


Abstract
Recent efforts in biological engineering have made detection of nucleic acids in samples more rapid, inexpensive and sensitive using CRISPR-based approaches. We expand one of these Cas13a-based methods to detect small molecules in a one-batch assay. Using SHERLOCK-based profiling of in vitrotranscription (SPRINT), in vitro transcribed RNA sequence-specifically triggers the RNase activity of Cas13a. This event activates its non-specific RNase activity, which enables cleavage of an RNA oligonucleotide labeled with a quencher/fluorophore pair and thereby de-quenches the fluorophore. This fluorogenic output can be measured to assess transcriptional output. The use of riboswitches or proteins to regulate transcription via specific effector molecules is leveraged as a coupled assay that transforms effector concentration into fluorescence intensity. In this way, we quantified eight different compounds, including cofactors, nucleotides, metabolites of amino acids, tetracycline and monatomic ions in samples. In this manner, hundreds of reactions can be easily quantified in a few hours. This increased throughput also enables detailed characterization of transcriptional regulators, synthetic compounds that inhibit transcription, or other coupled enzymatic reactions. These SPRINT reactions are easily adaptable to portable formats and could therefore be used for the detection of analytes in the field or at point-of-care situations.
INTRODUCTION
Transcription is regulated by diverse mechanisms that sense a broad spectrum of small molecules and other stimuli (1). Among this rich set of biosensory devices are transcription factors that enhance or repress the action of RNA polymerase. Bacteria have evolved several families of allosteric transcription factors (aTF) that alter their binding affinity for a DNA site upon binding a small molecule effector ligand (2–5) (Figure 1A). There is not only a large diversity of natural aTFs that respond to different effectors, but they can also be engineered to alter their ligand specificities (6–17). In bacteria, transcription is also often regulated at the mRNA level via riboswitches (18–21). Riboswitches are non-coding elements that are generally found in the leader region of mRNAs. The aptamer domain of a riboswitch specifically binds its cognate ligand, which leads to formation of alternative secondary RNA structures in the downstream expression platform. For transcriptional regulation, the formation of an RNA helix, called an intrinsic terminator, stalls and releases RNA polymerase thereby aborting mRNA synthesis (18,20) (Figure 1B). A plethora of natural riboswitches have been identified that regulate metabolic pathways in response to different metabolites or ions (18,20,21). Additionally, synthetic riboswitches with new ligand specificities have been designed (22–29) and several approaches and workflows have been described for making new riboswitches with desired ligand specificities and performance parameters (23,30–34).
Figure 1.

Transcription and nuclease activities constitute SPRINT assays. (A) Certain allosteric transcription factors release their DNA binding site when they are bound by ligand, therefore enabling transcript elongation. (B) An ON-riboswitch allows transcript elongation by RNA polymerase when the riboswitch is bound to its cognate ligand. (C) Cas13a detects RNA transcripts that contain the target sequence (yellow). After binding its target, Cas13a collaterally cleaves RNA oligonucleotides and de-quenches fluorophores. (D) Workflow of a typical SPRINT experiment. A master mix containing components such as RNA polymerase, Cas13a and DNA template is incubated and then added to wells that contain compounds that regulate transcription. The transcriptional output is then measured via the fluorescence signal.
While aTFs and riboswitches are capable of recognizing a broad array of compounds as effectors, transducing ligand binding into a robust and easy output remains challenging. RNA aptamers have been used for >20 years to develop various voltametric, fluorometric, and colorimetric diagnostic devices such as lateral flow assays or devices based on aggregation of nano-particles (35–39). However, aptamer-based diagnostics are still limited by a lack of efficient read-out methods. Aptamer-binding events can also be measured via their capacity to regulate transcription in vitro but this can be challenging, partly because highly sensitive detection methods are required to quantify the small amounts of RNA transcripts generated. For detection, RNA transcripts are typically radiolabeled by addition of 32P-labeled nucleotides and the products of the transcription reaction are resolved on an acrylamide gel (40,41). This approach requires specialized equipment, is time consuming, and low throughput, significantly restricting the scale of experiments that could be done. Alternatively, methods based on RT-qPCR have been developed (42,43), which circumvent the need for radioactive materials, but require each sample to be purified in several steps before the transcript can be quantified. Therefore, none of the reported methods enable biosensing via riboswitches and aTFs in a rapid and inexpensive way.
Recently, Specific High-Sensitivity Enzymatic Reporter UnLOCKing (SHERLOCK) was developed to sequence-specifically detect ssRNA in environmental and patient samples (44,45). The endonuclease Cas13a is programmed by a crRNA to sequence-specifically bind and cleave a 28-nucleotide target sequence of ssRNA (46). After target RNA cleavage, Cas13a changes its mode of action and collaterally cleaves short RNA oligonucleotides, which are labeled with a 5’-fluorophore and a 3’-quencher. Cleavage of the reporter RNA de-quenches the fluorophore and results in an increased fluorescent signal (Figure 1C). Therefore, SHERLOCK provides a rapid fluorescent signal for detection of RNA strands. Additionally, DNA sequences can be detected by coupling SHERLOCK with T7 in vitro transcription (44). By rapidly detecting nucleic acids via fluorescence, SHERLOCK was used to identify viral RNA in patient samples (47) and became the first CRISPR-based diagnostic tool that was approved by the FDA to detect SARS-CoV-2 (Satyanarayana, M. (2020) A COVID-19 diagnostic that uses CRISPR gets a nod from the FDA. Chemical & Engineering News). This technology has since been adapted to platforms that allow multiplexed processing of thousands of samples (48) and enable rapid detection of all 169 described human viruses in multiple samples simultaneously at a cost of $0.05 per test. These advantages make SHERLOCK a powerful, scalable diagnostic technology, although it remains limited to the detection of nucleic acids.
Similar technologies, called HOLMES (49) or DETECTR (50), use Cas12a to detect DNA via collateral DNA cleavage in a conceptually identical manner to SHERLOCK. Throughout 2019 and 2020, Cas12a-based small-molecule detection systems (51–53) have been published that were based on the DETECTR technology. These diagnostic systems couple ligand sensing by aptamers or transcription factors to the release of DNA strands, which are subsequently sensed by DETECTR. Similarly, a system called ROSALIND senses compounds by utilizing aTFs to regulate the transcription of fluorescent aptamers (54). While these approaches represent important advances in the field of small-molecule detection, they also come with their own specific disadvantages. They are either restricted to only aTFs (51,54) or only riboswitches (52,53), require several steps that limit sample throughput (51,53), or cannot be adapted to the detection of new molecules without changing components of the core technology (51).
In an effort to overcome the aforementioned challenges, we developed SHERLOCK-based profiling of in vitrotranscription (SPRINT). In this assay, RNA is transcribed from a DNA template and detected by Cas13a, which produces a fluorescent signal. Thereby, ligand-dependent transcription could be quantified, which enabled the measurement of the concentration of said ligands in samples (Figure 1D). Further, we demonstrate how this technology can not only be used to detect a diversity of compounds but also measure enzyme activities. In cases where the effector ligand is the product of an enzymatic reaction, SPRINT can be used for enzyme-coupled assays to indirectly measure the conversion of substrate to product. We demonstrate that the SPRINT method is easily adaptable to the detection of diverse classes of compounds and can be used in a rapid, high-throughput manner and represents a significant advance in CRISPR-based biosensing technology.
MATERIALS AND METHODS
Preparation of dsDNA templates for transcription reactions
Sequences encoding SPRINT transcription templates were cloned into pUC19 using the homology-based cloning method circular polymerase extension cloning (CPEC) (55). Plasmid backbones were generated using PCR (link to plasmid sequence is provided in Supplementary Table S1). When inserting a complete SPRINT template (tac promoter (56), riboswitch and target transcript) into the backbone, the backbone was linearized and amplified with the oligonucleotides ‘HA_rev’ and ‘CreateBBnogRNA2’ (sequences of oligonucleotides are provided in Supplementary Table S2). When generating backbones for the purpose of exchanging riboswitches within the constant regions of the SPRINT template, the oligonucleotides ‘e.coliProm_rev’ and ‘RiboswitchBB_fwd’ were used. In cases when only aptamer domains of the pbuE*6U expression platform were exchanged, the backbone was amplified with the oligonucleotides ‘pbuEBB_rev’ and ‘pbuEBB_fwd’. All plasmids were sequence-verified.
When replacing aTF operator sequences in a SPRINT template, the backbone was amplified by PCR with the oligonucleotides ‘RiboswitchBB_fwd’ and ‘pUC RiboBB_rev’. For generating linear dsDNA SPRINT templates with operator sequences for aTFs, the oligonucleotides ‘CreateSeq_fwd’ and ‘ssDNA1_rev’ were used for PCR.
Linear dsDNA SPRINT templates were amplified from sequence-verified plasmids via PCR unless stated otherwise. The oligonucleotides used for PCR to amplify templates with riboswitches were ‘InsulatorOligo_fwd’ and ‘ssDNA1_rev’. Sequences of SPRINT templates and their components are provided in Supplementary Table S1.
For amplification of the native metE riboswitch from Bacillus subtilis genomic DNA, the following protocol was used: colony PCR was performed using the oligonucleotides ‘metE_fwd’ and ‘metE_rev’ with an annealing temperature of 64°C with Q5 DNA polymerase (New England Biolabs). The constant 5′ and 3′ parts of the SPRINT transcription templates were also amplified via PCR reactions using a plasmid with the complete SPRINT sequence as template. The 5′ constant piece was amplified with oligonucleotides ‘InsulatorOligo_fwd’ and ‘e.coliProm_rev’ and the 3′ constant piece was amplified with oligonucleotides ‘RiboswitchBB_fwd’ and ‘ssDNA1_rev’. The three pieces (5′ constant, metE riboswitch, 3′ constant) were assembled and amplified in a single PCR reaction: 5 nM each of the 5′ region, the metE region, and the 3′ region and 500 nM each of the oligonucleotides ‘InsulatorOligo_fwd’ and ‘ssDNA1_rev’ were added to a PCR reaction with the Q5 DNA polymerase. Five amplification cycles were run with an annealing temperature of 61°C to assemble the three pieces and then 25 cycles were run with an annealing temperature of 70°C to amplify the assembled construct with the oligonucleotides. The PCR product was purified using a PCR cleanup kit (E.Z.N.A.® Cycle Pure Kit, Omega Bio-tek Inc.), quantified and used for a SPRINT reaction.
