

ABSTRACT
CACNA1 C, which codes for the Cav1.2 isoform of L-type Ca2+ channels (LTCCs), is a prominent risk gene in neuropsychiatric and neurodegenerative conditions. A role forLTCCs, and Cav1.2 in particular, in transcription-dependent late long-term potentiation (LTP) has long been known. Here, we report that elimination of Cav1.2 channels in glutamatergic neurons also impairs theta burst stimulation (TBS)-induced LTP in the hippocampus, known to be transcription-independent and dependent on N-methyl D-aspartate receptors (NMDARs) and local protein synthesis at synapses. Our expansion of the established role of Cav1.2channels in LTP broadens understanding of synaptic plasticity and identifies a new cellular phenotype for exploring treatment strategies for cognitive dysfunction.
KEYWORDS: Calcium, Cav1.2, long-term potentiation
Introduction
The role of the Cav1.2 isoform of L-type Ca2+ channels (LTCCs) is well known in hippocampal-mediated long-term memory and related behaviors [1–5].The clinical relevance of this relationship is bolstered by the fact thatCACNA1 C, the gene that encodesCav1.2, is a prominent risk gene for a wide array of neuropsychiatric [2,3,6,7]andneurodegenerative [8–11]disorders that manifest with cognitive impairment.
Hippocampal LTP at CA3 Schaffer collaterals to CA1 neurons is a common synaptic model of learning and memory [12,13], and it has long been known that Ca2+ influx from LTCCs plays an important role in N-methyl-D-aspartate receptor (NMDAR)-independent, transcription-dependent LTP [14–18]. This is also consistent with the critical role of LTCCs in activity-dependent transcription and long-term memory [19–22]. Recently, we have reported that Cav1.2 channels additionally regulate local synaptic protein synthesis through adjusting mTORC1protein translational machinery [23]. Since this same mechanism is also involved in NMDAR-dependent, transcription-independent LTP [24], we hypothesized that Cav1.2 channels might be required for this earlier form of LTP as well. NMDAR-dependent, transcription-independent LTP can be elicited by theta burst stimulation (TBS) at glutamatergic synapses in the hippocampus [25–27], and TBS closely mimics the natural rhythms of neuronal activity in the brain [25,28]. Importantly, TBS delivery through transcranial magnetic stimulation has been approved by the United States Food and Drug Administration for the treatment of major depression [29,30]. Thus, we examined whether conditional deletion of Cav1.2 in forebrain glutamatergic neurons (Cav1.2KO) would disrupt TBS-induced LTP at Schaffer collateral/CA1 synapses.
Materials and methods
Animals
All animal procedures were performed in accordance with the policies and regulations of the University of Iowa and Weill Cornell Medicine institutional animal care and use committees. Male CamK2-Cre Cav1.2KO mice and wild type (WT) littermates maintained on a C57Bl/6 J background were used. Mice were housed in temperature-controlled conditions, provided food and water ad libitum, and maintained on a 12-h light/dark cycle.
Preparation of acute hippocampal slices
Sagittalhippocampal slices (400 μm) from adult mice (> postnatal day 60) were cut using a Vibratome 1000 Plus (Vibratome, St. Louis, MO) in ice-cold slicing buffer (in mM: 127 NaCl, 26 NaHCO3, 1.2 KH2PO4, 1.9 KCl, 1.1 CaCl2, 2 MgSO4, 10 D-Glucose) bubbled with 95% O2 and 5% CO2. Slices were transferred to a holding chamber containing oxygenated artificial cerebrospinal fluid (ACSF; in mM: 127 NaCl, 26 NaHCO3, 1.2 KH2PO4, 1.9 KCl, 2.2 CaCl2, 1 MgSO4, 10 D-Glucose) for 30 min at 34°C and for another 30 min at 22°C for recovery. Slices were then transferred to a submersion recording chamber continually perfused with 32°C oxygenated artificial cerebrospinal fluid (ACSF) (rate: 2 ml/min). Slices were equilibrated for at least 15 min before each recording.
