

Abstract
Introduction:
Anticoagulation without bleeding is an ideal goal in treating thrombosis, however, this goal has not been achieved. All current anticoagulants are associated with significant bleeding which limits their safe use. Genetic and pharmacological findings indicate that factor XIa is a key player in thrombosis, yet it is a relatively marginal one in hemostasis. Thus, factor XIa and its zymogen offer a unique opportunity to develop anticoagulants with low bleeding risk.
Areas covered:
A survey of patent literature has retrieved more than 50 patents on the discovery of novel therapeutics targeting factor XI(a) since 2016. Small molecules, monoclonal antibodies, oligonucleotides, and polypeptides have been developed to inhibit factor XI(a). Many inhibitors are in early development and few have been evaluated in clinical trials.
Expert opinion:
Factor XI(a) is being actively pursued as a drug target for the development of effective and safer anticoagulants. Although many patents claiming factor XI(a) inhibitors were filed prior to 2016, recent literature reveals a moderately declining trend. Nevertheless, more agents have entered different levels of clinical trials. These agents exploit diverse mechanistic strategies for inhibition. Although further development is warranted, reaching one or more of these agents to the clinic will transform the anticoagulation therapy.
Keywords: Thrombosis, safe anticoagulants, factor XIa, active site inhibitors, pyridine-N-oxide, oxopyridine, macrocyclic, β-lactam, monoclonal antibodies, aptamers
1. Introduction
1.1. Current anticoagulants and the need for new ones
Thrombosis is the pathological formation of blood clots in blood vessels and/or heart. Venous thrombosis and arterial thrombosis are significantly associated with high morbidity and mortality rates in developing as well as developed countries [1,2]. Venous thrombosis is manifested as deep vein thrombosis or pulmonary embolism, whereas arterial thrombosis is manifested as ischemic heart disease or stroke. Atrial fibrillation, unstable angina, and disseminated intravascular coagulation are also examples of thrombosis-related diseases. Treatment of thrombotic diseases entails the use of antithrombotics i.e. anticoagulants and antiplatelets. Current anticoagulants include the indirect conventional drugs and the direct novel ones. In one hand, the indirect anticoagulants include vitamin K antagonists (war-farin) and antithrombin activators (fondaparinux, low molecular weight heparins, and unfractionated heparin). In the other hand, the direct novel anticoagulants include thrombin inhibitors (dabigatran etexilate and argatroban) and factor Xa (FXa) inhibitors (rivaroxaban, apixaban, edoxaban, and betrixaban) [3–5]. Although currently available anticoagulants are effective in treating and/or preventing thrombosis, yet they continue to be associated with a host of drug-specific and drug-nonspecific complications [3,4]. One of the most life-threatening complications of current anticoagulation therapy is bleeding [5,6].
All clinically used anticoagulants block the coagulation process by directly or indirectly inhibiting serine proteases in the common coagulation pathway i.e. thrombin and/or FXa, and thus, they affect the three stages of the coagulation process i.e. initiation, propagation, and amplification. In other words, current anticoagulants do not distinguish between the pathological coagulation i.e. thrombosis and the physiological coagulation i.e. hemostasis. As a result, they are clinically effective, yet they also induce serious bleeding complications which limit their safe use in a wide range of thrombotic patients [7–12]. In fact, the risk of bleeding with the use of anticoagulants is further increased in certain patient populations. For example, patients with chronic kidney diseases, who are considered as a high-risk group for thrombosis, have platelet abnormalities which put them at higher risk of bleeding. Another special group of patients is those with atrial fibrillation. Because of the risk of bleeding, more than 30% of these patients fail to receive anticoagulant prophylaxis and among patients receiving anticoagulants, up to 50% are ineffectively treated [7–12]. Thus, new anticoagulants that address the need of these patients are urgently needed.
In fact, proteins other than those of the common coagulation pathway have been considered to develop new anticoagulants with minimal risk of bleeding [13]. This includes coagulation protein of the extrinsic pathway i.e. factor VIIa (FVIIa) [14] and coagulation proteins of the intrinsic pathway and contact activation pathway i.e. factors XIIa, XIa, and IXa [15–17]. This review will focus on the drug development for molecules that interfere with the physiological function(s) of factor XI(a) (FXI(a)) which is considered as one of the most promising drug targets for effective and relatively safer anticoagulants [5,6].
1.2. Structure and function of human FXI(a): the promise of a new anticoagulant target
Factor XI (FXI) is the zymogen of factor XIa (FXIa), a coagulation serine protease of human plasma. Structurally, FXI is a disulfide-linked homodimer of two identical subunits with a total molecular weight of about 160 kD and approximate plasma concentration of 30 nM. Its biosynthesis happens largely in hepatic cells, and to some extent in pancreatic and renal tubule cells. Although not fully understood, platelets have also been reported to contribute to FXI expression/biosynthesis. The activation of the zymogen FXI to the protease FXIa takes place by cleaving the peptide bond R369-Ile370. This activation takes place in the presence of FXIIa or thrombin. Auto-activation of FXI may also take place in the presence of polyanions including polymeric inorganic polyphosphates [18–20].
Structurally, human FXIa possesses an N-terminal heavy chain and a C-terminal light domain (Figure 1). The former chain consists of four repeats known as the apple domains and labeled as A1 through A4. The latter chain is a trypsin-like catalytic domain. The X-ray crystal structure of the zymogen FXI revealed that the catalytic and the apple domains form a ‘cup and saucer’ relationship [18–20]. In one hand, the apple domains of FXIa contain several binding sites for macromolecules that interact with FXI(a) system during its course of physiological actions. Specifically, there is a binding site for thrombin in A1 domain [21], high molecular weight kininogen in A2 domain [22], factor IX (FIX) [23], platelet glycoprotein-Ib [24], and heparin/heparan sulfate in A3 domain [25], and FXIIa in A4 domain [26]. In the other hand, the catalytic domain contains the active site with the catalytic triad of H413, D462, and S557 (the chymotrypsin residue numbering: H57, D102, and S195). As with other proteases, the architecture of FXIa active site involves multiple subsites (S4–S3–S2–S1–S1′–S2′–S3′–S4′) with unique structural features so as to achieve substrate specificity. For instance, the S1 specificity pocket is known to bind to the basic arginine residue in the physiological substrate, FIX, and contains an aspartate residue (D189) at the bottom of the subsite [6]. Furthermore, the S2 specificity pocket engages with threonine residue in FIX whereas the S1` pocket prefers small and lipophilic residues involving alanine and/or valine. Subsites S3 and S2` commonly recognize leucine or phenylalanine and glutamate or valine, respectively. Additionally, the catalytic domain also has an anion-binding site which has been implicated in the protease regulation by either charge neutralization or allosteric modulation [27].
Figure 1.
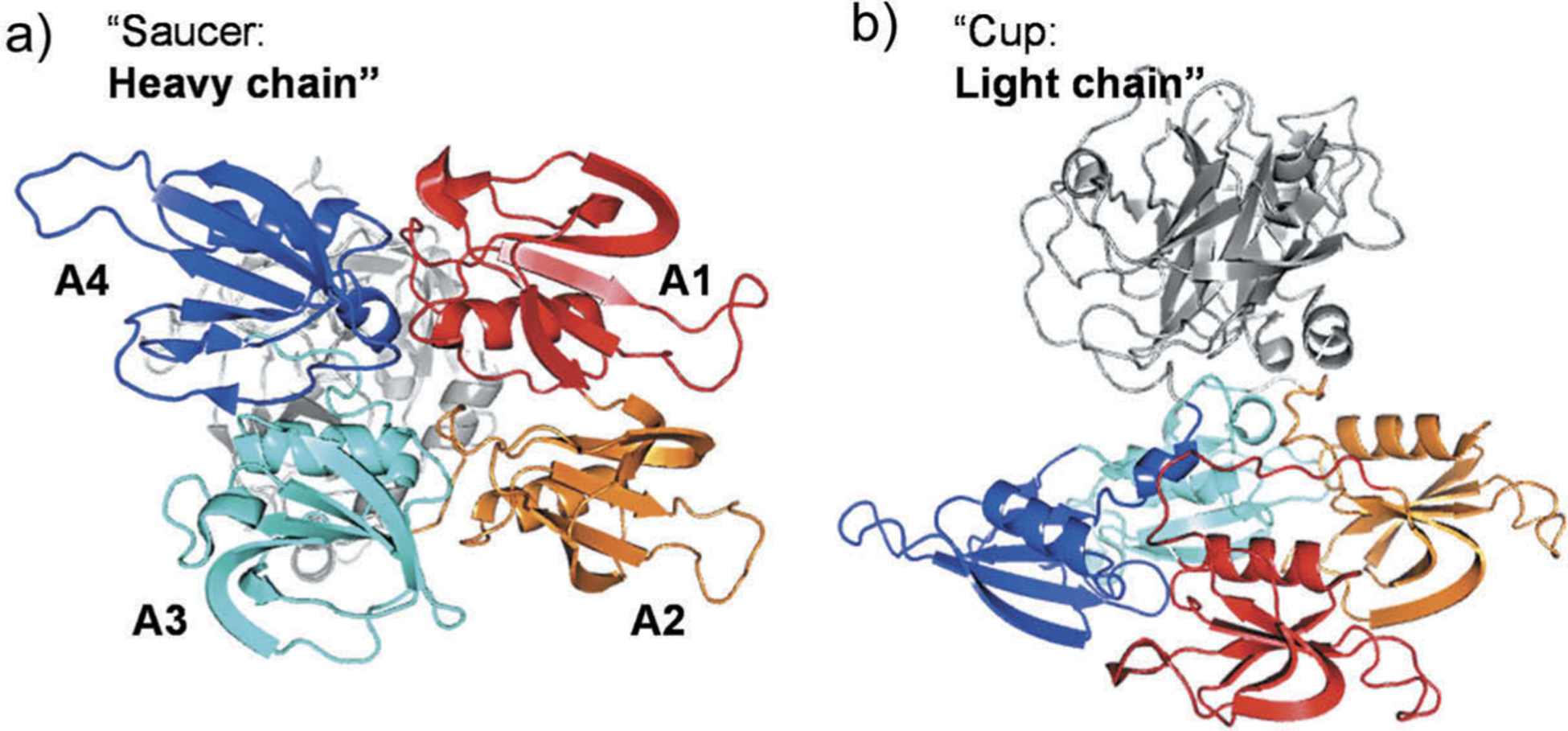
Cartoon depiction of monomeric human FXI structure (PDB ID: 2F83). (a) Back view showing the four apple domains: A1 (red) involving binding sites for thrombin and high molecular weight kininogen; A2 (orange) with some residues important for high molecular weight kininogen; A3 (cyan) involving binding sites for FIX, platelet glycoprotein-Ib, and heparin; and A4 (blue) involving FXIIa binding site. (b) Side view showing the cup-saucer relationship between the catalytic domain and the apple domains, respectively.
Physiologically, the protease FXIa has multiple substrates. FXIa activates FIX to factor IXa (FIXa) in the coagulation process. In the activation event, FIX binds to an exosite on the A3 apple domain of FXIa. The key amino acids in the active site of FXIa then cleave the peptide bond of R145-A146 and then the peptide bond of A180-V181 resulting in the formation of FIXaβ, which eventually promotes thrombin generation and blood clotting [28,29]. Furthermore, several studies indicate that FXIa can activate other proteases that are important for the coagulation process as well as the kallikrein-kinin system. Particularly, FXIa was reported to activate factors V, VIII, X, and XII [30–35]. FXIa also promotes clot formation/stability by inhibiting other coagulation regulators including tissue factor pathway inhibitor [36] and the von Willebrand factor-cleaving metalloproteinase [37]. Moreover, studies also indicate that FXI binds to platelets, and thus, contributes to platelet activation and accumulation. FXI localized to platelets was found to promote coagulation and inflammation in mice in a glycoprotein 1b-dependent manner [38]. FXI was also reported to bind to the platelet apolipoprotein E receptor-2 [39] suggesting that FXI binding to platelets may involve a complex process. FXI also interacts with the human recombinant laminin heterotrimers in the extracellular matrix of blood vessels and arteries [40]. The interaction was proposed to contribute to the FXIa-mediated activation of FIX and thrombin generation in the extracellular matrix following blood vessel injury [20]. Overall, given the multiple substrates that FXI(a) system interacts with, its role has been re-defined from a component in the intrinsic pathway in the waterfall hypothesis of coagulation to a key player that bridges the thrombin generation process via activating FIX and the host defense, inflammatory mechanism(s) via activating FXII (Figure 2).
Figure 2.
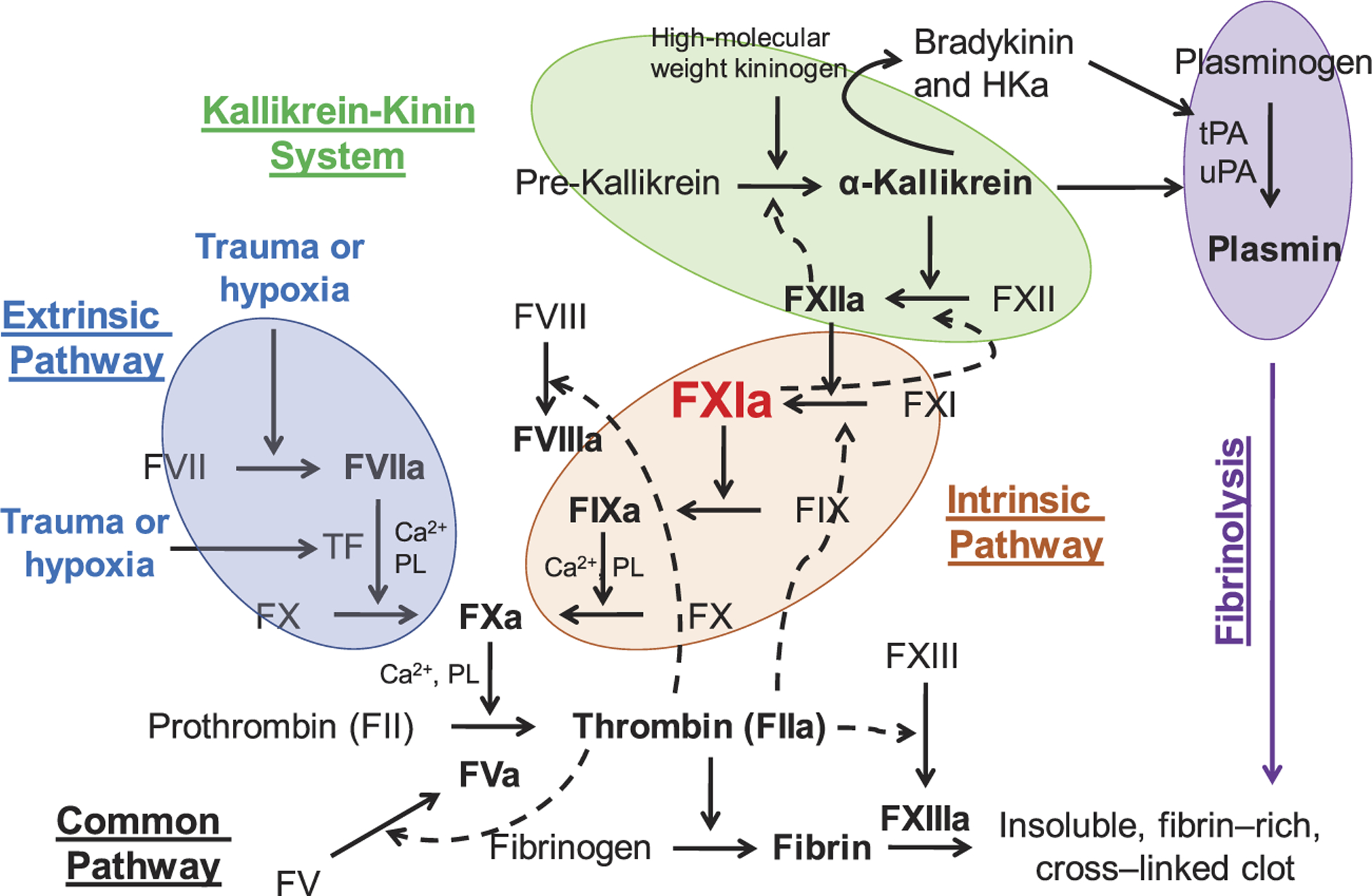
The key role of human FXIa. The role of FXIa has been re-defined from a component in the intrinsic pathway of coagulation in the waterfall hypothesis of coagulation to a key player that bridges the thrombin generation process via activating FIX and the host defense, inflammatory mechanism(s) via activating FXII. HKa is cleaved high molecular weight kininogen.
