


Abstract
Transcription termination defines accurate transcript 3′-ends and ensures programmed transcriptomes, making it critical to life. However, transcription termination mechanisms remain largely unknown in Archaea. Here, we reported the physiological significance of the newly identified general transcription termination factor of Archaea, the ribonuclease aCPSF1, and elucidated its 3′-end cleavage triggered termination mechanism. The depletion of Mmp-aCPSF1 in Methanococcus maripaludis caused a genome-wide transcription termination defect and disordered transcriptome. Transcript-3′end-sequencing revealed that transcriptions primarily terminate downstream of a uridine-rich motif where Mmp-aCPSF1 performed an endoribonucleolytic cleavage, and the endoribonuclease activity was determined to be essential to the in vivo transcription termination. Co-immunoprecipitation and chromatin-immunoprecipitation detected interactions of Mmp-aCPSF1 with RNA polymerase and chromosome. Phylogenetic analysis revealed that the aCPSF1 orthologs are ubiquitously distributed among the archaeal phyla, and two aCPSF1 orthologs from Lokiarchaeota and Thaumarchaeota could replace Mmp-aCPSF1 to terminate transcription of M. maripaludis. Therefore, the aCPSF1 dependent termination mechanism could be widely employed in Archaea, including Lokiarchaeota belonging to Asgard Archaea, the postulated archaeal ancestor of Eukaryotes. Strikingly, aCPSF1-dependent archaeal transcription termination reported here exposes a similar 3′-cleavage mode as the eukaryotic RNA polymerase II termination, thus would shed lights on understanding the evolutionary linking between archaeal and eukaryotic termination machineries.
INTRODUCTION
Transcription, the fundamental biological process for transforming the genetic information from DNA to RNA, must be properly terminated (1–3). The proper terminating transcription is essential to (i) guarantee accurate transcript 3′-ends, (ii) prevent read-through resulted undesired increase of downstream gene or antisense transcriptions, (iii) recycle RNA polymerase (RNAP) and (iv) minimize collision of biomacromolecular machineries (2–4). Consequently, organisms have evolved precise mechanisms to govern transcription termination (2–5). Bacteria primarily employ Rho-dependent and -independent (intrinsic) termination mechanisms, with the former relying on an RNA translocase Rho to dissociate the transcription elongation complex (TEC), while the latter merely depends on a nascent RNA structure harboring a 7–8 base-paired hairpin followed by a run of uridine residues (2,3,6,7). In eukaryotes, RNAP II, which transcribes mRNAs and non-coding RNAs, employs multifaceted termination strategies but usually with an involvement of transcript 3′-end processing event. In this event, the poly(A) site, one of the 3′-end processing/termination signals in nascent RNA, is recognized by a cleavage and polyadenylation complex (CPF/CPSF); thereafter, an endoribonuclease in CPF/CPSF, the human CPSF73 or the yeast Ysh1, cleaves the RNA downstream. This processing event is followed by polyadenylation for mRNA maturation, and triggers RNAP II dissociation for transcription termination (3,5). Additionally, efficient dismantling RNAP II requires other termination factors (3,5,8–12).
However, the transcription termination mechanisms in the third life form, Archaea, remain largely unknown (1,13). Archaea represent a primary domain of cellular life that consists of highly diverse prokaryotic phyla, and phylogenetically are closer to eukaryotes (14–16). Specifically, Archaea employ an eukaryotic RNAP II homolog, the archaeal RNAP (aRNAP) (17), but have compact genomes with short intergenic-regions (IGRs) and usually co-transcribed polycistronic operons. Therefore, transcription in Archaea must be properly terminated to prevent readthrough resulted transcription interference of the downstream gene. Earlier studies on individual genes have revealed that Archaea possess a bacteria-like but distinct intrinsic termination mode, by depending merely on a short uridine-rich sequence but not strictly requiring an upstream hairpin structure (1,18–22). Recently, the genome-level 3′-ends sequencing (Term-seq) analyses on three representative archaeal species, Methanosarcina mazei, Sulfolobus acidocaldarius and Haloferax volcanii, have identified similar over-representative uridine-rich tracts and without preceding hairpin structures in the transcripts 3′-ends (23,24). Term-seq also identified the eukaryotic-like alternative 3′-untranslated regions (3′-UTRs) in Archaea that are generated by consecutive termination sites, implying that Archaea could employ eukaryotic-like transcription termination modes and may also employ trans-acting (termination) factors (13). Eta has been reported to release stalled TECs from damaged DNA sites in Thermococcus kodakarensis (25), however it functions specially in response to DNA damages (1,25,26). Very recently, FttA, an archaeal ribonuclease aCPSF1 from T. kodakarensis, was reported in mediating transcription termination on a cytidine-rich sequence in an in vitro system (27), but the physiological significances of aCPSF1 mediated termination and the working mechanisms remain largely unknown.
In the present work, we report that the aCPSF1 ortholog, Mmp-aCPSF1 of Methanococcus maripaludis, depending on its ribonuclease activity, triggers a genome-wide transcription termination and ensures programmed transcriptome and optimal growth, so identifying aCPSF1 as a general transcription termination factor of Archaea. A phylogenetic analysis reveals that aCPSF1 is ubiquitously distributed in the archaeal phyla, and two orthologs from distant related Lokiarchaeota and Thaumarchaeota were experimentally verified to implement transcription termination in Methanococcus. Thus, the aCPSF1 dependent transcription termination could be widely employed in Archaea. Noticeably, aCPSF1 mediates the archaeal transcription termination via a transcript 3′-end cleavage mode, resembling its eukaryotic orthologs (yeast Ysh1 and human CPSF73) in eukaryotic RNAP II transcription termination.
MATERIALS AND METHODS
Strains, plasmids and culture conditions
Strains and plasmids used in this study are listed in Supplementary Table S1. M. maripaludis S2 and its derivatives were grown in pre-reduced McF medium under a gas phase of N2/CO2 (80:20) at 37 and 22°C as previously described (28), and 1.5% agar was used in the solid medium. Puromycin (2.5 μg/ml), neomycin (1.0 mg/ml) and 8-azahypoxanthine (0.25 mg/ml) were used for genetic modification selections unless indicated otherwise. Escherichia coli DH5α, BL21(DE3)pLysS and BW25113 were grown at 37°C in Luria-Bertani (LB) broth and supplemented with ampicillin (100 μg/ml) or streptomycin (50 μg/ml) when needed.
Construction of the Mmp-aCPSF1 gene depletion and complementary strains
Given the possible essentiality of Mmp-aCPSF1 (MMP0694) determined in M. maripaludis (29) and repeated failures in deletion of Mmp-aCPSF1, the Mmp-aCPSF1 expression depleted strain (▽aCPSF1) was constructed using a TetR-tetO repressor-operator system as follows.
First, a tetracycline responsive regulator (tetR) and operator (tetO) cassette from Bacillus subtilis was fused upstream to the N-terminal His6-tagged Mmp-aCPSF1, and integrated at the hpt locus of M. maripaludis S2 to construct a tetracycline regulated Mmp-aCPSF1 overexpression strain tetO-aCPSF1 by 8-azahypoxanthine sensitivity selection. To achieve gene expression in M. maripaludis, the promoter (Pmcr) and terminator (Tmcr) of the methyl-CoM reductase were respectively fused to the tetR gene at the 5′- and 3′-termini and inserted to pMD19-T (TAKARA) to obtain pMD19-T-Pmcr-tetR-Tmcr (Supplementary Table S1). The tetO operator was positioned downstream the nitrogen fixation gene nif promoter (Pnif) and fused upstream the amplified Mmp-aCPSF1 ORF to obtain a tetO fused Mmp-aCPSF1 fragment Pnif-tetO-His6-aCPSF1. For homologous recombination at the hpt site, two DNA fragments, each of approximately 800-bp respectively from the upstream and downstream hpt, was amplified from the S2 genomic DNA and fused with the Pmcr-tetR-Tmcr and Pnif-tetO-His6-aCPSF1 fragment at each end to obtain the plasmid of ptetR/tetO-His6-aCPSF1 (Supplementary Table S1) via the stepwise Gibson assembly using ClonExpress MultiS One Step Cloning Kit (Vazyme). The PCR fragment amplified from the plasmid ptetR/tetO-His6-aCPSF1 using primers Hptup-F/Hptdown-R (Supplementary Table S2) was transformed into S2 using the polyethylene glycol (PEG)-mediated transformation approach (30) to generate a tetracycline regulated Mmp-aCPSF1 overexpression strain (tetO-aCPSF1) via double cross homologous recombination. Transformants were selected by plating on McF agar containing 8-azahypoxanthine as described previously (31), and confirmed via PCR amplification using primers Hptup-F/Hptdown-R.
Next, an in-frame deletion of the indigenous Mmp-aCPSF1 gene was implemented in strain tetO-aCPSF1. Two ∼800-bp DNA fragments, each from the upstream and downstream the Mmp-aCPSF1 gene was PCR amplified and fused to the puromycin-resistance cassette pac gene amplified from the plasmid pIJA03 at each end to generate the plasmid pMD19T-ΔaCPSF1 via the stepwise Gibson assembly. The DNA fragment of aCPSF1up-pac-aCPSF1down amplified from pMD19T-ΔaCPSF1 was transformed into strain tetO-aCPSF1 to knockout the indigenous Mmp-aCPSF1. Transformants were selected on McF agar containing puromycin, and the Mmp-aCPSF1 gene deletion was validated by PCR amplification. Thus, a tetracycline regulated Mmp-aCPSF1 expression strain ▽aCPSF1 was obtained.
To obtain various ▽aCPSF1 complementary strains, the respective aCPSF1 orthologs or mutated gene were cloned into the XhoI/Bgl II sites of the replicative plasmid pMEV2 to produce pMEV2-aCPSF1(wt)/pMEV2-aCPSF1(mu) carrying the wild-type Mmp-aCPSF1 and its mutant, H243A/H246A, respectively, and pMEV2-Lokiarch44440/pMEV2-CENSYa1545, carrying two codon optimized Mmp-aCPSF1 orthologs from Ca. Lokiarchaeum sp. GC14_75 and Ca. Cenarchaeum symbiosum. The constructed plasmids were each transformed into ▽aCPSF1 via the PEG-mediated transformation approach to produce the complementary strains.
Construction of the in-frame deletion mutants of earA in strains S2 and ▽aCPSF1
The in-frame deletion mutants of earA were constructed similarly as above. Two DNA fragments, each of ∼800-bp respectively from the upstream and downstream earA (MMP1718), were amplified and fused to a neomycin-resistance cassette amplified from the plasmid pMEV2 at each end. The resulting earAup-neo-earAdown fragment was transformed into strains S2 and ▽aCPSF1, and via double cross homologous recombination, the earA deletion mutants ΔearA and ▽aCPSF1ΔearA (Supplementary Table S1) were obtained, respectively.
