. Chambers were incubated at 37°C/5% CO2 for 24 h to allow cells to invade. The following day, the inner membranes of the chambers were washed with DPBS (1x) and gently scraped with cotton swabs to remove any non-invaded cells and the layer of matrigel. Invaded cells located on the outer membrane were fixed with 100% methanol at room temperature for 10 min, rinsed with distilled water, and left to dry before being mounted with Vectashield mounting medium with DAPI (Cat#H-1200, Vector Laboratories). Using the EVOS FL microscope (ThermoFisher Scientific), five representative fields of view of the DAPI-stained invaded cells on each membrane were captured at 20x objective.
Wound-healing migration assay
Cells were seeded in 10 cm culture dishes and left to grow until 100% confluent. Once confluency was achieved, a sterile 200 μL pipette tip was used to induce a scratch on the monolayer of cells. Non-adherent cells were subsequently removed by washing twice with DPBS (1x). Complete medium containing vehicle (DMSO) (Cat#D128-500, ThermoFisher Scientific) or 10 μM pimozide (Cat#P1793, Sigma-Aldrich) was added to the cells. Images of the scratches were captured at T = 0 and T = 48 h using the transmitted setting on the EVOS FL microscope at 4x objective. Closure of the scratch gap was quantified by measuring the area contained within the scratch, using the “polygon selections” tool in ImageJ software (NIH).
Transwell migration assay
Briefly, 2.5 × 104 cells were resuspended in 500 μL of complete growth medium containing a reduced serum concentration of 0.5% FBS. Culture medium was supplemented with vehicle (DMSO) (Cat#D128-500, ThermoFisher Scientific) or 10 μM pimozide (Cat#P1793, Sigma-Aldrich). Cells were then seeded in transwell migration chambers with membranes of 8 μm pore size (Cat#08-771-21, ThermoFisher Scientific). Migration chambers were placed in 24 well plates (Cat#353047, Corning), with each well containing 750 μL of complete growth medium at a normal serum concentration of 10% FBS. Chambers were incubated at 37°C/5% CO2 for 16 h to allow cells to migrate. The inner membranes of the chambers were washed with DPBS (1x) and gently scraped with cotton swabs to remove any non-migratory cells. Migrated cells located on the outer membrane were fixed with 100% methanol at room temperature for 10 min, rinsed with distilled water, and left to dry before being mounted with Vectashield mounting medium with DAPI (Cat#H-1200, Vector Laboratories). Using the EVOS FL microscope (ThermoFisher Scientific), five representative fields of view of the DAPI-stained migrated cells on each membrane were captured at 20x objective.
Statistical analysis
Comparisons involving only two groups were analyzed using a two-tailed t-test. Comparisons between more than two groups were performed using a one-way ANOVA followed by post-hoc adjusted tests for multiple comparisons. Data were reported as mean ± standard error of the mean (SEM). Differences in fold change between groups were analyzed using a Wilcoxon two-sample test. Median and interquartile range (IQR) were reported. Statistical significance was reached at p < .05 (*)(#)(≠), p < .01 (**)(##)(≠≠), and p < .001 (***)(###)(≠≠≠). N = 3 is defined as three independent experiments performed using different passages of cells. Statistical analyses were performed using Excel and SAS version 9.4 (SAS Institute Inc.).
Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Ismail-Khan R, Bui MM.. A review of triple-negative breast cancer. Cancer Control. 2010;17:173–176. doi: 10.1177/107327481001700305. [DOI] [PubMed] [Google Scholar]
- 2.Uscanga-Perales GI, Santuario-Facio SK, Ortiz-Lopez R. Triple negative breast cancer: deciphering the biology and heterogeneity. Med Universitaria. 2016;18:105–114. doi: 10.1016/j.rmu.2016.05.007. [DOI] [Google Scholar]
- 3.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 4.Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429–4434. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 5.Wahba HA, El-Hadaad HA. Current approaches in treatment of triple-negative breast cancer. Cancer Biol Med. 2015;12:106–116. doi: 10.7497/j.issn.2095-3941.2015.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeichner SB, Terawaki H, Gogineni K. A review of systemic treatment in metastatic triple-negative breast cancer. Breast Cancer. 2016;10:25–36. doi: 10.4137/BCBCR.S32783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banerjee K, Resat H. Constitutive activation of STAT3 in breast cancer cells: a review. Int J Cancer. 2016;138:2570–2578. doi: 10.1002/ijc.v138.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galoczova M, Coates P, Vojtesek B. STAT3, stem cells, cancer stem cells and p63. Cell Mol Biol Lett. 2018;23:12. doi: 10.1186/s11658-018-0078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marotta LLC, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24-stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121:2723–2735. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carpenter RL, Lo H-W. STAT3 target genes relevant to human cancers. Cancers. 2014;6:897–925. doi: 10.3390/cancers6020897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alvarez JV, Febbo PG, Ramaswamy S, Loda M, Richardson A, Frank DA. Identification of a genetic signature of activated signal transducer and activator of transcription 3 in human tumors. Cancer Res. 2005;65:5054–5062. doi: 10.1158/0008-5472.CAN-04-4281. [DOI] [PubMed] [Google Scholar]
- 12.Walker SR, Xiang M, Frank DA. Distinct roles of STAT3 and STAT5 in the pathogenesis and targeted therapy of breast cancer. Mol Cell Endocrinol. 2014;382:616–621. doi: 10.1016/j.mce.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teng Y, Ross JL, Cowell JK. The involvement of JAK-STAT3 in cell motility, invasion, and metastasis. JAKSTAT. 2014;3:e28086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kamran MZ, Patil P, Gude RP. Role of STAT3 in cancer metastasis and translational advances. Biomed Res Int. 2013;2013:421821. doi: 10.1155/2013/421821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dechow TN, Pedranzini L, Leitch A, Leslie K, Gerald WL, Linkov I, Bromberg JF. Requirement of matrix metalloproteinase-9 for the transformation of human mammary epithelial cells by Stat3-C. Proc Natl Acad Sci USA. 2004;101:10602–10607. doi: 10.1073/pnas.0404100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Snyder M, Huang X-Y, JJ Z. Signal transducers and activators of transcription 3 (STAT3) directly regulates cytokine-induced fascin expression and is required for breast cancer cell migration. J Biol Chem. 2011;286:38886–38893. doi: 10.1074/jbc.M111.286245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson DE, O-Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15:234–248. doi: 10.1038/nrclinonc.2018.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furqan M, Akinleye A, Mukhi N, Mittal V, Chen Y, Liu D. STAT inhibitors for cancer therapy. J Hematol Oncol. 2013;6:90. doi: 10.1186/1756-8722-6-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miklossy G, Hilliard TS, Turkson J. Therapeutic modulators of STAT signalling for human diseases. Nat Rev Drug Discov. 2013;12:611–629. doi: 10.1038/nrd4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiong A, Yang Z, Shen Y, Zhou J, Shen Q. Transcription factor STAT3 as a novel molecular target for cancer prevention. Cancers. 