Importantly, mounting clinical, experimental, and epidemiological evidence indicates that the protease FXIa fundamentally contributes to the amplification phase of blood clotting, rather than the initiation phase. Accordingly, FXIa has been considered to mainly contribute to the pathological blood clotting i.e. thrombosis, and to play a relatively minor role in the physiological blood clotting i.e. hemostasis. Along these lines, patients with hemophilia C (FXI genetic deficiency in humans) are linked to a relatively mild-to-moderate risk of bleeding compared to those with hemophilia A or B. These patients show a normal prothrombin time (PT), yet a relatively prolonged activated partial thromboplastin time (APTT) which indicates a specific defect in the amplification phase of the coagulation process [41–43]. Furthermore, reports indicate that patients with severe FXI deficiency exhibit a reduced risk of deep vein thrombosis [44] and/or ischemic stroke [45]. Epidemiological studies also revealed that increased levels of FXI may increase the risk of deep vein thrombosis [46], stroke [47–49], myocardial infarction [50], and cardiovascular diseases in women [51]. Moreover, testing of antisense oligonucleotides, monoclonal antibodies, and small molecules in animal models (mice, rats, rabbits, and baboons) of arterial thrombosis and venous thrombosis as well as in humans strongly support that agents targeting FXI(a) do provide a clinically relevant protection against thrombosis with no significant impact on bleeding times [5,6]. In particular, the antisense oligonucleotide ISIS 416858 [52,53], the monoclonal antibodies MAA868 [54,55], BAY1213790 [56], and AB023 [57], and the small molecules EP-7041 [58] and BMS-986177 [59] have finished various levels of clinical trials (Phase I and, in some cases, Phase II) with very promising results as anticoagulants with no bleeding complications. Corresponding sections below will provide details about patents and reported results relevant to small molecules and monoclonal antibodies. Patent and reported results relevant to the antisense oligonucleotide ISIS 416858 were recently reviewed [5].
2. FXI(a) inhibitors
Given the promise of FXI(a) as a drug target for effective and safe anticoagulants, several small molecule active site inhibitors [58,59], glycosaminoglycan mimetic allosteric inhibitors [17,60,61], monoclonal antibodies [54–56], antisense oligonucleotides [52,53], aptamers, and polypeptides have been developed, and many were recently reviewed [5,6]. Furthermore, patents claiming FXI(a) inhibitors until early 2016 were also recently reviewed [5]. In this review, we describe the patents claiming molecular entities that target FXI(a) system starting 2016 to the date of this review publication. Given its promise, a survey of patents literature indicates that the number of published patents claiming molecular agents targeting FXI(a) system exceeds those reported to target any other coagulation factor including FIIa (thrombin), FXa, FIXa, FVIIa, or FXIIa (Figure 3(a)). Figure 3(b) shows the categories of agents targeting FXI(a) as well as the corresponding number of the patents that have been filed/approved since 2016. Parallel to the number of FXI(a)-related patents filed over the 2016–2019 period is the number of peer-reviewed publications pertaining to the same system (Figure 3(c)).
Figure 3.
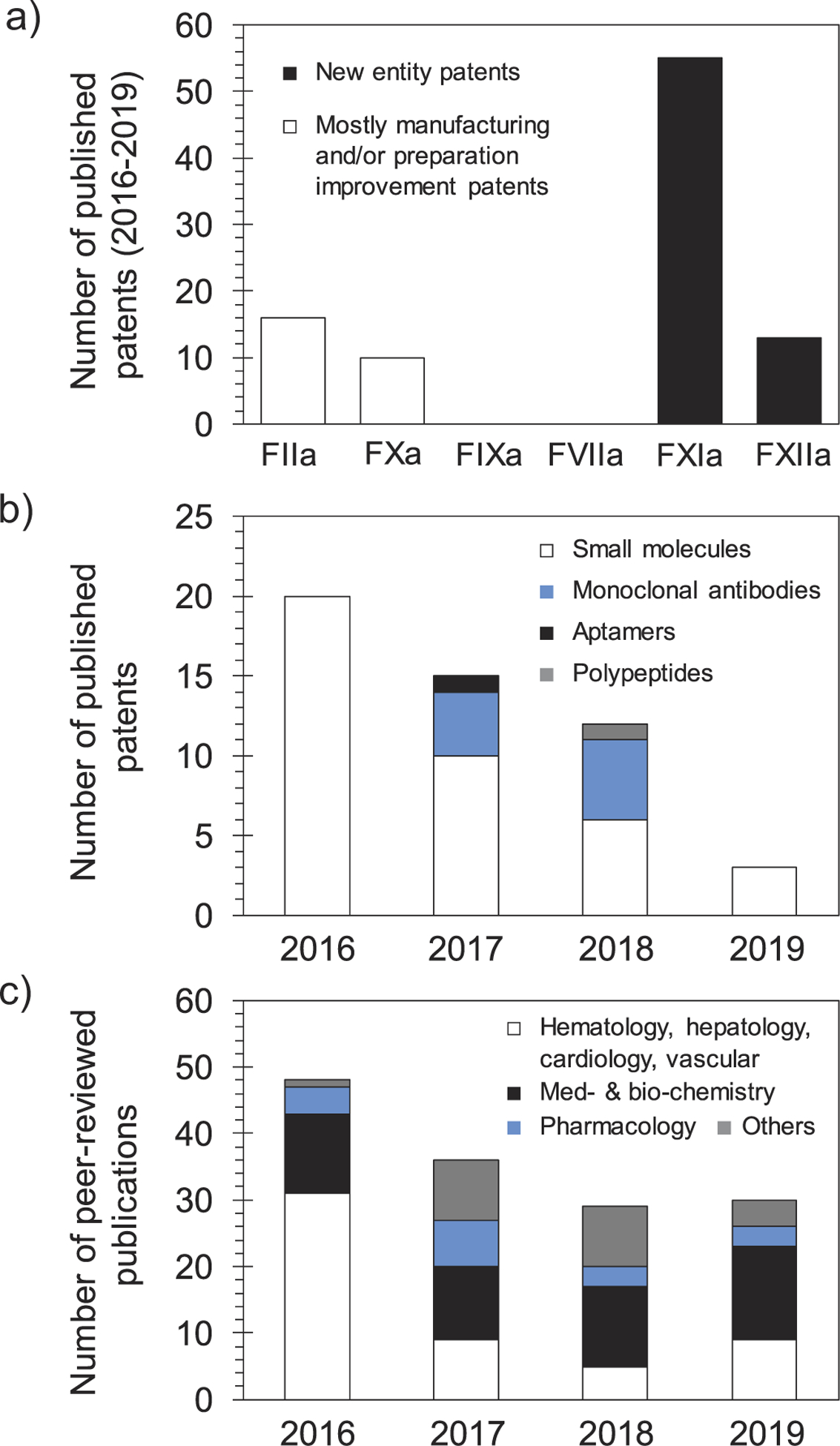
(a) A histogram shows the number of patents filed for molecules targeting different clotting factors over the 2016–2019 period. The highest number is for new molecular entities targeting FXI(a). Patents that involve FIIa and FXa inhibitors are generally to improve the manufacturing or the delivery method of approved drugs. No significant patents were retrieved for agents targeting FIXa or FVIIa. (b) A histogram shows the number of patents filed for molecular entities targeting the FXI(a) system per year starting 2016. The histogram also shows the type of molecular entities that have been claimed (small molecules, monoclonal antibodies, aptamers, and polypeptides). The claimed agents are predominantly small molecules. There is a declining trend in the number of patents filed over the 2016–2019 period, yet the number of FXI(a)-based agents that are being evaluated in clinical trials is increasing. (c) A histogram shows the number of peer-reviewed publications for topics involving FXI(a) system. The histogram highlights the most recognized areas of research. The overall declining trend in the number of FXIa publications is similar to that of patents, although publications in the areas of medicinal chemistry and biochemistry has remained steady. Data in histograms (a) and (b) were retrieved from SciFinder whereas data in histogram (c) were retrieved from Web of Science.
2.1. Small molecules
Several small molecules have been claimed by various pharmaceutical companies as selective, active site inhibitors of human FXIa or dual inhibitors of FXIa and plasma kallikrein. We chose to present the inhibitors according to their central structural domain which can be pyridine-N-oxide, oxypyridine, substituted phenylalanine, or β-lactam. Although macrocyclic FXIa inhibitors may include one of the above central domains, yet they are presented separately.
2.1.1. Pyridine N-oxide-containing inhibitors and their derivatives
In 2016, pyridine-N-oxide-containing molecules were claimed by Merck Co. to be FXIa inhibitors or dual inhibitors of FXIa and plasma kallikrein, and therefore, to be used singularly or in combination with other current anticoagulants to treat thromboses, embolisms, hypercoagulability, or fibrotic changes [62]. In the claimed inhibitors, the pyridine-N-oxide moiety is di-substituted at positions- 2 and −5 with the most frequent position-2 substituent is methine imidazole whereas position-5 is frequently substituted with chlorophenyl tetrazole moiety. Several derivatives were tested as racemic mixtures or pure enantiomers considering the stereochemistry of the methine carbon which was commonly substituted with benzyl or cyclopropyl methyl moiety. Example of molecules from this claim is inhibitor 1 (Figure 4) which inhibited FXIa and plasma kallikrein with IC50 values of 1.34 and 1.52 nM, respectively [62]. Potential binding mode is expected to be the active site where the chlorophenyl tetrazole group fits into the S1 subsite and the terminal methyl carbamate fits into the S2` subsite. In a similar claim by the same group, it was found that the replacement of the central imidazole moiety with a benzene ring, that is further substituted at position-3 with benzoic acid (carboxylic group is at position-3) as in inhibitor 2 (Figure 4), led to better inhibition potency to FXIa (IC50 = 0.46 nM) and better selectivity over plasma kallikrein (IC50 = 77.71 nM) [63]. Closer look at the structure-activity relationship of the above molecules indicated that using the (S)-stereochemistry may improve FXIa inhibition potency while changing the position of the carboxylic group of inhibitor 2 may decrease the potency [63].
Figure 4.
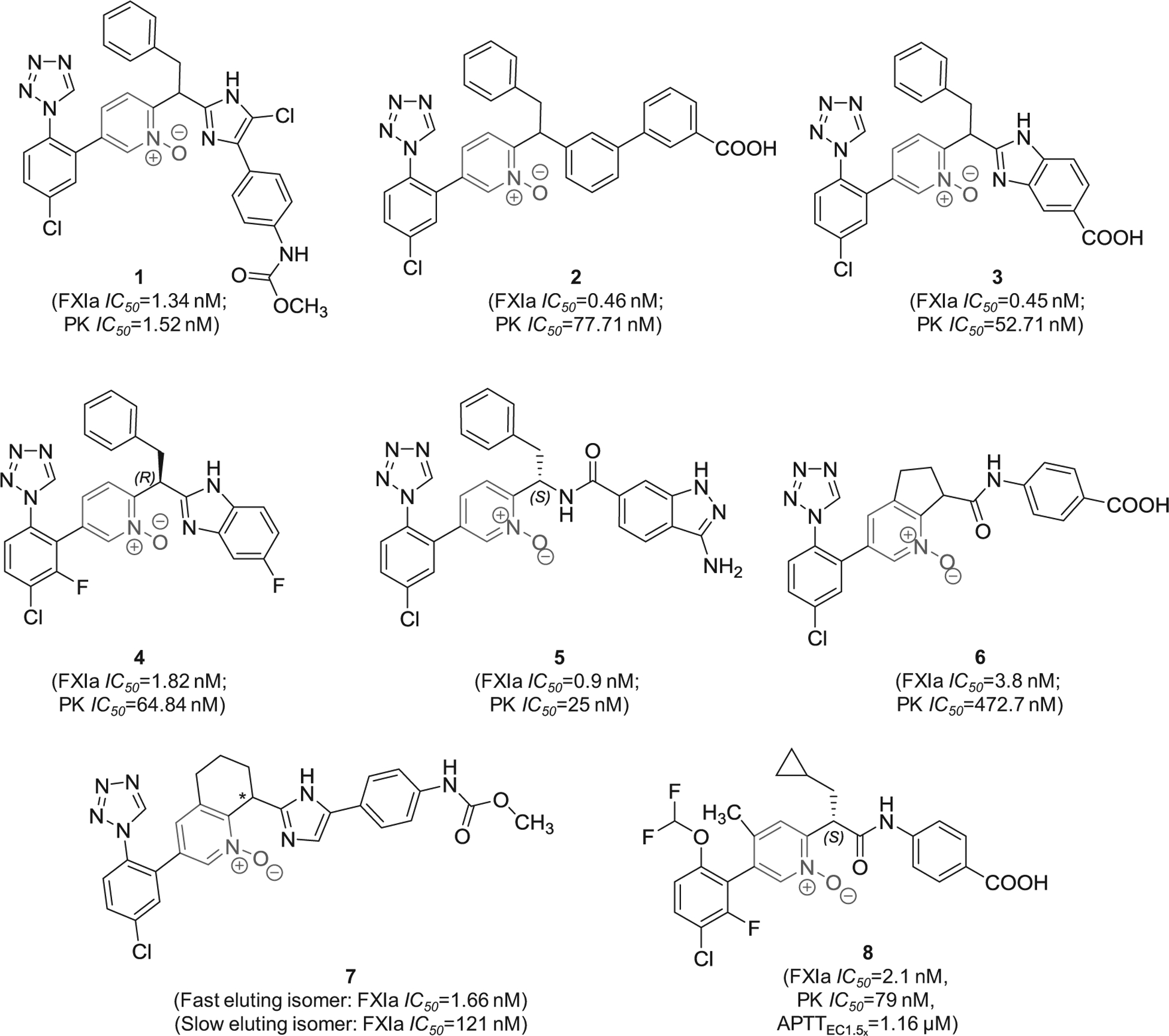
The chemical structures of pyridine-N-oxide-based inhibitors of FXIa (1–8) filed by Merck Sharp & Dohme Corp., US. Provided are the FXIa and plasma kallikrein (PK) IC50 values (nM) and the concentrations required to double clotting time in APTT assay (μM) for inhibitor 8.
In related patents, it was found that replacing the methyl (4-(5-chloro-1H-imidazol-4-yl)phenyl)carbamate moiety in inhibitor 1 with 1H-benzo[d]imidazole-5-carboxylic acid (inhibitor 3) maintained high potency toward FXIa and good selectivity over plasma kallikrein [64]. Furthermore, it appears that the inhibition potency and selectivity were also retained following the bioisosteric replacement of the carboxylic acid group with fluorine atom as shown by inhibitor 4 [64]. The methyl (4-(5-chloro-1H-imidazol-4-yl)phenyl)carbamate moiety in inhibitor 1 can also be replaced with 3-amino-1H-indazole-6-carboxamide as in inhibitor 5 which inhibited FXIa and plasma kallikrein with IC50 values of 0.9 and 25 nM, respectively [65]. The pyridine-N-oxide can also be rigidified in the form of cyclopenta[b]pyridine-N-oxide (inhibitor 6) [66] or in the form of 5,6,7,8-tetrahydroquinoline-N-oxide (inhibitor 7) [67], both of which possessed nanomolar inhibition potency toward FXIa and potentially significant selectivity over plasma kallikrein.