Construction of Flag-tagged Mmp-aCPSF1 and His-Flag-tagged Mmp-RpoL fusion strains
3 × Flag or His6 and 3 × Flag tags were firstly fused to the C termini of Mmp-aCPSF1 and Mmp-RpoL via PCR amplification and cloned into pMD19-T to construct the plasmids pMD19-T-aCPSF1–3Flag and pMD19-T-rpoL-3Flag-His6, respectively. The puromycin or neomycin cassette in the plasmid pIJA03 or pMEV2 was amplified and overlapping fused to the 800-bp fragment downstream of Mmp-aCPSF1 or Mmp-RpoL. The overlapped PCR fragments were then inserted into downstream of Mmp-aCPSF1–3Flag or Mmp-rpoL-3Flag-His6 to obtain pMD19-T-aCPSF1–3Flag-pac and pMD19-T-rpoL-3Flag-His6-neo (Supplementary Table S1), respectively. Next, DNA fragments were amplified from pMD19-T-aCPSF1–3Flag-pac and pMD19-T-rpoL-3Flag-His6-neo using primers aCPSF1Flag-F/aCPSF1Flagdw-R and RpoLFlag-F/RpoLFlagdw-R, respectively, and transformed into M. maripaludis S2 to obtain Flag-tagged strains of Mmp-aCPSF1-F and Mmp-RpoL-HF, respectively.
Extraction of genomic DNA and total RNA
The genomic DNA of M. maripaludis S2 and its derivatives was extracted and purified from the mid-exponential 37°C-grown cultures (OD600 = 0.5) using TIANamp Bacteria DNA Kit (TIANGEN Biotech, Beijing, China) by following the protocol instruction. Total RNA used for RNA-seq, Term-seq, northern blotting, and rapid amplification of cDNA 3′-ends was extracted from the mid-exponential cultures of 22°C-grown strains S2, Mmp-com(Mmp-C1), Mmp-com(Loki-C1) and Mmp-com(Csy-C1) (OD600 = 0.45), strains ▽aCPSF1 and Mmp-com(Mmp-C1mu)) (OD600 = 0.36) and 37°C-grown strains S2 and ▽aCPSF1 (OD600 = 0.5) using TRIzolTM reagent (Invitrogen). Quantification and qualification of the purified RNA were determined using the NanoPhotometer spectrophotometer (IMPLEN, CA, USA), Qubit 2.0 Flurometer (Life Technologies, CA, USA) and Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA).
RNA-seq and data analysis
Using the methods described previously (32), high-throughput strand-specific RNA-seq was performed for differential transcriptome analysis between strains S2 and ▽aCPSF1 with three biological replicates. The library quality of the RNA samples was assessed on an Agilent Bioanalyzer 2100 system. Library construction and Illumina sequencing was performed at Novogene Bioinformatics Technology Co., Ltd (Beijing, China). rRNA was depleted using Ribo-Zero™ rRNA Removal Kit (Epicentre). The raw RNA-seq reads were first subjected to adapter sequence trimming and then QC preprocessing to discard those meeting any of the following thresholds: truncated reads of ≤1/3 length of the original ones, reads containing ≥10% uncalled bases (Ns), or ≥50% low-quality bases (PHRED quality scores ≤ 5). Next, the QC filtered reads were mapped to the reference genome of M. maripaludis S2 (GCF_000011585) using Bowtie2 (v2.2.3) (-x refer.fa -1 1.clean.fq -2 2.clean.fq –fr –no-unal -S mapped.sam) (33) after removing the reads aligned to the reference rRNAs (Supplementary Table S3). HTSeq (v0.6.1) (-m union -s reverse -f bam -t exon) (34) was used to count the read numbers mapped to each gene. Differential expression analysis between strain S2 and ▽aCPSF1 was performed on un-normalized read counts using the DESeq2 (35) (v1.20.0) algorithm, and differentially expressed genes were defined for those having the Padj, an adjusted P-value, <0.05. The FPKM of each gene was calculated based on the fragment numbers per kilobase of transcript sequence per millions base pairs sequenced that mapped to the gene (36).
To calculate the genome-wide transcriptional read-through index (TRTindex), transcriptional units (TUs) were first predicted using Rockhopper (v2.0.3) (37) on the transcriptomic data of the wild-type strain S2. Intergenic regions (IGRs) were defined as the DNA region between the transcription termination site (TTS) of a TU to the transcription start site (TSS) of the tandem downstream TU on the same strand. Transcript abundances (FPKMs) of each TU and IGR were determined by HISAT2 (v2.0.5) (38) using parameters (–no-spliced –rna-strandness RF) and quantified using StringTie (v1.3.3) (39) with –e option, and the predicted TU and IGR regions were transformed to the GTF file format used as –G parameter in StringTie (40). TRTindex was calculated using the four criteria shown in Figure 2e and listed in Supplementary Dataset S1.
Figure 2.

The depletion of Mmp-aCPSF1 causes a genome-wide transcription read-through (TRT). (A, B) The strand-specific RNA-seq mapping profiles (left) show 3′-end extensions (dot magenta brackets) of MMP1100 (A) and MMP1147 (B) in ▽aCPSF1 (red) referenced to that in strain S2 (blue). Numbers on top indicate the genomic sites, and bullets show genes. Northern blot (middle) assayed TRTs using the respective probes (horizontal sticks in A), and 23S rRNA was used as an internal control. 3′RACE (right) assayed transcription termination sites (TTSs) and TRTs. (C) Pair-wise comparison of genome-wide Log2 FPKM ratios of transcription units (TUs) and the intergenic regions (IGRs) between strain S2 and ▽aCPSF1. The beneath ruler indicates the genomic location. (D) A metaplot diagram shows the average reads mapping pattern of TUs and the flanking IGRs in S2 and ▽aCPSF1 based on normalization of 750 TUs that have an IGR length >100 nt. (E) A flowchart depicts TRT identification and TRTindex calculation. (F) A diagram shows percentages of each TRT type occurred in ▽aCPSF1. Bent arrows and horizontal blue lines indicate TSS and transcript lengths in strain S2, respectively, and dot magenta arrows indicate transcripts that occur TRT in ▽aCPSF1. *, TU numbers occurring TRTs. (G) Boxplots show the FPKM fold changes of Type II TRT in (F) for the upstream TUs that generate TRTs (TU (Up)) and the tandem downstream TUs (TU (Down)), respectively.
Term-seq and data analysis
Term-seq libraries were constructed using the procedures described previously (23). Sequencing was performed on an Illumina HiSeq X-ten system with 150-bp paired-end reads. Raw Term-seq data were filtered using the similar QC steps as described above. Then, QC filtered reads (Supplementary Table S5) were aligned to the reference genome of M. maripaludis S2 (GCF_000011585) using Bowtie2 (33). The read counts of the 3′-ends mapped to each genomic position in strain S2 were recorded. The primary TTSs were assigned based on four criteria: (i) within 200 nt downstream the stop codon of a gene; (ii) >1.1 read ratio of -1 site (predicted TTS) to +1 site (1 nt downstream TTS); (iii) read-counts of −1 site minus +1 site > 5; (iv) upon (ii) and (iii) satisfied, the site that had the highest read count difference of −1 site minus +1 site and also occurred in the two biological replicates (Supplementary Dataset S2). Using WebLogo(v2.8.2) (41), the terminator logos were created on the sequences (Supplementary Dataset S2) flanking Term-seq predicted TTS. To compare the read differences flanking the primary TTSs in strains S2 and ▽aCPSF1, a metaplot analysis was performed on the average reads of each 20 nt upstream and downstream of all the primary TTSs, which were normalized by dividing the individual average read of each site from −21 to +20 by that of -21 site shown in Figure 4A.
Figure 4.
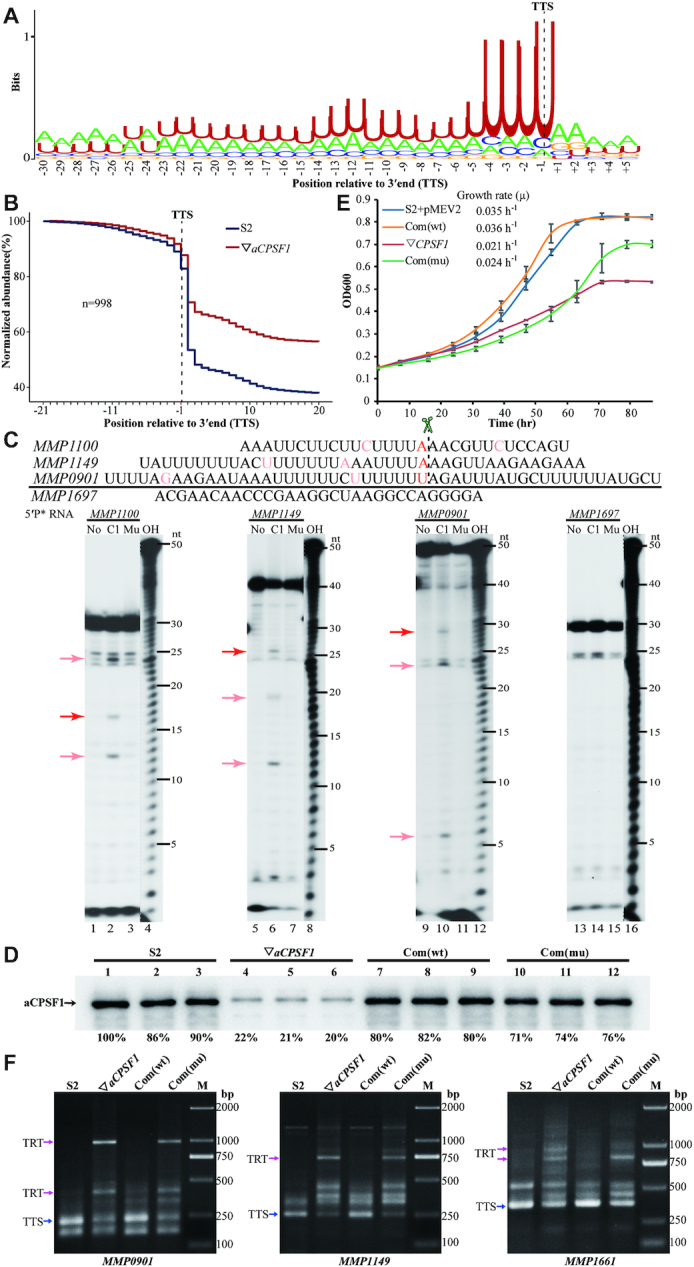
The terminator motif of M. maripaludis determined by Term-seq, and the ribonuclease activity of Mmp-aCPSF1 on the motif is essential to transcription termination. (A) Using WebLogo (v2.8.2) (41), a logo representation was generated for the sequence motif upstream of 998 primary TTSs that are defined by Term-seq. (B) A metaplot diagram shows the average reads of each 20 nt upstream and downstream of the 998 primary TTSs (dot black line) in 22°C-cultured S2 (blue line) and ▽aCPSF1 (red line). (C) The ribonuclease activity of Mmp-aCPSF1 was assayed on three representative uridine-rich RNAs derived from the TTSs (red bases) flanking sequences of MMP1100, MMP1149 and MMP0901 (upper). A uridine tract-less RNA fragment from MMP1697 was used as a control. A urea sequencing gel displays the enzymatic products (lower). No, C1, and Mu indicate the assays without, and with addition of Mmp-aCPSF1 and the catalytic inactive mutant (H243A/H246A), respectively. Red and pink arrows point to the presumed cleavage products at TTS and other sites downstream uridine-rich sequences. OH, a hydroxyl ladder indicates migrations of RNA products. (D) Western blot assays the Mmp-aCPSF1 protein abundances in three batches of 22°C-grown strains S2, ▽aCPSF1, Mmp-com(Mmp-C1) (Com(wt)) and Mmp-com(Mmp-C1mu) (Com(mu)). (E) Growth curves of the four strains were assayed on three batches of the 22°C-grown cultures, and the averages and standard deviations are shown. (F) 3′RACE assays the TRTs in 22°C-grown strains S2, ▽aCPSF1, Com(wt) and Com(mu). Blue and magenta arrows indicate the PCR products of normal terminations (TTSs) and TRTs, respectively. M, a DNA ladder indicates migrations of the PCR products.
