

ABSTRACT
Objective: Chimeric antigen receptor T (CAR-T) cell therapy has demonstrated an unprecedented therapeutic efficacy in hematological malignancies; however, its effectiveness in solid tumors remains elusive. In order to enable CAR-T cells more effective to solid tumors, a inverted chimeric cytokine receptor (ICR) was designed, which is consists of the TGF-β extracellular domain, IL-7 receptor intracellular domain, and co-expression on CAR-T cells.
Materials and Methods: We selected prostate specific membrane antigen (PSMA) as a target for CAR-T cells, constructed corresponding effector cells, and verified the anti-tumor activity of this enhanced PSMA-CAR-T cell by a series of repeated target cell stimulation experiments in vitro and the anti-tumor capabilities by using mice xenograft model in vivo.
Results: In vitro experiments showed that co-expression of ICR could significantly enhance sustained anti-tumor capabilities of PSMA-CAR-T cells. Moreover, in vivo experiments also confirmed that the enhanced PSMA-CAR-T cells exhibited significant superior anti-tumor capabilities and could prolong the survival time in the xenograft and PDX models of prostate cancer.
Conclusions: PSMA-CAR-T cells co-expressing ICR can be envisaged as a new therapeutic strategy for prostate cancer and support the translation of this enhanced approach in the clinical setting.
KEYWORDS: Chimeric antigen receptor T, prostate cancer, prostate specific membrane antigen, xenograft, effector cells
Introduction
Prostate cancer (PCa) represents the second most frequently diagnosed malignancy in men worldwide.1 For patients with locally advanced and metastatic prostate cancer, androgen-deprivation therapy (ADT) remains the mainstay palliative treatment and the majority of patients respond to initial therapy; however, recurrence in the lymph nodes and bones is inevitable, leading to castration-resistant prostate cancer (CRPC) and metastatic CRPC. Thus far, there is no recognized effective therapy for metastatic CRPC.2 Recently, adoptive immunotherapy with chimeric antigen receptor-modified T (CAR-T) cells has emerged as a preferred therapeutic modality to treating patients with refractory cancer.The success of CD19 CAR-T cell immunotherapy of B-cell malignancies has captured researchers’ attention in developing CAR-T cell therapy for malignant solid tumors including prostate cancer.3 Since prostate cancer expresses several antigens that have limited or no expression in normal tissues, these tissue-restricted antigens represent a putative target for CAR-T cell therapy.4 Apparently, there are two clinically relevant targets for treating prostate tumors, including prostate-specific membrane antigen (PSMA) and prostate stem cell antigen (PSCA),5 of which PSMA is considerably overexpressed antigen.6 Recently, studies have demonstrated that PSMA-CAR-T cells exhibited significant anti-tumor effects on PSMA-positive tumor cells in vitro and in animal models.7
Four generations of CAR have been investigated in preclinical and ongoing clinical studies. Recently, preclinical studies using the second-generation anti-PSMA CAR-T cells targeting the prostate cancer cells have demonstrated promising results. Nevertheless, tumor growth was inhibited; the tumor-bearing mice remained uncured, indicating that the high in vitro cytotoxicity of second-generation CAR-T cells might not be sufficient to reciprocate similar effects in vivo.8 This hindrance to the efficacy of CAR T-cell therapy for solid tumors might be attributed to a complex and dynamic inhibitory tumor microenvironment, which contains network of immunosuppressive factors, including cytokines (IL-4, 10, TGF-β),9 cells (such as tumor-associated macrophages (TAMs) and regulatory T cells (Tregs)10), and ligands (such as PD-L1) that limit the ability of T cells to eradicate tumor cells.11 Thus, strategies to optimize the efficacy of CAR-T cell immunotherapy including additional modification of CAR-T-cells and combinatorial approaches must be adopted in order to overcome these obstacles and enhance its efficacy. Most recently, it has been shown that the introduction of inverted chimeric cytokine receptor (ICR), which is consists of the IL-4 receptor extracellular domain and IL-7 receptor intracellular domain, can provide positive stimulation and enhance the anti-tumor activity by IL-4, thus allowing CAR-T cells to counter the inhibitory signals present within the tumor microenvironment.12
Accumulating evidence suggests that TGF-β inhibits CD8+ effector T cells and can transform the CD4+ helper T cell phenotype into Treg, thus preventing the anti-tumor immune responses.13 However, IL-7 has been recognized to prolong the in vivo survival of tumor-specific T cells.14 Moreover, in preclinical studies, the anti-tumor effects of T cells can be significantly enhanced by genetically modifying T cells to secrete IL-7 or overexpress IL-7 receptor.15 Therefore, in the present study, we designed and developed a signal transduction receptor, which comprised of the extracellular domain of the TGF-β receptor fused to the intracellular domain of the IL-7 receptor through genetic engineering. Furthermore, CAR-T cells targeted to PSMA were also designed, to facilitate PSMA-CAR-T cells to constitutively express ICR to substantiate the therapeutic effects of the enhanced PSMA-CAR-T cells on prostate cancer. The findings of the study indicated that PSMA-CAR-T cells that constitutively expressing ICR exhibited significant anti-tumor activities against prostate cancer, and the anti-tumor effects were significantly higher than that of the conventional PSMA-CAR-T cells. Consistently, in vivo experiments also demonstrated that PSMA-CAR-T cells constitutively expressing ICR exhibited longer survival time in mice, which could to some extent, improve the therapeutic effectiveness and reduce tumor recurrence. This study demonstrated that PSMA-CAR-T cells constitutively expressing ICR can overcome the limitations of conventional PSMA-CAR-T cell therapy for solid tumors and exhibited significantly enhanced and sustained anti-tumor functions against prostate cancer, thus this approach could provide a new effective strategy for the treatment of prostate cancer.
Materials and method
Cell lines and culture conditions
The study protocol was approved by the Ethics Committee of the First Affiliated Hospital of Xinjiang Medical University (number: 20190012) and written informed consent was obtained from each patient. Blood samples were collected from healthy volunteers. Peripheral blood mononuclear cells (PBMC) were isolated from whole blood samples by gradient centrifugation using LymphoprepTM (Axis-Shield, Norseland). Subsequently, T-cells were enriched through positive selection using human T cell subtype CD3+ sorting magnetic beads (Miltenyi Biotec Inc, Auburn, CA, USA). The isolated T cells were cultured in X-VIVO15 medium (Lonza, Switzerland) supplemented with 5% human AB serum (Valley Biomedical Inc, Winchester, VA, USA.), 10 mM N-acetyl L-cysteine (Sigma Aldrich, St. Louis, MO, USA) and 300 U/mL Human IL-2 (PeproTech, Rocky Hill, CT, USA).