Other modifications were also introduced as demonstrated by inhibitor 8 (Figure 4). This molecule inhibited FXIa and plasma kallikrein with IC50 values of 2.1 and 79 nM, respectively, and demonstrated APTTEC1.5x of 1.16 μM. Importantly, its (R)-enantiomer inhibited human FXIa only 47% at about 1000 μM. The molecule exhibited drug clearance of 5 ml/min/kg, mean residence time of 2.5 hrs, and bioavailability of 56%. In fact, inhibitor 8 indicated that the S1-binding group can be chloro-4-(difluoromethoxy)-2-fluoro-benzene moiety rather than the frequently recruited chlorophenyl tetrazole moiety whereas S1`-binding group can be cyclopropyl methyl rather than benzyl [68].
2.1.2. Oxopyridine-containing inhibitors and their derivatives
A series of patents by Bayer Pharma described 2-oxopyridine-containing molecules as inhibitors of human FXIa and plasma kallikrein. The inhibitors were claimed for treating and/or preventing thrombotic or thromboembolic diseases, edemas, and ophthalmological diseases [69–76]. Figure 5 provides examples from seven patents showing the chemical structures of 8 inhibitors with demonstrated inhibition potency toward FXIa and plasma kallikrein in the nanomolar range. Structural analysis revealed that the 2-oxopyridine moiety is always substituted at positions-1, −4, and −5. The most frequent substituent at position-5 is methoxy whereas the most frequent substituent at position-4, which likely binds into S1 subsite in the active site of FXIa, is 5-chloro-benzonitrile-2-yl. Position-1 is connected to the rest of the inhibitor’s chemical structure and bears two structural domains, one domain is frequently acyclic aliphatic and potentially binds to the S1` subsite (inhibitors 9–11 and 14–16) and the second is frequently carboxylated aromatic and potentially binds to the S2` subsite (inhibitors 9, 11, 15, and 16).
Figure 5.
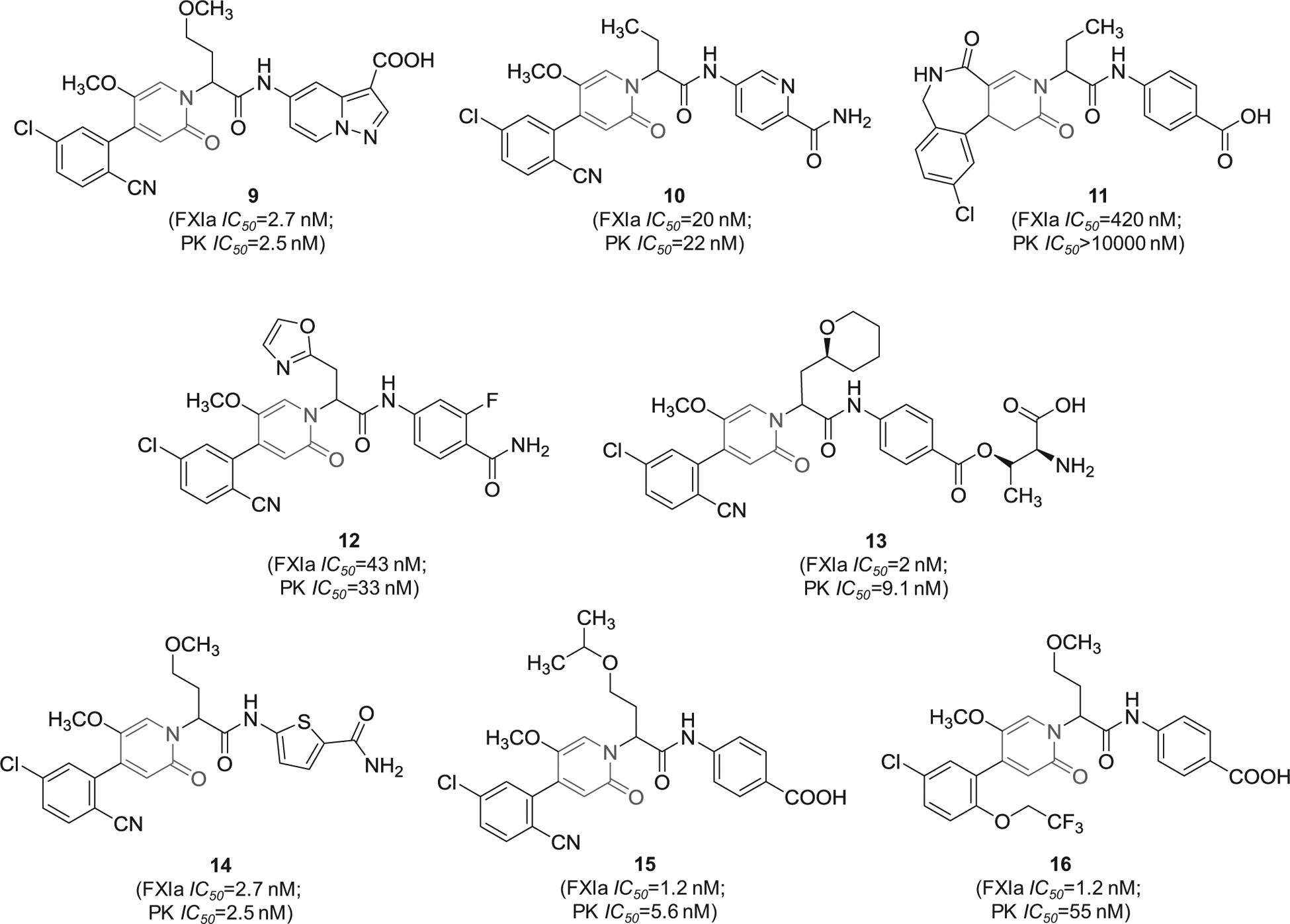
The chemical structures of 2-oxopyridine-based inhibitors of FXIa (9–16) filed by Bayer Pharma, Germany. Provided are the FXIa and PK IC50 values (nM).
Likewise, 2-oxopyridine-containing molecules 17 and 18 (Figure 6) were claimed by Chinese companies (Jiangsu Hengrui Medicine Co. and Shanghai Hengrui Pharmaceutical Co.). The two molecules inhibited FXIa with IC50 value of 34 nM [77] and 6.4 nM [78], respectively. Inhibitor 18 was also claimed as cyclo-butyl prodrug in another patent [79]. Inhibitor 19 was also claimed by the same Chinese companies. It inhibited FXIa with IC50 value of 92 nM [80]. Lastly, similar molecules that are oxo-pyrimidine derivatives were claimed by Bristol-Myers Squibb Co. as novel substituted glycine derivatives, and inhibitors of FXIa and/or plasma kallikrein for the prophylaxis or treatment of thromboembolic disorders, or for the treatment of retinal vascular permeability associated with diabetic macular edema and diabetic retinopathy [81]. Example of Bristol molecules is represented by molecule 20 (Figure 6), a pyrimidine-2-one derivative which inhibited FXIa and plasmin kallikrein with IC50 values of 2.5 and 189 nM, respectively, and affected the APTT at a concentration of 1.74 μM [81].
Figure 6.
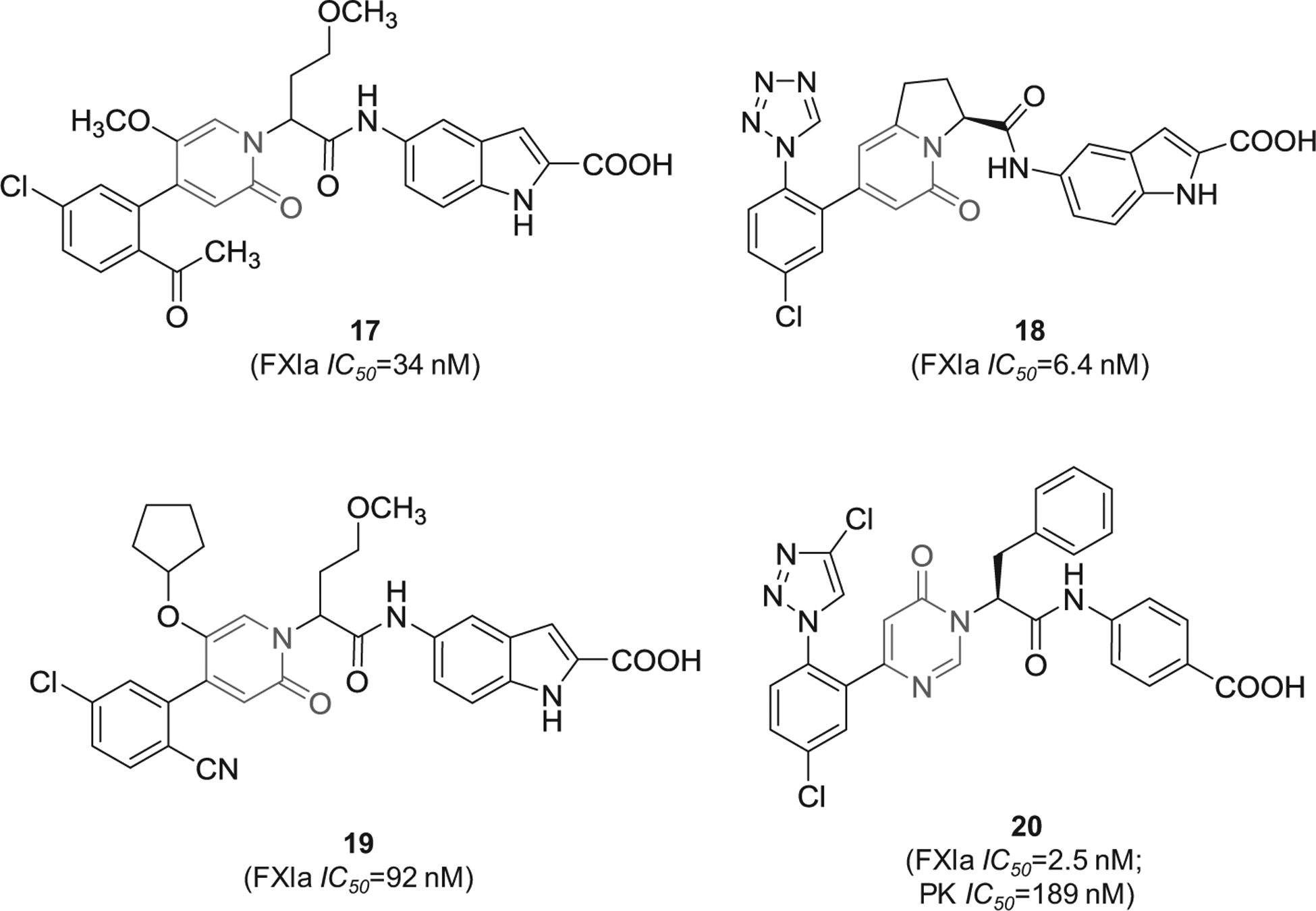
The chemical structures of 2-oxopyridine-based inhibitors of FXIa (17–19) filed by Shanghai Hengrui Pharmaceutical Co. and its partner, China, as well as the chemical structure of inhibitor 20 which was claimed by Bristol-Myers Squibb Co., US. Provided are the FXIa and PK IC50 values (nM).
2.1.3. Macrocyclic derivatives
Cyclization of peptide-based molecules is widely used strategy in the design of enzyme inhibitors. Potential gains are improved drug-like properties, particularly target affinity, metabolic stability, and oral bioavailability [82,83]. Following this concept, Bristol-Myers Squibb has filed several patents claiming macrocyclic FXIa inhibitors starting 2016 [84–90]. The macrocyclic molecules were claimed as inhibitors of FXIa and/or plasma kallikrein for the prophylaxis or treatment of thromboembolic disorders or for the treatment of retinal vascular permeability associated with diabetic macular edema and/or diabetic retinopathy. An example of these molecules is represented by the tri-amide macrocyclic inhibitor 21 (Figure 7) which was reported to inhibit FXIa in vitro with IC50 value of 17.89 nM. In this inhibitor, the p-chlorophenyl tetrazole moiety appears to bind to the S1 subsite in the active site of FXIa. Subsequently, inhibitor 22 (Figure 7) was claimed by the same company [84]. Rewarding modifications represented by introducing the pyrimidinone moiety to the P1 domain and by incorporating two heteroaromatic systems i.e. pyridine and N-substituted pyrazole into the macrocyclic domain led to FXIa IC50 value of 0.1 nM [85]. The molecule also inhibited plasma kallikrein with IC50 of 28 nM and increased the APTT in citrated normal human plasma by 1.5-fold at a concentration level of 0.5 μM. The solubility of its crystal form was claimed to be 6 μg/mL at pH = 6.5 and its free fraction of human plasma protein binding was about 9%. Likewise, inhibitor 23 inhibited FXIa and plasma kallikrein with Ki values of 2 and 179 nM, respectively [86]. Other examples include inhibitors 24 [87] and 25 [88], as well as the macrocyclic spiro-carbamate inhibitor 26 [89] (Figure 7).
Figure 7.
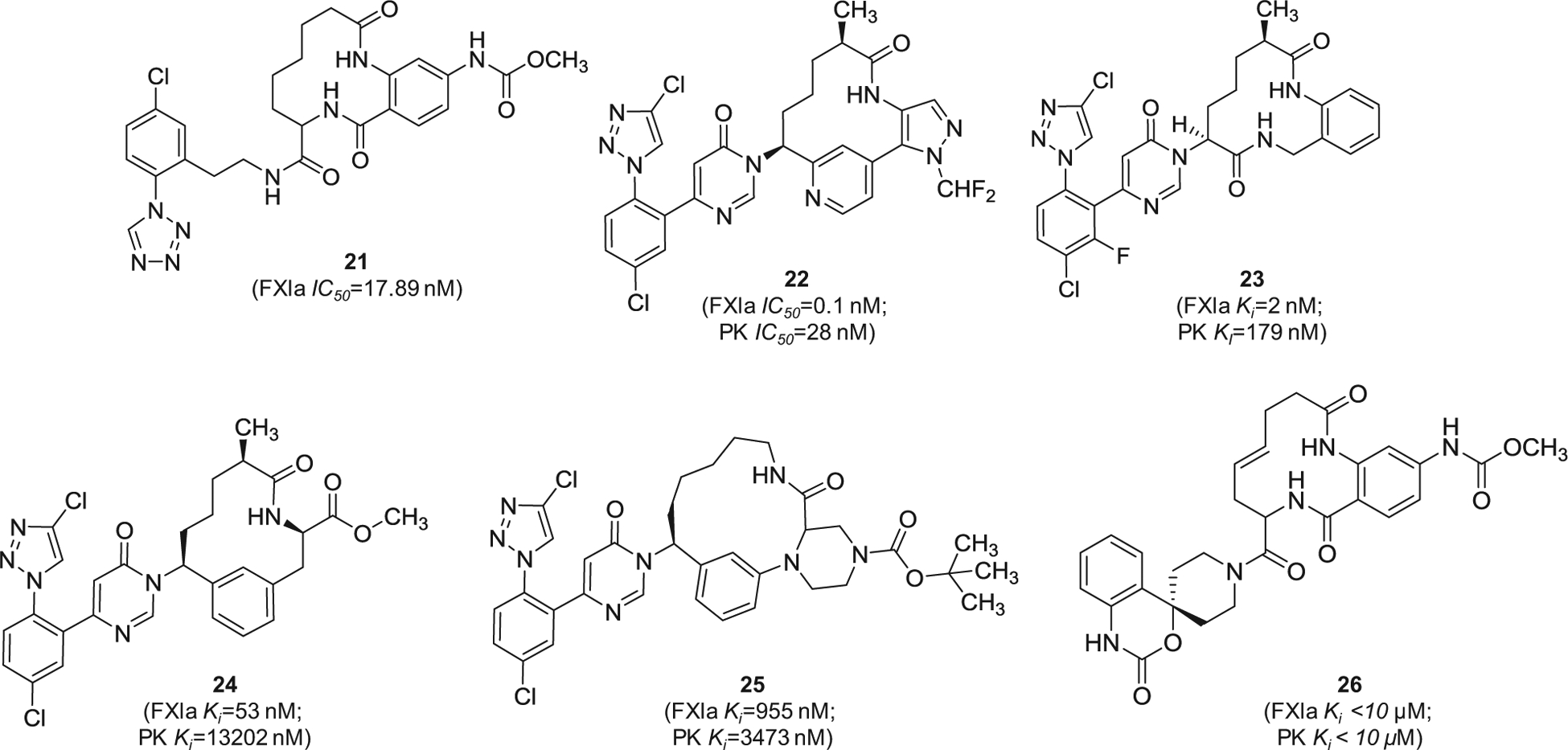
The chemical structures of macrocyclic inhibitors of FXIa (21–26) filed by Bristol-Myers Squibb Co., US. Provided are the FXIa and PK IC50 values (nM) and/or Ki values (nM).
