

There is a Blood Commentary on this article in this issue.
Key Points
Fetal liver hepcidin operates cell-autonomously to enable rapid buildup of fetal liver iron stores in the third trimester of gestation.
Placental ferroportin is not regulated by fetal hepcidin under normal physiological conditions.
Abstract
In the adult, the liver-derived hormone hepcidin (HAMP) controls systemic iron levels by blocking the iron-exporting protein ferroportin (FPN) in the gut and spleen, the sites of iron absorption and recycling, respectively. Impaired HAMP expression or FPN responsiveness to HAMP result in iron overload. HAMP is also expressed in the fetal liver but its role in controlling fetal iron stores is not understood. To address this question in a manner that safeguards against the confounding effects of altered maternal iron homeostasis, we generated fetuses harboring a paternally-inherited ubiquitous knock-in of the HAMP-resistant fpnC326Y. Additionally, to safeguard against any confounding effects of altered placental iron homeostasis, we generated fetuses with a liver-specific knock-in of fpnC326Y or knockout of the hamp gene. These fetuses had reduced liver iron stores and hemoglobin, and markedly increased FPN in the liver, but not in the placenta. Thus, fetal liver HAMP operates cell-autonomously to increase fetal liver iron stores. Our findings also suggest that FPN in the placenta is not actively regulated by fetal liver HAMP under normal physiological conditions.
Visual Abstract

Introduction
Iron is essential for growth and development. Suboptimal iron availability at birth is associated with cognitive, behavioral and motor skill deficits, and increased risk of developing anemia in infancy.1-7 Some of these developmental deficits are not reversible by oral iron therapy in the neonate.8-15 In the first 6 to 9 months of age, the neonate is not fully competent in regulating dietary iron absorption in response to iron needs16,17 and depends on liver iron stores to support its growth and development.18,19
In the adult, the liver-derived hormone hepcidin (HAMP) controls systemic iron availability, which in turn controls liver iron stores.20,21 HAMP operates by blocking the iron export protein ferroportin (FPN) in the gut and the spleen, the sites of iron absorption and recycling, respectively.20,21 Disruption of the HAMP/FPN axis, in a manner that reduces HAMP levels or FPN responsiveness to HAMP, leads to the iron-overload disease hemochromatosis. This is characterized by increased serum iron concentration, depletion of iron from the spleen, and excess deposition in the liver.22 The hemochromatosis phenotype is recapitulated in adult mice harboring ubiquitous knock-in of the HAMP-resistant fpnC326Y allele, or liver-specific knockout of the hamp gene.23,24
HAMP expression has also been detected in the fetal liver.25 In contrast to adult HAMP, the role of fetal HAMP in determining liver iron stores in utero is not entirely understood. One study reported that endogenous HAMP was not expressed in the mouse fetal liver but that transgenic ubiquitous overexpression of the hamp gene resulted in anemic fetuses.26 Another study found that ubiquitous homozygous loss of the matriptase-2 gene in fetuses (matriptase-2 is a negative regulator of hamp expression) resulted in reduced body nonheme iron levels.27 A third study comparing fetuses harboring a homozygous deletion of the hemochromatosis gene hfe (HFE is a protein that is necessary for hamp expression), with wild-type littermates did not find any difference in total liver iron stores, but reported higher levels of nonheme iron in hfe-ko fetuses when mothers were fed an iron-rich diet.28 Based on these studies, it has been hypothesized that fetal liver HAMP controls iron availability to the fetus by blocking FPN-mediated iron transport into the fetal capillaries of the placental syncytiotrophoblast.29 However, a more recent study found that ubiquitous deletion of the hamp gene did not affect fetal liver iron stores under either iron-replete or iron-deficient conditions.30
A common feature of the above studies is that the genetic variants in the fetuses were also present in the fetal part of the placenta, in the mother, and in the maternal part of the placenta. We sought to interrogate the role of fetal liver HAMP without these potentially confounding effects. To avoid those associated with altered iron control in the mother or maternal side of the placenta, we generated fetuses harboring a paternally inherited ubiquitous knock-in of the HAMP-resistant fpnC326Y allele. To avoid the additional potentially confounding effects of altered iron control in the fetal part of the placenta, we also generated fetuses harboring a liver-specific knock-in of the HAMP-resistant fpnC326Y allele, or liver-specific knockout of the hamp gene, through paternal inheritance of the hepatocyte-specific albumin-Cre recombinase transgene. For these models, we then compared the levels of liver iron stores and hemoglobin between the affected fetuses and their littermate controls.
Methods
Mice
All animal procedures were compliant with the UK Home Office Animals (Scientific Procedures) Act 1986 and approved by the University of Oxford Medical Sciences Division Ethical Review Committee. The conditional fpnC326Yfl allele was generated as described previously.23 Females were time-mated between 9 and 12 weeks of age and fetuses were harvested from first pregnancies.
Fetal tissue
Fetal tissues were harvested from phosphate-buffered saline (PBS)-perfused mothers at embryonic days e13.5 or e17.5, washed further in PBS then either snap-frozen for RNA and iron measurement studies or fixed in formalin for histological studies. Fetal blood was collected at e17.5 by decapitation.
Immunostaining
Formalin-fixed paraffin-embedded tissue sections were stained with rabbit polyclonal anti-mouse FPN antibody (NBP1-21502, Novus Biologicals) at 1/200 dilution. Alexa 488-conjugated anti-rabbit antibody (ab150073, Abcam) was then used as a secondary antibody at a dilution of 1/500.
Iron indices
Determination of total elemental iron in tissues from PBS-perfused animals was carried out by inductively coupled plasma mass spectrometry, as described previously.23 Concentrations were normalized to wet weight. Hemoglobin was recorded from fresh blood using the HemoCue Hb 201+ system. DAB-enhanced Perls’ iron stain was carried out in formalin-fixed paraffin-embedded sections, as described previously.23
Diet provision during pregnancy
Unless otherwise stated, mothers were fed a standard chow diet containing 200 ppm iron. In iron-loading studies, mothers were fed a diet containing 5000 ppm iron (Teklad TD.140464) as soon as mated. In iron-deficient studies, mothers were fed a diet containing 5 ppm iron (Teklad TD.99397) from weaning and throughout pregnancy.
Western blotting
Tissues were snap-frozen in liquid nitrogen, crushed then lysed using RIPA Lysis Buffer System (Santa Cruz sc-24948), according to the manufacturer’s instructions. Tissue lysates were cleared by centrifugation at 15 000g for 10 minutes at 4°C. Protein concentration in the lysates was measured by BCA Protein Assay (Pierce 23225) and normalized to the same concentration for each batch. Lysates were then diluted in nonreducing Laemmli sodium dodecyl sulfate sample buffer and heated at 95°C for 5 minutes. Thirty to 50 μg of protein were loaded onto Mini-PROTEAN TGX Gels (Biorad, 4561096). After electrophoresis, protein was transferred onto cellulose membrane using the BioRad Translotter system, and membranes were blocked for an hour in blocking buffer containing 5% bovine serum albumin. Membranes were then stained overnight at 4°C with rabbit polyclonal anti-mouse FPN antibody (NBP1-21502, Novus Biologicals) at 1/1000 or horseradish peroxidase-conjugated anti β-actin antibody (Proteintech HRP-60008) at 1/5000. Blots were developed using the ECL prime detection kit (RPN2232, VWR International). Signal intensities were quantified by ImageJ, and the ratio between the FPN and the β-actin signals calculated to produce normalized intensities.