Prostate cancer cell lines (DU145, LAPC-9, LNCaP, PC3, and CWR22RV1) were obtained from the American Type Culture Collection (ATCC). LNCaP cells and LAPC-9 cells were maintained in RPMI-1640 medium (Hyclone, Logan, UT, USA), while DU145 cells, PC3 cells, and CWR22RV1 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) medium (Hyclone). All cell culture media were supplemented with 10% fetal bovine serum (FBS), 2 mmol/L glutamine (Gibco, Gaithersburg, MD, USA), 100 U/mL penicillin and 100 μg/mL streptomycin (Sangong Biotech, Shanghai, China).
Lentiviral engineering of T cells and target cells
48 hours prior to transfection, the isolated T cells were activated using anti-human CD3-/CD28-coated beads (Invitrogen, Carlsbad, CA, USA) at a ratio of 2:1 magnetic bead to T-cells in the T-cell media. Activated T cells were transfected with the engineered virus particles at an MOI of 10, along with the addition of polybrene (Yeasen Biotech, Hong Kong, China) at a final concentration of 5 μg/mL. The cells were centrifuged at 1200 × g for 60 minutes and incubated overnight at 37°C under 5% CO2. 5 days post lentiviral transfection, the modified T cells were harvested; the expression of CAR was measured using flow cytometry and Western blot analysis.
Tumor cells (including PC3 cells, LNCaP cells, and LAPC-9 cells) were grown and harvested in the log-phase, and cells per were plated in a six-well plate containing fresh complete medium and 6 μg/mL polybrene. 50 μL of engineered virus particles was added to each well after the cell reached about 70% confluence. At 24 hours post incubation, the medium was replaced with 2 ml of fresh complete medium. At 5 days post-transduction, a red fluorescent protein (RFP)-positive cells were selected with 1.5 μg/ml puromycin (Beyotime, Beijing, China). The transfection was determined by flow cytometry analysis.
Flow cytometry
For flow cytometry analysis, cells were collected by centrifugation and washed three times with FACS wash buffer (1× PBS containing 0.5% BSA and 0.03% sodium azide). Cell surface staining was performed with an antibody against PSMA (Becton Dickinson (BD), San Jose, CA, USA). CAR expression on CD3 + T cells was detected by a goat-anti-human IgG F(ab′)2 antibody (Jackson ImmunoResearch) and phycoerythrin (PE)-streptavidin (BD Pharmingen). After three rounds of antigen-specific stimulation, the CAR-T cells were detected for the expression of PD-1, TIM-3 and LAG-3 using anti-human CD279 (BD, CA, USA,), anti-human CD366 (eBioscience, CA, USA) and anti-human CD223 (eBioscience, CA, USA) antibodies.
Enzyme-linked immunosorbent assays (ELISA)
For in vitro experiments, 1 × 104 target cells were mixed with effector cells at a ratio of 1:2 in a U-bottom 96-well plate. After 24 hours of incubation, the supernatant was harvested by centrifugation and the released IL-2, IFN-γ and TNF-α were assessed via ELISA assay according to the manufacturer’s instructions (MultiSciences, Beijing, China).
For in vivo experiments, 100 μL of peripheral blood was collected from mice in the experimental group 9 days after T-cell infusion. Cytokines in the serum, such as IFNγ and TNFα, were measured with ELISA kit according to the manufacturer’s protocol (MultiSciences).
Quantitation of T cell proliferation
Tumor cells were treated with 10 μg/mL Mitomycin C (Sigma Aldrich) for 2 hours, and 5 × 104 Mitomycin C-treated tumor cells were co-incubated with effector cells at indicated effector to target (E: T) ratios. The number of effector cells was 1 × 105 in all treatment groups, and the density of T cells was maintained at 5 × 105/mL. After 7 days of co-incubation, newly treated tumor cells were replaced in all groups for a total of three rounds of antigen-specific stimulations. Prior to the third stimulation, CAR-T cells from two groups were pre-stained with carboxy-fluorescein diacetate succinimidyl ester (CFSE) (Invitrogen), and the stained CAR-T cells were co-cultured with LAPC-9 at an effector to target ratio of 2:1 for 96 hours. The CFSE labeled cells in each group was measured by flow cytometry. No exogenous cytokines were added during the entire proliferation experiment.
Xenograft mouse models and living imaging assays
Female NOD-SCID IL-2 receptor gamma null (NSG) mice aged 5–7 weeks were procured from Shanghai Bioduro Biologics Co., Ltd. (China) and bred and kept in a specialized pathogen-free facility with daily monitoring in XX Hospital’s Laboratory Animal Research Center. The animals were cared for in accordance with the Guide for the care and use of laboratory animals. All procedures and animal experiments were approved by the Animal Care and Use Committee of. A xenograft model was established through a subcutaneous injection of LAPC-9-luc cells mixed with Matrigel into different groups of mice. Seven days after inoculation, 1 × 107 control T cells, PSMA-CAR-T cells or PSMA-CAR-T cells co-expressing ICR were administered through caudal veins, followed by the measurement of bioluminescence using Xenogen IVIS Spectrum System (Life Technologies, USA). Animals were monitored daily, tumor size and volume were measured once a week. Mice at a dying condition were euthanized. For the PDX model, tumor-bearing mice with a tumor size of approximately 100 mm3 in were randomized into control or treatment groups (n = 6). 1 × 107 of effector cells were administered through caudal veins of all mice. Body weight and tumor volume were measured every 3 days. When the weight loss exceeded 20% of the initial body weight, or when a mouse was unable to move or the tumor in the control group was ulcerated, the mouse was sacrificed.
Quantitation of T cell counts
To measure T-cells proliferation in vivo, 100 μl blood samples were collected from the mice (from the above mentioned experiment) on the 9th day after the inoculation of tumor cells. T cells were quantified in peripheral blood from each nude mouse, and the numbers of CD8+ and CD4+ T cells were determined. Furthermore, the number of circulating human T-cells in mice treated with CAR-T cells was determined using CD3-PerCP/CD4-FITC/CD8-PE TruCOUNT kit (BD Bioscience, Beijing, China). CD4 + and CD8+ T cells were quantified according to the manufacturer’s instructions. The analysis was performed on a FACS Calibur (BD Biosciences, Beijing, China) and analyzed using FlowJo Software (TreeStar, Ashland, OR).
Immunohistochemistry (IHC) assay
To assess the persistence of human T cell and Ki67 expressions in the treated xenograft models, immunostaining was performed in the formalin-fixed paraffin-embedded tumor tissue sections using mouse monoclonal anti-CD3 antibody (1:150, Thermo Scientific), and rabbit polyclonal anti-Ki67 antibody (1:100, Invitrogen). Human granzyme B was detected by staining for 45 minutes with rabbit anti-human granzyme B polyclonal antibody (1:100, Abcam). The slides were developed using 3, 3ʹ-diaminobenzidine (DAB) and counterstained with hematoxylin. The quantification of the number of CD3-positive and Ki67-positive cells and granzyme B staining was analyzed via integral optical density (IDO) measurement using software Image-Pro PLUS v6.0.