Similarly, Merck Sharp & Dohme Corp. has filed for multiple patents claiming the FXIa inhibitory activity of large number of macrocyclic derivatives (Figure 8). Specifically, inhibitors 27 [91] and 28 [92] were reported to inhibit both FXIa and plasma kallikrein. Other macrocyclic FXIa inhibitors were also claimed by Chinese companies. Inhibitor 29 [93] and inhibitor 30 [94] (Figure 9) were claimed by Sunshine Lake Pharma Co. and Sichuan Kelun-Biotech Biopharmaceutical Co., respectively. The former inhibited FXIa with IC50 value of 44.7 nM, whereas the latter inhibited FXIa with IC50 value of 0.59 nM.
Figure 8.
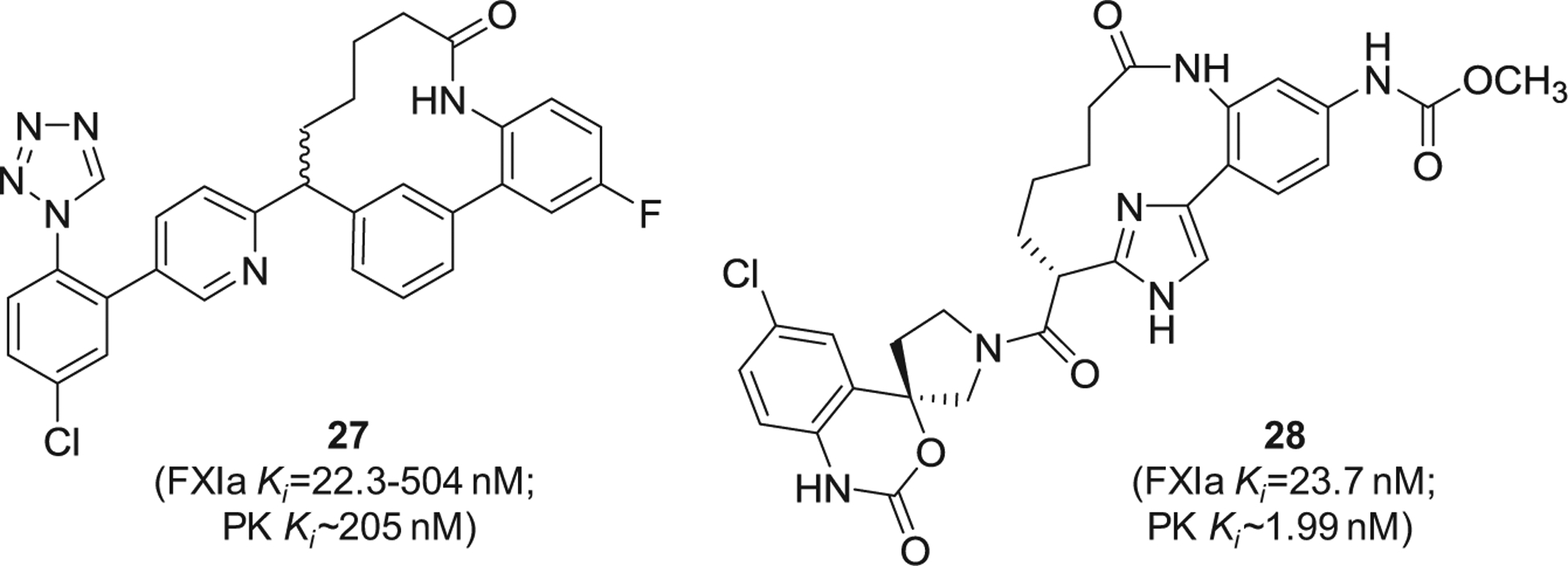
The chemical structures of macrocyclic inhibitors of FXIa (27 and 28) claimed by Merck Sharp & Dohme Corp, US. Provided are the FXIa and PK Ki values (nM).
Figure 9.
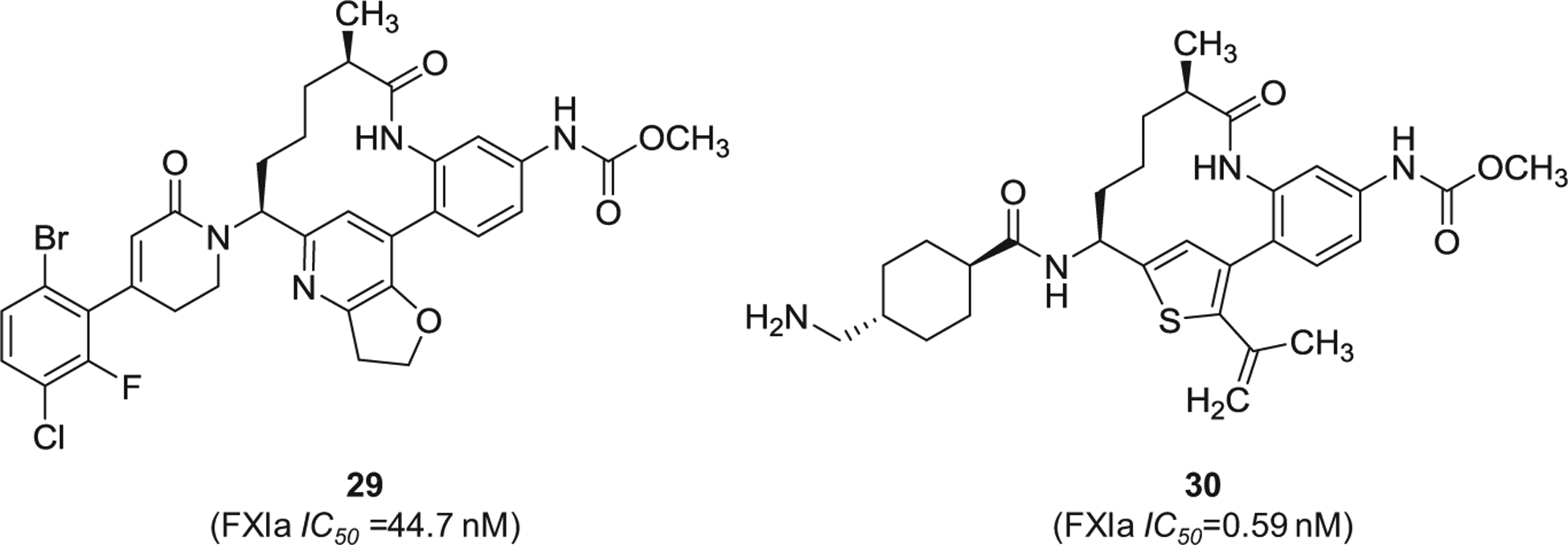
The chemical structures of macrocyclic inhibitors of FXIa (29 and 30) claimed by Sunshine Lake Pharma Co. and Sichuan Kelun-Biotech Biopharmaceutical Co., respectively, from China. Provided are the FXIa IC50 values (nM).
2.1.4. 4 -substituted phenylalanine derivatives
Several patent applications were filed by Bayer Pharma since 2016 in which substituted phenylalanine derivatives were claimed as FXIa inhibitors for the treatment and/or prophylaxis of fibrinolytic and thromboembolic diseases, in particular blood loss in the cases of hyperfibrinolytic states, organ transplants, or heart surgery interventions, or as a constituent of a fibrin adhesive [95–97]. The inhibitors 31–33 (Figure 10) have tripeptide-like structures in which the N-terminal residue is tranexamic acid which likely fits into S1 pocket of FXIa, the central residue is p-substituted phenylalanine which likely fits into S2 or S1′ pockets, and the C-terminal residue is variable and seems to bind to S2` pocket. In one hand, inhibitors 31 and 32 have the C-terminal residue of 5-phenyl-triazole that is substituted with tetra-fluorinated propionic acid at position-2. The two inhibitors exhibited IC50 values in the nanomolar range of 0.5 and 0.34 nM, respectively. Furthermore, inhibitor 32 was also reported to inhibit plasmin with IC50 value of 0.87 nM, to inhibit plasma kallikrein with IC50 value of 7.2 nM, and to prolong the APTT at 0.06 μM. The patents list other experiments establishing the inhibitors’ selectivity, effects on clot lysis, and their in vivo activity.
Figure 10.
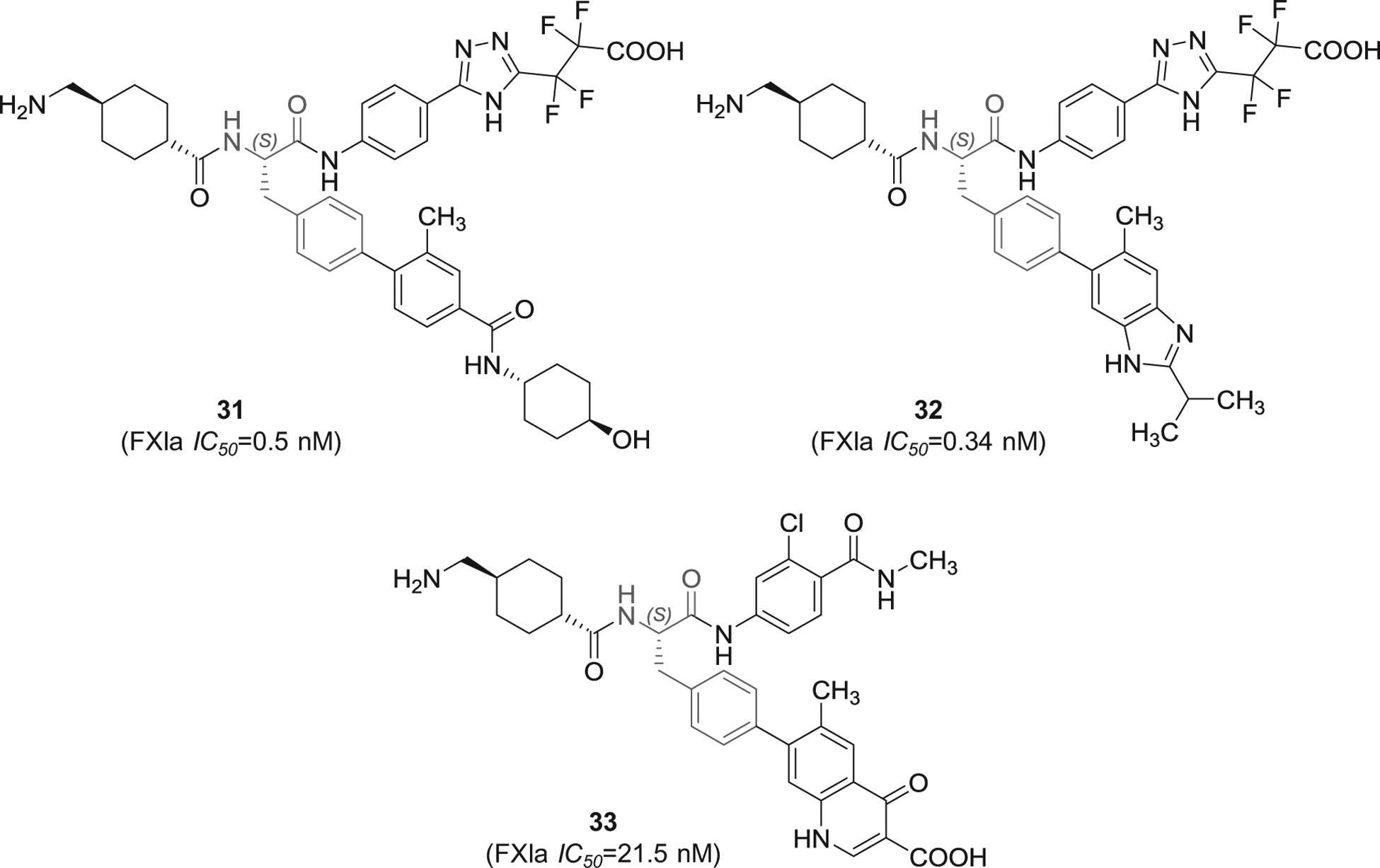
The chemical structures of 4-substituted phenylalanine-based inhibitors of FXIa (31–33) filed by Bayer Pharma, Germany. Provided are the FXIa IC50 values (nM).
Inhibitor 33 (Figure 10) is structurally different as the para-substituent of the phenylalanine moiety is 4-oxo-1,4-dihydroquinoline-3-carboxylic acid and the C-terminal is chlorinated benzamide. Inhibitor 33 was found to inhibit FXIa, plasmin, and plasma kallikrein with IC50 values of 21.5, 1.0, and 8.5 nM, respectively. The inhibitor prolonged the APTT at 0.7 μM. The patent lists other experiments establishing its selectivity, effect on clot lysis, and in vivo activity.
It is important to mention here that the patents [95–97] that claim 4-substituted phenylalanine derivatives as FXIa inhibitors provide the inhibitory activity for the (S)-enantiomer since the synthesis was started from L-phenylalanine. Generally speaking, the potency of the pure (S)-enantiomers appears to be superior to that of the racemic inhibitors or even those with pure (R)-stereochemistry.
2.1.5. β-lactam derivatives
Prior to 2016, multiple β-lactam-containing inhibitors (azetidi-none derivatives) were patented by Bristol Myers Squibb, Exithera Pharmaceuticals, and Daiamed [5,6]. In 2018 and 2019, more β-lactam-containing molecules were claimed by Exithera Pharmaceuticals to inhibit FXIa or plasma kallikrein for preventing or treating thrombosis or angioedema. Example of these molecules are represented by inhibitors 34 and 35 (Figure 11) [98,99]. Particularly, inhibitor 34 inhibited human FXIa with IC50 value of 10–100 nM and selectivity index of at least 100-fold over FXa, thrombin, and trypsin. The inhibitor’s solubility was reported to be 10–100 μM in PBS with significant Caco-2 permeability and plasma protein binding of >95% [98]. Mechanistically, some of β-lactam molecules may act as covalent inhibitors of FXIa as a result of the interaction between Ser195 of FXIa and the β-lactam moiety of the inhibitors, as reported previously [5,6]. Moreover, their guanidine group (or 2-aminopyridine group as in inhibitors 34 and 35) may bind into the S1 pocket of FXIa.
Figure 11.
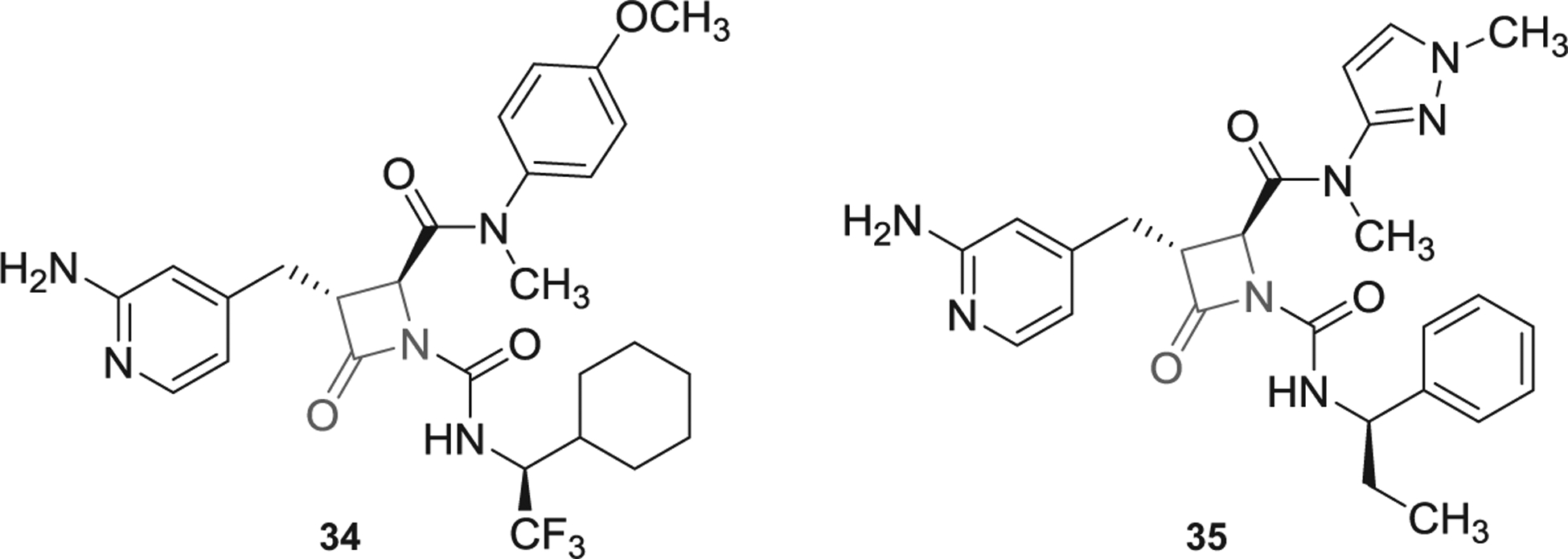
The chemical structures of β-lactam-based inhibitors of FXIa (34 and 35) filed by Exithera Pharmaceuticals, US.
