

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
Escape from epistasis of RNA splicing factor mutations occurs with specific mutant alleles and preservation of 1 wild-type allele.
Allele-specific differences in RNA binding and/or splicing account for exclusivity vs cooccurrence of splicing factor mutations.
Abstract
Large-scale sequencing studies of hematologic malignancies have revealed notable epistasis among high-frequency mutations. One of the most striking examples of epistasis occurs for mutations in RNA splicing factors. These lesions are among the most common alterations in myeloid neoplasms and generally occur in a mutually exclusive manner, a finding attributed to their synthetic lethal interactions and/or convergent effects. Curiously, however, patients with multiple-concomitant splicing factor mutations have been observed, challenging our understanding of one of the most common examples of epistasis in hematologic malignancies. In this study, we performed bulk and single-cell analyses of patients with myeloid malignancy who were harboring ≥2 splicing factor mutations, to understand the frequency and basis for the coexistence of these mutations. Although mutations in splicing factors were strongly mutually exclusive across 4231 patients (q < .001), 0.85% harbored 2 concomitant bona fide splicing factor mutations, ∼50% of which were present in the same individual cells. However, the distribution of mutations in patients with double mutations deviated from that in those with single mutations, with selection against the most common alleles, SF3B1K700E and SRSF2P95H/L/R, and selection for less common alleles, such as SF3B1 non-K700E mutations, rare amino acid substitutions at SRSF2P95, and combined U2AF1S34/Q157 mutations. SF3B1 and SRSF2 alleles enriched in those with double-mutations had reduced effects on RNA splicing and/or binding compared with the most common alleles. Moreover, dual U2AF1 mutations occurred in cis with preservation of the wild-type allele. These data highlight allele-specific differences as critical in regulating the molecular effects of splicing factor mutations as well as their cooccurrences/exclusivities with one another.
Visual Abstract

Introduction
The RNA splicing machinery is frequently mutated in subjects with clonal hematopoiesis and patients with myelodysplastic syndromes (MDS), acute myeloid leukemia (AML), and myeloproliferative neoplasms (MPNs).1-3 Splicing factor mutations are concentrated in the core RNA splicing factor SF3B1, the accessary splicing factor SRSF2, the small subunit of the U2AF heterodimer U2AF1, and the minor spliceosome component ZRSR2, each of which has an essential role in recognition of splicing signals to ensure faithful gene expression. Although heterozygous point mutations occur at highly conserved amino acid residues of SF3B1, SRSF2, and U2AF1, conferring a change of function,4-6 a variety of mutations, including frameshift, nonsense, and splice site changes occur throughout the X-chromosome–encoded ZRSR2, presumably resulting in loss of function.7
Intriguingly, although RNA splicing factor mutations occur in up to 45% to 85% of myeloid neoplasms, they consistently occur in a heterozygous state and mutually exclusive manner, such that it is rare to identify more than 1 RNA splicing factor mutation in an individual patient.1 Because of the discovery of these mutations, sequencing data from thousands of patients with myeloid neoplasms have reaffirmed mutual exclusivity of the mutations with great statistical significance, compared with the likelihood of their coexisting by chance.8-11 In parallel, efforts to identify the functional basis for epistasis of mutations in RNA splicing factors has shown that coexpression of the most common hotspot mutations in SF3B1 (SF3B1K700E) and SRSF2 (SRSF2P95H) in vivo in mice is intolerable to hematopoietic cells.12
Despite these observations, researchers in several studies have noted rare patients with concurrent mutations in 2 RNA splicing factors in the same sample.9,10,13,14 However, corresponding allelic frequencies of these mutations are absent from many series, precluding evaluation of whether both mutations are expressed within the same cell. Moreover, mutations in splicing factors may occur at multiple residues with potential distinct impacts on splicing,4,6,15 and prior studies have used different criteria for inclusion of variants as bona fide mutations. For these reasons, evaluation of known pathogenic mutant alleles (as opposed to mutant genes) where variant allele frequencies are available is necessary to clarify whether mutations in RNA splicing factors can actually coexist. Furthermore, the advent of single-cell genomic technologies now provides an opportunity to definitively assess the presence of these mutations in the same cells.
We provide the clinical, genetic, and molecular characteristics of patients with dual splicing factor mutations in bulk malignant cell populations and at the single-cell level. In so doing, we uncover enrichment of distinct alleles among patients with single vs dual splicing factor mutations and downstream effects on RNA splicing and binding, which provide a basis to explain the cooccurrence vs mutual exclusivity of mutant splicing factor alleles in patients with myeloid malignancies.
Methods
Patient samples
Studies were approved by the Institutional Review Boards of Memorial Sloan Kettering Cancer Center (MSK), the University of Manchester, and Mayo Clinic and were conducted in accordance with the protocol set forth by the Declaration of Helsinki. Patients with myeloid malignancies, including AML, MDS, and MPN, were identified from 4 public studies8,10,16,17 and in patients treated in clinical practices at MSK and Mayo Clinic. The overall frequency of splicing factor mutations and comparisons between individuals with single and double mutations were evaluated in this cohort of 4231 patients: 1319 from the German-Austrian AML Study Group,14 1242 from MSK, 644 from the United Kingdom, 608 from the Beat AML program,4 and 418 from the Internal Cancer Genome Consortium16 (supplemental Figure 1A; supplemental Table 1, available on the Blood Web site). For analysis specific to double mutations, 36 samples identified from this cohort were combined with an additional 22 samples contributed by Mayo Clinic (unpublished; supplemental Table 2).
