


Abstract
Retinoic acid receptors (RARs) as a functional heterodimer with retinoid X receptors (RXRs), bind a diverse series of RA-response elements (RAREs) in regulated genes. Among them, the non-canonical DR0 elements are bound by RXR–RAR with comparable affinities to DR5 elements but DR0 elements do not act transcriptionally as independent RAREs. In this work, we present structural insights for the recognition of DR5 and DR0 elements by RXR–RAR heterodimer using x-ray crystallography, small angle x-ray scattering, and hydrogen/deuterium exchange coupled to mass spectrometry. We solved the crystal structures of RXR–RAR DNA-binding domain in complex with the Rarb2 DR5 and RXR–RXR DNA-binding domain in complex with Hoxb13 DR0. While cooperative binding was observed on DR5, the two molecules bound non-cooperatively on DR0 on opposite sides of the DNA. In addition, our data unveil the structural organization and dynamics of the multi-domain RXR–RAR DNA complexes providing evidence for DNA-dependent allosteric communication between domains. Differential binding modes between DR0 and DR5 were observed leading to differences in conformation and structural dynamics of the multi-domain RXR–RAR DNA complexes. These results reveal that the topological organization of the RAR binding element confer regulatory information by modulating the overall topology and structural dynamics of the RXR–RAR heterodimers.
INTRODUCTION
Retinoids (retinoic acid (RA) and synthetic analogs) mediate organismal and cellular effects through activation of the retinoic acid receptors (RARs), members of the nuclear receptor (NR) superfamily (1,2). Both experimental and clinical studies have revealed that retinoids regulate a wide variety of essential biological processes, such as vertebrate embryogenesis and organogenesis, cell growth arrest, differentiation and apoptosis (3). The RARs exist as three paralogs (RARA, RARB and RARG) that originate from distinct genes, and function as heterodimers with one of the three retinoid X receptor paralogs (RXRA, RXRB, RXRG). The RAR and RXR paralogs have similar, but not identical, sequences and structures. They exert overlapping and unique functions in development and differentiation that cannot be replaced by the actions of the other paralogs.
RARs and RXRs are modular proteins composed of several domains, most notably a DNA-binding domain (DBD) and a ligand-binding domain (LBD) (4,5). Intensive studies of NR mediated gene transcription modulation led to a detailed comprehension of how the ligand, RA, constitutes a regulatory signal leading to communication with the general transcriptional machinery via conformational changes that affect the LBD of RARs and initiates a cascade of protein-protein interactions (3). The activity of RXR–RAR is modulated by distinct combinations of cofactors, depending on cell type, promoter, DNA-binding site, and the actions of various signalling pathways/ligands (6,7). An important, but poorly understood, determinant of NR-coregulator interaction is the allosteric influence of specific DNA-binding sites on the recruitment of coactivator or corepressor proteins. In the case of RXR–RAR heterodimers, positive or negative regulation have been shown to be strongly influenced by the spacing of the DNA elements to which they bind (8).
When heterodimerized with RXRs, RARs bind to specific DNA sequences, called RA response elements (RAREs), that are typically composed of two direct repeats (DR) of a core hexameric motif, (A/G)G(G/T)TCA and are located in the regulatory sequences of target genes. The number of spacer nucleotides between the half-sites was initially defined for various RXR heterodimers in a simplified manner by the ‘1–5 rule’: RXR–RXR and RAR-RXR (DR1), RXR–RAR (DR2), RXR-VDR (DR3), RXR-TR (DR4) and RXR–RAR (DR5) (9,10). RXR binds on the upstream half-site of DR elements (in the following, the first NR DBD named in a dimer defines the 5’ position within the promoter sequence upstream of the transcription start site, TSS), with the exception of DR1 complexes whose polarity is reversed (8,11,12). Genome-wide chromatin immunoprecipitation followed by sequencing (Chip-Seq) data allowed the characterization of RAR cistromes in different cell types (e.g. mESc, mEC, MCF-7, NB4 (13–17)) and highlighted several underestimated features of RXR–RAR binding sites (18,19). In addition to sites comprising the consensus direct repeat DR1, DR2 or DR5 elements, numerous sites comprising non-canonical sequences were detected, including DR1, 2 and 5 with non-canonical half-site sequences and novel spacing topology elements, the DR0 and DR8 elements. The non-canonical DR0 elements are observed with high prevalence in undifferentiated cells, while in differentiated cells the canonical DR5 motif dominates (19). Of particular interest are the DR0 elements since complexes of RXR–RAR bound to DR0s are not able to modulate gene expression in a transcriptional reporter assay (Supplementary Figure S1B) (18).
Understanding the allosteric effects of the DNA binding elements requires addressing the structure and organisation of the RXR–RAR dimer bound to the different DNA binding sites. Current understanding at the atomic level of DNA recognition by RXR–RAR is limited to the crystal structures of the RARA–RXRA DBDs (20) and RARB–RXRA ΔAB (21) bound to an idealized DR1 sequence with identical half-sites. In the present study, we analyse the structures of RXRA–RARA complexes on two types of natural RAREs (DR5 and DR0) in order to decipher how the binding of RXR–RAR to these DNA elements controls transcriptional activity.
MATERIALS AND METHODS
DNAs
The non-labelled single strand DNAs were purchased from Sigma Aldrich and annealed. The TAMRA5/6 labelled single strand DNAs and complementary non-labelled strands were purchased from IBA (GmbH, Germany) and annealed. Each single stranded oligonucleotide was re-suspended in 10 mM HEPES pH 7.0, 50 mM NaCl, to 1 mM concentration. The complementary strands were mixed in equimolar ratio, heated in a boiling water bath and slowly cooled overnight to room temperature. The final DNA concentration was measured by absorbance at 260 nm.
Cloning, protein expression and purification
In all experiments except for the crystal structure analysis, we used MmRXRA and MmRARA proteins truncated of their N-terminal region (Supplementary Figures S2 and S3). For X-ray structures determination, we used the HsRXRA and HsRARA DBDs that exhibit 100% and 98.7% identity with the mice sequences.
MmRARA ΔAB (84–462) and MmRARA ΔABF (84–421) were cloned into the pET15b vector containing an N-ter hexahistidine tag. MmRXRA ΔAB (135–467) was cloned into pET3a as a non-tagged protein. The recombinant proteins were expressed in the Escherichia coli BL21 (DE3) strain. For multidomain protein purification, a co-purification strategy was used as described in (22). 6L of MmRAR and 4L of MmRXR harvested cells were re-suspended in binding buffer (20 mM Tris pH8, 500 mM NaCl, 5 mM Imidazole, 5% (v/v) Glycerol, 2 mM CHAPS, cOmplete, EDTA-free protease inhibitor cocktail tablet (Roche Applied Science)), sonicated and centrifuged. The supernatant was loaded on a 5 ml HisTrap FF crude column (GE Healthcare). The proteins were eluted at 200 mM imidazole. 9cis retinoic acid (9cis RA (Sigma)) was added in a 2-fold excess and the fully liganded heterodimer was further purified by size exclusion chromatography (SEC) on Superdex 200 (16/60, GE Healthcare) column. The final buffer was 20 mM Tris pH7.5, 75 mM NaCl, 75 mM KCl, 5% (v/v) glycerol, 1 mM CHAPS, 4 mM MgSO4, 1 mM TCEP. Protein samples were concentrated using Amicon-Ultra centrifugal filter units (Millipore). MmRXR ΔAB-MmRAR ΔAB or MmRXR ΔAB-MmRAR ΔABF and DNA response elements were formed by adding a 1.1-fold molar excess of DNA to the receptor dimer followed by an additional SEC step.
The purity and homogeneity of the protein were assessed by SDS-PAGE (Supplementary Figure S4).
DBD purification, crystallization and structure resolution
The HsRXRA DBD (130–212) and HsRARA DBD (82–167) (Supplementary Figures S2 and S3) were expressed in fusion with thioredoxine and hexahistidine tags. The proteins were produced and purified as described in (23,24). Fusion tags were removed by thrombin proteolysis and the cleaved proteins were then purified by SEC on a Superdex 75 (16/60, GE Healthcare) in 10 mM HEPES pH 8, 50 mM NaCl, 5 mM MgCl2, 1 mM TCEP.
All crystallization experiments were carried out by sitting drop vapor diffusion at 293 K using a Mosquito Crystal nanolitre dispensing robot (SPT Labtech) in 96-well 2-drop MRC crystallization plates (Molecular Dimensions), and screening against commercially available (JCSG+, Morpheus (Molecular Dimensions), Nucleix, PACT, ProComplex, The PEGs (Qiagen)) and in-house crystallization screens.
For the complex with the Hoxb13 DR0, the HsRXR DBD, HsRAR DBD and DNA were mixed in an equimolar ratio and concentrated to a final concentration of 7 mg ml−1. Initial crystals of Hoxb13 complex obtained in 20% PEG 10 000, 0.1 M MES pH 6.5 were used as seeds. 0.4 μl of the complex were mixed with 0.35 μl of reservoir solution and 0.05 μl of seeds and equilibrated against 50 μl of reservoir solution. A single crystal was grown by seeding in condition 80 of the JCSG+ screen (20% PEG 3350, 0.15 M di-sodium dl-malonate). The crystal appeared after one day, reaching its maximum size (250 × 50 × 30 μm) after 5 days. The seed preparation was made using the 'seed-bead' kit from Hampton Research, as described by Luft and DeTitta (25).
For the Rarb2 DR5 complex, the HsRXR DBD, HsRAR DBD and DNA were mixed in a ratio 1:1:0.9 and concentrated to a final concentration of 8 mg ml−1. Equal volumes (0.1 μl) of protein–DNA complex and reservoir solution were mixed and equilibrated against 50 μl of reservoir solution. A single crystal appeared after 5 days in an in-house screen (25% PEG 3350, 0.2 M Li2SO4, 0.1 M bis–tris pH 6.5) reaching its full size (130 × 60 × 60 μm) after 5 weeks.
Crystals of the Hoxb13 DR0 complex were cryo-protected with Parabar 10312 (Hampton Research) before being flash cooled in liquid nitrogen. Data were collected on a Pilatus 6M Hybrid Pixel Detector (Dectris) at the Proxima 1 beamline of the synchrotron SOLEIL. 180° of data were collected using 0.1° rotation and 0.1 s exposure per image (50% transmission).