Importantly, EP-7041 (Exithera Pharmaceuticals) [100], a potentially β-lactam-based, parenterally acting FXIa inhibitor was recently tested in Phase I trial [58]. Although reports describing its structure and mechanism have not been published, yet EP-7041 appears to be active site inhibitor of FXIa. This inhibitor may structurally look similar to those listed in Figure 11. It may potentially have a carboxylic group at position-2 of the β-lactam ring and 2-aminopyridin-4-yl-methyl moiety at position-3. In Phase I trial, healthy individuals received IV injections of single ascending doses of EP-7041 (0.01 mg/kg up to 1.0 mg/kg) or continuous IV infusions for 5 days (0.01 mg/kg/h up to 0.6 mg/kg/h). The investigators indicated that there were no serious adverse events. Mild events of infusion site bruising and headache were reported in 7% and 23% of subjects, respectively. EP-7041 exhibited dose-proportional pharmacokinetics with rapid onset, fast off-set, and predictable dose-dependent increases of APTT with no effect on PT. The study concluded that selective inhibition of FXIa with EP-7041 promotes anticoagulant and profibrinolytic effect with minimal risk of bleeding.
2.1.6. Miscellaneous
Lastly, other chemotypes of FXIa inhibitors were reported by Chinese pharmaceutical companies. This includes pyrrolidine-amide derivative 36 [101] and 3-(4-fluorophenyl)-ethyl propano-ate derivative 37 [102] (Figure 12), both of which demonstrated nanomolar potency of FXIa inhibition.
Figure 12.
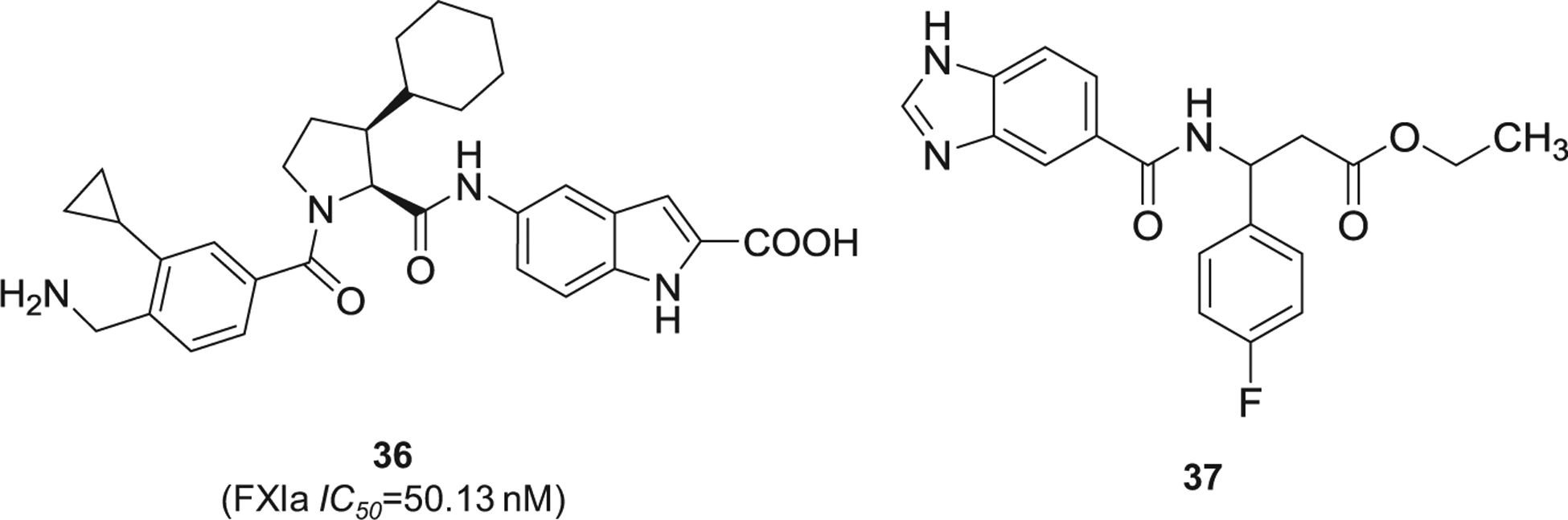
The chemical structures of miscellaneous inhibitors of FXIa (36 and 37) filed by international companies. Provided are the FXIa IC50 values (nM).
2.2. Macromolecules
2.2.1. FXI(a) monoclonal antibodies
Several companies and academic institutions reported on the preparation and/or the use of monoclonal antibodies to target FXI(a) system for the treatment and/or the prevention of thrombosis. This includes Novartis AG (2016 and 2018) [103–105], Bayer Pharma (2018) [106,107], the regents of the University of California (2017) [108], Prothix BV (2017) [109], Merck Sharp & Dohme Corp (2017) [110], and a couple of patents by Shanghai Benemae Pharmaceutical Corp [111,112]. Specific monoclonal antibodies that have been pharmacologically exploited to target FXI(a) and have shown promising results in clinical trials include MAA868 (Novartis) [54,55], BAY1213790 (Bayer) [56], and AB023 (Aronora) [57,113].
Considering MAA868, the phage display technology was initially exploited to generate a fully humanized antibody NOV1090 which, upon expression as Fab, showed a high binding affinity to the zymogen FXI with a KD value of 163 ± 24 pM and to the protease FXIa with a KD value of 244 ± 96 pM. NOV1090 Fab inhibited FXIa with an IC50 value of 2.8 nM, yet it did not affect other human serine proteases including thrombin, FVIIa, FIXa, FXa, FXIIa, and plasma kallikrein. MAA868 was subsequently produced by transforming NOV1090 Fab into a full-length human immunoglobulin G1 and by making its primary structure sequence similar to a human germline sequence. Mechanistically, MAA868 traps FXI(a) in an inactive, zymogen-like conformation. Binding affinity studies indicated that MAA868 binds to the catalytic domains of human FXI with a KD value of 1.3 ± 0.3 pM and human FXIa with a KD value of 4.7 ± 2.1 pM as well as to cynomolgus monkey FXI with KD values of 4–6 pM and cynomolgus monkey FXIa with KD values of 6–9 pM [54,55].
In APTT assay, MAA868 prolonged the clotting time of human plasma in a dose-dependent fashion (APTTEC2x = 0.015 μM). In thrombin generation assay, MAA868 decreased the amount of thrombin in human plasma in a concentration-dependent fashion with an IC50 value of 0.024 μM. MAA868 had no effect on the PT clotting time up to 1.3 μM. In a model of FXI knockout mice reconstituted with human FXI, the administration of 0.6 mg/kg MAA868 significantly decreased the formation of FeCl3-induced occlusive carotid artery thrombi. In a dose-escalation pharmacodynamic study in cynomolgus monkeys, both IV and SC administration of 3 mg/kg MAA868 led to a significant increase in APTT and ~90% decrease in free FXI levels with no bleeding or other adverse events up to 13 weeks including at the highest tested dose of 100 mg/kg/week [54,55].
In a Phase I, randomized, blinded, placebo-controlled trial (funded by Novartis), the safety, the pharmacokinetics, and the pharmacodynamics of MAA868 were examined in 61 healthy adult subjects by administering single ascending doses (5 mg up to 240 mg; SC). Vital signs, physical examination, blood biochemistry, hematology, and ECG were normal in placebo and MAA868 groups. No incidents of injection site reactions or hypersensitivity reactions were reported in those treated with MAA868. There were 9 adverse events considered to be potentially related to MAA868. Yet, the adverse events were of mild intensity with headache being the most common [54,55].
Considering the pharmacokinetic results, MAA868 exposures were dose-proportional with low-to-moderate intersubject variability. The median time-to-peak plasma concentrations was in the range of 7–21 days. Levels effective to prolong APTT by >2-fold were achieved by 24 hours. The terminal elimination half-life was from 20.1 to 28.6 days. With respect to the pharmacodynamic aspects, dose- and time-dependent prolongation of APTT and reduction in free FXI levels and FXI coagulation activity were observed. In fact, a statistically significant prolongation of APTT started with the 50 mg dose but was maintained >2-fold only at higher doses of 150 mg dose (up to Day 29) and of 240 mg dose (up to Day 57). Exposure-response modeling indicated that maximal APTT prolongations of ~2.5 fold were observed at plasma levels of >30 nM of MAA868. The maximal inhibition was achieved on Day 2 with a mean FXI coagulation activity of 3% of baseline in the 150 mg dose cohort and 240 mg dose cohort. Importantly, no changes were observed in PT or thrombin time (TT). Lastly, exposure to MAA868 and duration of FXI inhibition were moderately lower in obese individuals. This could be because of the relatively large volume of distribution of MAA868 and/or its marginally rapid clearance. Novoseven, a commercially available recombinant FVII, was found to be effective in reversing MAA868 anticoagulation effects [54,55].
The second monoclonal antibody of interest is BAY1213790. The phage display technology was exploited to produce a fully human immunoglobulin G1 monoclonal antibody which was named as BAY1213790 [56]. Mechanistic analysis indicated that BAY1213790 is allosteric inhibitor of FXIa. The antibody binds to a specific region in the catalytic domain of FXIa that is adjacent to the enzyme active site. The antibody’s binding event appears to cause a substantial disruption in the active site leading to enzyme inhibition. In animal studies, BAY1213790 reduced thrombosis in a mouse carotid artery FeCl3-induced thrombosis model and did not increase bleeding times in rabbit liver-incision model [114].
Given the initial in vitro activity of BAY1213790, its safety, pharmacokinetics, and pharmacodynamics were evaluated in a Phase 1, single-center, randomized, single-blind, parallel-group, placebo-controlled, dose-escalation trial (funded by Bayer) in 83 healthy Caucasian men [56]. Nine doses were used in the range of 0.015–10 mg/kg and the follow-up visits continued to Day 150 following administration.
The study revealed that single IV doses of BAY1213790 were well tolerated with 54 of 81 volunteers in the antibody group reported at least one treatment-emergent adverse event. The most frequently observed events were headache (18.5%) and nasopharyngitis (17.3%), yet no relationship was indicated between the antibody dose and causality or intensity of adverse events. All adverse events were of mild to moderate intensity. Likewise, no signs of potential hypersensitivity reactions, bleeding, or thrombocytopenia were detected during the course of the study. There were no treatment-related severe adverse events or deaths in any group. Vital signs, laboratory tests, ECG, and physical examinations were normal. Dose-dependent increases in APTT were significantly observed following all doses except the lowest dose of 0.015 mg/kg. Following the administration of the highest dose of 10 mg/kg, the maximal mean increases in APTT in relative to the baseline were 1.85–2.17-fold. For doses of 0.6 mg/kg or higher, APTT was elevated above the baseline for at least 20 days. Minimal interindividual variability was also observed for all doses [56]. Furthermore, a significant dose-dependent reduction in apparent FXI activity was recorded for doses of 0.15 mg/kg or higher. FXI activity was maximally inhibited directly after the drug administration and slowly returned to near baseline values in about 55 days, with a moderate level of interindividual variability. Interestingly, bleeding times did not increase and were similar across all study doses during the first 6 days. Moreover, ROTEM whole blood clotting time remained elevated in a dose-dependent fashion for at least 144 hours after all study doses. No significant increase in thrombosis biomarkers was observed throughout the study [56].
Considering BAY1213790 pharmacokinetics, there was a dose-dependent increase in measurable exposure to the antibody for all doses of 0.06 mg/kg or higher. The median time to maximum concentration of the antibody was between 1.05 hrs and 4 hrs, and the mean elimination half-life was ~30–44 days. The mean clearance and mean steady state volume of distribution were in the range of 0.13–0.29 L/day and 4–11 L, respectively [56].
The third monoclonal antibody of interest is AB023 (Xisomab 3G3) [57,113]. The antibody AB023 was produced by complementarity determining region-grafting technology from the monoclonal mouse anti-human FXI antibody 14E11. In previous studies, 14E11 was shown to significantly prolong carotid artery patency in mouse carotid artery FeCl3-induced thrombosis model as well as to significantly prolong the time to breathing cessation in a model of thromboplastin-induced pulmonary embolism. Prolongation of APTT was not accompanied with an increase in bleeding in those models [115].
AB023 is a humanized, immunoglobulin G2b monoclonal antibody which mechanistically binds to the A2 apple domain of FXI with a KD value of 3.66 nM. This binding event inhibits FXIIa-mediated activation of FXI. Likewise, AB023 binds the A2 apple domain of FXIa with a KD value of 1.38 nM which eventually blocks interactions between FXIa and high molecular weight kininogen [113]. Interestingly, the antibody does not appear to affect the active site of FXIa or thrombin-mediated activation of FXI. AB023 prolonged the APTT of plasma from normal rats, baboons, cynomolgus monkeys, and humans in a concentration-dependent fashion. In a baboon acute vascular graft thrombosis model, a single IV pre-treatment with 1.0 mg/kg of AB023 reduced total platelet accumulation and total fibrin deposition within the collagen-coated vascular grafts and in the region that is 10 cm downstream of the graft. APTT was prolonged following the antibody treatment at 30 and 60 minutes after injection and continued to be elevated beyond 24-hour post-treatment without any effect on PT [57].
Motivated by the in vivo pre-clinical results, the safety, the pharmacokinetics, and the pharmacodynamics of AB023 were evaluated in a Phase 1, randomized, double-blind, placebo-controlled, single ascending bolus dose study at a single site (funded in part by US National Heart Lung and Blood Institute) in 21 subjects. Doses used were 0.1, 0.5, 2.0, and 5.0 mg/kg. AB023 was found to be safe and well-tolerated in the healthy volunteers with no effect on mean bleeding time or PT. There were no serious adverse events with a total of 20 treatment-emergent adverse events. The most common treatment-emergent adverse event was a productive cough which was considered by the investigators as unrelated or unlikely treatment-related event [57].
In the pharmacokinetic studies, plasma exposure was found to increase with increasing the AB023 doses. Median values of time to maximum concentrations were in the range of 0.08–0.65 hrs. Mean elimination half-life values appeared to increase with increasing AB023 doses and ranged from ~1.3 hrs following the 0.1 mg/kg dose to ~121.5 hrs following the 5.0 mg/kg dose. The mean plasma clearance appeared to decrease with increasing AB023 doses and was in the range of 0.094–0.014 L/hrs. Steady state volume of distribution appeared to increase with increasing the antibody dose and was in the range of 2.5–4.3 L. The maximum APTT prolongation was achieved at doses of ≥0.5 mg/kg. Mean APTT values were highest or close to at 0.25–24 hrs post-administration at all concentrations tested. The APTT values returned to baseline by 24 hrs, 336 hrs, or 672 hrs for most or all subjects at dose levels of 0.1 mg/kg, 0.5 mg/kg, or 2.0–5.0 mg/kg, respectively [57].