In vitro transcription and purification of RNA with T7 RNAP
crRNA and ssRNA1 (target RNA) were transcribed in vitro and purified using standard protocols (40,41). DNA template for in vitro transcription was amplified in a 200 μl PCR reaction using the oligonucleotides ‘5′ gen’ and ‘crRNA1_rev’. RNA was synthesized in a 2.5 ml transcription reaction containing 200 μl unpurified PCR reaction in T7 transcription buffer. 1X transcription buffer contains 40 mM Tris–HCl, pH 8.0, 10 mM dithiothreitol (DTT), 8 mM MgCl2, 2 mM spermidine, 0.01% (v/v) Triton X-100. ATP, GTP, CTP and UTP were added to a final concentration of 4 mM each, inorganic pyrophosphatase (IPPase) was added to 160 mU/μl and T7 RNA polymerase was added to 320 nM. The reaction was incubated at 37°C for 2 h, followed by addition of 3 ml 100% ethanol to the reaction and precipitation of the RNA at −80°C for 1 h. The reaction was centrifuged at 4000 × g at 4°C for 15 min. The supernatant was discarded and the pellet was air-dried at 37°C for 3 h and then resuspended in 1 ml of 8 M urea, 500 μl 0.5 M EDTA, pH 8.0 and 1 ml of formamide loading dye (0.025% (w/v) bromophenol blue, 5 mM EDTA, pH 8.0, 0.025% (w/v) SDS dissolved in formamide). Transcripts were separated by electrophoresis using a denaturing polyacrylamide gel (10% 29:1 acrylamide/bisacrylamide, 1× TBE buffer (0.1 M Tris base, 80 mM boric acid, 1 mM Na2EDTA), and 8 M urea) and the RNA bands were visualized by UV shadowing. The correct length transcript was excised from the gel and the RNA extracted into 0.5× TE buffer (5 mM Tris–HCl, pH 8.0, 250 μM EDTA) by gentle agitation at 4°C overnight. The mixture was centrifuged at 4000 × g for 30 min and RNA from the supernatant was concentrated to ∼1 ml each using centrifugal concentrators with a 10 kDa molecular weight cutoff (Amicon Ultra, 0.5 ml) and buffer exchanged into 0.5× TE buffer. The concentrate was passed through a large-pored sepharose filter to remove remaining gel pieces. The concentration of RNAs was determined by their absorbance at 260 nm, and stored as concentrated stocks at −80°C. Prior to use, the crRNA was diluted to 2.25 μM and the ssRNA1 as 1 μM to 1 nM aliquots, which were stored at −20°C.
Radiolabeling transcription assays
Single turnover transcriptions were performed as previously described (57,58). 2 ng/μl template DNA was incubated at 37°C for 15 min in SPRINT buffer with 0.45 U/μl E. coli RNAP σ70 holoenzyme (NEB) and 0.5 μCi/μl of [α-32P] ATP. The reaction was initiated by adding rNTPs to 85 μM, heparin to 66 μg/ml, and the desired ligand concentration. After 20 min, 25 μl of 8 M urea was added to quench the reaction. Transcription products were resolved on a denaturing 29:1 acrylamide:bisacrylamide gel. The gel was dried and then exposed to a phosphor screen for at least one day. RNA band intensities were quantified using ImageJ 1.52a.
Purification of LwaCas13a protein
We followed a previously published purification protocol for purifying LwaCas13a with some modifications (44). Cas13a from Leptotrichia wadeii was used for all experiments in this study. The expression plasmid (pC013) (Addgene plasmid #90097) encodes LwaCas13a with an N-terminal histidine tag, followed by twinstrep and SUMO tags. The plasmid was transformed into BL21(DE3) Rosetta Escherichia coli cells. Protein expression in 2 l LB culture was induced by adding 0.5 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) at OD600 = 0.6. After 16 h at 20°C the cells were pelleted and resuspended in lysis buffer (0.5 M NaCl, 20 mM Tris–HCl, pH 8.0, and 1 mM DTT). All subsequent purification steps were carried out at 4°C. Cells were lysed using an Emulsiflex C3 homogenizer, cell debris was pelleted and the supernatant (∼35 ml) was added to a beaker and stirred rapidly while 250 μl lysis buffer containing 5% polyethyleneimine (PEI), pH 8.0, was added carefully and slowly. PEI was used to precipitate nucleic acid contaminants (59). The supernatant was further stirred for 15 minutes and then centrifuged at 12 000 × g for 20 min to pellet the precipitate. The supernatant was incubated with Ni-NTA sepharose beads (Qiagen) on an orbital shaker for 1 h at 4°C. Beads were centrifuged 2 min at 300 × g and washed 15 min with 40 ml lysis buffer (10 mM imidazole), again with 40 ml lysis buffer (50 mM imidazole) and 30 ml lysis buffer (250 mM imidazole) for the elution. 1 ml of SUMO protease (60) was added to the eluate and incubated at 4°C for 16 h while gently shaking. The protein was incubated with Ni-NTA beads to bind uncleaved protein and the supernatant was concentrated to 2 ml in S200 buffer (10 mM HEPES, 1 M NaCl, 5 mM MgCl2, 2 mM DTT, pH 7.0). Size exclusion purification was conducted on a Hiload 16/600 Superdex 200 column (AKTA Purifier system, GE Healthcare) in S200 buffer. The Cas13a-containing fractions were pooled and concentrated to ∼1.5 ml in Cas13a storage buffer (50 mM Tris–HCl, pH 7.5, 600 mM NaCl, 5% glycerol, 2 mM DTT). Protein concentration was determined using the absorbance at 280 nm and an extinction coefficient of 119 800 M−1 cm−1; 1.5 ml of 47 μM protein was obtained from 2 l of culture. Aliquots were diluted to 4.5 μM for use and stored at −20°C while the concentrated stock was stored at −80°C.
SPRINT reactions
Pentauridine RNA oligonucleotides (Integrated DNA Technologies) were labeled with carboxyfluorescein (FAM) or TEX 615 at the 5′-end and with Iowa Black® FQ at the 3′-end. Fluorescence measurements were taken at wavelengths 490/525 nm (excitation/emission) when the FAM fluorophore was used. In reactions with flavin-containing ligands (FMN or FAD), TEX-labeled RNA oligonucleotides were used at the wavelengths 576/615 (excitation/emission). For most SPRINT assays, a master mix was first prepared and then mixed with the remaining reaction components to yield a final reaction volume of 30 μl. Fluorescent readings of the reaction were performed in Corning 384 Flat Bottom Black Polystyrol plates covered with MicroAmp® Optical Adhesive Film to prevent evaporation of the sample. Fluorescence measurements were taken every 5 min.
For a riboswitch-regulated SPRINT reaction, SPRINT buffer was used (10x SPRINT buffer: 700 mM Tris–HCl, pH 8.0, 700 mM NaCl, 1 mM EDTA, 140 mM β-mercaptoethanol, 25 mM MgCl2). Aliquots of the 10× SPRINT buffer should be stored at −20°C and not undergo more than 10 freeze-thaw cycles to maintain activity. Master mix components were added in this order: water, 1× SPRINT buffer, 0.4 U/μl murine RNase Inhibitor (New England Biolabs, #M0314), 2.5 nM dsDNA template, 22.5 nM crRNA, 125 nM U5-RNA oligonucleotides, 45 nM Cas13a, 0.01 U/μl E. coli RNAP σ70 Holoenzyme (NEB, #M0551S). The master mix was gently mixed by pipetting up and down and incubated at 37°C for 15 min to allow binding of crRNA to Cas13a protein and binding of RNAP to its promoter sequence. The microwell plate was prepared by adding 3 μl of 10× ligand to the wells. The reaction was initiated by addition of rNTPs to the master mix to a concentration of 20 μM. For single turnover reactions, heparin needs to be added to 66 μg/ml in this last step. It is important to add heparin only after RNAP could bind its promoter sequence because heparin will prevent re-association of RNAP and DNA. Lastly, 27 μl of the complete master mix were added to each well. The plate was covered and inserted into a plate reader (Infinite® 200 PRO, Tecan) that was preheated to 37°C.
For SPRINT reactions with allosteric transcription factors (aTFs), ROSALIND (54) buffer was used (10x ROSALIND buffer: 400 mM Tris–HCl, pH 8.0, 200 mM NaCl, 20 mM spermidine, 100 mM DTT and 80 mM MgCl2). The buffer should be prepared fresh or stored as single-use aliquots at −80°C. The components are added in this order: water, 1× ROSALIND buffer, 0.4 U/μl murine RNase Inhibitor (New England Biolabs), 15 nM dsDNA template, 22.5 nM crRNA, 125 nM U5-RNA oligonucleotides, 45 nM Cas13a, 6.67 ng/μl T7 RNAP and aTF. The final concentration of the aTF monomers was either 2.5 μM TetR or 10 μM SmtB. The master mix was incubated for 15 min at 37°C to allow binding of crRNA to Cas13a protein, binding of RNAP to its promoter sequence, and binding of the aTFs to the operator sequences. Then, rNTPs were added to a concentration of 40 μM to initiate the reaction and the master mix was added to each well and pipetted up and down to mix with the ligand in the wells.
For the enzyme-coupled assay, buffer and reagents of a riboswitch-regulated SPRINT reaction were used as described above. Additionally, the following components were added: human purine nucleoside phosphorylase (Sigma-Aldrich, # 540221) was added to 0.01 U/μl, MgHPO4 was added to 1 mM, in some reactions Immucillin-H (also known as forodesine) (MedChemExpress, #HY-16209) was added to 10 μM.
