

Abstract
Neuroblastoma (NB) is the most common extracranial solid tumor in children. The outcomes for aggressive forms of NB remain poor. The aim of this study was to develop a new molecular-targeted therapy for NB using an antisense oligonucleotide (ASO) and superparamagnetic iron oxide (SPIO) nanoparticles (NPs), as a delivery vehicle, targeting the transcription regulator MAX dimerization protein 3 (MXD3). We previously discovered that MXD3 was highly expressed in high-risk NB, acting as an anti-apoptotic factor; therefore, it can be a good therapeutic target. In this study, we developed two ASO-NP complexes using electrostatic conjugation to polyethylenimine-coated SPIO NPs and chemical conjugation to amphiphilic polymers on amine-functionalized SPIO NPs. Both ASO-NP complexes demonstrated MXD3 knockdown, which resulted in apoptosis in NB cells. ASO chemically-conjugated NP complexes have the potential to be used in the clinic as they showed great efficacy with minimum NP-associated cytotoxicity.
Keywords: neuroblastoma, targeted therapy, antisense oligonucleotide, gene silencing, nanoparticle
Background
Neuroblastoma (NB) is a tumor of the sympathetic nervous system and is the most common extracranial solid tumor in children, representing 8–10% of pediatric cancers.1 The outcome of patients with a high-risk phenotype is poor, with a survival rate of about 50%.2,3 Therefore, there is an urgent need for more effective treatments for NB.
An antisense oligonucleotide (ASO) is a powerful tool to silence gene expression and has the potential for molecular-targeted therapy. An ASO is a 13–25 nucleotide-long oligodeoxynucleotide that binds to the complementary mRNA of the target sequence by base pairing, has high specificity, and is easy to modify chemically to improve its stability in vivo.4,5 Several ASOs have shown promising results in preclinical cancer models and phase I clinical trials, for prostate cancer6,7, lymphoma8 and lung cancer8. However, non-specific delivery and poor intracellular uptake into target cells are still challenges of ASOs for cancer therapy.9,10 To solve the challenge of intracellular delivery of ASOs, various nano-carriers have been studied.11,12
Several drug-delivery systems using nanoparticles (NPs) have been approved for cancer therapy and are currently in clinical trials or preclinical evaluations for several cancers, including breast cancer, pancreatic cancer, soft tissue sarcoma, and liver cancer.13–15 Superparamagnetic iron oxide (SPIO) NPs are promising nano-carriers due to their unique magnetic characteristics16,17, and they have been widely used for magnetic resonance imaging contrast enhancement and drug delivery.18,19 As a vehicle, SPIO NPs have been used to deliver drugs20, proteins21, antibodies22 and nucleotides.23,24 Taratula et al. reported that B-cell lymphoma 2 (BCL2)-specific small interfering RNA (siRNA) and poly (propyleneimine) generation 5 dendrimers conjugated with SPIO NP complexes showed effective siRNA delivery, gene silencing, and antitumor activity both in vitro and in vivo for lung cancer cells.25 Our group demonstrated effective in vitro intracellular delivery of siRNA using SPIO NPs for precursor B cell (preB) acute lymphoblastic leukemia (ALL)33 and for NB.26 Wen et al. reported that c-erbB2 oncogene-specific ASO-SPIO NP complexes were potentially useful as a magnetic resonance contrast-enhancing targeting agent to specifically diagnose tumors which had over-expression of the c-erbB2 oncogene in the in vitro studies.24,27 Therefore, SPIO NPs are a promising vehicle for successful delivery of nucleotides.
MXD3 (MAX dimerization protein 3) is a transcription regulator and a member of the MYC/MAX/MXD family of basic helix-loop-helix-leucine-zipper proteins.28 We discovered that MXD3 is highly expressed in NB cells compared to normal cells, and silencing MXD3 expression by siRNA induces cell apoptosis in NB cells.26,29 In this study, we developed novel therapeutic nanocomplexes using MXD3 ASO and SPIO NPs. We hypothesized that MXD3 ASO-SPIO NP complexes can be a potential new targeted therapeutic approach for NB. We showed successful formation and characterization of ASO-NP complexes, intracellular delivery of the nanocomplexes, MXD3 knockdown, and cytotoxicity in NB cell lines in vitro. This is the first report showing ASO-SPIO NP complexes as a potential therapy for NB.
Methods
Cell lines and cell culture
Two human NB cell lines, SK-N-DZ and SK-N-BE were maintained at 37 °C in a 5% CO2 incubator. SK-N-DZ cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) and 1% Minimum Essential Medium Non-Essential Amino Acids Solution. SK-N-BE cells were cultured in Eagle’s Minimum Essential Medium (EMEM) and Ham’s F-12 Nutrient Mix. Cells were used within the first 30 passages and were maintained at their logarithmic phase of growth prior to each experiment.
SPIO NPs
Iron oxide NPs with polyethylenimine (PEI) coating (SEI NPs) and amine iron oxide NPs (SHA NPs) were used in this study. SEI NPs (catalog No.SEI-15–05) are water-soluble iron oxide NPs with a monolayer of oleic acid, amphiphilic polymer and PEI coating (iron core diameter: 15nm). SHA NPs (catalog No.SHA-15–05) are water-soluble iron NPs with a monolayer of oleic acid, amphiphilic polymer and amine functionalization (iron core diameter: 15nm).
ASOs
ASOs were designed and synthesized using standard solid phase oligonucleotide synthetic methods as previously described.30 Briefly, the MXD3 ASO sequences are 5’-CACAGGGACGCATAAC-3’ for #632461 and cyclooctyne-modified MXD3 ASO, 5’-GGCCCTGGAGCGAACC-3’ for #632417. The negative control ASO sequence, which has no known homology to mammalian genes and has minimal nonspecific effects, is 5’-CCTTCCCTGAAGGTTCCTCC-3’. The 5’-end of each oligonucleotide was modified to comprise a cyclooctyne for subsequent click chemistry conjugation to an azide-labeled SHA NPs via 1,3-dipolar cycloaddition.31 The 5’-DBCO-TEG phosphoramidite was coupled to the 5’-end of each oligonucleotide using standard solid phase methods to form a phosphodiester linkage between the oligonucleotide and the 5’-DBCO-TEG moiety. Ammonia deprotection was completed at room temperature for a minimum of 48 hours.
Formulation and characterization of ASO-SPIO NP complexes
Fresh nanocomplexes were made for each experiment. For electrostatic binding, the SEI NPs were briefly vortexed with an amine-reactive succinimidyl ester labeled with Alexa Fluor 532 (A532) and incubated in the dark at 4°C for 3 hours. The molar ratio of SEI NP to succinimidyl ester was 1:1. After incubation, the labeled SEI-NPs were mixed briefly with ASO molecules by vortex. The ASO molecules were adsorbed on the surface of the SEI NPs based on electrostatic interactions between the negative backbone of ASO molecules and the amine-modified SEI NPs. The ratio of ASO:SEI NPs was determined based on our previous studies.26 The loading ratio was 1:1 by mass and 685:1 by mole.
For click chemistry conjugation, NHS-PEG4-Azide (azide) was dissolved in dimethyl sulfoxide (DMSO) at a final concentration of 10mM. SHA NPs were incubated with azide in phosphate buffered saline (PBS) at 4°C for 3 hours. The molar ratio of amine on the SHA NPs surface to azide was 1:100. After incubation, excess azide was removed using Slide-A-Lyzer MINI Dialysis Units. The azide-conjugated SHA NPs were then incubated with cyclooctyne-modified ASOs in water at 1:0.98 by mass and 1:600 by mole at 37°C for 30 minutes. The above reaction method was optimized by titrating the molar ratio of amine, azide, and ASO (Supplementary Table 1) and using an incubation time of SHA NPs-azide with ASO based on our previous studies.26 The ASO-SHA NP complexes were centrifuged at 9,000g for 10 minutes, and unbound ASOs in the supernatant were removed.
To measure the loading efficiency of ASO on SPIO NPs, the ASO-SPIO NP complexes were centrifuged at 9,000g for 4 to 30 minutes until the supernatant became clear. The pelleted nanocomplexes and supernatant, which contained unbound ASOs, were collected separately. SPIO NPs in the pellet were dissolved with 0.15% mercaptoacetic acid for 12–16 hours.32 The amount of ASOs in the supernatant and in the pellet (after dissolving SPIO NPs) was quantified using Nanodrop 2000. The hydrodynamic diameter and zeta potential of the nanocomplexes were measured using dynamic light scattering (DLS) on a Zetasizer Nano ZS. Briefly, 40 μg of SPIO NPs was combined with ASO as described above and diluted in 1 mL of ultrapure water. Measurements were performed three times in succession in triplicates. Zeta potentials were determined using the Smoluchowski model in three independent repeat experiments.33
In vitro treatment with ASO-SPIO NP complexes
Cells were plated at 60,000 cells in 500 μL medium per well in 48-well plates in triplicate for each treatment group 12 hours before treatment. The cells were treated with the control or MXD3 ASO-SPIO NP complexes at indicated concentrations, or left untreated. The cells were incubated with the ASO-NP complexes in Opti-MEM or serum-supplemented complete medium for 4 hours, then the medium was replaced with fresh complete growth medium. The cells were counted and assessed for MXD3 protein expression by immunocytochemistry at 4, 8, 24, 48 and 72 hours after treatment. MXD3 expression in five NB cell lines were determined in our previous study26 and two cell lines with high MXD3 expression levels, SK-N-DZ and SK-N-BE, were used in this study.