Erythroid progenitors in fetal livers
Quantitation of erythroid progenitors in fetal livers was performed according to a previously published method.31 Following harvest, fetal livers were dissociated in ice-cold PBS containing 3% FCS, then immediately costained with fluorescein isothiocyanate-conjugated rat anti-mouse cluster of differentiation CD71 antibody (BD Biosciences 553266) and phycoerythrin-conjugated rat anti-mouse TER-119 antibody (553673 BD Biosciences), both diluted at 1/1000. Cells were washed 3 times then subject to flow cytometry using the BDX20 system (BD Biosciences). Dot blot analysis was performed using FlowJo.
Gene expression
Gene expression was assessed by quantitative real-time polymerase chain reaction, using Applied Biosystems Taqman gene expression assay probes for fpn, transferrin receptor 1 (tfr1), (divalent metal transporter 1) dmt1, hamp, and house-keeping gene β-Actin (Life Technologies, Carlsbad, CA). The cycle threshold (CT) value for the gene of interest was first normalized by deducting the CT value for β-actin to obtain a Δ CT value. Δ CT values of test samples were further normalized to the average of the Δ CT values for control samples to obtain ΔΔ CT values. Relative gene expression levels were then calculated as 2-ΔΔCT.
Statistics
Values are shown as mean ± standard error of the mean (SEM). Paired comparisons were performed using Student t test. Multiple comparisons were drawn using analysis of variance. Post hoc tests used the Bonferroni correction. R values reported are Pearson correlation coefficients.
Results
Fetal liver iron concentration and hepcidin expression increase in the third trimester
In the mouse, the fetal liver forms in the second half of gestation. We found that the total concentration of elemental iron in the fetal liver increased by 2.75-fold between e13.5 and e17.5, to levels well above those in the maternal liver at e17.5 (and similar to those in nonpregnant females), consistent with increased liver iron stores (Figure 1A). Over the same period, fetal liver hamp messenger RNA (mRNA) expression increased by 24-fold (Figure 1B). Nevertheless, at e17.5, fetal liver hamp mRNA expression was ∼18-fold lower than that of maternal liver hamp (Figure 1B). There was a positive correlation between the concentration of iron and the expression of hamp mRNA in the fetal liver at e.17.5 (Figure 1C).
Figure 1.
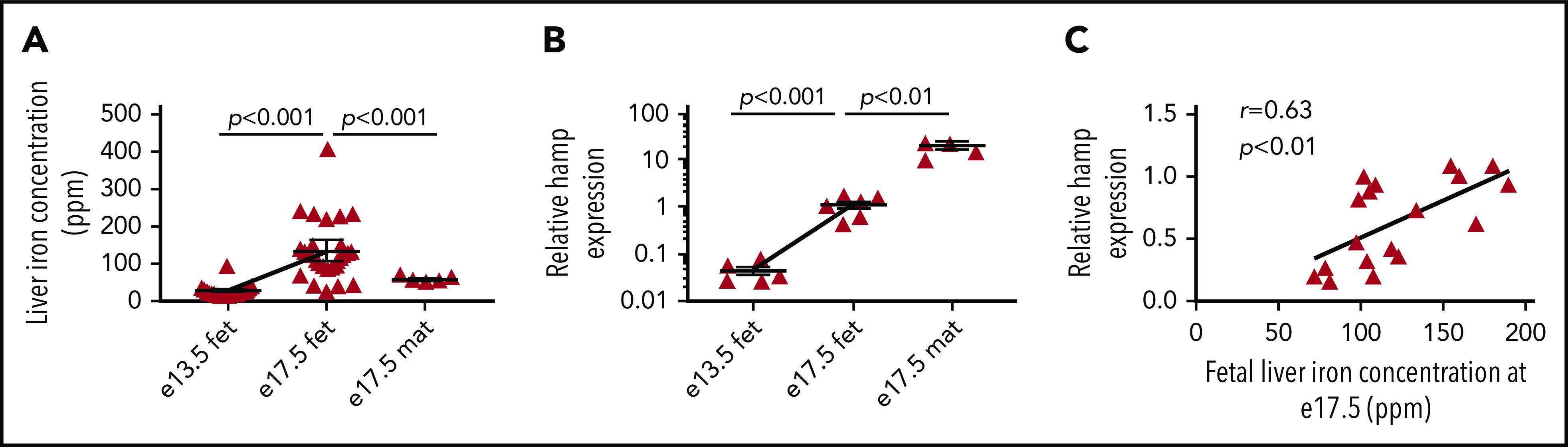
Fetal liver iron concentration and hepcidin expression increase in the third trimester. (A) Total iron concentration in fetal livers harvested at e13.5 (n = 27) and e17.5 (n = 28) and in maternal livers at e17.5 (n = 5). (B) Relative hamp mRNA expression in fetal livers harvested at e13.5 (n = 6) and e17.5 (n = 6) and in maternal livers at e17.5 (n = 5). (C) Correlation between relative hamp expression and total iron concentration in fetal livers at e17.5 (n = 18). Values are shown as mean ± SEM. P values are calculated using Student t test. r is Pearson’s correlation coefficient. fet, fetal; mat, maternal.
Ubiquitous loss of FPN responsiveness to hepcidin reduces fetal liver iron stores and hemoglobin
To probe the role of fetal HAMP in regulating fetal liver iron stores, we used mice harboring a ubiquitous knock-in of a HAMP-resistant FPN. To guard against a confounding effect of maternal iron overload, we set up matings using wild-type fpnwt/wt mothers and fpnwt/C326Y fathers heterozygous for the knock-in mutation of fpnC326Y, an isoform of FPN with intact iron export function but that is resistant to HAMP-mediated degradation. At e17.5, fpnwt/C326Y fetuses had lower fetal liver iron concentration compared with fpnwt/wt littermate controls (Figure 2A). DAB-enhanced Perls stain demonstrated iron staining in periportal hepatocytes in fpnwt/wt livers that was absent in fpnwt/C326Y livers (Figure 2B). The difference in liver iron stores between e17.5 fpnwt/C326Y fetuses and littermate controls disappeared within the first week of life (Figure 2A). As expected, by 12 weeks of age, fpnwt/C326Y mice had liver iron concentrations that were higher than those of fpnwt/wt littermate controls, consistent with the hemochromatosis phenotype reported in our previous studies.23 Extrahepatic iron concentration at e17.5 (measured in the carcass) was also reduced in fpnwt/C326Y mice compared with fpnwt/wt littermates, consistent with the ubiquitous expression of the fpnC326Y allele (Figure 2C). Extrahepatic iron concentration (measured as splenic iron) was comparable between mice of the 2 genotypes at birth (d0), and at 1 and 4 weeks of age, but lower in fpnwt/C326Y mice than in fpnwt/wt littermate controls at 12 weeks of age, consistent with the development of hemochromatosis at this later stage (Figure 2C).23 Hemoglobin levels at e17.5 and birth (d0) were lower in fpnwt/C326Y mice than in fpnwt/wt littermate controls (Figure 2D), although this difference disappeared by 1 week of age (Figure 2D). This finding suggests that limited fetal liver iron stores affect fetal hematopoiesis. In late gestation, the liver becomes a site of hematopoiesis, with erythroid progenitor cells transitioning through 5 stages of differentiation (R1-R5).31 We found that hemoglobin levels at e17.5 correlated most positively with the percentage of terminally differentiated cells (R5) and most negatively with the percentage of intermediate cells (R3) (supplemental Figure 1A-B, available on the Blood Web site). Importantly, reduced iron stores in fpnwt/C326Y fetuses were associated with lower percentage of R5 cells in their livers compared with livers of fpnwt/wt littermate controls (Figure 2E-F). These results confirm that reduced liver iron stores limit the later stages of erythroid differentiation in the fetal liver.