Crystals of the Rarb2 DR5 complex were transferred to 35% PEG 3350, 0.2 M Li2SO4, 0.1 M bis–tris pH 6.5 before flash cooling in liquid nitrogen. Data were collected on a Pilatus 6M Hybrid Pixel Detector at the ID23-1 beamline of the ESRF using the MxCuBE software (26). 180° of data were collected using 0.1° and 0.037s exposure per image (46% transmission).
All data were indexed, integrated and scaled using XDS (27) before performing anisotropic truncation and correction with the STARANISO server (http://staraniso.globalphasing.org). The crystals of the complex with the Hoxb13 DR0 diffracted anisotropically to 2.1 Å (2.7 Å in the worst direction) and belonged to the primitive monoclinic space group P21, with unit cell dimensions a = 45.7 Å, b = 62.5 Å, c = 53.6 Å, B = 104.84°. The crystals of the complex with the Rarb2 DR5 diffracted anisotropically to 2.4 Å (3.1 Å in the worst direction) and belonged to the primitive tetragonal space group P41212 or P43212, with unit cell dimensions a = b = 56.7 Å, c = 238.4 Å.
Both structures were solved by molecular replacement using PHASER (28) in the PHENIX suite (29). A monomer of the RXRA DBD bound to 6 bp of DNA from the structure of complex with the human Nr1d1 response element (PDB ID: 4CN5 (23)) was used as a search model. For the Hoxb13 complex, two copies of the RXR DBD plus 6 bp were found, showing that there is one copy of the RXR DBD homodimer complex in the asymmetric unit, with a Matthews’ coefficient (30) of 2.43 Å3 Da−1 (53.47% solvent). For the Rarb2 complex, two copies of the RXR DBD were found, but an examination of the electron density revealed that one copy was in fact RAR, showing that there is one heterodimer complex in the asymmetric unit, with a Matthews’ coefficient of 2.82 Å3 Da−1 (59.94% solvent). Refinement was performed using PHENIX (29) and BUSTER (31) followed by iterative model building in COOT (32). The quality of the final refined model was assessed using MOLPROBITY (33). Data collection and refinement statistics are given in Table S1. Structural figures were prepared using PyMOL (www.pymol.org). The coordinates and structure factors are deposited in the Protein Data Bank under the accession codes 6XWG (RXR–RAR DBD-Rarb2 DR5) and 6XWH (RXR DBD-RXR DBD-Hoxb13 DR0).
Small angle X-ray scattering
Synchrotron SAXS data were measured from samples of MmRXRA ΔAB-MmRARA ΔABF -F11r DR5 and -Hoxb13 DR0 utilising split-flow in-line SEC-SAXS coupled to a right-angle laser light scattering (RALLS), UV absorption spectroscopy and refractive index (RI) triple-detector array (Malvern Instruments Viscotek TDA 305) on the EMBL bioSAXS-P12 beam line (λ = 0.124 nm, 10 keV) at the PETRA III storage ring, Hamburg, Germany (34) employing BEQUEREL SEC-SAXS control software (35). Samples of RXR–RAR-F11r DR5 and RAR–RXR-Hoxb13 DR0 were made to an approximate injection concentrations of 4.8 and 5.5 mg/ml, respectively, in 20 mM Tris, 75 mM NaCl, 75 mM KCl, 5% (v/v) glycerol, 1 mM CHAPS, 4 mM MgSO4, 1 mM TCEP, pH 8 and 100 μl of each sample were successively injected onto a GE Healthcare Superdex 200 10/300 column (equilibrated in the same buffer) at a flow rate of 0.5 ml min−1. The SAXS data were collected every 1 s for a total of 2900 s for each sample elution profile. Data were processed using the SASFLOW pipeline (36) that included the identification of an appropriate solute-free blank for buffer subtraction purposes. Structural parameters and additional evaluations of the SAXS data were performed using the ATSAS 2.8 software package (37).The Rg was determined from the Guinier approximation (38) (ln I(s) versus s2, for sRg < 1.3) and from the p(r) profiles that were calculated using GNOM (39) which also provided estimates of Dmax. In all instances, PRIMUS (40) was used to manually evaluate the appropriate Guinier range and to calculate the Rg (Supplementary Figure 8) while the consistency of Rg of the individual SAXS data frames through the SEC-elution peaks for either complex were automatically determined using AUTORG (41). Those data frames with a consistent Rg through the SEC-peaks (Supplementary Figure 8) were scaled, checked for similarity using CorMap using a significance threshold of 0.01 (42) and then averaged to generate the final SAXS profiles. The SAXS profiles shown in Figure 5A correspond to the scaled and averaged SAXS data for both complexes. Additional data analysis steps included evaluating the number of Shannon channels (SHANUM (43)) from which the maximum working s and the final working smin – smax data range were determined taking into account the variance in the scattering intensities and the level of oversampling. The data were classified into shape categories (DATCLASS (44)) while the ambiguity of the SAXS profiles, with respect to 3D-shape reconstructions, were determined using AMBIMETER (45). The molecular mass of the complexes were evaluated from the TDA measurements – calibrated relative to monomeric bovine serum albumin – using the Malvern Omnisec software where the refractive index increment, dn/dc, was set to 0.185 mL g−1 (Supplementary Figure 8). Concentration-independent MW estimates extracted directly from the SAXS data were evaluated using Bayesian inference (46). The modelling program CORAL (47) was used to rigid-body refine the structures of the RXR–RAR-F11r DR5 and RAR–RXR-Hoxb13 DR0 complexes. The resulting models were constructed by combining the high-resolution models of the LDBs and DBDs (PDB 6XWG, 6XWH, 1XDK and 3A9E) with the respective response element and connecting the domains with appropriate length linkers, modeled as a string of dummy amino acids, and allowing the domains and linkers to refine their spatial position(s) against the SAXS data (47). The CORAL rigid-body modelling routines were run several times to assess the stability of the results in context of the ambiguity of the SAXS patterns (Table 1). The final fits of the model scattering to the experimental data were computed using CRYSOL (48) and the model/data similarity evaluated using the reduced χ2 test and CorMap P values. All data and models have been deposited to the SASBDB (SASBDB; www.sasbdb.org; (49)) with the accession codes SASDFT8 and SASDFU8, including the unsubstracted SEC-SAXS data frames, the RALLS/RI MW analysis and all CORAL models that fit the SAXS data.
Figure 5.

Solution structure models of multi-domain RXR–RAR–DNA complexes. (A) The final averaged SEC-SAXS data and the computed model fits to the data for RXRA ΔAB–RARA ΔABF-F11r DR5 and RARA ΔABF-RXRA ΔAB-Hoxb13 DR0. (B) The p(r) profiles calculated from the SAXS data showing changes in the distribution of real-space distances on comparing the DR5 or DR0 complexes. (C, D) Refined rigid-body models of RXR–RAR-F11r DR5 and RAR-RXR-Hoxb13 DR0 heterodimers showing changes in the position and tilt of the DR5 or DR0 response elements bound to the RAR–RXR DNA binding domains relative to the ligand binding domains. For each complex, three representative refined rigid-body models are shown in different colors (blue, pink and yellow for RXR–RAR–F11r DR5 and cyan, purple and green for RAR–RXR–Hoxb13 DR0). In all models, helices H12 of RXR and RAR are colored in red.
Table 1.
SAXS data evaluation summary. *(34). **The final working s-range was evaluated from the predicted Dmax and subsequent number of Shannon channels calculated using SHANUM. §IFT (indirect Fourier Transform) software, (39). §§Bayesian MW estimate and credibility interval (35). ***DATCLASS (44). ‡AMBIMETER (45). ‡‡CORAL (47). ‡‡‡CRYSOL (48). †The reduced χ2 assessments of the reciprocal-space p(r) and model fits to the data are higher than anticipated likely due to the underestimation of errors on the I(s), however CorMap P values are well above a 0.01 significance threshold (42)
| RAR-RXR-Hoxb13 DR0 | RXR-RAR-F11r DR5 | |
|---|---|---|
| EMBL-P12 bioSAXS beam line* | ||
| X-ray wavelength, nm (energy, keV) | 0.124 (10) | 0.124 (10) |
| Information content and working s-range | ||
| Measured s-range (nm−1) | 0.02–4.5 | 0.02–4.5 |
| Predicted Dmax (nm) | 13.2 | 13.1 |
| #Shannon channels | 10 | 10 |
| Final working s-range (smin– smax), (nm−1) ** | 0.07–2.5 | 0.08–2.5 |
| Guinier analysis | ||
| Rg Guinier (nm) | 3.78 ± 0.05 | 3.96 ± 0.06 |
| sRg range/(points used) | 0.28–1.3 (22–124) | 0.29–1.3 (21–117) |
| Linear correlation, R2 | 0.994 | 0.990 |
| p(r) analysis | ||
| R g p(r) (nm) | 3.96 ± 0.02 | 4.05 ± 0.02 |
| Dmax, (nm) | 14.5 | 13.5 |
| V p (nm3) | 132 | 130 |
| reciprocal-space fit to data (χ2, CorMap P)† | 1.2, 0.55 | 1.8, 0.79 |
| IFT software§ | GNOM 4.6 | GNOM 4.6 |
| MW analysis | ||
| Calculated MW, from amino acid and DNA sequence, 1:1 complex | 88.3 | 91.4 |
| Average MW from RALLS/RI, kDa (range) | 103 (87–114) | 105 (95–112) |
| MW from SAXS, kDa (Bayesian inference)§§ | 90 | 86 |
| MW from SAXS credibility Interval (kDa) | 84–92 | 81–93 |
| Shape classification and ambiguity | ||
| Classification/(predicted Dmax, nm)*** | Flat/modular (14.1) | Flat/modular (13.3) |
| Ambimeter score(sRgmax)‡ | 2.5 (4) | 2.9 (4) |
| #shape topologies | 330 | 853 |
| Uniqueness | Highly ambiguous | Highly ambiguous |
| Atomistic modelling | ||
| Method‡‡ | CORAL | CORAL |
| Final model fit evaluation‡‡‡ | CRYSOL | CRYSOL |
| Model Rg (nm) | 3.8 | 4.0 |
| Model fit to data (χ2, CorMap P)† | 1.2, 0.03–0.13 | 1.8, 0.36–0.59 |
| Small angle scattering biological data bank | ||
| Accession code | SASDFT8 | SASDFU8 |
Steady-state and time-resolved fluorescence spectroscopy
For the fluorescence experiments, MmRARA ΔAB-MmRXRA ΔAB was labelled through labelling the polyHis Nterminal tag of RAR with OG488-TrisNTA (50). SEC on Superdex 200 (16/60, GE Healthcare) column was performed to remove the free label. The molar extinction coefficients of the protein (ϵRAR–RXR λ = 280 nm = 40 000 M−1 cm−1) and of the dye (ϵOG488 λ = 490 nm = 87000 M−1 cm−1) were used to estimate the protein concentration and labeling efficiency, respectively. The DNA sequences labeled at the 5′ end with tetramethylrhodamine 5/6-isomer (TAMRA5/6) were purchased from IBA GmbH (DE) and annealed.