Lastly, University of California and Pfizer Inc. developed a series of human immunoglobulin Gs that inhibited FXIa by targeting its active site and did not affect its zymogen or other coagulation proteases [108,116]. The immunoglobulins were produced via phage display technology with the most potent being C24 and DEF. In FXIa fluorogenic substrate hydrolysis assay, C24 inhibited human, cynomolgus macaque, and rabbit FXIa with IC50 values of 0.22–0.26 nM [108,116]. The KD binding value to human FXIa was 0.0152 nM (C24 Fab). Furthermore, C24 did not bind to thrombin, FVIIa, FIXa, FXa, FXIIa, or plasma kallikrein, at concentrations 100-fold larger than those required to saturate FXIa in an ELISA-based assay. Likewise, C24 showed at least 80-fold inhibitory selectivity to small-substrate cleavage by FXIa over thrombin, FVIIa, FXa, FXIIa, plasma kallikrein, or activated protein C. C24 did not inhibit the proteolytic activation of FX by FIXa [108,116]. In human plasma, C24 inhibited the FXIIa-mediated peak thrombin generation with an IC50 value of 34 nM. In an intrinsic pathway-triggered clotting assay in human plasma and whole human blood, C24 led to half-maximal prolongations of clotting times at concentrations of ~69 nM and 21 nM, respectively. Using IV doses of 2–35 mg/kg, C24 inhibited FeCl3-induced arterial thrombosis in FXI-humanized mouse [108,116].
To improve C24’s drug-like properties, DEF monoclonal antibody was generated with similar activity, selectivity, and mechanism of action. In FXIa fluorogenic substrate hydrolysis assay, DEF inhibited human, cynomolgus macaque, and rabbit FXIa with IC50 values of 0.22–0.24 nM [108,116]. The KD binding value to human FXIa was 0.0339 nM (DEF Fab). DEF (0.1 and 1.0 mg/kg; IV) decreased venous thrombus formation in a rabbit model in a dose-dependent fashion. The effect correlated with APTT prolongation. DEF did not affect the extrinsic pathway or the common pathway functions as indicated by the lack of effect on PT at a dose as high as 10 mg/kg. Furthermore, treatment with DEF did not increase cuticle bleeding in rabbits (10 mg/kg; IV) or spontaneous bleeding in cynomolgus macaques (20, 75, and 266.5 mg/kg; IV), whereas APTT was maximally or near maximally prolonged and PT was unaffected. Interestingly, the DEF activity ex vivo in human plasma and in vivo in rabbits was rapidly reversed using a human immunoglobulin labeled as revC4 which exhibited an apparent KD value of 3 nM to DEF Fab [108,116].
2.2.2. DNA & RNA aptamers
Aptamers are single-stranded oligonucleotides. They are typically isolated from combinatorial libraries of nucleic acids using an in vitro selection process known as systematic evolution of ligands by exponential enrichment (SELEX) [117]. Generally speaking, aptamers can recognize a target protein or nucleic acid with high affinity and specificity which makes them very useful therapeutic, diagnostic, or research tools. Aptamers that target different coagulation factors have been developed including those targeting FIX(a), FXI(a), and FXII(a) [118–125].
Specifically, FXIa-targeting aptamers were claimed in the year of 2017 to be used in the prevention, treatment, or alleviation of the symptoms of thrombosis. The aptamers were also claimed to be used for the detection or purification of FXIa [123]. The patent provided information on the structural aspects of the aptamers and provided information regarding the aptamers’ anticoagulant activities. Briefly, the patent claimed aptamers with general formula of 5`−5W-C-3W-3` in which the 5`wing nucleotide sequence was 5`-GAATTCTAATACGACTCACTATA whereas the 3`wing nucleotide sequence was GCGTCCAACACATCG-3`. The core C represented the hypervariable central sequence [123].
In peer-reviewed reported results, a library of single-stranded DNA aptamers was screened for FXIa-binding using multiple cycles of negative and positive selection. Ultimately, 6 sequences out of 89 sequences were identified to inhibit the chromogenic substrate S2366 cleavage by FXIa (Figure 13). The most active aptamer was identified as Factor ELeven Inhibitory APtamer (FELIAP) which possessed a hypervariable central sequence of 5′-AACCTATCGGACTATTGTTAGTGATTTTTATAGTGT-3′ [124]. Kinetic studies indicated that FELIAP is a competitive inhibitor of FXIa-mediated hydrolysis of S-2366 with an estimated Ki value of 29 μM. Not only that, but 10 μM FELIAP also inhibited the FXIa-mediated activation of FIX to FIXa as well as the heparin-mediated interaction between FXIa and antithrombin. A KD value of 1.8 ± 0.1 nM was measured for FELIAP using surface plasmin resonance. Furthermore, 30 μM FELIAP prolonged the lag time of thrombin generation in recalcified diluted plasma. Likewise, FELIAP significantly reduced the endogenous thrombin potential by 3.2-fold as well as significantly increased the mean time to peak thrombin by 1.5-fold. These effects were eliminated when FXI-depleted plasma was exploited. Accordingly, FELIAP was put forward as a lead to develop DNA-based aptamers that target FXI(a) for clinical use [124].
Figure 13.

The nucleotide sequences of the hypervariable core domains of the six active aptamers identified in references 123 and 124. The most active aptamer was FELIAP. The positive signs on the right is indicative of activity against FXIa as determined in S-2366 chromogenic substrate hydrolysis assay. +++ = most active, ++ = moderately active, + = slightly active. Highlighted nucleotides indicate differences among the first four aptamers. ‘*’ in the fourth aptamer indicates that the last nucleotide is missing.
Along these lines, two RNA aptamers, identified as 12.7 and 11.16, were reported to inhibit FXIa with the former binding to both the protease FXIa and the zymogen FXI and the latter binding only to the protease FXIa [125]. Besides, the two aptamers did not bind to a panel of coagulation factors. In the S-2366 chromogenic substrate hydrolysis assay, the two aptamers were shown to be noncompetitive inhibitors targeting the anion-binding sites on the catalytic domain, the A3 domain, and/or the autolysis loop. The Ki values for the aptamers were 60 ± 20 and 63 ± 22 nM, respectively. The two aptamers also significantly delayed FXIa-mediated activation of FIX. Aptamer 12.7 increased the APTT from 28.4 ± 6.3 sec to 56.3 ± 14.6 sec, whereas aptamer 11.16 increased the APTT to only 36.4 ± 5.3 seconds. Accordingly, the two aptamers were proposed to serve as leads to further develop RNA-based aptamers that target FXI(a) for clinical use [125].
2.2.3. Polypeptides
Prior to 2016, few polypeptides were reported to influence FXIa activity. The most recognized ones were fasxiator, desmolaris, AcaNAP10, and Ir-CPI [5,6,126,127]. In 2018, a polypeptide named Sj13 from Schistosoma Japonicum, a parasite causing schistosomiasis, and its mutants were reported to exert anticoagulant activity by inhibiting FXIa [128]. The amino acid sequence of Sj13 polypeptide was identified. The recombinant Sj13 polypeptide and its mutants were also obtained by genetic engineering technology. The in vitro anticoagulant activity of the polypeptide and its mutant was established in APTT, PT, and TT tests. Their anticoagulant activities were attributed to inhibiting FXIa as well as FXa. They also inhibited the antifibrinolytic plasmin. In the mice model of FeCl3-induced carotid artery thrombosis, it was shown that Sj13, at doses of 1.0 mg/kg and 0.5 mg/kg, led to reductions in clot weights comparable to those achieved by heparin. The polypeptide was reported to be associated with low bleeding risk [128].
3. Conclusion
In the above discussion, we provided an overview of the most recent patents by industrial as well as academic institutions in which molecular entities that target FXIa or its zymogen FXI have been claimed to promote anticoagulant activity with minimal bleeding complications. In combination with our previous surveys of patent and peer-reviewed literatures [5,6], this patent update reveals that the agents that have been claimed and/or confirmed to inhibit FXI(a) are structurally and mechanistically diverse. These agents include small molecule inhibitors [5,6,17,60], monoclonal antibodies [54–57,113], antisense oligonucleotides [52,53], aptamers [119–121], and polypeptides [126,127]. Some of these agents target the protease FXIa whereas others target both the protease FXIa and its zymogen FXI. Some of these agents are competitive inhibitors and others are noncompetitive inhibitors. In addition, they are also diverse with respect to their pharmacokinetic aspects including the route of administration (oral vs parenteral), the onset of action (fast vs delayed), the duration of action (short vs extended), and the clearance (hepatic and/or renal dependent vs hepatic and/or renal independent). As a result, we expect the structural and the mechanistic diversity of molecular entities targeting FXI(a) system to increase the likelihood of one or more of these agents to entering the clinic.
4. Expert opinion
The anticoagulation therapy has significantly improved over the last decade. This can partially be attributed to the approval of the novel direct oral anticoagulants, particularly FXa inhibitors [129]. Furthermore, the approval of new reversal agents including idarucizumab for dabigatran [130] and andexanet alfa for FXa inhibitors [131] has permitted a better management of the anticoagulant-induced bleeding complications. Yet, none of the currently available anticoagulants fulfill the pursuit of an ‘ideal’ anticoagulant. In fact, all current anticoagulants continue to be associated with an elevated and variable risk of bleeding, which could become very serious in patients with moderate-to-severe renal dysfunction, atrial fibrillation [7–12], or those who are at the extremes of body weights [132–134], among others. From the standpoint of clinical practice, even minor bleeds may become problem because patients tend to stop their treatments; not only their anticoagulant treatment, but other life-saving therapies as well. Moreover, the clearance of most of the clinically used anticoagulants is dependent on hepatic and/or renal function which perhaps demands dose adjustment in patients with compromised hepatic and/or renal function [135,136]. Therefore, the search continues for an ideal anticoagulant that is (1) not associated with a risk of bleeding or severe toxicity in general; (2) safe in compromised patient populations such as those with chronic kidney failure, hepatic dysfunction, atrial fibrillation, or extreme body weights; (3) available in inexpensive oral and/or parenteral dosage forms; (4) exhibiting predictable pharmacokinetics; and (5) rapidly reversed by effective and inexpensive reversal agents [5].
Mechanistically, all anticoagulants in use today directly or indirectly inhibit thrombin and/or FXa, two serine proteases in the common coagulation pathway. This mechanistic aspect is the reason for their efficacy, but it is also the reason for the bleeding complications. Therefore, a rationale strategy to address the problem of bleeding has been to consider other proteins in the coagulation process. Nevertheless, with the advancement in our understanding of hemostasis and thrombosis as well as that of complications associated with the genetic deficiencies of clotting factors in humans, a compelling rationale for targeting FXI(a) to treat and/or prevent thromboembolism without inducing hemorrhagic complications has emerged and has been further advanced by many pre-clinical and clinical studies [5,6].
Although the number of patents claiming agents to inhibit/target FXI(a) has declined since the year of 2016 (Figure 1(b)), yet the number of agents advanced into clinical trials have significantly increased (Table 1). In Phase I trials, the tested agents (ISIS 416858, MAA868, BAY1213790, AB023, and EP-7041) demonstrated no serious adverse effects and prolonged APTT with no effect on PT. Furthermore, in a Phase II trial, the antisense oligonucleotide ISIS 416858 lowered FXI levels in patients undergoing elective unilateral total knee arthroplasty to a level that was sufficient to prevent postoperative venous thromboembolism without inducing bleeding complications [52,53]. Moreover, BMS-986177 (JNJ-70033093) completed multiple Phase I trials in healthy subjects, subjects with hepatic dysfunction, subjects with renal impairment, subjects with renal impairment on hemodialysis, subjects using aspirin, and subjects having potentially interacting medications. Thus far, the results of pre-clinical and clinical studies indicate that targeting FXI(a) of the intrinsic coagulation pathway is a powerful strategy toward achieving anticoagulation that is effective and significantly safer considering the bleeding complications.
Table 1.
Pharmacodynamic and pharmacokinetic characteristics of molecules targeting FXI(a).
| Small molecules | Polypeptides | Monoclonal antibodies | Aptamers | Antisense oligonucleotides | |
|---|---|---|---|---|---|
| Pharmacodynamics | |||||
| Mechanism of action | Competitive & noncompetitive inhibitors of FXIa | Competitive inhibitors of FXIa | Competitive & noncompetitive inhibitors of FXIa; some target FXI | Competitive & noncompetitive inhibitors of FXIa | FXI mRNA |
| Selectivity | Significant selectivity has been achieved by all classes | ||||
| Pharmacokinetics | |||||
| Route of administration | Oral and/or parenteral | Parenteral | |||
| Onset of action | Fast | Delayed | |||
| Duration | Relatively short (hours), but can be extended using specific formulation or drug delivery system | Relatively long (days or months) | |||
| Clearance | More likely to be dependent on renal and/or hepatic systems | Less likely to be dependent on renal and/or hepatic systems | |||
| Clinical trials | EP-7041 and BMS-986177 (Phase I) | None | MAA868, BAY1213790, and AB023 (Phase I) | None | ISIS 416858 (Phase II in postoperative VTE prevention) |
Further development for the above agents is undergoing. A Phase I trial is recruiting to examine the tolerability and safety of BAY1213790 in subjects with end-stage renal disease on hemodialysis (NCT03787368). A Phase II FOXTROT trial, to compare BAY1213790 to current therapies (enoxaparin or apixaban) for the prevention of blood clotting and safety in patients undergoing total knee arthroplasty, was completed (NCT03276143). A Phase II trial of the orally bioavailable BMS-986177 in combination with clopidogrel or aspirin is currently enrolling (NCT03766581) to determine the efficacy and the safety of the combination therapy in relative to the standard therapy in secondary ischemic stroke prevention. A study of BMS-986177 versus subcutaneous enoxaparin in participants undergoing elective total knee replacement surgery (AXIOMATIC-TKR) is also currently recruiting (NCT03891524). Furthermore, a subcutaneously administered ISIS 416858 is in a Phase II trial to examine its safety, pharmacokinetics, and pharmacodynamics in patients with end-stage renal disease on chronic hemodialysis (NCT03358030). Likewise, AB023 (xisomab 3G3) is under consideration for a Phase II trial in patients with end stage renal disease on chronic hemodialysis to evaluate its safety and efficacy for reducing dialysis circuit thrombosis (NCT03612856). A two-part study to assess the tolerability, safety, pharmacokinetics, and pharmacodynamics of ONO-7684 [137,138] (ONO-5450598; oral FXIa inhibitor that is being developed by Ono Pharmaceutical Co., Japan) in healthy adult volunteers is recruiting (NCT03919890).
Although further development is warranted, reaching one or more of the above FXI(a) targeting agents to the clinic will fundamentally transform the anticoagulation therapy. Potential indications for FXI(a) inhibitors include (a) prophylaxis against stroke in atrial fibrillation patients who are at a high bleeding risk; (b) cardiovascular events prevention in patients with chronic kidney disease or those undergoing hemodialysis; (c) thrombosis prophylaxis in patients with venous thromboembolism who are at increased risk of recurrent thrombosis following anticoagulant therapy cessation; (d) clotting prophylaxis in extracor-poreal membrane oxygenation circuits; (e) prevention of cardiac devices-triggered thromboembolic events; and (f) prevention of stent thrombosis in acute coronary syndrome patients who require anticoagulant therapy besides the antiplatelet therapy [5,6]. Overall, the new anticoagulant therapy is expected to benefit thrombotic patients for who current therapy is limited in safety and/or efficacy.
Article highlights.
Factor XIa is a homodimeric coagulation serine protease which continues to be an attractive drug target for effective and relatively safer anticoagulants.
Small molecules and monoclonal antibodies dominate the drug development arena for factor XI(a) inhibition.
The past 3 years have witnessed an increase in the number of clinical trials that evaluate the efficacy and safety of factor XI(a) inhibitors.
Small molecules offer the convenience of oral administration, whereas monoclonal antibodies provide the benefits of monthly dosing and the independence of the drug clearance process from hepatic and renal systems.
Funding
This work was supported by NIGMS under award number [SC3GM131986] and by IDeA program from NIGMS under grant number [P20GM103424]. The content is solely the responsibility of the author and does not necessarily represent the official views of NIH.