Figure 2.
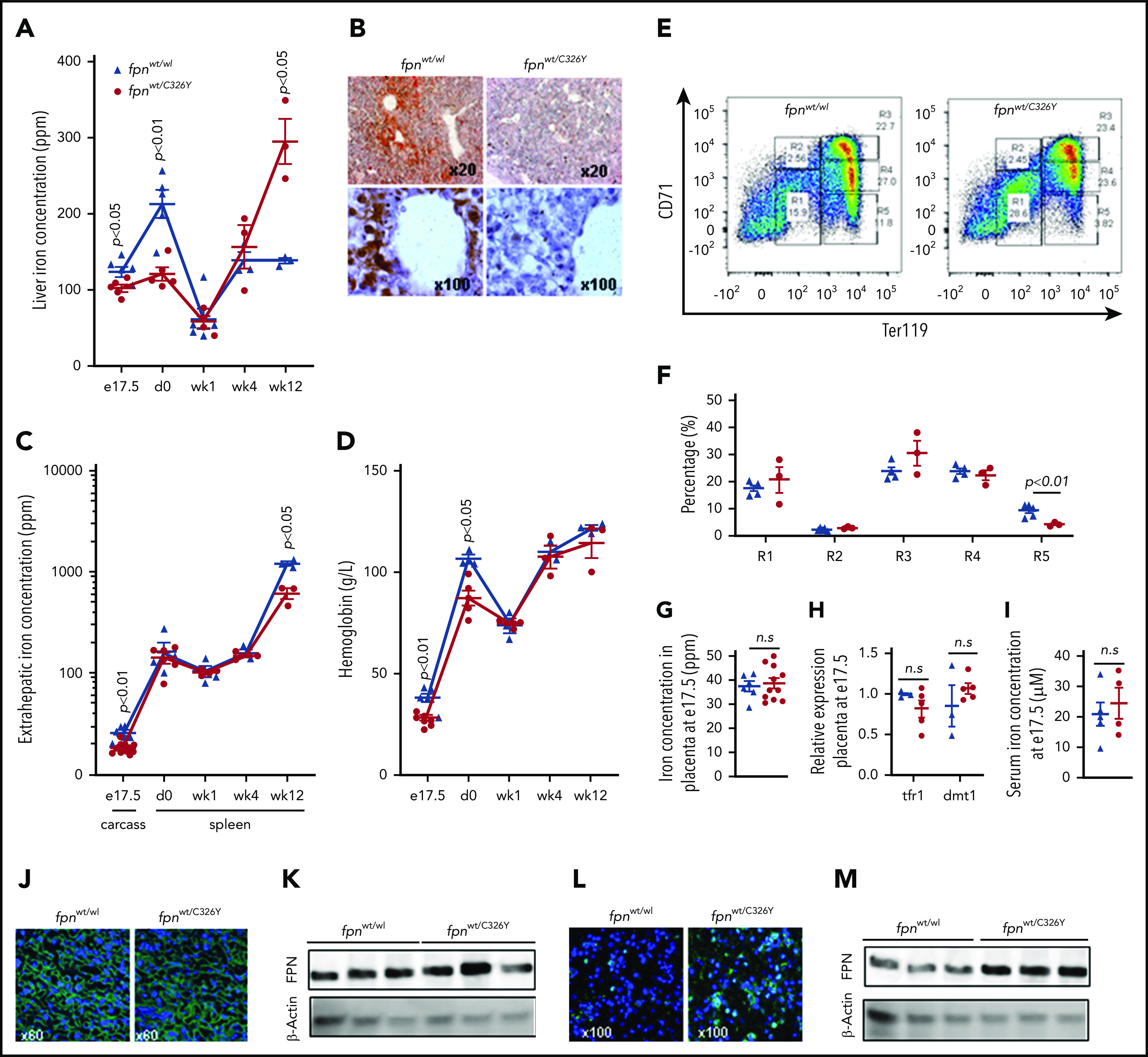
Ubiquitous loss of FPN responsiveness to hepcidin in the fetus reduces fetal liver iron stores and hemoglobin. (A) Liver iron concentration in fpnwt/wt animals and fpnwt/C326Y littermates at e17.5 (n = 6, 5, respectively), day 0 (n = 5, 5), week 1 (n = 5, 4), week 4 (n = 3, 3), and week 12 (n = 3, 3) of age. P values are shown relative to fpnwt/wt littermate controls at the respective timepoint. (B) Representative images of DAB-enhanced Perls’ iron stain in the livers of fpnwt/wt animals and fpnwt/C326Y littermates at e17.5. (C) Extrahepatic iron concentration in fpnwt/wt animals and fpnwt/C326Y littermates measured as carcass iron at e17.5 (n = 6, 11) and as splenic iron at day 0 (n = 4, 6), week 1 (n = 4, 4), week 4 (n = 3, 3), and week 12 (n = 3, 3) of age. P values are shown relative to fpnwt/wt littermate controls at the respective timepoint. (D) Hemoglobin concentration in fpnwt/wt animals and fpnwt/C326Y littermates at e17.5 (n = 6, 7), day 0 (n = 3, 5), week 1 (n = 3, 5), week 4 (n = 3, 3), and week 12 (n = 3, 3) of age. P values are shown relative to fpnwt/wt littermate controls at the respective timepoint. (E) Representative dot plots of erythroid progenitors from livers of fpnwt/wt animals and fpnwt/C326Y littermates at e17.5. (F) Quantitation of erythroid progenitor populations R1 through R5 from livers of fpnwt/wt animals and fpnwt/C326Y littermates at e17.5 (n = 5, 3). (G) Placental iron concentration in fpnwt/wt animals and fpnwt/C326Y littermates at e17.5 (n = 6, 11). (H) Relative expression of tfr1 and dmt1 mRNA transcripts in placentae of fpnwt/wt animals and fpnwt/C326Y littermates at e17.5 (n = 3, 5). (I) Iron concentration in the serum of fpnwt/wt animals and fpnwt/C326Y littermates at e17.5 (n = 5, 4). (J) Representative images of FPN immunostaining in the placentae of fpnwt/wt animals and fpnwt/C326Y littermates at e17.5. (K) Western blot for FPN in the placentae of fpnwt/wt animals and fpnwt/C326Y littermates at e17.5 (n = 3, 3). Normalized intensities are shown in supplemental Figure 5A. (L) Representative images of FPN immunostaining in the livers of fpnwt/wt animals and fpnwt/C326Y littermates at e17.5. (M) Western blot for FPN in the livers of fpnwt/wt animals and fpnwt/C326Y littermates at e17.5 (n = 3, 3). Normalized intensities are shown in supplemental Figure 5B. Values are shown as mean ± SEM. P values are calculated using Student t test. wk, week.