Fluorescence emission spectra were recorded with a Fluorolog or a Fluoromax-3 spectrophotometer (Horiba Jobin Yvon). Excitation and emission band widths were 2 nm. Spectra were recorded with an excitation wavelength of 470 nm (to limit the direct excitation of TAMRA5/6) at 20°C and corrected for lamp fluctuations as well as for wavelength dependence of the emission pathway. Experiments were performed in quartz cuvettes of 2 mm path length (115F – Micro cells, Hellma GmbH). Typically, the working concentration of OG488-labelled protein was 1 μM and titrations were performed by adding increasing quantities of TAMRA5/6-labeled DNA. In practice, successive additions of DNA stock solutions in the same cuvette allowed to reach [DNA]/[Protein] ratios of respectively 0, 1, 3, 5 and 10. Formation of the protein/DNA complexes led to a decrease of the donor emission at λ = 520 nm from which the FRET efficiency E was calculated using: E = 1 – IDA/ID, where ID and IDA correspond to the fluorescence intensity of the donor (OG488) in the absence and in the presence of the acceptor (TAMRA5/6), respectively. Control titrations of OG488-labelled protein with unlabeled DNAs were performed to check that no significant change in the fluorescence quantum yield and lifetime of OG488 was associated to the binding of the unlabeled DNAs (data not shown).
The changes in the transfer efficiency observed on titration of the OG488-labelled protein by TAMRA5/6-labelled DNA were fitted by the following rewritten Scatchard equation, assuming a unique binding site per DNA:
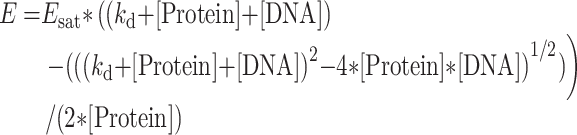 |
(1) |
where Esat is the FRET efficiency at saturation when all proteins are bound to their DNA target, kd is the dissociation constant, [Protein] and [DNA] are the concentrations of protein and DNA, respectively.
From the Esat value, an average interchromophore distance R between the donor and acceptor dyes was calculated using: R = R0(1/Esat – 1)1/6, where R0 is the Förster critical distance calculated by:
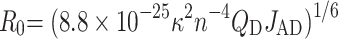 |
where n is the refractive index of the medium (n = 1.33), QD is the quantum yield of the donor and JAD is the overlap integral between the emission spectrum of the donor and the absorption spectrum of the acceptor. Since both OG488 and TAMRA5/6 dyes are covalently bound through flexible linkers to the termini of the DNA strand and the protein, both dyes likely undergo a complete dynamic isotropic orientational averaging, and thus a value of 2/3 can be taken for the orientation factor κ2.
Time-resolved fluorescence intensity measurements were performed using the time-correlated single-photon counting technique (TCSPC). Excitation pulses at 470 nm with a repetition rate of 4 MHz were generated by a pulse-picked frequency-doubled Titanium:Sapphire laser (Tsunami, Spectra Physics) pumped by a Millenia X laser (Spectra Physics). The instrumental response function (IRF) was recorded using a polished aluminum reflector, and its full-width at half-maximum was ∼30 ps. Fluorescence emission was recorded at 520 nm through a 4 nm band-pass monochromator (HORIBA Jobin-Yvon) on a micro-channel plate photomultiplier (Hamamatsu) coupled to a pulse pre-amplifier HFAC (Becker-Hickl GmbH). The single-photon events were recorded on a time correlated single photon counting board SPC-130 (Becker-Hickl GmbH). The software SPCM 9.75 (Becker-Hickl GmbH) was used to record the data and build the photon distribution over time.
Fluorescence decays were deconvoluted from the IRF and fitted by the maximum entropy method using the Pulse 5 software (51,52) to retrieve the most probable lifetime distributions. In all cases, the χ2 values were close to 1 and the weighted residuals, as well as the autocorrelation of the residuals were randomly distributed around zero, indicating an optimal fit.
Hydrogen deuterium exchange mass spectrometry experiments
Liganded MmRXRA ΔAB-MmRARA ΔAB, MmRXRA ΔAB-MmRARA ΔAB-Rarb2 DR5 and MmRXRA ΔAB-MmRARA ΔAB-Hoxb13 DR0 complexes were prepared in 20 mM Tris, 75 mM NaCl, 75 mM KCl, 2 mM CHAPS, 4 mM MgSO4, 1 mM TCEP, pH 8 buffer at a final concentration of 32 μM. Preparation and injection of the samples were automatically conducted using a LEAP HDX Automation Manager (Waters), while chromatography was carried out on Acquity UPLC system with HDX technology (Waters, Manchester, UK). Samples were incubated at 20°C for five deuteration timepoints including 0.5, 2, 10, 30 and 60 minutes in 95% of 20 mM Tris, 75 mM NaCl, 75 mM KCl, 2 mM CHAPS, 4 mM MgSO4, 1 mM TCEP, pD 8 deuterated buffer. The labelling reaction was then stopped by adding 1:1 (v/v) 2 M guanidine–HCl, 100 mM glycine, 100 mM TCEP, pH 2.5 quench buffer at 1°C during 0.5 min. Samples were then digested (40 pmol injections) through a pepsin-immobilized cartridge (Enzymate pepsin column, 300 Å, 5 μm, 2.1 × 30 mm, Waters, Manchester, UK) in 0.1% aqueous formic acid solution and generated peptides were trapped on UPLC pre-column (ACQUITY UPLC BEH C18 VanGuard pre-column, 2.1 mm I.D. × 5 mm, 1.7 μM particle diameter, Waters, Manchester, UK) at 200 μl min−1. Digested peptides were then separated on a UPLC column (ACQUITY UPLC BEH C18, 1.0 mm I.D. × 100 mm, 1.7 μM particle diameter, Waters) at 0.1 °C with a gradient elution of solvent A (0.1% formic acid aqueous) and solvent B (acetonitrile with 0.1% formic acid) [2–40% B (7 min), 40–85% B (0.5 min), and 85% B (1 min)] at a flow rate of 40 μl min−1. MS/MS analyses were acquired on a Synapt G2SI HDMS (Waters, Wilmslow, UK) in the positive ion mode using the resolution mode calibrated with glufibrinogen peptide and using a lock-mass correction (glufibrinogen). Data were acquired in MSE acquisition mode with the following parameters: capillary voltage, 3 kV; sampling cone voltage, 40 V; source temperature, 80°C; desolvation gas, 150°C and 600 l h−1; acquisition range, 50−2000 m/z; scan time, 0.3 s; trap collision energy, 15–40 eV. Data were processed using Waters ProteinLynx Global Server 2.5.3 (Waters) with a home-made protein sequence library containing MmRAR, MmRXR and pepsin sequences, where peptide and fragment tolerances were automatically adjusted by PLGS, and oxidized methionine was set as variable modification. Data were then filtered with DynamX 3.0 (Waters) as follows: each experiment was carried out in triplicate and only peptides identified in all replicates were kept with a minimum fragment of 0.2 per amino acid with a minimum intensity at 103, a length between 5 and 30 residues and a file threshold of 3. Deuterium uptakes for all identified peptides were manually checked and validated without correction for back-exchange, represented as relative. HDX-MS results were statistically validated using MEMHDX software (53) with a P value set to 0.01. HDX results were exported on RXR–RAR structures using PyMOL (www.pymol.org). The mass spectrometry proteomics data (including HDX uptake plots) have been deposited to the ProteomeXchange Consortium via the PRIDE (54) partner repository with the dataset identifier PXD018172.
SwitchSense experiments
The DNA sequences used are listed below; bold bases represent the RARE sequence and bases in italics represent the complementary sequence to nanolever NL-B48 immobilized on the chip surface:
Rarb2: 5′-TA GTG AAC TTT CGG TGA ACC CTG ATC AGC GTT CGA TGC TTC CGA CTA ATC AGC CAT ATC AGC TTA CGA CTA-3′
Hoxb13: 5′-TCT TGG CCT TGA CCT TCG ATC AGC GTT CGA TGC TTC CGA CTA ATC AGC CAT ATC AGC TTA CGA CTA-3′
All experiments were performed on a DRX24000 instrument (Dynamic Biosensors GmbH, Martinsried, DE) on standard multipurpose switchSENSE chips (MPC2-48-2-G1R1-S) using the static measurement mode (55). MmRXRA ΔAB-MmRARA ΔAB was dialyzed against 10 mM HEPES pH 7.5, 75 mM KCl, 4 mM MgCl2 and 0.05% tween 20 buffer. In the association measurement, 250 μl of RXR–RAR (concentrations of 75, 150 and 300 nM) were injected with a flowrate of 50 μl min−1 for a 3 min association phase. Dissociation was measured only for RXR–RAR at 300 nM concentration for 15 min by rinsing with a flowrate of 50 μl min−1 over the chip. All measurements were performed at 25°C. Analysis was performed with the switch-ANALYSIS software from Dynamic Biosensors. Normalized Dynamic Response values were obtained by subtracting the signal measured upon injection of a volume of buffer corresponding to injections performed in each titration measurement. The association and dissociation rate constants (kon and koff) of the RXR–RAR interaction with DNAs were derived from a single-exponential fit model. Measurements were performed in duplicate, and values reported represent the mean kon and koff of the datasets, leading to an average KD by: KD = koff,avg/kon,avg
RESULTS
Crystal structures of DBDs dimer on DR5 and DR0 elements
To characterize the structural features of the RXR–RAR dimer bound to DR5 and DR0 elements, we selected the classical Rarb2 DR5 sequence (11,12,18) and the DR0 sequence from Hoxb13 (18) mouse gene (Supplementary Figure S1A). We attempted to crystalize the RXR–RAR DBD heterodimer assembled on these two distinct elements, but crystals of the heterodimer could only be obtained in complex with Rarb2 DR5. For Hoxb13 DR0, crystals of the RXR–RXR DBD homodimer were only obtained. The difficulty to crystalize the heterodimer on DR0 is explained by the sample heterogeneity when mixing RAR and RXR DBDs, and DR0, with the presence of RAR and RXR monomer, homodimer and heterodimer DNA complexes observed by native mass spectrometry (56).