Statistical analysis
All statistical analyzes were performed using GraphPad Prism 6.0. For in vitro and in vivo comparisons, data were analyzed using Student’s t-test. P value of ≤ 0.05 was considered as statistically significant.
Results
Construction of antigen-specific effector cells and target cells
The second-generation CAR sequence targeted at PSMA was first constructed, with CD3ζ mediating T-cell activation, and 4-1BB serving as a co-stimulatory signal. The extracellular domain of the TGF-β receptor and the intracellular domain of the IL-7 receptor were selected as the inverted chimeric cytokine receptor (ICR) which was obtained by whole-gene synthesis. Based on the PSMA-specific second-generation CAR, an enhanced CAR sequence that co-expressed ICR was constructed (Figure 1a). Using flow cytometry, the transfection efficiency of CAR-T cells and the positive rate of CAR after lentiviral transfection were examined,(Figure 1b). Western blot analysis was performed to further verify the successful construction of CAR-T cells (Figure 1c).
Figure 1.
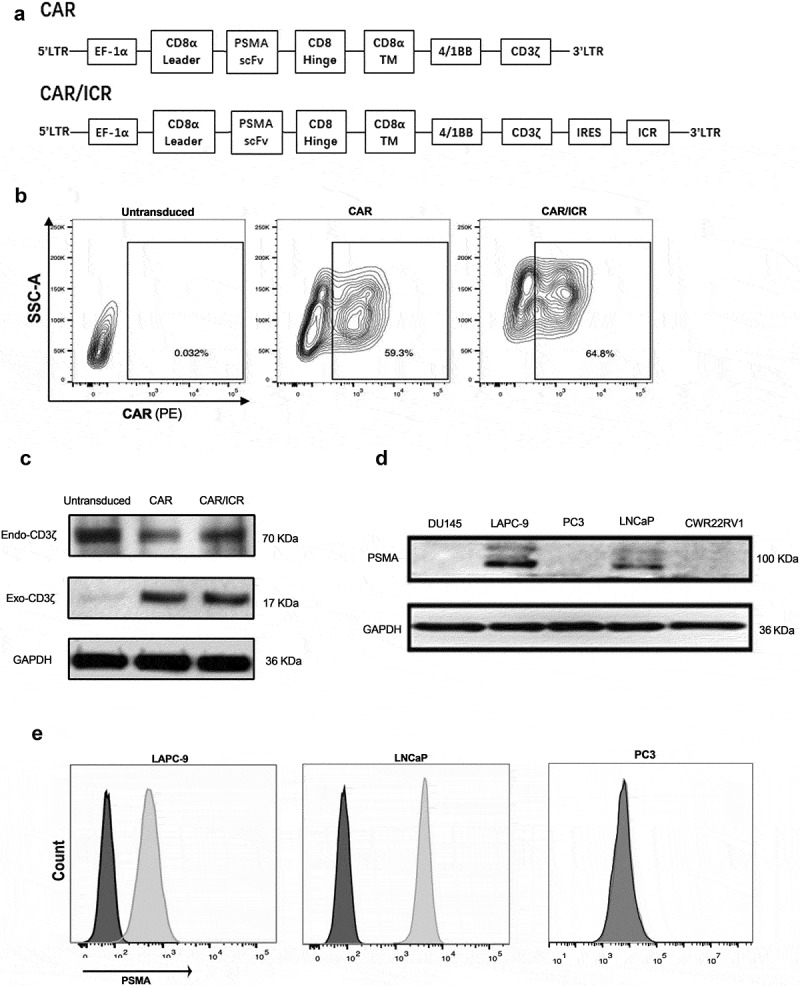
Selection of for target cells and construction of effector cells.
a. Gene structure of effector cell CAR. b. The positive rate of CAR-T cells measured by flow cytometry 5 days after T cell transfection. c. CD3ζ detected by western blot. Exogenous CD3ζ was presented as a 70 kD band only in the lanes of CAR-T cells, and endogenous CD3ζ was presented as a 17 kD band in the lanes of control T-cells and CAR-T cells. GAPDH was used as the internal reference. d. The expression of PSMA antigen in prostate cancer cells as assessed by Western blot. PSMA was around 100 kD. GAPDH was used as an internal reference. e. The expression of PSMA in LAPC-9,LNCaP and PC3 cells as analyzed by flow cytometry. f. Western blot was used to detect the content of TGF-β1 in the cell culture supernatant.
Western blot assay was used to verify PSMA protein in prostate cancer cells DU145, LAPC-9, PC3, LNCaP and CWR22RV1, and the results suggested that LAPC-9 and LNCaP cells significantly expressed PSMA (Figure 1d).Flow cytometry results confirmed the overexpression of PSMA in LAPC-9,LNCaP and PC3 cells (Figure1e). The PSMA-negative prostate cancer cells PC3 were used as a control. Since the ICR part contains the TGFβ receptor, we also detected TGF-β secreted by the three selected tumor cells by Western blot experiments (Figure 1f). For the subsequent in vivo and in vitro experiments, lentiviral transfection was adopted to confirm that the overexpression of RFP-tagged luciferase in target tumor cells. Cell lines overexpressing RFP including, LAPC-9-luc, LNCaP-luc and PC3-luc, were selected via antibiotic screening.
Determination of the optimal effector-target ratio of PSMA-CAR-T cells
In order to effectively characterize the activity of PSMA-CAR-T cells, the optimal effector-target ratio was first determined through in vitro cytotoxicity experiments. The xCELLigence RTCA DP instrument was used to monitor the cytotoxicity of PSMA-CAR-T cells against three prostate cancer cells, including LAPC-9, LNCaP, and PC3, at three different effector-target ratios (E: T ratio: 8:1, 4:1, 2:1). Based on the automatically generated tumor growth curve, PSMA-CAR-T cells exhibited rapid and potent cytotoxicity in LAPC-9 and LNCaP cells that overexpressed PSMA; however, PSMA-negative PC3 cells exhibited no cytotoxicity, indicating the PSMA-dependent cytotoxic effects of PSMA-CAR-T cells (Figure 2a). As cytotoxicity did not increase significantly with the increasing doses, an effector-target ratio of 2:1 was adopted for subsequent experiments.
Figure 2.