Rapid amplification of cDNA 3′-ends (3′RACE)
3′RACE was performed as described previously (42). A total of 20 μg of total RNA were ligated with 50 pmol 3′RACE Linker (5′-rAppCTGTAGGCACCATCAAT–NH2–3′; NEB) through a 16 h incubation at 16°C with 20 U T4 RNA ligase (Ambion). The 3′ linker-ligated RNA was recovered by isopropanol precipitation and one aliquot of 2 μg was mixed with 100 pmol 3′R-RT-P (5′-ATTGATGGTGCCTACAG-3′, complementary to the 3′RACE linker) by incubation at 65°C for 10 min and on ice for 2 min. Then, it was used in the reverse transcription (RT) reaction using 200 U SuperScript III reverse transcriptase (Invitrogen). After RT, nested PCR was conducted using primers (Supplementary Table S2) that target regions of <200 nt upstream the termination codons to obtain gene-specific products. Specific PCR products were excised from 2% agarose gel and cloned into pMD19-T (TaKaRa) and then sequenced. The 3′-end of a TU is defined as the nucleotide linked to the 3′RACE-linker.
Protein purification
For purification of the Mmp-aCPSF1 protein, a His-tagged SUMO-aCPSF1 fusion was first constructed and cloned into pSB1s using the ClonExpress MultiS One Step Cloning Kit. Plasmid pSB1s that carries His-SUMO-aCPSF1 was then transformed into E. coli BW25113 and the transformants were induced by 0.1% l-arabinose. After a 16-h induction at 22°C, the cells were harvested, and the protein was purified as described previously (43). Next, His-SUMO tags were removed by incubating with His-tagged Ulp1 protease at 30°C for 2 h, and the Mmp-aCPSF1 protein was purified through a His-Trap HP column to remove His-tagged SUMO and His-tagged Ulp1. The Rpo D/L protein of M. maripaludis S2 (Mmp-aRpoD/L) was overexpressed and purified as described previously (44). In brief, the ORFs of Mmp-rpoD (MMP1322) and Mmp-rpoL (MMP0262) were stepwise cloned into pGEX-4T-1 to obtain the plasmid pGEX-4T-1-rpoD/L, and transformed into E. coli BL21(DE3)pLysS for overexpression. Purified proteins were detected by SDS-PAGE, and the protein concentration was determined using a BCA protein assay kit (Thermo Scientific).
Nuclease activity assay
Four RNA fragments containing the termination sites and the flanking sequences of MMP1100, MMP1149, MMP0901 and MMP1697 were synthesized by Genscript (China). They were 5′-end labeled with [γ-32P] ATP (PerkinElmer) using T4 polynucleotide kinase (Thermo Scientific). A 10 μl nuclease reaction mixture contained 20 μM wild-type Mmp-aCPSF1 protein or its catalytic inactive mutant, 5 nM 5′-[γ-32P] labeled RNA, 20 mM HEPES, (pH 7.5), 150 mM NaCl, 5 mM MgCl2 and 5% (w/v) glycerol. Nucleolytic reactions were initiated by the addition of the protein at 37°C for 90 min and stopped by 15 min incubation with 10 μg/ml Proteinase K (Ambion) at 55°C. The reaction products were then mixed with formamide-containing dye and analyzed on 10% sequencing urea-PAGE. A nucleotide ladder was generated by alkaline hydrolysis of the labeled RNA substrates. The urea-PAGE gels were analyzed by autoradiography with X-ray film.
Western blot assay
Western blot was performed to determine the cellular Mmp-aCPSF1 or Mmp-Rpo D/L protein abundances in various genetic modified strains. A polyclonal rabbit antiserum against the purified Mmp-aCPSF1 or Mmp-Rpo D/L protein was raised by MBL International Corporation, respectively. The mid-exponential cells of M. maripaludis were harvested and resuspended in a lysis buffer [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 10 (w/v) glycerol, 0.05% (v/v) NP-40], and lysed by sonication. The cell lysate was centrifuged and proteins in the supernatant were separated on 12% SDS-PAGE and then transferred to a nitrocellulose membrane. The antisera of anti-Mmp-aCPSF1 (1: 8000) and anti-Mmp-aRpoD/L (1: 20 000) were diluted and used respectively, and a horseradish peroxidase (HRP)-linked secondary conjugate at 1:5000 dilutions was used for immunoreaction with the anti-Mmp-aCPSF1 or anti-Mmp-aRpoD/L antiserum. Immune-active bands were visualized by an Amersham ECL Prime Western blot detection reagent (GE Healthcare). Protein quantification was performed using Quantity One (Bio-Rad).
Northern blot and mRNA half-life analysis
Northern blot was performed to determine various cellular RNA species and contents as described previously (45). To measure the half-life of a specific transcript, a final concentration of 100 μg/ml actinomycin D (actD, MP Biomedicals) was added into the exponential cultures of M. maripaludis to stop transcription. At 0, 5, 10, and 15 min post-addition of actD, each 3 ml culture was collected and the RNA was isolated. The RNA content in each sample was quantified by northern blot using the corresponding labeled DNA probe. A linear regression plot based on the average RNA contents from three batches of culture plotted against the sampling time was used for calculation of the half-life of decay (t1/2) that was the time point when 50% RNA content was retained.
To determine the bulk mRNA half-life, M. maripaludis was cultured to the exponential phase, and 30 μCi [5,6–3H]-Uridine (PerkinElmer) was added to isotope label the nascent transcripts. After a 10 min-labeling, actD was added and each 600 μl of the culture were collected at 0, 2, 4, 6, 8 and 10 min post-addition of actD as described above. Then, 67 μl of 100% cold trichloroacetic acid (TCA) was added to precipitate the total RNA, and the mixture was incubated on ice for >30 min. A sample of 60 min post-addition of act D was used for counting the stable RNAs. RNA in each TCA mixture was precipitated by centrifuge at 12 000 g for 10 min and dissolved in 700 μl of Ecoscint A (National Diagnostics) for 3H isotope counting using liquid scintillator detector (PerkinElmer). The stable RNA 3H counts were first deducted from the total 3H ones at each sampling time point and the resultant count at 0 min post-addition of act D was recorded as 100% mRNA, and the residual mRNA content at each time point was plotted against the sampling time. The bulk mRNA half-life was calculated as described above.
Electronic microscopy and motility assay
The mid-exponential cells of M. maripaludis strains S2 and ▽aCPSF1 grown at 22°C or 37°C were loaded onto carbon-Formvar-coated copper grids and ddH2O washed, and stained with uranyl acetate. Cells and archaella were observed under an LKB-V Ultratome (Jel1400) transmission electronic microscope. For motility assay, cells were collected from a 5-ml stationary phase culture grown at 22°C or 37°C, and then resuspended gently in 200 μl of McF medium. 10 μl of cell suspension were inoculated in the semi-solid McF medium containing 0.25% (w/v) agar inside an anaerobic chamber. Plates were incubated at 22°C for 15 or 20 days or 37°C for 2 days in an anaerobic jar with Oxoid AnaeroGen (Thermo Scientific) to remove the oxygen.
Size exclusion chromatography
The exponential cells of M. maripaludis S2 were harvested and resuspended in the lysis buffer (50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 0.05% NP-40 and 10% (v/v) glycerol), and then lysed by sonication and centrifuged at 12 000g for 30 min at 4°C. The supernatant was incubated at 37°C for 30 min with and without treatment of DNase I (100 U), RNase A (100 U) or Benzonase Nuclease (100 U). A total of 500 μg of the pretreated supernatant was fractionated through the Superdex 200 10/300 GL column (GE Healthcare) and the molecular sizes of each fraction was estimated by gel filtration molecular weight markers (Kit No.: MWGF1000 of Sigma-Aldrich). Finally, each fraction was collected for protein identification by western blot.
Co-immunoprecipitation and tandem affinity chromatography (TAP)
The association of Mmp-aCPSF1 with Mmp-aRNAP was detected by co-immunoprecipitation and tandem affinity chromatography. To perform the anti-Flag co-immunoprecipitation, prewashed anti-FLAG M2 magnetic beads (Sigma) was added to 5 mg of pretreated cell extract and incubated 8 h at 4°C with gently shaking. The antigen bound magnetic beads were washed five times with the lysis buffer and eluted by 2.5 volumes of 3× FLAG peptide (150 ng/μl, Sigma) for 3 h at 4°C with gently shaking. For the His-tag affinity chromatography, about 20 mg of cell extract were purified through a His-Trap HP column as described above. For TAP, about 20 mg of cell extract were first purified by His-tagged affinity chromatography, and the product was concentrated and imidazole removed using Amicon Ultrafra-30 concentrators (Millipore). The concentrated product was then immunoprecipitated using the same procedure as for anti-Flag co-immunoprecipitation. All of the CoIP-, His-tagged affinity chromatography- and TAP-products were separated on a 12% SDS-PAGE gel and identified by Western blot.