Bulk DNA mutational analysis
The Ensembl Variant Effect Predictor (VEP) was used to systemically reannotate mutation data for known hotspot mutations in SF3B1, SRSF2, and U2AF1, as well as truncating mutations in ZRSR2.18 For sample sequences from MSK-IMPACT (Integrated Mutation Profiling of Actionable Cancer Targets), we used a variant allele fraction (VAF) threshold of 1% for hotspot mutations in SF3B1, SRSF2, and U2AF1 and of 5% for variants of unknown significance, consistent with normal clinical practice for MSK-IMPACT data.19,20 However, for the various techniques used for mutational calling from non-MSK data (including whole-exome sequencing and targeted DNA sequencing of various depths), the lowest VAF reported was 5%. Several functional alleles specific to myeloid diseases were added to a list derived primarily from solid tumors21 (supplemental Table 1). The frequencies of patients with 1 vs 2 splicing factor mutations were determined, and Fisher’s exact test was performed to assess for mutual exclusivity. Patients with single and double mutations were compared in terms of distribution of myeloid neoplasm subtypes and tumor mutation burden. The mutation burden was calculated by dividing the number of nonsynonymous mutations by the expected breadth of sequencing in the IMPACT Heme panel22 (1.21 Mb). For samples with double splicing factor mutations, the cancer cell fraction (CCF) of each mutation was summed to gauge the likelihood that it cooccurs in the same cells as opposed to different cells of a patient sample. Although 58 patient samples with double splicing factor mutations were identified as described, the CCF analysis could be performed in only 46 samples where VAFs of both splicing factor mutations were available. The CCF of hotspot mutations in SF3B1, SRSF2, and U2AF1 was assumed to be 2 times the VAF. For nonsense and frameshift mutations on ZRSR2, the ploidy was assumed to be 1 for males and 2 for females when this information was available. When it was not, VAFs >0.5 were taken to imply monoploidy. Cooccurrence of the splicing factor mutations in the same cells was substantiated by a combined CCF ≥1. Copy number analysis was performed on 600 patients in the MSK cohort sequenced with MSK-IMPACT, using previously published methods.19,20
Single-cell DNA sequencing analysis
Targeted single-cell DNA sequencing of cryopreserved bone marrow mononuclear cells (BM MNCs) from patients with double splicing factor mutations (supplemental Table 3) was performed on a microfluidic, droplet-based platform developed by Mission Bio, as previously described.23 Barcoded samples proceeded to targeted polymerase chain reaction amplification by a custom panel of 36 amplicons targeting 47 mutations across all 4 splicing factors, and 13 additional driver genes involved in myeloid malignancies (supplemental Tables 4 and 5). The identity and mutational profile of each cell were preserved through the process, as each amplicon was tagged with a unique cell barcode. Additional single-cell sequencing and analysis methods are described in the supplemental Methods: U2AF1 allele-specific sequencing; isothermal titration calorimetry; and genome annotations, RNA sequencing (RNA-seq) read mapping, and differential splicing analysis.
Results
Frequency of patients with 2 or more bona fide mutations in splicing factors
We systematically annotated primary genomic DNA sequencing data for known hotspot mutations in SF3B1, SRSF2, and U2AF1, as well as clearly deleterious mutations in ZRSR2 across 4231 patients with AML (n = 2822), MDS (n = 1050), or MPNs (n = 359), according to strict criteria for calling the mutation (Figure 1A; supplemental Tables 1 and 2; supplemental Methods). This information included previously unpublished data from 1242 patients at MSK, as well as from independent samples from 6 prior studies8,10,16,17,24 (supplemental Figure 1A). Mutations in these 4 RNA splicing factors were seen in 22.9% of patients overall and exhibited strong mutual exclusivity (q < .001). However, 0.85% of patients (n = 36) had 2 concurrent splicing factor mutations (supplemental Figure 1B). No patient had mutations in 3 or more splicing factors. Of note, the subtypes of myeloid neoplasms present among dual-mutation samples were reflective of the entire patient cohort (supplemental Figure 1C), suggesting that mutual exclusivity of splicing factor mutations is not related to any specific clinical subtype of myeloid neoplasm. The distribution of mutations in splicing factors across myeloid neoplasms was similar to that in prior studies,2,9,10,16,25 with an enrichment of SF3B1 K700 mutations in MDS over AML or MPNs and a slightly higher frequency of SRSF2 mutations in AML over MPN or MDS (supplemental Figure 1D). There was also no significant difference in the tumor mutational burden in patients with 1 vs 2 mutations in splicing factors (supplemental Figure 1E).