For reactions in the handheld illuminator, reactions were assembled as described above, except that the final concentration of FAM-labeled U5-RNA oligonucleotides was increased to 1.25 μM so that the fluorescence could be seen by eye. Pictures were taken with a Sony α6300 digital camera.
For the two-batch method, 30 μl in vitro transcription reactions were carried out as described above but without addition of Cas13a, crRNA, labeled RNA oligonucleotides, RNase inhibitor. Then, the reaction was washed three times with 500 μl ddH2O using an Amicon centrifugal filter (0.5 ml, 10 kDa cutoff). After concentrating the solution to approximately 20 μl, aliquots of 6 μl were taken and added to a 24 μl SHERLOCK reaction.
SHERLOCK reactions
SHERLOCK reactions with purified target RNA as input were carried out following the protocols by Gootenberg et al. (44,45). Concentration of the components, order of addition of components and measurement of fluorescence was carried out the same way as in the SPRINT reactions. 10x SHERLOCK buffer contained 200 mM HEPES, pH 6.8, 600 mM NaCl, 60 mM MgCl2.
Model fitting
A curve was fit to the data using the equation
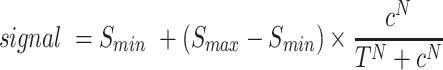 |
where signal is the fluorescent SPRINT signal or readthrough transcription, c is the concentration of ligand or target RNA input, Smax is the maximum signal, Smin is the minimal signal, T is the signal at half-maximal signal amplitude and N is a Hill coefficient. All data fitting was performed using Origin Pro 2020.
RESULTS
Coupling of transcription with RNA detection
Here, we present SHERLOCK-based profiling of in vitrotranscription (SPRINT) and show how this methodology can measure concentrations of various effector molecules and overcome limitations of existing methods. In an isothermal, one-step and one-batch reaction, Cas13a is targeted to an RNA site that is transcribed from a DNA template. As Cas13a does not cleave DNA, it will only detect its target sequence when it is transcribed as RNA. Cas13a subsequently cleaves fluorescently labeled pentauridine oligonucleotides, yielding a fluorescent signal. This assay was used to quantify the activation or repression of transcription under various conditions. Because transcription can be regulated by aTFs or riboswitches in a ligand-dependent manner, SPRINT can be used to quantify those ligands in samples with a fluorometric output (Figure 1D).
Optimization and benchmarking of SPRINT
As proof of principle, the well-described synthetic guanine-responsive riboswitch xpt/pbuE*6U (30) was used to establish reaction conditions for detecting transcription with Cas13a. The xpt/pbuE*6U contains the aptamer domain from the B. subtilis xpt-pbuX riboswitch which responds to the ligand guanine and a few related compounds such as hypoxanthine (61,62). The xpt aptamer is coupled to the pbuE*6U expression platform, which is derived from the B. subtilis pbuE adenine-responsive riboswitch (63). The nomenclature of aptamer/expression platform for chimeric riboswitches will be used throughout the manuscript. In the absence of guanine, the intrinsic terminator within the xpt/pbuE*6U riboswitch causes RNA polymerase to abort synthesis; in presence of guanine, full-length readthrough transcripts are synthesized that can be detected by Cas13a.
To couple in vitro transcription and Cas13a-mediated RNA detection, assay conditions had to be established for a reaction that is isothermal, one-step and one-batch. However, combining the reaction components at 37°C in SHERLOCK buffer (44) yielded a high background signal in the absence of ligand (Figure 2A). To overcome this issue, we tested a buffer that was used previously to assay synthetic riboswitches (30), referred to as SPRINT buffer. Unmodified SPRINT buffer reduced the background signal and enabled detection of the ligand with a 2.8-fold signal induction (Figure 2A). To further reduce the background signal, the contribution of Cas13a to the background signal was assessed. SHERLOCK reactions were run in SPRINT and SHERLOCK buffer (Figure 2B) and RNA could be reliably quantified in both buffers with negligible background signal (Supplementary Figure S1a). This indicates that the SHERLOCK components function efficiently in SPRINT buffer and instead in vitro transcription should be the focus for optimizing the dynamic range of the SPRINT assay.
Figure 2.

Optimization of conditions and benchmarking of signal. All fluorescence measurements were background corrected, bars indicate mean value and error bars indicate s.d. from the mean; n = 3. All measurements were normalized to 125 nM fluorescein. (A) SPRINT reactions were carried out with the guanine riboswitch xpt/pbuE*6U in SPRINT buffer or SHERLOCK buffer. Transcription of target RNA was regulated by the riboswitch in response to ±100 μM hypoxanthine. The measurements displayed were taken at 20 minutes reaction time. (B) SHERLOCK reactions were carried out in SHERLOCK buffer or SPRINT buffer at varying concentrations of added target RNA. The measurements displayed were taken at 20 min reaction time. (C, D) SPRINT reactions were carried out with the guanine riboswitch xpt/pbuE*6U with varying concentrations of rNTP and transcription template. Fluorescence was measured in response to ±10 μM guanine at 25 minutes reaction time. (E) SPRINT reactions were regulated by the guanine riboswitch xpt/pbuE*6U in response to ±10 μM guanine. Single turnover assays were conducted with 66 μg/ml heparin, multiple turnover assays were conducted without heparin.
The SPRINT buffer was systematically altered to identify improved conditions (Supplementary Figure S1B). SPRINT buffer is Tris-based, whereas SHERLOCK buffer is HEPES-based and when using HEPES instead of Tris buffer for SPRINT reactions, signal suppression in absence of ligand was abolished. This indicates that absence of HEPES in the SPRINT buffer is essential to enable transcription termination by the xpt/pbuE*6U riboswitch. Because bovine serum albumin (BSA) did not affect the assay, it was removed from the buffer (Supplementary Figure S1C) to decrease the risk of RNase contamination from BSA preparations. Since many hydrophobic small molecules of potential interest cannot be dissolved in water but can be dissolved in DMSO, the response of SPRINT reactions was also measured in increasing amounts of DMSO (Supplementary Figure S1D). Although DMSO slightly increases both background and on-signal, final concentrations of 10% DMSO did not inhibit the assay. However, most changes to the SPRINT buffer composition resulted either in no change or a decrease in detection of ligand-induced readthrough transcription (Supplementary Figure S1B).
To further optimize both signal intensity and reaction time, the concentration of NTPs and DNA template in the SPRINT reaction was systematically altered. The lowest concentrations of DNA and NTPs resulted in the largest fold change when transcription readthrough was induced with guanine (Figure 2C). However, because these conditions led to relatively weak signals (Figure 2D), we decided to compromise a higher fold induction for stronger signals and chose 2.5 nM DNA template and 20 μM NTP to enable reliable signal detection at 20 min reaction time. This modified SPRINT buffer (Figure 2A) is used in all experiments in this study, unless indicated otherwise. As the concentrations of NTPs and DNA template have a strong effect on inducibility and speed of the signal, they can be considered prime parameters for optimizing and adapting SPRINT assays.
The effect of heparin on SPRINT was assessed because in vitro transcription assays usually contain heparin to restrict RNA polymerase to a single turnover of transcription. SPRINT assays with the xpt/pbuE*6U guanine riboswitch were tested with and without heparin (Figure 2E). Repression of background signal is more efficient in the single turnover assays with heparin, but the induced signal in the multiple turnover assays was ∼35-fold larger and reached its maximum approximately twice as fast. This shows that multiple turnover transcription is mainly responsible for the strength of the fluorescent signal as opposed to the multiple turnover reactions of the Cas13a enzyme. This observation is consistent with previous results that showed amplification of the RNA input is necessary for a strong SHERLOCK signal (44,45). For this reason, most SPRINT assays were conducted without heparin but note that SPRINT can be run as a single turnover assay whenever a minimal background signal is preferred over a fast response.
To assess its sensitivity, the SPRINT reaction was compared to the gold standard approach of quantifying transcription products by separating 32P-labeled RNA transcripts using denaturing polyacrylamide gel electrophoresis (58). The transcriptional response of the guanine riboswitch xpt/pbuE*6U and the adenine riboswitch pbuE/pbuE‡ (64) to their respective ligand was measured by SPRINT and the radiolabeling method (Figure 3A, B). The T50 value, the ligand concentration at half-maximal activation of transcription, was obtained from a fit to the data and used to compare the methods. For both riboswitches, the T50 was similar between multiple turnover SPRINT, single turnover radiolabeling, and values from the literature that were also obtained with single turnover radiolabeling. This indicates that results obtained with SPRINT are comparable to those obtained with radiolabeling while increasing speed, ease, and throughput of transcription assays. Together, these data establish that we have effectively combined small-molecule dependent transcriptional regulation and Cas13a-mediated RNA detection into a single reaction.
Figure 3.

Dose-response of various riboswitches measured with SPRINT. All fluorescence measurements were background corrected, bars indicate mean value and error bars indicate s.d. from the mean; n = 3. (A, B) The responses of the guanine riboswitch xpt/pbuE*6U and the adenine riboswitch pbuE/pbuE‡ to their cognate ligands were measured. Results from SPRINT measurements are marked in blue, results from radiolabeling measurements are marked in red. The measurements displayed were taken at 20 min reaction time. Responses of various riboswitches to their cognate ligands and chemically similar compounds were measured: (C) a SAM sensing riboswitch, (D) a flavin mononucleotide sensing riboswitch, (E) a fluoride sensing riboswitch (signal measured at 60 minutes), and (F) a serotonin sensing riboswitch. (G) The native metE SAM sensing riboswitch is an OFF switch and attenuates transcription with increasing concentrations of ligand. Heparin was added to 66 μg/ml. Signal of the metE reaction was measured at 100 minutes reaction time. *(30), #(68), ‡(23), §(73), †(31).