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Wendelboe AM, Raskob GE. Global burden of thrombosis: epidemiologic aspects. Circ Res. 2016;118:1340–1347. [DOI] [PubMed] [Google Scholar]
- 2.Raskob GE, Angchaisuksiri P, Blanco AN, et al. Thrombosis: a major contributor to the global disease burden. J Thromb Haemost. 2014;12:1580–1590. [DOI] [PubMed] [Google Scholar]
- 3.Garcia DA, Baglin TP, Weitz JI, et al. Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: american college of chest physicians evidence-based clinical practice guidelines. Chest. 2012;141:e24S–43S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ageno W, Gallus AS, Wittkowsky A, et al. Oral anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American college of chest physicians evidence-based clinical practice guidelines. Chest. 2012;141:e44S–88S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Horani RA, Desai UR. Factor XIa inhibitors: a review of the patent literature. Expert Opin Ther Pat. 2016;26(3):323–345. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Thorough review of patent literature (−2016) for agents targeting FXI(a).
- 6.Al-Horani RA, Afosah DK. Recent advances in the discovery and development of factor XI/XIa inhibitors. Med Res Rev. 2018;38 (6):1974–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Thorough review of peer-reviewed literature for agents targeting FXI(a).
- 7.Alamneh EA, Chalmers L, Bereznicki LR. Suboptimal use of oral anticoagulants in atrial fibrillation: has the introduction of direct oral anticoagulants improved prescribing practices? Am J Cardiovasc Drugs. 2016;16:183–200. [DOI] [PubMed] [Google Scholar]
- 8.Barra ME, Fanikos J, Connors JM, et al. Evaluation of dose-reduced direct oral anticoagulant therapy. Am J Med. 2016;129:1198–1204. [DOI] [PubMed] [Google Scholar]
- 9.Hughes S, Szeki I, Nash MJ, et al. Anticoagulation in chronic kidney disease patients-the practical aspects. Clin Kidney J. 2014;7 (5):442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lutz J, Jurk K, Schinzel H. Direct oral anticoagulants in patients with chronic kidney disease: patient selection and special considerations. Int J Nephrol Renovasc Dis. 2017;10:135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Black-Maier E, Piccini JP. Oral anticoagulation in end-stage renal disease and atrial fibrillation: is it time to just say no to drugs? Heart. 2017;103(11):807–808. [DOI] [PubMed] [Google Scholar]
- 12.Heine GH, Brandenburg V. Anticoagulation, atrial fibrillation, and chronic kidney disease-whose side are you on? Kidney Int. 2017;91 (4):778–780. [DOI] [PubMed] [Google Scholar]
- 13.van Montfoort ML, Meijers JC. Anticoagulation beyond direct thrombin and factor Xa inhibitors: indications for targeting the intrinsic pathway? Thromb Haemost. 2013;110(2):223–232. [DOI] [PubMed] [Google Scholar]
- 14.Nagakura T, Tabata K, Kira K, et al. Selective tissue factor/factor VIIa Inhibitor, ER-410660, and its prodrug, E5539, have anti-venous and anti-arterial thrombotic effects with a low risk of bleeding. Thromb Res. 2013;132(2):271–279. [DOI] [PubMed] [Google Scholar]
- 15.Barbieri CM, Wang X, Wu W, et al. Factor XIIa as a novel target for thrombosis: target engagement requirement and efficacy in a rabbit model of microembolic signals. J Pharmacol Exp Ther. 2017;360(3):466–475. [DOI] [PubMed] [Google Scholar]
- 16.Ankrom W, Wood HB, Xu J, et al. Preclinical and translational evaluation of coagulation factor IXa as a novel therapeutic target. Pharmacol Res Perspect. 2016;4(1):e00207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Al-Horani RA, Abdelfadiel EI, Afosah DK, et al. A synthetic heparin mimetic that allosterically inhibits factor XIa and reduces thrombosis in vivo without enhanced risk of bleeding. J Thromb Haemost. 2019;17(12):2110–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]; • The in vivo anticoagulant activity of heparin-based, allosteric inhibitor of FXIa.
- 18.Emsley J, McEwan PA, Gailani D. Structure and function of factor XI. Blood. 2010;115:2569–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gailani D, Smith SB. Structural and functional features of factor XI. J Thromb Haemost. 2009;7(Suppl 1):75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohammed BM, Matafonov A, Ivanov I, et al. An update on factor XI structure and function. Thromb Res. 2018;161:94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Recent review of FXI(a) structure and function.
- 21.Baglia FA, Walsh PN. A binding site for thrombin in the apple 1 domain of factor XI. J Biol Chem. 1996;271:3652–3658. [DOI] [PubMed] [Google Scholar]
- 22.Renne T, Gailani D, Meijers JC, et al. Characterization of the H-kininogen-binding site on factor XI: a comparison of factor XI and plasma prekallikrein. J Biol Chem. 2002;277:4892–4899. [DOI] [PubMed] [Google Scholar]
- 23.Sun MF, Zhao M, Gailani D. Identification of amino acids in the factor XI apple 3 domain required for activation of factor IX. J Biol Chem. 1999;274:36373–36378. [DOI] [PubMed] [Google Scholar]
- 24.Baglia FA, Gailani D, Lopez JA, et al. Identification of a binding site for glycoprotein Ibalpha in the apple 3 domain of factor XI. J Biol Chem. 2004;279:45470–45476. [DOI] [PubMed] [Google Scholar]
- 25.Ho DH, Badellino K, Baglia FA, et al. A binding site for heparin in the apple 3 domain of factor XI. J Biol Chem. 1998;273:16382–16390. [DOI] [PubMed] [Google Scholar]
- 26.Baglia FA, Jameson BA, Walsh PN. Identification and characterization of a binding site for factor XIIa in the apple 4 domain of coagulation factor XI. J Biol Chem. 1993;268:3838–3844. [PubMed] [Google Scholar]
- 27.Yang L, Sun MF, Gailani D, et al. Characterization of a heparin-binding site on the catalytic domain of factor XIa: mechanism of heparin acceleration of factor XIa inhibition by the serpins antithrombin and C1-inhibitor. Biochemistry. 2009;48:1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolberg AS, Morris DP, Stafford DW. Factor IX activation by factor XIa proceeds without release of a free intermediate. Biochemistry. 1997;36:4074–4079. [DOI] [PubMed] [Google Scholar]
- 29.Geng Y, Verhamme IM, Messer A, et al. A sequential mechanism for exosite-mediated factor IX activation by factor XIa. J Biol Chem. 2012;287:38200–38209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matafonov A, Cheng Q, Geng Y, et al. Evidence for factor IX-independent roles for factor XIa in blood coagulation. J Thromb Haemost. 2013;11:2118–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whelihan MF, Orfeo T, Gissel MT, et al. Coagulation procofactor activation by factor XIa. J Thromb Haemost. 2010;8:1532–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi SH, Smith SA, Morrissey JH. Polyphosphate accelerates factor V activation by factor XIa. Thromb Haemost. 2015;113:599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Griffin JH. Role of surface in surface-dependent activation of Hageman factor (blood coagulation factor XII). Proc Natl Acad Sci USA. 1978;75:1998–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scott CF, Silver LD, Purdon AD, et al. Cleavage of human high molecular weight kininogen by factor XIa in vitro. Effect on structure and function. J Biol Chem. 1985;260:10856–10863. [PubMed] [Google Scholar]
- 35.Gailani D, Broze GJ Jr. Factor XI activation in a revised model of blood coagulation. Science. 1991;253(5022):909–912. [DOI] [PubMed] [Google Scholar]
- 36.Puy C, Tucker EI, Matafonov A, et al. Activated factor XI increases the procoagulant activity of the extrinsic pathway by inactivating tissue factor pathway inhibitor. Blood. 2015;125(9):1488–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Puy C, Garland KS, Shirai T, et al. Activated FXI regulates the catalytic activity of ADAMTS13 by removing the CUB domains. RPTH. 2017;1(Suppl. 1):98. [Google Scholar]
- 38.Kossmann S, Lagrange J, Jäckel S, et al. Platelet-localized FXI promotes a vascular coagulation-inflammatory circuit in arterial hypertension. Sci Transl Med. 2017;9:pii: eaah4923. [DOI] [PubMed] [Google Scholar]
- 39.White-Adams TC, Berny MA, Tucker EI, et al. Identification of coagulation factor XI as a ligand for platelet apolipoprotein E receptor 2 (ApoER2). Arterioscler Thromb Vasc Biol. 2009;29:1602–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White-Adams TC, Berny MA, Patel IA, et al. Laminin promotes coagulation and thrombus formation in a factor XII-dependent manner. J Thromb Haemost. 2010;8:1295–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.James P, Salomon O, Mikovic D, et al. Rare bleeding disorders—bleeding assessment tools, laboratory aspects and phenotype and therapy of FXI deficiency. Haemophilia. 2014;20(Suppl 4):71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seligsohn U Factor XI deficiency in humans. J Thromb Haemost. 2009;7(Suppl 1):84–87. [DOI] [PubMed] [Google Scholar]
- 43.Salomon O, Steinberg DM, Seligshon U. Variable bleeding manifestations characterize different types of surgery in patients with severe factor XI deficiency enabling parsimonious use of replacement therapy. Haemophilia. 2006;12:490–493. [DOI] [PubMed] [Google Scholar]
- 44.Salomon O, Steinberg DM, Zucker M, et al. Patients with severe factor XI deficiency have a reduced incidence of deep-vein thrombosis. Thromb Haemost. 2011;105:269–273. [DOI] [PubMed] [Google Scholar]
- 45.Salomon O, Steinberg DM, Koren-Morag N, et al. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood. 2008;111:4113–4117. [DOI] [PubMed] [Google Scholar]
- 46.Meijers JC, Tekelenburg WL, Bouma BN, et al. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med. 2000;342:696–701. [DOI] [PubMed] [Google Scholar]
- 47.Yang DT, Flanders MM, Kim H, et al. Elevated factor XI activity levels are associated with an increased odds ratio for cerebrovascular events. Am J Clin Pathol. 2006;126:411–415. [DOI] [PubMed] [Google Scholar]
- 48.Suri MF, Yamagishi K, Aleksic N, et al. Novel hemostatic factor levels and risk of ischemic stroke: the Atherosclerosis Risk in Communities (ARIC) study. Cerebrovasc Dis. 2010;29:497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siegerink B, Govers-Riemslag JW, Rosendaal FR, et al. Intrinsic coagulation activation and the risk of arterial thrombosis in young women: results from the Risk of Arterial Thrombosis in Relation to Oral Contraceptives (RATIO) case-control study. Circulation. 2010;122:1854–1861. [DOI] [PubMed] [Google Scholar]
- 50.Doggen CJ, Rosendaal FR, Meijers JC. Levels of intrinsic coagulation factors and the risk of myocardial infarction among men: opposite and synergistic effects of factors XI and XII. Blood. 2006;108:4045–4051. [DOI] [PubMed] [Google Scholar]
- 51.Berliner JI, Rybicki AC, Kaplan RC, et al. Elevated levels of factor XI are associated with cardiovascular disease in women. Thromb Res. 2002;107:55–60. [DOI] [PubMed] [Google Scholar]
- 52.Büller HR, Bethune C, Bhanot S, et al. Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med. 2015;372(3):232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Phase II clinical trial for ISIS-FXIRx in patients undergoing elective primary unilateral total knee arthroplasty.
- 53.Younis HS, Crosby J, Huh JI, et al. Antisense inhibition of coagulation factor XI prolongs APTT without increased bleeding risk in cynomolgus monkeys. Blood. 2012;119(10):2401–2408. [DOI] [PubMed] [Google Scholar]
- 54.Koch AW, Schiering N, Melkko S, et al. MAA868, a novel FXI antibody with a unique binding mode, shows durable effects on markers of anticoagulation in humans. Blood. 2019;133 (13):1507–1516. [DOI] [PubMed] [Google Scholar]; •• Phase I clinical trial for MAA868 in healthy volunteers.
- 55.Weitz JI, Chan NC. MAA868 locks factor XIa in a zymogen-like state. Blood. 2019;133(13):1393–1394. [DOI] [PubMed] [Google Scholar]
- 56.Thomas D, Thelen K, Kraff S, et al. BAY1213790, a fully human IgG1 antibody targeting coagulation factor XIa: first evaluation of safety, pharmacodynamics, and pharmacokinetics. Res Pract Thromb Haemost. 2019;3(2):242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Phase I clinical trial for BAY1213790 in healthy volunteers.
- 57.Lorentz CU, Verbout NG, Wallisch M, et al. Contact activation inhibitor and factor XI antibody, AB023, produces safe, dose-dependent anticoagulation in a Phase 1 first-in-human trial. Arterioscler Thromb Vasc Biol. 2019;39(4):799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Phase I clinical trial for AB023 in healthy volunteers.
- 58.Hayward NJ, Goldberg DI, Morrel EM, et al. Phase 1a/1b study of EP-7041: a novel, potent, selective, small molecule FXIa inhibitor. Circulation. 2017;136:A13747. [Google Scholar]; •• Phase I clinical trial for EP-7041 in healthy volunteers.