Electrophysiology
ACSF-filled glass electrodes (resistance <1 MΩ) were positioned in the stratum radiatum of area CA1 for extracellular recording. Synaptic responses were evoked by stimulating Schaffer collaterals with 0.2 ms pulses once every 15 s. The stimulation intensity was systematically increased to determine the maximal field excitatory post-synaptic potential (fEPSP) slope and then adjusted to yield 40–60% of the maximal (fEPSP) slope. Experiments with maximal fEPSPs of less than 0.5 mV, with large fiber volleys, or with substantial changes in the fiber volley during recording, were rejected. LTP was induced by 12TBS (12 bursts, each of 4 pulses at 100 Hz, with pulse duration of 0.2 ms and 5Hzinterburst frequency). Field EPSPs were recorded (AxoClamp 900A amplifier, Axon Instruments, Foster City, CA), filtered at 1 kHz, digitized at 10 kHz (Axon Digidata 1440), and stored for off-line analysis (Clampfit 10). Initial slopes of fEPSPs were expressed as percentages of baseline averages. In the summary graph of LTP, each point represents the average of four consecutive responses. Time-matched, normalized data were averaged across experiments and expressed as means±SEM.
Subcellular fractionation and immunoblotting
Synaptosomal fractions from adult (> P60) hippocampus were generated as previously described [23,31]and used for western blot analysis. Briefly, tissue was homogenized in 0.3 M sucrose/0.01 mM HEPES buffer containing protease and phosphatase inhibitors and centrifuged at 1000xg. The supernatant was then spun again at1000xg, with the subsequently obtained fresh supernatant spun at 12,000xg. The final pellet was resuspended in 4 mM HEPES/1 mM EDTA buffer and used as the synaptosome fraction. Protein concentrations were determined using the BCA assay, and protein lysates were separated on a 10% SDS gel along with a Kaleidoscope-prestained protein standard (Bio-Rad, Hercules, CA). Blots were blocked in 5% nonfat dry milk for 1 h and incubated in primary antibody (Table 1) for 12–48 h on a shaker at 4°C. Incubation in secondary antibody was performed at room temperature for 1 h in horseradish peroxidase-linked IgG conjugated antibody. Membranes were visualized using Western Lightning Chemiluminescence solution (Perkin Elmer Life Science, Boston, MA) and optical density was analyzed using NIHImage (NIH, Bethesda, MD). Immunoblot data were analyzed using an independent samples t-test, performed by Prism 8 Graphpad software. Proteins were normalized to GAPDH, which was used as a loading control. Western analyses were done using X-ray film.
Table 1.
List of antibodies used for immunoblots.
| Protein | Company | Catalog number | RRID | Antibody concentration | Molecular Weight (kDa) |
|---|---|---|---|---|---|
| GluN1 | Millipore | Ab9864 | 10,807,557 | 1:1000 | 120 |
| GluN2A | NeuroMab | 75–288 | 2,307,331 | 1:1000 | 170 |
| GluN2B | Millipore | 06–600 | 310,193 | 1:1000 | 180 |
| mTOR | Cell Signaling Technology | 2972 | 330,978 | 1:1000 | 250 |
| p-mTOR S2448 | Cell Signaling Technology | 2971 | 330,970 | 1:1000 | 250 |
| GAPDH | Abcam | Ab22555 | 447,153 | 1:10,000 | 36 |
Statistics
Electrophysiological data were time-matched, normalized, and averaged across experiments and expressed as mean±SEM. LTP was analyzed using a two-tailed unpaired t-test and significant differences were determined as a p value <0.05. Immunoblot data were analyzed using an independent samples t-test. Proteins were normalized to GAPDH, which was used as a loading control. All statistical analysis was performed by Prism 8 Graphpad software.