Chromatin Immunoprecipitation (ChIP) coupled quantitative PCR (ChIP-qPCR)
To obtain the chromosomal occupancy of Mmp-aCPSF1 and Mmp-aRpoL, ChIP assays were performed for strains Mmp-aCPSF1-F and Mmp-aRpoL-HF as described previously (46–48) with some modifications. Cells of M. maripaludis S2 were used as mock controls. About 0.5–1 × 109 middle exponential cells ml−1 were formaldehyde fixed and lysed. The chromosomal DNA in the lysed supernatant was sonicated to an average size of 200–500 bp using Bioruptor UCD300 (Diagenode). After overnight incubation with anti-FLAG M2 Magnetic Beads (Sigma) at 4°C, the beads were washed and the DNA-protein complexes were eluted by the ChIP elution buffer (10 mM Tris–HCl (pH 8.0), 1 mM EDTA, 1% SDS) at 65°C for 30 min with shaking. Reverse crosslinking was then performed by a treatment of 0.05 mg/ml RNase A and then 0.5 mg/ml proteinase K at 37°C for 2–4 h and followed by overnight incubation at 65°C. DNA fragments were purified using MiniElute columns (Qiagen) and quantified using the Qubit dsDNA HS kit (Life Technologies).
PCR and qPCR were performed to determine the DNA species and the enrichment by Mmp-aCPSF1 or Mmp-aRpoL as described previously (47,49). For PCR amplification, each 1 μl of input and IP or mock-IP DNA sample was used in a 25 μl reaction mix containing 400 nM of the corresponding oligonucleotide primer (Supplementary Table S2). For qPCR analysis, a similar reaction mixture of PCR except for 1× SYBR Green Real-time PCR Master Mix (Toyobo, Tokyo, Japan) was used and performed at Mastercycler EP realplex2 (Eppendorf, Germany). Enrichment folds between Mmp-aCPSF1 or Mmp-aRpoL and mork-IP samples were calculated as: ΔCt value (normalized to the input samples) for each sample was calculated according to the equation ΔCt = [Ct (sample) – adjusted Ct (input)]. Then, ΔΔCt was calculated through the equation ΔΔCt = (ΔCt (experimental sample) – ΔCt (mock sample)). Finally, the enrichment fold between experimental sample and mock sample was calculated using 2(–ΔΔCt). ChIP assays were performed for three biological triplicates, and PCR and qPCR were assayed for technical triplicates of each biological repeat.
Uridine-rich tracts searching
For searching uridine-rich tracts in the selected representative archaeal genomes (Supplementary Dataset S3), we downloaded the reference genomes and annotation files from the NCBI database (https://www.ncbi.nlm.nih.gov/search/). Samtools (version1.9) (50) faidx option was used to retrieve the sequence in length of 200 nt upstream and downstream the stop codons of the putative ORFs in the archaeal genomes, and then the U-rich tracts with successive five, or six or seven uridine bases (U5, U6 and U7) within the sequence were counted using an in-house script.
RESULTS
Depleted expression of Mmp-aCPSF1 causes growth reduction and a disordered transcriptome
The archaeal CPSF1 affiliates with the β-CASP family of ribonucleases, and the endoribonuclease activities were identified in three aCPSF1 orthologs (51–53), with one also exhibiting 5′–3′ exoribonuclease activity (52) via the in vitro assays. The aCPSF1 orthologs are universally distributed in all detected archaeal phyla (52), and Mmp-aCPSF1 was determined to be an essential gene in M. maripaludis by Sarmiento et al. (29) and by our repeated failures in deletion of the gene as well, highlighting the fundamental roles of aCPSF1 in Archaea. To investigate the physiological functions of aCPSF1, the Mmp-aCPSF1 expression depleted strain (▽aCPSF1) was constructed in M. maripaludis by using the TetR-tetO repressor-operator system (Supplementary Figure S1A). Compared with the wild-type strain S2, only 60% and 20% of the Mmp-aCPSF1 protein abundances were determined in the 37°C- and 22°C-grown ▽aCPSF1 (minus tetracycline), respectively, verifying a successful depletion of Mmp-aCPSF1 (Supplementary Figure S1B and Figure 1A). Although having a similar growth rate as the wild-type strain S2 at 37°C (Supplementary Figure S1C), ▽aCPSF1 exhibited markedly reduced growth at 22°C (μ = 0.035 h−1 versus 0.06 h−1 of S2, Figure 1B), corresponding to the three-fold lower protein abundance of Mmp-aCPSF1 in 22°C- than in 37°C-grown ▽aCPSF1. Concomitantly, an ∼2-fold longer bulk mRNA half-life was determined in the 22°C-cultured ▽aCPSF1 than that of S2 (8.4 min versus 4.7 min, Figure 1C), suggesting that aCPSF1 could have an effect on archaeal mRNA turnover.
Figure 1.

Depleted expression of Mmp-aCPSF1 results in reduced growth, prolonged RNA lifespan, and a disordered transcriptome of 22°C-cultured M. maripaludis. (A) Western blot assayed Mmp-aCPSF1 protein abundance (percentages referenced to lane 1) with (+) or without (–) 100 μg/ml of tetracycline in 22°C-grown S2 (wild-type), tetO-aCPSF1, and ▽aCPSF1. Averages and standard deviations of three replicates are shown. aCPSF1 and aCPSF1-His6 flanking the gel point the indigenous- and hpt-site inserted His6-Mmp-aCPSF1, respectively. The schematic depicting the construction of tetO-aCPSF1 and ▽aCPSF1 was shown in Supplementary Figure S1A. (B, C) Depletion of Mmp-aCPSF1 reduced 22°C-growth (B) and prolonged the bulk mRNA half-lives (C). Three batches of cultures of strains S2 and ▽aCPSF1 with or without tetracycline (Tc) were measured, and the averages and standard deviations are shown. Bulk mRNA half-lives were determined by quantifying the [3H]-uridine signal attenuation. (D) Hierarchical clustering (left) and functional category (right) analysis of the differential transcribed genes in three batches of 22°C-cultures of S2 and ▽aCPSF1. Heat plot representation of the differential expression ratio (log2) is shown with color intensity. Green and red represent the minima and maxima fold, respectively. The functional category enrichment ratio (a bar with gradient red intensity at lower right corner) was percentages of the up (orange)- and down (green)-regulated gene numbers (horizontal axis) in total genes of each arCOG categories (Supplementary Table S4). The significance statistical analysis was analyzed by Fisher's exact test with Benjamini-Hochberg multiple-testing correction to calculate P-values for each function category, and ** and * respectively mark the significant enriched categories with P < 0.01 and < 0.05 (Supplementary Table S4). (E) Hierarchical grouping of the differential transcribed genes caused by Mmp-aCPSF1 depletion. Based on the FPKMs in strain S2, transcripts were grouped into five ranks (<42, 43–103, 104–214, 215–573 and >573 of FPKMs), and then the up- and down-regulated and unchanged transcript numbers in each rank in ▽aCPSF1 were counted. (F) According to the gene essentiality in M. maripaludis defined by Sarmiento et al. (29), percentages of essential and non-essential genes falling in the up- and down-regulated and unchanged groups in ▽aCPSF1 were calculated, respectively.
Differential transcriptomic analysis based on strand-specific RNA-seq analysis revealed that the depletion of Mmp-aCPSF1 had changed transcriptions of about 69% genes (1188) in 22°C-grown cultures with each half up- and down-regulated (Figure 1D and Supplementary Dataset S4). Among the up-regulated genes, those belonging to the arCOGs of cell motility and transcription were significantly enriched, while the down-regulated genes fell mainly into the arCOGs of energy production and amino acid metabolism, conforming to the reduced growth of ▽aCPSF1. By hierarchically grouping all transcripts into five ranks based on their FPKMs in strain S2, we found that the transcript abundances of the highly and lowly expressed ranks were reversely changed in ▽aCPSF1, namely that the highly expressed genes were generally down-regulated, while the poorly expressed ones were mostly up-regulated (Figure 1E), behaving like ‘robbing the rich to feed the poor’ caused by Mmp-aCPSF1 depletion. In addition, about 40% of Sarmiento et al. (29) defined essential genes decreased expression and only 20% increased expression in ▽aCPSF1 (Figure 1F). This indicates that Mmp-aCPSF1 plays a role in the control of the ordered transcriptome.
Mmp-aCPSF1 depletion results in a genome-wide transcription read-through
Strikingly, by querying the strand-specific RNA-seq mapping files, we observed pervasive prolonged transcript 3′-ends, i.e. transcription read-throughs (TRTs) in ▽aCPSF1. Such as MMP1100, a predicated master regulator for methanogenesis (54), its 3′-end was markedly extended and multiple-reads mapped, and so resulted in an antisense transcription of MMP1099 on the opposite strand (Figure 2A, left); the 3′-end of the upstream MMP1147 was extended into the downstream MMP1146 on the same strand as well (Figure 2B, left). Northern blot (Figure 2A, B and Supplementary Figure S2, middle) and rapid amplification of the cDNA 3′-ends (3′RACE) (Figure 2 A, B and Supplementary Figure S2 right) verified the TRTs in ▽aCPSF1. The M. maripaludis genome comprises 1722 coding genes that are organized into 1079 predicted transcription units (TUs) with a median 200 bp of IGRs (Supplementary Dataset S1). A global pair-wise comparison was performed for the FPKMs of a TU and its IGR in S2 and ▽aCPSF1 and a metaplot diagram was visualized through normalizing those of 750 TUs, which displayed a genome-wide FPKM increase in IGRs but reduction in TU bodies upon aCPSF1 depletion (Figure 2C, D and Supplementary Dataset S1). Further statistical analysis on 1079 TUs in ▽aCPSF1 and S2 yielded higher folds of elevated transcription in IGRs (median: 4.36-fold) than in TU bodies (median: 1.21-fold) (Supplementary Figure S3). Thus, the Mmp-aCPSF1 depletion has caused a genome-wide TRT.
TRTs pervasively elevate expressions of the co-directional downstream genes and result in exuberant archaella along with enhanced motility of M. maripaludis
Next, a pipeline (Figure 2E) was developed to define the Mmp-aCPSF1 depletion caused TRTs via four criteria: (i) prediction of TUs based on the transcriptomic data of strain S2; (ii) delineation of an IGR from the upstream transcription termination site (TTS) to the downstream transcription start site (TSS) of TUs on the same DNA strand; (iii) calculation of TRTindex using the following equation,
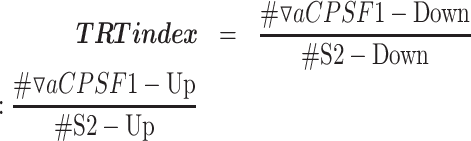 |
where #▽aCPSF1- and #S2-Down indicate the IGR FPKMs in ▽aCPSF1 and S2, respectively; #▽aCPSF1- and #S2-Up refer to the TU FPKMs in ▽aCPSF1 and S2, respectively; 4) defining a TRT by its TRTindex >1. Based on the criteria, TRT was identified in 965 TUs (89.5% of 1079 TUs) in ▽aCPSF1, of which 706 TUs (65.7% of 1079 TUs) had a TRTindex >2. According to TRTs’ locations and directions, four TRT types were classified, i.e. TRT of an upstream TU extending into the oppositely encoded one and producing antisense transcripts (type I, 45%), or into the downstream co-directional TU (type II, 42%), or into the downstream IGR (type III, 1%); and TRT of an IGR noncoding RNA extending into the downstream TU (type IV, 2%) (Figure 2F). Although type I TRTs did not markedly affect the abundances of convergently encoded TUs on genome-wide (Supplementary Figure S4), type II TRTs noticeably increased the transcriptions (median: 3.4-fold) of the tandem downstream TUs (Supplementary Dataset S1) compared to that of their own (median: 1.3-fold) in ▽aCPSF1 (Figure 2G), so can severely disturb downstream gene expression and likely bring physiological changes. This was exemplified as that the MMP1719 TRT increased transcription of the downstream archaella master regulator, earA (MMP1718) (Figure 3A and Supplementary Dataset S4).