Figure 1.
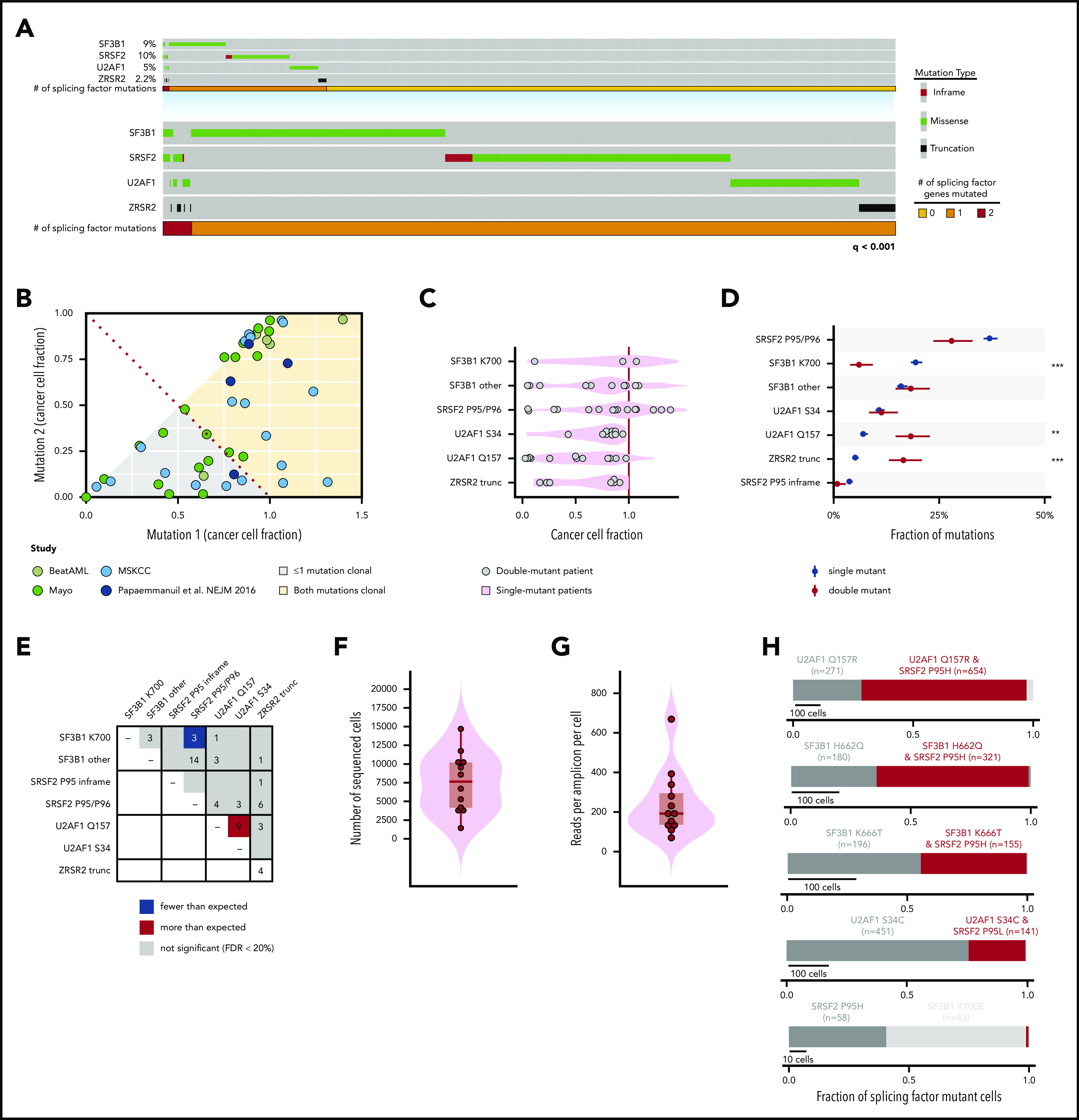
Genetic features of patients harboring 2 concomitant mutations in RNA splicing factors at the bulk and single-cell level. (A) Oncoprint of hotspot mutations in SF3B1, SRSF2, an U2AF1, as well as clearly deleterious mutations (nonsense or frameshift mutations) in ZRSR2 across 4231 patients. Each column represents 1 patient. The number of patients with 0, 1, or 2 splicing factor mutations is shown in yellow, orange, and red, respectively. Overall, mutations in each gene exhibited strong mutual exclusivity (q < .001; Fisher’s exact test). (B) CCF of each mutant splicing factor from genomic DNA sequencing of a cohort of 58 dual mutant splicing factor samples, including those from a single study.16 (C) CCF of mutations at SF3B1K700, other residues of SF3B1 (SF3B1 other), SRSF2P95/P96, U2AF1S34, and U2AF1Q157 as well as ZRSR2 truncation mutations (ZRSR2 trunc). (D) Percentage of patients with single or double splicing factor mutations (in black and red, respectively) with mutations in SRSF2, SF3B1, U2AF1, and ZRSR2. Error bars: 1 standard deviation, based on a binomial distribution. **P < .005; ***P < .0005 (Fisher’s exact test). (E) Plot describing the number of patients with coexisting mutant alleles in splicing factors. The expected number was based on the fraction of samples with exactly 2 mutations under the assumption of no mutual exclusivity and using a Poisson distribution. The distribution of the number of (F) total sequenced cells per patient and (G) reads per amplicon per cell from single-cell genomic DNA sequencing. Each point represents a sample from a unique patient. (H) Fraction of mutated cells with 1 or 2 mutations in RNA splicing factors within each patient with a dual splicing factor mutation. Red bar denotes fraction of individual cells where 2 splicing factor mutations were identified within the same cell. The number of cells containing each mutation is indicated.
Allele-specific mutual exclusivity and cooccurrence of splicing factor mutations
To address whether mutations in splicing factors coexist within the same cells in patients with 2 mutations, we calculated the CCF of each splicing factor mutation based on its corresponding VAF. For this analysis, we included a cohort of 22 additional patients identified in clinical practice as having >1 splicing factor mutation, for an overall cohort of 58 double-mutation samples (supplemental Table 2). Every possible combination of mutations in SF3B1, SRSF2, U2AF1, and ZRSR2 was observed, and there were also patients who harbored 2 distinct concomitant mutations in either U2AF1 or SF3B1 in the same sample (supplemental Figure 1F).
Interestingly, in 63% of these patients, the 2 splicing factor mutations had a combined CCF >1, suggesting that they coexisted within the same cell (Figure 1B). Overall, the CCF of mutations in SF3B1, SRSF2, U2AF1, and ZRSR2 within double-mutation samples mirrored that of single-mutation samples (Figure 1C). However, when we compared the frequency of mutations within each gene at the level of mutant alleles, we identified that SF3B1K700 and SRSF2P95/P96 substitutions, which represent the 2 most common splicing factor mutations among all patients, were significantly less common in double mutant than in single mutant alleles (Figure 1D; supplemental Figure 1G-H). For example, SF3B1K700 mutations accounted for 19% of all splicing factor mutations in patients with single mutations but only 3.9% of all splicing factor mutations in those with dual mutations. Consistent with this result, the specific combination of SF3B1K700 and SRSF2P95/P96 mutations occurred far less frequently than expected, based on the frequencies of these mutations in the overall cohort (P = .0013; Fisher’s exact test; Figure 1E). These data demonstrate selection against cooccurrence of the SF3B1K700 and SRSF2P95/P96 mutations, the most frequent mutant alleles among the splicing factors. At the same time, that less common mutations in SF3B1 and SRSF2 cooccur argues that the mutual exclusivity vs cooccurrence of these mutations occurs at the level of specific mutant alleles, rather than at a gene-specific level.
