

Abstract
Consumption of glucosinolates, pro-drug like metabolites abundant in brassica vegetables, has been associated with decreased risk of certain cancers. Gut microbiota have the ability to metabolize glucosinolates, generating chemopreventive isothiocyanates. Here, we identify a genetic and biochemical basis for the activation of glucosinolates to isothiocyanates by Bacteroides thetaiotaomicron, a prominent gut commensal species. Using a genome-wide transposon insertion screen, we identified an operon required for glucosinolate metabolism in B. thetaiotaomicron. Expression of BT2159-BT2156 in a non-metabolizing relative, Bacteroides fragilis, resulted in gain of glucosinolate metabolism. We show that isothiocyanate formation requires the action of BT2158 and either BT2156 or BT2157 in vitro. Monocolonization of mice with mutant BtΔ2157 showed reduced isothiocyanate production in the gastrointestinal tract. These data provide insight into the mechanisms by which a common gut bacterium processes an important dietary nutrient.
Graphical Abstract

The enzymatic mechanisms by which a common gut microbiome metabolizes plant-derived dietary glucosinolates to generate a class of small molecules with potential health benefits is elucidated.
INTRODUCTION
Diet is the largest source of plant-derived metabolites that influence human health. In addition to contributing fiber and micronutrients, dietary plants, including commonly consumed fruits and vegetables, contain secondary metabolites capable of eliciting pharmacological effects (Holst and Williamson, 2008; Martin et al., 2013). The diversity of these compounds has exceeded our understanding of their metabolic fates following consumption and resulting effect, limiting our ability to assess their impact on health. Commensal microbes in the gut have been implicated in the processing of many of these bioactive molecules, altering their reactivity, biochemical function, and propensity to be absorbed through the intestinal epithelium. However, these activities are often attributed to taxonomic groups and are rarely understood at the genetic or biochemical level (Bode et al., 2013; Clavel et al., 2006). A mechanistic understanding of how gut microbial metabolism impacts host biology will yield insight into how, from a common dietary input, the microbiome can drive divergent physiological outcomes based on which nutrient processing pathways dominate.
An example of diet-derived small molecules that influence the host involves edible vegetables such as broccoli, cabbage, and other plants in the family Brassicaceae. Epidemiological studies have linked diets rich in cruciferous vegetables to a decreased risk of gastrointestinal cancer, with isothiocyanates (ITCs) thought to be involved in the observed chemopreventive effects (Herr and Büchler, 2010). ITCs do not typically accumulate in crucifers; instead, they are hydrolysis products of biologically inert glucosinolates (GSs), thioglucosides with a sulfonated oxime moiety and an amino-derived side chain that are abundant in the plant tissue (Figure 1A). Efforts to take advantage of these protective effects have included the commercialization of Beneforte broccoli, a line bred to accumulate enhanced levels of the GS glucoraphanin (Mithen, 2013).
Figure 1. Activation of glucosinolates (GSs) to isothiocyanates (ITCs) by microbial myrosinases.

(A) Reaction scheme for the conversion of GSs to ITCs by myrosinases
(B) Metabolic fates of microbially produced ITCs (benzyl ITC (BITC) derived from glucotropaeolin (BGS), shown) in culture media or host urine. ITC in media was measured as ITC-cysteine, derived from in situ conjugation with cysteine in the media. ITC in urine was measured as the N-acetyl cysteine conjugate, an excreted product in mercapturic acid metabolism of ITC (Hwang and Jeffery, 2003).
(C) Benzyl ITC cysteine conjugate (BITC-cys) concentrations in culture supernatant, produced from BGS by Bacteroides strains grown in rich media for 24 hours. Bars represent the mean ± SD of three biological replicates, with individual replicates overlaid. Multiple reaction monitoring (MRM) by liquid chromatography with tandem mass spectrometry (LC-MS/MS) was used to track the transition of protonated BITC-cys with m/z 271.0 to a product ion with m/z 122.0.
ITCs are reactive electrophiles that have been studied in the context of a broad range of pharmacological effects using cell-based assays and in vivo models. For instance, the ITC sulforaphane (SFN), derived from hydrolysis of glucoraphanin, is an inducer of nuclear factor (erythroid-derived 2)-like 2 (Nrf2). Nrf2 activation by SFN upregulates xenobiotic-metabolizing and antioxidant-responsive phase II enzymes like quinone oxidoreductases and glutathione S-transferases (Traka and Mithen, 2009). Beyond xenobiotic metabolism, Nrf2 is a repressor of genes involved in hepatic lipid synthesis (Vomhof-DeKrey and Picklo, 2012), consistent with data from human intervention studies showing that consumption of Beneforte broccoli reduces plasma LDL-cholesterol to a greater extent than conventional broccoli (Armah et al., 2015). In addition to modulating plasma metabolite profiles and contributing to chemoprotection, SFN has been studied in the context of diabetes, with SFN treatment reducing fasting blood glucose levels in human patients with Type 2 diabetes and decreasing glucose production in rat hepatoma cell lines, mediated in part by Nrf2 regulation of gluconeogenesis (Axelsson et al., 2017). Beyond Nrf2 induction, ITCs have also been shown to interact with sensory and inflammatory pathways. Allyl ITC, derived from the GS sinigrin, is an agonist of the transient receptor potential ion channel TRPA1, which is involved in inflammatory pain signaling (Bellono et al., 2017), while SFN has been shown to modify lipopolysaccharide-activated Toll-like receptor 4, resulting in the reduced secretion of pro-inflammatory cytokines in human peripheral blood mononuclear cells (Folkard et al., 2014).
Plant and gut bacterial metabolism are two different routes for the conversion of GSs into biologically active ITCs. GSs in crucifers are involved in chemical defense in planta (Halkier and Gershenzon, 2006), and are stored in the plant tissue in an inert state before conversion into an active state by myrosinases, thio-specific glucoside hydrolases (Figure 1A). GSs are sequestered in vacuoles, while myrosinases are expressed in specialized myrosin cells (Kissen et al., 2009). Upon disruption of the plant tissue, myrosinases mediate the hydrolysis of GSs into a variety of possible products, including ITCs, nitriles, and epithionitriles, determined by environmental pH and the presence of specifier proteins (Halkier and Gershenzon, 2006).
In addition to being activated by plant enzymes, GSs can also be hydrolyzed to ITCs by the intestinal microbiota (Rabot et al., 1993). This route of GS activation plays an important role in a dietary context, since plant myrosinases are often denatured during cooking. Consumption of cooked broccoli by human volunteers following a regimen to reduce intestinal colonization resulted in diminished quantities of excreted ITC-derived metabolites compared to those who did not follow a microflora-reducing regimen (Shapiro et al., 1998). A consequence of the role that the gut microbiota plays in activating these prodrug-like molecules is variation in the level of bioactive ITCs produced following a meal of cooked crucifers between individuals (Navarro et al., 2014; Shapiro et al., 2001). The observed variability has been shown to be associated in part with the diverse metabolic capabilities represented in the intestinal microbiota, with ex vivo fecal bacterial cultures from high ITC-excreting individuals turning over more GSs than those from low ITC-excreting individuals (Li et al., 2011). Several gut bacteria are known to metabolize GSs, including Lactobacillus, Bifidobacterium, and Bacteroides strains (Cheng et al., 2004; Elfoul et al., 2001; Palop et al., 1995). As is the case with many interactions between dietary plant metabolites and the gut microflora, however, the bacterial genes encoding this conversion have not been identified.
Here, we investigate the genetic and biochemical basis behind the activation of dietary GSs by Bacteroides thetaiotaomicron (Bt), a prominent member of the human gut microbiota (Xu et al., 2003). Using insertional mutagenesis, we identified a previously uncharacterized operon, BT2160-BT2156, required for GS conversion to ITCs by Bt. While microbial genetics showed that BT2157 and BT2158 are required for activity in Bt, biochemical analyses revealed that the coordinated action of BT2158 with either BT2156 or BT2157 can promote GS transformation in vitro. Furthermore, mice monocolonized with mutant Bt lacking the complete operon (BtΔ2157) showed decreased ITC production when fed GSs. Collectively, these results increase our resolution of gut microbial processing of plant metabolites, a first step towards quantifying the impacts that these dietary plant molecules have on disease prevention.
RESULTS
GS metabolizing activity varies among Bt strains
To validate the ability of Bt strains to convert GSs to ITCs, we cultured human-associated isolates Bt VPI-5482, Bt 8736, Bt 7330, and Bt 3731 in rich medium containing glucotropaeolin (BGS), a GS with a benzyl moiety found in garden cress. Culture medium was supplemented with cysteine, included as a reucing agent (Bacic and Smith, 2008) and for in situ, reversible capture of any formed ITC (Angelino et al., 2015; Baillie and Slatter, 1991; Bruggeman et al., 1986). Conjugates of benzyl isothiocyanate with cysteine (BITC-cys) are detectable by liquid chromatography-tandem mass spectrometry (LC-MS/MS) and were measured in spent media (Figure 1B). The Bt strains displayed variable GS metabolism in rich media (Figure 1C), revealing that relative abundances of GS- and non-GS-metabolizing Bt strains may be a source of inter-individual variation in ITC production in the host. Other products of BGS hydrolysis, such as nitriles or epithionitriles, were not detected by liquid chromatography-mass spectrometry (LC-MS). To confirm that the formation of ITCs does not occur spontaneously in rich medium, including under the low pH conditions induced by Bt growth, we incubated BGS in sterile-filtered spent media from Bt VPI-5482 culture and did not observe any BITC-cys formation (Figure S1A). This result also suggests that the proteins responsible for GS metabolism are cell-associated.
A loss of function library screen yields an operon necessary for GS conversion to ITC in Bt
Because BLAST analysis revealed no Bt homologs to characterized plant myrosinases, we sought to identify candidate GS metabolizing genes in Bt with a genome-wide screen. We used the Mariner transposon to generate an insertion library in Bt VPI-5482 (Goodman et al., 2009) and selected ~7500 clones for screening using a coupled growth assay. While growth of Bt is not inhibited by ITC production (Figure S1B), ITCs exhibit bactericidal and bacteriostatic activity towards some strains (Dufour et al., 2015). We used this observation to develop an assay that links ITC production by Bt transposon mutants to growth inhibition of ITC-sensitive Escherichia coli (E. coli) inoculated into spent media from Bt cultures grown with BGS (Figure S1C). Bt mutants yielding spent media that did not inhibit E. coli growth were further evaluated for transposon insertions disrupting ITC-producing genes (Figure S1E). In order to avoid false positives from transposon insertions that hinder Bt growth, leading to low ITC production and reduced toxicity to E. coli, we monitored Bt growth and disregarded mutants with compromised growth (Figure S1F).
We next used semi-random PCR to identify the genes hit in the screen (Goodman et al., 2009), resulting in a list of 26 genes with several loci hit multiple times (Table S1). The most highly represented hits fell into two gene clusters, predicted to be operons. The most abundantly represented operon, BT1220-BT1222, contains genes involved in the pentose phosphate pathway. Because these genes encode a pathway in central carbon metabolism, we considered them unlikely to be directly responsible for the conversion of GS to ITC and excluded them from further evaluation.
The second most abundantly represented operon, BT2159-BT2156, included genes with predicted carbohydrate metabolizing activity and became our primary candidate for thioglucosidase function. The putative annotations ascribed to the genes in this operon include two nicotinamide-dependent oxidoreductases (BT2158 and BT2159), a glycoside hydrolase (BT2157), and a sugar-phosphate epimerase/isomerase (BT2156), under the control of transcriptional regulator BT2160 (Figure 2A). In silico analysis predicted that BT2160 spans the inner membrane, BT2159 is cytoplasmic, BT2158 is periplasmic, and BT2157 is an outer membrane lipoprotein (Figure S2A) (Käll et al., 2007; Petersen et al., 2011; Yu et al., 2010). While BT2156 contains a putative signal peptide and is predicted to be non-cytoplasmic, the localization could not be more specifically predicted.
Figure 2. An operon necessary for GS conversion to ITCs in B. thetaiotaomicron (Bt).
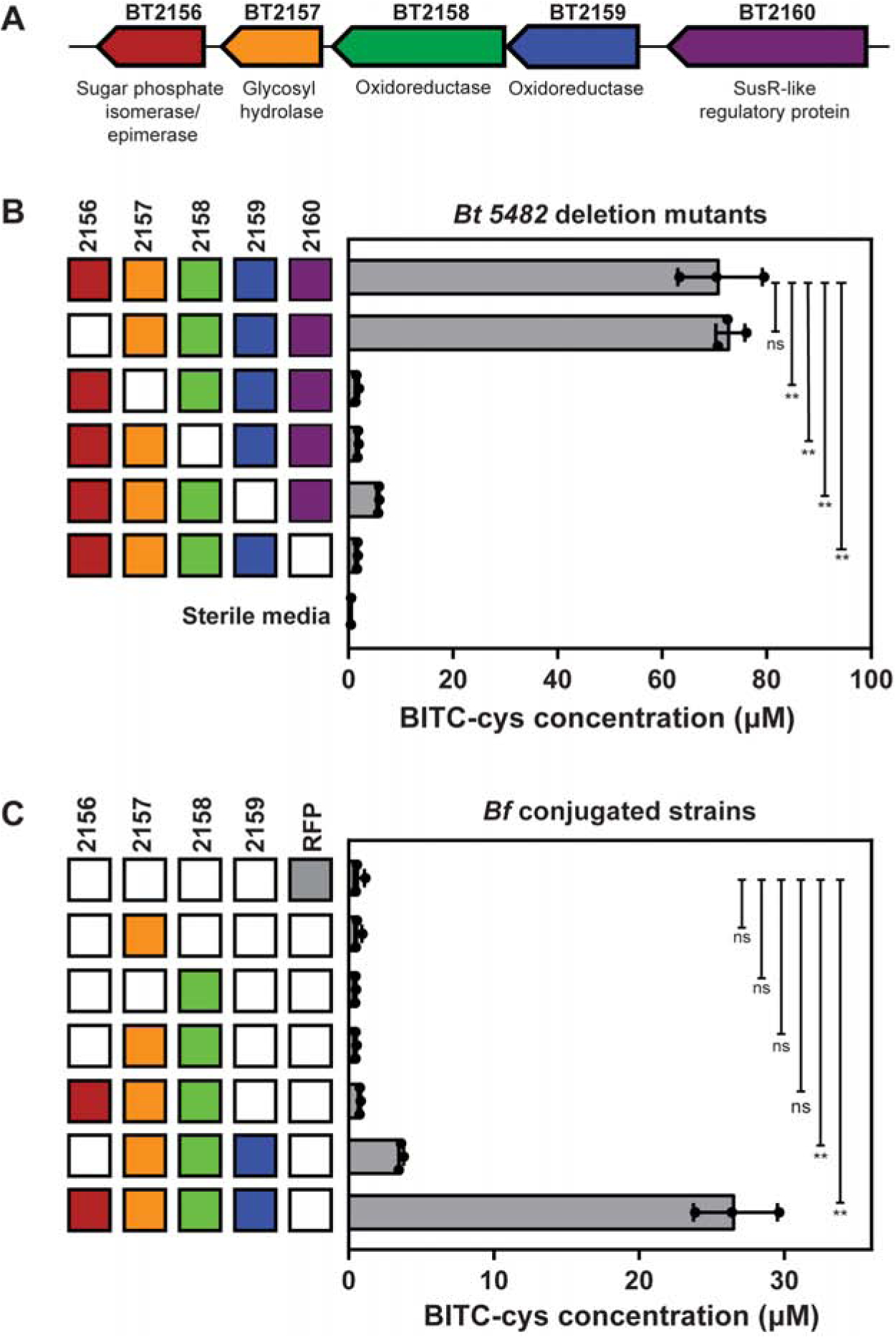
(A) The operon involved in GS metabolism in Bt. Predicted functions based on homology are annotated below each gene.