Immunocytochemistry and fluorescent image intensity quantification
Cells were collected and fixed with 10% buffered formalin and mounted on slides. Slides were incubated with anti-MXD3 monoclonal mouse antibody (Ab) at a final concentration of 2 μg/mL overnight at 4°C, then with a secondary goat anti-mouse Ab-Alexa488 at a final concentration of 4 μg/mL for 3 hours at room temperature in the dark as previously described.23 Nuclei were stained with DAPI (4’,6-diamidino-2-phenylindole). The MXD3 protein expression levels were quantified by mean fluorescent intensity (MFI) using the Image J as previously described.23,34 Briefly, individual cell boundaries were marked, and MFI was measured and calculated by determining the average pixel intensity per cell. The total cell counts were 25–50 cells per field, and one or two fields were measured for each treatment group. The background fluorescent signal, without cells, was subtracted from the MFI of each cell for correcting MFI. Data are an average of 2 independent experiments with technical triplicates.
Immunoblotting
Frozen whole cell pellets (1 million cells per group) were denatured and boiled with NuPAGE Sample Reducing agent in PBS at 100°C for 10 minutes before being run on a 4–20% Tris-Glycine SDS-PAGE gel. Protein was transferred to a nitrocellulose membrane using a wet transfer system at 100V for 1 hour. Blots were blocked with 5% non-fat dry milk in Tris-buffered saline (TBS) with 1% tween-20 (TBST) for 1 hour at room temperature, then incubated with primary antibodies in 5% non-fat dry milk in TBST overnight at 4°C. After the overnight incubation, blots were washed with TBST 3 times for 10 minutes each before incubation with secondary antibodies in 5% non-fat dry milk in TBST for 1 hour at room temperature. Blots were washed twice with TBST and once with TBS before incubation with SuperSignal™ West Pico Chemiluminescent kit according to the manufacturer’s instructions. Primary antibodies and incubation conditions were 1:1000 for mouse anti-MXD3 and 1:2000 for rabbit anti-histone antibodies. Secondary antibodies and incubation conditions were 1:1000 for goat anti-mouse horseradish peroxidase (HRP) and 1:2000 for goat anti-rabbit HRP antibodies.
Apoptosis assay
Cell apoptosis was measured by flow cytometry using annexin V (AV) conjugated to fluorescein-5-isothiocyanate (FITC) and propidium iodide (PI) as previously described.23 Briefly, cells were collected 4 and 8 hours after treatment. AV and PI were measured by flow cytometry using the FC500. Caspase activity was measured using the Caspase 3/7 Glo kit as previously described.23 Cells were treated with ASO-NP complexes (without labeling A532) as described above and collected 4 and 8 hours after treatment. Caspase level was measured by a Centro LB 960 Microplate Luminometer. Data are means of 2 independent experiments.
Statistical methods
Statistical analysis was performed using Prism6 to generate means and graphics and using standard linear models implemented in SAS® Version 9.4. Cell counts were log-transformed. Multiple comparisons of means used SAS® PROC MULTTEST, adjusted for multiple comparisons via the Hochberg-Benjamini approach.35 Significant findings in analysis of variance comparisons of means were further examined with Tukey’s studentized range test for experiment-wise control of Type I error. A P-value < 0.05 was considered significant for all statistical calculations. Error bars represent standard deviation.
Materials
SK-N-DZ, SK-N-BE, DMEM and EMEM were purchased from ATCC (Manassas, VA, USA). 1% Minimum Essential Medium Non-Essential Amino Acids Solution, Ham’s F-12 Nutrient Mix, A532, azide, Slide-A-Lyzer MINI Dialysis Units, Nanodrop 2000, Opti-MEM, Ab-Alexa488, DAPI, NuPAGE Sample Reducing agent and SuperSignal™ West Pico Chemiluminescent kit were all purchased from Thermo Fisher Scientific (Waltham, MA, USA). SPIO NPs were purchased from Ocean Nanotech (San Diego, CA, USA). ASOs were synthesized and provided by Ionis Pharmaceuticals (Carlsbad, CA, USA). 5’-DBCO-TEG phosphoramidite, Zetasizer Nano ZS, FC500 and Centro LB 960 Microplate Luminometer were from Glen Research (Sterling, VA, USA), Malvern (UK), Beckman Coulter (Brea, CA, USA) and Berthold Technologies (Oakridge, TN, USA), respectively. Anti-MXD3 monoclonal mouse Ab, rabbit anti-histone Ab, AV conjugated to FITC, PI and Caspase 3/7 Glo kit were purchased from Neuromab (Davis, CA, USA), Abcam (Cambridge, UK), BD Biosciences (San Jose, CA, USA), Roche (Nutley, NJ, USA) and Promega (Madison, WI, USA), respectively. Image J was from NIH (Bethesda, MD, USA).
Results
Characterization of MXD3 ASO-SEI NP complexes
The average diameter of the SEI NPs was 36.5 nm. After ASO binding, the size of ASO-SEI NP complexes was 51.3 nm in diameter (Table 1). Surface charges of the SEI NPs decreased from an initial net positive charge of +58.9 mV to +34.2 mV after complexation with ASOs (Table 1). The binding efficiency of ASO on the SEI NPs was 94% (= 643.9 molecules per NP) (Table 1).
Table 1.
characterization of MXD3 ASO-SEI NP complexes
| size and zeta potential | Size [nm] (average ± SD) | Zeta potential [mV] (average ± SD) | |
|---|---|---|---|
| SEI NP | 36.5 ± 1.2 | 58.9 ± 2.2 | |
| MXD3 ASO-SEI NP complexes | 51.3 ± 1.3 | 34.2 ± 3.2 | |
| ASO loading | MXD3 ASO [μg] | [%] of total | Bound ASO molecules per nanoparticle |
| MXD3 ASO-SEI NP complex pellet (bound MXD3 ASO) | 28.2 | 94.0 | 643.9 |
| Supernatant (unbound MXD3 ASO) | 1.5 | 5.0 |
SD: standard deviation
MXD3 ASO-SEI NP complexes show MXD3 protein knockdown and cell count reduction in SK-N-DZ cells
SK-N-DZ cells were treated with the control or MXD3 ASO-SEI NP complexes, or left untreated. The final concentration of ASO and NP in the complexes was 34.8 nM and 0.054 nM, respectively (Table 2). The concentrations used were determined based on our previous studies.26 The MXD3 ASO-SEI NP complex treatment showed significant MXD3 protein knockdown, confirmed by immunocytochemistry (Figure 1A), at all the tested times: 4, 8, 24, 48 and 72 hours (Figure 1A and B). Knockdown seemed to peak at 8 hours and continued until 72 hours. Relative percent knockdown of MXD3 level in treatment groups compared to untreated or control ASO-SEI NP complex-treated cells was 53% for both (***p<0.001) at 4 hours, 68% (****p<0.0001) and 63% (***p<0.001) at 8 hours, 68% (****p<0.0001) and 66% (***p<0.001) at 24 hours, 72% and 71% (both ****p<0.0001) at 48 hours, and 73% (****p<0.0001) and 60% (**p<0.01) at 72 hours after treatment, respectively. Of note, MXD3 protein knockdown was also confirmed by immunoblotting (Supplementary Figure 1). Based on similar results between immunoblotting and immunocytochemistry measurement, immunocytochemistry was used in the subsequent studies.
Table 2.
summary of ASO molecules and SPIO NP amounts per well in each treatment
| Treatment | NP conc. [nM] (per well) | ASO conc. [nM] (per well) | ASO molecules per NP | MXD3 reduction [%] at 8 hours |
|---|---|---|---|---|
| MXD3 ASO*-SEI NP complexes | 0.054 | 34.8 | 643.9 | 59.1 |
| MXD3 ASO**-SHA NP complexes | 0.054 | 3.3 | 60.6 | 48.6 |
| three times amount of MXD3 ASO**-SHA NP complexes | 0.16 | 9.8 | 60.6 | 95.8 |
Molecular weight (MW) MXD3 ASO for ASO-SEI NP complex is 5448.
MW of cyclooctyne-modified MXD3 ASO for ASO-SHA NP complex is 6018.
Figure 1. MXD3 ASO-SEI NP complexes show efficient MXD3 protein knockdown and cytotoxicity from 4 to 72 hours after treatment in SK-N-DZ cells.