2014;6:926–957. doi: 10.3390/cancers6020926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, Doig A, Guilliams T, Latimer J, McNamee C, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2018;18:41–58. doi: 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- 22.Gupta SC, Sung B, Prasad S, Webb LJ, Aggarwal BB. Cancer drug discovery by repurposing: teaching new tricks to old dogs. Trends Pharmacol Sci. 2013;34:508–517. doi: 10.1016/j.tips.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 23.Oprea TI, Bauman JE, Bologa CG, Buranda T, Chigaev A, Edwards BS, Jarvik JW, Gresham HD, Haynes MK, Hjelle B, et al. Drug repurposing from an academic perspective. Drug Discov Today Ther Strateg. 2011;8:61–69. doi: 10.1016/j.ddstr.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sleire L, Forde-Tislevoll HE, Netland IA, Leiss L, Skeie BS, Enger PO. Drug repurposing in cancer. Pharmacol Res. 2017;124:74–91. doi: 10.1016/j.phrs.2017.07.013. [DOI] [PubMed] [Google Scholar]
- 25.Telleria CM. Drug repurposing for cancer therapy. J Cancer Sci Ther. 2012;4:ix–xi. doi: 10.4172/1948-5956.1000e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sultana A, McMonagle T. Pimozide for schizophrenia or related psychoses. Cochrane Database Syst Rev. 2000;3:CD001949. [DOI] [PubMed] [Google Scholar]
- 27.Strange PG, Neve K. Dopamine receptors. Tocris Biosci Sci Rev Ser. 2013; 1–11. Available from: http://www.komabiotech.co.kr/www/techniques/pdf/tocris_review_dopamine-receptors-review-2019-web.pdf. [Google Scholar]
- 28.Regeur L, Pakkenberg B, Fog R, Pakkenberg H. Clinical features and long-term treatment with pimozide in 65 patients with Gilles de la Tourette’s syndrome. J Neurol Neurosurg Psychiatry. 1986;49:791–795. doi: 10.1136/jnnp.49.7.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quezada J, Coffman KA. Current approaches and new developments in the pharmacological management of Tourette syndrome. CNS Drugs. 2018;32:33–45. doi: 10.1007/s40263-017-0486-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fako V, Yu Z, Henrich CJ, Ransom T, Budhu AS, Wang XW. Inhibition of wnt/β-catenin signaling in hepatocellular carcinoma by an antipsychotic drug pimozide. Int J Biol Sci. 2016;12:768–775. doi: 10.7150/ijbs.14718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jandaghi P, Najafabadi HS, Bauer AS, Papadakis AI, Fassan M, Hall A, Monast A, von Knebel Doeberitz M, Neoptolemos JP, Costello E, et al. Expression of DRD2 is increased in human pancreatic ductal adenocarcinoma and inhibitors slow tumor growth in mice. Gastroenterology. 2016;151:1218–1231. doi: 10.1053/j.gastro.2016.08.040. [DOI] [PubMed] [Google Scholar]
- 32.Chen J, Dexheimer TS, Ai Y, Liang Q, Villamil MA, Inglese J, Maloney DJ, Jadhav A, Simeonov A, Zhuang Z. Selective and cell-active inhibitors of the USP1/UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem Biol. 2011;18:1390–1400. doi: 10.1016/j.chembiol.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wiklund ED, Catts VS, Catts SV, Ng TF, Whitaker NJ, Brown AJ, Lutze-Mann LH. Cytotoxic effects of antipsychotic drugs implicate cholesterol homeostasis as a novel chemotherapeutic target. Int J Cancer. 2010;126:28–40. doi: 10.1002/ijc.v126:1. [DOI] [PubMed] [Google Scholar]
- 34.Strobl JS, Kirkwood KL, Lantz TK, Lewine MA, Peterson VA, Worley III JF. Inhibition of human breast cancer cell proliferation in tissue culture by the neuroleptic agents pimozide and thioridazine. Cancer Res. 1990;50:5399–5405. [PubMed] [Google Scholar]
- 35.Strobl JS, Peterson VA. Tamoxifen-resistant human breast cancer cell growth: inhibition by thioridazine, pimozide and the calmodulin antagonist, W-13. J Pharmacol Exp Ther. 1992;263:186–193. [PubMed] [Google Scholar]