SPRINT can be implemented with various riboswitches
To test the versatility and specificity of SPRINT, detection of other small molecules and ions via riboswitches was assessed. The adenine riboswitch pbuE/pbuE‡ responded selectively to adenine but not guanine (Figure 3B). The S-adenosylmethionine (SAM)-responsive riboswitch yitJ/pbuE*6U detected SAM but not the related compound S-adenosylhomocysteine (SAH), as expected (30). The T50 value for SAM was comparable to prior measurements using the 32P-labeling assay (Figure 3C). The flavin mononucleotide (FMN) riboswitch ribD/pbuE*7U riboswitch was observed to have a high-affinity response to FMN with a T50 value around 1.01 ± 0.03 μM, a low-affinity response to the closely related flavin adenine dinucleotide (FAD) (30,65), and no response to ribocil, which is a selective agonist of the related ribB riboswitch (66,67) (Figure 3D). This is consistent with published results (30,65–67). All four riboswitches (xpt/pbuE*6U, pbuE/pbuE‡, yitJ/pbuE*6U and ribD/pbuE*7U) are synthetic with variations of the pbuE expression platform (30), highlighting the utility of artificial riboswitches as robust biosensors.
Occasionally, reaction conditions needed to be optimized to accommodate new riboswitches. The ligand-dependent induction of the FMN riboswitch was initially very weak. To improve this, the magnesium concentration in the SPRINT reaction was increased. Of the four riboswitches that were tested at higher MgCl2 concentrations, only the ribD riboswitch showed a significant increase in fold induction (Supplementary Figure S2). Therefore, SPRINT reactions with the ribD/pbuE*7U riboswitch were conducted at 10 mM MgCl2 instead of 2.5.
Some ligands inhibited the SPRINT assay when added to high concentrations. The fluoride riboswitch crcB turns on transcription readthrough by selectively responding to fluoride. Using the SPRINT assay, we measured a T50 value of 11 ± 1 μM (Figure 3E), which is, surprisingly, lower than previously observed KD values of ∼60 μM resulting from in-line probing (68). This may be explained in part by an observed inhibition of the assay at fluoride concentrations above 100 μM. To understand whether this inhibitory effect stems from inhibition of the E. coli RNAP or inhibition of Cas13a, SHERLOCK reactions were conducted at varying concentrations of sodium fluoride. Cas13a was gradually inhibited with increasing fluoride concentrations and the signal sharply dropped ∼600 μM (Supplementary Figure S3A). To measure the response of E. coli RNAP to fluoride, a two-batch protocol was developed (Supplementary Figure S3B) to first transcribe RNA in presence of fluoride, then wash out the fluoride and add the washed transcripts to a SHERLOCK reaction. Fluoride induced transcription from the fluoride riboswitch in a linear manner up to concentrations of 100 μM, but above concentrations of 125 μM fluoride drastically reduced overall transcriptional activity (Supplementary Figure S3C). This suggests that E. coli RNAP itself is the most fluoride-sensitive component in the SPRINT reactions. Studies with crcB knockout strains of E. coli support these findings by showing significant growth inhibition when the fluoride concentrations in the media exceed 100 μM (68). These results show how the two-step assay can be used to separate in vitro transcription reactions from Cas13a reactions or potentially to wash out Cas13a-inhibiting compounds after transcription that would otherwise interfere with the assay.
Novel RNA aptamers are routinely generated via systematic evolution of ligands by exponential enrichment (SELEX) against compounds of interest (23,69–71) and the incorporation of such synthetic aptamers into SPRINT can greatly expand its biosensing repertoire. A synthetic aptamer that binds 5-hydroxytryptophan (5HTP) (23) was previously incorporated into the expression platform pbuE’ to generate an ON-riboswitch that responds to both 5HTP and serotonin (72). This riboswitch, called P1/pbuE’7U, was used with SPRINT to successfully detect serotonin. The T50 value was measured as 5 ± 3 μM for serotonin and the riboswitch discriminated against the related compound tryptamine (Figure 3F). Surprisingly, serotonin inhibited both the transcription and Cas13a reaction at concentrations >100 μM, whereas similar compounds such as 5HTP did not cause inhibitory effects (Supplementary Figure S3D–F). For this reason, the displayed dose-response curve should be interpreted with caution as inhibitory effects distort the signal at concentrations ∼100 μM. Despite the idiosyncratic inhibition by serotonin, these results demonstrate how expression platforms can be combined with synthetic aptamers in a plug-and-play manner to create novel biosensors for the SPRINT platform.
SPRINT can rapidly assess native riboswitch function
All riboswitches described in this study thus far are ‘ON’ switches that induce transcription in presence of ligands (30,31,68,72) and are thus predisposed to work well in the context of the SPRINT assay, whereas most native riboswitches are ‘OFF’ switches. To assess whether SPRINT can robustly monitor OFF switches, we examined the metE riboswitch from B. subtilis that aborts transcription in response to SAM (73). The native riboswitch sequence was amplified from the B. subtilis genome via PCR and in a second PCR step the tac promoter was appended to the 5′-end and the Cas13a target transcript to the 3′-end so that the resulting PCR product could be used directly as DNA template for SPRINT (Supplementary Figure S4A). Initially, very high concentrations of SAM were required to repress the signal (Supplementary Figure S4B). To circumvent this issue, heparin was added to the SPRINT assay to restrict the RNA polymerase to a single turnover of transcription. Although the reaction was slowed down, the sensitivity of the assay was greatly improved. Titrating SAM yielded a T50 value of around 1 μM (Figure 3G), which is similar to previously established values of 0.5 and 2 μM (31,73). This demonstrates that both, ON and OFF riboswitches can be integrated into a DNA template for SPRINT.
SPRINT can sense small molecules via transcriptional repressor proteins
Previously developed methods use allosteric transcription factors (aTFs) to detect compounds. Jung et al. (54) developed a system called ROSALIND that senses compounds with aTFs to regulate T7 RNAP-driven transcription of a fluorophore-binding aptamer (74) and thereby yielding a fluorometric output (Figure 1A). In this manner, 16 diverse compounds, including tetracyclines, macrolides, small molecules and metal ions were detected. To incorporate the broad repertoire of aTF-biosensors into SPRINT, we explored conditions that enable an isothermal, one-batch and one-step reaction that detects compounds with aTFs and generates a signal via Cas13a.
The ROSALIND buffer was adopted for aTF-based SPRINT reactions, although some modifications were made to the buffer in order to optimize sensitivity and speed of the reaction. The inorganic pyrophosphatase (IPPase) enzyme was deemed unnecessary and removed from the ROSALIND buffer. Further adaptions include reducing the concentrations of rNTP from 2.85 mM to 40 μM, DNA template from 25 to 15 nM, and T7 RNAP from 10 to 6.7 ng/μl.
Using the adapted buffer and conditions, the responses of the repressors TetR and SmtB were measured with SPRINT. The repressor SmtB enabled transcription in a zinc-dependent manner with a T50 of 4.8 ± 0.3 μM and showed a high selectivity against copper (Figure 4A). The repressor TetR responded to increasing concentrations of anhydrotetracycline with a T50 of 1.9 ± 0.2 μM which is within the range of 1–2.5 μM previously measured (54) (Figure 4B). This shows that SPRINT can not only measure regulation of E. coli RNAP by riboswitches, but also regulation of T7 RNAP by transcription factors. Combining these two mechanisms of detecting ligands greatly expands the scope of molecules that can be detected with SPRINT.
Figure 4.

Transcription regulation by transcription factors. Fluorescent signal of the SPRINT reactions was measured at 20 minutes reaction time. Background was subtracted and signal normalized to 125 nM fluorescein. Bars indicate mean value, error bars indicate s.d. from the mean. n = 3. (A) SmtB de-represses transcription in response to zinc, but not copper. (B) TetR de-represses transcription in response to anhydrotetracycline. *(54).
SPRINT can screen compounds in a high-throughput format
There is a growing interest in drugging RNA structures (75–79) including targets such as the HIV TAR element (80), human expansion repeats (75,81), or riboswitches (82–85) but developing platforms for high-throughput screening of RNA-drug interactions remains challenging. As a proof of concept, the response of the guanine riboswitch xpt/pbuE*6U to 30 different small organic molecules was measured in a high-throughput screening format (Figure 5A, Supplementary Figure S5A). For all compounds, the SPRINT signal at high concentration was plotted against the signal at low concentration to visualize the dose-response relation for each compound. Exact concentrations and full names of the compounds can be found in Supplementary Table S3. Compounds such as guanine that elicited an equally strong transcriptional activation at low and high concentrations are found in the upper right quadrant of the plot and are expected to be strong agonists of the target. Compounds such as N2-methylguanine that are found in the upper left quadrant are expected to have less agonistic activity towards the RNA target. Non-binding compounds such as N6-methyladenine are in the lower left quadrant. Compounds such as 7-deazaguanine that showed some activation of transcription at low concentrations but reduced or no activation at high concentrations are suspected to inhibit the assay at higher concentrations. Notably, the results of this screen are consistent with prior reports on binding of individual compounds to the xpt aptamer domain using ITC or footprinting approaches (61,86–89). Therefore, the compound screen with SPRINT enabled us to quickly assess the effect of various compounds on the RNA target to find compounds of interest.
Figure 5.

SPRINT can be used for compound screens and enzyme-coupled assays. Fluorescent signal of the SPRINT reactions was background subtracted and normalized to 125 nM fluorescein. (A) The guanine riboswitch xpt/pbuE*6U regulated transcription in response to 30 different compounds. Signal with solvent only was subtracted from signal with ligand to correct for differences in solvents of ligands. Dots represent mean value, n = 3. (B) The effect of the compounds on transcription from a constitutive promoter was measured. Signal with solvent only was subtracted from signal with ligand to correct for differences in solvents of ligands. Dots represent one biological replicate. (C) SPRINT measured constitutive transcription of Cas13a target as transcription was inhibited at increasing concentrations of rifampicin. Bars indicate mean value, error bars indicate s.d. from the mean. n = 3. (D) Diagram of an enzyme-coupled assay. The enzyme hPNP converts inosine to hypoxanthine, which is detected by the guanine riboswitch xpt/pbuE*6U and triggers the SPRINT signal. (E) The enzyme-coupled assay was used to measure enzymatic activity of hPNP. Concentration of inosine was 1 mM, hPNP was added to an activity of 10 mU/μl, concentration of Immucillin-H was 10 μM. Bars indicate mean value, error bars indicate s.d. from the mean. n = 3. p-value was calculated using a two-tailed t-test. (F) The enzyme-coupled assay was used to measure concentration-dependent substrate conversion. hPNP was added to an activity of 1 mU/μl. Bars indicate mean value, error bars indicate s.d. from the mean. n = 3.
