

Abstract
BACKGROUND:
Infigratinib (BGJ398) is a potent and selective fibroblast grown factor receptor 1 to 3 (FGFR1–3) inhibitor with significant activity in patients with advanced or metastatic urothelial carcinoma bearing FGFR3 alterations. Given the distinct biologic characteristics of upper tract urothelial carcinoma (UTUC) and urothelial carcinoma of the bladder (UCB), the authors examined whether infigratinib had varying activity in these settings.
METHODS:
Eligible patients had metastatic urothelial carcinoma with activating FGFR3 mutations and/or fusions. Comprehensive genomic profiling was performed on formalin-fixed, paraffin-embedded tissues. Blood was collected for cell-free DNA analysis using a 600-gene panel. Patients received infigratinib at a dose of 125 mg orally daily (3 weeks on/1 week off) until disease progression or intolerable toxicity occurred. The overall response rate (ORR; partial response [PR] plus complete response [CR]) and disease control rate (DCR; CR plus PR plus stable disease [SD]) were characterized.
RESULTS:
A total of 67 patients were enrolled; the majority (70.1%) had received ≥2 prior antineoplastic therapies. In 8 patients with UTUC, 1 CR and 3 PRs were observed (ORR, 50%); the remaining patients achieved a best response of SD (DCR, 100%). In patients with UCB, 13 PRs were observed (ORR, 22%), and 22 patients had a best response of SD (DCR, 59.3%). Notable differences in genomic alterations between patients with UTUC and those with UCB included higher frequencies of FGFR3-TACC3 fusions (12.5% vs 6.8%) and FGFR3 R248C mutations (50% vs 11.9%), and a lower frequency of FGFR3 S249C mutations (37.5% vs 59.3%).
CONCLUSIONS:
Differences in the cumulative genomic profile were observed between patients with UTUC and those with UCB in the current FGFR3-restricted experience, underscoring the distinct biology of these diseases. These results support a planned phase 3 adjuvant study predominantly performed in this population.
Keywords: FGFR3, genomic profile, lower tract urothelial carcinoma, upper tract urothelial carcinoma
INTRODUCTION
Although cisplatin-based chemotherapy has represented the cornerstone of therapy for patients with metastatic urothelial cancer (mUC) for several decades, multiple studies have shown a role for checkpoint inhibitors (CPIs) beyond platinum-based chemotherapy and in patients who are ineligible for treatment with cisplatin.1,2 CPIs work by blocking programmed cell death protein 1 (PD-1) or its cognate ligand, programmed death–ligand 1 (PD-L1).3–7 Although these agents undoubtedly represent a major milestone in therapy for patients with mUC, response rates generally are <30% in unselected populations. In particular, patients with immunologically “cold” tumors are less likely to derive benefit from PD-1/PD-L1 inhibition.8–10
With these data in mind, there is great interest in the development of targeted therapies for mUC. Guiding this are several seminal studies related to the genomics of UC. In one study of 295 patients with stage III to IV UC who were assessed using comprehensive genomic profiling (CGP), mutations in cyclin-dependent kinase inhibitor 2A (CDKN2A), fibroblast growth factor receptor 3 (FGFR3), and phosphatidylinositol 3-kinase catalytic subunit alpha (PIK3CA) were noted in 34%, 21%, and 20% of patients, respectively.11 FGFR3-altered tumors are especially attractive for targeted drug development in patients with UC because they are associated with immune exclusion; there is conflicting evidence surrounding their responsiveness to treatment with immunotherapy, especially CPIs (eg, atezolizumab, pembrolizumab). A composite analysis of the ImVigor210 and CheckMate 275 studies evaluating atezolizumab and nivolumab, respectively, in patients with mUC demonstrated no difference in response among patients harboring FGFR3 mutations.12 However, a recent analysis by Santiago-Walker et al9 demonstrated that FGFR-altered UCs are poorly receptive to immune CPIs. More specifically, for patients treated with second-line anti–PD-1/PD-L1 therapy, the median overall survival (OS) was 3.09 months (95% confidence interval [95% CI], 0.26 months to not estimable) in patients with FGFR-altered disease versus 6.11 months (95% CI, 4.76–10.09 months) in those with FGFR-unaltered disease. A similar result was observed in patients treated with first-line anti–PD-1/PD-L1 therapy in this study.
It also recently has been noted that upper tract urothelial carcinoma (UTUC) and urothelial carcinoma of the bladder (UCB) may have distinct biologic underpinnings. Moss et al performed whole-exome sequencing in a series of 31 patients with UTUC.13 Most notably, their results suggested alterations in FGFR3 in approximately 74% of cases, with 94% of low-grade UTUCs harboring FGFR3 alterations.13 In what to our knowledge is the largest comparison of UTUC versus UCB genomics published to date, Audenet et al14 assessed 195 patients with UTUC and 454 with UCB. In their study, FGFR3 and HRAS were more frequently altered in UTUC compared with UCB (40% vs 25% and 12% vs 4%, respectively).14
Collectively, these studies have suggested that patients with UC harbor a broad array of potentially actionable mutations. Furthermore, FGFR3 appears to be a target that is enriched in patients with UTUC. We previously reported data related to infigratinib (BGJ398), a potent and selective FGFR1–3 tyrosine kinase inhibitor, in 67 patients with mUC bearing alterations in FGFR3.15 In this cohort of mostly heavily pretreated patients (70.1% had received ≥2 prior anticancer therapies), an overall response rate (ORR) of 25.4% was observed, with an additional 38.8% of patients achieving stable disease (SD) (disease control rate [DCR], 64.2%). In the current study, we sought to determine whether patients with UTUC and UCB had a distinct pattern of response. In addition, given the availability of tumor tissue and blood from the study participants, we sought to confirm biologic distinctions in this biomarker-selected cohort.