Having observed that ubiquitous loss of FPN responsiveness to hepcidin in fetal tissues resulted in reduced fetal liver iron stores and lower hemoglobin levels, we sought to determine whether these effects could be attributed to changes in iron transfer across the placenta. However, we found that placental iron concentration (Figure 2G), placental expression of iron-regulated transcripts tfr1 and dmt1 (Figure 2H), and the concentration of iron in fetal serum (Figure 2I) were all comparable between fpnwt/C326Y mice and fpnwt/wt littermates, suggesting that placental iron flux is not different between animals of the 2 genotypes. Additionally, FPN levels were not different between placentae of fpnwt/C326Y mice and placentae of fpnwt/wt littermates (Figure 2J-K), indicating that FPN at this site is not actively regulated by hepcidin in the iron-replete state. To explore whether placental FPN was regulated by hepcidin in the setting of iron overload, we provided fpnwt/wt mothers carrying fpnwt/wt litters with iron-loaded diet containing 5000 ppm iron. Provision of iron-loaded diet raised the concentration of iron and the expression of hepcidin in the fetal and maternal livers and in the placenta itself (supplemental Figure 2A-B). FPN was decreased in the fetal liver but increased in the placenta in response to the iron-loaded diet (supplemental Figure 2C-F). Consistent with this increase in placental FPN, the concentration of iron in the fetal serum was also raised by the iron-loaded diet (supplemental Figure 2G). These data further confirm that fetal liver but not placental FPN is subject to regulation by HAMP.
Apart from the placenta, the other major site of FPN expression is the fetal liver itself. Therefore, we examined whether FPN expression in the livers of fpnwt/C326Y fetuses was altered, and found that it was markedly increased compared with livers of fpnwt/wt littermates at e17.5 (Figure 2L-M). Therefore, we hypothesized that this marked increase in FPN expression is the cause of decreased fetal liver iron stores seen in fpnwt/C326Y fetuses.
Liver-specific loss of FPN responsiveness to hepcidin reduces fetal liver iron stores and hemoglobin
To test the hypothesis that increased FPN in the fetal liver leads to a reduction in iron stores, we generated fetuses harboring a liver-specific knock-in of the fpnC326Y allele, by mating fpnC326Yfl/fl mothers with fpnC326Yfl/fl fathers transgenic for Cre recombinase driven by the hepatocyte-specific albumin promoter (fpnC326Yfl/fl, Alb.Cre+). Liver iron concentration in fpnC326Yfl/fl, Alb.Cre+ livers at e17.5 and at birth was reduced relative to fpnC326Yfl/fl littermates (Figure 3A). This was confirmed histologically by DAB-enhanced Perls’ iron stain (Figure 3B). Interestingly, at 12 weeks of age, fpnC326Yfl/fl, Alb.Cre+ mice had a reduction in liver iron concentration suggesting a possible autocrine role for HAMP in regulating liver iron stores in the adult (Figure 3A). Unlike fpnwt/C326Y mice, extrahepatic iron concentration at e17.5 (measured in the carcass) and at later stages (measured as splenic iron) were not reduced in fpnC326Yfl/fl, Alb.Cre+ mice relative to the respective controls. This confirms that iron loss in this setting is limited to the liver, in line with the specificity conferred by the Alb.Cre+ transgene (Figure 3C). fpnC326Yfl/fl, Alb.Cre+ fetuses had lower hemoglobin levels at e17.5, although this recovered to control levels in the first week of life (Figure 3D). Lower hemoglobin levels at e17.5 were also associated with a reduction in the percentage of terminally differentiated erythroblasts in the fetal liver (Figure 3E-F). The findings that hemoglobin levels and erythroid differentiation were reduced even when loss of HAMP responsiveness was confined to hepatocytes is consistent with a direct role for hepatocytes iron stores per se in supporting hematopoiesis in the fetal liver. The reduction in fetal liver iron concentration could not be attributed to changes in placental iron flux, because placental iron concentration (Figure 3G), placental expression of iron-regulated transcripts tfr1 and dmt1 (Figure 3H) and iron concentration in the fetal serum (Figure 3I) were not different between fpnC326Yfl/fl, Alb.Cre+ animals and their littermate controls. Consistent with this, placental FPN protein levels were similar between animals of the 2 genotypes (Figure 3J-K). In contrast, hepatic FPN expression was higher in fpnC326Yfl/fl, Alb.Cre+ fetuses that in fpnC326Yfl/fl littermates at e17.5 (Figure 3L-M).These results confirm that FPN in fetal hepatocytes is subject to regulation by HAMP and that this regulation is important for the control of fetal liver iron stores.
Figure 3.