The RXR–RAR DBD-Rarb2 DR5 complex crystallized in the P43212 space group, with 1 dimer–DNA complex per asymmetric unit. Anisotropic data were collected to 2.4 Å resolution (3.1 Å in the worst direction, Supplementary Table S1). The DBD monomers lie in a head to tail orientation with non-equivalent protein-protein interactions from each monomer and with the expected polarity with RXR bound to the 5’ half-site and RAR bound to the 3’ half-site (Figure 1 and Supplementary Figure S5A). The tertiary structures of RAR and RXR DBDs are similar to previously solved DBD structures (20) and are composed of an N-terminal β-hairpin, two α-helices (helix I and II) followed by a single turn of 310 helix, and a C-terminal extension that contains the T-box subdomain (Supplementary Figures S2 and S3). The N-terminal helix I directly interacts with the DNA half-site in the major groove, and helix II is perpendicular to helix I stabilizing the core of the DBD. Each DBD forms sequence-specific direct and water mediated base contacts that involve highly conserved residues: Glu153, Lys156 and Arg161 for RXR, and Glu106 and Lys109, for RAR (Figure 1C and D). Interestingly, in the heterodimer complex, RAR forms additional interaction with the backbone sugar of the second nucleotide of the 3′ half-site, and RXR Arg172 interacts with the phosphate backbone of the last nucleotide of the spacer. The dimerization of RXR–RAR (Figure 1E and Supplementary Figure S5B) with a buried surface of 190 Å2, involves residues from the second Zn module of the RXR DBD and the N-terminal region of RAR which was defined as the pre-finger region (11). Indeed, RXR Arg172 forms a stacking interaction with RAR Tyr98, as well as a hydrogen bond with RAR Lys86. In addition RXR Arg186 interacts with RAR Arg83.
Figure 1.

Crystal structure of RXR–RAR DBDs-Rarb2 DR5. (A) Overall structure. The spheres indicate the Zn atoms. RXR is shown in blue and RAR in purple. (B) Sequence of Rarb2 DR5. (C) Specific interactions of RXR and RAR DBDs to Rarb2 DR5. Left: View along the DNA-recognition helix of RXR showing residues Glu153, Lys156, Arg161 and their direct base contacts. Right: corresponding view of Rarb2 DR5 recognition by RAR DBD showing residues Glu106, Arg109 and Arg113. Hydrogen-bonds and water molecules are shown as red dotted lines and red spheres, respectively. (D) Schematic view of the RXR–RAR DBDs-Rarb2 DR5 contacts calculated with NUCPLOT with a 3.9 Å distance cutoff. Bridging water molecules are shown as red circles. (E) Dimerization interface that involves residues from the second Zn module of RXR and the pre-finger region of RAR. Hydrogen bond and Van der Waals interactions are shown by red and grey dashed lines, respectively. Right: Electrostatic surface representation of the complex (red, negative; blue, positive; light gray, neutral).
The RXR–RXR-Hoxb13 DR0 crystallized in the P21 space group, with 1 dimer-DNA complex per asymmetric unit and anisotropic data were collected to 2.1 Å (2.7 Å in the worst direction, Supplementary Table S1). The two RXR DBDs are bound to opposite sides of the DNA in a head-to-tail fashion (Figure 2 and Supplementary Figure S5C). Each half-site makes specific contacts through 4 base pairs for the first half-site and 3 bp for the second half-site (Figure 2B-C and Supplementary Figure S5D). Both RXR DBDs also form extensive contacts with the phosphate backbone along 9 bp. Residues of the T-box make backbone interactions with the two first bases of the first half-site and the first base of the second half-site (Figure 2D). No interaction between the RXR monomers is observed, consistent with the binding to Hoxb13 DR0 as two monomers at equivalent sites. This is in agreement with the analysis of the free-energy gains calculated by the PISA server for the interaction of each monomer with its half site, which indicates similar values; ΔG of −9 kcal mol−1 for the first 5′ motif and ΔG of −8 kcal mol−1 for the 3′ motif.
Figure 2.

Crystal structure of RXR–RXR DBDs-Hoxb13 DR0. (A) Overall structure of RXR–RXR–DNA complex. The spheres indicate the Zn atoms. (B) Sequence of Hoxb13 DR0. (C) Specific interactions of RXR homodimer DBDs to Hoxb13 DR0. Left: View along the DNA-recognition helix of 5′ RXR. Right: The corresponding view of 3′ RXR. Hydrogen-bonds and water molecules are shown as red dotted lines and red spheres, respectively. (D) Schematic view of the RXR–RXR DBDs-Hoxb13 DR0 contacts calculated with NUCPLOT with a 3.9 Å distance cutoff. Bridging water molecules are shown as red circles.
Polarity of RXR–RAR on DR0 response elements
To determine the polarity of the DBDs of RXR–RAR dimer bound to DR0 response elements, we performed fluorescence resonance energy transfer (FRET) experiments using a TAMRA5/6-labeled DNA and an acceptor-labeled polyHis-N-ter-RAR with OG488-TriNTA (50). We used the purified multi-domain proteins that form a homogeneous heterodimer (Supplementary Figure S4). Due to instability of the NTDs of RAR and RXR, which are easily prone to degradation, we choose to work with proteins truncated of their NTDs (57). The RXR–RAR ΔAB heterodimer was co-purified using a histidine-tagged RAR ΔAB and a non-tagged RXR ΔAB. We used two different DR0 sequences from Socs3 and Hoxb13 mouse genes (Supplementary Figure S1) and Rarb2 DR5 and Ramp2 DR1 as controls, for which the polarities are known (11,12). All studied DRs are well characterized RAREs (18). Titrations of RXR–RAR by the TAMRA5/6-labeled DNA sequences are gathered in Figure 3. Upon addition of increasing concentrations of DNA, the strong decrease of the fluorescence of the donor at 520 nm together with the strong increase of the of TAMRA5/6 band at 585 nm clearly show the FRET between OG488 and TAMRA5/6. As seen in the insets of Figure 3, the dependence of the FRET efficiency on the DNA concentration can be well fitted by Equation (1), assuming a single binding site per DNA. The protein showed a similar affinity for the Ramp2 DR1 (kd = 0.5 μM) and Rarb2 DR5 (kd = 0.8 μM) sequences, whereas a significantly lower affinity was measured for the Hoxb13 DR0 (kd = 1.8 μM) and the Socs3 DR0 (kd = 2.7 μM). Concerning the FRET efficiency at saturation, the high value obtained for Ramp2 DR1 and Hoxb13 DR0 (Esat ∼ 0.8) suggests a binding mode that favors a close proximity between the donor and acceptor dyes. In contrast, experiments with Rarb2 DR5 gave a significantly lower value (Esat = 0.46) indicating that on the average the distance between the two dyes is longer, in line with a different binding mode as compared to Ramp2 DR1 and Hoxb13 DR0 sequences. Interestingly, an intermediate Esat value (0.61) was obtained for Socs3 DR0.
Figure 3.

Polarity of DNA bound RXR–RAR complexes. Fluorescence spectra of OG488-RARA ΔAB-RXRA ΔAB in the absence (black curve) and in the presence of increasing concentrations of DNA-TAMRA5/6 (red, green, blue and cyan curves correspond respectively to DNA/protein ratios of 1, 3, 5 and 10). In each panel, the inset represents the evolution of the FRET efficiency as a function of DNA concentration. The FRET efficiency (square marks) was deduced from the decrease of the donor emission. The red curve represents the best fit to Equation (1). (A) [RAR-RXR] = 0.73 μM titrated by Ramp2 DR1 (kd = 0.5 μM, Esat = 0.75), (B) [RAR–RXR] = 0.81 μM titrated by Rarb2 DR5 (kd = 0.8 μM, Esat = 0.46), (C) [RAR-RXR] = 0.78 μM titrated by Hoxb13 DR0 (kd = 1.8 μM, Esat = 0.81) and (D) [RAR-RXR] = 0.78 μM titrated by Socs3 DR0 (kd = 2.7 μM, Esat = 0.61).
In order to gain more insight into the binding modes of the different DNAs by RXR–RAR, we measured and analyzed the fluorescence decays of the proteins in the absence and in the presence of saturating concentrations of DNA. Supplementary Table 2 revealed that the decay of RXR–RAR labeled at the N-terminus of RAR by OG488 is characterized by three lifetime components (0.31, 1.43 and 3.31 ns) of nearly equal amplitude, giving an average lifetime of 1.69 ns. Since the three lifetimes are shorter than the single lifetime value (4.09 ns, (58)) of the free OG488 dye, it is likely that the flexible linker attaching the dye to the protein N-terminus allows dynamic quenching of the dye by the protein side chains and backbone. The appearance of multiple lifetimes as a result of conformation- and context-dependent quenching of fluorescent probes in proteins is a common feature that has notably be well illustrated for intrinsic tryptophan residues (59).
Upon addition of a saturating concentration of Ramp2-TAMRA5/6, we observed the disappearance of the long-lived lifetime as well as a strong decrease of the population associated with the 1.43 ns component mainly to the benefit of dark species (a0 ≈ 0.51). The dark species are endowed with lifetimes shorter than the time resolution of our set-up (20 ps) and are deduced from the comparison of the quantum yields and mean lifetime values. Moreover, addition of Ramp2-TAMRA5/6 was found to decrease the lifetime of the short-lived component from 0.31 ns to 0.18 ns and to induce the appearance of a new short-lived (0.59 ns) component. Altogether the sub-nanosecond species represented 90% of the overall species in solution. This dramatic increase in the population of short-lived lifetimes is clearly not associated to a quenching of the OG488 fluorescence by the DNA backbone or bases, since addition of unlabeled DNA to RXR-OG488-labeled RAR had no impact on the fluorescence quantum yield or lifetime (data not shown). Therefore, the short-lived lifetimes are obviously the consequence of a high FRET efficiency between OG488 and TAMRA5/6. As the population of the two long-lived components (1.47 and 3.28 ns) decreased from 63% to 10%, it can be reasonably deduced that the major part of the sub-nanosecond species observed in the presence of the labeled DNA originate from FRET with these components. Assuming an upper lifetime value of 20 ps for the dark species and a Förster distance R0 of 56 Å, then the observed FRET is associated with inter-chromophore distances ranging from 24 to 48 Å. These values are in line with the short distance between the FRET pairs (∼20 Å) calculated from the structural data of the RAR–RXR–DR1 structure (20) and for a binding orientation of the protein on the DNA sequence with RAR bound to the 5′ site and RXR bound to the 3′ site. For the Hoxb13-TAMRA5/6 sequence, the observed changes were very similar to those with Ramp2-TAMRA5/6, strongly suggesting that RAR–RXR adopts the same binding mode on the two sequences.