When screening for target inhibitor drugs, it is essential to reliably identify pan-assay interference compounds (PAINS) (90) that might show up as false positives. For this purpose, the drug panel was tested in SPRINT reactions that constitutively transcribe a Cas13a target without regulatory components such as riboswitches or aTFs. This setup only measures effects of compounds on the core assay itself. In this way, compounds such as 7-deazaguanine, N2-methylguanine, 2,5,6-triaminpyrimidin-4-one (2,5,6-TAP) and to a lesser extent 2-fluoroadenine could successfully be identified as assay-interfering compounds (Figure 5B, Supplementary S5B, Supplementary Table S3).
Previous studies were aimed at identifying novel antibiotics by targeting the transcriptional machinery of bacteria (91,92). This can include targeting riboswitches (66,82–84,93), transcription factors (94,95) or the RNA polymerase itself (96,97). Rifampicin inhibits the bacterial RNAP (98) by blocking elongation (99) and is the leading drug to treat mycobacterial infections such as tuberculosis (100). We measured the inhibitory effect of rifampicin on the RNAP with SPRINT. A constitutive transcription reaction was titrated with different concentrations of rifampicin (Figure 5C). A T50 value of 5.9 ± 0.6 nM was obtained, which is very close to the established KD of 3 nM (101). This demonstrates how SPRINT can be used to screen for drugs that target transcription and rapidly quantify drug efficacy.
Enzyme-coupled assay
SPRINT can generate fast fluorescent signals in response to various compounds and therefore could be used to detect products of enzymatic reactions. Most enzyme coupled assays are limited to the detection of one particular metabolite such as ADP generated in a kinase reaction or NADH generated in a redox reaction (102). Detecting enzymatic products with a riboswitch or transcription factor is an attractive alternative because of the large diversity of compounds that can be detected with these systems. We examined human purine nucleoside phosphorylase (hPNP) (103,104), part of the purine salvage pathway, which catalyzes the phosphorylation of the ribose moiety of various purine nucleosides and concurrent removal of the sugar from the nucleobase and assessed conditions for an isothermal, one-batch and one-step enzyme-coupled SPRINT assay.
The conversion of inosine to hypoxanthine by hPNP was coupled to the synthetic guanine riboswitch xpt/pbuE*6U (Figure 5D). The guanine riboswitch responds to hypoxanthine (30) but does not bind nucleosides such as inosine. This enzyme-coupled assay enabled the observation of enzymatic activity by the hPNP enzyme (Figure 5E) without changing any components of the SPRINT buffer, except adding the substrates inosine and phosphate (Supplementary Figure S6). Titration of the coupled reaction with inosine showed a concentration-dependent increase of hypoxanthine production by the hPNP enzyme (Figure 5F). The conversion of deoxyguanosine to guanine by hPNP can be inhibited with nucleoside analogs such as Immucillin-H (forodesine) which leads to accumulation of deoxyguanosine and consequent apoptosis in activated T-cells. Therefore, hPNP is an important drug target for the treatment of leukemia, arthritis, multiple sclerosis and transplant rejection (105–111). Adding the competitive inhibitor Immucillin-H at 100-fold lower concentration than the substrate inosine caused a significant reduction in the enzymatic activity as measured in the SPRINT assay (Figure 5E). Together, these results demonstrate how SPRINT can be used to assess the activity of enzymes, measure the inhibition of such enzymes with drugs and indirectly detect compounds such as inosine that are not bound by aTFs or riboswitches.
Portable assay formats
To address the need for point-of-care diagnostic devices, SPRINT needs to be adaptable to portable devices such as lateral flow assays (LFA) (45,112) or portable fluorometric devices (37,54,113,114). An inexpensive, hand-held device that can be made with a 3D-printer was previously developed for detecting water contaminants via fluorescence (54). This portable device illuminates reaction tubes with blue light ∼470 nm and fluorescent probes can be seen through a yellow film used as a long-pass optical filter. Fluorescence from SPRINT reactions was easily visible to the human eye using the device and no adaptations were required except increasing the concentration of FAM-labeled RNA in SPRINT reactions to 1.25 μM (Supplementary Figure S7). Using the fluoride riboswitch, different fluoride concentrations could be differentiated by eye after 20 minutes of reaction time (Figure 6A) at 30°C which can be easily achieved by tightly holding the tubes in the hand. Further, the zinc-responsive transcription factor SmtB was used to detect zinc in municipal water samples that were collected, filtered and shipped from Paradise, California where zinc levels were affected by 2018 Camp Fire (54) (Figure 6B). By comparing the fluorescence of the samples with a calibration curve, the zinc concentration of the samples could be approximated by eye to be around 0–1 μM (I), 2.5–5 μM (II) and ∼5 μM (III), respectively. The zinc concentration in the municipal water samples was previously determined by flame atomic absorption spectroscopy (FAAS) (54) for comparison. These results demonstrate that SPRINT can be adapted to inexpensive portable formats and shows how the assay could be used for field testing.
Figure 6.

Detection of analytes with a handheld device. SPRINT assays were assembled and added to water samples containing varying concentrations of the analyte. The concentrations indicated refer to the final concentration of analyte after mixing sample with the assay components. After sample and assay components were mixed, the tubes were placed in the LED device and photographs were taken right afterwards (0 min). The different appearances of the fluorescent samples in A and B are due to variations of the ambient light when the pictures were taken. (A) SPRINT reactions were run with fluoride riboswitch crcB. (B) SPRINT reactions were run with the transcription factor SmtB that responds to zinc. The five reactions on the left contained ddH2O with added ZnCl2. The three reactions to the right labeled I, II, and III contained municipal water samples from Paradise, California.
DISCUSSION
Here, we show that the transcriptional response to various small molecules can be coupled to a fast and convenient fluorescent output. This allows the rapid detection and quantification of compounds in laboratory settings or with portable devices, leveraging the broad biosensory repertoire of transcription factors and riboswitches. Furthermore, we demonstrate how enzymatic reactions can be coupled to SPRINT to report enzymatic activity, measure the effect of clinically relevant drugs, or detect compounds that are not directly bound by available biosensors. Performing all of those functions in a fast, inexpensive, isothermal and single-step reaction overcomes issues associated with previous technologies and provides a significant advancement in the field of small-molecule detection.
A critical factor for any diagnostic platform is its adaptability to the detection of new and different compounds. SPRINT has the potential to detect a broad range of compounds such as metabolites, ions and other small molecules because it can leverage both riboswitches and transcription factors. Furthermore, modifying the specificity of aTFs (6–17) or riboswitches (23–25,27–34) can be used to detect new compounds of interest. When necessary, we have shown how the buffer or reaction conditions could be adapted to a specific analyte by rational optimization. For example, the FMN riboswitch ribD required increased magnesium concentrations to function, which is not surprising, given that ribD is the largest and structurally most complex riboswitch that was tested in this study and magnesium is essential to riboswitch folding and stability (115). Similarly, NaHPO4 sequestered magnesium from the buffer, but hPNP requires inorganic phosphate as a substrate so MgHPO4 was simply added instead of NaHPO4 to avoid sequestering of magnesium by the phosphate. Additionally, the robustness of the key enzyme Cas13a probably contributed to the adaptability of SPRINT, because Cas13a has been demonstrated to function in three different buffer systems (SHERLOCK, ROSALIND, SPRINT) and at different temperatures (30°C, 37°C). Given the diversity of conditions and biosensors used in this work, we anticipate that small and rational adjustments should suffice to adapt SPRINT to new reactions or analytes.
Future research into riboswitches could be significantly accelerated by the method presented here. Importantly, SPRINT measures the transcriptional response to a compound as opposed to binding of the ligand at equilibrium. This is particularly important, given that many riboswitches rely on the kinetics of co-transcriptional ligand binding (116) and because methods such as ITC or in-line or chemical probing that measure ligand binding to RNA are laborious and time consuming (117). The increased throughput has the potential to greatly facilitate identification of ligands for ‘orphan’ riboswitches (118) whose effector molecule is not yet known. Similarly, riboswitches could be screened for responses to drugs to identify potential antibiotics. Previous studies used phenotypic screens to assess the efficacy of potential riboswitch-targeting antibiotics (66,85,119). However, during unbiased screens for antibiotics the potential drug targets are initially unknown, which requires the subsequent identification of the drug targets (119,120). SPRINT combines advantages of these different approaches by being a target-based, yet functional screen.
Apart from riboswitches, SPRINT might also be used for drug screens against other RNA targets. It has been shown previously that well-characterized expression platforms can be combined with new aptamer domains through rational design (23,30,31) and likewise it might be possible to combine expression platforms with RNA structures of clinical interest so that drug binding to the target RNA elicits a transcriptional response. While some previous efforts have been successful in identifying small molecule inhibitors of functional human or viral RNAs (75–81), most approaches require chemical modification and immobilization of the drug (75) which prevents a complete exploration of possible drug-target interactions. Developing a high-throughput platform with SPRINT could overcome these issues and would be a great advancement towards drugging RNA.