Results
We determined the effect of forebrain-specific deficiency of Cav1.2 onTBS-induced LTP in adult Cav1.2KO mice and wild type littermates. Recordings were obtained for 60 minutes. Stimulating pulses were delivered to the Schaffer collateral fibers projecting from CA3 to CA1 pyramidal cells, with recording electrodes placed in the CA1 region of the hippocampus (Figure 1(a)). LTP was quantified as the field excitatory postsynaptic potential (fEPSP) slope as a percentage of baseline.Cav1.2KOmicedemonstrated significantly reduced LTP immediately after TBS, and the reduction was sustained for 60 minutes (Figure 1(b)). Measurement of paired-pulse facilitation, an index of presynaptic probability of release, showed that impaired TBS-induced LTP was independent of altered presynaptic machinery, as there was no difference across a wide range of inter-stimulus intervals between Cav1.2KOand WT littermate controls (Figure 1(c)). In addition, no difference in baseline synaptic transmission was observed, based on input/output curves from Cav1.2KOand WT littermate controls (Figure 1(d)).
Figure 1.
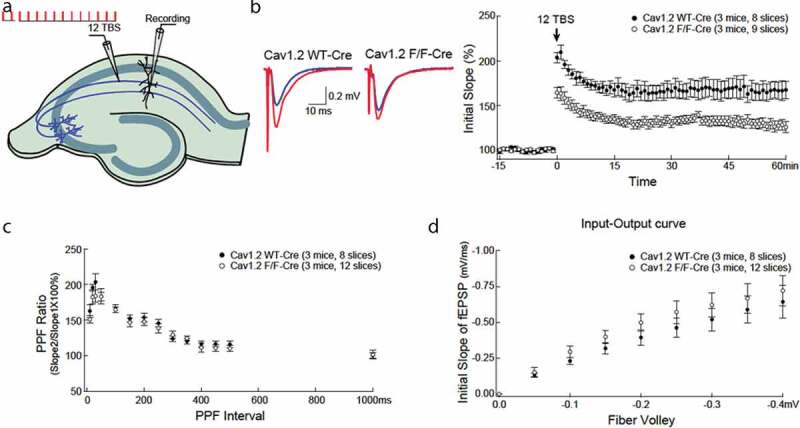
Long-term potentiation (LTP) is significantly impaired in male CamK2Cre, Cav1.2KO mice without apparent alteration in paired-pulse facilitation or input-output curve. (a) Illustration of the recording scheme. The Schaffer collateral pathway projecting to CA1 neurons was stimulated with bipolar stimulating electrodes. LTP was induced by theta burst stimulation (12TBS; 12 bursts, each of 4 pulses at 100 Hz). (b) Example traces before (blue) and after (red) TBS. LTP (at time 60 min) is substantially reduced in CamK2Cre, Cav1.2KO mice compared to wildtype (WT) littermate controls (166 ± 10% vs. 128 ± 6%, t(15) = 3.549, p = 0.0029). Post-stimulation potentiation (at time 0 min) was also significantly reduced in CamK2Cre, Cav1.2KO mice (180 ± 7% vs. 145 ± 5%, t(15) = 4.220, p = 0.0007). (c)There was no difference in paired pulse facilitation between CamK2Cre, Cav1.2KO mice and WT littermate controls over a wide range of inter-stimulus intervals, indicating intact presynaptic machinery in CamK2Cre, Cav1.2KO mice. (d) Input-output curves with the postsynaptic response (initial slope of field excitatory postsynaptic potential (fEPSP)) plotted as a function of the presynaptic fiber volley amplitude were indistinguishable between CamK2Cre, Cav1.2KO mice and WT mice, indicating intact baseline synaptic transmission.