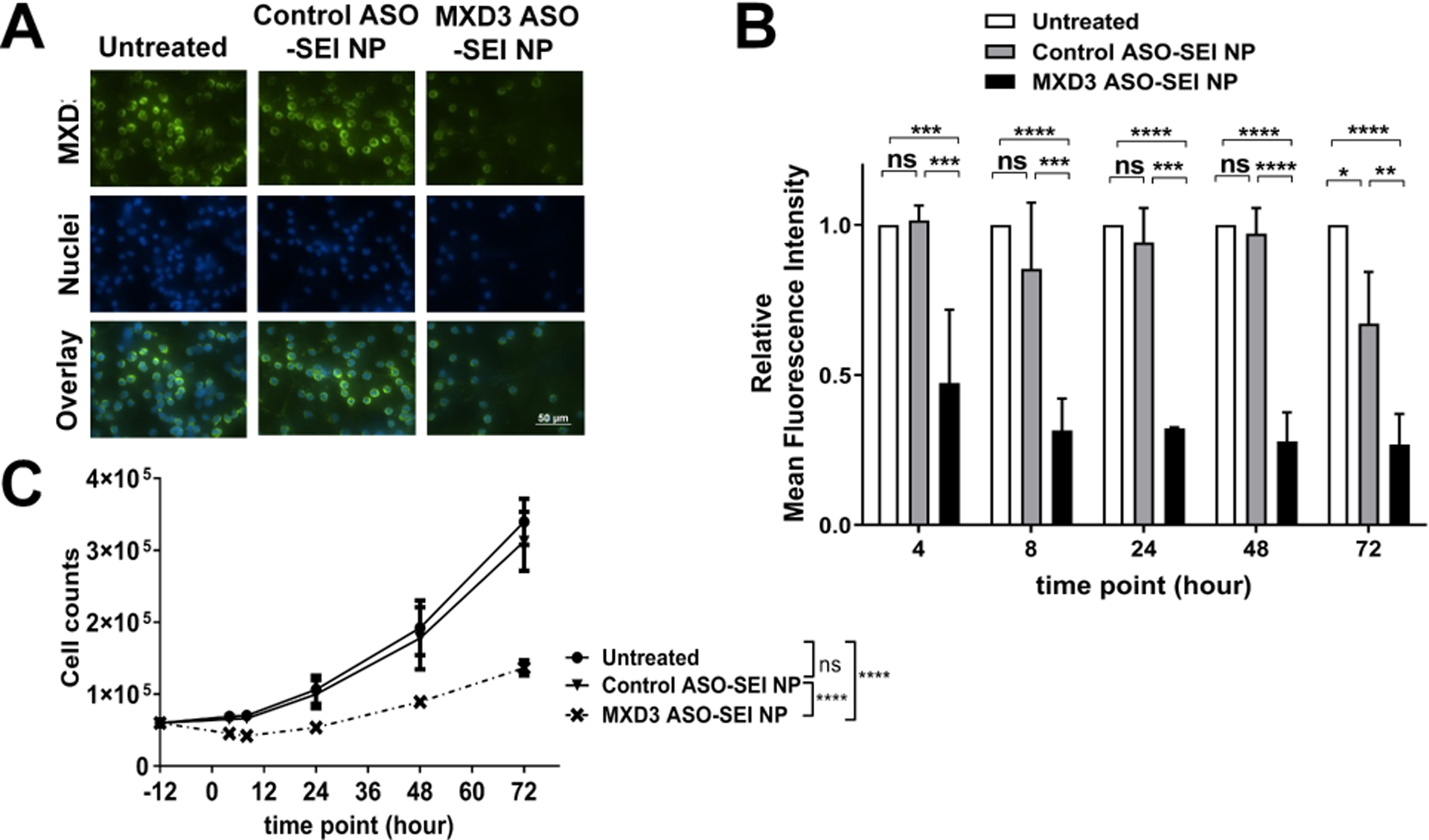
(A) Immunocytochemistry 4 hours after treatment. (B) Relative mean fluorescence intensity (MFI) and (C) living cell count 4, 8, 24, 48 and 72 hours after treatment (n=6, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
MXD3 ASO-SEI NP treatment resulted in significant cell count reduction compared to no treatment or control ASO-SEI NP treatment (Figure 1C). The maximum MXD3 knockdown and cell count reduction were at 8 hours after treatment among the 5 time points; therefore, we used 8-hour time point data for the outcome in subsequent studies.
We also tested three different concentrations of MXD3 ASO: 3.5 nM, 17.4 nM, and 34.8 nM. The percentage MXD3 protein knockdown was dose-dependent, at 13% and 14% (3.5 nM), both 79% (17.4 nM), both 82% (34.8 nM), compared with untreated cells or control ASO-SEI NP complex-treated cells, respectively, 8 hours after treatment (Supplementary Figure 2). The latter two conditions showed similar MXD3 knockdown, suggesting saturation of ASO molecules. To achieve maximum therapeutic efficacy, we chose the NPs with the highest ASO molecules (34.8 nM) for subsequent studies.
Furthermore, we tested one additional MXD3 ASO which targets a different site (#632417) on the MXD3 gene. SK-N-DZ cells treated with the MXD3 ASO (#632417)-SEI NP complexes also showed significant reductions in MXD3 protein expression of 39.9% (*p<0.05), compared to untreated or control ASO NP complex-treated cells at 8 hours after treatment (Supplementary Figure 3). MXD3 ASO-SEI NP complexes #632461 showed slightly higher knockdown (59.1%) than #632417 (39.9%); however, there were no statistical differences between the two MXD3 ASOs.
MXD3 ASO-SEI NP complex treatment induces cell apoptosis in SK-N-DZ cells
MXD3 knockdown induces apoptosis in NB cells and other cancers.23,26,29,30 Apoptosis was measured as an outcome using AV, PI and caspase.26 At 4 hours after treatment, untreated or control ASO-SEI NP complex-treated cells showed few AV positive cells (average 1.6% or 1.9%, respectively) (Figure 2B) or AV and PI double positive cells (average 3.5% or 3.8%, respectively) (Figure 2C). However, the MXD3 ASO-SEI NP complex-treated cells showed significantly more AV positive cells (average 6.9%, both *p<0.05) (Figure 2B) or double positive cells (average 7.9%, both *p<0.05) (Figure 2C) than the controls. At 8 hours after treatment, both untreated and control ASO-SEI NP complex-treated cells continued to have few AV positive cells (average 1.0% or 1.6%, respectively) (Figure 2B) or double positive cells (average 2.8% or 3.6%, respectively) (Figure 2C). The MXD3 ASO-SEI NP complex-treated cells showed significantly more AV positive cells (average 4.2%) (Figure 2B) or double positive cells (average 11.35%, vs. untreated ***p<0.001, vs. control ASO-SEI NP complexes **p<0.01) (Figure 2C). Caspase 3/7 activity also showed a significant increase in the MXD3 ASO-SEI NP complex-treated cells compared to controls at both 4 and 8 hours after treatment (average luminescence signal 90217 at 4 hours or 115901 at 8 hours, both ****p<0.0001) (Figure 2D). These results indicate that the MXD3 ASO-SEI NP complex treatment successfully delivered sufficient ASOs to the target MXD3 molecule, knocked down the protein, and resulted in cell apoptosis, as expected.23 Of note, the control ASO-SEI NP complex-treated cells showed significantly increased caspase activity compared to untreated cells (average luminescence signal 48949 at 4 hours or 39287 at 8 hours, both *p<0.05) (Figure 2D), suggesting non-specific toxicities of NPs.
Figure 2. MXD3 ASO-SEI NP complex treatment induces cell apoptosis in SK-N-DZ cells.
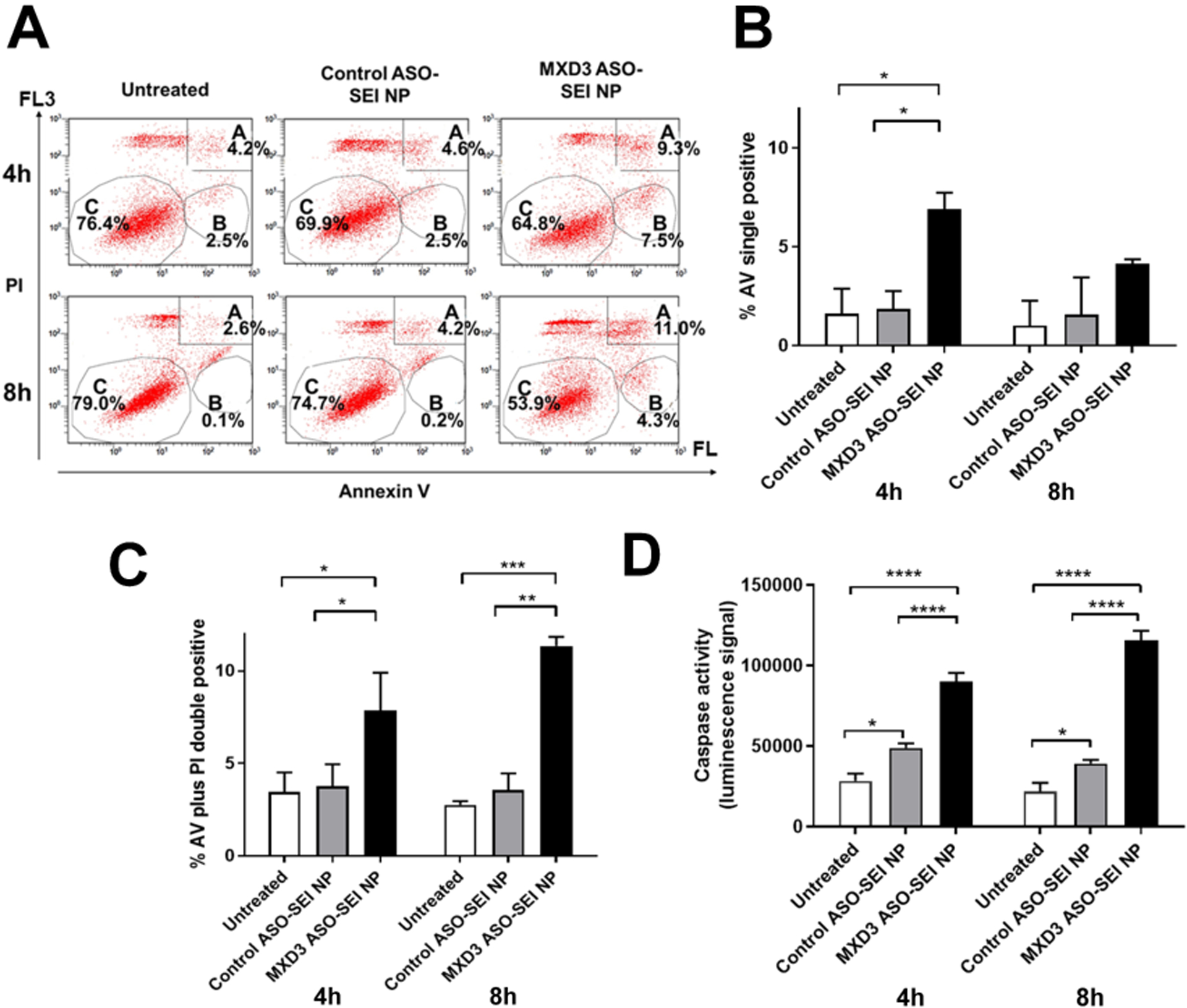
(A) Annexin V (AV) and propidium iodide (PI) 4 and 8 hours after treatment. Area A, B and C indicate AV and PI double positive, AV single positive, and double negative population, respectively. (B) Quantification of AV single positive cells and (C) AV and PI double positive cells (n=2, *p<0.05, **p<0.01, ***p<0.001). (D) Caspase 3 and 7 activity levels 4 and 8 hours after treatment (n=6 for each time point, ****p<0.0001).