Figure 3.

TRT upregulates the archaella regulator earA and results in exuberant archaella along with elevated motility in 22°C-cultured ▽aCPSF1. (A) A diagram shows the RNA-seq reads mapped to earA (MMP1718) and the adjacent genes in S2 (blue) and ▽aCPSF1 (red). Dot magenta brackets show that Mmp-aCPSF1 depletion caused 3′-end extension. Numbers on top indicate the mapped genomic regions, and bullets represent the corresponding genes. (B) Northern blot assays MMP1719 TRT into earA and MMP1717 by using DNA probes P1, P2 and P3 shown in (A). (C) 3′RACE amplified fragments contain the transcription termination site (TTS) in S2 and TRT of MMP1719 in ▽aCPSF1, respectively. (D) Northern blot assays the earA transcript stability in S2 and ▽aCPSF1 (upper panel) using probe P2. Hybridization signal intensities were estimated using Quantity One (v4.6.6) and the residual percentages at indicated times referenced to that at 0 min were shown at beneath. Half-lives (t1/2) were calculated from the regression curve of remnant mRNA contents along time (lower panel). Experiments were performed on three batches of cultures, and the averages and standard deviations are shown. (E) A schematic depicts gene organization of the fla operon and its activator EarA (upper). TTS is located 104 nt downstream the stop codon (nt 1618850) of fla J. The histograms (lower) show the FPKMs of the fla operon genes in S2 and ▽aCPSF1. (F) Northern blot assays the flaB2 transcript abundances in strains S2, ▽aCPSF1, earA deletion (ΔearA), and the double mutation of Mmp-aCPSF1 depletion and earA deletion (▽aCPSF1ΔearA). 23S rRNA was used as the sample control. (G) Representative electron microscopic images show more archaella (arrows pointed) at ▽aCPSF1 than S2 cells (upper panels) and in the magnified regions (dot framed, lower panels). Archaella were observed for each 20 cells, and additional representative images are shown in Supplementary Figure S5. (H) Mobility assay displays larger lawn and more bubbles in ▽aCPSF1 than that in S2 when grown on 0.25% agar.
Using three probes that are respectively complementary to MMP1717, earA and MMP1719, northern blot detected a long transcript encompassing MMP1717-earA-MMP1719 in 22°C-cultured ▽aCPSF1, but only a co-transcribed TU of MMP1717-earA in S2 (Figure 3B). 3′RACE also verified the MMP1719 TRT into earA and MMP1717 (Figure 3C). Moreover, a longer mRNA half-life (53.3 min) was determined for the long transcript of MMP1717-earA-MMP1719 in ▽aCPSF1 than that of the short MMP1717-earA (11.2 min) in S2 (Figure 3D). Thereby, the markedly increased abundance of the earA transcript in ▽aCPSF1 was likely an overlay effect of MMP1719 TRT upregulated transcription and the elevated stability of the extended transcript. Consistently, the EarA regulated fla operon genes (55) were markedly upregulated in ▽aCPSF1 (Figure 3E and Supplementary Dataset S4), and the increased transcription of flaB2 disappeared in the double mutation of Mmp-aCPSF1 depletion and earA deletion but occurred in ▽aCPSF1 (Figure 3F), verifying that TRT resulted upregulation of earA increased the fla operon transcript abundances in ▽aCPSF1. Accordingly, exuberant archaella and more active motility were observed in the ▽aCPSF1 cells compared with S2 (Figure 3G, H and Supplementary Figure S5). Moreover, 3′RACE also verified the MMP1719 TRT into earA and Northern blot also detected the increased transcription of flaB2 at 37°C cultured ▽aCPSF1 stain, and consequently the exuberant archaella and more active motility were also detected in ▽aCPSF1 cells than S2 at 37°C (Supplementary Figure S6). This determines that the Mmp-aCPSF1 dependent transcription termination assures, in addition to optimal growth and ordered transcriptome (Figure 1), the flagellation and motility of the methanoarchaeon.
Term-seq reveals a uridine-rich terminator motif and verifies a genome-wide TRT caused by Mmp-aCPSF1 depletion
Next, Term-seq was employed to sequence the transcript 3′-ends and determine the terminator characteristics of M. maripaludis, as well as the function of Mmp-aCPSF1 in transcription termination. Based on a stringent filtration workflow for TTS definition as described in the Materials and Methods, 998 primary TTSs were identified for 960 TUs and 38 non-coding RNAs (Supplementary Dataset S2) representing 67.7% (730) of the predicted 1,079 and newly identified 230 TUs in S2. Analyzing the sequences flanking TTSs revealed a distinct upstream terminator motif, which is characterized by a 23 nt uridine-tract with the TTS most proximal 4 nt having the highest uridine match (Figure 4A and Supplementary Dataset S2). Reads abundance normalization on each 20 nt upstream and downstream of the 998 primary TTSs determined a >50% reads decrease between bases -2 and +2 flanking a TTS in S2, so defined as transcription termination. However, in ▽aCPSF1, only <20% of reads abundance decrease between –2 and +2, and markedly higher abundant reads were mapped at the 20 nts downstream TTS (Figure 4B), verifying that aCPSF1 depletion caused a genome-wide TRT at a single-base resolution, i.e. a genome-wide termination defect at the uridine-rich tract. Next, 3′RACE amplification and sequencing of the 3′-extensions in 19 TUs in ▽aCPSF1 (Figure 2A and Supplementary Figures S2 and S7) validated Term-seq-defined TTSs and the upstream uridine-rich tracts in a single-base accuracy (Supplementary Figure S8). Similar but slighter remarkable TRTs and transcription abundance changes were also detected in selected TU pairs in 37°C-cultured than 22°C-cultured ▽aCPSF1 strains by northern blot and 3′RACE assays (Supplementary Figure S9). These results collectively demonstrated that the archaeal ribonuclease, aCPSF1, is involved in genome-wide transcription termination.
Mmp-aCPSF1 depends on its ribonuclease activity to trigger transcription termination
Based on the nuclease nature of Mmp-aCPSF1, its nucleolytic activity on Term-seq defined terminator motif was then assayed using three representative uridine-rich RNA substrates (5′-[32P]-labeled) that are derived from the 3′-ends of MMP1100 (30 nt), MMP1149 (40 nt) and MMP0901 (50 nt) (Figure 4C, upper). Addition of the recombinant Mmp-aCPSF1 protein, but not the activity-inactive mutant H243A/H246A (Supplementary Figure S10), produced the cleavage fragments of ∼17 nt, ∼26 nt and ∼29 nt, matching the predicted sizes of products cleaved at downstream the uridine-tract motif in the three synthetic RNAs. However, no cleaving products were found from the non-uridine-tract containing MMP1697 RNA (Figure 4C). This confirmed the endoribonucleolytic activity of Mmp-aCPSF1 at the uridine-tract downstream TTSs. Additional products cleaved downstream other uridine-tracts were also observed (Figure 4C). Nevertheless, Mmp-aCPSF1 only exhibited weak in vitro ribonuclease activity, implying that interactions with other proteins possibly promote its in vivo activity.
To evaluate the contribution of the aCPSF1 ribonuclease activity to transcription termination in vivo, the wild-type Mmp-aCPSF1 and its catalytic-inactive mutant, H243A/H246A, were expressed in ▽aCPSF1 to construct two complementary strains, Com(wt): Mmp-com(Mmp-C1) and Com(mu): Mmp-com(Mmp-C1mu) (Supplementary Table S1) respectively. Although comparable cellular Mmp-aCPSF1’s abundances were detected (Figure 4D), the Com(mu) was incapable of restoring the 22°C-growth defect of ▽aCPSF1, but the Com(wt) can (Figure 4E). Accordingly, 3′RACE determined similar TRTs from MMP0901, MMP1149, and MMP1661 in Com(mu) as in ▽aCPSF1, but no TRTs were found in Com(wt) (Figure 4F). This collectively determined that Mmp-aCPSF1, depending on its ribonuclease activity, triggers transcription termination.
Mmp-aCPSF1 exhibits the in vivo association with archaeal RNA polymerase and a chromosomal occupancy at the transcript 3′-ends
A transcription termination factor would associate with RNAPs in vivo. We first investigated the association of Mmp-aCPSF1 with Mmp-aRNAP by means of size exclusion chromatography-based cell-extract fractionation and coupled with western blot. Co-occurrence of Mmp-aCPSF1 and Mmp-RpoD, one of the 13 aRNAP subunits of M. maripaludis, was found in a protein complex of ∼200–660 kD. Although ribonuclease digestion resulted in some increase of free Mmp-aCPSF1 (∼140 kD), the majority of the protein retained in the aRNAP co-occurred complex fractions (Figure 5A), indicating a nucleic-acid independent association of Mmp-aCPSF1 with Mmp-aRNAP. Furthermore, Mmp-aCPSF1 was immunoprecipitated from strain Mmp-aCPSF1-F (Supplementary Table S1) that carries the 3× Flag fused Mmp-aCPSF1, and it was also detected in the His-affinity purification, co-immunoprecipitated, and tandem affinity chromatography products of strain Mmp-RpoL-HF that carries the His6–3× Flag fused Mmp-RpoL (Figure 5B). RNase, DNase, and Benzonase (digesting both DNA and RNA) treatments did not alter the co-immunoprecipitation results (Figure 5C), further verifying the in vivo association of Mmp-aCPSF1 and Mmp-aRNAP. Moreover, by quantitative western blot, a comparative cellular content of aCPSF1 and RNA polymerase was determined, with aCPSF1 and aRpoL being 0.125 ± 0.009 pmol and 0.072 ± 0.008 pmol at mid-exponential phase and 0.14 ± 0.017 pmol and 0.086 ± 0.006 pmol at stationary phase per μg total cell proteins in 37°C cultures, respectively (Supplementary Figure S11).
Figure 5.