Liver-specific loss of FPN responsiveness to hepcidin reduces fetal liver iron stores and hemoglobin. (A) Liver iron concentration in fpnC326Yfl/fl animals and fpnC326Yfl/fl, Alb.Cre+ littermates at e17.5 (n = 3, 3, respectively), day 0 (n = 3, 4), week 1 (n = 4, 3), week 4 (n = 4, 4), and week 12 (n = 3, 3) of age. P values are shown relative to fpnC326Yfl/fl littermate controls at the respective timepoint. (B) Representative images of DAB-enhanced Perls’ iron stain in the livers of fpnC326Yfl/fl animals and fpnC326Yfl/fl, Alb.Cre+ littermates at e17.5. (C) Extrahepatic iron concentration in fpnC326Yfl/fl animals and fpnC326Yfl/fl, Alb.Cre+ littermates measured as carcass iron at e17.5 (n = 3, 5) and as splenic iron at day 0 (n = 3, 4), week 1 (n = 4, 3), week 4 (n = 4, 4), and week 12 (n = 3, 3) of age. P values are shown relative to fpnC326Yfl/fl littermate controls at the respective timepoint. (D) Hemoglobin concentration in fpnC326Yfl/fl animals and fpnC326Yfl/fl, Alb.Cre+ littermates at e17.5 (n = 12, 6), day 0 (n = 4, 3), week 1 (n = 4, 3), week 4 (n = 4, 4), and week 12 (n = 3, 3) of age. P values are shown relative to fpnC326Yfl/fl littermate controls at the respective timepoint. (E) Representative dot plots of erythroid progenitors from livers of fpnC326Yfl/fl animals and fpnC326Yfl/fl, Alb.Cre+ littermates at e17.5. (F) Quantitation of erythroid progenitor populations R1 through R5 from livers of fpnC326Yfl/fl animals and fpnC326Yfl/fl, Alb.Cre+ littermates at e17.5 (n = 5, 4). (G) Placental iron concentration in fpnC326Yfl/fl animals and fpnC326Yfl/fl, Alb.Cre+ littermates at e17.5 (n = 11, 6). (H) Relative expression of tfr1 and dmt1 mRNA transcripts in placentae of fpnC326Yfl/fl animals and fpnC326Yfl/fl, Alb.Cre+ littermates at e17.5 (n = 3, 5). (I) Iron concentration in the serum of fpnC326Yfl/fl animals and fpnC326Yfl/fl, Alb.Cre+ littermates at e17.5 (n = 11, 6). (J) Representative images of FPN immunostaining in the placentae of fpnC326Yfl/fl animals and fpnC326Yfl/fl, Alb.Cre+ littermates at e17.5. (K) Western blot for FPN in the placentae of fpnC326Yfl/fl animals and fpnC326Yfl/fl, Alb.Cre+ littermates at e17.5 (n = 3, 3). Normalized intensities are shown in supplemental Figure 5C. (L) Representative images of FPN immunostaining in the livers of fpnC326Yfl/fl animals and fpnC326Yfl/fl, Alb.Cre+ littermates at e17.5. (M) Western blot for FPN in the livers of fpnC326Yfl/fl animals and fpnC326Yfl/fl, Alb.Cre+ littermates at e17.5 (n = 3, 3). Normalized intensities are shown in supplemental Figure 5D. Values are shown as mean ± SEM. P values are calculated using Student t test.
Fetal liver hepcidin regulates fetal liver FPN and iron stores
Next, we set out to identify the source of HAMP that regulates fetal liver FPN and consequently the liver’s iron stores. The previous observations that fetal liver hamp mRNA increased 24-fold between e13.5 and e17.5 and that it correlated positively with fetal liver iron stores (Figure 1B,D) suggest that the fetal liver is the source of HAMP that regulates fetal liver FPN and iron concentration. To test this hypothesis, we deleted the hamp gene specifically in the fetal liver by mating hampfl/fl mothers with hampfl/fl, Alb.Cre+ fathers. We found that at e17.5, hamp mRNA expression was reduced by 80% in the livers of hampfl/fl, Alb.Cre+ fetuses relative to hampfl/fl littermates, confirming the activity of the Cre allele at this gestational stage (supplemental Figure 3A). Loss of fetal liver HAMP resulted in decreased fetal liver iron concentration compared with hampfl/fl littermates (Figure 4A), and this was further confirmed histologically by DAB-enhanced Perls’ iron stain (Figure 4B). The difference in liver iron stores between hampfl/fl, Alb.Cre+ fetuses and hampfl/fl littermates disappeared within the first week of life, with mice of both genotypes having comparable liver iron stores at 1 and 4 weeks of age (Figure 4A). As expected, 12-week-old hampfl/fl, Alb.Cre+ mice had markedly increased liver iron concentrations consistent with the well-recognized role for hepatic HAMP in controlling liver iron stores in the adult (Figure 4A). Extrahepatic iron concentration at e17.5 (measured in the carcass) was not different between hampfl/fl, Alb.Cre+ fetuses and hampfl/fl littermates, consistent with the notion that fetal liver HAMP regulates liver iron primarily in an autocrine manner (Figure 4C). In contrast, extrahepatic iron concentration (measured as splenic iron) was markedly reduced in 12-week-old hampfl/fl, Alb.Cre+ mice, consistent with the redistribution of iron from the spleen to the liver (Figure 4C). The hampfl/fl, Alb.Cre+ fetuses also had reduced hemoglobin levels at e17.5 (Figure 4D), associated with a reduction in the percentage of late differentiation stages R4 and R5 erythroid cells in the fetal liver (Figure 4E-F). The reduction in fetal liver iron stores could not be attributed to changes in placental iron flux because placental iron concentration (Figure 4G), placental expression of iron-regulated transcripts tfr1 and dmt1 (Figure 4H), and the concentration of iron in the fetal serum (Figure 4I) were not different between hampfl/fl, Alb.Cre+ fetuses and their hampfl/fl littermate controls. Consistent with this, placental FPN protein levels were comparable between animals of the 2 genotypes (Figure 4J-K). In contrast, FPN levels were markedly increased in the livers of hampfl/fl, Alb.Cre+ fetuses compared with littermate controls (Figure 4L, M).