As continued progress is being made in the development of SHERLOCK, a core component of SPRINT, those developments can be incorporated into SPRINT to advance its utility. SHERLOCK has already been adapted to portable paper strip format (45), which could likewise be applied to a paper strip format for SPRINT. Also, a protocol was recently published that describes how clinical samples can be quickly prepared for SHERLOCK tests by heat-inactivating endogenous RNases (121). This protocol was initially applied to detect nucleic acids in clinical samples with SHERLOCK, but it would be conceivably easy to apply this protocol to SPRINT and thus detect specific metabolites in patient samples. Also, a massively multiplexed droplet-based version of SHERLOCK has been published (48) that can process thousands of different samples at once, which could be applied to vastly increase the throughput of SPRINT. Even though SPRINT was already developed to be a flexible, fast, and easy-to-use biosensor, this abundance of interfaceable technology shows that there are still many avenues available to improve and expand its capabilities.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Meagan Nakamoto for gifting the SUMO protease and Julius B. Lucks, Kirsten J. Jung and Walter Thavarajah for gifting the purified transcription factors TetR and SmtB, plasmids encoding the respective operator sequences, the illuminator device, the California water samples and for providing insightful discussions.
Author contributions: R.S.I. and R.T.B. conceived the project and wrote the manuscript. R.S.I. carried out all experiments and data analyses.
Contributor Information
Roman S Iwasaki, Department of Biochemistry, University of Colorado, Boulder, CO 80309-0596, USA.
Robert T Batey, Department of Biochemistry, University of Colorado, Boulder, CO 80309-0596, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R01 GM073850 to R.T.B.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. The authors R.T.B. and R.S.I. declare the following competing financial interests: R.T.B. and R.S.I. have submitted a patent application related to the use of SPRINT for direct or indirect detection and quantification of substances.
REFERENCES
- 1. Bervoets I., Charlier D.. Diversity, versatility and complexity of bacterial gene regulation mechanisms: opportunities and drawbacks for applications in synthetic biology. FEMS Microbiol. Rev. 2019; 43:304–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weickert M.J., Adhya S.. A family of bacterial regulators homologous to Gal and Lac repressors. J. Biol. Chem. 1992; 267:15869–15874. [PubMed] [Google Scholar]
- 3. Gallegos M.T., Schleif R., Bairoch A., Hofmann K., Ramos J.L.. Arac/XylS family of transcriptional regulators. Microbiol. Mol. Biol. Rev. 1997; 61:393–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ramos J.L., Martinez-Bueno M., Molina-Henares A.J., Teran W., Watanabe K., Zhang X., Gallegos M.T., Brennan R., Tobes R.. The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 2005; 69:326–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fernandez-López R., Ruiz R., de la Cruz F., Moncalián G.. Transcription factor-based biosensors enlightened by the analyte. Front. Microbiol. 2015; 6:648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Taylor N.D., Garruss A.S., Moretti R., Chan S., Arbing M.A., Cascio D., Rogers J.K., Isaacs F.J., Kosuri S., Baker D. et al.. Engineering an allosteric transcription factor to respond to new ligands. Nat. Methods. 2016; 13:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Paepe B., Peters G., Coussement P., Maertens J., De Mey M.. Tailor-made transcriptional biosensors for optimizing microbial cell factories. J. Ind. Microbiol. Biotechnol. 2017; 44:623–645. [DOI] [PubMed] [Google Scholar]
- 8. Voyvodic P.L., Pandi A., Koch M., Conejero I., Valjent E., Courtet P., Renard E., Faulon J.-L., Bonnet J.. Plug-and-play metabolic transducers expand the chemical detection space of cell-free biosensors. Nat. Commun. 2019; 10:1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu J., Jiang P., Chen W., Xiong D., Huang L., Jia J., Chen Y., Jin J.-M., Tang S.-Y.. Design and application of a lactulose biosensor. Sci. Rep. 2017; 7:45994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Raman S., Taylor N., Genuth N., Fields S., Church G.M.. Engineering allostery. Trends Genet. 2014; 30:521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Juárez J.F., Lecube-Azpeitia B., Brown S.L., Johnston C.D., Church G.M.. Biosensor libraries harness large classes of binding domains for construction of allosteric transcriptional regulators. Nat. Commun. 2018; 9:3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Machado LF.M., Currin A., Dixon N.. Directed evolution of the PcaV allosteric transcription factor to generate a biosensor for aromatic aldehydes. J. Biol. Eng. 2019; 13:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang J., Wang Z., Su T., Sun H., Zhu Y., Qi Q., Wang Q.. Tuning the binding affinity of heme-responsive biosensor for precise and dynamic pathway regulation. iScience. 2020; 23:101067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chang H.-J., Mayonove P., Zavala A., De Visch A., Minard P., Cohen-Gonsaud M., Bonnet J.. A modular receptor platform to expand the sensing repertoire of bacteria. ACS Synth. Biol. 2018; 7:166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hanko E.K.R., Minton N.P., Malys N.. A transcription factor-based biosensor for detection of itaconic acid. ACS Synth. Biol. 2018; 7:1436–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grazon C., Baer R.C., Kuzmanović U., Nguyen T., Chen M., Zamani M., Chern M., Aquino P., Zhang X., Lecommandoux S. et al.. A progesterone biosensor derived from microbial screening. Nat. Commun. 2020; 11:1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Snoek T., Chaberski E.K., Ambri F., Kol S., Bjørn S.P., Pang B., Barajas J.F., Welner D.H., Jensen M.K., Keasling J.D.. Evolution-guided engineering of small-molecule biosensors. Nucleic Acids Res. 2020; 48:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Breaker R.R. Riboswitches and the RNA World. Cold Spring Harb. Perspect. Biol. 2012; 4:a003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Breaker R.R. Riboswitches and translation control. Cold Spring Harb. Perspect. Biol. 2018; 10:a032797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garst A.D., Edwards A.L., Batey R.T.. Riboswitches: structures and mechanisms. Cold Spring Harb. Perspect. Biol. 2011; 3:a003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pavlova N., Kaloudas D., Penchovsky R.. Riboswitch distribution, structure, and function in bacteria. Gene. 2019; 708:38–48. [DOI] [PubMed] [Google Scholar]
- 22. Robinson C.J., Medina-Stacey D., Wu M.-C., Vincent H.A., Micklefield J.. Rewiring riboswitches to create new genetic circuits in bacteria. Methods in Enzymology. Elsevier. 2016; 575:319–348. [DOI] [PubMed] [Google Scholar]
- 23. Porter E.B., Polaski J.T., Morck M.M., Batey R.T.. Recurrent RNA motifs as scaffolds for genetically encodable small-molecule biosensors. Nat. Chem. Biol. 2017; 13:295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boussebayle A., Torka D., Ollivaud S., Braun J., Bofill-Bosch C., Dombrowski M., Groher F., Hamacher K., Suess B.. Next-level riboswitch development-implementation of capture-SELEX facilitates identification of a new synthetic riboswitch. Nucleic Acids Res. 2019; 47:4883–4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weigand J.E., Sanchez M., Gunnesch E.-B., Zeiher S., Schroeder R., Suess B.. Screening for engineered neomycin riboswitches that control translation initiation. RNA. 2008; 14:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wachsmuth M., Findeiss S., Weissheimer N., Stadler P.F., Morl M.. De novo design of a synthetic riboswitch that regulates transcription termination. Nucleic Acids Res. 2013; 41:2541–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Davidson M.E., Harbaugh S.V., Chushak Y.G., Stone M.O., Kelley-Loughnane N.. Development of a 2, 4-dinitrotoluene-responsive synthetic riboswitch in E. coli cells. ACS Chem. Biol. 2013; 8:234–241. [DOI] [PubMed] [Google Scholar]
- 28. Xiu Y., Jang S., Jones J.A., Zill N.A., Linhardt R.J., Yuan Q., Jung G.Y., Koffas M.A.G.. Naringenin-responsive riboswitch-based fluorescent biosensor module for Escherichia coli co-cultures. Biotechnol. Bioeng. 2017; 114:2235–2244. [DOI] [PubMed] [Google Scholar]
- 29. Dwidar M., Seike Y., Kobori S., Whitaker C., Matsuura T., Yokobayashi Y.. Programmable artificial cells using histamine-responsive synthetic riboswitch. J. Am. Chem. Soc. 2019; 141:11103–11114. [DOI] [PubMed] [Google Scholar]
- 30. Ceres P., Trausch J.J., Batey R.T.. Engineering modular ‘ON’ RNA switches using biological components. Nucleic Acids Res. 2013; 41:10449–10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ceres P., Garst A.D., Marcano-Velázquez J.G., Batey R.T.. Modularity of select riboswitch expression platforms enables facile engineering of novel genetic regulatory devices. ACS Synth. Biol. 2013; 2:463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Findeiß S., Etzel M., Will S., Mörl M., Stadler P.. Design of artificial riboswitches as biosensors. Sensors. 2017; 17:1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu M.J., Andreasson J.O.L., Kladwang W., Greenleaf W., Das R.. Automated design of diverse Stand-Alone riboswitches. ACS Synth. Biol. 2019; 8:1838–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martini L., Ellington A.D., Mansy S.S.. An in vitro selection for small molecule induced switching RNA molecules. Methods. 2016; 106:51–57. [DOI] [PubMed] [Google Scholar]