(B) BITC-cys concentrations in culture supernatant of Bt mutants with single deletions in the BT2160-BT2156 operon grown in rich media with BGS for 30 hours. Filled boxes represent natively expressed genes, while empty boxes represent deleted genes. Bars represent the mean±SD of three biological replicates, with individual replicates overlaid. BITC-cys background in sterile media is shown on the bottom-most row. Significant differences from wild-type Bt are marked by ns (not significant) or ** (p<0.0001), as determined using Dunnett’s multiple comparison test.
(C) BITC-cys concentrations in culture supernatant of B. fragilis (Bf) strains expressing subsets of BT2159-BT2156 after 30 hours of growth in rich media with BGS. Filled boxes represent extra-chromosomally complemented genes. Bars represent the mean±SD of three biological replicates, with individual replicates overlaid. Significant differences from the RFP expressing negative control strain are marked by ns or **(p<0.0001). See also Figures S1–S2 and Table S1.
This operon is one of many carbohydrate metabolizing loci in Bt, including 261 glycoside hydrolases and polysaccharide lyases, involved in the ability of Bt to utilize a spectrum of plant glycans (Martens et al., 2009). To assess how widely the BT2159-BT2156 operon is represented across bacterial strains, we performed a BLAST search for homologs with >60% identity on the amino acid level and >60% coverage of the query sequence. The results of this analysis revealed that this operon is highly conserved across Bacteroides strains (Figure S2B). To explore the relationship between presence of the gene cluster and ITC production, we profiled a subset of the strains containing homologs of BT2160-BT2156 for BGS metabolism. Some of the strains containing the complete gene cluster showed GS metabolizing activity, including B. sp 1_1_6, B. sp 2_1_22, B. sp D2, and B. ovatus ATCC 8483. Strains lacking a homolog of the regulatory protein BT2160 but containing homologs of BT2159-BT2156, B. uniformis ATCC 8492 and B. intestinalis DSM17393, were unable to metabolize GS to ITC. Presence of the complete gene cluster, however, was not predictive of ITC production; B. caccae ATCC 43185 and B. xylanisolvens SD CC 1b both contained homologs to the full operon but did not show GS metabolism under the tested conditions. Chemical genomics studies have identified this operon as being important for catabolism of trehalose, leucrose, palatinose, and raffinose (Liu et al., 2019). Gene expression data from chemostat-grown monocultures of Bt (GEO dataset GPL1821) showed that the operon genes are strongly upregulated by glucose-containing media relative to pig mucin glycan-containing media (Sonnenburg et al., 2006). Disruption of this operon also lowered fitness of Bt in mice when Bt was co-colonized with Eubacterium rectale or with a community of Firmicutes but not when in a mono-colonized context or in a community of other Bacteroides strains (Goodman et al., 2009). Despite their abundant representation in Bacteroides genomes and their importance for Bt fitness, however, none of these proteins have been biochemically characterized.
Two enzymes are necessary for GS transformation in Bt
To identify which genes in the BT2159-BT2156 operon are directly involved in GS hydrolysis, we generated targeted genomic deletions by replacing each open reading frame in the operon with an in-frame start-stop codon pair (Koropatkin et al., 2008). We found the growth of the knockout strains to be consistent with wild-type (WT) Bt growth in rich media with glucose (Figure S2C). Deletion mutants were cultured in rich medium supplemented with BGS and assayed for BITC-cys in spent media using LC-MS/MS (Figure 2B). As anticipated, knockout of transcriptional regulator BT2160 resulted in loss of GS metabolism. Of the genes that encode enzymes, BT2157 was necessary for ITC production in Bt and deletion of BT2158 resulted in only trace production of BITC-cys. Deletion of BT2159 was observed to be detrimental to GS metabolism but detectable levels of ITC were still produced, while knockout of BT2156 did not significantly alter GS metabolism.
From simultaneous monitoring of Bt growth and BITC production in rich medium containing glucose, we observed that GS metabolism became significant only in late stages of growth (Figure S2E), suggesting that GS was being used as a late-stage carbon source following depletion of glucose. To ensure that the Bt mutants maintained consistent GS metabolizing behavior across different carbon sources, we cultured BtΔ2157 in rich medium with a panel of carbohydrates and BGS. For each of the carbon sources surveyed, BtΔ2157 lacked the ability convert GSs to ITCs (Figure S2F).
To support the role of BT2157 and BT2158 in the conversion of GSs to ITCs, we used a plasmid-based expression system (Smith et al., 1992) to complement BtΔ2157 and BtΔ2158 with their respective deleted genes. Expression of the deleted gene rescued GS metabolism in both strains, confirming that ITC production is only observed when both BT2157 and BT2158 are present in Bt (Figure S2G).
Expression of BT2159-BT2156 in an inactive Bacteroides strain results in gain of activity
We sought to determine if BT2157 and BT2158 represent the minimal unit required for GS hydrolysis in Bacteroides fragilis (Bf), which we determined is unable to metabolize GS in culture media (Figure 1C). BLAST analysis also confirmed the absence of close homologs of BT2160-BT2156 in the Bf genome. Using a plasmid-based expression system (Smith et al., 1992), we expressed subsets of the BT2159-BT2156 operon in Bf, cultured these strains in rich medium supplemented with BGS, and assayed for BITC-cys in spent media using LC-MS/MS. Growth of these Bf strains was also monitored to ensure that the constitutive expression of these genes did not result in growth defects relative to a strain expressing RFP (Figure S2D). We found that simultaneous expression of BT2157 and BT2158 was not sufficient for gain of GS metabolizing activity (Figure 2C). In contrast, expression of either BT2156 or BT2159 in addition to BT2157 and BT2158 resulted in measurable production of ITCs. Furthermore, expression of all four enzymes results in gain of GS metabolizing function in Bf.
In vitro conversion of GS to ITC requires BT2158 and either BT2156 or BT2157
Given that BT2157 and BT2158 are important but not sufficient for activity in vivo, we next aimed to test the function of these proteins in vitro. We used E. coli as a heterologous host for the production of each of the proteins encoded by BT2159-BT2156. While three of the proteins were produced with sufficient yields to permit biochemical assays, we achieved only low yields of BT2158 (Figure S3A). To improve the yield of soluble protein, we turned to Bt as an expression host. We used a Bf phage promoter sequence to drive overproduction of His-tagged BT2158 with its native ribosome binding site in the BtΔ2158 mutant (Whitaker et al., 2017). This approach resulted in increased yields of soluble protein (Figure S3B). To ensure that native Bt proteins were not co-eluting or being pulled down with the His-tagged BT2158, we analyzed the purified protein fraction with SDS-PAGE and observed a single protein band corresponding to the mass of BT2158 (Figure S3C).
Before initiating biochemical assays, we considered the cofactors that might be required for protein activity. InterPro analysis revealed domains in BT2159 and BT2158 that are unique to the MocA/Idh/Gfo family of proteins (Figure S2A), members of which are known to bind nicotinamide adenine dinucleotide (phosphate) (NAD(P)) (Taberman et al., 2016). Characterized members of this protein family include the myo-inositol 2-dehydrogenase from Bacillus subtilis (BsIDH). Sequence alignment of BT2158 with BsIDH showed that Lys-133, Asp-248, and His-252 of BT2158 correspond to Lys-97, Asp-172, and His-176 of BsIDH, conserved residues that form a triad involved in hydride transfer (van Straaten et al., 2010). Based on this analysis, we chose to include NAD+ in our initial experiments.
We next incubated different combinations of purified BT2159-BT2156 in the presence of BGS and NAD+ to identify the minimal set of proteins required for in vitro GS conversion. After quenching the reactions with acetonitrile and adding cysteine for ITC conjugation, we measured BITC-cys formation and BGS depletion using LC-MS (Figure 3A). As with the complemented Bf strains, individual proteins alone did not mediate BGS conversion in vitro. When pairs of proteins were tested, however, measurable activity was observed for BT2158 when combined with either BT2156 or BT2157. In contrast to the in vivo analysis of Bf strains expressing subsets of these genes, however, addition of BT2156 and BT2159 to BT2157 and BT2158, representing the complete set of proteins encoded by the operon, did not seem to enhance BGS turnover. Taken together, our data indicate that BT2158 is necessary but not sufficient for BGS hydrolysis in vitro.
Figure 3. In vitro GS conversion by recombinant proteins.

(A) Extracted ion chromatograms (EIC) of BGS (m/z 408.0428) and BITC-cys (m/z 271.0570) produced by different combinations of BT2159-BT2156 proteins in vitro, shown on the same scale. The first row of chromatograms represents BGS and BITC-cys standards. Data shown represent the mean of three replicates, with the shaded regions representing one standard deviation.
(B) Direct injection mass spectrometry (MS) EIC showing glucose release (m/z 179.0561), substrate consumption (glucoraphanin (GRP): m/z 436.0411; glucobrassicin (GBR): m/z 447.0538), and ITC (sulforaphane (SFN): m/z 178.0355) production over time by different combinations of BT2159-BT2156 proteins. The ion count corresponding to a known reference SFN concentration is represented by a dotted line. No corresponding ITC was observed for GBR. Substrate ion count was normalized against the initial ion count for each combination of proteins. Data shown is one replicate, representative of triplicate results.
(C) Direct injection MS EIC showing glucose release (m/z 179.0561) and substrate consumption (m/z 341.1089) over time by different combinations of BT2159-BT2156 proteins with cellobiose and maltose. Substrate ion count was normalized against the initial ion count for each combination of proteins. Data shown is one replicate, representative of triplicate results. See also Figure S3.
To further investigate the cofactor requirements of the Bt proteins, we tested BT2157 and BT2158 with a panel of nicotinamide cofactors (Figure S3D). While the presence of any of the four nicotinamide cofactors improved BGS conversion by BT2157 and BT2158, NAD+ resulted in the most significant increase in activity by the protein pair. However, basal levels of activity were still observed in the absence of added cofactor, suggesting either that nicotinamide cofactor is not necessary for activity or that low levels of cofactor were co-purified with the proteins.
We also tested the activity of BT2159-BT2156 towards GSs with non-aromatic side groups, including glucoraphanin (GRP), which has been directly associated with the chemopreventive effects of brassicas (Jeffery and Araya, 2009), and glucobrassicin (GBR), another GS found in broccoli (Figure 3B). GS conversion and glucose production were tracked by hourly direct injections with time-of-flight mass spectrometry. While SFN production from GRP was also simultaneously measured, mass features corresponding to the ITC derived from GBR were not observed, likely due to the instability of indole ITC (Agerbirk et al., 2009). As observed with BGS, BT2158 with either BT2156 or BT2157 represent the minimal set of proteins required for activity on GRP or GBR. With the evidence from knockout Bt strains and complemented Bf strains, these in vitro results indicate that BT2158 is necessary for GS hydrolysis to ITCs, but the addition of other proteins, namely BT2156 or BT2157, is required for activity.
BT2159-BT2156 metabolize a selection of disaccharides in vitro
Considering the carbohydrate-metabolizing annotations ascribed to BT2160-BT2156, we hypothesized that other dietary carbohydrates might also serve as substrates. We incubated different combinations of purified protein with NAD+ and a panel of carbohydrates, measuring substrate consumption and glucose production by direct injection mass spectrometry. Subsets of the proteins were found to be active on both cellobiose and maltose, with BT2156 and BT2158 representing the minimal set required for cellobiose hydrolysis, and the combination of BT2157 and BT2158 needed for maltose hydrolysis (Figure 3C). Carbohydrates that were not significantly hydrolysable by any protein combination tested include raffinose and lactose (Figure S3E), potentially indicating that a substrate with glucose on the non-reducible end is required for activity. In contrast to the redundant activity we observed with different pairs of enzymes on GSs, it is notable that these disaccharides was acted on by only one pair of proteins. The pair of BT2157 and BT2158 did not exhibit activity on cellobiose and BT2156 with BT2158 did not act on maltose. Maltose and cellobiose are both composed of two glucose units linked by an α - or β -glycosidic bond, respectively, suggesting that BT2157 or BT2156 may dictate α-versus β-O-glucosidase activity when combined with BT2158. For both disaccharides, addition of the remaining proteins encoded by the operon did not significantly improve activity. Comparison of the metabolic rates of these proteins on the different substrates revealed a preference by each active protein pair for its respective disaccharide over GSs (Figure S3F).
ITC production is reduced in gnotobiotic mice colonized with a Bt mutant deficient in GS metabolism
To determine if this locus is required for GS processing by Bt in the context of gut colonization, we tested GS metabolism in mice mono-colonized with WT Bt or BtΔ2157 (Figure 4A). Relative colonization densities of WT Bt and BtΔ2157 were similar throughout the duration of the dietary intervention (Figures S5B and S5C). Mice were fed a polysaccharide-deficient chow, supplemented with two consecutive daily doses of pure BGS. We observed higher levels of BITC N-acetyl cysteine (BITC-NAC) and BITC-cys, established biomarkers of crucifer intake and ITC exposure (Hwang and Jeffery, 2003), in the urine of WT Bt-colonized mice compared to BtΔ2157-colonized or germ-free mice (Figure S4E). These data provided initial evidence that BT2159-BT2156 play a role in the metabolism of GSs in vivo, corroborating our genetic and in vitro biochemical findings. Although we observed higher levels of BITC-derived metabolites in the urine and feces of Bt-colonized mice following BGS doses, this effect was inconsistent; moreover, we detected background GS hydrolysis in germ-free mice, as noted in a previous report (Budnowski et al., 2015).
Figure 4. Monocolonization of gnotobiotic mice with mutant Bt.

(A) Schematic for colonization, dosing schedule, and collection of urine and feces. Each mark in the dosing schedule corresponds to an interval of 24 hours. Numbers in the unfilled shapes represent the dose within that series. Sample collection timepoints for colonization density measurements (Fig. S5B) are marked with red arrows.
(B) Sulforaphane N-acetyl cysteine (SFN-NAC), SFN-cys mercapturic acid conjugates, and GRP excreted in urine six hours after broccoli meals containing GRP. Creatinine-normalized quantities are shown in Fig. S5D. The ion counts corresponding to a known reference concentrations are represented by dotted lines. Metabolites were measured by LC-MS/MS and quantified by MRM (SFN-NAC: m/z 341.1→ m/z 177.9; SFN-cys: m/z 299.1→ m/z 136; GRP: m/z 436.0→ m/z 372). n=5 for each colonized group and time point unless otherwise noted on the graph. ** represents significance with p<0.01 from the germ-free control at the corresponding time point. * represents significance with p<0.05. Statistical groups were determined using the Tukey test.
(C) SFN-cys and GRP in feces collected six hours after broccoli meals containing GRP. SFN-cys was the product of in situ conjugation of SFN with exogenous cysteine. Bars represent the mean values for each group, with individual data overlaid (different shapes correspond to individual mice). See also Figure S4.
Reasoning that both issues might stem from administration of purified GS outside of the context of a food matrix, we administered GS in the form of reconstituted broccoli meals, followed by one bolus of a GS-free broccoli meal supplemented with pure BGS the following week (Figure 4A). Though the broccoli meals contained a variety of GS including neoglucobrassicin, hydroxyglucobrassicin, and erucin (Figure S4D), we limited our analysis of urine and fecal samples to BGS, GRP, and the products of their metabolism, due to the established biological activities of their ITCs. Six hours after the broccoli meals, higher levels of SFN-NAC and SFN-cys were consistently observed in the urine and feces, respectively, of mice colonized by Bt versus BtΔ2157 (Figure 4B). Consistent with pure compound feedings, higher levels of SFN-cys, the upstream metabolite of SFN-NAC in the mercapturic acid pathway, and free SFN were also observed in the urine of mice colonized with WT Bt compared to BtΔ2157 or germ-free mice (Figure S4F). Taken together, these experiments indicate that the BT2159-BT2156 operon plays an important role in GS hydrolysis in vivo.