Mmp-aCPSF1 associates with Mmp-aRNAP and the chromosomal DNA. (A) Co-occurrence of Mmp-aCFSF1 and Mmp-RpoD was assayed through size exclusion chromatography (upper) based cell extract fractionation and coupled with western blot (lower). +/–RNase, with or without RNase A treatment. (B, C) Association of Mmp-aCPSF1 with Mmp-aRNAP was determined through co-immunoprecipitation (anti-Flag CoIP), Ni column affinity chromatography (His-affinity), and tandem affinity purification (TAP) using strains Mmp-RpoL-HF and Mmp-aCPSF1-F that carry His6–3× Flag fused Mmp-RpoL and 3× Flag fused Mmp-aCPSF1, respectively. Strain S2 was included as a mock control. Western blot detected Mmp-aCPSF1 in captured products. -, RNase, DNase, and Benzonase indicate treatments without and with the corresponding nucleases of the Mmp-RpoL-HF cell extract before immunoprecipitation. (D) PCR products (arrows pointed) of the indicated genes were amplified from the ChIP DNAs of strains S2 (mock), Mmp-RpoL-HF (LF), and Mmp-aCPSF1-F (C1F), respectively. M, DNA marker. (E) qPCR assayed the enriched genes in ChIP products, and the enrichment fold of a given gene in the ChIP eluates of strains Mmp-RpoL-HF and Mmp-aCPSF1-F over that in S2 (mock control) was calculated as described in the Materials and Methods.
Next, the chromosomal DNA occupancy of Mmp-aCPSF1 was detected via chromatin immunoprecipitation (ChIP). The chromatin DNAs captured by Flag-tagged Mmp-aCPSF1 or aRNAP were immunoprecipitated by ChIP (Supplementary Figure S12) and used as PCR templates. Abundant PCR products in length of 130–150 bp were amplified for 11 highly expressed TUs (Figure 5D), and quantitative PCR (qPCR) determined ∼6- and 6- to 12-fold enrichments of the tested TU 3′-end regions in Flag-tagged Mmp-aCPSF1- and Mmp-RpoL-captured DNAs, respectively, comparing to that from the mock ChIP of strain S2 (Figure 5E). No PCR products were amplified from the poorly expressed MMP0791 (Figure 5D, E), and the TTS downstream regions of MMP0137, MMP0180, MMP0183 and MMP1100 (Supplementary Figure S12C). Collectively, Mmp-aCPSF1 associates with aRNAP as well as chromosomal DNA in vivo, the key features of a transcription termination factor.
aCPSF1 and the uridine-rich motif within IGRs are widely distributed among Archaea
A previous phylogenomic analysis on the aCPSF1 orthologs retrieved from 110 complete archaeal genomes has indicated that this protein is strictly conserved and appear being inherited vertically (52). To explore the universality of aCPSF1-mediated transcription termination among Archaea, the aCPSF1 orthologs in 31 species representing all the four recently defined archaeal superphyla (Euryarchaeota, TACK, Asgard and DPANN) (56), were subjected to phylogenetic analysis in comparative with that of a protein concatenation of aRNAP subunits A, B and E, and the conserved transcription elongation factor Spt5 retrieved from the same species (Figure 6A). It was found that the aCPSF1 phylogeny exhibited an equivalent clustering topology as that of the concatenated sequences of 26 universally conserved proteins (57), and also a convergent clustering pattern as aRNAPs (Figure 6A). Thus, aCPSF1 appear displaying a similar conservative evolution as the RNA transcription machinery in Archaea.
Figure 6.

The ubiquitous distribution of the aCPSF1 orthologs among Archaea and the aCPSF1 dependent archaeal transcription termination reveals a similar 3′-end cleavage mode as the eukaryotic RNAP II termination. (A) The Maximum Likelihood phylogenetic trees based on aCPSF1 proteins (left) and concatenated RNA polymerase subunits A, B and E, and transcription elongation factor Spt5 (right). Protein sequences were retrieved from genomes or metagenome-assembled genomes of the representative archaeal lineages that have been described thus far, including Euryarchaeota (E), Crenarchaeota (C), Thaumarchaeota (T), Aigarchaeota (A), Korarchaeota (K), Nanoarchaeota (N), and Lokiarchaeota (Loki). The unrooted tree was constructed using Maximum-Likelihood methods in MEGA 7.0 software package. Bootstrap values are shown at each node. (B) Protein abundances of the aCPSF1 orthologs in complemented ▽aCPSF1 strains were examined by western blot using the Mmp-aCPSF1 polyclonal antiserum. The percentiles of protein content referenced to that in S2 are shown beneath the gel. (C) Ectopic expression of the aCPSF1 orthologs from Lokiarchaeum GC14_75 (com(Loki-C1)) and Cenarchaeum symbiosum (com(Csy-C1)) restored the 22°C-growth defect of ▽aCPSF1 similarly as the complementation of itself (com(Mmp-C1)) and wild-type S2 carrying an empty vector (S2-pMEV2). The averages of three replicates and standard deviations are shown. (D) 3′RACE assays the TRT transcripts of MMP0901 and MMP0155 in the strains numbered as in panel (B). (E) A proposed mechanism of aCPSF1 cleavage triggering the archaeal transcription termination exposes a homolog of the eukaryotic RNAP II termination mode (3–5). The bacterial intrinsic termination is included for comparison. LUCA means last universal common ancestor.
Next, distribution of the terminator motif, uridine-rich sequence, was searched in each 200 nt upstream and downstream the putative stop codons of the annotated ORFs among 34 representative archaeal species. The result indicated that uridine-rich tracts with successive 5, 6 and 7 uridines (U5, U6 and U7) were significantly enriched in IGRs compared to the upstream gene-bodies in most searched archaeal genomes (Supplementary Figure S13 and Dataset S3). This indicates that the prevalent IGR uridine-rich tract could be a conserved terminator sequence of Archaea. The highly conservation of aCPSF1 and the prevalence of the 3′-end uridine-rich motif together suggest that the aCPSF1 dependent transcription termination could be universally employed in Archaea.
Two phylogenetic distant aCPSF1 orthologs implement transcription termination in M. maripaludis
To verify if the aCPSF1 orthologs all function in transcription termination, two orthologs, one from Ca. Lokiarchaeum sp. GC14_75 belonging to Lokiarchaeota and the other from Ca. Cenarchaeum symbiosum affiliating with Thaumarchaeota, were selected. The two share 48% and 40% amino acid sequence identities with Mmp-aCPSF1 respectively (Supplementary Figure S14), and after codon optimized (Supplementary Figure S15), were ectopically expressed in ▽aCPSF1 (Figure 6B). Behaving as Mmp-aCPSF1, the two orthologs not only restored the growth defect of ▽aCPSF1 at 22°C (Figure 6C), but also eliminated the TRTs of convergent (MMP0901, type I) and co-directional TUs (MMP0155, type II) (Figure 6D). This demonstrated that the aCPSF1 orthologs from various archaeal phyla play a same role in transcription termination.
DISCUSSION
Transcription termination mechanisms remain largely unknown in Archaea thus far (1,13,25), while very recently the ribonuclease aCPSF1, FttA, of T. kodakarensis was reported to function as a transcription terminate factor of Archaea mainly via an in vitro system (27). Almost at the same time, this work, through intensive genetic, molecular and biochemical experiments, and high-throughput RNA-seq and Term-seq, has revealed that the aCPSF1 ortholog of M. maripaludis, by cleaving at the transcript 3′-end uridine-rich sequences (Figure 4), triggers a genome-wide transcription termination (Figure 2), so guarantees the programmed transcriptome and an optimal growth as well as flagellation and motility (Figures 1 and 3) of the methanoarchaeon. aCPSF1, via associations with archaeal RNAP as well as the chromosome DNA (Figure 5), plays an essential role in archaeal genome-wide transcription termination. Therefore, aCPSF1 functions as a general transcription termination factor of Archaea, and the indispensable role in transcription termination reported here deciphers its universality and essentiality in Archaea.
Given the short IGRs distributed in the prokaryotic compact genomes, transcription termination could be more vital to an ordered transcriptome. This is verified by that an approximate 80% depleted expression of Mmp-aCPSF1 caused a genome-wide TRT and disordered transcriptome with inversely changed expression of the highly- and poorly-transcribed genes (Figure 1E, F) and concomitant defective growth (Figure 1). This might be due to a deficient termination sequestering RNAP for new rounds of transcription. In addition, transcription readthrough particularly increases transcription of the downstream genes (type II TRT, 42%) (Figures 1 and 2), and has caused physiological changes, such as increased flagellation and cellular motility (Figure 3). This highlights the significance of aCPSF1 dependent transcription termination in maintaining an ordered transcriptome and optimal physiology of Archaea. Similar disordered transcriptomes derived from defective transcription termination are also found in Bacteria (58,59), in which the absence or depletion of the transcription termination factors caused genome-wide abnormal antisense, and intragenic and stable RNA transcriptions. In Eukaryotes, defective termination generates prevalent transcriptions of cryptic unstable RNA and the transcript 3′ flanking region, which disturbs ordered transcriptomes as well (4,8,60,61).
Term-seq analysis determines a terminator motif of two consecutive uridine-tracts in transcripts of M. maripaludis (Figure 4A). Similar, but slightly different, uridine-rich motifs have also been observed in three representative Euryarchaeota and Crenarchaeota species, M.mazei, S.acidocaldarius (13) and H.volcanii (24). Uridine-rich sequences were considered as the intrinsic termination signal based on the earlier and primarily in vitro studies (18,19,22), however, whether this sequence merely pauses or also dismantles aRNAP for termination has not been clarified. This work not only determined that aCPSF1 depletion causes an overall termination deficiency at the transcript 3′-end uridine-tract, but also demonstrated that Mmp-aCPSF1 performs an in vitro endoribonucleolytic cleavage at the putative TTSs downstream the uridine-rich sequences, and the in vivo endoribonuclease activity is determined to be essential to transcription termination (Figure 4). Thus, we suppose that both the uridine-rich termination signal and aCPSF1 are required by aRNAP for efficient transcription termination. Based on these results, a model shown in Figure 6E was proposed. The intrinsic uridine-rich termination signal directs the progressing aRNAP to pause or slowdown, which could buy time for aCPSF1 to recognize and cleave at downstream of the signal sequence. This event in turn triggers aRNAP dissociating from the DNA templates and nascent RNA, and results in transcription termination. An interaction with aRNAP (Figure 5A–D) warrants the recruitment of aCPSF1 to the transcription machinery and the uridine-rich motif in nascent RNAs. The high conservation of aCPSF1 (Figure 6A) and the prevalent uridine-rich motif in archaeal genome IGRs (Supplementary Figure S13 and Dataset S3) together suggest that the aCPSF1 mediated termination mechanism could be widely employed among Archaea. Supportively, two aCPSF1 orthologs from the distant related Lokiarchaeota and Thaumarchaeota were capable of implementing transcription termination in M. maripaludis (Figure 6B–D). Therefore, it is hypothesized that aCPSF1 dependent transcription termination could have an ancient origin predating the divergence of various archaeal phyla and is still widely employed in modern Archaea. Differently, based on a lower termination efficiency on cytidine-less than on guanine-less templates in an in vitro system, the T. kodakarensis aCPSF1 ortholog, FttA, was proposed to work on cytidine-rich sequences in a mode akin to the bacterial Rho protein (27). Whereas, Term-seq has revealed overrepresented uridine-rich sequences but not cytidine-rich upstream the transcript 3′-ends in four Archaea. Thus, the in vivo mechanism of FttA needs to be verified.