Cellular-level mutual exclusivity and cooccurrence of splicing factor mutations
Next, we sought to investigate whether mutual exclusivity of the most common splicing factor mutations and possible coexistence of less common alleles holds true at the single-cell level. We performed high-throughput single-cell DNA sequencing (scDNA-seq) to quantitatively assess the clonal architecture of myeloid neoplasms harboring dual splicing factor mutations in the same sample (Tapestri platform; Mission Bio). Using a custom panel of 36 amplicons across 17 genes intended to capture all mutations detected by bulk DNA sequencing in each patient sample with a double mutation, we sequenced a total of 98 932 BM MNCs from 11 patients (supplemental Tables 3, 4, and 5), each of whom bore 2 bona fide mutations in RNA splicing factors (supplemental Table 4). We sequenced a median of 7 643 BM MNCs per patient (interquartile range [IQR], 4 152-10 201; Figure 1F). The median coverage was 192× per amplicon per cell (IQR, 135-295; Figure 1G). The VAFs from bulk and scDNA-seq correlated significantly (R2 = 0.32; P = .001), suggesting that despite variability in the data, the single-cell sequencing results were representative of bulk sequencing (supplemental Figure 2A). Target loci were genotyped in the majority of cells (median, 92%; IQR, 86-9%), with the notable exception of SRSF2P95, a locus prone to allele dropout (ADO)26 (supplemental Figure 2B; supplemental Methods). We attempted to address ADO at SRSF2 by jointly estimating the cooccurrence of SRSF2, SF3B1, and U2AF1 mutations and ADO at both loci under the assumption that hotspot mutations in these genes are diploid and either wild type (WT) or heterozygous in all cells. These are reasonable assumptions, because <5% of patients had copy number alterations at regions of hotspot mutations in SF3B1, SRSF2, and U2AF1 mutations in >500 patients with myeloid neoplasm (supplemental Figure 2C-D), and bulk RNA-seq data consistently reveal simultaneous expression of both SRSF2 WT and mutant alleles.8,24,27
Our scDNA-seq analysis showed true cooccurrence of concomitant mutations in RNA splicing factors at the single-cell level in multiple patients, including combined SF3B1K666T and SRSF2P95H, SF3B1H662Q and SRSF2P95H, SF3B1G740R and U2AF1Q157R, and U2AF1Q157R and U2AF1S34F mutations, among others (Figures 1H and 2A-C; supplemental Figures 2E and 3). However, consistent with their exclusivity in bulk sequencing data, SF3B1K700E and SRSF2P95H mutations remained mutually exclusive at the level of individual cells (Figures 1H and 2A-B). Moreover, in a male patient with 2 ZRSR2 frameshift mutations (ZRSR2E118Dfs*28 and ZRSR2R290*), the mutations existed in distinct cells, a result probably explained by the location of ZRSR2 on chromosome X and the presumed convergent effects of loss-of-function mutations in the same gene (Figure 2C). In contrast, ZRSR2 mutations clearly coexisted within the same cells as hotspot mutations in other splicing factors (Figure 2C). We accounted for ADO under the aforementioned assumptions and found that adjusted estimates for mutant splicing factor clones closely resembled unadjusted estimates in each case (supplemental Figure 4). These data reaffirm the mutual exclusivity of SF3B1K700E and SRSF2P95H mutations, while highlighting the potential for cooccurrence of other splicing mutant alleles at the single-cell level.
Figure 2.
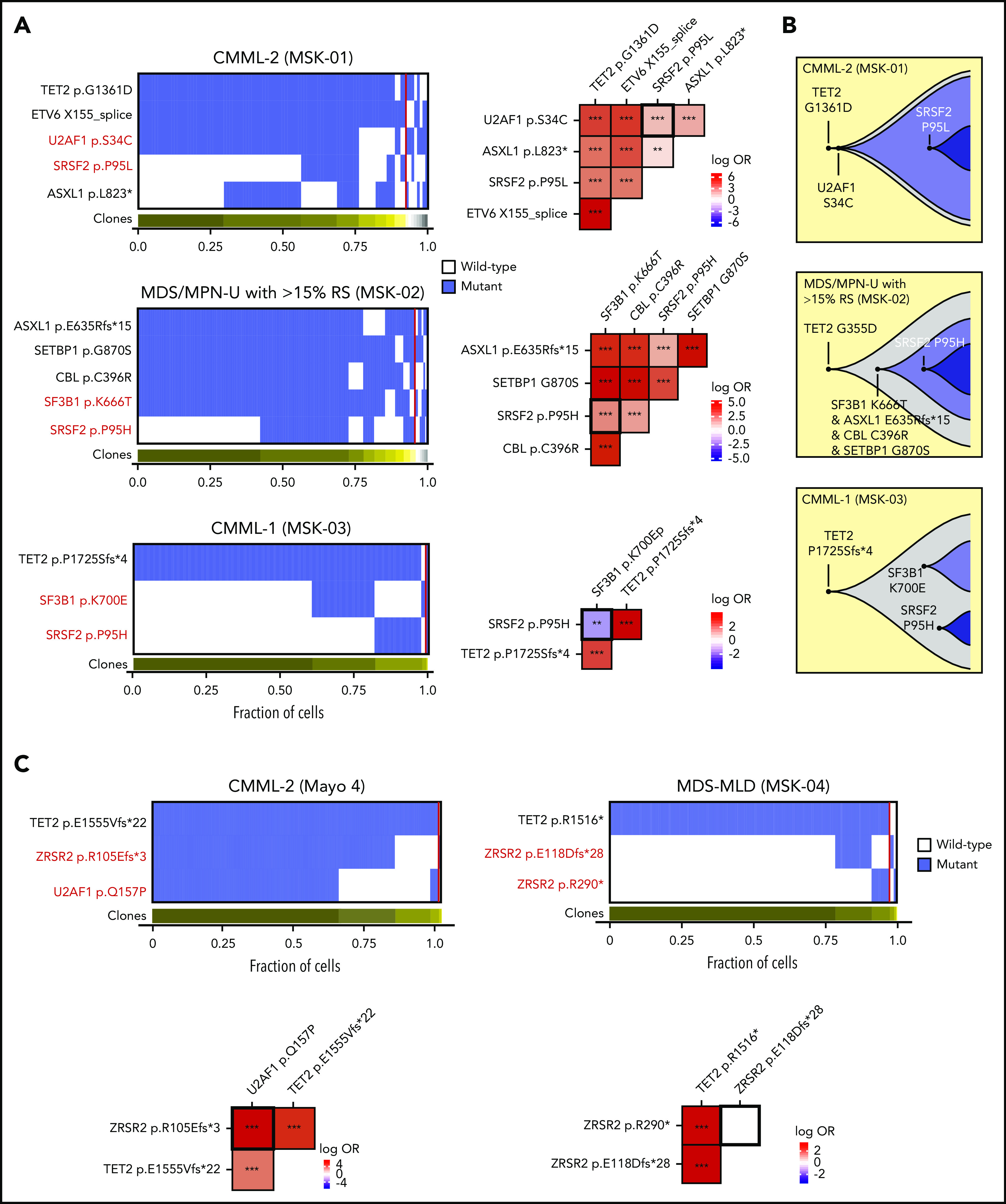
Allele-specific mutational cooccurrences in RNA splicing factor mutations. (A) Oncoprint indicating cellular-level cooccurrence of mutations in select cases of double splicing factor mutations (the clinical diagnosis and sample ID is listed above each Oncoprint). Each column in the heat map (left) depicts an individual cell, with the genotype of each sequenced cell for each variant. Clustering is based on the genotypes of driver mutations, and the horizontal bar depicts the detected clones in each case. Mutant and wild-type cells are indicated in blue and white, respectively. The subclones located to the right of the red line comprised <1% of the total sequence cells, because such small subclones can represent false-positive or -negative genotypes as a result of allele dropout or multiplets. The figures on the right show the pairwise association of mutations. The color and size of each panel represent the degree of the logarithmic odds ratio (log OR). The vertical bar indicates the association of the colors with the log OR. Cooccurrence and mutual exclusivity are indicated by red and blue, respectively. The statistical significance of the associations based on the false discovery rate (FDR) is indicated by the asterisks. *FDR < 0.1; **FDR < 0.05; ***FDR < 0.001. (B) Fish plots showing the inferred clonal hierarchy based on the single-cell genotype data for the 3 patients in panel A. (C) Oncoprint, as in panel A, but evaluating cellular cooccurrence or mutual exclusivity of deleterious ZRSR2 mutations with mutations in other splicing factors (left) or with one another (right). CMML, chronic myelomonocytic leukemia; MDS-MLD, myelodysplastic syndrome with multilineage dysplasia; MDS/MPN-U with >15% RS, MDS/myeloproliferative neoplasm unclassified with >15% ring sideroblasts.