Characterization and loading efficiency of MXD3 ASO-SHA NP complexes
We next investigated SHA NPs as an alternative vehicle. The net surface charge of SHA NPs is close to neutral and these particles are expected to be less cytotoxic than SEI NPs. The diameter of the SHA NPs was 28.2 nm. Alkyne-modified ASOs were conjugated to the azide-functionalized SHA NPs. After conjugation, the size of the ASO-SHA NP complexes was 38.9 nm in diameter (Table 3). Surface charges of the SHA NPs alone and the ASO-SHA NP complexes were −9.6 mV and −14.8 mV, respectively (Table 3). Only 10.1% (60.6 molecules per NP) of ASO molecules (that were added in the reaction) were bound on the SHA NPs (Table 3). However, there are approximately 50–60 amine molecules per SHA NP (personal communication with Ocean Nanotech); therefore, almost all the amine molecules on SHA NPs were saturated with ASO.
Table 3.
Characterization of MCD3 ASO-SHA NP nanocomplexes
| size and zeta potential | Size [nm] (average ± SD) | Zeta potential [mV] (average ± SD) | |
|---|---|---|---|
| SHA NP | 28.2±1.9 | −9.6 ± 0.2 | |
| SHA NP (+azide) | 30.8 ± 1.7 | −6.4 ± 0.7 | |
| MXD3 ASO-SHA NP nanocomplexes | 38.9 + 1.3 | 14.8 ±0.4 | |
| ASO loading | MXD3 ASO[μg] | [%] of total | Bound ASO molecules per nanoparticle |
| MXD3 ASO-SHA NP nanocomplex pellet (bound MXD3 ASO) | 3.0 | 10.1 | 60.6 |
| Supernatant (unbound MXD3 ASO) | 26.0 | 88.9 |
SD: standard deviation
MXD3 ASO-SHA NP complexes show efficient MXD3 protein knockdown and cell count reduction in high MXD3 NB cell lines
We tested the MXD3 ASO-SHA NP complexes at the same NP concentrations as SEI NP complexes: NP 0.054 nM and ASO 3.3 nM, which showed MXD3 reduction at 48.6% compared to the controls at 8 hours after treatment (**p<0.01) (Figure 3A and B, and Table 2). We then tested the nanocomplexes at three times higher concentration than above: NP 0.16 nM and ASO 9.8 nM, which showed MXD3 reduction at 95.8% compared to untreated cells at 8 hours after treatment (***p<0.001) (Figure 3A and B, and Table 2). MXD3 ASO-SHA NP complexes also showed dose-dependent cytotoxicity (Figure 3C). The MXD3 ASO-SHA NP complex treatment, using the higher concentration (NP 0.16nM and ASO 9.8nM), showed highly significant MXD3 protein knockdown, at all the tested times: 4, 8, 24, 48 and 72 hours, after treatment compared with controls (Figure 4A and B), resulting in cell count reduction (Figure 4C). Relative percent knockdown of MXD3 level in treatment sets compared to untreated or control ASO-SHA NP complex-treated cells was 97% and 96% at 4 hours, 95% and 96% at 8 hours, 95% for both at 24 hours, 95% for both at 48 hours, and 97% for both at 72 hours after treatment, respectively (all ****p<0.0001) (Figure 4B). The MXD3 ASO-SHA NP complexes showed much higher knockdown efficiency than the MXD3 ASO-SEI NP complexes, despite a much lower ASO molecule content than in SEI NP complexes (60.6 vs. 643.9) per NP (Tables 1 and 3).
Figure 3. MXD3 ASO-SHA NP complexes show a dose-dependent MXD3 knockdown in SK-N-DZ cells.
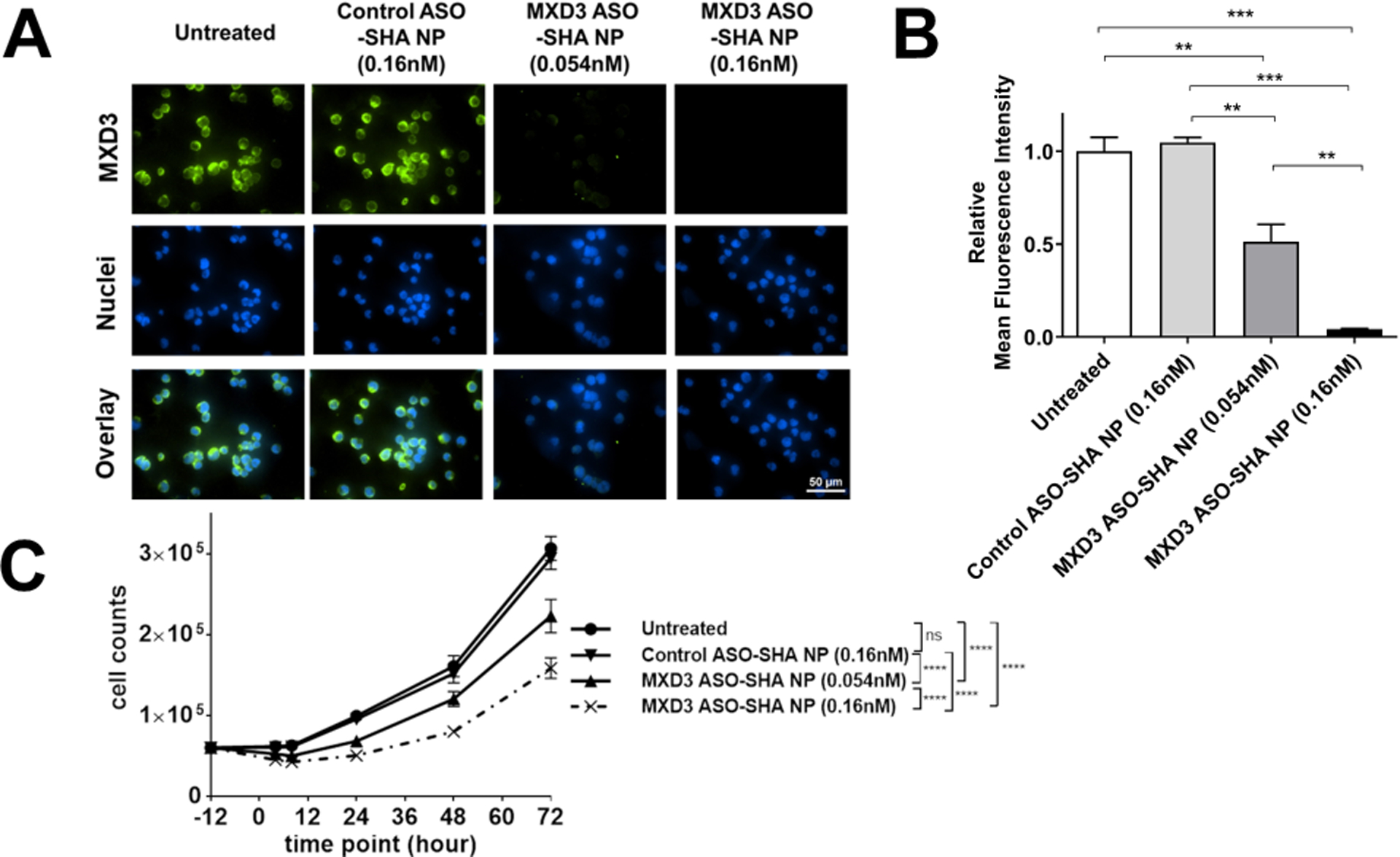
(A) Immunocytochemistry and (B) relative MFI 8 hours after treatment (n=6, **p<0.01, ***p<0.001).
(C) Living cell count 4, 8, 24, 48 and 72 hours after treatment. (n=6, ****p<0.0001)
Figure 4. MXD3 ASO-SHA NP complexes show efficient MXD3 protein knockdown and cytotoxicity from 4 to 72 hours after treatment in SK-N-DZ cells.