Figure 4.
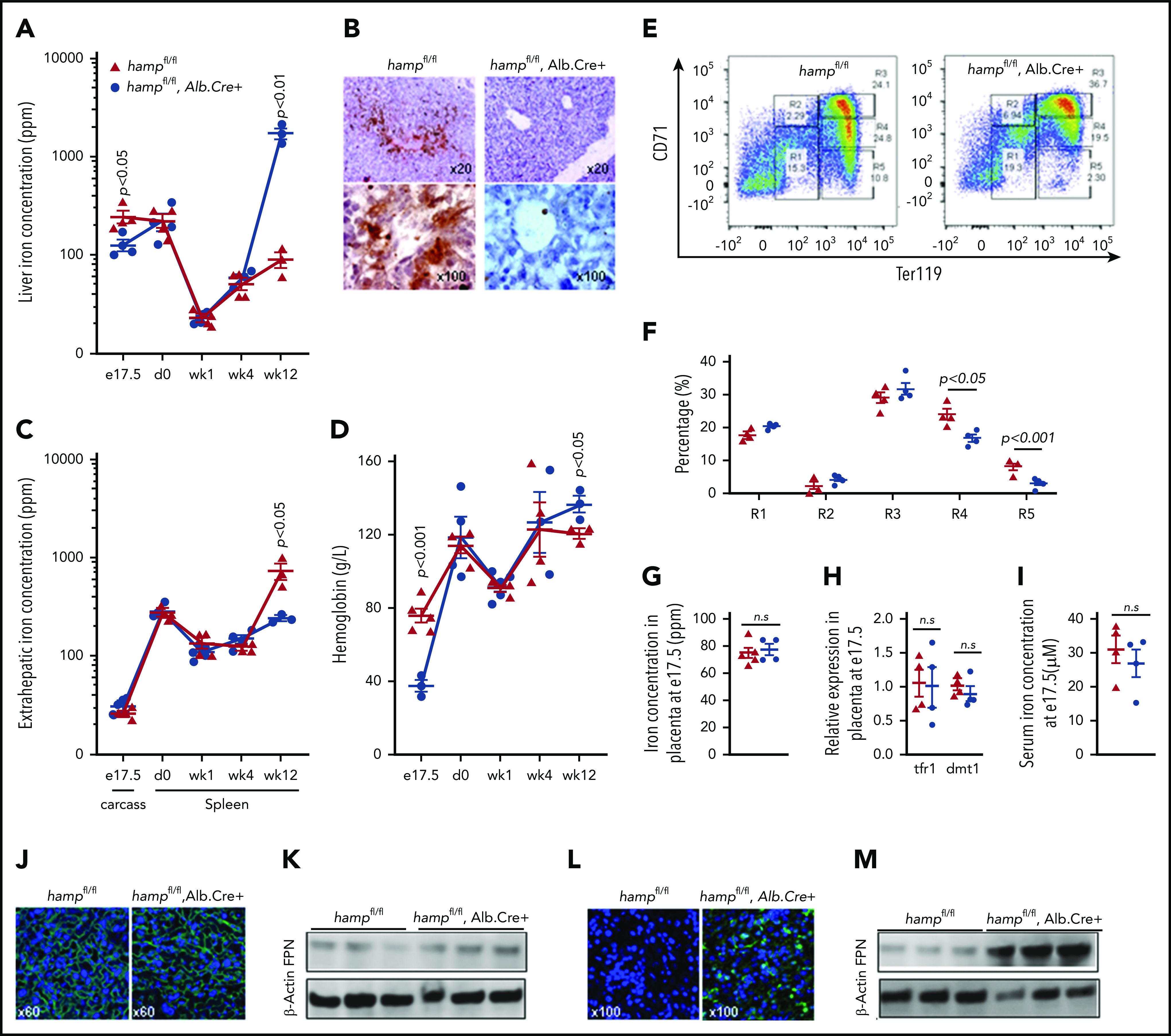
Liver-specific loss of hepcidin reduces fetal liver iron stores and hemoglobin. (A) Liver iron concentration in hampfl/fl animals and hampfl/fl, Alb.Cre+ littermates at e17.5 (n = 4, 4, respectively), day 0 (n = 4, 4), week 1 (n = 4, 4), week 4 (n = 4, 3), and week 12 (n = 3, 3) of age. P values are shown relative to hampfl/fl littermate controls at the respective timepoint. (B) Representative images of DAB-enhanced Perls’ iron stain in the livers of hampfl/fl animals and hampfl/fl, Alb.Cre+ littermates at e17.5. (C) Extrahepatic iron concentration in hampfl/fl animals and hampfl/fl, Alb.Cre+ littermates measured as carcass iron at e17.5 (n = 4, 5) and as splenic iron at day 0 (n = 4, 4), week 1 (n = 4, 5), week 4 (n = 3, 4), and week 12 (n = 3, 3) of age. P values are shown relative to hampfl/fl littermate controls at the respective timepoint. (D) Hemoglobin concentration in hampfl/fl animals and hampfl/fl, Alb.Cre+ littermates at e17.5 (n = 5, 3), day 0 (n = 4, 4), week 1 (n = 3, 5), week 4 (n = 4, 3), and week 12 (n = 3, 3) of age. P values are shown relative to hampfl/fl littermate controls at the respective timepoint. (E) Representative dot plots of erythroid progenitors from livers of hampfl/fl animals and hampfl/fl, Alb.Cre+ littermates at e17.5. (F) Quantitation of erythroid progenitor populations R1 through R5 from livers of hampfl/fl animals and hampfl/fl, Alb.Cre+ littermates at e17.5 (n = 4, 4). (G) Placental iron concentration in hampfl/fl animals and hampfl/fl, Alb.Cre+ littermates at e17.5 (n = 5, 4). (H) Relative expression of tfr1 and dmt1 mRNA transcripts in placentae of hampfl/fl animals and hampfl/fl, Alb.Cre+ littermates at e17.5 (n = 4, 4). (I) Iron concentration in the serum of hampfl/fl animals and hampfl/fl, Alb.Cre+ littermates at e17.5 (n = 4, 4). (J) Representative images of FPN immunostaining in the placentae of hampfl/fl animals and hampfl/fl, Alb.Cre+ littermates at e17.5. (K) Western blot for FPN in the placentae of hampfl/fl animals and hampfl/fl, Alb.Cre+ littermates at e17.5 (n = 3, 3). Normalized intensities are shown in supplemental Figure 5E. (L) Representative images of FPN immunostaining in the livers of hampfl/fl animals and hampfl/fl, Alb.Cre+ littermates at e17.5. (M) Western blot for FPN in the livers of hampfl/fl animals and hampfl/fl, Alb.Cre+ littermates at e17.5 (n = 3, 3). Normalized intensities are shown in supplemental Figure 5F. Values are shown as mean ± SEM. P values are calculated using Student t test.
These results demonstrate that fetal liver HAMP operates to control fetal liver iron stores in the iron replete setting. Interestingly, fetal liver HAMP does not appear to protect fetal liver iron stores in the setting of iron deficiency. Indeed, both hampfl/fl, Alb.Cre+ fetuses and their hampfl/fl littermates became severely iron-depleted when mothers were fed an iron-deficient diet from weaning and throughout pregnancy (supplemental Figure 3B). Consistent with this, provision of an iron-deficient diet to the mother did not affect the expression of hepcidin in the fetal liver (supplemental Figure 4A), despite causing a marked reduction in fetal liver iron stores (supplemental Figure 4B).
Discussion
The importance of liver iron stores for neonatal growth is well recognized.1-7 As demonstrated in the present study, these stores also support hematopoiesis in the fetal liver. To ensure adequate liver iron endowment in late gestation and at birth, there is a need for a rapid buildup of iron levels within fetal hepatocytes.
The first important finding from this study is that fetal liver HAMP operates in a cell-autonomous manner to facilitate the buildup of fetal liver iron stores. The findings that fetal liver HAMP expression is significantly lower than that of maternal liver HAMP, and that loss of fetal liver HAMP does not affect extrahepatic iron levels, are consistent with an autocrine rather than endocrine mode of action. This autocrine mode of action is further supported by the findings of a previous study, in which raised HAMP in the livers of neonates with early-onset neonatal sepsis were associated with decreased FPN and increased iron sequestration in the fetal liver.32 In the present study, we found that ubiquitous, but not liver-specific, loss of HAMP responsiveness was associated with a transient decrease in extrahepatic iron in fetuses. This finding suggests that iron levels in other fetal tissues may well be subject to regulation by nonhepatic HAMP.
The second important finding from the present study is that fetal liver HAMP does not exert endocrine effects on FPN in the placenta under normal physiological conditions. In the setting of early-onset neonatal sepsis, it has been reported that cord blood HAMP levels are raised and associated with decreased FPN levels in the placenta.32 However, maternal HAMP is also known to be raised in inflammatory conditions, and cross-contamination of cord blood with maternal blood is a well-documented phenomenon that cannot be excluded.33-36 Nonetheless, it would be important to determine if, under certain pathological conditions, fetal liver HAMP levels are sufficiently raised to exert endocrine effects on the placenta.