The BT2160-BT2156 gene cluster is prevalent in metagenomic stool cohorts
To assess how widely this operon is represented in the human gut, the metagenomic prevalence of BT2160-BT2156 was examined in stool cohorts collected from healthy subjects in the US (denoted Abu-Ali, HMP1–2, and HMP1) (Abu-Ali et al., 2018; Lloyd-price et al., 2017; The Human Microbiome Project Consortium, 2012), Spain, Denmark (Nielsen et al., 2014), China (Wang et al., 2012), and Fiji (Brito et al., 2016). With the exception of the Fijian cohort, the full gene cluster was present (defined as covering ≥50% of each gene in the operon) in >40% of subjects in all cohorts (Figure 5; Figure S5). In addition to metagenomic distribution, we also queried for expression of BT2159-BT2156 in one cohort of 96 subjects (Abu-Ali et al., 2018) for which paired metatranscriptomic data had also been collected. Of this cohort, the BT2159-BT2156 gene cluster was found in the metagenomic data of 77 subjects, with transcripts of BT2159-BT2156 present in samples from 15% of these individuals. These data suggest that this gene cluster is widely distributed but may be conditionally expressed.
Figure 5. Metagenomic and transcriptomic prevalence of BT2160-BT2156.

Solid bars represent the fraction of subjects in metagenomic stool cohorts containing all five genes in the operon, with gene presence defined as a coverage of ≥50%. Cross-hatched bars represent the fraction of subjects in the metatranscriptomic cohort containing BT2159-BT2156, of the total subjects with the BT2160-BT2156 in the paired metagenome. The total number of subjects in a cohort is denoted on the right of each bar. See also Figure S5.
DISCUSSION
Despite the impact of dietary plant metabolites on human health and the role of the gut microbiome in modulating their activity, few microbial genes required for conversion of these molecules have been described. Our understanding of this metabolism has often been limited to identification of the involved strains (Clavel et al., 2006; Tsuchihashi et al., 2008). One of the few studies that report specific bacterial genes and mechanisms acting on bioactive plant molecules identified a cytochrome-encoding operon responsible for inactivation of the plant-derived cardiac drug digoxin by the gut bacterium Eggerthella lenta (Haiser et al., 2013). The lack of knowledge regarding the genetic basis for production of reactive ITCs by gut microbiota limits our ability to understand inter-individual differences in GS hydrolysis and use quantitative approaches to connect GS exposure to their effects on the host.
Our efforts to identify a genetic basis for GS conversion to ITCs by gut microbiota have led to the identification of an operon in Bt that is necessary and sufficient for GS activation and is also able to metabolize selected disaccharides. An interesting implication that arises from the identification of this pathway is its potential involvement in mediating microbe-microbe interactions in the gut. Certain bacterial strains are more susceptible to growth inhibition by ITC (Dufour et al., 2015). In this study, we examined growth inhibition of Bt and E. coli, as well as of the pathogen Salmonella typhimurium (Figure S1D), in the presence of ITCs generated by Bt. While growth of E. coli and S. typhimurium was inhibited by ITCs in spent Bt media, Bt growth in the presence of GSs was uninhibited. Though the mechanisms underlying susceptibility and resistance to ITC are not well understood, these data suggest that ITC production mediated by this metabolic pathway has the potential to influence relative species abundances in the microbiome. The extent to which this gene cluster may affect community profiles in the gut, however, is yet to be determined.
While this operon plays an important role in mediating ITC production by a specific gut commensal, there are other mechanisms through which GS activation may occur in the host. Beyond Bacteroides strains, other human isolates have been associated with GS metabolism, including Lactobacillus agilis R16 (Palop et al., 1995) and E. coli VL8 (Luang-In et al., 2016). Because they lack homologs to BT2160-BT2156, these strains likely perform this metabolism through different mechanisms. Based on the prevalence of this gene cluster in public stool data sets, however, the pathway for GS activation encoded by BT2160-BT2156 is both widely represented and expressed (Figure 2B). These distinct pathways for GS activation, differences in metabolic rate, conditional expression of the involved genes (Figure 5), and the relative abundance of metabolizing strains may contribute to the inter-individual variability in ITC production observed in human feeding studies (Li et al., 2011). With the genetic and biochemical basis responsible for one method of gut microbe-mediated GS activation now identified, we can begin to distinguish between the relative contributions of these mechanisms towards host exposure to ITC.
To date, identified bacterial glycosidases capable of cleaving the thio-linked sugar of GS include a glycoside hydrolase family 3 β-O-glucosidase in the soil-isolate Citrobacter WyE1(Albaser et al., 2016); after confirmation of GS degradation by cell-free protein extract, activity-guided purification was used to isolate the active protein. A 6-phospho-β-glucosidase was also found to be associated with GS metabolism in the pathogenic E. coli strain 0157:H7, identified through homology to plant myrosinases (Cordeiro et al., 2015); however, deletion of the identified genes did not abolish the ability of the bacteria to hydrolyze the aliphatic GS sinigrin. While, to our knowledge, there have not been any reports of gut bacterial myrosinase-encoding genes that are both necessary and sufficient for GS hydrolysis, the sax operon in certain pathovars of the plant pathogen Pseudomonas syringae has been identified as being necessary and sufficient for overcoming ITC toxicity (Fan et al., 2011). Putative myrosinase activity by the 6-phospho-β-glucosidase from E. coli 0157:H7, in conjunction with GS-induced activation of a glucose phosphotransferase system in E. coli VL8, has led to the proposal that phosphorylation of the glucose moiety may be prerequisite for GS hydrolysis in these strains (Narbad and Rossiter, 2018). While the annotation of BT2156 as a sugar phosphate epimerase may suggest that BT2159-BT2156 act on a phosphorylated substrate, our time course in vitro reactions were performed in phosphate-free buffers and in the absence of externally supplied ATP (Figures 3B and C). In addition, features with masses corresponding to phosphorylated GS or phosphorylated hexose were not detected in either reactions with purified proteins or in the spent media of Bt strains. An alternative mechanistic hypothesis involves a two-step transformation for GS hydrolysis; initial oxidation of the GS sugar using NAD+ is followed by hydrolysis and concomitant reduction of the sugar back to glucose using the generated NADH (Taberman et al., 2016). We searched extensively for modified forms of GS, including GS bearing an oxidized sugar, in our assays, but did not observe putative intermediates. The lack of observable intermediates, however, does not necessarily preclude a redox mechanism for GS hydrolysis; these intermediates may be immediately processed by an enzyme complex and therefore not accumulate.
While Bacteroidetes are well known metabolizers of plant glycans, the question remains whether the BT2160-BT2156 operon was evolved to specifically process GS. Bacteroides spp. have been found to metabolize a variety of plant molecules, including the lignan secoisolariciresinol diglucoside (SDG) (Clavel et al., 2006) and the flavonoid glycoside rutin (Bokkenheuser et al., 1987). The β-glucosidase involved in SDG hydrolysis in Bacteroides uniformis ZL1 was also found to exhibit activity on other plant glycosides, including astragalin, rutin, and isoquercetin (Tao et al., 2014). Similarly, an α-L-rhamnosidase from Bacteroides JY-6 was isolated for its activity on different rhamnoglucosides, including rutin and hesperidin (Jang and Kim, 1996). BT2159-BT2156 exhibit similarly broad activity towards cellobiose and maltose in vitro, which suggests that these proteins may have evolved to process other glycosides in addition to GSs. The native substrate of these proteins, however, is yet to be determined.
Of the 88 polysaccharide utilizing loci (PUL) in Bt, the starch utilization system (Sus) is the most well-characterized and is comprised of eight proteins that work in a concerted manner to bind and degrade starch (Martens et al., 2009). Sus is regulated by the membrane protein SusR, which is activated upon binding with maltose or higher oligomers. Efforts to identify similarly regulated loci have revealed four SusR paralogs in Bt, including BT2160 (Ravcheev et al., 2013). Though BT2160 is annotated as SusR-like, there are a few differences between Sus and BT2160-BT2156. BT2160-BT2156 is not classified as a PUL because it lacks homologs to SusC or SusD, involved in substrate binding and transport (Foley et al., 2016). Our in vitro studies suggest that coordinated action by the periplasmic BT2158 with either BT2156 or BT2157 is required for hydrolytic activity. In contrast, SusA and SusB are involved in hydrolytic activity in the periplasm, but do not require coordinated activity with another enzyme. In addition, single deletions of susA or susB were not deleterious to growth on starch (D’Elia and Salyers, 1996), whereas single deletions of BT2157 and BT2158 both resulted in a loss of GS conversion. SusG is annotated as an outer membrane-bound glycosyl hydrolase and was found to be necessary but by itself insufficient for growth on starch, similarly to BT2157 (Shipman et al., 1999). In Sus, outer membrane proteins SusC-G form a complex that is required for starch binding and transport. SusC and SusD individually are insufficient for starch binding, but together are capable of binding 60% of the starch bound by WT Bt, with the addition of SusE and SusF providing the remaining affinity (Shipman et al., 2000). Though BT2157 and BT2158 together are sufficient for GS hydrolysis in vitro, presence of this pair in the inactive Bf did not result in gain of activity. Additional expression of BT2156 or BT2159 increased activity to 3% and 14%, respectively, of the activity of the Bf strain expressing the full operon, suggestive that these proteins may similarly require formation of a complex for GS hydrolysis. Interestingly, the in vitro hydrolysis of cellobiose and maltose require different combinations of proteins. While the mechanism behind GS and disaccharide hydrolysis by these proteins is yet to be determined, the combinations of proteins required for different substrates may provide insight into the catalytic roles that each of the proteins play.
In this study, we have identified a genetic and biochemical basis for the activation of a class of dietary plant metabolite, GSs, by a widely conserved bacterial species in the human gut microbiota. GSs have been extensively studied for the role of the activated product, ITCs, on human health. Despite their importance, genes in the gut microbiota that contribute to activation have remained elusive. Using a genome-wide screen, we identified the operon BT2159-BT2156 as a key player in GS conversion to ITCs by Bt. Discovery of the microbial pathways that produce bioactive compounds such as ITCs, in conjunction with recent developments in genetic tools for cellular therapeutics (Mimee et al., 2015; Whitaker et al., 2017) can contribute to engineering gut commensals for therapeutic benefit. On a more fundamental level, this work provides a critical advance in our understanding of the many interrelated mechanisms through which the gut microbiota and plant-derived metabolites in our diet interact to influence health and disease.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Elizabeth Sattely (sattely@stanford.edu). All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbe strains
Bacteroides strains were streaked from glycerol stocks on either brain heart infusion agar supplemented with 10% horse blood (BHI-BA) plates or brain heart infusion agar supplemented with 5 μg/mL hemin and 0.5 μg/mL menadione (BHIS) plates. Plates were supplemented, if requiring antibiotic selection, with 200 μg/mL gentamycin and 5 μg/mL erythromycin. Streaked plates were incubated at 37°C for 24–30 hours for non-selective plates and 48–54 hours for plates containing antibiotics, under anaerobic conditions using the GasPak anaerobic system (BD).
Following incubation, colonies from plates were inoculated into tryptone-yeast extract medium, prepared as described previously (Whitaker et al., 2017) and freshly supplemented with 0.5 mg/mL cysteine as a reducing agent, 0.5% glucose (TYG), and 5 μg/mL erythromycin if requiring antibiotic selection. Per liter of TYG: 10 g tryptone, 5 g yeast extract, 100 mL potassium phosphate buffer (1 M, pH 7.2), 40 mL TYG salt solution (per liter: 0.5 g MgSO4·7H2O, 10 g NaHCO3, 2 g NaCl in water), 1 mL CaCl2 solution (0.8%), and 1 mL FeSO4 solution (0.4 mg/mL) were combined, brought up to 1 L with distilled water, and then autoclaved. After tempering to 55°C, 1 mL menadione solution (1 mg/mL in ethanol) and 1 mL hematin-histidine solution (0.2 M, pH 8) were added. Liquid cultures were grown anaerobically at 37°C for 17–24 hours using the GasPak anaerobic system (BD) or an anaerobic chamber (Coy, 10% CO2, 5% H2). For glucosinolate metabolism assays and coupled growth assays, Bacteroides liquid cultures were then subcultured into fresh TYG supplemented with 0.5 mM glucotropaeolin (BGS) and 5 μg/mL erythromycin, if required for plasmid selection and incubated anaerobically at 37°C for 24–30 hours.
To screen for disruption in glucosinolate (GS) metabolizing genes in the Bacteroides thetaiotaomicron VPI-5482 (Bt) transposon insertion library, the isothiocyanate (ITC)-sensitive E. coli strain TOP10 harboring the irrelevant plasmid pENTR-sGFP (Life Technologies), included to permit growth on kanamycin-containing media, was inoculated from a glycerol stock into LB containing 50 μg/mL kanamycin and incubated aerobically with shaking at 37°C for 16–18 hours. This E. coli culture was diluted 1:10 into LB with kanamycin in preparation for use as the indicator strain for the coupled growth bioassay, as described below.
In vitro studies
For heterologous production of Bt proteins, E. coli BL21 DE3 strains were grown for 18 hours at 37°C from glycerol stocks streaked on LB agar plates containing 50 μg/mL kanamycin. Single colonies were inoculated into 20 mL of LB with 50 μg/mL kanamycin. Following growth at 37°C for 18 hours, strains were subcultured into 2L of LB with kanamycin and grown at 37°C with agitation until an OD600 of 0.6 was reached. Cultures were induced with 0.1 mM ispropyl β-D-1 thiogalactopyranoside (IPTG), after which they were incubated at 30°C for 6 hours, and then harvested for protein purification, as described below.
For improved soluble production of BT2158, the single deletion mutant BtΔ2158, BtΔ2158 was streaked onto BHIS agar plate and grown for 30 hours at 37°C under standard anaerobic conditions, described above. At least three colonies were inoculated into 12 mL of TYG and grown at 37°C for 24 hours under anaerobic conditions. Following growth, the strain was subcultured into 1 L of TYG, grown at 37°C for 24 hours under anaerobic conditions, and harvested for protein purification, as described below.
Mice
Mouse studies were performed in strict accordance with a Protocol for Care and Use of Laboratory Animals, approved by the Stanford University Institutional Animal Care and Use Committee. Germ-free Swiss Webster mice (Taconic) were maintained in gnotobiotic isolators under aseptic conditions on a 12 hour light cycle and fed ad libitum with a standard autoclaved chow diet (LabDiet 5K67). Groups of five 10–13 week old male mice were randomly assigned to experimental groups and colonized with either wild-type Bt or BtΔ2157 by oral gavage of overnight cultures (Marcobal et al., 2011), or maintained germ-free as a negative control. To confirm colonization and maintenance of WT or mutant Bt status, fecal pellets were collected six days after gavage. Briefly, 1 μL of fecal pellet was serially diluted, plated on BHI-BA, and grown anaerobically for 24 hours at 37°C. Colonization density was recorded as the maximum and minimum numbers of cfu per L of plated fecal pellet for each colonization group. Cultures were inoculated into TYG supplemented with 0.5 mM BGS and grown anaerobically for 24 hours at 37°C. Cells were pelleted, and spent supernatant was assayed for conversion of glucosinolate to isothiocyanate by LC-MS/MS, as described below (Figure S4A). Five days following colonization, the mice were switched to a polysaccharide deficient (PD) diet (Bio-Serv S5805) and maintained on this diet for the remainder of the mouse experiment. Six hours following the final glucosinolate dose (Figure 4A), mice were sacrificed using CO2 asphyxiation in accordance with approved protocols, and tissue was immediately harvested and processed as described below.