We next evaluated the clonal architecture of the mutations in splicing factors and other driver genes implicated in myeloid malignancies. In samples with true cooccurrence of splicing factor mutations, we most frequently detected a dominant single splicing factor mutant clone followed by acquisition of a second splicing factor mutation within the same cells (Figure 1H; supplemental Figure 2E), most commonly consisting of an SF3B1non-K700 or U2AF1 mutant clone followed by acquisition of an SRSF2 mutation. Quantitative analysis of the precise clonal architecture in patients with the dual splicing factor mutation including additional driver genes showed that, in each case, pathogenic mutations in epigenetic modifiers such as TET2, ASXL1, KDM6A, or DNMT3A nearly always preceded acquisition of an RNA splicing factor mutation (Figure 2; supplemental Figure 3).
Functional basis for cooccurrence of splicing factor mutations
To understand the allele-specific basis for the rare cooccurrence of splicing factor mutations, we next evaluated the functional impact of splicing factor mutations selected in samples from patients with double mutations in more detail. The most common mutational cooccurrence among splicing factors in dual-mutation samples was the combination of SRSF2P95/P96 and SF3B1 mutations in residues other than K700 (Figure 1E). Even in these cases, the specific mutant alleles in SRSF2 deviated from the most common SRSF2 mutant allele substitutions. For example, we identified 1 case of an SRSF2P95A mutation in a patient with an SF3B1K666N mutation (Figure 3A). Previous work has identified that mutations at proline 95 in SRSF2 alter the ability of SRSF2 to physically interact with exonic splicing enhancer sequences.6,28 Although WT SRSF2 recognizes C- and G-rich sequences equally well, SRSF2P95H/L/R mutations have enhanced binding avidity to C-rich RNA sequences, a biochemical hallmark of pathologic substitutions at SRSF2P95. However, the effect of SRSF2 P95A mutations (which are extremely rare overall) on RNA binding has not been tested in a assay similar to the one used for SRSF2P95H/L/R mutations. We therefore purified the RNA recognition motif domain of SRSF2, as previously described6,29 (also in supplemental Methods), with or without P95H, L, R, or A substitutions and performed isothermal titration calorimetry with the RNA ligand 5′-uCCAGu-3′, a previously demonstrated optimal SRSF2 target according to the SSNG consensus sequence.29 In contrast to the nearly fivefold increase in RNA binding affinity seen with SRSF2 P95H, P95L, and P95R substitutions relative to WT SRSF2 (Figure 3B), P95A substitutions did not influence RNA binding affinity (Figure 3B). These data indicate the importance of evaluating the exact allelic substitution in splicing factor mutations and suggest that the specific mutations present in patients with double mutations may allow for escape from epistasis as a result of the mitigating effects on RNA binding and/or splicing.
Figure 3.

Allele-specific effects on RNA binding and splicing in splicing factor mutations seen in patients harboring 2 concomitant mutations in splicing factors. (A) Pie chart of SRSF2 P95 amino acid substitutions across the entire cohort. (B) Binding affinities of WT vs P95H/L/R/A mutant SRSF2 peptides to UCCAGU RNA oligonucleotides as absolute Kd values. The column labeled “change in affinity” provides the Kd ratio of the mutant:WT peptide. (C) Comparison of the quantitative effects of SF3B1K700E and SF3B1K666N mutations on splicing, stratified by mutant allele fraction. Each point illustrates the absolute change in isoform usage (ΔPSI) for 1 of the top 20 most misspliced events associated with each mutation. For each panel, the top 20 most misspliced events were computed using only samples with SF3B1K700E or SF3B1K666N mutations. Missplicing of those 20 events was then computed for all samples, irrespective of mutation, and plotted as illustrated. SF3B1K700E and SF3B1K666N mutations cause missplicing of similar sets of genes, but SF3B1K700E mutations cause more dramatic changes. Lines, shading, and equations indicate the best-fit linear regressions and corresponding 95% confidence intervals. (D) As in panel C, but computed using the top 5 most misspliced events for each mutation (SF3B1K700E; SF3B1K666N). (E) Box plot illustrating the data from panel C and associated P-value, computed using a 2-sided Wilcoxon rank-sum test. (F) Fraction of U2AF1S34F, U2AF1Q157R, or dual U2AF1S34F/Q157R mutated cells from a patient harboring both U2AF1S34F/WT and U2AF1Q157R/WT mutations. Red bar indicates fraction of U2AF1S34F/Q157R dual mutant cells. (G) Clonal hierarchy of mutations in the patient from panel E. Each column represents a cell at the indicated scale, as in panel A. Cells with mutations and WT cells are indicated in blue and white, respectively. (H) Fish plots showing the inferred clonal hierarchy based on the single-cell genotype data from panel G. (I) Sanger sequencing electropherograms from representative single cell clones from the patient in panel F. As enumerated on the right, all colonies were either U2AF1 dual WT or U2AF1S34F/Q157R dual mutant, indicating that these mutations always occur in cis with preservation of the WT allele. (J) Heat map of percentage spliced in values of cassette exons in patients with U2AF1S34, Q157, and U2AF1S34/Q157 dual mutations displaying cassette exon splicing events specific to the U2AF1S34 or Q157 single-mutant state. Standard deviation of <0.2 among single mutants and mean(U2AF1 S34) − mean (U2AF1 Q157) < 0.32. Each row is a unique patient, and each column is a single splicing event.