The role of fetal liver HAMP has been explored before. One study reported that transgenic ubiquitous overexpression of the hamp gene resulted in anemic fetuses.26 Another study found that ubiquitous homozygous loss of the matriptase-2 gene in fetuses resulted in reduced body nonheme iron levels.27 A third study using fetuses harboring a homozygous deletion of the hemochromatosis gene hfe did not find any difference in total liver iron stores, but reported higher levels of nonheme iron in hfe-ko fetuses when mothers were fed an iron-rich diet.28 A more recent study found that ubiquitous deletion of the hamp gene did not affect fetal liver iron stores either under iron-replete or under iron-deficient conditions.30 Along with the present study, these studies diverge from one another in terms of the conclusions drawn as to the importance of fetal liver HAMP. However, there are 2 very important aspects within the experimental approaches that may underlie this divergence. First, when the deletion of the hamp, matriptase-2, or hfe genes is present throughout all stages of pregnancy, it can potentially affect FPN in early gestation, before the placenta has been formed. Indeed, before it is ever expressed in the placenta, FPN is present in the visceral endoderm and yolk sac, where it plays a nonredundant role in iron supply to the fetus (explaining why fpn null embryos are lethal by e7.5).37,38 In the present study, the use of the Alb-Cre transgene means that fetal liver HAMP or HAMP responsiveness is only removed once the albumin promoter becomes activated in the developing liver. Second, when the deletion of the hamp, matriptase-2, or hfe genes is achieved through maternal inheritance, it will also alter iron homeostasis in the mother and in the placenta. In this context, both maternal iron status and placental iron homeostasis are known to be important determinants of fetal liver iron stores.28,30 These potentially confounding factors have been eliminated in the present study by the use of paternally inherited alleles. Nonetheless, the divergent outcomes of these studies indicate the complexity of the role of the HAMP/FPN axis in pregnancy, which involves HAMP from potentially 3 different sources, pre- and postplacental functions of FPN, and the modifying effects of maternal and placental iron homeostasis. Another interesting observation is that, under conditions of early and sustained iron deficiency (as with the provision of iron-deficient diet to the mothers throughout pregnancy), fetal liver iron stores are reduced to a similar extent regardless of the presence or absence of fetal liver HAMP. This apparent lack of modifying effect by HAMP is most likely explained by the profound suppression of the fpn transcript that we observed in this setting (supplemental Figure 4C), with the implication that the levels of fetal liver HAMP are of little consequence in the absence of FPN. Under conditions of limited iron availability, iron-regulatory proteins (IRPs) act to reduce FPN levels, normally through translational rather than transcriptional suppression.39 Therefore, it will be important in the future to identify the mechanisms mediating suppression of fpn transcript by iron deficiency and to determine if these mechanisms protect fetal liver iron stores in the setting of severe iron deficiency. The present study raises further questions as to the role of IRPs in regulating fetal liver iron stores. Indeed, despite a reduction in liver iron concentration, the expression of iron-regulated genes tfr1 and dmt1 were not raised in the livers of fpnwt/C326Y hampfl/fl, Alb.Cre+ mice and fpnC326Yfl/fl, Alb.Cre+ fetuses compared with their respective littermate controls (supplemental Figure 4D-F). Additionally, provision of iron-deficient diet that produced an even more profound reduction in fetal liver iron concentration (supplemental Figure 4B) failed to change the expression of tfr1 and dmt1 in fetal livers (supplemental Figure 4G-H), while markedly increasing them in the placenta (supplemental Figure 4G-H). These findings are consistent with a recent study reporting that IRP1 in the placenta is important for regulating placental iron homeostasis.30 In that study, IRP1−/− fetuses did not differ from IRP1+/+ fetuses in terms of liver iron concentration, and this is also consistent with the notion that IRP1 may not be important in the fetal liver at this stage.
The present study also determined the effect of the reduction in liver iron stores on fetal hematopoiesis. The findings that hemoglobin levels and erythroid differentiation were reduced in the absence of any change in fetal serum iron levels and even when loss of HAMP responsiveness was confined to hepatocytes, are consistent with a paracrine role for hepatocytes in supporting hematopoiesis in the fetal liver. The current consensus is that erythroid progenitors acquire iron from nurse macrophages.40 One possibility is that fetal hepatocytes support hematopoiesis indirectly by supplying iron to these nurse macrophages. Another possibility is that fetal hepatocytes supply iron directly to erythroid progenitors. In the future, it would be important to establish whether hepatocyte iron stores are transferred to erythroid progenitors directly or indirectly and to identify the form(s) (transferrin, ferritin, heme) in which this iron transferred.
In conclusion, this study demonstrates that buildup of hepatic iron stores in late gestation requires the autocrine action of the HAMP/FPN axis in fetal hepatocytes. Formal interrogation of the roles of the HAMP/FPN and of the IRP axes in the placenta depends on the development of genetic tools that can target specific cell populations within the placenta without affecting iron control in the fetus or the mother.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
This work was supported by a Medical Research Council project Grant (MR/L010054/1) (S.L.-L. and P.A.R.) and a British Heart Foundation Intermediate Basic Science Postdoctoral Fellowship (FS/12/63/29895) (S.L.-L.).
Footnotes
For original data, please e-mail the corresponding author.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.L.-L. and P.A.R. conceived the study and sought funding; L.K., G.M., M.W., and S.L.-L. collected data and analyzed results; S.L.-L. drafted the manuscript; and all authors were involved in reviewing and revising the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Samira Lakhal-Littleton, Department of Physiology, Anatomy and Genetics, University of Oxford, Parks Rd, Oxford OX1 3PT, United Kingdom; e-mail: samira.lakhal-littleton@dpag.ox.ac.uk.