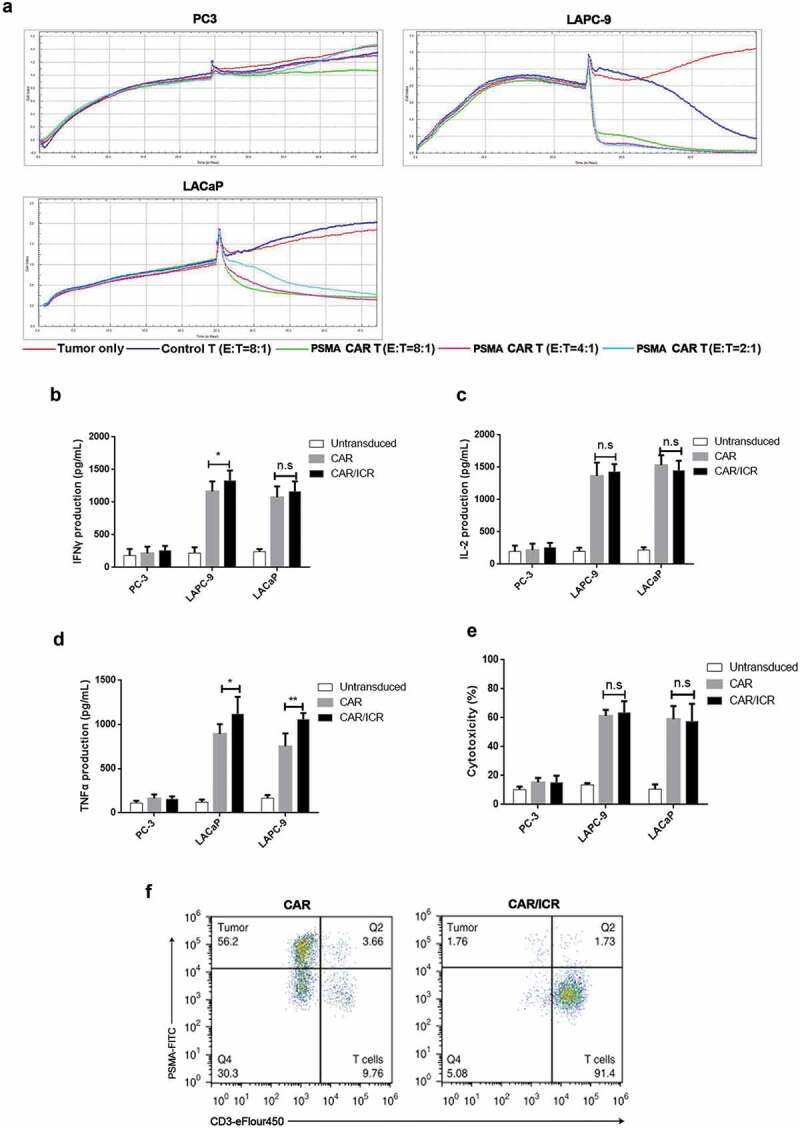
Effects of ICR on in vitro activation and cytotoxicity of PSMA-CAR-T cells.
a. Real-time cytotoxicity assay showed the proliferation of adherent cancer cells; each curve represented a well of cancer cells. Three different effector-target ratios were selected for PSMA-CAR-T cells (E: T ratio: 8:1, 4:1 and 2:1), while only one E: T ratio was chosen for the control T cells (8:1). At the initial stage, 1 × 104 tumor cells were inoculated into each well for 24 hours of incubation. In the second stage, T cells or the media were added to corresponding wells for 24 hours of further incubation. b. 1 × 104 target cells were co-incubated with effector cells at an effector-target ratio of 2:1 for 24 h, followed by the determination of secretion level of IFN-γ. (n = 3, bar value represents the dispersion degree, *, p < .05, **, p < .01). c. Determination of the secretion level of IL-2. (n = 3, bar value represents the dispersion degree, *, p < .05, **, p < .01). d. Determination of the secretion level of TNF-α. (n = 3, bar value represents the dispersion degree, *, p < .05, **, p < .01). e. 1 × 104 tumor cells were co-incubated with effector cells at an effector-target ratio of 2:1 for 6 h, followed by measurement of the level of LDH in the supernatant to characterize cytotoxicity (n = 3, bar value represents the dispersion degree, *, p < .05). F. Representative flow cytometry dot diagram indicated the lysis of target cells induced by CAR-T cells after 3 rounds of tumor cell stimulation.
Effect of ICR on in vitro activation and cytotoxicity of PSMA-CAR-T cells
To investigate the effects of ICR on PSMA-CAR-T cell activation, three experimental groups were selected for validation: untransfected T-cell group (Untransduced group), PSMA-CAR-T cell group (CAR group), and PSMA-CAR-T cell co-transfected with ICR group (CAR/ICR group). The previously selected PSMA-positive LAPC-9 cells, LNCaP cells, and PSMA-negative PC3 cells were selected as target cells. The cells were co-incubated for 24 hours at an effector-target ratio of 2:1, and the release level of cytokines (IL-2, IFN-γ, TNF-α) was measured (Figure 2b–d) to identify the activation condition of CAR-T cells. Notably, the release level of LDH in the supernatant was also examined to determine the cytotoxicity of CAR-T cells against target cells after 6 hours of cell co-incubation (Figure 2e). The results indicated that compared with PSMA-negative PC3 cells, the PSMA-positive prostate cancer cells including LAPC-9 and LNCaP were the target cells, and the cytokine release levels in CAR and CAR/ICR groups were significantly enhanced, together with considerably improved cytotoxicity; however no significant difference was noted between the two groups for cytokine release level or cytotoxicity. Conceivably, in PSMA-negative target cell group, PSMA-CAR-T cells were not activated, and its cytokine release level showed no significant difference with that of the untransduced group, and no cytotoxicity was observed against the target cells. In contrast, in the PSMA-positive target cell group, PSMA-CAR-T cells were activated, and its cytokine release level was significantly higher compared with the control group, and the cytotoxicity was remarkably enhanced. These results indicated that PSMA-CAR-T cells could be activated in vitro and kill tumor cells directed against a specific antigen. However, with the short-term co-incubation, co-expressing ICR did not exhibit enhanced activation and cytotoxicity levels.