A very different behavior was observed for the binding of Rarb2-TAMRA5/6. The values of both long-lived lifetimes were observed to drop by 12–15%, suggesting a FRET with a distance of ∼75 Å between the dyes. A similar drop may also occur for the short-lived lifetime, but is probably obscured by the accuracy of the measurements at these low values. From this limited FRET, it may be concluded that RXR–RAR proteins binds with an opposite polarity on Rarb2 DR5 as compared to Ramp2 DR1 and Hoxb13 DR0. In addition, the amplitude of the long-lived lifetime was observed to decrease (from 0.31 to 0.09) and redistribute into the two other lifetimes as well as into the dark species (a0 ≈ 0.15), indicating that a limited population of complexes with high FRET and thus, with a binding polarity similar to Ramp2 DR1 or Hoxb13 DR0 also form.
For the sequence Socs3-TAMRA5/6, the changes in the time-resolved fluorescence parameters are similar but less pronounced than those observed with Ramp2 DR1 or Hoxb13 DR0. For instance, the summed amplitudes of the long-lived lifetimes decreased only by 2-fold (from 0.63 to 0.30), while the amplitude of the dark species reached only 0.4 (as compared to 0.51 for Ramp2 DR1 or Hoxb13 DR0). Moreover, no change in the value of the short-lived lifetime as well as no additional sub-ns component were observed. This suggests that only part of the RAR–RXR proteins bind to Socs3 DR0 in the same orientation as Ramp2 DR1 or Hoxb13 DR0.
Altogether these data indicate that the polarity of the RAR–RXR bound to DR0 is rather complex, most probably due to the non-cooperative binding of the dimer to DR0. While it is clear that on Hoxb13 DR0, RAR DBD binds to the 5′ half site and RXR DBD to the 3’half site similarly to DR1 elements, for Socs3 DR0 only a part of the heterodimer binds with this polarity (Figure 3). Note that the dissociation constants obtained in the FRET titrations are weaker than in our previous measurements performed by isothermal titration calorimetry (18), due to differences in buffer and experimental conditions. However, lower affinities for DR0 compared to DR5 and DR1 were obtained by both methods.
We also investigated the binding kinetics of RXR–RAR to Rarb2 DR5 and Hoxb13 DR0 using switchSENSE technology (55) as illustrated in Figure 4. The switching speed of the association phase was recorded for different protein concentrations while the dissociation measurement was assessed only for the highest protein concentration. For the Hoxb13 DR0 the association and dissociation rates are slower and faster, respectively, compared to the Rarb2 DR5, thus leading to an apparent higher dissociation constant for the DR0 response element.
Figure 4.

Kinetic analysis of RXRA ΔAB-RARA ΔAB interacting with (A) Rarb2 DR5 and (B) Hoxb13 DR0 using the switchSENSE technology. The raw data are superimposed by global exponential fits. The kon, koff and KD values are indicated.
Architecture of multi-domain RXR–RAR–DR5 and RAR–RXR–DR0 complexes
To characterize the in solution architecture of the multidomain RXR–RAR–DR5 and –DR0 complexes, we performed size-exclusion chromatography small-angle X-ray scattering (SEC-SAXS) coupled to a triple detector array (TDA) (36) using the RARA and RXRA proteins truncated of their NTDs and of the disordered F domain for RAR, RXR ΔAB-RAR ΔABF (Supplementary Figures S2-S3). We have previously characterized the solution structures of the RXRA ΔAB-RARA ΔAB and RXRΔAB–RARA ΔABF complexes with the Rarb2 DR5 (60) and have shown that the presence of the RARA F domain leads to an increase in structural parameters indicating an extended non-interacting F domain. We now have selected another DR5 element from the F11r gene (Supplementary Figure S1) which has been identified as a regulated RARE (13,61) and which exhibits similar binding as the Rarb2 DR5 (56) and Hoxb13 DR0 for which the polarity is defined as unique. The use of the coupled SEC-SAXS-TDA and the measurement of their subsequent SAXS intensity profiles (measured as I(s) versus s, where s = 4πsin θ/λ, 2θ is the scattering angle) and the validation of the corresponding molecular weights (MWs) from the TDA that were compared to the MWs obtained directly from the SAXS data (Supplementary Figure S6). The experimentally determined MWs of the two complexes are consistent with those calculated from the protein and DNA sequences of RXR–RAR heterodimers bound to either one F11r DR5 or one Hoxb13 DR0 response elements (Table 1). The SAXS profiles are shown in Figure 5A. The structural parameters of the complexes including the radius of gyration, Rg, the maximum particle dimension, Dmax, and the Porod volume, Vp, are reported in Table 1 and are in agreement with those parameters previously determined for the related RXR–RAR–Rarb2 DR5 complex (60). The probable distribution of real-space scattering pair distances, or p(r) profiles, are shown in Figure 5B. The two complexes are generally comparable with respect to their Rg (ca. 4 nm), Dmax (13.5–14.5 nm) and Vp (ca. 130 nm3). However, the p(r) profiles indicate that structural rearrangements occur when the RAR–RXR heterodimer interacts with its corresponding response element. Changes in the overall conformation for RXR–RAR–F11r DR5 are evidenced by the increased frequency of mid-to-long range distances and the development of ‘humps’ in the p(r) profile between 7 and 10 nm that are characteristic of more-defined separated regions of mass within the modular complex (on average) as compared to RAR–RXR–Hoxb13 DR0.
Rigid-body refined atomistic model representations of the RXR–RAR–F11r DR5 and RAR–RXR–Hoxb13 DR0 complexes are shown in Figure 5C and D. The fits of the computed model scattering patterns by CRYSOL (48) to the experimental data (Figure 5A) are in good agreement, falling in the χ2 range of 1.2–1.8 and with Correlation Map (CorMap) P values between 0.03 and 0.6 (42). The RXR–RAR LBD dimer appears asymmetrically positioned with respect to the two-half-sites while the linkers connecting the LBDs to the DBDs are likely adaptable allowing for spatial reorientation(s) of the LDBs relative to DBDs. A comparison between the RXR–RAR-F11r DR5 and RAR-RXR-Hoxb13 DR0 models indicates that the DR0 response element is reoriented and tilted more toward the LBDs compared to DR5 and occupies different spatial position(s). These differences are consistent with the p(r) profiles. The computed models of RXR–RAR-F11r DR5 fit with good agreement (χ2 range of 1.7–1.8 as computed with CRYSOL) the experimental SAXS curves of RXR–RAR–Rarb2 DR5 (not shown) indicating similar structures on both DR5 elements. Taken together, the SAXS data and models suggest that the linkers connecting the LDBs to the DBDs must be sufficiently flexible to allow for the alternative positioning and engagement of the DBDs onto the distinct response elements with different topology and polarity of the bound proteins. Ultimately this flexibility results in global changes of conformation in the respective RXR–RAR heterodimer complexes.
Effect of DNA binding on the conformational dynamics of RXR–RAR complexes
The characterization of local in solution dynamics of the RXR–RAR complexes on the two types of DNA elements was then investigated using hydrogen/deuterium exchange coupled with mass spectrometry (HDX-MS). Here, two differential HDX-MS experiments were carried out to characterize the effect of Rarb2 DR5 and Hoxb13 DR0 binding to the RXR–RAR heterodimer. We used proteins truncated of their NTDs, RXRA ΔAB-RARA ΔAB.
Firstly, we investigated the impact of Rarb2 DR5 binding to the RXR–RAR complex by comparing the deuterium incorporation of RXR–RAR heterodimer and the heterodimer bound to Rarb2 DR5. Interestingly, a major protection effect upon DR5 binding was observed for regions 80–142 and 146–153 of RAR, which span the DBD of the NR and includes helices HI and HII (Figure 6 and Supplementary Figure S7). In addition, residues 456–462 of the C-terminal region of the RAR (part of the F domain) was strongly deprotected in the presence of Rarb2 DR5. Concerning RXR, identified peptides covering regions 141–194 and 201–225 of RXR presented strong protection from H/D exchange upon Rarb2 DR5 binding. These regions encompass the entire DBD as well as the C-terminal DBD extension of RXR and the C-terminal part of the linker region. Interestingly, several regions of the LBD of RXR were also affected upon ternary complex formation. Indeed, deprotection was observed for regions 276–284, 325–335 and 436–454 of the LBD, corresponding to the N-terminal part of helix H3, the β sheet and helix H11, respectively.
Figure 6.

Conformational dynamics of RXR–RAR upon DNA binding. Export of RFU differences on RXRA ΔAB-RARA ΔAB heterodimer structure upon Rarb2 DR5 (A) and Hoxb13 DR0 (B) bindings determined by HDX-MS. Export is performed for 0.5 min deuteration experiments. RFU differences are color scaled from red (deprotection) to blue (protection) upon DNA binding (–20% to 20% range of RFU difference), while white regions are non-affected or non-covered. Secondary structures exhibiting statistically validated differences for the magnitude are indicated in green for RAR and in purple for RXR. (C) Deuterium uptake of selected perturbed peptides upon DR0 and DR5 binding plotted as a function of deuteration time. Blue, red and orange curves correspond to RXR–RAR, RAR–RXR–Hoxb13 DR0 and RXR–RAR–Rarb2 DR5 states respectively. The corresponding secondary structures are indicated for each plot.