Remarkably, the working mechanism of aCPSF1 resembles the eukaryotic 3′-end processing/cleavage triggered RNAP II termination mode, in which the aCPSF1 homologs, the yeast Ysh1 or the human CPSF73, in the multi-subunits 3′-end processing machinery execute the 3′-end cleavage (3,5,8,62,63), and the accessory cleavage factors IA and IB (CFIA and CFIB) recognize the flanking uridine-rich sequences and facilitate the 3′-end cleavage of Ysh1 (62). However, aCPSF1 may rely on its N-terminal K homology (KH) domain, a characteristic RNA binding domain (52,53), to recognize the uridine-rich signals and implement the cleavage by itself. Interestingly, Lokiarchaeota that belongs to the Asgard superphylum, a supposed archaeal ancestor of Eukaryotes (15,64) appears using the aCPSF1 dependent termination mechanism as well. This is evidenced not only by that Loki-aCPSF1 is capable of terminating the methanococcal transcription (Figure 6B–D), but also the pervasive IGR uridine-rich sequences (Supplementary Dataset S3). Therefore, based on the homologous core components of the termination machineries, CPSF73 and aCPSF1, and the similar 3′-end cleavage triggered termination mechanisms, we proposed that the eukaryotic RNAP II termination might have evolved from the aCPSF1 dependent mode reported here, i.e. the aCPSF1 (CPSF73 homologous) mode exposed an archaeal precursory system of the eukaryotic transcription termination machinery. Distinctly, the eukaryotic CPSF73 cleavage downstream the 3′-end poly(A) site not only terminates transcription but also provides a polyadenylation site for transcript maturation (63,65), while no 3′ terminus polyadenylation follows aCPSF1 3′-end cleavage in Archaea. Moreover, very few homologs of the eukaryotic multi-subunit 3′-end processing complex (66,67) and termination factors (8–11) have been identified in Archaea, implying that Archaea could employ a simplified termination strategy. Additionally, the eukaryotic 5′-3′ exoribonuclease, like yeast Rat1 or human Xrn2, is involved in transcription termination by rapidly ‘chewing’ Ysh1/CPSF73 cleaved RNA products so to dismantle the associated RNAPs, noted as the ‘torpedo’ model (8,9,12,68). Although no homologs of Rat1 or Xrn2 have been found in Archaea, exoribonucleases aRNase J, aCPSF2 and even aCPSF1, all possessing the similar 5′-3′ exoribonucleolytic activities (43,51,52,69), could function comparatively in Archaea (Figure 6E). Therefore, the aCPSF1-dependent archaeal transcription termination could expose a mode akin to the eukaryotic RNAP II termination mechanism.
Interestingly, the 3′-end proximal uridine-rich tract likely represents a shared termination signal in the archaeal aCPSF1 termination system and the bacterial intrinsic termination (Figure 6E), as both could cause a pausing of the progressing RNAP and facilitate the upstream hairpin formation and aCPSF1 cleavage at nascent RNAs to trigger RNAP dissociation in bacteria and Archaea, respectively. Thus, compared to the simple intrinsic termination of bacteria, aCPSF1 and its eukaryotic homologs’ cleavage triggered terminations could expose complexity-upgraded transcription termination mechanisms in Archaea and Eukaryotes (Figure 6E), while the eukaryotic 3′ polyadenylation adds more complexity. Therefore, insight into aCPSF1 dependent archaeal transcription termination would shed light on understanding both the complex eukaryotic transcription termination and the evolutionary trajectory of life's transcription process. In conclusion, this work describes the first archaeal general transcription termination factor aCPSF1 and its cleavage dependent termination mechanism, which might be a precursory system of the eukaryotic RNAP II termination mode.
DATA AVAILABILITY
Both the Term-seq and the strand-specific RNA-seq data have been deposited at the NCBI GEO Submission (GSE141346) [NCBI tracking system #20481779]. Custom codes used in this study are accessible at https://github.com/DXZbioinfor/aCPSF1-dependent-genome-wide-transcription-termination-of-Archaea.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Prof. William B. Whitman at the University of Georgia providing strain Methanococcus maripaludis S2 and plasmids pIJA03 and pMEV2, as well as Prof. Tao Yong at Institute of Microbiology, CAS for providing Escherichia coli BW25113 and plasmid pSB1s. The authors also thank Jingnan Liang for electron microscopic images and Accdon (www.letpub.com) for linguistic assistance on the manuscript preparation.
Author contributions: L.Y. and J.L. designed and performed the genetic and biochemical experiments. J.L. and X.Z.D. conceptualized the experiments and acquired funding. L.Y. and L.Q. performed the transcriptomics assays. Z.H.L. performed the experiments of the 37°C cultures and protein content quantification. B.Z. and F.Q.Z. performed the bioinformatics analyses. L.L. cultured strains. X.W.Z. performed the phylogenetic analysis. J.L. and X.Z.D. wrote the manuscript and all of the authors approved the final manuscript.
Contributor Information
Lei Yue, State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China; University of Chinese Academy of Sciences, No. 19A Yuquan Road, Shijingshan District, Beijing 100049, China.
Jie Li, State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China.
Bing Zhang, University of Chinese Academy of Sciences, No. 19A Yuquan Road, Shijingshan District, Beijing 100049, China; Beijing Institutes of Life Science, Chinese Academy of Sciences, Beijing 100101, China.
Lei Qi, State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China.
Zhihua Li, State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China; University of Chinese Academy of Sciences, No. 19A Yuquan Road, Shijingshan District, Beijing 100049, China.
Fangqing Zhao, University of Chinese Academy of Sciences, No. 19A Yuquan Road, Shijingshan District, Beijing 100049, China; Beijing Institutes of Life Science, Chinese Academy of Sciences, Beijing 100101, China.
Lingyan Li, State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China.
Xiaowei Zheng, State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China.
Xiuzhu Dong, State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China; University of Chinese Academy of Sciences, No. 19A Yuquan Road, Shijingshan District, Beijing 100049, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key R&D Program of China [2020YFA0906800, 2018YFC0310800, 2019YFA0905500]; National Natural Science Foundation of China [91751203, 31670049]. Funding for open access charge: National Key R&D Program of China [2020YFA0906800, 2018YFC0310800, 2019YFA0905500]; National Natural Science Foundation of China [91751203].
Conflict of interest statement. None declared.
REFERENCE
- 1. Maier L.K., Marchfelder A.. It's all about the T: transcription termination in archaea. Biochem Soc. T. 2019; 47:461–468. [DOI] [PubMed] [Google Scholar]
- 2. Ray-Soni A., Bellecourt M.J., Landick R.. Mechanisms of bacterial transcription termination: all good things must end. Annu. Rev. Biochem. 2016; 85:319–347. [DOI] [PubMed] [Google Scholar]
- 3. Porrua O., Boudvillain M., Libri D.. Transcription termination: variations on common themes. Trends Genet. 2016; 32:508–522. [DOI] [PubMed] [Google Scholar]
- 4. Porrua O., Libri D.. Transcription termination and the control of the transcriptome: why, where and how to stop. Nat. Rev. Mol. Cell Bio. 2015; 16:190–202. [DOI] [PubMed] [Google Scholar]
- 5. Kuehner J.N., Pearson E.L., Moore C.. Unravelling the means to an end: RNA polymerase II transcription termination. Nat. Rev. Mol. Cell Bio. 2011; 12:283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gusarov I., Nudler E.. The mechanism of intrinsic transcription termination. Mol. Cell. 1999; 3:495–504. [DOI] [PubMed] [Google Scholar]
- 7. Peters J.M., Vangeloff A.D., Landick R.. Bacterial transcription terminators: the RNA 3′-end chronicles. J. Mol. Biol. 2011; 412:793–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eaton J.D., Davidson L., Bauer D.L.V., Natsume T., Kanemaki M.T., West S.. Xrn2 accelerates termination by RNA polymerase II, which is underpinned by CPSF73 activity. Gene Dev. 2018; 32:127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baejen C., Andreani J., Torkler P., Battaglia S., Schwalb B., Lidschreiber M., Maier K.C., Boltendahl A., Rus P., Esslinger S. et al.. Genome-wide analysis of RNA polymerase II termination at protein-coding genes. Mol. Cell. 2017; 66:38–49. [DOI] [PubMed] [Google Scholar]
- 10. Larochelle M., Robert M.A., Hebert J.N., Liu X.C., Matteau D., Rodrigue S., Tian B., Jacques P.E., Bachand F.. Common mechanism of transcription termination at coding and noncoding RNA genes in fission yeast. Nat. Commun. 2018; 9:4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grzechnik P., Gdula M.R., Proudfoot N.J.. Pcf11 orchestrates transcription termination pathways in yeast. Gene Dev. 2015; 29:849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim M., Krogan N.J., Vasiljeva L., Rando O.J., Nedea E., Greenblatt J.F., Buratowski S.. The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature. 2004; 432:517–522. [DOI] [PubMed] [Google Scholar]
- 13. Dar D., Prasse D., Schmitz R.A., Sorek R.. Widespread formation of alternative 3′ UTR isoforms via transcription termination in archaea. Nat. Microbiol. 2016; 1:16143. [DOI] [PubMed] [Google Scholar]
- 14. Eme L., Spang A., Lombard J., Stairs C.W., Ettema T.J.G.. Archaea and the origin of eukaryotes. Nat. Rev. Microbiol. 2017; 15:711–723. [DOI] [PubMed] [Google Scholar]
- 15. Zaremba-Niedzwiedzka K., Caceres E.F., Saw J.H., Backstrom D., Juzokaite L., Vancaester E., Seitz K.W., Anantharaman K., Starnawski P., Kjeldsen K.U. et al.. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature. 2017; 541:353–358. [DOI] [PubMed] [Google Scholar]
- 16. Williams T.A., Cox C.J., Foster P.G., Szollosi G.J., Embley T.M.. Phylogenomics provides robust support for a two-domains tree of life. Nat. Ecol. Evol. 2020; 4:138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Werner F., Grohmann D.. Evolution of multisubunit RNA polymerases in the three domains of life. Nat. Rev. Microbiol. 2011; 9:85–98. [DOI] [PubMed] [Google Scholar]
- 18. Santangelo T.J., Reeve J.N.. Archaeal RNA polymerase is sensitive to intrinsic termination directed by transcribed and remote sequences. J. Mol. Biol. 2006; 355:196–210. [DOI] [PubMed] [Google Scholar]