SF3B1K700E exhibits strong mutual exclusivity with other splicing factor mutations, a phenomenon not recapitulated by other hotspot mutations in SF3B1. We therefore characterized differences in RNA splicing between the most common mutant alleles in SF3B1 found in myeloid neoplasms, SF3B1K700E and SF3B1K666 substitutions, by performing RNA-seq on blood or BM MNCs from a new cohort of 24 patients with MDS or AML bearing the following mutations: 10, SF3B1 WT; 6, SF3B1K666; and 8, SF3B1K700E (supplemental Table 6). To differentiate whether the 2 groups of mutant alleles have distinct influences on splicing or whether the SF3B1K666 mutant alleles have a less potent response, we compared the quantitative extent of missplicing of the events that responded most strongly to each type of SF3B1 mutation. Interestingly, this analysis revealed that, although both SF3B1K700E and SF3B1K666 mutations cause missplicing of a similar sets of genes, SF3B1K700E mutations result in quantitatively more dramatic changes in splicing than do SF3B1K666 mutations. SF3B1K666 mutations drive only modest changes in missplicing of the top SF3B1K700E mutation-responsive events; conversely, SF3B1K700E mutations are just as capable as SF3B1K666 mutations of driving strong missplicing of the top SF3B1K666-responsive events (Figure 3C-E; supplemental Figure 5A). These data demonstrate important allele-specific differences in splicing among the distinct SF3B1 mutations seen in myeloid neoplasm that may underlie the cooccurrence and exclusivity of SF3B1 mutations with mutations in other splicing factors.
In contrast to significant exclusivity of the most common mutant alleles in SRSF2 and SF3B1, U2AF1S34, and U2AF1Q157 hotspot mutations cooccurred at a higher rate than expected by chance alone (Figure 1E). Moreover, scDNA-seq analysis of >6000 cells from a patient harboring both U2AF1S34F and U2AF1Q157 mutations clearly revealed that both mutations occurred in the same cells. Although we hypothesized that the U2AF1 mutations occurred at about the same time, given that virtually all of the U2AF1 mutant cells carried both mutations (Figure 3F), it is also conceivable dual mutant cells had a significant proliferative advantage over single mutant cells, such that they became the only detectable clone over time. Evaluation of the clonal architecture of this patient based on sequencing of the 5 mutations identified in bulk DNA showed that initiating mutations in KDM6B followed by DNMT3A were followed by acquisition of the dual U2AF1 mutant clone (Figure 3G-H).
Cooccurrence of multiple U2AF1 mutations is surprising, given prior data suggesting the requirement for expression of a WT allele in the presence of a hotspot mutation in U2AF1 for cell survival.30 We therefore evaluated whether cooccurring U2AF1S34F and U2AF1Q157R mutations were present on the same allele or different alleles using long-range reverse-transcription-polymerase chain reaction spanning exons 2 to 7 of U2AF1 (which encode the S34 and Q157 residues of U2AF1), followed by subcloning and sequence analysis of individual clones. All clones that emerged from this analysis were either entirely WT for U2AF1 or contained both the U2AF1S34F and U2AF1Q157 mutations (Figure 3I). In addition, there are distinct mRNA isoforms of U2AF1, 1 of which includes a premature termination codon that targets the resulting U2AF1 mRNA for nonsense-mediated mRNA decay.31 However, U2AF1S34F/Q157 double-mutant clones did not differ from U2AF1 WT clones in their usage of U2AF1 isoforms (supplemental Figure 5B). Consistent with these findings, patients with U2AF1S34F/Q157 double mutations harbored differential cassette exon splicing events, characteristic of both the U2AF1S34F and U2AF1Q157 single-mutation states (Figure 3J). Although these data indicate that dual U2AF1 hotspot mutations occurred in cis with preservation of 1 WT allele in this sample, further evaluation of additional samples is important for understanding if coexisting U2AF1S34F/Q157 hotspot mutations are tolerated in trans.
Discussion
Genomic analyses of patients with myeloid malignancies have successfully identified, not only individual genes that are recurrently mutated, but also groups of genes that have important genetic interactions with one another. Notable groups of mutations that display statistically significant mutual exclusivity in patients with MDS and AML include mutations in the TET2 and IDH1/2 enzymes,32 cohesin subunits, and RNA splicing factors (SF3B1, SRSF2, U2AF1, and ZRSR2).1 The most common explanation for such epistatic interactions is that mutations are mutually exclusive because of either redundant effects of each mutation and/or intolerability of coexpression of these mutations within the same cell. Studying the basis for these genetic interactions has elucidated novel disease biology. For example, discovery of the mutual exclusivity of TET2 and IDH1/2 mutations in AML led in part to the identification of the convergent effects of these mutations on DNA cytosine modification.32 We focused here on evaluating the clinical and genetic characteristics of patients with ≥2 simultaneous mutations in RNA splicing factors to understand the basis for rare coexistence of these mutations. Such patients have been anecdotally noted in several studies but the frequency, characteristics, and basis for the existence of such cases have not been systematically studied.
The existence of patients with double splicing factor mutations appears counter to the mutual exclusivity of these mutations in most patients,1 prior functional data demonstrating intolerability of the most common splicing factor mutations,12 and the known requirement of WT splicing in cells carrying hotspot mutations in splicing factors.12,30,33,34 However, our data revealed that mutual exclusivity of mutations in RNA splicing factors occurs at the level of specific mutant alleles, dependent on the precise amino acid substitutions in SF3B1, SRSF2, and U2AF1, rather than at the gene level. In other words, mutations in RNA splicing factors can and do coexist in the same individuals and even in the same cells, but only combinations involving less common mutant alleles are identified in such individuals.