We next characterized the binding of the Hoxb13 DR0 to RXR–RAR by comparing the deuterium incorporation of RXR–RAR heterodimer and the heterodimer bound to Hoxb13 DR0 (Figure 6 and Supplementary Figure S8). Firstly, similar regions of RAR were impacted upon Hoxb13 binding compared to Rarb2 DR5 binding: a major effect was observed for peptides spanning the DBD of RAR, which showed protection upon Hoxb13 DR0 binding. The additional 456–462 C-terminal region of the RAR construct (part of the F domain) was also deprotected upon Hoxb13 DR0 binding. Concerning RXR, only two peptides covering helix HI of the DBD (region 151–168) exhibited protection upon Hoxb13 DR0 binding, and particularly the 151–159 region. Moreover, the 271–279 region in the LBD of RXR, encompassing the N-terminal part of helix H3 was observed as deprotected upon Hoxb13 DR0 binding.
HDX-MS allowed to highlight a differential allosteric effect of Rarb2 DR5 and Hoxb13 DR0 binding to RXR–RAR heterodimer. Indeed, a similar protection effect on the RAR-DBD upon DNA binding was observed for Rarb2 DR5 and Hoxb13 DR0. The RAR-DBD impacted region involves the highly conserved residues Glu106 and Lys109, which form direct and water mediated base contacts with Rarb2 DR5 as described in the crystal structure (Figure 1). Furthermore, for both DR5 and DR0 binding, the first Zn domain of RAR was protected from H/D exchange upon DNA binding, in agreement with its known involvement in DNA recognition of DR5 and DR0 complexes as well as in the heterodimerization interface of DBDs with DR5. However, in the case of RXR, the second Zn module involved in the dimerization in DR5 complex showed protection only upon DR5 binding. For the dimerization interface between RXR and RAR LBDs, helix 11 of RXR that is part of the interface was deprotected in complex with DR5, not with DR0. While no effect for helix 12 was observed, part of the F domain of RARA was deprotected upon binding to DR0 or DR5, suggesting a strong allosteric effect for the F domain upon DNA binding by liganded RXR–RAR.
Finally, we observed a reduced effect in RXR in presence of Hoxb13 DR0 compared to Rarb2 DR5. While Hoxb13 DR0 binding only impacted helix HI of the DBD and the C-terminal part of helix H3 of the LBD, the entire DBD of RXR, the C-terminal part of the linker region H3, the β sheet and H11 of the RXR LBD were affected upon Rarb2 DR5 binding. Altogether, these HDX-MS data show that RXR–RAR binding to DR5 directly influences the conformation of RXR LBD whereas binding to DR0 does not.
DISCUSSION
RXR–RAR heterodimer binds to a large repertoire of sites with diverse spacing and topology of the DNA binding elements that are composed of two direct repeats separated by 0 to 8 nucleotides spacer or of inverted repeat IR0 (3,18). DR0 element is an important motif in NR-associated with pluripotent cell decision processes (19). Several NRs that bind to DR0 are either involved in pro-differentiation such as GCNF, COUP-TFI or COUP-TFII or in the maintenance of undifferentiated pluripotent state such as ESRRB or LRH1 (62). RXR–RAR has been shown to be bound to DR0 elements in undifferentiated pluripotent cells and during the early phase of RA-induced differentiation (19). A drastic reorganization of RXR–RAR binding repertoire occurs during RA-induced differentiation with a relocation from DR0 prevalent sites to DR5 elements (19). Functional data showed that RXR–RAR bound to DR0s are not able to modulate gene expression in a transcriptional reporter assay (18). The complementary approaches reported here provide important information for understanding the structural basis of DNA recognition of DR5 and DR0 elements and the allosteric control of RXR–RAR activity.
In this study, we determined the crystal structures of the RXR–RAR DBDs with Rarb2 DR5 and of the RXR–RXR DBDs with Hoxb13 DR0 response elements. The only previously reported structures of RXR–RAR DNA complex was obtained with the idealized DR1 element (20,21). In the DR5 complex reported here, although most of the residues involved in DNA binding are conserved, additional specific contacts are observed. More importantly, binding to the Rarb2 DR5 induces a DNA curvature that allows the two DBDs to interact. The dimerization differs from the previously reported structure and involves the second Zn module of RXR and the N-terminal region preceding the first Zn module of RAR. These interactions explain the highly cooperative heterodimer formed on the DR5 element. A cooperative effect, although involving other regions of the proteins, was also observed for RXR–RAR binding to DR1 compared to the RXR homodimer (20). With Hoxb13 DR0, due to heterogeneity of DNA–DBD complexes, only crystals of the RXR DBD homodimer were obtained. The structure reveals that the two DBDs bind on opposite sides of the DNA, and form a non-cooperative dimer with no protein-protein interactions. The determined polarity of the heterodimer on DR0 by FRET, indicates that on the Hoxb13 DR0, RAR binds to the 5’half-site and RXR to the 3’half-site which is a reverse polarity compared to the Rarb2 DR5. The situation is more complex on the Socs3 DR0, showing both populations with a similar polarity and a reversed polarity. Modelling of RAR-RXR on the Hoxb13 DR0 indicates a similar to RXR–RXR, non-cooperative dimer (Supplementary Figure S9A). Some of the NRs that bind DR0 elements have also been crystallized, LRH-1 has been shown to bind as a monomer (63,64) whereas GCNF binds as a homodimer (64) in a similar way as the present structure of RXR with the Hoxb13 DR0 (Supplementary Figure S9B). Non-cooperative binding was also demonstrated with GR on negative GRE (nGRE), where GR binds DNA in a conformation that prevents DNA-mediated dimerization, which has been suggested to explain the repressed activity of GR on nGRE (65). The FRET and switchSENSE data demonstrate that RXR–RAR binds to DR0 non-cooperatively with faster kinetics compared to the cooperative and slower kinetics of the binding to DR5, suggesting that the binding mode and dynamics of RXR–RAR are crucial parameters of the allosteric regulation. The importance of the kinetics and cooperative binding properties to modulate the transcriptional output have been demonstrated for other transcription factors, such as Msn2 (66) or NF-κB (67).
We also characterized the architecture in solution and the structural dynamics of RXR–RAR multidomain proteins with DR5 and DR0 elements. RXR–RAR adopts an asymmetric elongated conformation when bound to DR5 elements in agreement with our previous solution structural study (60). The architecture of the RXR–RAR complex with DR0 is also elongated, but as a consequence of a non-cooperative DBD dimer on the DR0 element, the DNA bound complex shows larger structural variability and flexibility. The hinge domains connecting the DBDs to the LBDs play an essential role in establishing and maintaining the integrity of the functional structures. This is particularly true for the RXR–RAR heterodimer that bind with high affinity to diverse DRs with different polarities and less defined preferential spacing compared to other heterodimers (68). The high flexibility of the hinges of RAR and RXR facilitates heterodimer formation with various RAREs, allowing the LBD dimer to rotate around both the pseudo two-fold axis and DNA. They control the relative spatial position of the domains and hence the orientation of the cofactors.
Our HDX-MS data show significant differences in the protection of structural elements between the multi-domain RXR–RAR complex bound either to DR5 or DR0 elements. RXR–RAR binding to DR5 directly influences the conformation of the RXR LBD whereas binding to DR0 does not. These differences may lead to differential coregulator interactions as suggested by pull-down assays showing that various natural and synthetic RAREs defined the relative affinity of liganded RXR–RAR for the coactivators (69,70). Allosteric role of DNA sequence has been described for the heterodimers RXR-VDR (71,72) and PPAR-RXR (73). Interestingly, a strong deprotection was observed for the F domain of liganded RARA upon DNA binding. The F domain is known to enhance the repression of RARA in absence of hormone (74,75) by stabilizing helix H12. The HDX data suggest a strong allosteric effect for the F domain upon binding of liganded RXR–RAR to both DR0 and DR5 elements, while no such effect was observed for helix 12.
The protein constructs used in this study were lacking the A/B domains that are very variable in length and sequence (76). The intrinsic disorder state of this N-terminal domain (77,78) prevents a proper description of its role. As it may modulate transcriptional function in a promoter-specific context (79), its role in transcriptional modulation on DR0 elements remains to be determined.
In summary, our work reveal differences in cooperativity and kinetics of DNA binding between DR5 and DR0 by RXR–RAR leading to difference in flexibility and dynamics of the multidomain RXR–RAR complexes (Figure 7). Alternative conformations and dynamics driven by DNA binding could be expected to lead to large differences in coregulator binding and controlled transcriptional output, explaining the inability of the RAR heterodimer to activate transcription when bound to DR0 compared to DR5. However, additional factors such as increased corepressor association or cooperation with neighboring DNA-bound transcription factors that are often bound in proximity to RXR–RAR-heterodimer bound to DR0 sequences (19), may also be involved in the inactive state of RXR–RAR bound to DR0. These new structural insights contribute to our understanding of the structural role of the DNA in allosteric control of NR activation profile.
Figure 7.

Differences in the binding mode of the RXR–RAR to DR5 or DR0 response elements.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD018172. All data and models have been deposited to the SASBDB (www.sasbdb.org) with the accession codes SASDFT8 and SASDFU8.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Jacob Piehler (University of Osnabrück, DE) who kindly provided OG488-TrisNTA, Guillaume Bec and Eric Ennifar (IBMC, France) for the Switchsense experiments. We thank the staff of Proxima 1 at synchrotron SOLEIL and of ID23–1 at ESRF for assistance in using the beamlines.
Contributor Information
Judit Osz, Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), Illkirch, France; Institut National de La Santé et de La Recherche Médicale (INSERM) U1258, Illkirch, France; Centre National de Recherche Scientifique (CNRS) UMR 7104, Illkirch, France; Université de Strasbourg, Illkirch, France.
Alastair G McEwen, Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), Illkirch, France; Institut National de La Santé et de La Recherche Médicale (INSERM) U1258, Illkirch, France; Centre National de Recherche Scientifique (CNRS) UMR 7104, Illkirch, France; Université de Strasbourg, Illkirch, France.
Maxime Bourguet, Laboratoire de Spectrométrie de Masse BioOrganique, Université de Strasbourg, CNRS UMR 7178, IPHC, Strasbourg, France.
Frédéric Przybilla, Laboratoire de Bioimagerie et Pathologies, CNRS UMR 7021, Faculté de Pharmacie, Université de Strasbourg, Illkirch, France.
Carole Peluso-Iltis, Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), Illkirch, France; Institut National de La Santé et de La Recherche Médicale (INSERM) U1258, Illkirch, France; Centre National de Recherche Scientifique (CNRS) UMR 7104, Illkirch, France; Université de Strasbourg, Illkirch, France.