METHOD DETAILS
Transposon mutagenesis of Bt VPI-5482
The transposon library was constructed using the Mariner transposon vector pSAM_Bt to mutagenize Bt VPI-5482 (Goodman et al., 2009). Bt VPI-5482 was inoculated from glycerol stocks into TYG and cultured anaerobically at 37°C for 16–18 hours. Escherichia coli (E. coli) harboring pSAM_Bt was similarly inoculated into LB with 50 μg/mL ampicillin and grown aerobically with shaking at 37°C for 16–18 hours. Bt and E. coli were subcultured 1:20 into TYG and 1:100 into LB with ampicillin, respectively, and grown to exponential phase as determined by OD600 values between 0.4 and 0.6. Cells were pelleted by centrifugation at 17,000 g for 20 minutes. For the conjugation reaction, the Bt and E. coli pellets were resuspended in 1 mL of TYG, combined, plated as three 100 μL spots on BHI-BA, and then incubated aerobically for 8 hours at 37°C. Conjugation reactions were pooled and resuspended in phosphate buffered saline (PBS). Pooled resuspensions were then plated as 100 μL aliquots onto each of 50 BHI-BA plates supplemented with 25 μg/mL erythromycin and 200 μg/mL gentamicin and grown anaerobically at 37°C for 20–30 hours. Following selection, single colonies were inoculated into 200 μL TYG containing 25 μg/mL erythromycin and 200 μg/mL gentamicin and grown anaerobically at 37°C for 48 hours. Glycerol stocks of individual mutants were prepared by supplementing the liquid cultures with an equal volume of 60% glycerol in TYG (30% glycerol final concentration) and stored at −80°C until screening for glucosinolate metabolizing activity.
Bioassay for loss of GS metabolizing activity
To understand growth inhibition of bacterial strains by Bt-produced isothiocyanate (ITC), wild-type Bt was inoculated from glycerol stock into TYG and incubated anaerobically at 37°C for 22–30 hours. Liquid Bt culture was subcultured 1:50 into fresh TYG supplemented with 0.5 mM glucotropaeolin (BGS) and incubated anaerobically at 37°C for an additional 24 hours, followed by pelleting the cells by centrifugation at 5000 g for 15 minutes. In parallel, E. coli TOP10 harboring the irrelevant plasmid pENTR-sGFP (Life Technologies, included to permit growth on kanamycin-containing media) and Salmonella typhimurium SL1344 were inoculated from glycerol stocks into LB containing 50 μg/mL kanamycin and 50 μg/mL streptomycin, respectively, and incubated with shaking at 37°C for 16–18 hours. Liquid E. coli and S. typhimurium cultures were diluted 1:10 into LB with kanamycin or streptomycin, as appropriate, and then further diluted 1:10 into spent Bt supernatant. Cultures were incubated aerobically at 37°C with shaking, with growth monitored by OD600 measurements taken every 10 minutes using an Epoch 2 plate reader (Biotek) held at 37°C.
Similarly, to screen for disruption in candidate GS metabolizing genes in the Bt transposon insertion library, ITC-sensitive E. coli TOP10 harboring the plasmid pENTR-sGFP (Life Technologies) was inoculated from a glycerol stock into LB containing 50 μg/mL kanamycin and incubated aerobically with shaking at 37°C for 16–18 hours. E. coli culture was diluted 1:10 into LB with kanamycin in preparation for use as the indicator strain for the bioassay (described below). In parallel, Bt transposon mutants were inoculated from glycerol stocks into TYG in microwell plates and incubated anaerobically at 37°C for 22–30 hours. The Bt mutants were subcultured 1:50 into TYG supplemented with 0.5 mM glucotropaeolin (BGS) and incubated anaerobically at 37°C for an additional 23–25 hours. The endpoint OD600 values of the Bt cultures were measured, and then the cells were pelleted by centrifugation at 5,000 g for 15 minutes. Next, 90 μL of supernatant was transferred to microwell plates containing 10 μL of the diluted E. coli culture described above (1:100 overall E. coli dilution). Microplates were incubated aerobically with agitation at 37°C. Growth of E. coli was monitored by measuring OD600 at the following timepoints: 0 h, 3 h, 6 h, 9 h, and 16 h.
Wild-type Bt VPI-5482 and Bt 8736 were included on each microplate as positive and negative control strains, respectively, for glucosinolate metabolizing activity. Approximately 2% of mutants (152 mutants) from the high-throughput screen were chosen for rescreening based on the following two criteria: 1) uncompromised Bt growth relative to wild-type Bt VPI-5482 (Bt mutant OD600 ≥ average − 1 standard deviation of Bt VPI-5482 OD600), and 2) loss of ability to inhibit subsequent E. coli growth (E. coli OD600 ≥ average − 1 standard deviation of E. coli OD600 grown in supernatant spent by Bt 8736). Of this top 2% of mutants, 77 were chosen for sequencing based on the consistency of their performance in a second and third round of rescreening, using the same criteria employed for the primary screen.
The following instrumentation was used at the High-Throughput Bioscience Center at the Stanford School of Medicine: Infinite M1000 plate reader (Tecan) to measure OD600, Matrix Wellmate (Thermo Scientific) to dispense media, and Bravo Liquid Handler (Agilent) to transfer spent media.
Identification of transposon mutation sites
Transposon insertion sites in Bt VPI-5482 mutants were identified using a semi-random PCR approach, adapted from Goodman et al. (Goodman et al., 2009). Genomic regions adjacent to transposon insertion sites were amplified by PCR using the transposon-specific primer 5’-ACGTACTCATGGTTCATCCCGATA-3’ and one of three degenerate primers 5’-GGCCACGCGTCGACTAGTACNNNNNNNNNNGATGC-3’,
5’-GGCCACGCGTCGACTAGTACNNNNNNNNNNGGCCG-3’,
5’-GGCCACGCGTCGACTAGTACNNNNNNNNNNGTAAT-3’, where N represents either A, T, G, or C synthesized using the hand-mix option from Integrated DNA Technologies. Products from the first PCR were purified (QIAquick kit, Qiagen) and used as template for a second round of PCR, using the nested transposon specific primer 5’-GCGTATCGGTCTGTATATCAGCAA-3’ and primer 5’-GGCCACGCGTCGACTAGTAC-3’. Following agarose gel electrophoresis, amplicons were purified from an agarose gel (Zymoclean kit, Zymo Research) and submitted for Sanger sequencing using the nested transposon-specific primer 5’-TCTATTCTCATCTTTCTGAGTCCAC-3’. Loci corresponding to the genomic regions adjacent to transposon insertions were identified using BLAST.
In this manner, transposon insertion sites were identified for 65 Bt mutants that displayed a loss of GS metabolizing activity. More than two thirds of these mutations occurred in two putative operons. One operon (BT1220-BT1222) encodes genes annotated as the pentose phosphate pathway and was deemed likely to eliminate glucosinolate metabolizing activity via pleiotropic effects. The other operon (BT2156-BT2160) carried annotations suggestive of carbohydrate metabolizing activity was selected for further molecular genetic and biochemical characterization.
Gene cluster homology search
To search for homologs to the BT2160-BT2156 gene cluster, microbial genomes containing potential homologs were identified using a combination of JGI IMG neighborhood search (Chen et al., 2018) and MegaBlast. BlastX was then used to search this set of genomes (downloaded from NCBI on 5 July 2019) with default parameters, using individual genes in BT2160-BT2156 as queries. Strains containing homologs to the gene cluster were defined as having homologs to at least BT2159-BT2156, with greater than 60% identity on the amino acid level and greater than 60% coverage of the query sequence. A subset of these strains was profiled for glucosinolate metabolizing activity, as described below.
Bacterial assays for glucosinolate metabolism
Bacteroides strains were streaked from glycerol stocks onto BHIS plates and incubated anaerobically at 37°C for 22–26 hours. For strains harboring the pFD340 plasmid, 5 μg/mL of erythromicin and 200 μg/mL of gentamicin were included. Single colonies were inoculated into 200 μL of TYG, containing 5 μg/mL erythromycin if harboring a pFD340 plasmid, and grown anaerobically at 37°C without shaking. After 17–24 hours of growth, strains were subcultured into 200 μL fresh TYG supplemented with 0.5 mM glucotropaeolin (BGS) and 5 μg/mL erythromycin, if required for plasmid selection. Following anaerobic incubation at 37°C for 24 hours, culture density was measured by OD600 using an Epoch 2 plate reader (Biotek) to confirm relative growth, and cells were pelleted by centrifugation at 1400 g for 20 minutes. The supernatant was diluted 1:10 with [10% (v/v) acetonitrile and 90% (v/v) water with 0.1% (v/v) formic acid], filtered through 0.45 μm PTFE filters, and analyzed by either liquid chromatography-mass spectrometry (LC-MS) or liquid chromatography-tandem mass spectrometry (LC-MS/MS), as described below.
Targeted mutagenesis of Bt VPI-5482
All targeted deletion mutants in Bt were constructed in a 5-fluoro-2’deoxyuridine (FUdR) resistant Bt VPI-5482 mutant lacking thymidine kinase (BtΔtdk) using counterselectable allelic exchange (Koropatkin et al., 2008). Genomic sequences spanning 1.5 kb upstream and downstream of the gene to be deleted were amplified from Bt VPI-5482 genomic DNA by PCR. Amplicons were then assembled using fusion PCR into an in-frame start-stop codon pair flanked by the ~1.5 kb genomic regions surrounding the target gene. Purified inserts were cloned into the pExchange-tdk suicide vector digested with PstI and NotI using the Gibson assembly method (Gibson et al., 2009)and transformed into E. coli S17–1 λpir chemically competent cells (Strand et al., 2014). Plasmids from several transformants were miniprepped and sequenced to confirm correct assembly, and then conjugated into BtΔtdk. For conjugation, E. coli S17–1 λpir harboring the gene-targeting pExchange shuttle vectors were grown on LB plates containing 100 μg/mL ampicillin and inoculated into LB containing 100 μg/mL ampicillin and grown for 17–20 hours at 37°C with agitation. In parallel, BtΔtdk was grown anaerobically on BHI-BA plates, and then inoculated into TYG and grown anaerobically for 17–20 hours at 37°C. Cells were pelleted by centrifugation at 1400 g for 15 minutes and washed with LB. Pelleted strains containing the donor plasmid were resuspended in LB and then used to resuspend BtΔtdk. Mating mixtures were spotted onto BHI-BA plates and incubated aerobically at 37°C for 24 hours. To select for the first round of recombination, mating cultures were streaked onto BHI-BA plates containing 200 μg/mL gentamycin and 5 μg/mL erythromycin and grown anaerobically for 48 hours. To counter-select for the second recombination event, colonies from the first round were streaked onto BHI-BA plates containing 200 μg/mL FUdR and grown for 48–72 hours. PCR amplification of genomic DNA flanking the gene was used to confirm deletion, using the following sets of primers:
BT2159_F: 5’-GCTAGTTTTGCGATATCAGTTTTCG-3’
BT2159_R: 5’-GGCAACTTCCATCCTTCACG-3’
BT2158_F: 5’-AGCTAATGGATAAAATACTTTCTTCATAATAATAACTATTTAATTTTTCT-3’
BT2158_R: 5’-ATCCGTGCTGCAGCCAATATAAACGAATTTGTGCCA-3’
BT2157_F: 5’-AACAAGACACTGGAATGGGAC-3’
BT2157_R: 5’- CACCGCGGTGGCGGCCGCTCTAGCAGTATAGCTGTCGATTAGTATG-3’
BT2156_F: 5’-CGATAAGCTTGATATCGAATTCCTGCAGCAGTATAGCTGTCGATTAGTATG-3’
BT2156_R: 5’-ATCCGTGGATCCCACGATAAAAGTAAGTTAAGA-3’
PCR amplification of the gene was used to confirm that the gene had not been inserted into another region of the genome using the following sets of primers:
BT2159_F: 5’-TGCGATACAGATCCTACCACGC-3’
BT2159_R: 5’-CAATGTGAAGAGCCCGACAACC-3’
BT2158_F: 5’-CGAAACAATTTGCAGCCGAAC-3’
BT2158_R: 5’-GGCAACTTCCATCCTTCACG-3’
BT2157_F: 5’-TGCAAGCCAGCAAATTCAGC-3’
BT2157_R: 5’-CAGTCCAGAACTTTCACGCG-3’
BT2156_F: 5’-CTGCCGGGCTGAAGGTTTTATC-3’
BT2156_R: 5’-TCAGCAATACACTGGTCCCACC-3’
Heterologous expression in B. fragilis
Expression of subsets of BT2159-BT2156 in the heterologous host Bacteroides fragilis (B. fragilis) was achieved using the extra-chromosomal Bacteroides expression vector, pFD340 (Smith et al., 1992). Target gene sequences, including the ~50 bp region upstream of the gene to encapsulate the native ribosome binding site, were amplified from Bt VPI-5482 genomic DNA by PCR. Amplicons encoding sequences for BT2159-BT2157 and BT2158-BT2156 were purified, and then inserted into pFD340 plasmid digested with BamHI using the Gibson assembly method. All other single and multi-gene amplicons were purified, digested with SacI and BamHI, and then ligated with T4 DNA ligase into pFD340 plasmid digested with the same enzymes. An empty vector control was generated by amplifying a sequence encoding RFP, digesting the purified amplicons with BamHI and PstI, and then ligating it using T4 DNA ligase into pFD340 digested with the same enzymes. Assembled vectors were transformed into chemically competent E. coli TOP10 and conjugated into B. fragilis via triparental mating, similarly to the procedure described above, but with the inclusion of helper strain E. coli RK231 in the mating mix (Smith et al., 1992). Selection was performed on BHI-BA plates containing 200 μg/mL gentamycin and 5 μg/mL erythromycin.
Bacteroides growth curves and ITC time course
For growth curves of Bt and Bf mutants, strains were cultured using the same method as described for glucosinolate metabolism assays above. Following subculturing in TYG supplemented in 0.5 mM BGS, cultures were transferred into an anaerobic chamber and OD600 measurements were taken every 10 minutes using an Epoch 2 plate reader (Biotek) held at 37°C. For simultaneous measurements of growth and ITC production over time, 12 mL of wild-type Bt was cultured in the presence of 0.5 mM BGS as previously described for glucosinolate metabolism assays, in an anaerobic chamber at 37°C. Growth of Bt cultures was measured over a course of 24 hours by sampling 700 μL of culture and measuring OD600 using a Genesys 20 spectrophotometer (Thermo Scientific). After OD600 measurement at each sampled time point, 500 μL of culture was frozen in liquid nitrogen and stored at −80°C. After completion of the time course, frozen samples were thawed and centrifuged at 13,000 g for 10 minutes, and spent media was diluted and analyzed by LC-MS as described above.
Recombinant protein production
Sequences encoding BT2156, BT2157, and BT2159 were amplified from Bt VPI-5482 genomic DNA by PCR. Purified amplicons for BT2156, BT2157, and BT2159 were digested with BamHI and XhoI and ligated using T4 DNA ligase into the C-terminal His-tagged expression vector pET-24b digested with the same enzymes. Plasmids were transformed into E. coli TOP10 chemically competent cells, selected, miniprepped, and verified by Sanger sequencing (ELIM Biopharm). For protein production, purified plasmid was subsequently transformed into E. coli BL21 DE3 chemically competent cells.
E. coli BL231 DE3 strains were grown for 18 hours at 37°C from glycerol stocks streaked on LB agar plates containing 50 μg/mL kanamycin. Single colonies were inoculated into 20 mL of LB with 50 μg/mL kanamycin. Following growth at 37°C for 18 hours, strains were subcultured into 2L of LB with kanamycin and grown at 37°C with agitation until an OD600 of 0.6 was reached. Cultures were induced with 0.1 mM ispropyl β-D-1 thiogalactopyranoside (IPTG), after which they were incubated at 30°C for 6 hours. Cells were pelleted by centrifugation at 5500 g for 5 mins, and supernatant was discarded. Cell pellets were then frozen in liquid nitrogen and stored at −80°C.