Enhanced viability of PSMA-CAR-T cells by ICR after repeated stimulation of tumor cells in vitro
To assess the anti-tumor capability of CAR-T cells in vivo, we analyzed the cytotoxicity and proliferative activity of PSMA-CAR-T cells and PSMA-CAR-T cells co-expressing ICR after repeated stimulation of tumor cells in vitro. The effector cells were co-cultured with LAPC-9 at an effector-target ratio of 2:1, and LAPC-9 cells were added into the co-incubation system at the 7th, 14th and 21st day for stimulation. It was observed that PSMA-CAR-T cells lost their capabilities of amplifying and clearing tumor cells after the third stimulation (Figure 2f). In contrast, PSMA-CAR-T cells expressing ICR responded to all three consecutive tumor cell stimulation and exhibited high anti-tumor activity with a significantly higher proliferation rate than that of conventional PSMA-CAR-T cells, (Figure 3a,b). The expression of PD-1, TIM-3, and LAG-3 on T cells was also determined (Figure 3c,d). There were no significant differences between PSMA-CAR-T cells and PSMA-CAR-T cells co-expressing ICR with respect to the level of TIM-3 after three rounds of antigen-specific stimulation with LAPC-9. However, after antigen-specific stimulation, compared with PSMA-CAR-T cells co-expressing ICR, PSMA-CAR-T cells showed an up-regulated expression of PD-1 and LAG-3. These data indicated a stronger immunosuppressive effect in PSMA-CAR-T cells following repeated antigen activation. Furthermore, since PSMA-CAR-T cells co-expressing ICR showed higher proliferation ability than PSMA-CAR-T cells, it was speculated that ICR could protect PSMA-CAR-T cells from apoptosis induced by repeated antigen stimulation. Thus, PI3K/Akt activation serves as key downstream signaling for T cell activation. After the third stimulation, we evaluated the AKT phosphorylation level and also the expression level of anti-apoptotic protein Bcl-xL in PSMA-CAR-T cells and PSMA-CAR-T cells co-expressing ICR. As expected, the levels of AKT phosphorylation and Bcl-xL expression were significantly higher in PSMA-CAR-T cells co-expressing ICR than that of PSMA-CAR-T cells (Figure 3e,f). Taken together, these findings suggested that ICR could promote proliferation, and enhance the cytotoxic activity of PSMA-CAR-T cells after repeated stimulation of tumor cells and thus exhibiting better therapeutic potentials.
Figure 3.
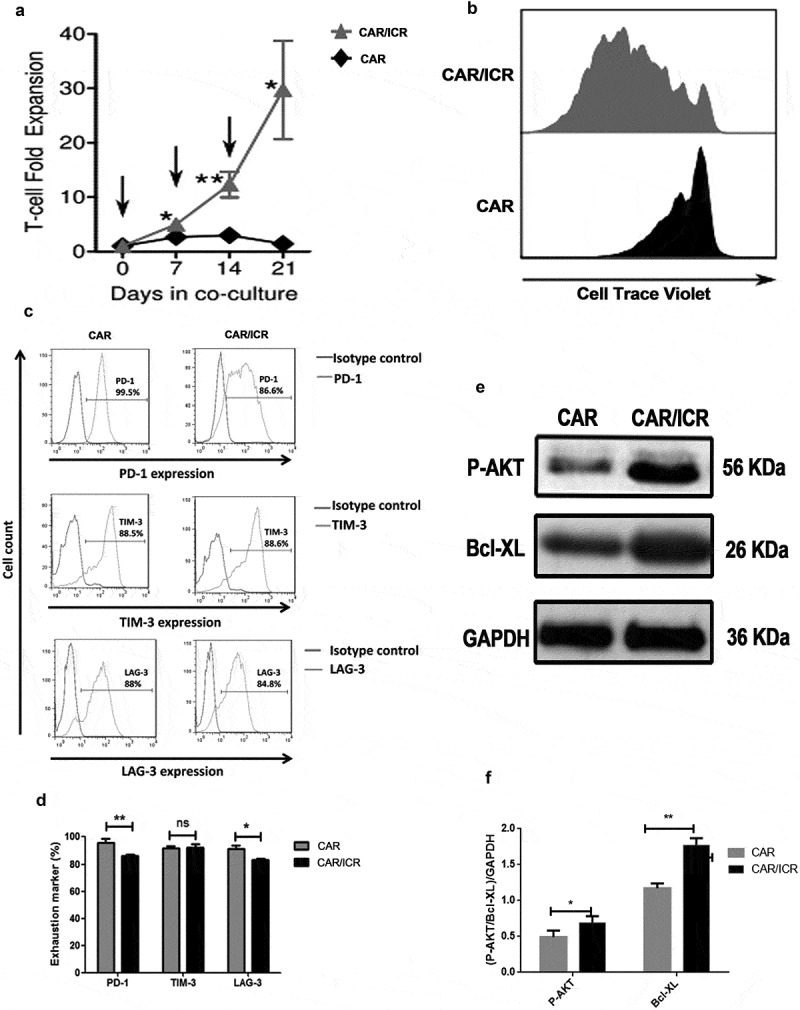
ICR enhanced the viability of PSMA-CAR-T cells after repeated in vitro stimulation of tumor cells.
a. Cumulative amplification of PSMA-CAR-T cells or PSMA-CAR-T cells co-expressing ICR during continuous co-culture. The arrow indicated the time at which T cells were re-stimulated by tumor cells. b. For proliferation analysis, PSMA-CAR-T cells and PSMA-CAR-T cells co-expressing ICR were collected at the end of the second tumor cell stimulation and labeled with Cell Trace Violet, at the 4th day, cells were collected after the third stimulation for flow cytometric analysis. c. After three rounds of antigen-specific stimulation with LAPC-9 cells, flow cytometry was performed to determine the expression of biomarkers representing cell depletion on the surface of T-cells, including PD-1, TIM-3, and LAG-3 antibodies. d. Bar graph of PD-1, TIM-3 and LAG-3 expression (n = 3, bar value represents the dispersion degree, ns not significant, p > .05, * p < .05, ** p < .01, * ** p < .001). e. After three rounds of antigen-specific stimulation with LAPC-9 cells, T cells were isolated and subjected to Western blot for measurement of the expression levels of p-Akt and Bcl-xL. F. The bar graph showed the quantitative densitometry results of the protein levels of phosphorylated Akt and Bcl-xL. (n = 3, bar value represents the dispersion degree, * p < .05, ** p < .01).
PSMA-CAR-T cells co-expressing ICR significantly enhanced the anti-tumor functions in xenograft models
To further compare the cytotoxicity of PSMA-CAR-T cells and PSMA-CAR-T cells co-expressing ICR in vivo, we used NOD/SCID mice bearing subcutaneous LAPC-9-luc xenograft model. To monitor tumor regression in mice, the fluorescence intensity of tumor cells was detected by in vivo imaging. The results of in vivo imaging suggested that both types of PSMA-CAR-T cells were effective in killing tumors at an early stage (Figure 4a,b). No apparent changes concerning body weight were observed during the treatment (Figure 4c). Previous studies have suggested that the degree of infiltration of tumor tissues by CAR-T cells in vivo was highly associated with the efficacy of CAR-T cells. Therefore, at the end of the experiment, immunohistochemical analyzes of different tumor site of mice were performed. The results indicated that the number of infiltrating T cells in the tumor tissues of mice in the PSMA-CAR-T cells co-expressing ICR group was significantly higher than that in the PSMA-CAR-T cells group. Furthermore, immuno-staining with Ki67 in tumor tissues was also performed, and the results indicated that the group treated with PSMA-CAR-T cells co-expressing ICR exhibited significantly lower staining intensity than the other two groups. Moreover, the PSMA-CAR-T cells co-expressing ICR exhibited the highest expression level of granzyme B among the three groups (Figure 4d). Collectively, the results of the in vivo experiments were consistent with those of the in vitro, indicating that PSMA-CAR-T cells co-expressing ICR and conventional PSMA-CAR-T cells both exerted significant anti-tumor effects against prostate cancer.