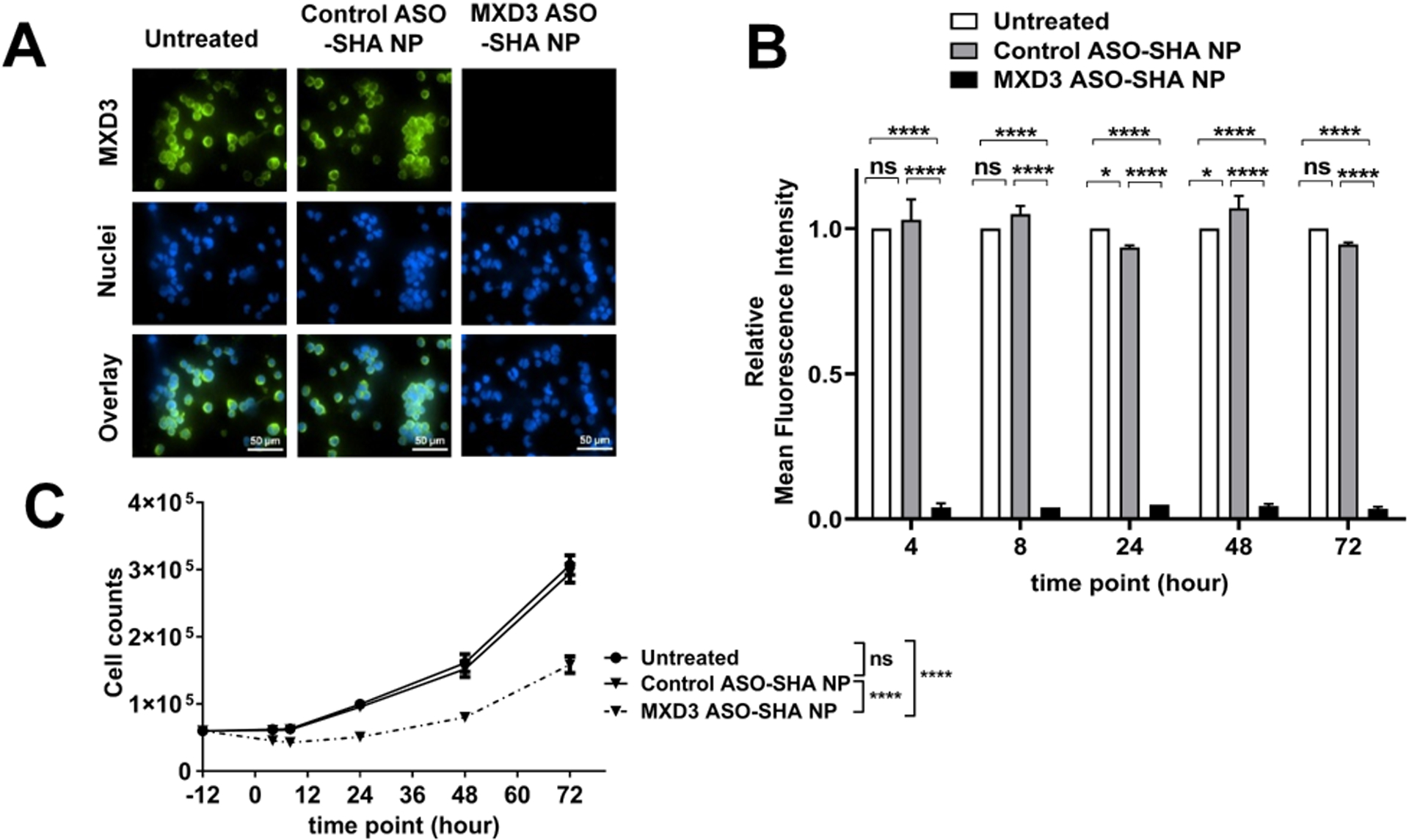
(A) Immunocytochemistry 4 hours after treatment. (B) Relative mean fluorescence intensity (MFI) and (C) living cell count 4, 8, 24, 48 and 72 hours after treatment (n=6, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
To confirm efficient delivery of ASOs via SHA NPs, the MXD3 ASO-SHA NP complexes were compared with free MXD3 ASO plus free SHA NPs at equimolar amounts in nanocomplexes. Free MXD3 ASO plus free SHA NPs treated cells showed only 15.0% MXD3 knockdown whereas the MXD3 ASO-SHA NP complexes showed 90% knockdown (Supplementary Figure 4A and B). These results suggest that although some fraction of free MXD3 ASOs could enter the cells by themselves, SHA NPs can deliver them much more efficiently, leading to MXD3 knockdown.
We next evaluated the MXD3 ASO-SHA NP complexes in another NB cell line, SK-N-BE cells, which is known to have high MXD3 expression similar to SK-N-DZ.26 SK-N-BE cells were treated with MXD3 ASO-SHA NP complexes the same way as the SK-N-DZ cells, which showed a 92.6% MXD3 protein knockdown compared to untreated cells at 8 hours after treatment (***p<0.001) (Figures 5A and B).
Figure 5. MXD3 ASO-SHA NP complexes also show efficient MXD3 protein knockdown in SK-N-BE cells.
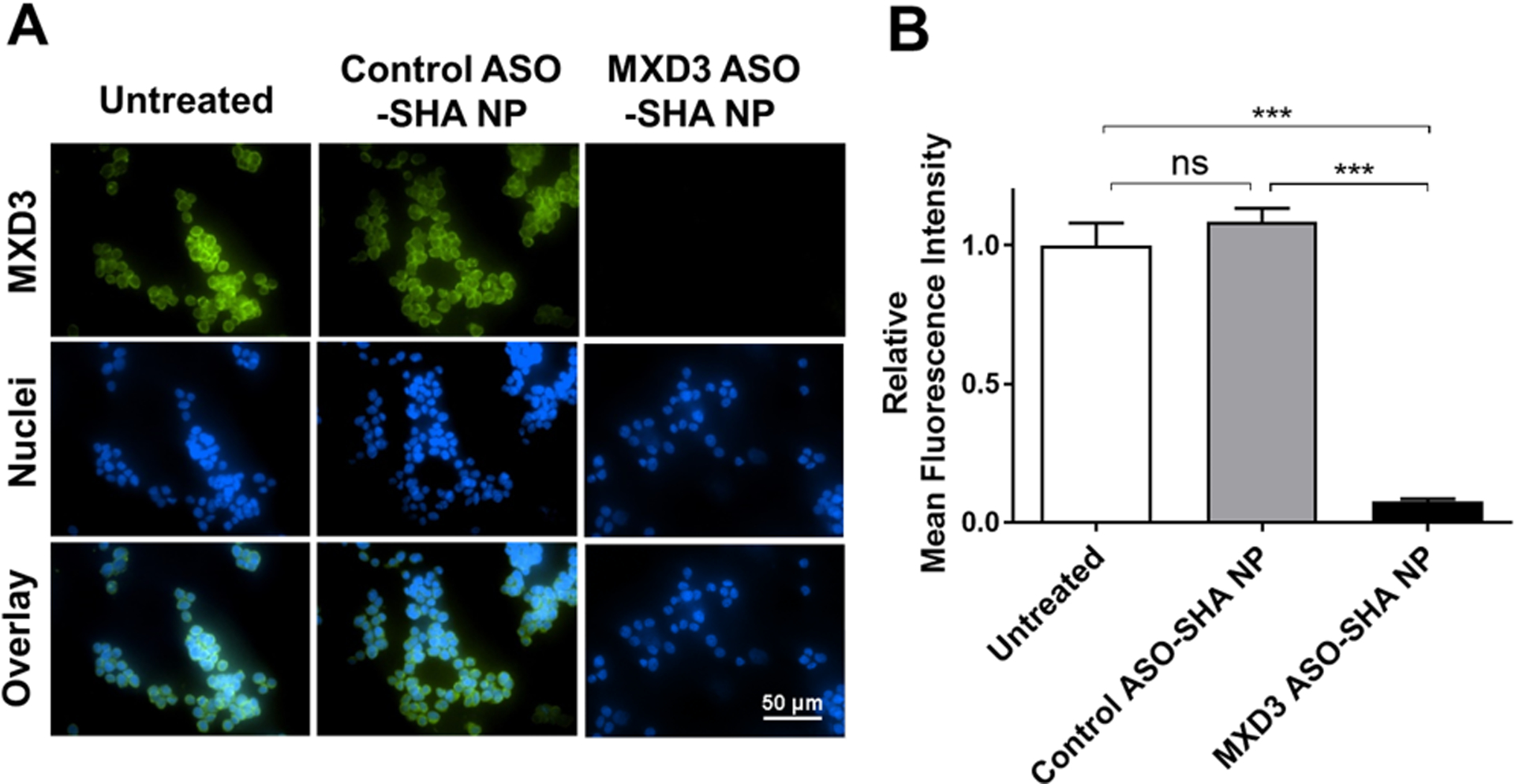
(A) Immunocytochemistry and (B) relative MFI 8 hours after treatment (n=6, ***p<0.001).
MXD3 ASO-SHA NP complex treatment also induces cell apoptosis in SK-N-DZ cells
MXD3 ASO-SHA NP complexes induced apoptosis in SK-N-DZ cells (Figure 6A to C). At 4 hours after treatment, both untreated and control ASO-SHA NP complex-treated cells showed few AV positive cells (average 4.5% or 4.8%, respectively) (Figure 6B) or AV and PI double positive cells (average 4.0% or 4.3%, respectively) (Figure 6C). The MXD3 ASO-SHA NP complex-treated cells showed more AV positive cells (average 7.0%) (Figure 6B) or double positive cells (average 10.4%) (Figure 6C) than controls. At 8 hours after treatment, both untreated and control ASO-SHA NP complex-treated cells continued to have few AV cells (average 2.9% or 3.1%, respectively) or double positive cells (average 4.0% or 4.3%, respectively). However, the MXD3 ASO-SHA NP complex-treated cells showed significantly more AV positive cells (average 7.4%, **p<0.01) (Figure 6B) or double positive cells (average 13.4%, *p<0.05) (Figure 6C) than the untreated cells or the control ASO-SHA NP complex-treated cells. MXD3 ASO-SHA NP complexes showed more MXD3 reduction than MXD3 ASO-SEI NP complexes, although the amount of ASO for MXD3 ASO-SHA NP complexes (9.8nM) was lower than for MXD3 ASO-SEI NP complexes (34.8nM) (Table 2). In addition, control ASO-SHA NP complexes, even at the higher concentration (0.16 nM of SHA NP), showed no toxicity compared with control ASO-SEI NP complexes (0.054 nM of SEI NP) (Supplementary Figure 5A and B). These results suggest that SHA NPs were an efficient tool for ASO delivery with minimum non-specific toxicity.