Given that TBS-induced LTP is NMDAR- and mTORC1-dependent [24,27], we next questioned whether the impaired TBS-induced LTP in Cav1.2KOmice might be related to altered levels of NMDAR subunits and phosphorylated mTOR at serine 2448, a marker of active mTORC1 [32].Western blots using synaptosomal fractions of dorsal hippocampus (Figure 2(a)) revealed no difference in levels of NMDAR subunitsGRIN1, GRIN2A, or GRIN2B (Figure 2(b-d)), but did show lower levels of S2448phospho-mTOR (Figure 2(e)), without any difference in total mTOR protein (Figure 2(f)).
Figure 2.
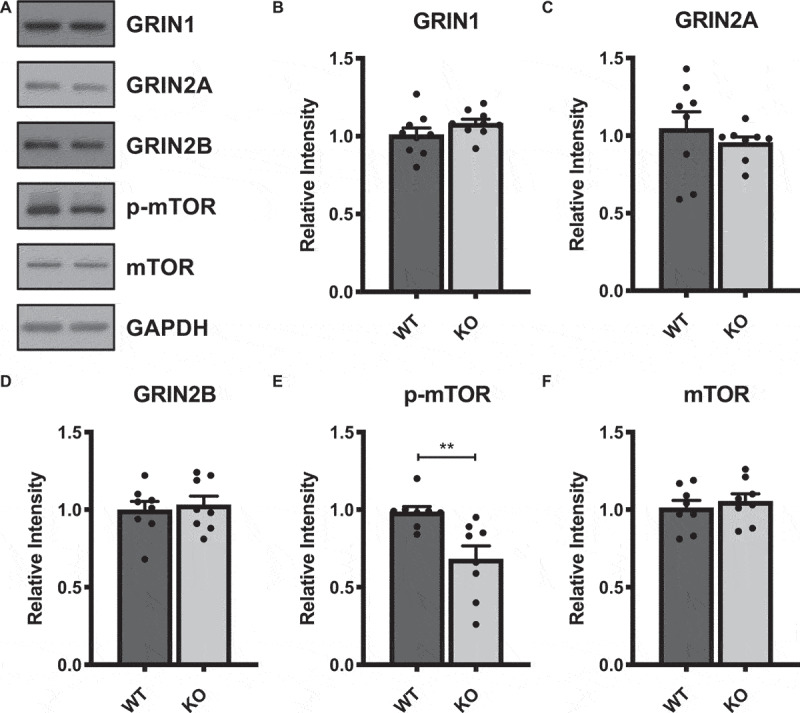
Western blots from isolated synaptosomal fractions of dorsal hippocampus of CamK2Cre, Cav1.2KO mice and WT littermates reveal a decrease on phosphorylated mTOR expression. (a) Representative bands of NMDAR subunits (GRIN1, GRIN2A, and GRIN2B), S2448 phosphorylated mTOR (p-mTOR), and total mTOR protein levels taken from the same blot and adjacent lanes. (b-f) Quantification of relative intensities of respective bands, normalized to GAPDH expression levels.P-mTOR, t(14) = 3.201**p = 0.0064. Data are displayed as mean ± SEM.