Figure 4.
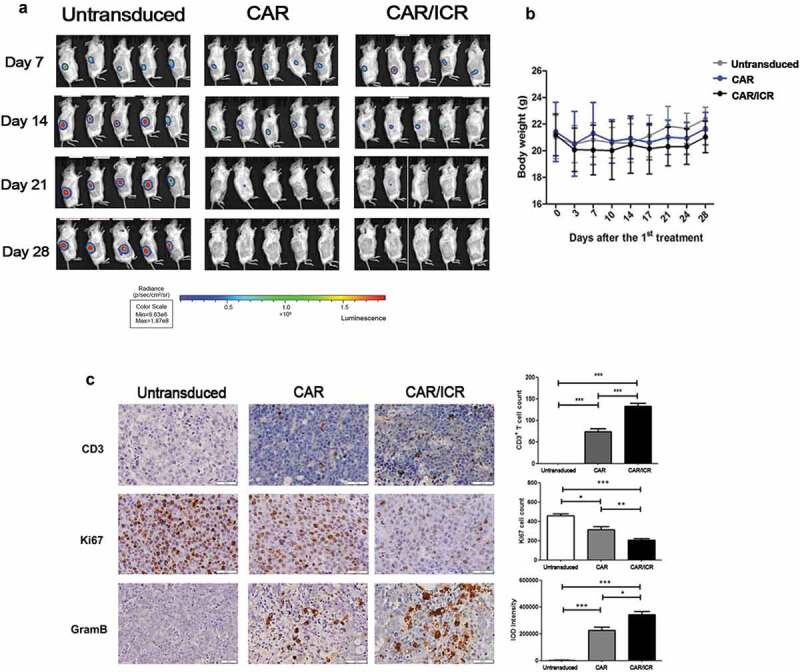
PSMA-CAR-T cells co-expressing ICR significantly enhanced the anti-tumor effects in the xenograft model.
a. In vivo image of LAPC-9-luc xenograft model. b. Quantitative value of fluorescence intensity of tumors in vivo in each treatment group. c. Variations in the body weight of the xenograft model inoculated with tumor cells and after treatment (n = 5, bar value represents the dispersion degree). d. Immunohistochemical expression of CD3, Ki67, and granzyme B in tumor cells. Each datum represents three independent experiments (n = 3, bar value represent the dispersion degree, ns not significant, p > .05, * p < .05, ** p < .01, *** p < .001).
PSMA-CAR-T cells co-expressing ICR significantly enhanced anti-tumor effects in PDX model
To further investigate the anti-tumor effects of PSMA-CAR-T cells, a mouse PDX model of human prostate cancer was established. The immunohistochemical analysis of tumor tissue specimens from a patient with metastatic prostate cancer showed a high expression of PSMA antigen (Figure 5a). 15 days after PDX inoculation, the mice were intravenously injected with Untransduced cells, PAMA-CAR-T cells or PSMA-CAR-T cells co-expressing ICR. Subsequently, tumor volume was measured every 3 days (Figure 5b). On the 41st day after PDX inoculation, the tumor weight was measured (Figure 5c). The tumor volume and tumor weight of mice in the CAR/ICR group was significantly reduced compared with the UTD group, and furthermore, were also substantially decreased relative to the CAR group. Moreover, we measured the level of T cells in the peripheral blood of mice 9 days after T cell injection. We found that both types of CAR-T cells exhibited better durability in vivo than the untransduced T cells. Besides, significantly higher CAR-T cells were detected in the CAR/ICR group than in the CAR group (Figure 5d). At the same time, the levels of IFN-γ and TNF-α in the CAR/ICR group were considerably higher than those in the CAR group (Figure 5e,f). Finally, hematoxylin and eosin (H&E) staining showed no significant difference between all the treatment groups, suggesting that PSMA-CAR-T cells and PSMA-CAR-T cells co-expressing ICR had no adverse effects on any of the major organs, confirming the safety of this reconstruction (Figure 5g). Taken together, the results from the PDX model indicated that PSMA-CAR-T cells co-expressing ICR exhibited better anti-tumor effects than conventional PSMA-CAR-T cells.
Figure 5.
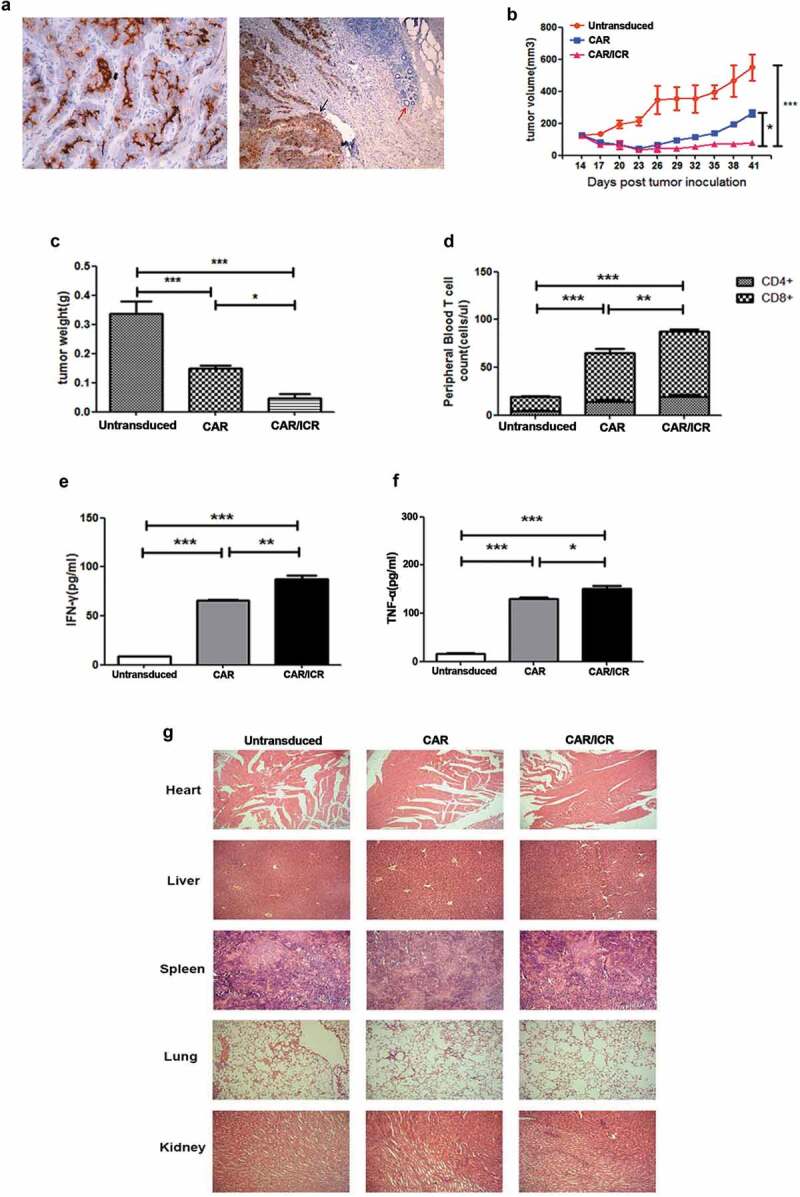
PSMA-CAR-T cells co-expressing ICR significantly enhanced the anti-tumor effects in the PDX model.