Figure 6. MXD3 ASO-SHA NP complex treatment also induces cell apoptosis in SK-N-DZ cells.
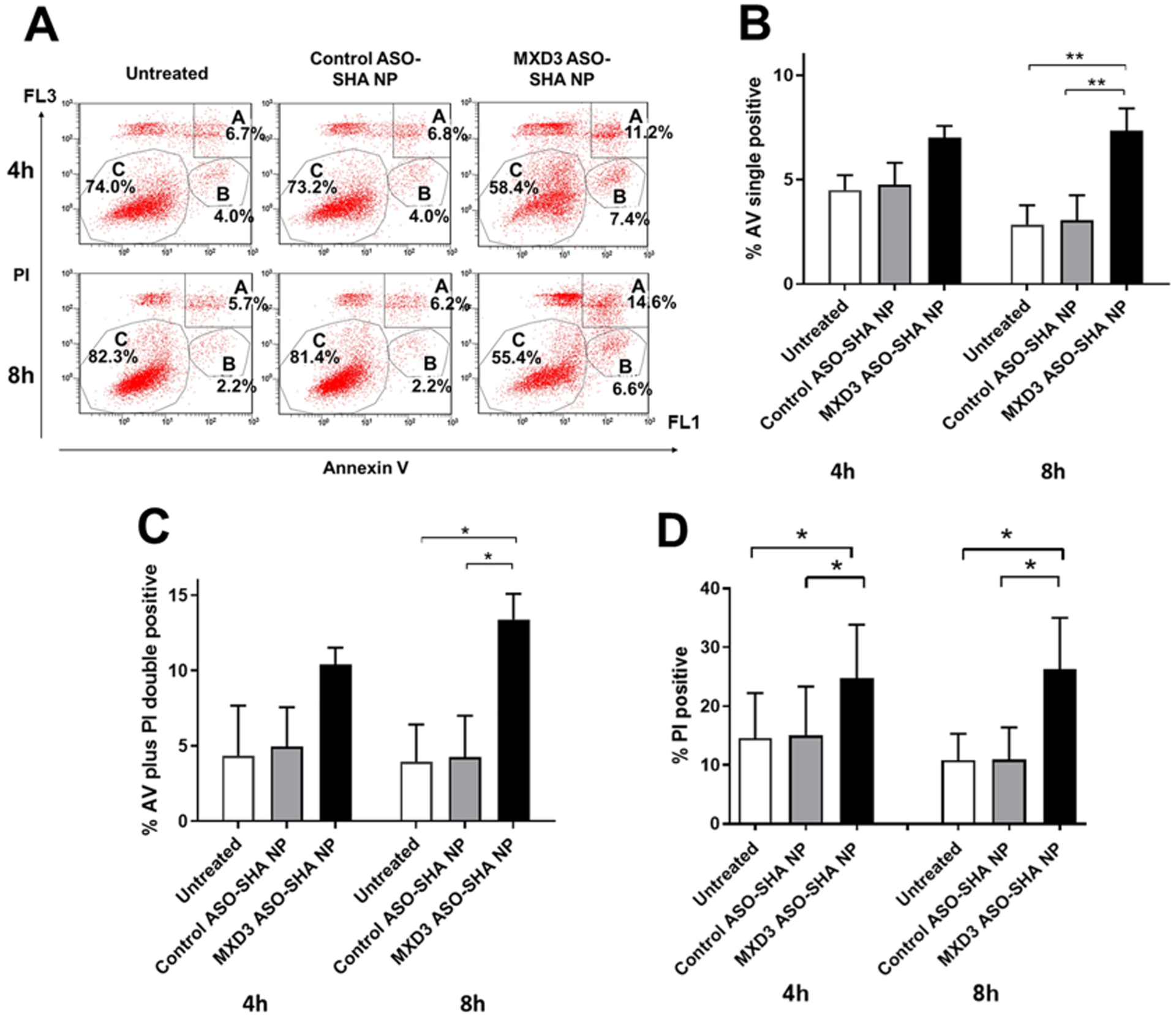
(A) AV and PI 4 and 8 hours after treatment. Area A, B and C indicate the same as in Figure 2. (B) Quantification of AV single positive cells and (C) AV and PI double positive cells and (D) PI single positive cells (n=2, *p<0.05, **p<0.01).
To confirm the stability of nanocomplexes in serum-supplemented condition, MXD3 ASO-SHA NP complexes were tested in 10% heat inactivated serum-supplemented complete SK-N-DZ medium. The nanocomplex-treated cells showed similar MXD3 knockdown in serum-supplemented medium (89.7%, Figure 7A and B) compared to serum-free medium (95.8%, Figure 4A and B) at 8 hours after treatment. On the other hand, SEI NP complex-treated cells showed less MXD3 knockdown in serum-supplemented medium (34%) compared to serum-free medium (59.1%) at 8 hours after treatment (data not shown). These results suggest that MXD3 ASO-SHA NP complexes were more stable in serum-supplemented condition than MXD3 ASO-SEI NP complexes, which should be an advantage for in vivo use.
Figure 7. MXD3 ASO-SHA NP complexes also show MXD3 protein knockdown in serum-supplemented medium for SK-N-DZ cells.

(A) Immunocytochemistry and (B) relative MFI 8 hours after treatment (n=6, **p<0.01).
Discussion
During the last decade, new molecular targets and therapeutic approaches have been discovered based on emerging knowledge regarding the biology and genetic basis of NB.2,36,37 We demonstrated the potential of ASO NP complexes targeting a transcription factor MXD3 in NB. We used SPIO NPs as a delivery platform for ASOs based on their unique magnetic properties, biocompatibility and ease of physical and chemical conjugation of various biological agents.38,39 Regarding the fate of iron oxide NPs, potential cellular degradation of iron oxide NPs has been reported.40–43 In these studies, NP degradation was measured based on the release of59Fe from the NPs and its incorporation into hemoglobin in blood. Rate of degradation depends on the type of cells in the body. The degradation of NPs by liver cells is relatively rapid as compared to the perivascular endothelial cells. They also suggest some fraction of NPs or their remnants may remain in some cells, and this may induce chronic toxicity or inflammatory response. These potential side effects of NP treatments need to be experimentally and clinically validated as these results can be significantly influenced by both the size and nature of coatings on the NPs.43
We first developed MXD3 ASO-SEI NP complexes, using an electrostatic interaction, resulting in MXD3 knockdown (Figure 1 and Supplementary Figure 1) in a dose-dependent manner (Supplementary Figure 2), leading to an increase in cell apoptosis (Figure 2A–D). To date, there are no reports of ASO-PEI coated SPIO NP complexes for gene silencing; however, we have demonstrated successful knockdown of MXD3 using siRNA-SEI NP complexes, in NB26 and preB ALL cell lines.23 Veiseh et al. also reported successful development of siRNA-PEI coated SPIO NP complexes, leading to gene silencing in a glioblastoma cell line in vitro.44 Although ASO-PEI-SPIO complexes were effective in reducing the protein expression of MDX3 (over 80%), they were cytotoxic as observed in the control cells, possibly due to the high cationic charge on the PEI polymer. Similar NP-associated toxicity was also observed in our previous study using siRNA-antiCD22 antibody-SEI NP complexes in Reh preB ALL cells.23 Based on the charge and its structure, PEI is well known to be toxic to cells.45 The cytotoxicity may be reduced by modification of the PEI polymer, including its molecular weight and structure. In addition to the cytotoxicity, the knockdown efficiency of ASO-PEI-SPIO complexes was significantly reduced in the presence of serum, indicating potential non-specific interactions of PEI SPIO particles with serum components, including proteins. These interactions may inhibit entry of these particles into cells.46,47 To alleviate these challenges, we evaluated the chemically-conjugated ASOs with amine-functionalized SHA NPs using a click chemistry approach.
Chemically-conjugated ASOs on SHA NPs knocked down the target protein MXD3 (over 90% compared to the controls), with minimum NP-associated cytotoxicity and improved serum stability (Figure 7A and B). The antisense efficiency of SHA NPs was significantly higher than SEI NPs despite the loaded ASO being higher in the SEI NPs. The difference in antisense activity of the NP-ASO complexes may be attributed to the stability of the complexes, internalization of the complexes, and their intracellular interactions with target RNA and other cellular machinery. Our results also show that ASO-SHA NPs are relatively stable in serum in contrast to ASO-SEI NPs (Figure 7), which may enhance their cellular uptake in the presence of serum. The net positive charge of the ASO-SEI NPs may favor their binding to the cell surface and potential internalization in cells; however, due to the cytotoxicity of SEI-NPs, their intracellular fate may be significantly different than the SHA-NPs. It has been reported that positively charged surfaces may be more readily covered with diverse proteins48 or targeted to specific compartments in the cells such as lysosomes49 that can inhibit or delay the activity of the ASO-NP complexes. Further studies are required to validate these differences for the complexes evaluated in this study.