- 35. Du Y., Dong S.. Nucleic acid biosensors: recent advances and perspectives. Anal. Chem. 2017; 89:189–215. [DOI] [PubMed] [Google Scholar]
- 36. Yan S.-R., Foroughi M.M., Safaei M., Jahani S., Ebrahimpour N., Borhani F., Rezaei Zade Baravati N., Aramesh-Boroujeni Z., Foong L.K.. A review: recent advances in ultrasensitive and highly specific recognition aptasensors with various detection strategies. Int. J. Biol. Macromol. 2020; 155:184–207. [DOI] [PubMed] [Google Scholar]
- 37. Dhiman A., Kalra P., Bansal V., Bruno J.G., Sharma T.K.. Aptamer-based point-of-care diagnostic platforms. Sens. Actuators B Chem. 2017; 246:535–553. [Google Scholar]
- 38. Ilgu M., Nilsen-Hamilton M.. Aptamers in analytics. Analyst. 2016; 141:1551–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cho E.J., Lee J.-W., Ellington A.D.. Applications of aptamers as sensors. Annu. Rev. Anal. Chem. 2009; 2:241–264. [DOI] [PubMed] [Google Scholar]
- 40. Edwards A.L., Garst A.D., Batey R.T.. Mayer G. Nucleic Acid and Peptide Aptamers: Methods and Protocols. 2009; NY: Humana. [Google Scholar]
- 41. Beckert B., Masquida B.. Nielsen H. Synthesis of RNA by in vitro transcription. RNA: Methods and Protocols. 2011; Totowa: Humana Press; 29–41. [DOI] [PubMed] [Google Scholar]
- 42. Voss C., Schmitt B., Werner-Simon S., Lutz C., Simon W., Anderl J.. A novel, non-radioactive eukaryotic in vitro transcription assay for sensitive quantification of RNA polymerase II activity. BMC Mol. Biol. 2014; 15:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang J., Zhao S., Zhou Y., Wei Y., Deng W.. Establishment and validation of a non-radioactive method for in vitro transcription assay using primer extension and quantitative real time PCR. PLoS One. 2015; 10:e0135317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gootenberg J.S., Abudayyeh O.O., Lee J.W., Essletzbichler P., Dy A.J., Joung J., Verdine V., Donghia N., Daringer N.M., Freije C.A. et al.. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017; 356:438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gootenberg J.S., Abudayyeh O.O., Kellner M.J., Joung J., Collins J.J., Zhang F.. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science. 2018; 360:439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Abudayyeh O.O., Gootenberg J.S., Konermann S., Joung J., Slaymaker I.M., Cox D.B.T., Shmakov S., Makarova K.S., Semenova E., Minakhin L. et al.. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016; 353:aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Joung J., Ladha A., Saito M., Segel M., Bruneau R., Huang M.W., Kim N.-G., Yu X., Li J., Walker B.D. et al.. Point-of-care testing for COVID-19 using SHERLOCK diagnostics. 2020; medrXiv doi:08 May 2020, preprint: not peer reviewed 10.1101/2020.05.04.20091231. [DOI]
- 48. Ackerman C.M., Myhrvold C., Thakku S.G., Freije C.A., Metsky H.C., Yang D.K., Ye S.H., Boehm C.K., Kosoko-Thoroddsen T.-S.F., Kehe J. et al.. Massively multiplexed nucleic acid detection using Cas13. Nature. 2020; 582:277–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li S.-Y., Cheng Q.-X., Wang J.-M., Li X.-Y., Zhang Z.-L., Gao S., Cao R.-B., Zhao G.-P., Wang J.. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 2018; 4:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen J.S., Ma E., Harrington L.B., Da Costa M., Tian X., Palefsky J.M., Doudna J.A.. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science. 2018; 360:436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liang M., Li Z., Wang W., Liu J., Liu L., Zhu G., Karthik L., Wang M., Wang K.-F., Wang Z. et al.. A CRISPR-Cas12a-derived biosensing platform for the highly sensitive detection of diverse small molecules. Nat. Commun. 2019; 10:3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xiong Y., Zhang J., Yang Z., Mou Q., Ma Y., Xiong Y., Lu Y.. Functional DNA regulated CRISPR-Cas12a sensors for point-of-care diagnostics of non-nucleic-Acid targets. J. Am. Chem. Soc. 2020; 142:207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li J., Yang S., Zuo C., Dai L., Guo Y., Xie G.. Applying CRISPR-Cas12a as a signal amplifier to construct biosensors for non-DNA targets in ultralow concentrations. ACS Sens. 2020; 5:970–977. [DOI] [PubMed] [Google Scholar]
- 54. Jung J.K., Alam K.K., Verosloff M.S., Capdevila D.A., Desmau M., Clauer P.R., Lee J.W., Nguyen P.Q., Pastén P.A., Matiasek S.J. et al.. Cell-free biosensors for rapid detection of water contaminants. Nat. Biotechnol. 2020; doi:10.1038/s41587-020-0571-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Quan J., Tian J.. Circular polymerase extension cloning for high-throughput cloning of complex and combinatorial DNA libraries. Nat. Protoc. 2011; 6:242–251. [DOI] [PubMed] [Google Scholar]
- 56. de Boer H.A., Comstock L.J., Vasser M.. The tac promoter: a functional hybrid derived from the trp and lac promoters. Proc. Natl. Acad. Sci. U.S.A. 1983; 80:21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wostenberg C., Ceres P., Polaski J.T., Batey R.T.. A highly coupled network of tertiary interactions in the SAM-I Riboswitch and their role in regulatory tuning. J. Mol. Biol. 2015; 427:3473–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Artsimovitch I., Henkin T.M.. In vitro approaches to analysis of transcription termination. Methods. 2009; 47:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Burgess R.R. Use of polyethyleneimine in purification of DNA-Binding proteins. Methods Enzymol. 1991; 208:3–10. [DOI] [PubMed] [Google Scholar]
- 60. Mossessova E., Lima C.D.. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in Yeast. Mol. Cell. 2000; 5:865–876. [DOI] [PubMed] [Google Scholar]
- 61. Mandal M., Boese B., Barrick J.E., Winkler W.C., Breaker R.R.. Riboswitches control fundamental biochemical pathways in bacillus subtilis and other bacteria. Cell. 2003; 113:577–586. [DOI] [PubMed] [Google Scholar]
- 62. Batey R.T., Gilbert S.D., Montange R.K.. Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature. 2004; 432:411–415. [DOI] [PubMed] [Google Scholar]
- 63. Mandal M., Breaker R.R.. Adenine riboswitches and gene activation by disruption of a transcription terminator. Nat. Struct. Mol. Biol. 2004; 11:29–35. [DOI] [PubMed] [Google Scholar]
- 64. Drogalis L.K., Batey R.T.. Requirements for efficient cotranscriptional regulatory switching in designed variants of the Bacillus subtilis pbuE adenine-responsive riboswitch. 2018; biorXiv doi:19 July 2018, preprint: not peer reviewed 10.1101/372573. [DOI] [PMC free article] [PubMed]
- 65. Winkler W.C., Cohen-Chalamish S., Breaker R.R.. An mRNA structure that controls gene expression by binding FMN. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:15908–15913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Howe J.A., Wang H., Fischmann T.O., Balibar C.J., Xiao L., Galgoci A.M., Malinverni J.C., Mayhood T., Villafania A., Nahvi A. et al.. Selective small-molecule inhibition of an RNA structural element. Nature. 2015; 526:672–677. [DOI] [PubMed] [Google Scholar]
- 67. Howe J.A., Xiao L., Fischmann T.O., Wang H., Tang H., Villafania A., Zhang R., Barbieri C.M., Roemer T.. Atomic resolution mechanistic studies of ribocil: a highly selective unnatural ligand mimic of the E. coli FMN riboswitch. RNA Biol. 2016; 13:946–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Baker J.L., Sudarsan N., Weinberg Z., Roth A., Stockbridge R.B., Breaker R.R.. Widespread genetic switches and toxicity resistance proteins for fluoride. Science. 2012; 335:233–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ellington A.D., Szostak J.W.. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990; 346:818–822. [DOI] [PubMed] [Google Scholar]
- 70. Tuerk C., Gold L.. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990; 249:505–510. [DOI] [PubMed] [Google Scholar]
- 71. McKeague M., DeRosa M.C.. Challenges and opportunities for small molecule aptamer development. J. Nucleic Acids. 2012; 2012:748913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Porter E.B. 2015; The Use of Riboswitches as Scaffolds for Selection and Novel RNA Devices, Ph.D. Dissertation Thesis. [Google Scholar]
- 73. Tomsic J., McDaniel B.A., Grundy F.J., Henkin T.M.. Natural variability in S-adenosylmethionine (SAM)-dependent riboswitches: S-box elements in Bacillus subtilis exhibit differential sensitivity to SAM in vivo and in vitro. J. Bacteriol. 2008; 190:823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Alam K.K., Tawiah K.D., Lichte M.F., Porciani D., Burke D.H.. A fluorescent split aptamer for visualizing RNA–RNA assembly in vivo. ACS Synth. Biol. 2017; 6:1710–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Disney M.D., Dwyer B.G., Childs-Disney J.L.. Drugging the RNA World. Cold Spring Harb. Perspect. Biol. 2018; 10:a034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rizvi N.F., Smith G.F.. RNA as a small molecule druggable target. Bioorg. Med. Chem. Lett. 2017; 27:5083–5088. [DOI] [PubMed] [Google Scholar]