- 19. Santangelo T.J., Cubonova L., Skinner K.M., Reeve J.N.. Archaeal intrinsic transcription termination in vivo. J. Bacteriol. 2009; 191:7102–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hirtreiter A., Grohmann D., Werner F.. Molecular mechanisms of RNA polymerase-the F/E (RPB4/7) complex is required for high processivity in vitro. Nucleic Acids Res. 2010; 38:585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thomm M., Hausner W., Hethke C.. Transcription factors and termination of transcription in Methanococcus. Syst. Appl. Microbiol. 1994; 16:648–655. [Google Scholar]
- 22. Spitalny P., Thomm M.. A polymerase III-like reinitiation mechanism is operating in regulation of histone expression in archaea. Mol. Microbiol. 2008; 67:958–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dar D., Shamir M., Mellin J.R., Koutero M., Stern-Ginossar N., Cossart P., Sorek R.. Term-seq reveals abundant ribo-regulation of antibiotics resistance in bacteria. Science. 2016; 352:6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Berkemer S.J., Maier L.K., Amman F., Bernhart S.H., Wortz J., Markle P., Pfeiffer F., Stadler P.F., Marchfelder A.. Identification of RNA 3′ ends and termination sites in Haloferax volcanii. RNA. Biol. 2020; 17:663–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Walker J.E., Luyties O., Santangelo T.J.. Factor-dependent archaeal transcription termination. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E6767–E6773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Blombach F., Matelska D., Fouqueau T., Cackett G., Werner F.. Key concepts and challenges in Archaeal transcription. J. Mol. Bio. 2019; 431:4184–4201. [DOI] [PubMed] [Google Scholar]
- 27. Sanders T.J., Wenck B.R., Selan J.N., Barker M.P., Trimmer S.A., Walker J.E., Santangelo T.J.. FttA is a CPSF73 homologue that terminates transcription in Archaea. Nat. Microbiol. 2020; 5:545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sarmiento B.F., Leigh J.A., Whitrnan W.B.. Genetic systems for hydrogenotrophic methanogens. Method Enzymol. 2011; 494:43–73. [DOI] [PubMed] [Google Scholar]
- 29. Sarmiento F., Mrazek J., Whitman W.B.. Genome-scale analysis of gene function in the hydrogenotrophic methanogenic archaeon Methanococcus maripaludis. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:4726–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tumbula D.L., Makula R.A., Whitman W.B.. Transformation of Methanococcus Maripaludis and identification of a Psti-like restriction system. FEMS Microbiol. Lett. 1994; 121:309–314. [Google Scholar]
- 31. Moore B.C., Leigh J.A.. Markerless mutagenesis in Methanococcus maripaludis demonstrates roles for alanine dehydrogenase, alanine racemase, and alanine permease. J. Bacteriol. 2005; 187:972–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li J., Zhang B., Zhou L., Qi L., Yue L., Zhang W., Cheng H., Whitman W.B., Dong X.. The archaeal RNA chaperone TRAM0076 shapes the transcriptome and optimizes the growth of Methanococcus maripaludis. PLos Genet. 2019; 15:e1008328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Anders S., Pyl P.T., Huber W.. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L.. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010; 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McClure R., Balasubramanian D., Sun Y., Bobrovskyy M., Sumby P., Genco C.A., Vanderpool C.K., Tjaden B.. Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res. 2013; 41:e140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim D., Landmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pertea M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., Salzberg S.L.. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015; 33:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pertea M., Kim D., Pertea G.M., Leek J.T., Salzberg S.L.. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016; 11:1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang J., Li E.H., Olsen G.J.. Protein-coding gene promoters in Methanocaldococcus (Methanococcus) jannaschii. Nucleic Acids Res. 2009; 37:3588–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zheng X., Feng N., Li D.F., Dong X.Z., Li J.. New molecular insights into an archaeal RNase J reveal a conserved processive exoribonucleolysis mechanism of the RNase J family. Mol. Microbiol. 2017; 106:351–366. [DOI] [PubMed] [Google Scholar]
- 44. Smollett K., Blombach F., Werner F.. Transcription in Archaea: preparation of Methanocaldococcus jannaschii transcription machinery. Methods Mol. Biol. 2015; 1276:291–303. [DOI] [PubMed] [Google Scholar]
- 45. Qi L., Yue L., Feng D., Qi F., Li J., Dong X.. Genome-wide mRNA processing in methanogenic archaea reveals post-transcriptional regulation of ribosomal protein synthesis. Nucleic Acids Res. 2017; 45:7285–7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smollett K., Blombach F., Reichelt R., Thomm M., Werner F.. A global analysis of transcription reveals two modes of Spt4/5 recruitment to archaeal RNA polymerase. Nat. Microbiol. 2017; 2:17021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li J., Zheng X., Guo X.P., Qi L., Dong X.Z.. Characterization of an archaeal two-component system that regulates methanogenesis in Methanosaeta harundinacea. PLoS One. 2014; 9:e95502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wilbanks E.G., Larsen D.J., Neches R.Y., Yao A.I., Wu C.Y., Kjolby R.A.S., Facciotti M.T.. A workflow for genome-wide mapping of archaeal transcription factors with ChIP-seq. Nucleic Acids Res. 2012; 40:e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Song N.N., Duc T.N., van Oeffelen L., Muyldermans S., Peeters E., Charlier D.. Expanded target and cofactor repertoire for the transcriptional activator LysM from Sulfolobus. Nucleic Acids Res. 2013; 41:2932–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Proc G.P.D.. The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Levy S., Portnoy V., Admon J., Schuster G.. Distinct activities of several RNase J proteins in methanogenic archaea. RNA Biol. 2011; 8:1073–1083. [DOI] [PubMed] [Google Scholar]
- 52. Phung D.K., Rinaldi D., Langendijk-Genevaux P.S., Quentin Y., Carpousis A.J., Clouet-d’Orval B.. Archaeal beta-CASP ribonucleases of the aCPSF1 family are orthologs of the eukaryal CPSF-73 factor. Nucleic. Acids. Res. 2013; 41:1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Silva A.P.G., Chechik M., Byrne R.T., Waterman D.G., Ng C.L., Dodson E.J., Koonin E.V., Antson A.A., Smits C.. Structure and activity of a novel archaeal beta-CASP protein with N-terminal KH domains. Structure. 2011; 19:622–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yoon S.H., Turkarslan S., Reiss D.J., Pan M., Burn J.A., Costa K.C., Lie T.J., Slagel J., Moritz R.L., Hackett M. et al.. A systems level predictive model for global gene regulation of methanogenesis in a hydrogenotrophic methanogen. Genome Res. 2013; 23:1839–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ding Y., Nash J., Berezuk A., Khursigara C.M., Langelaan D.N., Smith S.P., Jarrell K.F.. Identification of the first transcriptional activator of an archaellum operon in a euryarchaeon. Mol. Microbiol. 2016; 102:54–70. [DOI] [PubMed] [Google Scholar]
- 56. Spang A., Caceres E.F., Ettema T.J.G.. Genomic exploration of the diversity, ecology, and evolution of the archaeal domain of life. Science. 2017; 357:eaaf3883. [DOI] [PubMed] [Google Scholar]
- 57. Guy L., Ettema T.J.G.. The archaeal ‘TACK’ superphylum and the origin of eukaryotes. Trends Microbiol. 2011; 19:580–587. [DOI] [PubMed] [Google Scholar]
- 58. Peters J.M., Mooney R.A., Grass J.A., Jessen E.D., Tran F., Landick R.. Rho and NusG suppress pervasive antisense transcription in Escherichia coli. Gene Dev. 2012; 26:2621–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Peters J.M., Mooney R.A., Kuan P.F., Rowland J.L., Keles S., Landick R.. Rho directs widespread termination of intragenic and stable RNA transcription. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:15406–15411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Thiebaut M., Kisseleva-Romanova E., Rougemaille M., Boulay J., Libri D.. Transcription termination and nuclear degradation of cryptic unstable transcripts: a role for the nrd1-nab3 pathway in genome surveillance. Mol. Cell. 2006; 23:853–864. [DOI] [PubMed] [Google Scholar]
- 61. Grzechnik P., Gdula M.R., Proudfoot N.J.. Pcf11 orchestrates transcription termination pathways in yeast. Genes Dev. 2015; 29:849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hill C.H., Boreikaite V., Kumar A., Casanal A., Kubik P., Degliesposti G., Maslen S., Mariani A., von Loeffelholz O., Girbig M. et al.. Activation of the endonuclease that defines mRNA 3′ ends requires incorporation into an 8-subunit core cleavage and polyadenylation factor complex. Mol. Cell. 2019; 73:1217–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dominski Z. The hunt for the 3′ endonuclease. Wires RNA. 2010; 1:325–340. [DOI] [PubMed] [Google Scholar]
- 64. Spang A., Saw J.H., Jorgensen S.L., Zaremba-Niedzwiedzka K., Martijn J., Lind A.E., van Eijk R., Schleper C., Guy L., Ettema T.J.G.. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature. 2015; 521:173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yang X.C., Sullivan K.D., Marzluff W.F., Dominski Z.. Studies of the 5′ exonuclease and endonuclease activities of CPSF-73 in histone pre-mRNA processing. Mol. Cell. Biol. 2009; 29:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Casanal A., Kumar A., Hill C.H., Easter A.D., Emsley P., Degliesposti G., Gordiyenko Y., Santhanam B., Wolf J., Wiederhold K. et al.. Architecture of eukaryotic mRNA 3′-end processing machinery. Science. 2017; 358:1056–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shi Y.S., Di Giammartino D.C., Taylor D., Sarkeshik A., Rice W.J., Yates J.R., Frank J., Manley J.L.. Molecular architecture of the human pre-mRNA 3′ Processing Complex. Mol. Cell. 2009; 33:365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jimeno-Gonzalez S., Haaning L.L., Malagon F., Jensen T.H.. The yeast 5′-3′ exonuclease Rat1p functions during transcription elongation by RNA polymerase II. Mol. Cell. 2010; 37:580–587. [DOI] [PubMed] [Google Scholar]
- 69. Clouet-d’Orval B., Rinaldi D., Quentin Y., Carpousis A.J.. Euryarchaeal beta-CASP proteins with homology to bacterial RNase J have 5′- to 3′-exoribonuclease activity. J. Biol. Chem. 2010; 285:17574–17583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Both the Term-seq and the strand-specific RNA-seq data have been deposited at the NCBI GEO Submission (GSE141346) [NCBI tracking system #20481779]. Custom codes used in this study are accessible at https://github.com/DXZbioinfor/aCPSF1-dependent-genome-wide-transcription-termination-of-Archaea.