Previous studies have used ASOs conjugated to inorganic nanoparticles, such as Rosi et al.50, which evaluated the knockdown of green fluorescent protein (EGFP) using ASOs conjugated to gold nanoparticles, and Kim et al.51, which demonstrated the knockdown of p53 protein in HeLa cells using ssDNA-functionalized gold nanoparticles. Similar to the results of our study, these studies demonstrate the effectiveness of chemically-conjugated ASOs to knockdown the target gene expression without significant cytotoxicity. However, the knockdown efficiency in these previously-reported studies were limited to less than 40%, while we observed a dose-dependent increase in knockdown efficiency of up to 90% knockdown of the target protein expression.50–52 Better knockdown in our study could be attributed to improved nuclease stability of the ASOs compared to unmodified DNA molecules used in previous studies. Previous studies have noted improvement in knockdown efficiency with increased stability of nucleic acid molecules.52,53 The effect of nuclease stability of ASOs may be enhanced as these ASOs are non-specifically delivered to the cytoplasm of target cells, often involving endocytic uptake of the particles. Late Endocytic vesicles enriched with nucleases can degrade non-chemically modified DNA molecules and thus reduce the efficiency of the knockdown of gene expression.54,55 In addition, enhanced knockdown may also depend on the stability of the target protein. MXD3 has a short half-life, for instance 0.37 hours in SK-N-DZ cells (Supplementary Figure 6) and 0.25 hours in Reh cells (data not shown) compared to EGFP (26 hours)56 and P53 proteins (0.6 hours)57. Since the measurement of knockdown efficiency is focused on protein expression levels, it is likely that the less stable proteins may provide higher knockdown efficiency. In addition, the nucleotide sequence targeting the MXD3 RNA sequence may also have an effect on the knockdown efficiency as prior antisense studies have shown sequence-dependent effects on the knockdown of target gene expression.30 However, in this study, we varied the target sequences and did not observe significant differences in the knockdown efficiency of the target protein, suggesting that the dosage and stability of the ASOs were the leading factors influencing knockdown efficiency of MXD3 protein.
The biological effect of MXD3 knockdown on cell physiology was measured based on the increase in number of apoptotic cells compared to negative controls. Early (AV positive) and late (AV and PI positive) apoptosis were measured by flow cytometry.58 Our data, showing more double positive cells and less AV positive cells at 8 hours than at 4 hours (Figures 2A, B, and C and 6A and B), suggest cells undergoing apoptosis. For SEI NP complexes, apoptosis was also confirmed using caspase activity (figure 2D). The potential for apoptosis induced by SEI NPs is also supported by another study.43 This study illustrated that PEI-coated iron oxide NPs have significantly higher toxicity than neutral charged PEG-coated iron oxide NPs. This higher toxicity of PEI-NPs was attributed to both ROS generation and induction of apoptosis. Previously, we demonstrated that MXD3 knockdown in Reh cells, using a small hairpin RNA, resulted in decreased cell numbers with no change in G0/G1, S or G2/M population.29 These results indicate that MXD3 knockdown causes clonogenic cell death, but not cell senescence. In addition to apoptosis, necrosis is a possibility, which will be determined in future studies. MXD3 knockdown induced cell apoptosis was observed as quickly as 4 hours and peaked at 8 hours after ASO nanocomplex treatment. This fast phenotype change is most likely due to the short half-life of MXD3 protein.
Current standard therapy for high-risk NB, such as conventional chemotherapy and radiation therapy, has significant limitations and systemic toxicities for young children. A few NB-targeted treatments, such as 131-iodine metaiodobenzylguanidine (MIBG)59 and anti-Ganglioside GD2 antibody60, have been introduced with some success. However, better and safer treatments are still needed. Our ASO-SHA NP complexes have significant potential as a future therapy, because our target molecule MXD3 is preferentially expressed in high-risk NB26, ASO conjugated NP complexes can be delivered intracellularly without major toxicities, and the complexes led to NB cell apoptosis. Because of these unique properties, our nanocomplexes can be included with conventional chemotherapy, and replace or reduce the doses of some of the chemotherapy drugs, to lessen side effects. Further studies are required to understand the function of MXD3 in cancer and mechanism of apoptosis induction when it is knocked down to develop effective MXD3 targeted therapeutics.
Supplementary Material
Financial support:
This work was supported by research funding from the Keaton’s Child Cancer Alliance, The Hartwell Foundation, Hyundai Hope on Wheels, Academic Senate Research Grant, National Center for Advancing Translational Sciences, NIH, through Grant #UL1 TR000002, and CTSC-MCRTP (Satake), and Children’s Cancer Association of Japan (Yoshida). Statistical support was provided through the Biostatistics Shared Resource, UC Davis Comprehensive Cancer Center Support Grant P30CA093373-04.
References
- 1.Schleiermacher G, Janoueix-Lerosey I, Delattre O. Recent insights into the biology of neuroblastoma. Int J Cancer 2014; 135: 2249–61. [DOI] [PubMed] [Google Scholar]
- 2.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet 2007; 369: 2106–20. [DOI] [PubMed] [Google Scholar]
- 3.Moreno L, Vaidya SJ, Schrey D, Pinkerton CR, Lewis IJ, Kearns PR, et al. Long-term analysis of children with metastatic neuroblastoma treated in the ENSG5 randomised clinical trial. Pediatric blood & cancer 2019; 66: e27565. [DOI] [PubMed] [Google Scholar]
- 4.Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov 2012; 11: 125–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sardone V, Zhou H, Muntoni F, Ferlini A, Falzarano MS. Antisense Oligonucleotide-Based Therapy for Neuromuscular Disease. Molecules (Basel, Switzerland) 2017; 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chowdhury S, Burris HA III, Patel M, Infante JR, Jones SF, Voskoboynik M, et al. A phase I dose escalation, safety and pharmacokinetic (PK) study of AZD5312 (IONIS-ARRx), a first-in-class Generation 2.5 antisense oligonucleotide targeting the androgen receptor (AR). European Journal of Cancer 2016; 69: S145. [Google Scholar]
- 7.Yamamoto Y, Loriot Y, Beraldi E, Zhang F, Wyatt AW, Al Nakouzi N, et al. Generation 2.5 antisense oligonucleotides targeting the androgen receptor and its splice variants suppress enzalutamide-resistant prostate cancer cell growth. Clinical cancer research : an official journal of the American Association for Cancer Research 2015; 21: 1675–87. [DOI] [PubMed] [Google Scholar]
- 8.Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med 2015; 7: 314ra185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castanotto D, Stein CA. Antisense oligonucleotides in cancer. Current opinion in oncology 2014; 26: 584–9. [DOI] [PubMed] [Google Scholar]
- 10.Moreno PM, Pego AP. Therapeutic antisense oligonucleotides against cancer: hurdling to the clinic. Frontiers in chemistry 2014; 2: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding Y, Jiang Z, Saha K, Kim CS, Kim ST, Landis RF, et al. Gold nanoparticles for nucleic acid delivery. Molecular therapy : the journal of the American Society of Gene Therapy 2014; 22: 1075–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu XA, Choi CH, Zhang C, Hao L, Mirkin CA. Intracellular fate of spherical nucleic acid nanoparticle conjugates. Journal of the American Chemical Society 2014; 136: 7726–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Misra R, Acharya S, Sahoo SK. Cancer nanotechnology: application of nanotechnology in cancer therapy. Drug Discovery Today 2010; 15: 842–50. [DOI] [PubMed] [Google Scholar]
- 14.Sun T, Zhang YS, Pang B, Hyun DC, Yang M, Xia Y. Engineered nanoparticles for drug delivery in cancer therapy. Angewandte Chemie (International ed in English) 2014; 53: 12320–64. [DOI] [PubMed] [Google Scholar]
- 15.Jung KH, Kim K-P, Yoon DH, Hong YS, Choi C-M, Ahn J-H, et al. A phase I trial to determine the maximum tolerated dose and evaluate the safety and pharmacokinetics (PK) of docetaxel-PNP, polymeric nanoparticle formulation of docetaxel, in subjects with advanced solid malignancies. JOURNAL OF CLINICAL ONCOLOGY; 2012: AMER SOC CLINICAL ONCOLOGY 2318 MILL ROAD, STE 800, ALEXANDRIA, VA 22314 USA; 2012. [Google Scholar]
- 16.Li L, Jiang W, Luo K, Song H, Lan F, Wu Y, et al. Superparamagnetic iron oxide nanoparticles as MRI contrast agents for non-invasive stem cell labeling and tracking. Theranostics 2013; 3: 595–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neri M, Maderna C, Cavazzin C, Deidda-Vigoriti V, Politi LS, Scotti G, et al. Efficient In Vitro Labeling of Human Neural Precursor Cells with Superparamagnetic Iron Oxide Particles: Relevance for In Vivo Cell Tracking. STEM CELLS 2008; 26: 505–16. [DOI] [PubMed] [Google Scholar]
- 18.Duan J, Dong J, Zhang T, Su Z, Ding J, Zhang Y, et al. Polyethyleneimine-functionalized iron oxide nanoparticles for systemic siRNA delivery in experimental arthritis. Nanomedicine (London, England) 2014; 9: 789–801. [DOI] [PubMed] [Google Scholar]
- 19.Shah V, Taratula O, Garbuzenko OB, Patil ML, Savla R, Zhang M, et al. Genotoxicity of different nanocarriers: possible modifications for the delivery of nucleic acids. Current drug discovery technologies 2013; 10: 8–15. [PMC free article] [PubMed] [Google Scholar]