Discussion
Until now, LTCCs in neurons have been predominantly studied in the context of their requisite role in induction of NMDAR-independent, transcription-dependent LTP [14–18,33]. With the data presented here, however, we have now established an additional role for Cav1.2 in TBS-induced LTP, which is known to be transcription-independent and dependent on N-methyl D-aspartate receptors (NMDARs) and local protein synthesis at synapses [24]. Specifically, we observed that mice lacking Cav1.2 channels in glutamatergic neurons are impaired inTBS-induced LTPat Schaffer collateral to CA1 synapses, while basal synaptic transmission and presynaptic function are intact. We find no change in NMDAR subunit (GRIN1, GRIN2A, or GRIN2B) levels in hippocampal synaptosomal fractions (generated from the entire hippocampus) as a consequence of Cav1.2 knockout, but do observe reduced levels of active mTORC1, a marker for local protein synthesis. This is reflected by a small decrease in phosphorylated mTOR. It is important to note, however, that these associative results do not prove a causal relationship to diminished TBS-induced LTP. Future studies will address this question at the Schaffer collateral/CA1 synapse. It is also important to note that deletingCav1.2 could conceivably affect NMDAR activity without affecting overall expression levels of NMDAR subunits. Future work to directly measure NMDAR currents will be required to definitively address this possibility. Further investigation will also be necessary to parse the specific mechanisms that relate to our findings. There are several well-studied pathways downstream of Cav1.2 channels that regulate LTP, such as BDNF signaling [34–36], which can be locally translated at the synapse to contribute to synaptic plasticity. Likewise, the CaM Kinase II pathway, which is activated by Cav1.2 [37,38] and enriched in dendritic spines during LTP [39], could result in increased α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) conductance at synapses via CaMKII-mediated phosphorylation of AMPAR subunits, a key mechanism for induction of LTP and synaptic plasticity [40]. In conclusion, our data show that Cav1.2 is required forTBS-induced LTP, which may depend on local synaptic mechanisms in the adult hippocampus.
At first glance, our results appear to contradict a previous report of no role for Cav1.2 in this form of LTP [17]. This difference is likely due to the different promoters that were used to drive Cre recombinase expression during the creation of the two different strains of Cav1.2KO mice. In the previous report, Moosmang et al. [17]used theNex promoter, which is activated during development at embryonic day 12 [41]. By contrast, here we used thealpha-CamK2 promoter, which is not activated until postnatal day 18 [42]. Since Cav1.2calcium signaling is a critical regulator of early neuronal, dendritic and synaptic development [43–47], very early elimination of Cav1.2 via theNexpromoter could lead to developmental adaptations that might allow sufficient synaptic strengthening for the maturation of the embryonic brain necessary for viability. This adaptation could then result in an adult brain deficient in Cav1.2that is still able to execute TBS-induced LTP. Our results here, in which Cav1.2has been selectively eliminated at a much later date (~postnatal day 21), likely represent more faithfully the role of Cav1.2channels in the adult brain. In addition, a role of Cav1.2 channels in TBS-induced LTP is compatible with previous reports of Cav1.2 channel-mediated NMDAR-signaling [48,49] as well as our recent discovery that loss of Cav1.2 results in decreased activation of mTORC1 [23], which is required for TBS-induced LTP [24].Future identification of the molecular adaptations in the Nex promoter-driven versus CaMK2 promoter-driven Cav1.2 KO mouse models could provide insight into early versus later Cav1.2neurodevelopmental processes.
We also note that a critical role for Cav1.2in TBS-induced LTP could be related to the neurocognitive deficits that we and others have previously observed in these same mice [23,50–52], and this form of LTP could result from Cav1.2-mediated hippocampal phenotypes [35,50]. Interestingly, this impairment in LTP is reminiscent of our previously reported findings in the methyl-CpG binding protein 2 (MECP2)-deficient mouse model of Rett syndrome [53], and MECP2 is a downstream target of LTCCs [54]. Conceivably, deficits in TBS-induced LTP may represent a commonality across neuropsychiatric disorders with dysregulated local protein synthesis and cognitive deficits [55,56].In conclusion, this previously unknown role of Cav1.2 in TBS-induced LTPprovidesnew direction for studying Cav1.2 channel mechanisms in this form of LTP and developing potential therapeutics in neuropsychiatric and neurodegenerative disease.
Acknowledgments
We thank Dr. Elisa Na for her critical review of the manuscript.
Funding Statement
This work was supported by grants to A.M.R. by the National Institute of Drug Abuse, The Hartwell Foundation and the Paul Fund, and to A.A.P. by the Brockman Foundation, the Elizabeth Ring Mather & William Gwinn Mather Fund, the S. Livingston Samuel Mather Trust, the G.R. Lincoln Family Foundation, the Leonard Krieger Fund of the Cleveland Foundation, the Maxine and Lester Stoller Parkinson’s Research Fund, the Louis Stokes VA Medical Center resources and facilities, and Gordon & Evie Safran. Some of this material is the result of work supported with resources and the use of facilities at the Louis Stokes VA Medical Center in Cleveland.