- 36.Dakir E-H, Pickard A, Srivastava K, McCrudden CM, Gross SR, Lloyd S, Zhang S-D, Margariti A, Morgan R, Rudland PS, et al. The anti-psychotic drug pimozide is a novel chemotherapeutic for breast cancer. Oncotarget. 2018;9:34889–34910. doi: 10.18632/oncotarget.v9i79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mistry H, Hsieh G, Buhrlage SJ, Huang M, Park E, Cuny GD, Galinsky I, Stone RM, Gray NS, D’Andrea AD, et al. Small molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol Cancer Ther. 2013;12:2651–2662. doi: 10.1158/1535-7163.MCT-13-0103-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nelson EA, Walker SR, Weisberg E, Bar-Natan M, Barrett R, Gashin LB, Terrell S, Klitgaard JL, Santo L, Addorio MR, et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood. 2011;117:3421–3429. doi: 10.1182/blood-2009-11-255232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nelson EA, Walker SR, Xiang M, Weisberg E, Bar-Natan M, Barrett R, Liu S, Kharbanda S, Christie AL, Nicolais M, et al. The STAT5 inhibitor pimozide displays efficacy in models of acute myelogenous leukemia driven by FLT3 mutations. Genes Cancer. 2012;3:503–511. doi: 10.1177/1947601912466555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simpson HM, Furusawa A, Sadashivaiah K, Civin CI, Banerjee A. STAT5 inhibition induces TRAIL/DR4 dependent apoptosis in peripheral T-cell lymphoma. Oncotarget. 2018;9:16792–16806. doi: 10.18632/oncotarget.v9i24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nelson EA, Sharma SV, Settleman J, Frank DA. A chemical biology approach to developing STAT inhibitors: molecular strategies for accelerating clinical translation. Oncotarget. 2011;2:518–524. doi: 10.18632/oncotarget.v2i6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou W, Chen M-K, Yu H-T, Zhong Z-H, Cai N, Chen G-Z, Zhang P, Chen -J-J. The antipsychotic drug pimozide inhibits cell growth in prostate cancer through suppression of STAT3 activation. Int J Oncol. 2016;48:322–328. doi: 10.3892/ijo.2015.3229. [DOI] [PubMed] [Google Scholar]
- 43.Chen -J-J, Cai N, Chen G-Z, Jia -C-C, Qiu D-B, Du C, Liu W, Yang Y, Long Z-J, Zhang Q. The neuroleptic drug pimozide inhibits stem-like cell maintenance and tumorigenicity in hepatocellular carcinoma. Oncotarget. 2017;8:17593–17609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cai N, Zhou W, Ye -L-L, Chen J, Liang Q-N, Chang G, Chen -J-J. The STAT3 inhibitor pimozide impedes cell proliferation and induces ROS generation in human osteosarcoma by suppressing catalase expression. Am J Transl Res. 2017;9:3853–3866. [PMC free article] [PubMed] [Google Scholar]
- 45.Lehmann BD, Pietenpol JA, Tan AR. Triple-negative breast cancer: molecular subtypes and new targets for therapy. Am Soc Clin Oncol Educ Book. 2015:e31–9. doi: 10.14694/EdBook_AM.2015.35.e31 [DOI] [PubMed] [Google Scholar]
- 46.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiu H-W, Lin H-Y, Tseng I-J, Lin Y-F. OTUD7B upregulation predicts a poor response to paclitaxel in patients with triple-negative breast cancer. Oncotarget. 2018;9:553–565. doi: 10.18632/oncotarget.23074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Merdad A, Karim S, Schulten H-J, Dallol A, Buhmeida A, Al-Thubaity F, Gari MA, Chaudhary AG, Abuzenadah AM, Al-Qahtani MH. Expression of Matrix Metalloproteinases (MMPs) in primary human breast cancer: MMP-9 as a potential biomarker for cancer invasion and metastasis. Anticancer Res. 2014;34:1355–1366. [PubMed] [Google Scholar]
- 49.Mehner C, Hockla A, Miller E, Ran S, Radisky DC, Radisky ES. Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer. Oncotarget. 2014;5:2736–2749. doi: 10.18632/oncotarget.v5i9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jang SY, Kim A, Kim JK, Kim C, Cho Y-H, Kim J-H, Kim CH, Lee J-Y. Metformin inhibits tumor cell migration via down-regulation of MMP9 in tamoxifen-resistant breast cancer cells. Anticancer Res. 2014;34:4127–4134. [PubMed] [Google Scholar]