- 20.Butoescu N, Jordan O, Burdet P, Stadelmann P, Petri-Fink A, Hofmann H, et al. Dexamethasone-containing biodegradable superparamagnetic microparticles for intra-articular administration: physicochemical and magnetic properties, in vitro and in vivo drug release. European journal of pharmaceutics and biopharmaceutics : official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik eV 2009; 72: 529–38. [DOI] [PubMed] [Google Scholar]
- 21.Hogemann-Savellano D, Bos E, Blondet C, Sato F, Abe T, Josephson L, et al. The transferrin receptor: a potential molecular imaging marker for human cancer. Neoplasia (New York, NY) 2003; 5: 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suwa T, Ozawa S, Ueda M, Ando N, Kitajima M. Magnetic resonance imaging of esophageal squamous cell carcinoma using magnetite particles coated with anti-epidermal growth factor receptor antibody. Int J Cancer 1998; 75: 626–34. [DOI] [PubMed] [Google Scholar]
- 23.Satake N, Duong C, Chen C, Barisone GA, Diaz E, Tuscano J, et al. Targeted therapy with MXD3 siRNA, anti-CD22 antibody and nanoparticles for precursor B-cell acute lymphoblastic leukaemia. British journal of haematology 2014; 167: 487–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wen M, Li B, Ouyang Y, Luo Y, Li S. Preparation and quality test of superparamagnetic iron oxide labeled antisense oligodeoxynucleotide probe: a preliminary study. Annals of biomedical engineering 2009; 37: 1240–50. [DOI] [PubMed] [Google Scholar]
- 25.Taratula O, Garbuzenko O, Savla R, Wang YA, He H, Minko T. Multifunctional nanomedicine platform for cancer specific delivery of siRNA by superparamagnetic iron oxide nanoparticles-dendrimer complexes. Current drug delivery 2011; 8: 59–69. [DOI] [PubMed] [Google Scholar]
- 26.Duong C, Yoshida S, Chen C, Barisone G, Diaz E, Li Y, et al. Novel targeted therapy for neuroblastoma: silencing the MXD3 gene using siRNA. Pediatric research 2017; 82: 527–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wen M, Li B, Bai W, Li S, Yang X. Application of atomic force microscopy in morphological observation of antisense probe labeled with magnetism. Molecular vision 2008; 14: 114–7. [PMC free article] [PubMed] [Google Scholar]
- 28.Grandori C, Cowley SM, James LP, Eisenman RN. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annual review of cell and developmental biology 2000; 16: 653–99. [DOI] [PubMed] [Google Scholar]
- 29.Barisone GA, Satake N, Lewis C, Duong C, Chen C, Lam KS, et al. Loss of MXD3 induces apoptosis of Reh human precursor B acute lymphoblastic leukemia cells. Blood cells, molecules & diseases 2015; 54: 329–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Satake N, Duong C, Yoshida S, Oestergaard M, Chen C, Peralta R, et al. Novel Targeted Therapy for Precursor B Cell Acute Lymphoblastic Leukemia: anti-CD22 Antibody-MXD3 Antisense Oligonucleotide Conjugate. Molecular medicine (Cambridge, Mass) 2016; 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agard NJ, Prescher JA, Bertozzi CR. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. Journal of the American Chemical Society 2004; 126: 15046–7. [DOI] [PubMed] [Google Scholar]
- 32.Nitin N, LaConte LE, Zurkiya O, Hu X, Bao G. Functionalization and peptide-based delivery of magnetic nanoparticles as an intracellular MRI contrast agent. Journal of biological inorganic chemistry : JBIC : a publication of the Society of Biological Inorganic Chemistry 2004; 9: 706–12. [DOI] [PubMed] [Google Scholar]
- 33.Hunter R Zeta Potential in Colloid Science Academic. New York 1981: 69. [Google Scholar]
- 34.Burgess A, Vigneron S, Brioudes E, Labbe JC, Lorca T, Castro A. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proceedings of the National Academy of Sciences of the United States of America 2010; 107: 12564–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Statistics in medicine 1990; 9: 811–8. [DOI] [PubMed] [Google Scholar]
- 36.Nicolai S, Pieraccioli M, Peschiaroli A, Melino G, Raschella G. Neuroblastoma: oncogenic mechanisms and therapeutic exploitation of necroptosis. Cell death & disease 2015; 6: e2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luksch R, Castellani MR, Collini P, De Bernardi B, Conte M, Gambini C, et al. Neuroblastoma (Peripheral neuroblastic tumours). Critical reviews in oncology/hematology 2016; 107: 163–81. [DOI] [PubMed] [Google Scholar]
- 38.Yang Z, Duan J, Wang J, Liu Q, Shang R, Yang X, et al. Superparamagnetic iron oxide nanoparticles modified with polyethylenimine and galactose for siRNA targeted delivery in hepatocellular carcinoma therapy. International journal of nanomedicine 2018; 13: 1851–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mok H, Zhang M. Superparamagnetic iron oxide nanoparticle-based delivery systems for biotherapeutics. Expert opinion on drug delivery 2013; 10: 73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bargheer D, Nielsen J, Gebel G, Heine M, Salmen SC, Stauber R, et al. The fate of a designed protein corona on nanoparticles in vitro and in vivo. Beilstein J Nanotechnol 2015; 6: 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Freund B, Tromsdorf UI, Bruns OT, Heine M, Giemsa A, Bartelt A, et al. A simple and widely applicable method to 59Fe-radiolabel monodisperse superparamagnetic iron oxide nanoparticles for in vivo quantification studies. ACS nano 2012; 6: 7318–25. [DOI] [PubMed] [Google Scholar]
- 42.Bargheer D, Giemsa A, Freund B, Heine M, Waurisch C, Stachowski GM, et al. The distribution and degradation of radiolabeled superparamagnetic iron oxide nanoparticles and quantum dots in mice. Beilstein J Nanotechnol 2015; 6: 111–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng Z, Yuan J, He G, Hu W, Lin Z, Li D, et al. Tunable critical temperature for superconductivity in FeSe thin films by pulsed laser deposition. Sci Rep 2018; 8: 4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Veiseh O, Kievit FM, Liu V, Fang C, Stephen ZR, Ellenbogen RG, et al. In vivo safety evaluation of polyarginine coated magnetic nanovectors. Molecular pharmaceutics 2013; 10: 4099–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hao F, Li Y, Zhu J, Sun J, Marshall B, Lee RJ, et al. Polyethylenimine-based formulations for delivery of oligonucleotides. Current medicinal chemistry 2018. [DOI] [PubMed] [Google Scholar]
- 46.Douat C, Bornerie M, Antunes S, Guichard G, Kichler A. Hybrid Cell-Penetrating Foldamer with Superior Intracellular Delivery Properties and Serum Stability. Bioconjugate chemistry 2019; 30: 1133–9. [DOI] [PubMed] [Google Scholar]
- 47.Douat C, Aisenbrey C, Antunes S, Decossas M, Lambert O, Bechinger B, et al. A cell-penetrating foldamer with a bioreducible linkage for intracellular delivery of DNA. Angewandte Chemie (International ed in English) 2015; 54: 11133–7. [DOI] [PubMed] [Google Scholar]
- 48.Chen L, Simpson JD, Fuchs AV, Rolfe BE, Thurecht KJ. Effects of Surface Charge of Hyperbranched Polymers on Cytotoxicity, Dynamic Cellular Uptake and Localization, Hemotoxicity, and Pharmacokinetics in Mice. Molecular pharmaceutics 2017; 14: 4485–97. [DOI] [PubMed] [Google Scholar]
- 49.Chen Z, Zhang L, He Y, Shen Y, Li Y. Enhanced shRNA delivery and ABCG2 silencing by charge-reversible layered nanocarriers. Small 2015; 11: 952–62. [DOI] [PubMed] [Google Scholar]
- 50.Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AK, Han MS, Mirkin CA. Oligonucleotide-modified gold nanoparticles for intracellular gene regulation. Science (New York, NY) 2006; 312: 1027–30. [DOI] [PubMed] [Google Scholar]
- 51.Kim JH, Jang HH, Ryou SM, Kim S, Bae J, Lee K, et al. A functionalized gold nanoparticles-assisted universal carrier for antisense DNA. Chemical communications (Cambridge, England) 2010; 46: 4151–3. [DOI] [PubMed] [Google Scholar]
- 52.Jiwaji M, Sandison ME, Reboud J, Stevenson R, Daly R, Barkess G, et al. Quantification of functionalised gold nanoparticle-targeted knockdown of gene expression in HeLa cells. PloS one 2014; 9: e99458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo S, Huang Y, Jiang Q, Sun Y, Deng L, Liang Z, et al. Enhanced gene delivery and siRNA silencing by gold nanoparticles coated with charge-reversal polyelectrolyte. ACS nano 2010; 4: 5505–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ponnuswamy N, Bastings MMC, Nathwani B, Ryu JH, Chou LYT, Vinther M, et al. Oligolysine-based coating protects DNA nanostructures from low-salt denaturation and nuclease degradation. Nature communications 2017; 8: 15654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun X, Liu C, Liu D, Li P, Zhang N. Novel biomimetic vectors with endosomal-escape agent enhancing gene transfection efficiency. International journal of pharmaceutics 2012; 425: 62–72. [DOI] [PubMed] [Google Scholar]
- 56.Corish P, Tyler-Smith C. Attenuation of green fluorescent protein half-life in mammalian cells. Protein Engineering, Design and Selection 1999; 12: 1035–40. [DOI] [PubMed] [Google Scholar]
- 57.Baresova P, Musilova J, Pitha PM, Lubyova B. p53 tumor suppressor protein stability and transcriptional activity are targeted by Kaposi’s sarcoma-associated herpesvirus-encoded viral interferon regulatory factor 3. Molecular and cellular biology 2014; 34: 386–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods 1995; 184: 39–51. [DOI] [PubMed] [Google Scholar]
- 59.Trieu M, DuBois SG, Pon E, Nardo L, Hawkins RA, Marachelian A, et al. Impact of Whole-Body Radiation Dose on Response and Toxicity in Patients With Neuroblastoma After Therapy With 131 I-Metaiodobenzylguanidine (MIBG). Pediatric blood & cancer 2016; 63: 436–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sait S, Modak S. Anti-GD2 immunotherapy for neuroblastoma. Expert Rev Anticancer Ther 2017; 17: 889–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.