Author contribution
P.S.S. interpreted data and wrote the manuscript. Y.L. conducted electrophysiology experiments and R.C.R conducted molecular experiments. A.A.P. and A.M.R. designed experiments, interpreted data, and wrote the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Kabir ZD, Martínez-Rivera A, Rajadhyaksha AM.. From gene to behavior: L-type calcium channel mechanisms underlying neuropsychiatric symptoms. Neurotherapeutics. 2017;14:588–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kabir ZD, Lee AS, Rajadhyaksha AM. L-type Ca2+channels in mood, cognition and addiction: integrating human and rodent studies with a focus on behavioural endophenotypes: L-type Ca2+channels in mood, cognition and addiction. J Physiol. 2016;594:5823–5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Moon AL, Haan N, Wilkinson LS, et al. CACNA1C: association with psychiatric disorders, behavior, and neurogenesis. Schizophrenia Bull. 2018;44:958–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zamponi GW, Striessnig J, Koschak A, et al. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev. 2015;67:821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Nanou E, Catterall WA. Calcium channels, synaptic plasticity, and neuropsychiatric disease. Neuron. 2018;98:466–481. [DOI] [PubMed] [Google Scholar]
- [6].Heyes S, Pratt WS, Rees E, et al. Genetic disruption of voltage-gated calcium channels in psychiatric and neurological disorders. Prog Neurobiol. 2015;134:36–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Liao X, Li Y. Genetic associations between voltage-gated calcium channels and autism spectrum disorder: a systematic review. Mol Brain. 2020;13:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yang T, Wang J, Sun Q, et al. Detecting genetic risk factors for Alzheimer’s disease in whole genome sequence data via Lasso screening. 2015 Ieee 12th Int Symposium Biomed Imaging Isbi, New York, USA. 2015; 985–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hohman TJ, Bush WS, Jiang L, et al. Discovery of gene-gene interactions across multiple independent data sets of late onset Alzheimer disease from the Alzheimer disease genetics consortium. Neurobiol Aging. 2016;38:141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Koran MEI, Hohman TJ, Thornton-Wells TA. Genetic interactions found between calcium channel genes modulate amyloid load measured by positron emission tomography. Hum Genet. 2013;133:85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang L, Maldonado L, Beecham GW, et al. DNA variants in CACNA1C modify Parkinson disease risk only when vitamin D level is deficient. Neurol Genet. 2016;2:e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Granger AJ, Nicoll RA. Expression mechanisms underlying long-term potentiation: a postsynaptic view, 10 years on. Philos Trans R Soc Lond B Biol Sci. 2013;369:20130136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Malenka RC. Synaptic plasticity and AMPA receptor trafficking. Ann Ny Acad Sci. 2003;1003:1–11. [DOI] [PubMed] [Google Scholar]
- [14].Morgan SL, Teyler TJ. VDCCs and NMDARs underlie two forms of LTP in CA1 hippocampus in vivo. J Neurophysiol. 1999;82:736–740. [DOI] [PubMed] [Google Scholar]
- [15].Grover LM, Teyler TJ. N-methyl-d-aspartate receptor-independent long-term potentiation in area CA1 of rat hippocampus: input-specific induction and preclusion in a non-tetanized pathway. Neuroscience. 1992;49:7–11. [DOI] [PubMed] [Google Scholar]
- [16].Grover LM, Teyler TJ. Activation of NMDA receptors in hippocampal area CA1 by low and high frequency orthodromic stimulation and their contribution to induction of long-term potentiation. Synapse. 1994;16:66–75. [DOI] [PubMed] [Google Scholar]
- [17].Moosmang S, Haider N, Klugbauer N, et al. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci [Internet]. 2005; 25: 9883–9892. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6725564/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Huber KM, Mauk MD, Kelly PT. Distinct LTP induction mechanisms: contribution of NMDA receptors and voltage-dependent calcium channels. J Neurophysiol. 1995;73:270–279. [DOI] [PubMed] [Google Scholar]