Pierre Poussin-Courmontagne, Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), Illkirch, France; Institut National de La Santé et de La Recherche Médicale (INSERM) U1258, Illkirch, France; Centre National de Recherche Scientifique (CNRS) UMR 7104, Illkirch, France; Université de Strasbourg, Illkirch, France.
Yves Mély, Laboratoire de Bioimagerie et Pathologies, CNRS UMR 7021, Faculté de Pharmacie, Université de Strasbourg, Illkirch, France.
Sarah Cianférani, Laboratoire de Spectrométrie de Masse BioOrganique, Université de Strasbourg, CNRS UMR 7178, IPHC, Strasbourg, France.
Cy M Jeffries, European Molecular Biology Laboratory, Hamburg Outstation, Hamburg, Germany.
Dmitri I Svergun, European Molecular Biology Laboratory, Hamburg Outstation, Hamburg, Germany.
Natacha Rochel, Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), Illkirch, France; Institut National de La Santé et de La Recherche Médicale (INSERM) U1258, Illkirch, France; Centre National de Recherche Scientifique (CNRS) UMR 7104, Illkirch, France; Université de Strasbourg, Illkirch, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Agence Nationale de la Recherche [ANR-11-BSV8-023]; Fondation ARC [SFI20121205585]; French Infrastructure for Integrated Structural Biology (FRISBI) [ANR-10-INBS-05]; Instruct-ERIC; Agence Nationale de la Recherche under the program Investissements d’Avenir [ANR-10-LABX-0030-INRT, ANR-10-IDEX-0002-02]; French Proteomic Infrastructure ProFI [ANR-10-INBS-08-03]; Portions of the research received funding from the European Community's Seventh Framework Programme [FP7/2007-2013] under BioStruct-X [283570 to D.I.S.] and from the Bundesministerium für Bildung und Forschung (D.I.S. award nos. BIOSCAT [05K12YE1]); Horizon 2020 programme of the European Union, iNEXT [653706 to D.I.S.]; the authors thank GIS IBiSA and Région Alsace for financial support in purchasing a Synapt G2 HDMS instrument; M.B. was supported by a fellowship from the Région Alsace; Y.M. is grateful to the ‘Institut Universitaire de France (IUF)’ for support and providing additional time to be dedicated to research. Funding for open access charge: Fondation ARC pour la Recherche sur le Cancer.
Conflict of interest statement. None declared.
REFERENCES
- 1. Chambon P. The nuclear receptor superfamily: a personal retrospect on the first two decades. Mol. Endocrinol. 2005; 19:1418–1428. [DOI] [PubMed] [Google Scholar]
- 2. Benbrook D.M., Chambon P., Rochette-Egly C., Asson-Batres M.A.. History of retinoic acid receptors. Subcell. Biochem. 2014; 70:1–20. [DOI] [PubMed] [Google Scholar]
- 3. di Masi A., Leboffe L., De Marinis E., Pagano F., Cicconi L., Rochette-Egly C., Lo-Coco F., Ascenzi P., Nervi C.. Retinoic acid receptors: from molecular mechanisms to cancer therapy. Mol. Aspects Med. 2015; 41:1–115. [DOI] [PubMed] [Google Scholar]
- 4. Gronemeyer H., Gustafsson J.A., Laudet V.. Principles for modulation of the nuclear receptor superfamily. Nat. Rev. Drug Discov. 2004; 3:950–964. [DOI] [PubMed] [Google Scholar]
- 5. Perissi V., Rosenfeld M.G.. Controlling nuclear receptors: the circular logic of cofactor cycles. Nat. Rev. Mol. Cell Biol. 2005; 6:542–554. [DOI] [PubMed] [Google Scholar]
- 6. Wei L.N. Retinoid receptors and their coregulators. Annu. Rev. Pharmacol. Toxicol. 2003; 43:47–72. [DOI] [PubMed] [Google Scholar]
- 7. le Maire A., Bourguet W.. Retinoic acid receptors: structural basis for coregulator interaction and exchange. Subcell. Biochem. 2014; 70:37–54. [DOI] [PubMed] [Google Scholar]
- 8. Kurokawa R., DiRenzo J., Boehm M., Sugarman J., Gloss B., Rosenfeld M.G., Heyman R.A., Glass C.K.. Regulation of retinoid signalling by receptor polarity and allosteric control of ligand binding. Nature. 1994; 371:528–531. [DOI] [PubMed] [Google Scholar]
- 9. Umesono K., Murakami K.K., Thompson C.C., Evans R.M.. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell. 1991; 65:1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mangelsdorf D.J., andEvans R.M.. The RXR heterodimers and orphan receptors. Cell. 1995; 83:841–850. [DOI] [PubMed] [Google Scholar]
- 11. Zechel C., Shen X.Q., Chen J.Y., Chen Z.P., Chambon P., Gronemeyer H.. The dimerization interfaces formed between the DNA binding domains of RXR, RAR and TR determine the binding specificity and polarity of the full-length receptors to direct repeats. EMBO J. 1994; 13:1425–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zechel C., Shen X.Q., Chambon P., Gronemeyer H.. Dimerization interfaces formed between the DNA binding domains determine the cooperative binding of RXR/RAR and RXR/TR heterodimers to DR5 and DR4 elements. EMBO J. 1994; 13:1414–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Delacroix L., Moutier E., Altobelli G., Legras S., Poch O., Choukrallah M.A., Bertin I., Jost B., Davidson I.. Cell-specific interaction of retinoic acid receptors with target genes in mouse embryonic fibroblasts and embryonic stem cells. Mol. Cell. Biol. 2010; 30:231–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ross-Innes C.S., Stark R., Holmes K.A., Schmidt D., Spyrou C., Russell R., Massie C.E., Vowler S.L., Eldridge M., Carroll J.S.. Cooperative interaction between retinoic acid receptor-alpha and estrogen receptor in breast cancer. Genes Dev. 2010; 24:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Martens J.H., Brinkman A.B., Simmer F., Francoijs K.J., Nebbioso A., Ferrara F., Altucci L., Stunnenberg H.G.. PML-RARalpha/RXR alters the epigenetic landscape in acute promyelocytic leukemia. Cancer Cell. 2010; 17:173–185. [DOI] [PubMed] [Google Scholar]
- 16. Mahony S., Mazzoni E.O., McCuine S., Young R.A., Wichterle H., Gifford D.K.. Ligand-dependent dynamics of retinoic acid receptor binding during early neurogenesis. Genome Biol. 2011; 12:R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mendoza-Parra M.A., Walia M., Sankar M., Gronemeyer H.. Dissecting the retinoid-induced differentiation of F9 embryonal stem cells by integrative genomics. Mol. Syst. Biol. 2011; 7:538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moutier E., Ye T., Choukrallah M.A., Urban S., Osz J., Chatagnon A., Delacroix L., Langer D., Rochel N., Moras D., Benoit G., Davidson I.. Retinoic acid receptors recognize the mouse genome through binding elements with diverse spacing and topology. J. Biol. Chem. 2012; 287:26328–26341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chatagnon A., Veber P., Morin V., Bedo J., Triqueneaux G., Sémon M., Laudet V., d’Alché-Buc F., Benoit G.. RAR/RXR binding dynamics distinguish pluripotency from differentiation associated cis-regulatory elements. Nucleic Acids Res. 2015; 43:4833–4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rastinejad F., Wagner T., Zhao Q., Khorasanizadeh S.. Structure of the RXR–RAR DNA-binding complex on the retinoic acid response element DR1. EMBO J. 2000; 19:1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chandra V., Wu D., Li S., Potluri N., Kim Y., Rastinejad F.. The quaternary architecture of RARβ-RXRα heterodimer facilitates domain-domain signal transmission. Nat. Commun. 2017; 8:868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peluso-Iltis C., Osz N., Rochel N.. DNA recognition by retinoic acid nuclear receptors. Methods Enzymol. 2020; 637:235–260. [DOI] [PubMed] [Google Scholar]
- 23. Osz J., McEwen A.G., Poussin-Courmontagne P., Moutier E., Birck C., Davidson I., Moras D., Rochel N.. Structural basis of natural promoter recognition by the retinoid X nuclear receptor. Sci. Rep. 2015; 5:8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Osz J., McEwen A.G., Wolf J., Poussin-Courmontagne P., Peluso-Iltis C., Chebaro Y., Kieffer B., Rochel N.. Modulation of RXR-DNA complex assembly by DNA context. Mol. Cell. Endocrinol. 2019; 481:44–52. [DOI] [PubMed] [Google Scholar]
- 25. Luft J.R., DeTitta G.T.. A method to produce microseed stock for use in the crystallization of biological macromolecules. Acta Crystallogr. D. Biol. Crystallogr. 1999; 55:988–993. [DOI] [PubMed] [Google Scholar]
- 26. Gabadinho J., Beteva A., Guijarro M., Rey-Bakaikoa V., Spruce D., Bowler M.W., Brockhauser S., Flot D., Gordon E.J., Hall D.R. et al.. MxCuBE: a synchrotron beamline control environment customized for macromolecular crystallography experiments. J. Synchrotron Radiat. 2010; 17:700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kabsch W. Xds. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Matthews B.W. Solvent content of protein crystals. J. Mol. Biol. 1968; 33:491–497. [DOI] [PubMed] [Google Scholar]
- 31. Smart O.S., Womack T.O., Flensburg C., Keller P., Paciorek W., Sharff A., Vonrhein C., Bricogne G.. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. D. Biol. Crystallogr. 2012; 68:368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr D Biol Crystallog. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen V.B., Arendall W.B., Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C.. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blanchet C.E., Spilotros A., Schwemmer F., Graewert M.A., Kikhney A., Jeffries C.M., Franke D., Mark D., Zengerle R., Cipriani F. et al.. Versatile sample environments and automation for biological solution X-ray scattering experiments at the P12 beamline (PETRA III, DESY). J. App. Cryst. 2015; 48:431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hajizadeh N.R., Franke D., Svergun D.I.. Integrated beamline control and data acquisition for small-angle X-ray scattering at the P12 BioSAXS beamline at PETRAIII storage ring DESY. J. Synchrotron Radiat. 2018; 25:906–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Graewert M.A., Franke D., Jeffries C.M., Blanchet C.E., Ruskule D., Kuhle K., Flieger A., Schäfer B., Tartsch B., Meijers R. et al.. Automated pipeline for purification, biophysical and x-ray analysis of biomacromolecular solutions. Sci.Rep. 2015; 5:10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Franke D., Petoukhov M.V., Konarev P.V., Panjkovich A., Tuukkanen A., Mertens H.D.T., Kikhney A.G., Hajizadeh N.R., Franklin J.M., Jeffries C.M. et al.. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Cryst. 2017; 50:1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guinier A. La diffraction des rayons X aux très petits angles : application à l’etude de phénomènes ultramicroscopiques. Ann. Phys. (Paris). 1939; 12:161–237. [Google Scholar]