For expression of His-tagged BT2158 in Bt VPI-5482, the gene was amplified from genomic DNA, including 50 bp upstream of the start codon to encapsulate the native RBS, as well as an appended sequence encoding a C-terminal 6xHis tag. Purified amplicon was digested with BamHI and XhoI and ligated using T4 DNA ligase into pFD340 digested with the same enzymes. To achieve higher levels of soluble protein yield, the IS4351 promoter sequence in the pFD340 vector was replaced with the phage promoter P_BfP1E6 sequence (Whitaker et al., 2017) using the PstI and BamHI restriction sites. Ligation products were transformed into E. coli TOP10 chemically competent cells and miniprepped plasmids were confirmed using Sanger sequencing. The construct was expressed in single deletion mutant BtΔ2158 via triparental conjugation with helper strain RK231, as described above. The transformed strain was streaked onto BHIS plates containing 200 μg/mL gentamycin and 5 μg/mL erythromycin and incubated at 37°C for 48 hours under anaerobic conditions. Single colonies were inoculated into 24 mL of TYG containing 5 μg/mL erythromycin and grown at 37°C for 24 hours. The strain was subcultured into 1 L of TYG containing the same antibiotics and incubated anaerobically at 37°C for another 20 hours. Cells were pelleted by centrifugation at 6000 g for 5 minutes, and supernatant was discarded. Cell pellets were frozen in liquid nitrogen and stored at −80°C.
For protein purification, E. coli and BtΔ2158 cell pellets were thawed by resuspension in 25 mL of lysis buffer (50 mM potassium phosphate, 300 mM NaCl, 10 mM imidazole, pH 7.8) and lysed with six 20 second bursts of sonication at 60% amplitude. Cell debris was pelleted by centrifugation at 38,000 g for 30 minutes at 4°C and discarded. All subsequent processes were performed at 4°C. Lysate was equilibrated with Ni-NTA agarose resin (Thermo Scientific) for 1 hour with rocking and loaded onto a 1.5×10 cm Econo-column chromatography column (Biorad). The resin was washed with 20 mL lysis buffer and 20 mL wash buffer (50 mM potassium phosphate, 300 mM NaCl, 20 mM imidazole, pH 7.8), followed by elution with successively higher concentrations of imidazole (50 mM potassium phosphate, 300 mM NaCl, 50/100/200/500 mM imidazole, pH 7.8). Elution fractions were analyzed by SDS-PAGE and the fractions with the desired protein were combined. Pooled fractions were buffer exchanged into storage buffer (50 mM potassium phosphate, pH 7.8) and concentrated using Amicon Ultra-4 centrifugal filter units with 10 kDa molecular weight cut off (Millipore). Glycerol was added to a final concentration of 10% (v/v), after which fractions were aliquoted, frozen in liquid nitrogen, and stored at −80°C. Total protein concentration was quantified using absorbance at 280 nm, measured using a Nanodrop 1000 spectrophotometer (Thermo Scientific).
In vitro endpoint biochemical assays
Standard in vitro endpoint protein reactions consisted of 0.5 μL of 100 mM BGS substrate, 1 μL of 50 mM NAD+, 200 pmol of each protein, and reaction buffer (10 mM potassium phosphate buffer, pH 7.2) up to a total volume of 50 μL. Each reaction contained final concentrations of 1 mM each of BGS and NAD+, and 4 pmol/μL of each protein. Reactions were initiated by the addition of substrate and incubated at 37°C for 15 hours, prior to quenching with 90 μL acetonitrile and 10 μL 15 mg/mL cysteine, to promote conjugation of free ITC and enable detection of ITC-cysteine conjugates by MS. Quenched reactions were centrifuged at 15,000 g for 5 minutes to pellet denatured protein. Supernatant was filtered through a 0.45 μm PTFE filter prior to analysis by LC-MS, as described below.
In vitro time course biochemical assays
All proteins were buffer exchanged using Amicon ultra-4 centrifugal filters with 10 kDa molecular weight cut off (Millipore) into volatile reaction buffer (10 mM N-methyl morpholinium acetate, pH 7) to ensure MS compatibility. Reactions monitoring a time course of in vitro protein activity consisted of 1 μL of 100 mM substrate, 2 μL of 50 mM NAD+, 450 pmol of each protein, and reaction buffer up to a total volume of 100 μL. Each reaction contained final concentrations of 1 mM each of substrate and NAD+, and 4.5 pmol/μL of each protein. Reactions were initiated by the addition of substrate, incubated at room temperature in an Agilent 1290 Infinity II multisampler, and sampled hourly for MS analysis for 17 hours, using the conditions described below.
Mouse study
Animal studies were performed under a protocol approved by the Stanford University Institutional Animal Care and Use Committee. Germ-free Swiss Webster mice (Taconic) were maintained in gnotobiotic isolators under aseptic conditions and fed ad libitum with a standard autoclaved chow diet (LabDiet 5K67). Groups of five 10–13 week old male mice were colonized with either wild-type Bt or BtΔ2157 by oral gavage of overnight cultures (Marcobal et al., 2011), or maintained germ-free as a negative control. To confirm colonization and maintenance of WT or mutant Bt status, fecal pellets were collected six days after gavage. Briefly, 1 μL of fecal pellet was serially diluted, plated on BHI-BA, and grown anaerobically for 24 hours at 37°C. Colonization density was recorded as the maximum and minimum numbers of cfu per L of plated fecal pellet for each colonization group. Cultures were inoculated into TYG supplemented with 0.5 mM BGS and grown anaerobically for 24 hours at 37°C. Cells were pelleted, and spent supernatant was assayed for conversion of glucosinolate to isothiocyanate by LC-MS/MS, as described below (Figure S4A). Five days following colonization, the mice were switched to a polysaccharide deficient (PD) diet (Bio-Serv S5805).
Pure glucosinolate dosing.
Two days following introduction of the PD diet, all mice received two daily doses of 200 μL of 45 mM sterile-filtered BGS by gavage, corresponding to a total dosage of 9 μmol per animal per day. BGS doses were sterile filtered to prevent contamination of the gnotobiotic environment. Urine and fecal samples were collected immediately prior to each treatment, as well as 3 and 6 hours after the first dose and 3, 6, 12, and 24 hours after the second dose. Urine and feces were both stored at −80°C until analysis and analyzed for BGS and BITC content by a combination of LC-MS and LC-MS/MS. Previous studies have shown that the primary metabolic fate of ITCs is conjugation to glutathione, followed by sequential metabolism via the mercapturic acid pathway to ITC-cysteinylglycine and ITC-cysteine (Shapiro et al., 2001). ITC-cysteine conjugates are acetylated to N-acetyl cysteine conjugates, which are then excreted in the urine. Previous studies have established ITC-NAC excretion in urine as a biomarker for crucifer intake and ITC exposure (Hwang and Jeffery, 2003). Urine samples were specifically analyzed for N-acetyl cysteine conjugates of ITC (ITC-NAC) (Figure 1C), as well as ITC-cysteine conjugates, the metabolite upstream of ITC-NAC conjugates in the mercapturic acid pathway. Metabolites from fecal samples were extracted, incubated with an excess of cysteine for reversible thiol conjugation, and analyzed for ITC-cys conjugates by LC-MS/MS.
Broccoli meal feeding.
Five days following the second pure BGS dose, mice received four daily doses of broccoli meals containing broccoli glucosinolates in a food matrix. To maintain gnotobiotic conditions, broccoli meals were comprised of a mixture of autoclaved, glucosinolate-free broccoli slurry and sterile-filtered, concentrated broccoli extract.
Broccoli meals were prepared from broccoli florets purchased from a local grocery store. To prepare concentrated broccoli extract, florets were microwaved for 3 minutes to inactivate native plant myrosinase, frozen in liquid nitrogen, and then lyophilized to dryness. Lyophilized florets were ground using a ball mill homogenizer (Retsch) and then reconstituted in water (13 mL/g dry weight) at 75°C for 20 minutes. Plant material was collected by centrifugation at 1875 g for 15 minutes and then discarded. Supernatant was filtered through a 5 μm nylon filter to remove remaining plant material. To concentrate the extract, filtered supernatant, totaling about 485 mL, was frozen in liquid nitrogen, and then lyophilized to dryness. Lyophilized extract was resuspended in 33 mL of deionized water, resulting in about a ~15 fold concentration of the original extract. Concentrated extract was sterile filtered through a 0.2 μm regenerated cellulose filter, aliquoted, frozen in liquid nitrogen, and then stored at −20°C. To quantify glucosinolates in the concentrated extract, 90 μL of extract was diluted into 180 μL of hydrophilic interaction liquid chromatography (HILIC) acetonitrile mobile phase, filtered through a 0.45 μm PTFE filter, and then analyzed by LC-MS, as described below.
For the glucosinolate-free broccoli slurry, raw florets were frozen in liquid nitrogen and then lyophilized to dryness. Lyophilized florets were ground using a ball mill homogenizer, reconstituted in water (8 mL/g dry weight), and then autoclaved at 128°C for 20 minutes, a process that resulted in thermal degradation of glucosinolates. Immediately prior to gavage, 1.8 mL of thawed concentrated extract was added to 1.2 mL of the glucosinolate-free slurry to constitute broccoli meals for a given treatment. To prevent degradation of glucosinolates in the extract, concentrated extracts were stored at −20°C and transferred daily into the gnotobiotic incubators.
All mice received daily doses of 200 μL of broccoli meal by gavage for four consecutive days, corresponding to a quantified dosage of 1.4 μmol of glucoraphanin and 0.13 μmol of glucobrassicin, as well as unquantified amounts of hydroxyglucobrassicin, methoxyglucobrassicin, and glucoerucin. Urine and feces were collected immediately prior to, as well as 3 and 6 hours after the first and third doses. Urine and feces samples were both stored at −80°C until analysis.
Twenty four hours following the final broccoli meal, all mice received one dose of 200 μL of BGS mixed with the glucosinolate-free broccoli slurry, providing a total dose of 4.5 μmol of BGS per mouse. Urine and feces were collected immediately prior to the feeding, as well as 3 and 6 hours following the feeding. Urine and feces were both stored at −80°C until analysis. Mice were sacrificed six hours following the feeding by CO2 asphyxiation in accordance with approved protocols, blood samples were collected in Microtainer SST tubes (BD), and serum and cecal contents were harvested. Serum and cecum samples were frozen in liquid nitrogen and stored at −80°C. Microbial DNA from 0.1 g of cecal contents from wild-type Bt and BtΔ2157-colonized mice was purified using the PureLink Microbiome DNA Purification Kit (Invitrogen). Colonization density of cecal samples was measured using quantitative PCR (qPCR) with Bt-specific primers 5’-GGGGGTATCTTCACCTTCGT-3’ and 5’-ATTCGGTTGAACGCTTGTCT-3’. qPCR reactions were performed using SensiMix SYBR No-ROX kit (Bioline), with measurements taken on the QuantStudio 3 Real-Time PCR system (ThermoFisher).
Urine samples were thawed and centrifuged at 14,000g for 5 minutes. For samples collected following pure BGS treatments, 30 μL of sample was diluted into 30 μL acetonitrile and 240 μL water with 0.1% (v/v) formic acid, and then filtered through a 0.45 μm PTFE filter. For samples in which less than 30 μL of urine was collected, deionized water was added to make up the difference and the dilution was accounted for in corresponding peak area calculations. Filtered samples were analyzed using LC-MS with an Agilent 6520 qTOF, as described below. For urine samples collected from broccoli meal feedings, 15 μL of urine was diluted into 135 μL of HILIC acetonitrile mobile phase. For urine samples with less than 15 μL volume, the quantity of HILIC mobile phase used as diluent was adjusted to maintain a 10-fold dilution factor. Diluted samples were filtered through a 0.45 μm PTFE filter, and then analyzed by LC-MS/MS, as described below.
Fecal pellets were thawed, resuspended in 5 μL/mg potassium phosphate buffer (pH 7), and then vortexed until homogenized. An equal volume of methanol containing 40 mM cysteine was added to promote conjugation of free, extracted isothiocyanate and incubated with rocking for 2 hours at room temperature. Samples were centrifuged at 14,000 g for 5 minutes, and then supernatant was filtered through a 0.45 μm PTFE filter for analysis by LC-MS/MS, as described below.
For analysis of serum samples, 50 μL of serum was mixed with 150 μL of acetonitrile to precipitate serum proteins. Samples were centrifuged at 10,000 g for 3 minutes, and then filtered through a 0.45 μm PTFE filter prior to analysis by LC-MS, as described below.
LC-MS and LC-MS/MS analysis
GS metabolism in spent bacterial media.
Glucosinolate conversion to ITC by Bacteroides strains, Bt deletion mutants, and complemented Bf strains, as well as in vitro protein activity, were analyzed by reversed phase liquid chromatography on an Agilent 1290 Infinity II HPLC, using a 1.8 μm, 2.1 × 50 mm Zorbax RRHD Eclipse Plus C18 column (Agilent). For all samples, a volume of 1 μL was injected. Water with 0.1% (v/v) formic acid and acetonitrile with 0.1% (v/v) formic acid were used as mobile phase solvents, with a flow rate of 0.6 mL/min. Chromatographic separation was achieved using the following linear gradient (with percentages indicating levels of water with formic acid): 95% to 5%, 4.2 min; 5% to 0%, 1 min; 0% to 95%, 0.4 min. Coupled mass spectrometry data was collected with an Agilent 6470 triple quadrupole (QQQ) mass spectrometer. Parameters for the 6470 QQQ MS were as follows: gas temperature, 250C; gas flow rate, 12 L/min; nebulizer, 25 psig. Glucotropaeolin was detected using monitored transitions with the following parameters: polarity, negative; precursor ion, 408.04; product ions, 166 and 96.9*; dwell, 100 ms; fragmentor, 138 V; collision energy, 21 V and 25 V for respective product ions; cell accelerator, 4 V. BITC-cys was detected using monitored transitions with the following parameters: polarity, positive; precursor ion, 271.1; product ions, 254 and 122*; dwell, 100 ms; fragmentor, 100 V; collision energy, 6 V and 10 V for respective product ions; cell accelerator, 4 V. * denotes the product ion used for quantification of the respective metabolite, with the unmarked product ion used as a qualifier.
Standards for quantification of BITC-cys concentration in spent media were generated by combining 0.5 mM BITC with 0.5 mg/mL cysteine in 50 mM potassium phosphate buffer, pH 7., to mimic culturing conditions. Reactions were incubated at room temperature for 17 hours, followed by dilution for a standard curve.
Time course in vitro assays.
In vitro protein reactions were incubated at room temperature in the Agilent 1290 Infinity II autosampler and directly sampled with no quenching hourly by injection onto an Agilent 1290 Infinity II HPLC coupled to an Agilent 6545 qTOF MS. Injection volumes were 0.20 μL for all samples. No chromatography column was used. Water with 0.1% (v/v) formic acid and acetonitrile with 0.1% (v/v) formic acid were used as solvents, with the following isocratic flows (percentages indicate level of water with formic acid): 95% for 1 minute, 5% for 0.2 minutes. Mass spectrometry data was collected in fast polarity switching mode, with the following parameters: mass range, 50–700 m/z; drying gas temperature, 250C; drying gas flow rate, 12 L/min; nebulizer, 20 psig; fragmentor, 100V; skimmer, 50V.
Urine, feces, and serum sample analysis.
Urine samples collected from mice following pure BGS doses were analyzed using reversed phase liquid chromatography on an Agilent 1260 Infinity HPLC, using a 5 μm, 2 × 100 mm Gemini NX-C18 column (Phenomenex). For all samples, a volume of 5 μL was injected. Water with 0.1% (v/v) formic acid and acetonitrile with 0.1% (v/v) formic acid were used as mobile phase solvents, with a flow rate of 0.4 mL/min. Chromatographic separation was achieved using the following linear gradient (with percentages indicating levels of water with formic acid): 97% to 50%, 10 min; 50% to 5%, 1 min; 5%, 2 min; 5% to 97%, 1 min; 97%, 3 min. Coupled mass spectrometry data was collected with a Agilent 6520 quadrupole time-of-flight (qTOF) ESI mass spectrometer in positive polarity. Parameters for the 6520 qTOF MS were as follows: mass range, 50–1200 m/z; gas temperature, 350°C; drying gas flow rate, 11 L/min; nebulizer, 35 psig; fragmentor, 150 V; skimmer, 65 V.