- 77. Warner K.D., Hajdin C.E., Weeks K.M.. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018; 17:547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Matsui M., Corey D.R.. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2017; 16:167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Brown J.A. Unraveling the structure and biological functions of RNA triple helices. WIREs RNA. 2020; e1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chavali S.S., Bonn-Breach R., Wedekind J.E.. Face-time with TAR: Portraits of an HIV-1 RNA with diverse modes of effector recognition relevant for drug discovery. J. Biol. Chem. 2019; 294:9326–9341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Benhamou R.I., Angelbello A.J., Andrews R.J., Wang E.T., Moss W.N., Disney M.D.. Structure-Specific cleavage of an RNA repeat expansion with a dimeric small molecule is advantageous over Sequence-Specific recognition by an oligonucleotide. ACS Chem. Biol. 2020; 15:485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lünse C.E., Schüller A., Mayer G.. The promise of riboswitches as potential antibacterial drug targets. Int. J. Med. Microbiol. 2014; 304:79–92. [DOI] [PubMed] [Google Scholar]
- 83. Matzner D., Mayer G.. (Dis)similar analogues of riboswitch metabolites as antibacterial lead Compounds: Miniperspective. J. Med. Chem. 2015; 58:3275–3286. [DOI] [PubMed] [Google Scholar]
- 84. Mehdizadeh Aghdam E., Hejazi M.S., Barzegar A.. Riboswitches: from living biosensors to novel targets of antibiotics. Gene. 2016; 592:244–259. [DOI] [PubMed] [Google Scholar]
- 85. Mulhbacher J., Brouillette E., Allard M., Fortier L.-C., Malouin F., Lafontaine D.A.. Novel riboswitch ligand analogs as selective inhibitors of guanine-related metabolic pathways. PLoS Pathog. 2010; 6:e1000865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gilbert S.D., Stoddard C.D., Wise S.J., Batey R.T.. Thermodynamic and kinetic characterization of ligand binding to the purine riboswitch aptamer domain. J. Mol. Biol. 2006; 359:754–768. [DOI] [PubMed] [Google Scholar]
- 87. Gilbert S.D., Reyes F.E., Edwards A.L., Batey R.T.. Adaptive ligand binding by the purine riboswitch in the recognition of guanine and adenine analogs. Structure. 2009; 17:857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Yan L.-H., Le Roux A., Boyapelly K., Lamontagne A.-M., Archambault M.-A., Picard-Jean F., Lalonde-Seguin D., St-Pierre E., Najmanovich R.J., Fortier L.-C. et al.. Purine analogs targeting the guanine riboswitch as potential antibiotics against Clostridioides difficile. Eur. J. Med. Chem. 2018; 143:755–768. [DOI] [PubMed] [Google Scholar]
- 89. Matyjasik M.M., Hall S.D., Batey R.T.. High affinity binding of N2-modified guanine derivatives significantly disrupts the ligand binding pocket of the guanine riboswitch. Molecules. 2020; 25:2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Aldrich C., Bertozzi C., Georg G.I., Kiessling L., Lindsley C., Liotta D., Merz K.M., Schepartz A., Wang S.. The ecstasy and agony of assay interference compounds. J. Chem. Inf. Model. 2017; 57:387–390. [DOI] [PubMed] [Google Scholar]
- 91. Blount K.F., Breaker R.R.. Riboswitches as antibacterial drug targets. Nat. Biotechnol. 2006; 24:1558–1564. [DOI] [PubMed] [Google Scholar]
- 92. Ma C., Yang X., Lewis P.J.. Bacterial transcription as a target for antibacterial drug development. Microbiol. Mol. Biol. Rev. 2016; 80:139–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lee E.R., Blount K.F., Breaker R.R.. Roseoflavin is a natural antibacterial compound that binds to FMN riboswitches and regulates gene expression. RNA Biol. 2009; 6:187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Gonzalez A., Fillat M.F., Lanas A.. Transcriptional regulators: valuable targets for novel antibacterial strategies. Future Med. Chem. 2018; 10:541–560. [DOI] [PubMed] [Google Scholar]
- 95. Soheili V., Tajani A.S., Ghodsi R., Bazzaz B.S.F.. Anti-PqsR compounds as next-generation antibacterial agents against Pseudomonas aeruginosa: a review. Eur. J. Med. Chem. 2019; 172:26–35. [DOI] [PubMed] [Google Scholar]
- 96. Yang X., Ma C., Lewis P.J.. Identification of inhibitors of bacterial RNA polymerase. Methods. 2015; 86:45–50. [DOI] [PubMed] [Google Scholar]
- 97. Mosaei H., Harbottle J.. Mechanisms of antibiotics inhibiting bacterial RNA polymerase. Biochem. Soc. Trans. 2019; 47:339–350. [DOI] [PubMed] [Google Scholar]
- 98. Sippel A., Hartmann G.. Mode of action of rifamycin on the RNA polymerase reaction. Biochim. Biophys. Acta. 1967; 157:218–219. [DOI] [PubMed] [Google Scholar]
- 99. McClure W.R., Cech C.L.. On the mechanism of rifampicin inhibition of RNA synthesis. J. Biol. Chem. 1978; 253:8949–8956. [PubMed] [Google Scholar]
- 100. Glaziou P., Floyd K., Raviglione M.. Global epidemiology of tuberculosis. Semin. Respir. Crit. Care Med. 2018; 39:271–285. [DOI] [PubMed] [Google Scholar]
- 101. Yarbrough L.R., Wu F.Y.H., Wu C.-W.. Molecular mechanism of the rifampicin-RNA polymerase interaction. Biochemistry. 1976; 15:2669–2676. [DOI] [PubMed] [Google Scholar]
- 102. Acker M.G., Auld D.S.. Considerations for the design and reporting of enzyme assays in high-throughput screening applications. Perspect. Sci. 2014; 1:56–73. [Google Scholar]
- 103. Agarwal R.P., Parks R.E. Jr. Purine nucleoside phosphorylase from human erythrocytes. J. Biol. Chem. 1969; 244:644–647. [PubMed] [Google Scholar]
- 104. Schomburg D., Schomburg I., Chang A.. Purine-nucleoside phosphorylase. Class 2 · Transferases VI: EC 2.4.2.1–2.5.1.30. 2007; Berlin, Heidelberg: Springer; 1–33. [Google Scholar]
- 105. Giblett E.R., Ammann A.J., Wara D.W., Sandman R., Diamond L.K.. Nucleoside-phosphorylase deficiency in a child with severely defective T-cell immunity and normal B-cell immunity. Lancet Lond. Engl. 1975; 1:1010–1013. [DOI] [PubMed] [Google Scholar]
- 106. Mitchell B.S., Mejias E., Daddona P.E., Kelley W.N.. Purinogenic immunodeficiency diseases: selective toxicity of deoxyribonucleosides for T cells. Proc. Natl. Acad. Sci. U.S.A. 1978; 75:5011–5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ullman B., Gudas L.J., Clift S.M., Martin D.W.. Isolation and characterization of purine-nucleoside phosphorylase-deficient T-lymphoma cells and secondary mutants with altered ribonucleotide reductase: Genetic model for immunodeficiency disease. Proc. Natl. Acad. Sci. U.S.A. 1979; 76:1074–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Sircar J.C., Suto M.J., Scott M.E., Dong M.K., Gilbertsen R.B.. Inhibitors of human purine nucleoside phosphorylase. Synthesis, purine nucleoside phosphorylase inhibition, and T-cell cytotoxicity of 2, 5-diaminothiazolo[5, 4-d]pyrimidin-7(6H)-one and 2, 5-diaminothiazolo[4, 5-d]pyrimidin-7(6H)-one. Two thioisosteres of 8-aminoguanine. J. Med. Chem. 1986; 29:1804–1806. [DOI] [PubMed] [Google Scholar]
- 109. Schramm V.L. Development of transition state analogues of purine nucleoside phosphorylase as anti-T-cell agents. Biochim. Biophys. Acta BBA - Mol. Basis Dis. 2002; 1587:107–117. [DOI] [PubMed] [Google Scholar]
- 110. Korycka A., Blonski J.Z., Robak T.. Forodesine (BCX-1777, immucillin H) - a new purine nucleoside analogue: mechanism of action and potential clinical application. Mini.-Rev. Med. Chem. 2007; 7:976–983. [DOI] [PubMed] [Google Scholar]
- 111. Robak P., Robak T.. Older and new purine nucleoside analogs for patients with acute leukemias. Cancer Treat. Rev. 2013; 39:851–861. [DOI] [PubMed] [Google Scholar]
- 112. Reid R., Chatterjee B., Das S.J., Ghosh S., Sharma T.K.. Application of aptamers as molecular recognition elements in lateral flow assays. Anal. Biochem. 2020; 593:113574. [DOI] [PubMed] [Google Scholar]
- 113. Thavarajah W., Silverman A.D., Verosloff M.S., Kelley-Loughnane N., Jewett M.C., Lucks J.B.. Point-of-Use detection of environmental fluoride via a Cell-Free Riboswitch-Based biosensor. ACS Synth. Biol. 2020; 9:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Katzmeier F., Aufinger L., Dupin A., Quintero J., Lenz M., Bauer L., Klumpe S., Sherpa D., Dürr B., Honemann M. et al.. A low-cost fluorescence reader for in vitro transcription and nucleic acid detection with Cas13a. PLoS One. 2019; 14:e0220091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bowman J.C., Lenz T.K., Hud N.V., Williams L.D.. Cations in charge: magnesium ions in RNA folding and catalysis. Curr. Opin. Struct. Biol. 2012; 22:262–272. [DOI] [PubMed] [Google Scholar]
- 116. Wickiser J.K., Cheah M.T., Breaker R.R., Crothers D.M.. The kinetics of ligand binding by an Adenine-Sensing riboswitch. Biochemistry. 2005; 44:13404–13414. [DOI] [PubMed] [Google Scholar]
- 117. Regulski E.E., Breaker R.R.. Wilusz J. In-line probing analysis of riboswitches. Post-Transcriptional Gene Regulation. 2008; Totowa: Humana Press; 53–67. [DOI] [PubMed] [Google Scholar]
- 118. Greenlee E.B., Stav S., Atilho R.M., Brewer K.I., Harris K.A., Malkowski S.N., Mirihana Arachchilage G., Perkins K.R., Sherlock M.E., Breaker R.R.. Challenges of ligand identification for the second wave of orphan riboswitch candidates. RNA Biol. 2018; 15:377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Blount K.F., Wang J.X., Lim J., Sudarsan N., Breaker R.R.. Antibacterial lysine analogs that target lysine riboswitches. Nat. Chem. Biol. 2007; 3:44–49. [DOI] [PubMed] [Google Scholar]
- 120. Ataide S.F., Wilson S.N., Dang S., Rogers T.E., Roy B., Banerjee R., Henkin T.M., Ibba M.. Mechanisms of resistance to an amino acid antibiotic that targets translation. ACS Chem. Biol. 2007; 2:819–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Myhrvold C., Freije C.A., Gootenberg J.S., Abudayyeh O.O., Metsky H.C., Durbin A.F., Kellner M.J., Tan A.L., Paul L.M., Parham L.A. et al.. Field-deployable viral diagnostics using CRISPR-Cas13. Science. 2018; 360:444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.