Allele-specific differences in RNA splicing factor mutations make sense in light of the fact that distinct hotspot mutations in RNA splicing factors are known to have unique disease manifestations and, in some cases, are known to have unique effects on RNA splicing and gene expression. For example, mutations at the S34 and Q157 residues of U2AF1 have distinct sequence-specific effects on cassette exon usage and misspliced target genes.4 Consistent with these nonoverlapping effects of U2AF1 S34 and Q157 mutations, we uncovered rare significant coenrichment of U2AF1 S34 and Q157 mutations in patients, and this co-mutation was enriched, even within individual cells. This finding suggests potential cooperative interaction of the U2AF1 S34 and Q157 mutations, a hypothesis that would be interesting to examine in future studies. Nonetheless, U2AF1S34/Q157 dual mutations occurred in cis with preservation of the WT allele, in line with prior work demonstrating requirement of the WT allele in U2AF1 mutant cells.30
In accordance with the concept of allele-specific regulation of mutational cooccurrences, specific combinations of splicing factor mutations exhibit significant mutual exclusivity. In fact, the most common mutation substitutions in SF3B1 (SF3B1K700E) and SRSF2 (SRSF2P95H/L/R) were substantially selected against at the bulk cellular level, as well as within individual cells. In a particularly illustrative example, we found that 1 patient who ostensibly appeared to have coexisting SRSF2P95 and SF3B1 mutations, actually had selection for an extremely rare mutation in SRSF2 (SRSF2P95A), which although located at the commonly mutated residue in SRSF2, did not alter the ability of SRSF2 to bind RNA. In contrast, SF3B1 hotspot mutations outside of K700 frequently coexisted with other splicing factor mutations, probably caused by the allele-specific effects of distinct SF3B1 mutations on RNA splicing and gene expression, as has been demonstrated for mutant U2AF1.
SF3B1 mutations have been suspected to have allele-specific differences, given the enrichment of specific SF3B1 mutations in distinct subtypes of cancer. For example, SF3B1R625 substitutions are common in melanomas but rare in other cancers,35 and SF3B1G742 mutations are most common in chronic lymphocytic leukemia.36 These findings suggest allele-specific differences across distinct SF3B1 mutant hotspots, but these differences have not been elucidated. In fact, a recent study identified adverse outcome among patients with MDS with SF3B1K666 mutations specifically37; however, global analyses of splicing were absent from this study (and the few splicing changes suggested to be specific to SF3B1K666N genotype are actually evident in SF3B1K700E samples). We found that K700E and K666 mutations in SF3B1, which represent the 2 most commonly mutated SF3B1 residues in myeloid neoplasms, have significant differences in their global effects on splicing. Compared with SF3B1K700E, SF3B1K666 mutations have less dramatic effects on splicing, which may account for the intolerability of the former, and the tolerability of the latter, with other splicing factor mutations. Although both K666 and K700 residues are located in the HEAT domain of SF3B1, recent structural analyses of human SF3B1 highlighted key functional differences between these locations.38 The K666 residue is involved in intramolecular hydrogen bonds within SF3B1 to maintain its tertiary structure. In contrast, the K700 residue is exposed on the surface of SF3B1 and appears to be involved in the interaction of SF3B1 with other U2 snRNP components.38 Thus, mutations in K666 and K700 are expected to create distinct structural disturbances in SF3B1 which likely account for their subsequent different effects on RNA splicing. Strong data on the clinical and morphologic associations of individual mutations in RNA splicing factors in myeloid malignancies necessitate a closer future evaluation of the potential effects of combined mutations of RNA splicing factors on disease characteristics, such as morphologic features, blood counts, and outcome.
The discovery that cells containing hotspot mutations in RNA splicing factors are intolerant of additional genetic perturbations in splicing led to a therapeutic effort to modulate splicing as a novel form of therapy for splicing factor mutant leukemias.31 However, the allele-specific effects of splicing factor mutations on vulnerability to additional genetic perturbations to splicing identified in our study suggest the possibility that response to such therapies may vary based on the exact mutant allele present. For example, cells bearing SF3B1K700E and SRSF2P95H/L/R substitutions may be more sensitive to splicing modulatory drugs than U2AF1S34 or U2AF1Q157 mutations or rarer mutant alleles in SRSF2 or SF3B1, which have less prominent effects on RNA splicing than K700E or P95H/L/R substitutions. It is also possible that ZRSR2 mutant cells may be less responsive to such therapies, given that ZRSR2 differs from other leukemia-associated mutant splicing factors, in that it functions in the minor spliceosome. These hypotheses should be considered in future preclinical as well as clinical studies testing drugs that globally perturb RNA splicing.
One frequent reason for cooccurrence of mutations that are normally mutually exclusive is distribution of such mutations to distinct subclones. Studying the clonal architecture of such cases has recently been instrumental in understanding novel mechanisms of resistance to mutationally targeted therapies. For example, although mutations in IDH1 and IDH2 typically do not cooccur, resistance to IDH2 inhibitors can develop because of clonal outgrowth of IDH1 subclones.39 Similarly, FLT3 inhibitor resistance has been shown in some cases to arise from clonal outgrowth of RAS mutant cells. One can envision, then, that prospective therapies targeting specific RNA splicing factor mutations could select for outgrowth of cells bearing other splicing factor mutations that have convergent effects on cell survival.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the Mt Sinai School of Medicine Technology Development Laboratory for help with the MissionBio Tapestri platform and sequencing, the Manchester Cancer Research Centre Biobank for help with samples, and Mission Bio for providing assistance with data generation, analysis, and interpretation.
This work was supported by the Conquer Cancer Foundation of the American Society of Clinical Oncology, the American Association for Cancer Research, the American Society of Hematology (ASH) and the ASH HONORS program (X.M.), the Robert Wood Johnson Foundation, and the National Institutes of Health (NIH), National Cancer Institute (NCI) (1K08CA230319-01) (J.T.); by the Regenerative Medicine Research Fund, State of Connecticut, the NIH, National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (R01DK102792), and the Frederick A. Deluca Foundation (S.H.); by the Evans MDS Foundation (S.H., O.A.-W., R.K.B.); by the NIH, National Heart, Lung and Blood Institute (R01HL128239), the NIH, NCI (1R01CA251138-01, 1R01CA251138, 1R01CA242020), the Department of Defense Bone Marrow Failure Research Program (W81XWH-12-1-0041), and the Leukemia and Lymphoma Society (O.A.-W., R.K.B); and by the NIH, NIDDK (R01DK103854), the Henry and Marilyn Taub Foundation, and the NIH, NCI (R01CA242020-01A1) (R.K.B.).