a. Immunohistochemical analysis of PSMA antigen expression in patient-derived tissues. b. After the establishment of the PDX model, CAR-T cells were administered through the caudal vein for the experiment groups and untransduced T cells for the control group. Tumor volume was measured with a vernier calliper every 3 days. c. On the 41st day, mice were sacrificed and tumor weight was measured. d. 100 μL of peripheral blood of the mice was collected on the 7th day after the infusion of effector cells, and the number of CD4+ and CD8+ T cells was counted. e. Level of IFN-γ in the peripheral blood of mice. f. Levels of TNF-α in the peripheral blood of mice. Each datum represents 3 independent experiments (n = 6, bar value represent the dispersion degree, * p < .05, ** p < .01, *** p < .001). g. Safety analysis of major organs after CAR-T cell treatment. The hearts, livers, spleens, lungs, and kidneys of each group were embedded in paraffin and cut into sections, which were stained with hematoxylin and eosin. Images were taken at 10 × magnification.
Discussion
With the unprecedented therapeutic efficacy of CAR-T cell therapy among hematological malignancies, it has become the major focus in the field of immunotherapy to extend the application of CAR-T cells in the treatment of solid cancers.16 As several tumor antigens,17 including PSMA, are widely overexpressed in prostate cancer,18 CAR-T cell therapy directed at PSMA could have a wide-ranging prospect in the malignant or refractory tumor.19 However, CAR T-cell therapy is subjected to several additional constraints in patients with solid tumors, which is mainly attributed to the inhibition of CAR-T cells due to the presence of inhibitory influences in the tumor microenvironment.20 To enable CAR-T cells to resist the inhibitory factors present in the microenvironment, we constructed an ICR by combining the extracellular domain of the TGF-β receptor and the intracellular domain of the IL-7 receptor. When ICR is bound to TGF-β in the tumor microenvironment, no inhibitory signals against T cells would be produced,21 instead the IL-7 receptor signaling pathway was activated, thus, augmenting the proliferation of CAR-T cells and preventing it from apoptosis.22 Therefore, CAR-T cells co-expressing this engineered receptor are expected to provide an effective therapeutic modality for solid tumors.
In this study, prostate cancer cells overexpressing PSMA were selected, and conventional second-generation PSMA-CAR-T cells and PSMA-CAR-T cells co-expressing ICR were constructed to validate if the engineered CAR-T cells could exert high anti-tumor efficacies against prostate cancer. Through a series of experiments, we observed that PSMA-CAR-T cells could effectively kill PSMA-positive prostate cancer cells in vitro and in vivo, and ICR could remarkably enhance the proliferation of PSMA-CAR-T cells and reinforced the cytotoxicity of PSMA-CAR-T cells after repeated stimulation by tumor cells. Through the established mouse xenograft model, we demonstrated that PSMA-CAR-T cells could mediate tumor elimination in vivo; and co-expression of ICR prolonged the survival time of PSMA-CAR-T cells in mice, thus, promoting the infiltration of PSMA-CAR-T cells in tumor tissues and also the release of cytokines. Moreover, histopathological analysis of tumor tissue sections indicated that the enhanced CAR-T cells exhibited no significant cytotoxicity to normal tissues, confirming the safety and effectiveness of this therapy.
Furthermore, PSMA-CAR-T cells co-expressing ICR exhibited significant anti-tumor activities against prostate cancer would pave the way for developing further treatment strategy, directed at potential targets including prostate cancer stem cell antigen (PSCA)23,24 and epithelial cell adhesion molecule (EpCAM),25,26 to extend the application of this enhanced CAR-T cells in the treatment of prostate cancer. In conclusion, through a series of in vitro and in vivo experiments, we have demonstrated that PSMA-CAR-T cells co-expressing ICR exhibited superior anti-tumor activities against prostate cancer. However, there are some limitations to the present study. CAR-T cells targeted at single PSMA antigen remain at the risk of off-target toxicities.27 Therefore, in the next step, we consider adding switch molecules on CAR-T cells to prevent its toxicity on time.28
Conclusion
The enhanced CAR-T cells exhibit significantly enhanced and sustained anti-tumor activities functions than the second-generation CAR-T cells, this approach can be envisaged as a new therapeutic strategy for prostate cancer and support the translation of this enhanced approach in the clinical setting.
Funding Statement
This work was supported by the The Natural Science Foundation of Xinjiang Uygur Autonomous Region [2018D01C177].