Urine samples collected from mice following broccoli meals were analyzed by hydrophilic interaction liquid chromatography (HILIC) on an Agilent 1290 Infinity II HPLC, using a 1.7 μm, 2.1 × 50 mm Acquity BEH Amide column (Waters). For all samples, a volume of 1 μL was injected. Water with 0.125% (v/v) formic acid and 10 mM ammonium formate and acetonitrile were used as mobile phase solvents, with a flow rate of 0.6 mL/min. Chromatographic separation was achieved using the following linear gradient (with percentages indicating levels of water with formic acid and ammonium formate): 5%, 1.5 min; 5% to 62%, 4.5 min; 62%, 2 min; 62% to 71.5%, 0.5 min; 71.5% to 5%, 1 min, 5%, 2.5 min. Coupled mass spectrometry data was collected with an Agilent 6470 QQQ MS in both positive and negative polarities. Parameters for the 6470 QQQ MS were as follows: gas temperature, 250°C; gas flow rate, 12 L/min; nebulizer, 25 psig. Glucobrassicin was detected using monitored transitions with the following parameters: polarity, negative; precursor ion, 447.1; product ions, 250.9 and 96.9; dwell, 50 ms; fragmentor, 156 V; collision energy, 22 V and 26 V for respective product ions; cell accelerator, 4 V. Glucoraphanin was detected using monitored transitions with the following parameters: polarity, negative; precursor ion, 436.0; product ions, 372* and 96.9; dwell, 50 ms; fragmentor, 152 V; collision energy, 18 V and 26 V for respective product ions; cell accelerator, 4 V. Sulforaphane N-acetyl cysteine conjugate (SFN-NAC) was detected using monitored transitions with the following parameters: polarity, positive; precursor ion, 341.1; product ions, 177.9* and 114; dwell, 50 ms; fragmentor, 160 V; collision energy, 10 V and 22 V for respective product ions; cell accelerator, 4 V. Sulforaphane cysteine conjugate (SFN-cys) was detected using monitored transitions with the following parameters: polarity, positive; precursor ion, 299.1; product ions, 136* and 114; dwell, 50 ms; fragmentor, 100 V; collision energy, 6 V and 22 V for respective product ions; cell accelerator, 4V. Sulforaphane (SFN) was detected using monitored transitions with the following parameters: polarity, positive; precursor ion, 178.0; product ions, 114.0* and 72.0; dwell, 25 ms; fragmentor, 80 V; collision energy, 10 V and 34 V for respective product ions; cell accelerator, 4 V. Benzyl isothiocyanate N-acetyl cysteine conjugate (BITC-NAC) was detected using monitored transitions with the following parameters: polarity, positive; precursor ion, 313.1; product ions, 164* and 91.1; dwell, 50 ms; fragmentor, 100V; collision energy, 6 V and 42 V for respective product ions; cell accelerator, 4V. Glucotropaeolin and BITC-cys were detected using monitored transitions as described above for detection in spent bacterial media. Creatinine was detected using monitored transitions with the following parameters: polarity, positive; precursor ion, 114.1; production ions, 86.2* and 72.1; dwell, 25 ms; fragmentor, 132 V; collision energy, 9 V and 13 V for respective product ions; cell accelerator, 5 V.
Fecal samples collected from mice following both pure glucotropaeolin doses and broccoli meals were analyzed by reversed phase chromatography, using the same chromatography column and method used to determine glucosinolate metabolism in spent bacterial media. Coupled mass spectrometry data was collected with an Agilent 6470 QQQ MS in both positive and negative polarities, with the same parameters as described above. BITC-cys and BGS were detected using monitored transitions as described above for detection in spent bacterial media. Following analysis, fecal samples resulting from broccoli meals were diluted 10 times in HILIC acetonitrile mobile phase, filtered through a 0.45 μm PTFE filter, and further analyzed using HILIC, using the same chromatography column and method used to separate metabolites in urine samples resulting from broccoli meals. Coupled MS data was collected with an Agilent 6470 QQQ MS in both positive and negative polarities, with the same parameters as described above. BITC-cys, SFN-cys, glucotropaeolin, and glucoraphanin were detected using monitored transitions as described above. Mouse serum samples collected post-sacrifice were analyzed using HILIC, with the same chromatography column and method used to separate metabolites in urine samples resulting from broccoli meals. Coupled MS data was collected using an Agilent 6545 qTOF MS in both positive and negative polarities, with the same parameters as described above.
Data analysis.
Extracted ion chromatograms (EIC) were extracted from raw data files using MassHunter Qualitative Data Analysis software. For MS data collected using the Agilent 6545 qTOF, EICs were extracted using a 40 ppm window centered on the exact m/z value. For data collected using the Agilent 6520 qTOF, a 20 ppm window was used. Exact m/z values for the different substrates and products are as follows, with (+) or (−) indicating detection of the [M+H]+ or [M-H]− species in positive or negative polarity, respectively: glucotropaeolin, (−) 408.0428; glucoraphanin, (−) 436.0411; cellobiose and maltose, (−) 341.1089; glucobrassicin, (−) 447.0538; hexose, (−) 179.0561; sulforaphane, (+) 178.0355; BITC-cys, (+) 271.0570; BITCNAC, (+) 313.0675; SFN-cys, (+) 299.0552; SFN-NAC, (−) 341.0658.
For LC-MS/MS data, multiple reaction monitoring (MRM) chromatograms were extracted from raw data files and integrated using MassHunter Quantitative Data Analysis software. The product ions used for the quantification and qualification of different metabolites are as described above in the LC-MS/MS parameters section. Quantification product ions chromatograms were integrated if the corresponding qualifier were present.
Metagenomic and metatranscriptomic analysis
To determine the presence of the BT2160-BT2156 gene cluster in stool cohorts, filtered fastq reads from each of seven metagenomic stool cohorts (Abu-Ali et al., 2018; Brito et al., 2016; Lloyd-price et al., 2017; Nielsen et al., 2014; The Human Microbiome Project Consortium, 2012; Wang et al., 2012) were mapped to the gene cluster using Bowtie2 (Langmead and Salzberg, 2012; Langmead et al., 2019) with settings minscore L, 0.6, −0.3, which for a 100 bp read allows at most 5 mismatches in alignment. When relevant, results were averaged over several visits for a given subject and weighted according to the number of reads in each visit. ORFs were defined for each gene in BT2160-BT2156, and coverage and RPKM per subject per gene were calculated. A given gene was considered present if coverage was 50% or greater. To determine expression of the gene cluster, a similar analysis was performed on a metatranscriptomic cohort (Abu-Ali et al., 2018). From 96 total subjects, we selected 77 subjects with BT2160-BT2156 present in the paired metagenomic data. ORFs were defined for each gene in BT2159-BT2156 to account for independent regulation of the BT2160 transcription factor.
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical details of the experiments, including the statistical tests used, the value and definition of n, and precision measures, can be found in the figures and corresponding figure legends. Statistical significance for Bacteroides glucosinolate metabolism assays was determined using Dunnett’s multiple comparison test, with n of three cell culture replicates. Statistical significance for excretion of ITC-derived metabolites in mouse experiments was determined using the Tukey multiple comparison test with n of five, unless otherwise indicated in the figure. Statistical analyses were performed in GraphPad Prism.
DATA AND CODE AVAILABILITY
The results of the metagenomic and metatranscriptomic analyses used to generate Figure 5 are included as a supplementary file. All other relevant data are available from the authors upon request.
Supplementary Material
Figure S1. High throughput screen to identify the genes necessary for glucosinolate metabolism. Related to Figure 2.(A) BITC-cys production 24 hours after addition of BGS to 1) wild-type Bt cultured for 24 hours in TYG media, and 2) filtered cell-free spent supernatant from wild-type Bt cultured for 24 hours in TYG media. Data shown is the mean ± SD of three biological replicates. Extracted ion count (m/z 271.0570) with LC-MS was used to track quantities of protonated BITC-cys.
(B) Growth of Bt in TYG media (red) and TYG media supplemented with glucotropaeolin (blue). Bt growth was not inhibited by glucosinolate conversion to isothiocyanate.
(C) Growth of E. coli in media spent by wild-type Bt cultured for 24 hours in TYG media (red solid line) or TYG media supplemented with glucotropaeolin (blue dashed line). E. coli growth was inhibited by glucosinolate conversion to isothiocyanate in Bt spent media.
(D) Growth of S. typhimurium in media spent by wild-type Bt cultured for 24 hours in TYG media (red solid line) or TYG media supplemented with glucotropaeolin (blue dashed line). S. typhimurium growth was inhibited by glucosinolate conversion to isothiocyanate in Bt spent media.
(E) Schematic of the high throughput screen developed to identify the genes involved in glucosinolate metabolism, using a transposon insertion library in Bt VPI-5482. About 7500 members of the insertion library were screened for loss of glucosinolate metabolizing activity. The bottom left figure shows growth curves for E. coli grown in media spent by a Bt mutant deficient in glucosinolate metabolizing activity (BtΔ2157, red line) relative to E. coli grown in media spent by a Bt strain with glucosinolate metabolizing activity (wild type Bt, blue line).
(F) Representative results of one microplate from high-throughput screen of Bt mutant library. Light gray bars in foreground represent the terminal optical densities of Bt grown to stationary phase (25 hours). Dark blue bars in background represent the optical density of E. coli after 8 hours of growth in spent Bt media. Averaged optical densities of sterile and wild-type controls of Bt VPI-5482 (WT5) and Bt VPI-8736 (WT8), a strain identified with limited glucose metabolizing activity, are shown as the left-most three bars. Mutants with reduced glucosinolate metabolizing activity (marked with *) were identified based on the following two criteria: first, uncompromised growth relative to WT5 (Bt mutant OD ≥ average − 1 standard deviation of WT5 OD, “Bt cutoff”) and second, loss of the ability to inhibit subsequent E. coli growth (E. coli OD ≥ average − 1 standard deviation of E. coli OD grown in the supernatant of WT8, “Ec cutoff”). Approximately 2.9% (218 mutants) of the top hits from the high-throughput screen were chosen for rescreening based on these criteria. Fifty nine mutants were successfully sequenced based on the consistency of their performance in a second and third round of rescreening, using the same criteria employed for the primary screen.
Figure S2. An operon necessary for GS conversion to ITCs in Bt. Related to Figure 2. (A) Annotations and predicted domains in the BT2160-BT2156 operon. Detected domains in the operon sequence are shown below the gene arrows. Predicted localization and secretion domains are shown above.
(B) Homologs of BT2160–2156 conserved across various Bacteroides strains. Homologs were identified using a BLAST search for genes with greater than 60% amino acid similarity and greater than 60% coverage relative to the Bt VPI-5482 gene. Percent identity on the amino acid level is shown in white for each gene analyzed. Select strains were surveyed for glucosinolate conversion to isothiocyanate following 24 hours of growth in rich media containing glucose. The corresponding concentration of BITC-cys in culture supernatant is shown on the right of each surveyed strain, and was quantified using LC-MS/MS to track the transition of the protonated BITC-cys molecule with m/z 271.0 to a product ion with m/z 122.0. Within Bt VPI-5482, BT4449–4446 are 69.5% to 99.6% identical at the amino acid sequence level; however, none of the genes encoding these proteins were identified in the transposon screen and do not appear to be necessary for GS metabolism to ITC.
(C) Growth curves of Bt single deletion mutants in TYG media with BGS, measured by OD at 600 nm. Data shown is the mean of three biological replicates ± SD.
(D) Growth curves of Bf strains expressing subsets of the BT2159-BT2156 operon in TYG media with BGS, measured by OD at 600 nm. Data shown is the mean ± SD of three biological replicates.
(E) Time courses of growth and glucosinolate conversion for wild-type Bt in TYG media supplemented with BGS, measured by optical density (OD) at 600 nm (black), BITC-cys production in the media (red), and BGS depletion (blue). BITC-cys and BGS were monitored by LC-MS and quantified using EIC (BITC-cys: m/z 271.0570; BGS: m/z 168.0478, corresponding to the -SO4 -glucose in-source fragment). Data shown is the mean ± SD of three biological replicates.
(F) Growth (top) and BITC-cys production (bottom) of wild-type Bt (light grey bars) and BtΔ2157 (dark grey bars) in TY media supplemented with different disaccharides and monosaccharides (0.5% w/v) and 0.5 mM BGS after 24 hours. Data shown is the mean ± SD of three biological replicates.
(G) BITC-cys produced from BGS by BtΔ2157 and BtΔ2158 mutants (bottom) and BtΔ2159–57 mutant (top), complemented with subsets of the operon and grown in TYG media for 24 hours. Each row in the grid of boxes represents the genes expressed in each complemented strain; cross-hatched boxes represent constitutive extra-chromosomal expression from plasmid, while solid boxes represent natively expressed genes. Half solid-half cross hatched boxes represent genes that are both expressed natively and extra-chromosomally.
Figure S3. In vitro reactions with purified recombinant Bt proteins. Related to Figure 3. (A) Coomassie stained SDS-PAGE gels for the Ni-NTA purification of His-tagged BT2159-BT2156 proteins recombinantly produced in E. coli BL21 DE3. Ladder markings show protein sizes in kDa. Sol: total soluble fraction; insol: total insoluble fraction, FT: column flow-through. Total culture volumes were 2 L for BT2156, BT2157, and BT2159. Culture volume was 6 L for BT2158. Less than 1 mg of BT2158 was purified per liter of E. coli culture. Despite attempts to optimize expression, most of the BT2158 that was produced remained in the insoluble fraction of the E.coli lysate.
(B) Coomassie stained SDS-PAGE gel for the Ni-NTA purification of His-tagged BT2158 produced in BtΔ2158, with a yield of ~10 mg of purified protein per liter of Bt culture. The total culture volume was 1 L.
(C) Silver stained SDS-PAGE gel of purified fractions of BT2158 produced in BtΔ2158, Bf, and E. coli. Purified BT2157 produced in E. coli provided as a reference.
(D) In vitro conversion of BGS by the combination of purified BT2157 and BT2158 in the presence of 1 mM different nicotinamide cofactors, quantified using EIC peak area of BITC-cys (m/z 271.0570), detected by LC-MS. Data shown is the mean ± SD of two replicates.
(E) Proteins produced in E. coli (BT2156, BT2157, BT2159) or BtΔ2158 (BT2158) were purified and combined in N-methyl morpholine (NMM) buffer with NAD and di- or trisaccharide substrate. Integrated extracted ion count peaks areas corresponding to hexose release (top row; m/z 179.0561) and substrate consumption, normalized against initial substrate ion count, (bottom row; m/z 341.1089 for disaccharides and m/z 503.1618 for raffinose) were measured. Data shown is one replicate, representative of triplicate experiments.
(F) Initial reaction rates of combinations of proteins purified from E. coli (BT2156, BT2157, BT2159) or BtΔ2158 (BT2158) on disaccharide (cellobiose and maltose) and glucosinolate (glucoraphanin and glucobrassicin) substrates. Rates were calculated as the initial linear rate of change of integrated extracted ion peak areas corresponding to hexose release (m/z 179.0561) over time. Data shown is the mean±SD of three replicates, with individual replicates shown as filled circles.
Figure S4. Monocolonization of gnotobiotic mice by mutant Bt. Related to Figure 4. (A) Feces from mice colonized with wild-type Bt (light grey bar) or BtΔ2157 colonized mice (dark grey bar) were collected prior to glucosinolate dosing, plated at limiting dilutions, and single colonies were cultured in TYG medium with BGS. MRM by LC-MS/MS was used to track the transition of the protonated BITC-cys molecule with m/z 271.0 to a product ion with m/z 122.0. Data shown are the mean ± SD of five biological replicates.
(B) Range of fecal colonization densities of wild-type Bt and BtΔ2157 in gnotobiotic mice prior to dietary glucosinolate intervention. Feces from mice colonized with wild-type wild-type Bt and BtΔ2157 were collected prior to the first glucotropaeolin dose and plated at limiting dilutions. Data shown is the maximum and minimum colony forming units (cfu) per microliter of feces of five biological replicates.
(C) Cecal colonization densities of wild-type Bt and BtΔ2157 in gnotobiotic mice following two weeks of dietary glucosinolate intervention. Cecal contents from mice were collected six hours following the final glucotropaeolin dose and assessed for Bt abundance using quantitative PCR. Data shown are the mean ± SD of five biological replicates.