- 59.NCT02608970, NCT03362437, NCT03196206, NCT03000673, NCT02959060, and NCT03341390. [cited 2019 Dec 10]. Available from: https://clinicaltrials.gov
- 60.Al-Horani RA, Desai UR. Designing allosteric inhibitors of factor XIa. Lessons from the interactions of sulfated pentagalloyl glucopyranosides. J Med Chem. 2014;57(11):4805–4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Afosah DK, Al-Horani RA. Sulfated non-saccharide glycosaminoglycan mimetics as novel drug discovery platform for various pathologies. Curr Med Chem. 2018;25 DOI: 10.2174/0929867325666181120101147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu W, Edmondson SD, Guo Z, et al. ; Merck Sharp & Dohme Corp., US. Pyridine N-oxides as factor XIa inhibitors and their preparation. WO2016015593A1. 2016. [Google Scholar]
- 63.Mertz E, Edmondson SD, Guo Z, et al. ; Merck Sharp & Dohme Corp., US. Preparation of 5-[5-chloro-2-(1H-tetrazol-1-yl)phenyl]-2-[1-substituted 2-(phenyl)ethyl] pyridine 1-oxide derivatives as factor XIa inhibitors. WO2016018702A1. 2016. [Google Scholar]
- 64.Liu W, Edmondson SD, Guo Z, et al. ; Merck Sharp & Dohme Corp., US. Preparation of oxopyridinyl benzimidazole derivatives as factor XIa inhibitors. WO2016018701A1. 2016. [Google Scholar]
- 65.Ogawa AK, Brockunier LL, Tata J, et al. ; Merck Sharp & Dohme Corp., US. Pyridine N-oxides as factor XIa and plasma kallikrein inhibitors and their preparation. WO2017095760A1. 2017. [Google Scholar]
- 66.Mertz E, Liu W, Edmondson SD, et al. ; Merck Sharp & Dohme Corp., US. Preparation of substituted cyclopenta-pyridine-carboxamides as factor XIa inhibitors. WO2016118403A1. 2016. [Google Scholar]
- 67.Mertz E, Edmondson SD, So S, et al. ; Merck Sharp & Dohme Corp., US. Cyclopenta[b]pyridine 1-oxides and 5,6,7,8-tetrahydroquinoline-1-oxides as factor XIa inhibitors and their preparation. WO2016168098A1. 2016. [Google Scholar]
- 68.Shen DM, Kuang R, Kumar P, et al. ; Merck Sharp & Dohme Corp., US. Preparation of arylamide pyridine oxides as factor XIa inhibitors for the treatment and prevention of thrombosis and related disease. US20180050022A1. 2018. [Google Scholar]
- 69.Roehrig S, Hillisch A, Strassburger J, et al. ; Bayer Pharma, Germany. Preparation of substituted oxopyridine derivatives as factor XIa and plasma kallikrein inhibitors. WO2016046158A1. 2016. [Google Scholar]
- 70.Roehrig S, Jimenez-Nunez E, Schlemmer K-H, et al. ; Bayer Pharma, Germany. Preparation of substituted oxopyridine derivatives as factor XIa and plasma kallikrein inhibitors. WO2016046166A1. 2016. [Google Scholar]
- 71.Roehrig S, Hillisch A, Heitmeier S, et al. ; Bayer Pharma, Germany. Preparation of factor XIa-inhibiting pyridobenzazepine and pyridobenzazocine derivatives. WO2016046157A1. 2016. [Google Scholar]
- 72.Roehrig S, Jimenez NE, Schlemmer K-H, et al. ; Bayer Pharma, Germany. Preparation of substituted oxopyridine derivatives as factor XIa and plasma kallikrein inhibitors. WO2016046164 A1. 2016. [Google Scholar]
- 73.Roehrig S, Hillisch A, Heitmeier S, et al. ; Bayer Pharma, Germany. Preparation of substituted oxopyridine derivatives as factor XIa and plasma kallikrein inhibitors. WO2016046159A1. 2016. [Google Scholar]
- 74.Roehrig S, Jimenez NE, Schlemmer K-H, et al. ; Bayer Pharma, Germany. Preparation of substituted oxopyridine derivatives as factor XIa and plasma kallikrein inhibitors. WO2016046156A1. 2016. [Google Scholar]
- 75.Roehrig S, Teller H, Heitmeier S, et al. ; Bayer Pharma, Germany. Preparation of oxopyridine derivatives as factor XIa inhibitors for the treatment of thrombosis. WO2016146606A1. 2016. [Google Scholar]
- 76.Jimenez NE, Ackerstaff J, Ellerbrock P, et al. ; Bayer Pharma, Germany. Preparation of substituted oxopyridine derivatives as factor XIa and plasma kallikrein inhibitors. WO2017037051A1. 2017. [Google Scholar]
- 77.Yang F, Wang W, Li X, et al. ; Shanghai Hengrui Pharmaceutical Co., China. Process for preparation of oxopicolinamide derivative and pharmaceutical use WO2018041122A1. 2018. [Google Scholar]
- 78.Yang F, Qu J, Wang C, et al. ; Shanghai Hengrui Pharmaceutical Co., China. Indole-amide derivatives as XIa factor inhibitors and their preparation, pharmaceutical compositions and use in the treatment of cardiovascular diseases. WO2016011940A1. 2016. [Google Scholar]
- 79.Yang F, Wang W, Ying Y, et al. ; Shanghai Hengrui Pharmaceutical Co., China. Preparation of oxazoloindole derivatives useful as XIa factor inhibitors for the treatment of cardiovascular disease. CN106496249A. 2017. [Google Scholar]
- 80.Yang F, Chi J, He F, et al. ; Shanghai Hengrui Pharmaceutical Co., China. Epoxy-substituted oxopyridine derivative as blood coagulation factor XIa inhibitor and its preparation. CN107793396A. 2018. [Google Scholar]
- 81.Corte JR, Ewing WR, Pinto DJP, et al. ; Bristol-Myers Squibb Co., US. Preparation of novel substituted glycine derivatives as factor XIa and/or plasma kallikrein inhibitors. WO2017023992A1. 2017. [Google Scholar]
- 82.Cummings MD, Sekharan S. Structure-based macrocycle design in small-molecule drug discovery and simple metrics to identify opportunities for macrocyclization of small-molecule ligands. J Med Chem. 2019;62(15):6843–6853. [DOI] [PubMed] [Google Scholar]
- 83.Mallinson J, Collins I. Macrocycles in new drug discovery. Future Med Chem. 2012;4(11):1409–1438. [DOI] [PubMed] [Google Scholar]
- 84.Shi J, Ewing WR, Nielsen L, et al. ; Bristol-Myers Squibb Co., US. Preparation of diamide macrocycles as factor XIa inhibitors for treatment of thromboembolic disorders. WO2016036893A1. 2016. [Google Scholar]
- 85.Dilger AK, Corte JR, De Lucca I, et al. ; Bristol-Myers Squibb Co., US. Pyrimidinones as factor XIa inhibitors and their preparation. WO2016053455A1. 2016. [Google Scholar]
- 86.Smith LM II, Pinto DJP, Corte JR, et al. ; Bristol-Myers Squibb Co., US. Preparation of diamide macrocycles as factor XIa inhibitors. WO2016205482A1. 2016. [Google Scholar]
- 87.Pabbisetty KB, Corte JR, Dilger AK, et al. ; Bristol-Myers Squibb Co., US. Preparation of factor XIa macrocyclic inhibitors bearing alkyl or cycloalkyl p2′ moieties. WO2017019819A1. 2016. [Google Scholar]
- 88.Zhu Y, Dilger AK, Ewing WR, et al. ; Bristol-Myers Squibb Co., US. Preparation of factor XIa macrocyclic inhibitors bearing alkyl or cycloalkyl p2′ moieties. WO2017019821A1. 2017. [Google Scholar]
- 89.Shi J, Ewing WR, Pinto DJP; Bristol-Myers Squibb Co., US. Preparation of diamide macrocycles having factor XIa inhibiting activity. WO2017151746A1. 2017. [Google Scholar]
- 90.Corte JR, Dilger AK, Ewing WR, et al. ; Bristol-Myers Squibb Co., US. Preparation of macrocycles with novel P1 groups as selective factor XIa inhibitors or dual inhibitors of FXIa and plasma kallikrein. US20180162821A1. 2018. [Google Scholar]
- 91.Xu J, Ali A, Zhou W, et al. ; Merck Sharp & Dohme Corp., US. Macrocyclic compounds as factor XIa inhibitors and their preparation. WO2017074832A1. 2017. [Google Scholar]
- 92.Ali A, Lim Y-H, Xu J, et al. ; Merck Sharp & Dohme Corp., US. Preparation of macrocyclic spirocarbamate derivatives as factor XIa inhibitors, pharmaceutically acceptable compositions and their use. WO2017074833A1. 2017. [Google Scholar]
- 93.Zuo Y, Wang J, Lin J, et al. ; Sunshine Lake Pharma Co., China. Preparation of macro-heterocyclic compounds as blood coagulation factor XIa inhibitors. WO2018133793A1. 2018. [Google Scholar]
- 94.Zhu J, Song Z, Song L, et al. ; Sichuan Kelun-Biotech Biopharmaceutical Co., China. Preparation of macrocyclic amide compounds for preventing or treating thromboembolic diseases. WO2019011166A1. 2019. [Google Scholar]
- 95.Ackerstaff J, Ellermann M, Platzek J, et al. ; Bayer Pharma, Germany. Method for producing phenylalanine derivatives as blood-coagulation factor XIa inhibitors. WO2016146599A1. 2016. [Google Scholar]
- 96.Roehn U, Ellermann M, Roehrig S, et al. ; Bayer Pharma, Germany. Preparation of substituted phenylalanine derivatives as blood-coagulation factor XIa inhibitors. WO2016146605A1. 2016. [Google Scholar]
- 97.Roehn U, Ellermann M, Roehrig S, et al. ; Bayer Pharma, Germany. Preparation of 7-{4-[(2S)-2-({[trans-4-(aminomethyl) cyclohexyl]car-bonyl}amino)-3-(phenylamino)-3-oxopropyl]phenyl}−6-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylic acid derivatives as factor XIa inhibitors for the treatment of fibrinolytic and thromboembolic diseases. WO2016146604A1. 2016. [Google Scholar]
- 98.Chenard BL, Xu Y, Stassen FL, et al. ; Exithera Pharmaceuticals Inc., US. Preparation of substituted azetidinedicarboxamides as factor XIa or kallikrein inhibitors. WO2018118705A1. 2018. [Google Scholar]
- 99.Chenard BL, Xu Y, Stassen FL, et al. ; Exithera Pharmaceuticals Inc., US. Preparation of β-lactam derivatives that inhibit Factor XIa or kallikrein (2019) WO2019156929A1. 2019. [Google Scholar]
- 100.Chrusciel RA, Gadwood RC, Hayward NJ, et al. ; Exithera Pharmaceuticals Inc., US. Preparation of βLactam derivatives as inhibitors of factor Xia or kallikrein. WO2015120062. 2015. [Google Scholar]
- 101.Yang F, Wang W, Xue T, et al. Preparation of pyrrolidine-amide derivatives as factor XIa inhibitor for treatment of thromboembolism. CN2017–10156222. 2017. [Google Scholar]
- 102.Li S, Xiao SZ, Yipeng S, et al. Anticoagulant small molecule compound and application thereof and medicament containing same. CN109627218A. 2019. [Google Scholar]
- 103.Eder J, Ewert S, Hassiepen U, et al. ; Novartis AG, Switzerland. Factor XI antibodies for coagulation therapy. WO2016207858A1. 2016. [Google Scholar]
- 104.Aigner M, Koch AW, Waldhuber M; Novartis AG, Switzerland. Factor XI antibodies for treatment of thrombosis or thromboembolic disease. WO2018116255A1. 2018. [Google Scholar]
- 105.Friedman C, Khder Y, Martin L; Novartis AG, Switzerland. Methods of treatment with anti-factor XI/XIa antibodies. WO2018116267A2. 2018. [Google Scholar]
- 106.Olbrich C, Trill T, Marieke V; Bayer Pharma, Germany. Stable formulations for anti-FXIa antibodies WO2018134184A1. 2018. [Google Scholar]
- 107.Wilmen A, Buchmueller A, Tucker E; Bayer Pharma, Germany. Preparation of novel chimeric or humanized monoclonal anti-human factor XI and/or XIa antibodies for treatment and prophylaxis of thrombotic or thromboembolic disorders. WO2018054813A1. 2018. [Google Scholar]
- 108.Mikita T, Ely LK, Gao H, et al. ; The Regents of The University of California, US. Antibodies to coagulation factor XIa. WO2017015619A1. 2017. [Google Scholar]
- 109.Boross P, Yildiz C, Hack CE; Prothix BV, Netherlands. Monoclonal antibodies against the active site of factor XI. WO2017162791A1. 2017. [Google Scholar]
- 110.Chen Z, Ellsworth K, Milligan J, et al. ; Merck Sharp & Dohme Corp., US. Anti-coagulation factor XI antibodies. US20170355780A1. 2017. [Google Scholar]
- 111.Wang W, Yu Q, Liu X; Shanghai Benemae Pharmaceutical CO., China. Anti-coagulation factor XI antibodies for treating and/or preventing coagulation related diseases. WO2018145533A1. 2018. [Google Scholar]
- 112.Wang W, Yu Q, Liu X; Shanghai Benemae Pharmaceutical CO., China. Preparation of anti-coagulation factor XI and/or XIa antibodies for prevention and therapy of thrombosis or sepsis. CN108409863A. 2018. [Google Scholar]
- 113.Chan NC, Weitz JI. AB023, a novel antibody that binds factor XI and blocks its activation by factor XIIa. Arterioscler Thromb Vasc Biol. 2019;39(4):533–535. [DOI] [PubMed] [Google Scholar]
- 114.Tillman BF, Gruber A, McCarty OJT, et al. Plasma contact factors as therapeutic targets. Blood Rev. 2018;32(6):433–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cheng Q, Tucker EI, Pine MS, et al. A role for factor XIIa-mediated factor XI activation in thrombus formation in vivo. Blood. 2010;116 (19):3981–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.David T, Kim YC, Ely LK, et al. Factor XIa-specific IgG and a reversal agent to probe factor XI function in thrombosis and hemostasis. Sci Transl Med. 2016;8(353):353ra112. [DOI] [PubMed] [Google Scholar]
- 117.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. [DOI] [PubMed] [Google Scholar]
- 118.Rusconi CP, Scardino E, Layzer J, et al. RNA aptamers as reversible antagonists of coagulation factor IXa. Nature. 2002;419:90–94. [DOI] [PubMed] [Google Scholar]
- 119.Woodruff RS, Xu Y, Layzer J, et al. Inhibiting the intrinsic pathway of coagulation with a factor XII-targeting RNA aptamer. J Thromb Haemost. 2013;11:1364–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Woodruff RS, Sullenger BA. Modulation of the coagulation cascade using aptamers. Arterioscler Thromb Vasc Biol. 2015;35:2083–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Excellent review of anticoagulant aptamers.
- 121.Soule EE, Bompiani KM, Woodruff RS, et al. Targeting two coagulation cascade proteases with a bivalent aptamer yields a potent and antidote-controllable anticoagulant. Nucleic Acid Ther. 2016;26:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sullenger BA, Nair S. From the RNA world to the clinic. Science. 2016;352:1417–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sheffield WP, Donkor DA Structure and therapeutic use of human factor xia-specific aptamers. EP3241904A1. 2017. [Google Scholar]
- 124.Donkor DA, Bhakta V, Eltringham-Smith LJ, et al. Selection and characterization of a DNA aptamer inhibiting coagulation factor XIa. Sci Rep. 2017;7(1):2102. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Describes FELIAP as the first FXIa-inhibitory aptamer.
- 125.Woodruff RS, Ivanov I, Verhamme IM, et al. Generation and characterization of aptamers targeting factor XIa. Thromb Res. 2017;156:134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ma D, Mizurini DM, Assumpção TC, et al. Desmolaris, a novel factor XIa anticoagulant from the salivary gland of the vampire bat (Desmodus rotundus) inhibits inflammation and thrombosis in vivo. Blood. 2013;122(25):4094–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen W, Carvalho LP, Chan MY, et al. Fasxiator, a novel factor XIa inhibitor from snake venom, and its site-specific mutagenesis to improve potency and selectivity. J Thromb Haemost. 2015;13 (2):248–261. [DOI] [PubMed] [Google Scholar]
- 128.Chen Z, Ding L, Luo X, et al. Application of Sj13 polypeptide in preparing antithrombotic. CN108517009A. 2018. [Google Scholar]
- 129.Gómez-Outes A, García-Fuentes M, Suárez-Gea ML. Discovery methods of coagulation-inhibiting drugs. Expert Opin Drug Discov. 2017;12(12):1195–1205. [DOI] [PubMed] [Google Scholar]
- 130.Pollack CV Jr, Reilly PA, van Ryn J, et al. Idarucizumab for dabigatran reversal - full cohort analysis. N Engl J Med. 2017;377 (5):431–441. [DOI] [PubMed] [Google Scholar]
- 131.Heo YA. Andexanet alfa: first global approval. Drugs. 2018;78 (10):1049–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Moll S, Crona DJ, Martin K. Direct oral anticoagulants in extremely obese patients: OK to use? Res Pract Thromb Haemost. 2018;3 (2):152–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Park CS, Choi EK, Kim HM, et al. Increased risk of major bleeding in underweight patients with atrial fibrillation who were prescribed non-vitamin K antagonist oral anticoagulants. Heart Rhythm. 2017;14(4):501–507. [DOI] [PubMed] [Google Scholar]
- 134.Talamo L, Hillary S, Maitland HS, et al. Safety of doacs in patients of extreme weight. Blood. 2016;128(22):1450. [Google Scholar]
- 135.Ribic C, Crowther M. Thrombosis and anticoagulation in the setting of renal or liver disease. Hematology Am Soc Hematol Educ Program. 2016;2016(1):188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ingrasciotta Y, Crisafulli S, Pizzimenti V, et al. Pharmacokinetics of new oral anticoagulants: implications for use in routine care. Expert Opin Drug Metab Toxicol. 2018;14(10):1057–1069. [DOI] [PubMed] [Google Scholar]
- 137.Kouyama S, Ono T, Hagio T, et al. Discovery of ONO-5450598, a highly orally bioavailable small molecule factor XIa inhibitor: the pharmacokinetic and pharmacological profiles. Res Pract Thromb Haemost. 2017;1(Suppl.1):PB2139 [abstract]. 99. [Google Scholar]; • Emerging orally active FXIa inhibitor; ONO-5450598 and its pharmacokinetics.
- 138.Kouyama S, Ono T, Sakimoto S, et al. Investigation of pharmacodynamic biomarkers and neutralizers for ONO- 7684 (ONO- 5450598), an oral FXIa inhibitor. Res Pract Thromb Haemost. 2019;3(Suppl.1):PB1246 [abstract]. 172. [Google Scholar]; • Emerging orally active FXIa inhibitor; ONO-5450598 and its pharmacodynamics.