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Vaidyanathan V, Karunasinghe N, Jabed A, Pallati R, Kao C, Wang A, Marlow G, Ferguson L.. Prostate cancer: is it a battle lost to age? Geriatrics (Basel). 2016;1(4):E27. doi: 10.3390/geriatrics1040027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roudi R, Nemati H, Rastegar Moghadam M, Sotoudeh M, Narouie B, Shojaei A. Association of homeobox B13 (HOXB13) gene variants with prostate cancer risk in an Iranian population. Med J Islam Repub Iran. 2018;32:97. doi: 10.14196/mjiri.32.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schuster SJ. CD19-directed CAR T cells gain traction. Lancet Oncol. 2019;20(1):2–3. doi: 10.1016/S1470-2045(18)30900-8. [DOI] [PubMed] [Google Scholar]
- 4.Alayed Y, Cheung P, Chu W, Chung H, Davidson M, Ravi A, Helou J, Zhang L, Mamedov A, Commisso A, et al. Two StereoTactic ablative radiotherapy treatments for localized prostate cancer (2STAR): results from a prospective clinical trial. Radiother Oncol. 2019;135:86–90. doi: 10.1016/j.radonc.2019.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Dierks T, Heijnsdijk EAM, Korfage IJ, Roobol MJ, de Koning HJ. Informed decision-making based on a leaflet in the context of prostate cancer screening. Patient Educ Couns. 2019;102(8):1483–1489. doi: 10.1016/j.pec.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidt LH, Heitkötter B, Schulze AB, Schliemann C, Steinestel K, Trautmann M, Marra A, Hillejan L, Mohr M, Evers G, et al. Prostate specific membrane antigen (PSMA) expression in non-small cell lung cancer. PLoS One. 2017;12(10):e0186280. doi: 10.1371/journal.pone.0186280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Junghans RP, Ma Q, Rathore R, Gomes EM, Bais AJ, Lo ASY, Abedi M, Davies RA, Cabral HJ, Al-Homsi AS, et al. Phase I trial of anti-PSMA designer CAR-T cells in prostate cancer: possible role for interacting interleukin 2-T cell pharmacodynamics as a determinant of clinical response. Prostate. 2016;76(14):1257–1270. doi: 10.1002/pros.v76.14. [DOI] [PubMed] [Google Scholar]
- 8.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richards RM, Sotillo E, Majzner RG. CAR T cell therapy for neuroblastoma. Front Immunol. 2018;9:2380. doi: 10.3389/fimmu.2018.02380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verma A, Warner SL, Vankayalapati H, Bearss DJ, Sharma S. Targeting Axl and Mer kinases in cancer. Mol Cancer Ther. 2011;10(10):1763–1773. doi: 10.1158/1535-7163.MCT-11-0116. [DOI] [PubMed] [Google Scholar]
- 11.Zhao J, Lin Q, Song Y, Liu D. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol. 2018;11(1):132. doi: 10.1186/s13045-018-0677-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim WA, June CH. The principles of engineering immune cells to treat cancer. Cell. 2017;168(4):724–740. doi: 10.1016/j.cell.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huber M, Lohoff M. Change of paradigm: CD8+ T cells as important helper for CD4+ T cells during asthma and autoimmune encephalomyelitis. Allergo J Int. 2015;24(1):8–15. doi: 10.1007/s40629-015-0038-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Melchionda F, Fry TJ, Milliron MJ, McKirdy MA, Tagaya Y, Mackall CL. Adjuvant IL-7 or IL-15 overcomes immunodominance and improves survival of the CD8+ memory cell pool. J Clin Invest. 2005;115(5):1177–1187. doi: 10.1172/JCI200523134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown MH, Dustin ML. Steering CAR T cells into solid tumors. N Engl J Med. 2019;380(3):289–291. doi: 10.1056/NEJMcibr1811991. [DOI] [PubMed] [Google Scholar]
- 16.Forsberg EMV, Lindberg MF, Jespersen H, Alsén S, Bagge RO, Donia M, Svane IM, Nilsson O, Ny L, Nilsson LM, et al. HER2 CAR-T cells eradicate uveal melanoma and T-cell therapy-resistant human melanoma in IL2 transgenic NOD/SCID IL2 receptor knockout mice. Cancer Res. 2019;79(5):899–904. doi: 10.1158/0008-5472.CAN-18-3158. [DOI] [PubMed] [Google Scholar]
- 17.Liu L, Li E, Luo L, Zhao S, Li F, Wang J, Luo J, Zhao Z. PSCA regulates IL-6 expression through p38/NF-κB signaling in prostate cancer. Prostate. 2017;77(14):1389–1400. doi: 10.1002/pros.v77.14. [DOI] [PubMed] [Google Scholar]
- 18.Yin C, Ho B, Chan L, Emmett L. Asymptomatic prostate cancer brain metastases on 68Ga-PSMA PET/CT. Clin Nucl Med. 2019;44(6):e382–e384. doi: 10.1097/RLU.0000000000002548. [DOI] [PubMed] [Google Scholar]
- 19.Wei X, Lai Y, Li J, Qin L, Xu Y, Zhao R, Li B, Lin S, Wang S, Wu Q, et al. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology. 2017;6(3):e1284722. doi: 10.1080/2162402X.2017.1284722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hotchkiss KA, Basile CM, Spring SC, Bonuccelli G, Lisanti MP, Terman BI. TEM8 expression stimulates endothelial cell adhesion and migration by regulating cell-matrix interactions on collagen. Exp Cell Res. 2005;305(1):133–144. doi: 10.1016/j.yexcr.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 21.Loubaki L, Chabot D, Bazin R. Involvement of the TNF-α/TGF-β/IDO axis in IVIg-induced immune tolerance. Cytokine. 2015;71(2):181–187. doi: 10.1016/j.cyto.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 22.Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, Yi Z, Sauer T, Liu D, Parihar R, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. 2017;7(11):1238–1247. doi: 10.1158/2159-8290.CD-17-0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X, Hu Q, Chen Y, Li M, Yin H, Zhou C, Li G, Hou J. PSCA rs1045531 polymorphism and the risk of prostate cancer in a Chinese population undergoing prostate biopsy. Technol Cancer Res Treat. 2017;16(6):1168–1172. doi: 10.1177/1533034617740264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cui H, Tang M, Zhang M, Liu S, Chen S, Zeng Z, Shen Z, Song B, Lu J, Jia H, et al. Variants in the PSCA gene associated with risk of cancer and nonneoplastic diseases: systematic research synopsis, meta-analysis and epidemiological evidence. Carcinogenesis. 2019;40(1):70–83. doi: 10.1093/carcin/bgy151. [DOI] [PubMed] [Google Scholar]
- 25.Lee S, Ahn HJ. Anti-EpCAM-conjugated adeno-associated virus serotype 2 for systemic delivery of EGFR shRNA: its retargeting and antitumor effects on OVCAR3 ovarian cancer in vivo. Acta Biomater. 2019;91:258–269. doi: 10.1016/j.actbio.2019.04.044. [DOI] [PubMed] [Google Scholar]
- 26.Yamada-Kanazawa S, Tasaki Y, Kajihara I, Sakamoto R, Maeda-Otsuka S, Ihn H. The expression of EpCAM in extramammary Paget’s disease. Intractable Rare Dis Res. 2019;8(1):20–23. doi: 10.5582/irdr.2019.01010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Migliorini D, Dietrich PY, Stupp R, Linette GP, Posey AD Jr, June CH. CAR T-cell therapies in glioblastoma: a first look. Send to Clin Cancer Res. 2018;24(3):535–540. doi: 10.1158/1078-0432.CCR-17-2871. [DOI] [PubMed] [Google Scholar]
- 28.Martinez M, Moon EK. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol. 2019;10:128. doi: 10.3389/fimmu.2019.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]