(D) Annotated representative extracted ion chromatogram of glucosinolates identified in broccoli extract. Glucosinolates corresponding to the extracted exact masses are shown in boxes.
(E) BITC-NAC and BITC-cys mercapturic acid conjugates and unconverted BGS excreted in urine and feces after doses of pure glucotropaeolin (top), and BITC-cys and unconverted BGS in feces collected after doses of pure glucotropaeolin (bottom). BITC-cys in feces was the product of in situ conjugation of free BITC with exogenously applied cysteine. Bars represent the mean values for each group, with individual data overlaid (different shapes correspond to individual mice). Metabolites were measured by LC-MS/MS and quantified by MRM (BITC-NAC: m/z 313.1→ m/z 164; BITC-cys: m/z 271.1→ m/z 122.0; BGS: m/z 408.0→ m/z 96.9). Mice mono-colonized with mutant BtΔ2157 compared to wild-type Bt showed increased glucosinolate concentration in fecal samples (Fig. S6B), which may be indicative of indirect impacts of glucosinolate conversion, other physiological activities encoded by this operon (Liu et al., 2019), or effects specific to monocolonized mouse models.
F) Free SFN, SFN-NAC and SFN-cys mercapturic acid conjugates, and unconverted glucoraphanin excreted in urine six hours following doses of broccoli meal containing glucoraphanin. Bars represent the mean values for each group, with individual data overlaid (different shapes correspond to individual mice). Metabolite quantities were measured by LC-MS/MS, quantified by MRM (SFN-NAC: m/z 341.1→ m/z 177.9; SFN-cys: m/z 299.1→ m/z 136; glucoraphanin: m/z 436.0→ m/z 372, SFN: m/z 178.0→ m/z 114.0), and converted to concentrations. Metabolite levels were normalized to urinary creatinine concentrations, as quantified by LC-MS/MS using the transition from protonated creatinine (m/z 114.1) to a product ion with m/z 86.2.
Figure S5. Metagenomic and transcriptomic prevalence of BT2160-BT2156. Related to Figure 5. A) Metagenomic and metatranscriptomic RPKM abundance of each of the genes in BT2160-BT2156 in the stool cohorts examined. Each row in the heatmap corresponds to a subject in the cohort, with each column corresponding to a gene in the gene cluster.
B) Metagenomic and metatranscriptomic coverage of each of the genes in BT2160-BT2156 in the stool cohorts examined.
Table S3. Metagenomic and metatranscriptomic abundance of BT2160-BT2156. Related to Figure 5.
HIGHLIGHTS.
A B. thetaiotaomicron operon is required for activation of plant glucosinolates
Operon expression in inactive B. fragilis results in glucosinolate metabolism
Glucosinolate metabolism encoded by BT2159-BT2156 requires multiple enzymes
Monoassociation of mice with BtΔ2157 reduces host exposure to isothiocyanates
ACKNOWLEDGEMENTS
This work was supported by an HHMI and Simons Foundation Grant 55108565 and NIH DP2AT00832101 (to E.S.S.), a Stanford Bio-X Bowes (to C.S.L.), and ChEM-H seed grant funding (to J.L.S. and E.S.S.). We thank M. Fischbach, E. Carlson, and M. Voges (Stanford) for comments on the manuscript, and K. Smith, R. de la Pena, and other members of the Sattely lab for helpful discussions. We thank K. Ng, D. Dodd, and E. Shepherd (Stanford) for assistance with and advice on gnotobiotic mouse studies, and W. Whitaker (Stanford) for providing the Bacteroides phage promoter sequence. We thank A. Wawro (Stanford) for assistance with methods for creatinine measurements. We thank M. Wang (Stanford) for providing Bacteroides strains. We thank C. Miller, S. Hull, and S. Lensch (Stanford) for their efforts on in vitro biochemical characterization. We thank S. Mazmanian (California Institute of Technology) for providing plasmid pFD340. We thank the Stanford High-Throughput Bioscience Center for technical assistance with the Bt mutant screen.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abu-Ali GS, Mehta RS, Lloyd-Price J, Mallick H, Branck T, Ivey KL, Drew DA, Dulong C, Rimm E, Izard J, et al. (2018). Metatranscriptome of human faecal microbial communities in a cohort of adult men. Nat. Microbiol 3, 356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agerbirk N, De Vos M, Kim JH, and Jander G (2009). Indole glucosinolate breakdown and its biological effects. Phytochem. Rev 8, 101–120. [Google Scholar]
- Albaser A, Kazana E, Bennett MH, Cebeci F, Luang-In V, Spanu PD, and Rossiter JT (2016). Discovery of a Bacterial Glycoside Hydrolase Family 3 (GH3) β-Glucosidase with Myrosinase Activity from a Citrobacter Strain Isolated from Soil. J. Agric. Food Chem 64, 1520–1527. [DOI] [PubMed] [Google Scholar]
- Angelino D, Dosz EB, Sun J, Hoeflinger JL, Van Tassell ML, Chen P, Harnly JM, Miller MJ, and Jeffery EH (2015). Myrosinase-dependent and -independent formation and control of isothiocyanate products of glucosinolate hydrolysis. Front. Plant Sci 6, 831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armah CN, Derdemezis C, Traka MH, Dainty JR, Doleman JF, Saha S, Leung W, Potter JF, Lovegrove JA, and Mithen RF (2015). Diet rich in high glucoraphanin broccoli reduces plasma LDL cholesterol: Evidence from randomised controlled trials. Mol. Nutr. Food Res 59, 918–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson AS, Tubbs E, Mecham B, Chacko S, Nenonen HA, Tang Y, Fahey JW, Derry JMJ, Wollheim CB, Wierup N, et al. (2017). Sulforaphane reduces hepatic glucose production and improves glucose control in patients with type 2 diabetes. Sci. Transl. Med 9, 4477. [DOI] [PubMed] [Google Scholar]
- Bacic MK, and Smith CJ (2008). Laboratory Maintenance and Cultivation of Bacteroides Species. Curr. Protoc. Microbiol 9, 13C – 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie TA, and Slatter JG (1991). Glutathione: A Vehicle for the Transport of Chemically Reactive Metabolites in Vivo. Acc. Chem. Res 24, 264–270. [Google Scholar]
- Bellono NW, Bayrer JR, Leitch DB, Castro J, Zhang C, O’Donnell TA, Brierley SM, Ingraham HA, and Julius D (2017). Enterochromaffin Cells Are Gut Chemosensors that Couple to Sensory Neural Pathways. Cell 170, 185–198.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode LM, Bunzel D, Huch M, Cho G-S, Ruhland D, Bunzel M, Bub A, Franz CM, and Kulling SE (2013). In vivo and in vitro metabolism of trans-resveratrol by human gut microbiota. Am. J. Clin. Nutr 97, 295–309. [DOI] [PubMed] [Google Scholar]
- Bokkenheuser VD, Shackletont CHL, and Winter J (1987). Hydrolysis of dietary flavonoid glycosides by strains of intestinal Bacteroides from humans. Biochem. J 248, 953–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito IL, Yilmaz S, Huang K, Xu L, Jupiter SD, Jenkins AP, Naisilisili W, Tamminen M, Smillie CS, Wortman JR, et al. (2016). Mobile genes in the human microbiome are structured from global to individual scales. Nature 535, 435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggeman IM, Temmink JHM, and van Bladeren PJ (1986). Glutathione- and cysteine-mediated cytotoxicity of allyl and benzyl isothiocyanate. Toxicol. Appl. Pharmacol 83, 349–359. [DOI] [PubMed] [Google Scholar]
- Budnowski J, Hanske L, Schumacher F, Glatt H, Platz S, Rohn S, and Blaut M (2015). Glucosinolates Are Mainly Absorbed Intact in Germfree and Human Microbiota-Associated Mice. J. Agric. Food Chem 63, 8418–8428. [DOI] [PubMed] [Google Scholar]
- Chen IMA, Chu K, Palaniappan K, Pillay M, Ratner A, Huang J, Huntemann M, Varghese N, White JR, Seshadri R, et al. (2018). IMG/M v.5.0: An integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 47, D666–D677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng DL, Hashimoto K, and Uda Y (2004). In vitro digestion of sinigrin and glucotropaeolin by single strains of Bifidobacterium and identification of the digestive products. Food Chem. Toxicol 42, 351–357. [DOI] [PubMed] [Google Scholar]
- Clavel T, Henderson G, Engst W, Doré J, and Blaut M (2006). Phylogeny of human intestinal bacteria that activate the dietary lignan secoisolariciresinol diglucoside. FEMS Microbiol. Ecol 55, 471–478. [DOI] [PubMed] [Google Scholar]
- Cordeiro RP, Doria JH, Zhanel GG, Sparling R, and Holley RA (2015). Role of glycoside hydrolase genes in sinigrin degradation by E. coli O157: H7. Int. J. Food Microbiol 205, 105–111. [DOI] [PubMed] [Google Scholar]
- D’Elia JN, and Salyers AA (1996). Contribution of a neopullulanase, a pullulanase, and an alpha-glucosidase to growth of Bacteroides thetaiotaomicron on starch. J. Bacteriol 178, 7173–7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour V, Stahl M, and Baysse C (2015). The antibacterial properties of isothiocyanates. Microbiol. (United Kingdom) 161, 229–243. [DOI] [PubMed] [Google Scholar]
- Elfoul L, Rabot S, Khelifa N, Quinsac A, Duguay A, and Rimbault A (2001). Formation of allyl isothiocyanate from sinigrin in the digestive tract of rats monoassociated with a human colonic strain of Bacteroides thetaiotaomicron. FEMS Microbiol. Lett 197, 99–103. [DOI] [PubMed] [Google Scholar]
- Fan J, Crooks C, Creissen G, Hill L, Fairhurst S, Doerner P, and Lamb C (2011). Pseudomonas sax genes overcome aliphatic isothiocyanate-mediated non-host resistance in Arabidopsis. Science (80-.). 331, 1185–1189. [DOI] [PubMed] [Google Scholar]
- Foley MH, Cockburn DW, and Koropatkin NM (2016). The Sus operon: a model system for starch uptake by the human gut Bacteroidetes. Cell. Mol. Life Sci 73, 2603–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkard DL, Melchini A, Traka MH, Al-Bakheit A, Saha S, Mulholland F, Watson A, and Mithen RF (2014). Suppression of LPS-induced transcription and cytokine secretion by the dietary isothiocyanate sulforaphane. Mol. Nutr. Food Res 58, 2286–2296. [DOI] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, and Gordon JI (2009). Identifying Genetic Determinants Needed to Establish a Human Gut Symbiont in Its Habitat. Cell Host Microbe 6, 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, and Turnbaugh PJ (2013). Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science (80-.). 335, 1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halkier BA, and Gershenzon J (2006). Biology and Biochemistry of Glucosinolates. Annu. Rev. Plant Biol 57, 303–333. [DOI] [PubMed] [Google Scholar]
- Herr I, and Büchler MW (2010). Dietary constituents of broccoli and other cruciferous vegetables: Implications for prevention and therapy of cancer. Cancer Treat. Rev 36, 377–383. [DOI] [PubMed] [Google Scholar]
- Holst B, and Williamson G (2008). Nutrients and phytochemicals: from bioavailability to bioefficacy beyond antioxidants. Curr. Opin. Biotechnol 19, 73–82. [DOI] [PubMed] [Google Scholar]
- Hwang ES, and Jeffery EH (2003). Evaluation of urinary N-acetyl cysteinyl allyl isothiocyanate as a biomarker for intake and bioactivity of Brussels sprouts. Food Chem. Toxicol 41, 1817–1825. [DOI] [PubMed] [Google Scholar]
- Jang I-S, and Kim D-H (1996). Purification and Characterization of a-L-Rhamnosidase from Bacteroides JY-6, a Human Intestinal Bacterium. Chem. Pharm. Bull 19, 1546–1549. [DOI] [PubMed] [Google Scholar]
- Jeffery EH, and Araya M (2009). Physiological effects of broccoli consumption. Phytochem. Rev 8, 283–298. [Google Scholar]
- Käll L, Krogh A, and Sonnhammer ELL (2007). Advantages of combined transmembrane topology and signal peptide prediction-the Phobius web server. Nucleic Acids Res. 35, 429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissen R, Rossiter JT, and Bones AM (2009). The “mustard oil bomb”: Not so easy to assemble?! Localization, expression and distribution of the components of the myrosinase enzyme system. Phytochem. Rev 8, 69–86. [Google Scholar]
- Koropatkin NM, Martens EC, Gordon JI, and Smith TJ (2008). Starch catabolism by a prominent human gut symbiont is directed by the recognition of amylose helices. 16, 1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Charles R, Wilks C, and Antonescu V (2019). Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 35, 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Hullar MAJ, Beresford SAA, and Lampe JW (2011). Variation of glucoraphanin metabolism in vivo and ex vivo by human gut bacteria. Br. J. Nutr 106, 408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Price MN, Carlson HK, Chen Y, Ray J, and Anthony L (2019). Large-scale chemical-genetics of the human gut bacterium Bacteroides thetaiotaomicron. BioRxiv 10.1101/573055 [DOI] [Google Scholar]
- Lloyd-price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, Hall AB, Brady A, Creasy HH, Mccracken C, Giglio MG, et al. (2017). Strains , functions and dynamics in the expanded Human Microbiome Project. Nat. Publ. Gr 550, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luang-In V, Albaser AA, Nueno-Palop C, Bennett MH, Narbad A, and Rossiter JT (2016). Glucosinolate and Desulfo-glucosinolate Metabolism by a Selection of Human Gut Bacteria. Curr. Microbiol 73, 442–451. [DOI] [PubMed] [Google Scholar]
- Marcobal A, Barboza M, Sonnenburg ED, Pudlo N, Martens EC, Desai P, Lebrilla CB, Weimer BC, Mills DA, German JB, et al. (2011). Bacteroides in the infant gut consume milk oligosaccharides via mucus-utilization pathways. Cell Host Microbe 10, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens EC, Koropatkin NM, Smith TJ, and Gordon JI (2009). Complex glycan catabolism by the human gut microbiota: The bacteroidetes sus-like paradigm. J. Biol. Chem 284, 24673–24677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C, Zhang Y, Tonelli C, and Petroni K (2013). Plants, Diet, and Health. Annu. Rev. Plant Biol 64, 19–46. [DOI] [PubMed] [Google Scholar]
- Mimee M, Tucker AC, Voigt CA, and Lu TK (2015). Programming a Human Commensal Bacterium, Bacteroides thetaiotaomicron, to Sense and Respond to Stimuli in the Murine Gut Microbiota. Cell Syst. 1, 62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mithen RF (2013). Development and commercialisation of “Beneforte” broccoli and potential health benefits. Acta Hortic. 1005, 67–70. [Google Scholar]
- Narbad A, and Rossiter JT (2018). Gut Glucosinolate Metabolism and Isothiocyanate Production. Mol. Nutr. Food Res 1700991, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro SL, Schwarz Y, Song X, Wang C-Y, Chen C, Trudo SP, Kristal AR, Kratz M, Eaton DL, and Lampe JW (2014). Cruciferous vegetables have variable effects on biomarkers of systemic inflammation in a randomized controlled trial in healthy young adults. J. Nutr 144, 1850–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen HB, Almeida M, Juncker AS, Rasmussen S, Li J, Sunagawa S, Plichta DR, Gautier L, Pedersen AG, Le Chatelier E, et al. (2014). Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol 32, 822–828. [DOI] [PubMed] [Google Scholar]
- Palop M.Ll., Smiths JP, and Brink B. ten (1995). Degradation of sinigrin by Lactobacillus agilis strain R16. Int. J. Food Microbiol 26, 219–229. [DOI] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, Von Heijne G, and Nielsen H (2011). SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. [DOI] [PubMed] [Google Scholar]
- Rabot S, Nugon-Baudon L, Raibaud P, and Szylit O (1993). Rape-seed meal toxicity in gnotobiotic rats: influence of a whole human faecal flora or single human strains of Escherichia coli and Bacteroides vulgatus. Br. J. Nutr 70, 323. [DOI] [PubMed] [Google Scholar]
- Ravcheev DA, Godzik A, Osterman AL, and Rodionov DA (2013). Polysaccharides utilization in human gut bacterium Bacteroides thetaiotaomicron: Comparative genomics reconstruction of metabolic and regulatory networks. BMC Genomics 14, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro TA, Fahey JW, Wade KL, Stephenson KK, and Talalay P (1998). Human metabolism and excretion of cancer chemoprotective glucosinolates and isothiocyanates of cruciferous vegetables. Cancer Epidemiol. Biomarkers Prev 7. [PubMed] [Google Scholar]