Footnotes
New genomic sequencing data have been deposited to cbioportal.org and are available for use at https://www.cbioportal.org/study/summary?id=mds_mskcc_2020. In addition, the RNA sequencing data have been uploaded to dbGAP.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.T., X.M., K.N., M.B., A.P., M.M.P., R.K.B., and O.A.-W. designed the study; J.T., M.B., T.L., K.K., M.H., D.W., A.T., M.M.P., and O.A.-W. provided patient samples; J.T., X.M., M.B., A.P., J.P., S.B., D.W., M.M.P., and O.A.-W. performed computational analyses of mutational data; K.N., A.P., and R.K.B. performed computational analyses of the RNA-seq data; X.M. and B.L. performed cloning and colony assays; S.H. and Y.L. performed the isothermal titration calorimetry assays; and J.T., K.N., X.M., A.P., R.K.B., and O.A.-W. wrote the manuscript with approval by all coauthors.
Conflict-of-interest disclosure: O.A.-W. has served as a consultant for H3B Biomedicine, Foundation Medicine Inc, Merck, and Janssen; is on the Scientific Advisory Board of Envisagenics Inc; and has received prior research funding from H3B Biomedicine that was unrelated to the current study. The remaining authors declare no competing financial interests.
Correspondence: Omar Abdel-Wahab, Memorial Sloan Kettering Cancer Center, 1275 York Ave, New York, NY 10065; e-mail: abdelwao@mskcc.org.
REFERENCES
- 1.Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64-69. [DOI] [PubMed] [Google Scholar]
- 2.Papaemmanuil E, Cazzola M, Boultwood J, et al. ; Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium . Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graubert TA, Shen D, Ding L, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2011;44(1):53-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ilagan JO, Ramakrishnan A, Hayes B, et al. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015;25(1):14-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darman RB, Seiler M, Agrawal AA, et al. Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell Rep. 2015;13(5):1033-1045. [DOI] [PubMed] [Google Scholar]
- 6.Kim E, Ilagan JO, Liang Y, et al. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell. 2015;27(5):617-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Madan V, Kanojia D, Li J, et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun. 2015;6(1):6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tyner JW, Tognon CE, Bottomly D, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562(7728):526-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papaemmanuil E, Gerstung M, Malcovati L, et al. ; Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium . Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616-3627, quiz 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Makishima H, Yoshizato T, Yoshida K, et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet. 2017;49(2):204-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee SC, North K, Kim E, et al. Synthetic lethal and convergent biological effects of cancer-associated spliceosomal gene mutations. Cancer Cell. 2018;34(2):225-241 e228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shiozawa Y, Malcovati L, Gallì A, et al. Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat Commun. 2018;9(1):3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bondu S, Alary AS, Lefèvre C, et al. A variant erythroferrone disrupts iron homeostasis in SF3B1-mutated myelodysplastic syndrome. Sci Transl Med. 2019;11(500):eaav5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pangallo J, Kiladjian JJ, Cassinat B, et al. Rare and private spliceosomal gene mutations drive partial, complete, and dual phenocopies of hotspot alterations. Blood. 2020;135(13):1032-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, Baran J, Cros A, et al. International Cancer Genome Consortium Data Portal--a one-stop shop for cancer genomics data. Database (Oxford). 2011;2011(0):bar026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLaren W, Gil L, Hunt SE, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17(1):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17(3):251-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients [published correction appears in Nat Med. 2017;23:1004]. Nat Med. 2017;23:703-713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang MT, Bhattarai TS, Schram AM, et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov. 2018;8(2):174-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Durham BH, Getta B, Dietrich S, et al. Genomic analysis of hairy cell leukemia identifies novel recurrent genetic alterations. Blood. 2017;130(14):1644-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pellegrino M, Sciambi A, Treusch S, et al. High-throughput single-cell DNA sequencing of acute myeloid leukemia tumors with droplet microfluidics. Genome Res. 2018;28(9):1345-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ley TJ, Miller C, Ding L, et al. ; Cancer Genome Atlas Research Network . Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia [published correction appears in N Engl J Med. 2013;369(1):98]. N Engl J Med. 2013;368(22):2059-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grinfeld J, Nangalia J, Baxter EJ, et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med. 2018;379(15):1416-1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morita K, Wang F, Jahn K, et al. Clonal evolution of acute myeloid leukemia revealed by high throughput single-cell genomics. bioRxiv. 2020. doi: 10.1101/2020.02.07.925743. [DOI] [PMC free article] [PubMed]
- 27.Yoshimi A, Lin KT, Wiseman DH, et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature. 2019;574(7777):273-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J, Lieu YK, Ali AM, et al. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci USA. 2015;112(34):E4726-E4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daubner GM, Cléry A, Jayne S, Stevenin J, Allain FH. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J. 2012;31(1):162-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fei DL, Motowski H, Chatrikhi R, et al. Wild-type U2AF1 antagonizes the splicing program characteristic of U2AF1-mutant tumors and is required for cell survival. PLoS Genet. 2016;12(10):e1006384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pacheco TR, Gomes AQ, Barbosa-Morais NL, et al. Diversity of vertebrate splicing factor U2AF35: identification of alternatively spliced U2AF1 mRNAS. J Biol Chem. 2004;279(26):27039-27049. [DOI] [PubMed] [Google Scholar]
- 32.Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee SC, Dvinge H, Kim E, et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins [published correction appears in Nat Med. 2016;22(6):692.]. Nat Med. 2016;22(6):672-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Q, Derti A, Ruddy D, et al. A chemical genetics approach for the functional assessment of novel cancer genes. Cancer Res. 2015;75(10):1949-1958. [DOI] [PubMed] [Google Scholar]
- 35.Seiler M, Peng S, Agrawal AA, et al. Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep. 2018;23(1):282-296 e284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Lawrence MS, Wan Y, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365(26):2497-2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dalton WB, Helmenstine E, Pieterse L, et al. The K666N mutation in SF3B1 is associated with increased progression of MDS and distinct RNA splicing. Blood Adv. 2020;4(7):1192-1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cretu C, Schmitzová J, Ponce-Salvatierra A, et al. Molecular architecture of SF3b and structural consequences of its cancer-related mutations. Mol Cell. 2016;64(2):307-319. [DOI] [PubMed] [Google Scholar]
- 39.Harding JJ, Lowery MA, Shih AH, et al. Isoform switching as a mechanism of acquired resistance to mutant isocitrate dehydrogenase inhibition. Cancer Discov. 2018;8(12):1540-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.