- [19].Wu G-Y, Deisseroth K, Tsien RW. Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc Natl Acad Sci. 2001;98:2808–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wheeler DG, Groth RD, Ma H, et al. CaV1 and CaV2 channels engage distinct modes of Ca2+ signaling to control CREB-dependent gene expression. Cell. 2012;149:1112–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wheeler DG, Barrett CF, Groth RD, et al. CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation–transcription coupling. J Cell Biol. 2008;183:849–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dolmetsch RE, Pajvani U, Fife K, et al. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP Kinase pathway. Science. 2001;294:333–339. [DOI] [PubMed] [Google Scholar]
- [23].Kabir ZD, Che A, Fischer DK, et al. Rescue of impaired sociability and anxiety-like behavior in adult cacna1c-deficient mice by pharmacologically targeting eIF2α. Mol Psychiatry. 2017;22:1096–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Panja D, Bramham CR. BDNF mechanisms in late LTP formation: a synthesis and breakdown. Neuropharmacology. 2014;76:664–676. [DOI] [PubMed] [Google Scholar]
- [25].Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. [DOI] [PubMed] [Google Scholar]
- [26].Kleppisch T, Pfeifer A, Klatt P, et al. Long-term potentiation in the hippocampal CA1 region of mice lacking cGMP-dependent kinases is normal and susceptible to inhibition of nitric oxide synthase. J Neurosci. 1999;19:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Huang Y-Y, Kandel ER. Theta frequency stimulation induces a local form of late phase LTP in the CA1 region of the hippocampus. Learn Memory. 2005;12:587–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Otto T, Eichenbaum H, Wible CG, et al. Learning‐related patterns of CA1 spike trains parallel stimulation parameters optimal for inducing hippocampal long‐term potentiation. Hippocampus. 1991;1:181–192. [DOI] [PubMed] [Google Scholar]
- [29].Chung SW, Hoy KE, Fitzgerald PB. Theta-burst stimulation: a new form of TMS treatment for Depression? Depress Anxiety. 2014;32:182–192. [DOI] [PubMed] [Google Scholar]
- [30].Somani A, Kar SK. Efficacy of repetitive transcranial magnetic stimulation in treatment-resistant depression: the evidence thus far. Gen Psychiatry. 2019;32:e100074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Knackstedt LA, Moussawi K, Lalumiere R, et al. Extinction training after cocaine self-administration induces glutamatergic plasticity to inhibit cocaine seeking. J Neurosci. 2010;30:7984–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Acosta-Jaquez HA, Keller JA, Foster KG, et al. Site-specific mTOR phosphorylation promotes mTORC1-mediated signaling and cell growth. Mol Cell Biol. 2009;29:4308–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Raymond CR, Redman SJ. Spatial segregation of neuronal calcium signals encodes different forms of LTP in rat hippocampus: spatial Ca2+signals underlying LTP. J Physiol. 2005;570:97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Leal G, Comprido D, Duarte CB. BDNF-induced local protein synthesis and synaptic plasticity. Neuropharmacology. 2013;76(Pt):C: 639–56. [DOI] [PubMed] [Google Scholar]
- [35].Lee AS, Jesús-Cortés HD, Kabir ZD, et al. The neuropsychiatric disease-associated Gene cacna1c mediates survival of young hippocampal neurons. Eneuro [Internet]. 2016; 3. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4819284/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ghosh A, Carnahan J, Greenberg M. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. [DOI] [PubMed] [Google Scholar]