- Shapiro TA, Fahey JW, Wade KL, Stephenson KK, and Talalay P (2001). Chemoprotective Glucosinolates and Isothiocyanates of Broccoli Sprouts : Metabolism and Excretion in Humans. Cancer Epidemiol. Biomarkers Prev 10, 501–508. [PubMed] [Google Scholar]
- Shipman JA, Cho KH, Siegel HA, and Salyers AA (1999). Physiological Characterization of SusG, an Outer Membrane Protein Essential for Starch Utilization by Bacteroides thetaiotaomicron Physiological Characterization of SusG , an Outer Membrane Protein Essential for Starch Utilization by Bacteroides thetaiota. J. Bacteriol 181, 7206–7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipman JA, Berleman JE, and Salyers AA (2000). Characterization of four outer membrane proteins involved in binding starch to the cell surface of Bacteroides thetaiotaomicron. J. Bacteriol 182, 5365–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, Rogers MB, and Mckee ML (1992). Heterologous gene expression in Bacteroides fragilis. Plasmid 27, 141–154. [DOI] [PubMed] [Google Scholar]
- Sonnenburg JL, Chen CTL, and Gordon JI (2006). Genomic and metabolic studies of the impact of probiotics on a model gut symbiont and host. PLoS Biol. 4, 2213–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand TA, Lale R, Degnes KF, Lando M, and Valla S (2014). A new and improved host-independent plasmid system for RK2-based conjugal transfer. PLoS One 9, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberman H, Parkkinen T, and Rouvinen J (2016). Structural and functional features of the NAD(P) dependent Gfo/Idh/MocA protein family oxidoreductases. Protein Sci. 25, 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao YL, Yang DH, Zhang YT, Zhang Y, Wang ZQ, Wang YS, Cai SQ, and Liu SL (2014). Cloning, expression, and characterization of the β-glucosidase hydrolyzing secoisolariciresinol diglucoside to secoisolariciresinol from Bacteroides uniformis ZL1. Appl. Microbiol. Biotechnol 98, 2519–2531. [DOI] [PubMed] [Google Scholar]
- The Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traka M, and Mithen R (2009). Glucosinolates, isothiocyanates and human health. Phytochem. Rev 8, 269–282. [Google Scholar]
- Tsuchihashi R, Sakamoto S, Kodera M, Nohara T, and Kinjo J (2008). Microbial metabolism of soy isoflavones by human intestinal bacterial strains. J. Nat. Med 62, 456–460. [DOI] [PubMed] [Google Scholar]
- van Straaten KE, Zheng H, Palmer DRJ, and Sanders DAR (2010). Structural investigation of myo -inositol dehydrogenase from Bacillus subtilis : implications for catalytic mechanism and inositol dehydrogenase subfamily classification. Biochem. J 432, 237–247. [DOI] [PubMed] [Google Scholar]
- Vomhof-DeKrey EE, and Picklo MJ (2012). The Nrf2-antioxidant response element pathway: Aism target for regulating energy metabolism. J. Nutr. Biochem 23, 1201–1206. [DOI] [PubMed] [Google Scholar]
- Wang J, Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, et al. (2012). A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60. [DOI] [PubMed] [Google Scholar]
- Whitaker WR, Shepherd ES, and Sonnenburg JL (2017). Tunable Expression Tools Enable Single-Cell Strain Distinction in the Gut Microbiome. Cell 169, 538–546.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, and Gordon JI (2003). A Genomic View of the Human-Bacteroides thetaiotaomicron Symbiosis. Science (80-.). 299, 2074–2076. [DOI] [PubMed] [Google Scholar]
- Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Cenk Sahinalp S, Ester M, Foster LJ, et al. (2010). PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. High throughput screen to identify the genes necessary for glucosinolate metabolism. Related to Figure 2.(A) BITC-cys production 24 hours after addition of BGS to 1) wild-type Bt cultured for 24 hours in TYG media, and 2) filtered cell-free spent supernatant from wild-type Bt cultured for 24 hours in TYG media. Data shown is the mean ± SD of three biological replicates. Extracted ion count (m/z 271.0570) with LC-MS was used to track quantities of protonated BITC-cys.
(B) Growth of Bt in TYG media (red) and TYG media supplemented with glucotropaeolin (blue). Bt growth was not inhibited by glucosinolate conversion to isothiocyanate.
(C) Growth of E. coli in media spent by wild-type Bt cultured for 24 hours in TYG media (red solid line) or TYG media supplemented with glucotropaeolin (blue dashed line). E. coli growth was inhibited by glucosinolate conversion to isothiocyanate in Bt spent media.
(D) Growth of S. typhimurium in media spent by wild-type Bt cultured for 24 hours in TYG media (red solid line) or TYG media supplemented with glucotropaeolin (blue dashed line). S. typhimurium growth was inhibited by glucosinolate conversion to isothiocyanate in Bt spent media.
(E) Schematic of the high throughput screen developed to identify the genes involved in glucosinolate metabolism, using a transposon insertion library in Bt VPI-5482. About 7500 members of the insertion library were screened for loss of glucosinolate metabolizing activity. The bottom left figure shows growth curves for E. coli grown in media spent by a Bt mutant deficient in glucosinolate metabolizing activity (BtΔ2157, red line) relative to E. coli grown in media spent by a Bt strain with glucosinolate metabolizing activity (wild type Bt, blue line).
(F) Representative results of one microplate from high-throughput screen of Bt mutant library. Light gray bars in foreground represent the terminal optical densities of Bt grown to stationary phase (25 hours). Dark blue bars in background represent the optical density of E. coli after 8 hours of growth in spent Bt media. Averaged optical densities of sterile and wild-type controls of Bt VPI-5482 (WT5) and Bt VPI-8736 (WT8), a strain identified with limited glucose metabolizing activity, are shown as the left-most three bars. Mutants with reduced glucosinolate metabolizing activity (marked with *) were identified based on the following two criteria: first, uncompromised growth relative to WT5 (Bt mutant OD ≥ average − 1 standard deviation of WT5 OD, “Bt cutoff”) and second, loss of the ability to inhibit subsequent E. coli growth (E. coli OD ≥ average − 1 standard deviation of E. coli OD grown in the supernatant of WT8, “Ec cutoff”). Approximately 2.9% (218 mutants) of the top hits from the high-throughput screen were chosen for rescreening based on these criteria. Fifty nine mutants were successfully sequenced based on the consistency of their performance in a second and third round of rescreening, using the same criteria employed for the primary screen.
Figure S2. An operon necessary for GS conversion to ITCs in Bt. Related to Figure 2. (A) Annotations and predicted domains in the BT2160-BT2156 operon. Detected domains in the operon sequence are shown below the gene arrows. Predicted localization and secretion domains are shown above.
(B) Homologs of BT2160–2156 conserved across various Bacteroides strains. Homologs were identified using a BLAST search for genes with greater than 60% amino acid similarity and greater than 60% coverage relative to the Bt VPI-5482 gene. Percent identity on the amino acid level is shown in white for each gene analyzed. Select strains were surveyed for glucosinolate conversion to isothiocyanate following 24 hours of growth in rich media containing glucose. The corresponding concentration of BITC-cys in culture supernatant is shown on the right of each surveyed strain, and was quantified using LC-MS/MS to track the transition of the protonated BITC-cys molecule with m/z 271.0 to a product ion with m/z 122.0. Within Bt VPI-5482, BT4449–4446 are 69.5% to 99.6% identical at the amino acid sequence level; however, none of the genes encoding these proteins were identified in the transposon screen and do not appear to be necessary for GS metabolism to ITC.
(C) Growth curves of Bt single deletion mutants in TYG media with BGS, measured by OD at 600 nm. Data shown is the mean of three biological replicates ± SD.
(D) Growth curves of Bf strains expressing subsets of the BT2159-BT2156 operon in TYG media with BGS, measured by OD at 600 nm. Data shown is the mean ± SD of three biological replicates.
(E) Time courses of growth and glucosinolate conversion for wild-type Bt in TYG media supplemented with BGS, measured by optical density (OD) at 600 nm (black), BITC-cys production in the media (red), and BGS depletion (blue). BITC-cys and BGS were monitored by LC-MS and quantified using EIC (BITC-cys: m/z 271.0570; BGS: m/z 168.0478, corresponding to the -SO4 -glucose in-source fragment). Data shown is the mean ± SD of three biological replicates.
(F) Growth (top) and BITC-cys production (bottom) of wild-type Bt (light grey bars) and BtΔ2157 (dark grey bars) in TY media supplemented with different disaccharides and monosaccharides (0.5% w/v) and 0.5 mM BGS after 24 hours. Data shown is the mean ± SD of three biological replicates.
(G) BITC-cys produced from BGS by BtΔ2157 and BtΔ2158 mutants (bottom) and BtΔ2159–57 mutant (top), complemented with subsets of the operon and grown in TYG media for 24 hours. Each row in the grid of boxes represents the genes expressed in each complemented strain; cross-hatched boxes represent constitutive extra-chromosomal expression from plasmid, while solid boxes represent natively expressed genes. Half solid-half cross hatched boxes represent genes that are both expressed natively and extra-chromosomally.
Figure S3. In vitro reactions with purified recombinant Bt proteins. Related to Figure 3. (A) Coomassie stained SDS-PAGE gels for the Ni-NTA purification of His-tagged BT2159-BT2156 proteins recombinantly produced in E. coli BL21 DE3. Ladder markings show protein sizes in kDa. Sol: total soluble fraction; insol: total insoluble fraction, FT: column flow-through. Total culture volumes were 2 L for BT2156, BT2157, and BT2159. Culture volume was 6 L for BT2158. Less than 1 mg of BT2158 was purified per liter of E. coli culture. Despite attempts to optimize expression, most of the BT2158 that was produced remained in the insoluble fraction of the E.coli lysate.
(B) Coomassie stained SDS-PAGE gel for the Ni-NTA purification of His-tagged BT2158 produced in BtΔ2158, with a yield of ~10 mg of purified protein per liter of Bt culture. The total culture volume was 1 L.
(C) Silver stained SDS-PAGE gel of purified fractions of BT2158 produced in BtΔ2158, Bf, and E. coli. Purified BT2157 produced in E. coli provided as a reference.
(D) In vitro conversion of BGS by the combination of purified BT2157 and BT2158 in the presence of 1 mM different nicotinamide cofactors, quantified using EIC peak area of BITC-cys (m/z 271.0570), detected by LC-MS. Data shown is the mean ± SD of two replicates.
(E) Proteins produced in E. coli (BT2156, BT2157, BT2159) or BtΔ2158 (BT2158) were purified and combined in N-methyl morpholine (NMM) buffer with NAD and di- or trisaccharide substrate. Integrated extracted ion count peaks areas corresponding to hexose release (top row; m/z 179.0561) and substrate consumption, normalized against initial substrate ion count, (bottom row; m/z 341.1089 for disaccharides and m/z 503.1618 for raffinose) were measured. Data shown is one replicate, representative of triplicate experiments.
(F) Initial reaction rates of combinations of proteins purified from E. coli (BT2156, BT2157, BT2159) or BtΔ2158 (BT2158) on disaccharide (cellobiose and maltose) and glucosinolate (glucoraphanin and glucobrassicin) substrates. Rates were calculated as the initial linear rate of change of integrated extracted ion peak areas corresponding to hexose release (m/z 179.0561) over time. Data shown is the mean±SD of three replicates, with individual replicates shown as filled circles.
Figure S4. Monocolonization of gnotobiotic mice by mutant Bt. Related to Figure 4. (A) Feces from mice colonized with wild-type Bt (light grey bar) or BtΔ2157 colonized mice (dark grey bar) were collected prior to glucosinolate dosing, plated at limiting dilutions, and single colonies were cultured in TYG medium with BGS. MRM by LC-MS/MS was used to track the transition of the protonated BITC-cys molecule with m/z 271.0 to a product ion with m/z 122.0. Data shown are the mean ± SD of five biological replicates.
(B) Range of fecal colonization densities of wild-type Bt and BtΔ2157 in gnotobiotic mice prior to dietary glucosinolate intervention. Feces from mice colonized with wild-type wild-type Bt and BtΔ2157 were collected prior to the first glucotropaeolin dose and plated at limiting dilutions. Data shown is the maximum and minimum colony forming units (cfu) per microliter of feces of five biological replicates.
(C) Cecal colonization densities of wild-type Bt and BtΔ2157 in gnotobiotic mice following two weeks of dietary glucosinolate intervention. Cecal contents from mice were collected six hours following the final glucotropaeolin dose and assessed for Bt abundance using quantitative PCR. Data shown are the mean ± SD of five biological replicates.
(D) Annotated representative extracted ion chromatogram of glucosinolates identified in broccoli extract. Glucosinolates corresponding to the extracted exact masses are shown in boxes.
(E) BITC-NAC and BITC-cys mercapturic acid conjugates and unconverted BGS excreted in urine and feces after doses of pure glucotropaeolin (top), and BITC-cys and unconverted BGS in feces collected after doses of pure glucotropaeolin (bottom). BITC-cys in feces was the product of in situ conjugation of free BITC with exogenously applied cysteine. Bars represent the mean values for each group, with individual data overlaid (different shapes correspond to individual mice). Metabolites were measured by LC-MS/MS and quantified by MRM (BITC-NAC: m/z 313.1→ m/z 164; BITC-cys: m/z 271.1→ m/z 122.0; BGS: m/z 408.0→ m/z 96.9). Mice mono-colonized with mutant BtΔ2157 compared to wild-type Bt showed increased glucosinolate concentration in fecal samples (Fig. S6B), which may be indicative of indirect impacts of glucosinolate conversion, other physiological activities encoded by this operon (Liu et al., 2019), or effects specific to monocolonized mouse models.
F) Free SFN, SFN-NAC and SFN-cys mercapturic acid conjugates, and unconverted glucoraphanin excreted in urine six hours following doses of broccoli meal containing glucoraphanin. Bars represent the mean values for each group, with individual data overlaid (different shapes correspond to individual mice). Metabolite quantities were measured by LC-MS/MS, quantified by MRM (SFN-NAC: m/z 341.1→ m/z 177.9; SFN-cys: m/z 299.1→ m/z 136; glucoraphanin: m/z 436.0→ m/z 372, SFN: m/z 178.0→ m/z 114.0), and converted to concentrations. Metabolite levels were normalized to urinary creatinine concentrations, as quantified by LC-MS/MS using the transition from protonated creatinine (m/z 114.1) to a product ion with m/z 86.2.
Figure S5. Metagenomic and transcriptomic prevalence of BT2160-BT2156. Related to Figure 5. A) Metagenomic and metatranscriptomic RPKM abundance of each of the genes in BT2160-BT2156 in the stool cohorts examined. Each row in the heatmap corresponds to a subject in the cohort, with each column corresponding to a gene in the gene cluster.
B) Metagenomic and metatranscriptomic coverage of each of the genes in BT2160-BT2156 in the stool cohorts examined.
Table S3. Metagenomic and metatranscriptomic abundance of BT2160-BT2156. Related to Figure 5.
Data Availability Statement
The results of the metagenomic and metatranscriptomic analyses used to generate Figure 5 are included as a supplementary file. All other relevant data are available from the authors upon request.