- 39. Svergun D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst. 1992; 25:495–503. [Google Scholar]
- 40. Konarev P.V., Volkov V.V., Sokolova A.V., Koch M.H.J., Svergun D.I.. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Cryst. 2003; 36:1277–1282. [Google Scholar]
- 41. Petoukhov M.V., Konarev P.V., Kikhney A.G., Svergun D.I.. ATSAS 2.1 - towards automated and web-supported small-angle scattering data analysis. J. Appl. Cryst. 2007; 40:223–228. [Google Scholar]
- 42. Franke D., Jeffries C.M., Svergun D.I.. Correlation Map, a goodness-of-fit test for one-dimensional X-ray scattering spectra. Nat. Methods. 2015; 12:419–422. [DOI] [PubMed] [Google Scholar]
- 43. Konarev P.V., Svergun D.I.. A posteriori determination of the useful data range for small-angle scattering experiments on dilute monodisperse systems. IUCrJ. 2015; 2:352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Franke D., Jeffries C.M., Svergun D.I.. Machine learning methods for X-Ray scattering data analysis from biomacromolecular solutions. Biophys. J. 2018; 114:2485–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Petoukhov M.V., Svergun D.I.. Ambiguity assessment of small-angle scattering curves from monodisperse systems. Acta Crystallogr. D. 2015; 71:1051–1058. [DOI] [PubMed] [Google Scholar]
- 46. Hajizadeh N.R., Franke D., Jeffries C.M., Svergun D.I.. Consensus Bayesian assessment of protein molecular mass from solution X-ray scattering data. Sci. Rep. 2018; 8:7204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Petoukhov M.V., Franke D., Shkumatov A.V., Tria G., Kikhney A.G., Gajda M., Gorba C., Mertens H.D., Konarev P.V., Svergun D.I.. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Cryst. 2012; 45:342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Svergun D.I., Barberato C., Koch M.H.J.. CRYSOL - a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 1995; 28:768–773. [Google Scholar]
- 49. Valentini E., Kikhney A.G., Previtali G., Jeffries C.M., Svergun D.I.. SASBDB, a repository for biological small-angle scattering data. Nucleic Acids Res. 2015; 43:357–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lata S., Piehler J.. Synthesis of a multivalent chelator lipid for stably tethering histidine-tagged proteins onto membranes. Nat. Protoc. 2006; 1:2104–2109. [DOI] [PubMed] [Google Scholar]
- 51. Brochon J.C. Maximum entropy method of data analysis in time-resolved spectroscopy. Methods Enzymol. 1994; 240:262–311. [DOI] [PubMed] [Google Scholar]
- 52. Livesey A.K., Brochon J.C.. Analyzing the distribution of decay constants in pulse-fluorimetry using the maximum entropy method. Biophys. J. 1987; 52:693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hourdel V., Volant S., O’Brien D.P., Chenal A., Chamot-Rooke J., Dillies M.A., Brier S.. MEMHDX: an interactive tool to expedite the statistical validation and visualization of large HDX-MS datasets. Bioinformatics. 2016; 32:3413–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M. et al.. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019; 47:D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cléry A., Sohier T.J.M., Welte T., Langer A., Allain F.H.T.. SwitchSENSE: A new technology to study protein-RNA interactions. Methods. 2017; 118–119:137–145. [DOI] [PubMed] [Google Scholar]
- 56. Nguyen-Huynh N.T., Osz J., Peluso-Iltis C., Rochel N., Potier N., Leize-Wagner E.. Monitoring of the retinoic acid receptor-retinoid X receptor dimerization upon DNA binding by native mass spectrometry. Biophys. Chem. 2016; 210:2–8. [DOI] [PubMed] [Google Scholar]
- 57. Chen Z.P., Shemshedini L., Durand B., Noy N., Chambon P., Gronemeyer H.. Pure and functionally homogeneous recombinant retinoid X receptor. J. Biol. Chem. 1994; 269:25770–25776. [PubMed] [Google Scholar]
- 58. Gustafsson N. Experimental investigation of the Photophysics of Oregon Green 488 and its suitability to biological research applications. Advanced ITPL Practical Report. 2014; [Google Scholar]
- 59. Lakowicz J.R. Principles of Fluorescence Spectroscopy. 2006; 3rd ednSpringer. [Google Scholar]
- 60. Rochel N., Ciesielski F., Godet J., Moman E., Roessle M., Peluso-Iltis C., Moulin M., Haertlein M., Callow P., Mély Y. et al.. Common architecture of nuclear receptor heterodimers on DNA direct repeat elements with different spacings. Nat. Struct. Mol. Biol. 2011; 18:564–570. [DOI] [PubMed] [Google Scholar]
- 61. Edey C., Hess B., Mears A.J.. Identification of novel retinoic acid target genes. Dev. Biol. 2014; 395:199–208. [DOI] [PubMed] [Google Scholar]
- 62. Benoit G., Cooney A., Giguere V., Ingraham H., Lazar M., Muscat G., Perlmann T., Renaud J.P., Schwabe J., Sladek F. et al.. International Union of Pharmacology. LXVI. Orphan nuclear receptors. Pharmacol. Rev. 2006; 58:798–836. [DOI] [PubMed] [Google Scholar]
- 63. Solomon I.H., Hager J.M., Safi R., McDonnell D.P., Redinbo M.R., Ortlund E.A.. Crystal structure of the human LRH-1 DBD-DNA complex reveals Ftz-F1 domain positioning is required for receptor activity. J. Mol. Biol. 2005; 354:1091–1102. [DOI] [PubMed] [Google Scholar]
- 64. Weikum E.R., Tuntland M.L., Murphy M.N., Ortlund E.A.. A structural investigation into Oct4 regulation by orphan nuclear receptors, Germ Cell Nuclear Factor (GCNF), and Liver Receptor Homolog-1 (LRH-1). J. Mol. Biol. 2016; 428:4981–4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hudson W.H., Youn C., Ortlund E.A.. The structural basis of direct glucocorticoid-mediated transrepression. Nat. Struct. Mol. Biol. 2013; 20:53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hao N., O'Shea E.K.. Signal-dependent dynamics of transcription factor translocation controls gene expression. Nat. Struct. Mol. Biol. 2011; 19:31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Giorgetti L., Siggers T., Tiana G., Caprara G., Notarbartolo S., Corona T., Pasparakis M., Milani P., Bulyk M.L., Natoli G.. Noncooperative interactions between transcription factors and clustered DNA binding sites enable graded transcriptional responses to environmental inputs. Mol. Cell. 2010; 37:418–428. [DOI] [PubMed] [Google Scholar]
- 68. Penvose A., Keenan J.L., Bray D., Ramlall V., Siggers T.. Comprehensive study of nuclear receptor DNA binding provides a revised framework for understanding receptor specificity. Nat. Commun. 2019; 10:2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. La Vista-Picard N., Hobbs P.D., Pfahl M., Dawson M.I., Pfahl M.. The receptor-DNA complex determines the retinoid response: a mechanism for the diversification of the ligand signal. Mol. Cell. Biol. 1996; 16:4137–4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mouchon A., Delmotte M.H., Formstecher P., Lefebvre P.. Allosteric regulation of the discriminative responsiveness of retinoic acid receptor to natural and synthetic ligands by retinoid X receptor and DNA. Mol. Cell. Biol. 1999; 19:3073–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhang J., Chalmers M.J., Stayrook K.R., Burris L.L., Wang Y., Busby S.A., Pascal B.D., Garcia-Ordonez R.D., Bruning J.B., Istrate M.A. et al.. DNA binding alters coactivator interaction surfaces of the intact VDR-RXR complex. Nat. Struct. Mol. Biol. 2011; 18:556–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zheng J., Chang M.R., Stites R.E., Wang Y., Bruning J.B., Pascal B.D., Novick S.J., Garcia-Ordonez R.D., Stayrook K.R., Chalmers M.J. et al.. HDX reveals the conformational dynamics of DNA sequence specific VDR co-activator interactions. Nat. Commun. 2017; 8:923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. de Vera I.M.S., Zheng J., Novick S., Shang J., Hughes T.S., Brust R., Munoz-Tello P., Gardner W.J. Jr, Marciano D.P., Kong X. et al.. Synergistic regulation of Coregulator/Nuclear receptor interaction by ligand and DNA. Structure. 2017; 25:1506–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Farboud B., Privalsky M.L.. Retinoic acid receptor-alpha is stabilized in a repressive state by its C-terminal, isotype-specific F domain. Mol. Endocrinol. 2004; 18:2839–2853. [DOI] [PubMed] [Google Scholar]
- 75. Patel S.R., Skafar D.F.. Modulation of nuclear receptor activity by the F domain. Mol. Cell. Endocrinol. 2015; 418:298–305. [DOI] [PubMed] [Google Scholar]
- 76. Al Tanoury Z., Piskunov A., Rochette-Egly C.. Vitamin A and retinoid signaling: genomic and nongenomic effects. J. Lipid Res. 2013; 54:1761–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Martinez-Zapien D., Delsuc M.A., Travé G., Lutzing R., Rochette-Egly C., Kieffer B.. Production and characterization of a retinoic acid receptor RARγ construction encompassing the DNA binding domain and the disordered N-terminal proline rich domain. Protein Expr. Purif. 2014; 95:113–120. [DOI] [PubMed] [Google Scholar]
- 78. Belorusova A., Osz J., Petoukhov M.V., Peluso-Iltis C., Kieffer B., Svergun D.I., Rochel N.. Solution behavior of the intrinsically disordered N-Terminal domain of retinoid X receptor α in the context of the Full-Length protein. Biochemistry. 2016; 55:1741–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nagpal S., Friant S., Nakshatri H., Chambon P.. RARs and RXRs: evidence for two autonomous transactivation functions (AF-1 and AF-2) and heterodimerization in vivo. EMBO J. 1993; 12:2349–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD018172. All data and models have been deposited to the SASBDB (www.sasbdb.org) with the accession codes SASDFT8 and SASDFU8.