- [37].Schierberl K, Hao J, Tropea TF, et al. Cav1.2 L-type Ca2+ channels mediate cocaine-induced GluA1 trafficking in the nucleus accumbens, a long-term adaptation dependent on ventral tegmental area Ca(v)1.3 channels. J Neurosci Official J Soc Neurosci. 2011;31:13562–13575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Burgdorf CE, Schierberl KC, Lee AS, et al. Extinction of contextual cocaine memories requires Cav1.2 within D1R-Expressing cells and recruits hippocampal Cav1.2-dependent signaling mechanisms. J Neurosci. 2017;37:11894–11911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lee S-JR, Escobedo-Lozoya Y, Szatmari EM, et al. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458:299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lee HK, Takamiya K, He K, et al. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol. 2010;103:479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Goebbels S, Bormuth I, Bode U, et al. Genetic targeting of principal neurons in neocortex and hippocampus of NEX‐Cre mice. Genesis. 2006;44:611–621. [DOI] [PubMed] [Google Scholar]
- [42].Tsien JZ, Chen DF, Gerber D, et al. Subregion- and cell type–restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. [DOI] [PubMed] [Google Scholar]
- [43].Horigane S, Hamada S, Kamijo S, et al. Development of an L-type Ca2+ channel-dependent Ca2+ transient during the radial migration of cortical excitatory neurons. Neurosci Res. 2020June;26:S0168-0102(20)30391–6. [DOI] [PubMed] [Google Scholar]
- [44].Horigane S-I, Ozawa Y, Zhang J, et al. A mouse model of Timothy syndrome exhibits altered social competitive dominance and inhibitory neuron development. Febs Open Bio. 202010(8):1436–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kamijo S, Ishii Y, Horigane S, et al. A critical neurodevelopmental role for L-type voltage-gated calcium channels in neurite extension and radial migration. J Neurosci. 2018;38:5551–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Panagiotakos G, Haveles C, Arjun A, et al. Aberrant calcium channel splicing drives defects in cortical differentiation in timothy syndrome. Elife. 2019;8:e51037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Krey JF, Paşca SP, Shcheglovitov A, et al. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat Neurosci. 2013;16:201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Rajadhyaksha A, Barczak A, Macías W, et al. L-type Ca 2+ channels are essential for glutamate-mediated CREB phosphorylation and c-fos gene expression in striatal neurons. J Neurosci. 1999;19:6348–6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Li B, Tadross MR, Tsien RW. Sequential ionic and conformational signaling by calcium channels drives neuronal gene expression. Science. 2016;351:863–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Temme SJ, Bell RZ, Fisher GL, et al. Deletion of the mouse homolog of CACNA1C disrupts discrete forms of hippocampal-dependent memory and neurogenesis within the dentate gyrus. Eneuro. 2016;3:ENEURO.0118–16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dedic N, Pöhlmann ML, Richter JS, et al. Cross-disorder risk gene CACNA1C differentially modulates susceptibility to psychiatric disorders during development and adulthood. Mol Psychiatry. 2017;23:533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lee AS, Ra S, Rajadhyaksha AM, et al. Forebrain elimination of cacna1c mediates anxiety-like behavior in mice. Mol Psychiatr [Internet]. 2012; 17: 1054–1055. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3481072/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Na ES, Jesús-Cortés HD, Martinez-Rivera A, et al. D-cycloserine improves synaptic transmission in an animal mode of Rett syndrome. Plos One. 2017;12:e0183026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chen WG, Chang Q, Lin Y, et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. [DOI] [PubMed] [Google Scholar]
- [55].Costa-Mattioli M, Monteggia LM. mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat Neurosci. 2013;16:1537–1543. [DOI] [PubMed] [Google Scholar]
- [56].Costa RM, Federov NB, Kogan JH, et al. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature. 2002;415:526–530. [DOI] [PubMed] [Google Scholar]