- 51.Battaglia RA, Delic S, Herrmann H, Snider NT. Vimentin on the move: new developments in cell migration. F1000Res. 2018: 7. F1000 Faculty Rev-1796. doi: 10.12688/f1000research.15967.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu C-Y, Lin -H-H, Tang M-J, Wang Y-K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget. 2015;6:15966–15983. doi: 10.18632/oncotarget.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68:3033–3046. doi: 10.1007/s00018-011-0735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tester AM, Ruangpanit N, Anderson RL, Thompson EW. MMP-9 secretion and MMP-2 activation distinguish invasive and metastatic sublines of a mouse mammary carcinoma system showing epithelial-mesenchymal transition traits. Clin Exp Metastasis. 2000;18:553–560. doi: 10.1023/A:1011953118186. [DOI] [PubMed] [Google Scholar]
- 55.Yam C, Mani SA, Moulder SL. Targeting the molecular subtypes of triple negative breast cancer: understanding the diversity to progress in the field. Oncologist. 2017;22:1086–1093. doi: 10.1634/theoncologist.2017-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Du FL, Eckhardt BL, Lim B, Litton JK, Moulder S, Meric-Bernstam F, Gonzalez-Angulo AM, Ueno NT. Is the future of personalized therapy in triple-negative breast cancer based on molecular subtype? Oncotarget. 2015;6:12890–12908. doi: 10.18632/oncotarget.3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lehmann BD, Pietenpol JA. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J Pathol. 2014;232:142–150. doi: 10.1002/path.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Attili I, Karachaliou N, Bonanno L, Berenguer J, Bracht J, Codony-Servat J, Codony-Servat C, Ito M, Rosell R. STAT3 as a potential immunotherapy biomarker in oncogene-addicted non-small cell lung cancer. Ther Adv Oncol. 2018;10:1758835918763744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beniey M, Haque T, Hassan S. Translating the role of PARP inhibitors in triple-negative breast cancer. Oncoscience. 2019;6:287–288. doi: 10.18632/oncoscience.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McCann KE, Hurvitz SA. Advances in the use of PARP inhibitor therapy for breast cancer. Drugs Context. 2018;7:212540. doi: 10.7573/dic.212540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nakai K, Hung MC, Yamaguchi H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am J Cancer Res. 2016;6:1609–1623. [PMC free article] [PubMed] [Google Scholar]
- 62.Deng X-S, Wang S, Deng A, Liu B, Edgerton SM, Lind SE, Wahdan-Alaswad R, Thor AD. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle. 2012;11:367–376. doi: 10.4161/cc.11.2.18813. [DOI] [PubMed] [Google Scholar]
- 63.Thomas DW, Burns J, Audette J, Carroll A, Dow-Hygelund C, Hay M. Clinical development success rates 2006-2015. BIO Ind Anal. 2016; 1–26. Available from https://www.bio.org/sites/default/files/legacy/bioorg/docs/Clinical%20Development%20Success%20Rates%202006-2015%20-%20BIO,%20Biomedtracker,%20Amplion%202016.pdf. [Google Scholar]
- 64.Ranjan A, Gupta P, Srivastava SK. Penfluridol: an antipsychotic agent suppresses metastatic tumor growth in triple negative breast cancer by inhibiting integrin signaling axis. Cancer Res. 2016;76:877–890. doi: 10.1158/0008-5472.CAN-15-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McDaniel JM, Varley KE, Gertz J, Savic DS, Roberts BS, Bailey SK, Shevde LA, Ramaker RC, Lasseigne BN, Kirby MK, et al. Genomic regulation of invasion by STAT3 in triple negative breast cancer. Oncotarget. 2017;8:8226–8238. doi: 10.18632/oncotarget.v8i5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qin JJ, Yan L, Zhang J, Zhang WD. STAT3 as a potential therapeutic target in triple negative breast cancer: a systematic review. J Exp Clin Cancer Res. 2019;38:195. doi: 10.1186/s13046-019-1206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Amar S, Fields GB. Potential clinical implications of recent MMP inhibitor design strategies. Expert Rev Proteomics. 2015;12:445–447. doi: 10.1586/14789450.2015.1069190. [DOI] [PMC free article] [PubMed] [Google Scholar]