REFERENCES
- 1.Hernández-Martínez C, Canals J, Aranda N, Ribot B, Escribano J, Arija V. Effects of iron deficiency on neonatal behavior at different stages of pregnancy. Early Hum Dev. 2011;87(3):165-169. [DOI] [PubMed] [Google Scholar]
- 2.Beard J. Recent evidence from human and animal studies regarding iron status and infant development. J Nutr. 2007;137(2):524S-530S. [DOI] [PubMed] [Google Scholar]
- 3.Lozoff B, Beard J, Connor J, Barbara F, Georgieff M, Schallert T. Long-lasting neural and behavioral effects of iron deficiency in infancy. Nutr Rev. 2006;64(5 Pt 2):S34-S43, NaN-S91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piñero DJ, Li NQ, Connor JR, Beard JL. Variations in dietary iron alter brain iron metabolism in developing rats. J Nutr. 2000;130(2):254-263. [DOI] [PubMed] [Google Scholar]
- 5.Shafir T, Angulo-Barroso R, Jing Y, Angelilli ML, Jacobson SW, Lozoff B. Iron deficiency and infant motor development. Early Hum Dev. 2008;84(7):479-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirata M, Kusakawa I, Ohde S, Yamanaka M, Yoda H. Risk factors of infant anemia in the perinatal period. Pediatr Int (Roma). 2017;59(4):447-451. [DOI] [PubMed] [Google Scholar]
- 7.Freeman VE, Mulder J, van’t Hof MA, Hoey HM, Gibney MJ. A longitudinal study of iron status in children at 12, 24 and 36 months. Public Health Nutr. 1998;1(2):93-100. [DOI] [PubMed] [Google Scholar]
- 8.Grantham-McGregor S, Ani C. A review of studies on the effect of iron deficiency on cognitive development in children. J Nutr. 2001;131(2 2S-2):649S-666S, discussion 666S-668S. [DOI] [PubMed] [Google Scholar]
- 9.Lozoff B, Brittenham GM, Viteri FE, Wolf AW, Urrutia JJ. Developmental deficits in iron-deficient infants: effects of age and severity of iron lack. J Pediatr. 1982;101(6):948-952. [DOI] [PubMed] [Google Scholar]
- 10.Lozoff B, Brittenham GM, Viteri FE, Wolf AW, Urrutia JJ. The effects of short-term oral iron therapy on developmental deficits in iron-deficient anemic infants. J Pediatr. 1982;100(3):351-357. [DOI] [PubMed] [Google Scholar]
- 11.Walter T, De Andraca I, Chadud P, Perales CG. Iron deficiency anemia: adverse effects on infant psychomotor development. Pediatrics. 1989;84(1):7-17. [PubMed] [Google Scholar]
- 12.Aukett MA, Parks YA, Scott PH, Wharton BA. Treatment with iron increases weight gain and psychomotor development. Arch Dis Child. 1986;61(9):849-857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lozoff B, Brittenham GM, Wolf AW, et al. . Iron deficiency anemia and iron therapy effects on infant developmental test performance. Pediatrics. 1987;79(6):981-995. [PubMed] [Google Scholar]
- 14.Walter T, Pino P, Pizarro F, Lozoff B. Prevention of iron-deficiency anemia: comparison of high- and low-iron formulas in term healthy infants after six months of life. J Pediatr. 1998;132(4):635-640. [DOI] [PubMed] [Google Scholar]
- 15.Idjradinata P, Pollitt E. Reversal of developmental delays in iron-deficient anaemic infants treated with iron. Lancet. 1993;341(8836):1-4. [DOI] [PubMed] [Google Scholar]
- 16.Leong WI, Bowlus CL, Tallkvist J, Lönnerdal B. DMT1 and FPN1 expression during infancy: developmental regulation of iron absorption. Am J Physiol Gastrointest Liver Physiol. 2003;285(6):G1153-G1161. [DOI] [PubMed] [Google Scholar]
- 17.Collard KJ. Iron homeostasis in the neonate. Pediatrics. 2009;123(4):1208-1216. [DOI] [PubMed] [Google Scholar]
- 18.Dallman PR. Changing iron needs from birth through adolescence In: Fomon SJ, Zlotkin S, eds.. Nutritional Anemias, Vevey, Switzerland: Raven Press; 1992:29-36. [Google Scholar]
- 19.Heath AL, Tuttle CR, Simons MS, Cleghorn CL, Parnell WR. Longitudinal study of diet and iron deficiency anaemia in infants during the first two years of life. Asia Pac J Clin Nutr. 2002;11(4):251-257. [DOI] [PubMed] [Google Scholar]
- 20.Nemeth E, Tuttle MS, Powelson J, et al. . Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090-2093. [DOI] [PubMed] [Google Scholar]
- 21.Ganz T. Cellular iron: ferroportin is the only way out. Cell Metab. 2005;1(3):155-157. [DOI] [PubMed] [Google Scholar]
- 22.Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106(12):3710-3717. [DOI] [PubMed] [Google Scholar]
- 23.Lakhal-Littleton S, Crosby A, Frise MC, et al. . Intracellular iron deficiency in pulmonary arterial smooth muscle cells induces pulmonary arterial hypertension in mice. Proc Natl Acad Sci USA. 2019;116(26):13122-13130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zumerle S, Mathieu JR, Delga S, et al. . Targeted disruption of hepcidin in the liver recapitulates the hemochromatotic phenotype. Blood. 2014;123(23):3646-3650. [DOI] [PubMed] [Google Scholar]
- 25.Pigeon C, Ilyin G, Courselaud B, et al. . A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276(11):7811-7819. [DOI] [PubMed] [Google Scholar]
- 26.Nicolas G, Bennoun M, Porteu A, et al. . Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci USA. 2002;99(7):4596-4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Willemetz A, Lenoir A, Deschemin JC, et al. . Matriptase-2 is essential for hepcidin repression during fetal life and postnatal development in mice to maintain iron homeostasis. Blood. 2014;124(3):441-444. [DOI] [PubMed] [Google Scholar]
- 28.Balesaria S, Hanif R, Salama MF, et al. . Fetal iron levels are regulated by maternal and fetal Hfe genotype and dietary iron. Haematologica. 2012;97(5):661-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ganz T. Hepcidin and iron regulation, 10 years later. Blood. 2011;117(17):4425-4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sangkhae V, Fisher AL, Wong S, et al. . Effects of maternal iron status on placental and fetal iron homeostasis. J Clin Invest. 2020;130(2):625-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koulnis M, Pop R, Porpiglia E, Shearstone JR, Hidalgo D, Socolovsky M. Identification and analysis of mouse erythroid progenitors using the CD71/TER119 flow-cytometric assay. J Vis Exp. 2011;5(54):2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tabbah SM, Buhimschi CS, Rodewald-Millen K, et al. . Hepcidin, an iron regulatory hormone of innate immunity, is differentially expressed in premature fetuses with early-onset neonatal sepsis. Am J Perinatol. 2018;35(9):865-872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toldi G, Stenczer B, Molvarec A, et al. . Hepcidin concentrations and iron homeostasis in preeclampsia. Clin Chem Lab Med. 2010;48(10):1423-1426. [DOI] [PubMed] [Google Scholar]
- 34.Cardaropoli S, Todros T, Nuzzo AM, Rolfo A. Maternal serum levels and placental expression of hepcidin in preeclampsia. Pregnancy Hypertens. 2018;11:47-53. [DOI] [PubMed] [Google Scholar]
- 35.Dao MC, Sen S, Iyer C, Klebenov D, Meydani SN. Obesity during pregnancy and fetal iron status: is hepcidin the link? J Perinatol. 2013;33(3):177-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morin AM, Gatev E, McEwen LM, et al. . Maternal blood contamination of collected cord blood can be identified using DNA methylation at three CpGs. Clin Epigenetics. 2017;9(1):75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mao J, McKean DM, Warrier S, Corbin JG, Niswander L, Zohn IE. The iron exporter ferroportin 1 is essential for development of the mouse embryo, forebrain patterning and neural tube closure. Development. 2010;137(18):3079-3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Donovan A, Lima CA, Pinkus JL, et al. . The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1(3):191-200. [DOI] [PubMed] [Google Scholar]
- 39.Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol. 2006;2(8):406-414. [DOI] [PubMed] [Google Scholar]
- 40.Bessis MC, Breton-Gorius J. Iron metabolism in the bone marrow as seen by electron microscopy: a critical review. Blood. 1962;19(6):635-663. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.