MATERIALS AND METHODS
Patient Selection
Adult patients with mUC who had received prior platinum-based chemotherapy or were deemed intolerant of or ineligible for platinum-based chemotherapy were screened for FGFR3 alterations using comprehensive genomic profiling of 324 genes in a Clinical Laboratory Improvement Amendments (CLIA)–certified laboratory (Foundation Medicine, Cambridge, Massachusetts). These patients were enrolled in an expansion cohort of the phase 1 study of infigratinib.16
Patients possessing somatic alterations in FGFR3 with suspected functional significance were eligible for the current study. For inclusion in the study, patients also had to demonstrate a World Health Organization performance status of 0 to 2, adequate bone marrow function, and adequate hepatic and renal function. Prior therapy with FGFR or MEK inhibitors was not allowed. In this international, multi-institutional study, the protocol and consent were approved by each institutional ethics board. All patients enrolled provided separate consent for screening for FGFR3 alterations (unless genomic testing was performed as per standard of care) and for therapy with infigratinib.
Treatment Regimen
Patients received oral infigratinib for 21 days in a 28-day cycle at a starting dose of 125 mg/day. Dose reductions to 100 mg/day followed by 75 mg/day were permitted, with further dose reductions allowed on an individual basis. Patients underwent baseline imaging including brain imaging (using either computed tomography [CT] or magnetic resonance imaging); CT scans of the chest, abdomen, and pelvis; and technetium bone scan. CT scans of the chest, abdomen, and pelvis (along with bone scan if indicated) were repeated at 8-week intervals while the patient was receiving treatment. The study drug was continued until disease progression or intolerable toxicity occurred.
Genomic Assessment of Tissue and Blood Specimens
The methods for CGP used in the current study have been described previously.11 CGP was performed for patients with available formalin-fixed, paraffin-embedded tissue derived from primary and/or metastatic site biopsy, transurethral resection of the bladder tumor, or cystectomy and/or nephroureterectomy. The functional significance of mutations in FGFR3 was determined through interrogation of the Catalogue Of Somatic Mutations In Cancer (COSMIC) database (https://cancer.sanger.ac.uk/cosmic)17 and review of the published literature. Ultimately, these included mutations in exon 7 (R248C and S249C); exon 10 (G372C, A393E, and Y375C); exon 15 (K652M/T and K652E/Q); and FGFR3 fusions, including but not limited to the FGFR3-TACC fusion. Amino acid numbers are in reference to functional FGFR3 isoform 3 (RefSeq database identification NP_001156685.1).
Plasma was collected at baseline and at multiple prespecified timepoints during treatment (screening and/or at baseline, day 1 of cycle 2, day 28 of cycle 2, day 28 of cycles 3 and beyond, and at the end of treatment). Cell-free DNA (cfDNA) was extracted from plasma specimens using the QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany), and the Illumina TruSeq Nano DNA Library Prep Kit (Illumina, San Diego, California) was used to construct whole-genome sequencing libraries. A 600-gene pan-cancer panel was used; sequencing was performed on an Illumina HiSeq 2500 System (Illumina) with a median coverage of ×775. Detailed methods have been published previously.15
Statistical Analysis
Response was assessed in all patients using Response Evaluation Criteria in Solid Tumors (RECIST; version 1.0). The ORR (partial response [PR] plus complete response [CR]) and DCR (CR plus PR plus SD [≥6 weeks]) then were calculated. In the current analysis, the ORR was presented separately by the UTUC and UCB cohorts. Descriptive statistics were provided for mutation frequencies in these cohorts.
RESULTS
Patient Characteristics and Clinical Efficacy
A total of 67 patients were enrolled between August 6, 2014, and July 29, 2016, across 27 centers in the United States, Europe, and Asia. Tables 1 and 2 show the baseline characteristics for the patients; the majority (70%) had received ≥2 prior anticancer therapies. Overall, 41 patients (61.2%) had visceral disease of the lung and 25 patients (37.3%) had visceral disease of the liver. Bellmunt risk scores of 0, 1, 2, and 3 were characterized in 12 patients (17.9%), 27 patients (40.3%), 25 patients (37.3%), and 3 patients (4.5%), respectively. Patients with UTUC predominantly were receiving second-line treatment (62.5%); 25% had a response to previous treatment (Table 2). In contrast, patients with UCB predominantly were receiving second-line or third-line treatment (45.7%), and 16.9% had a response to prior therapy. Thirteen patients in the series received prior immunotherapy. Of these, 1 patient had UTUC whereas 12 patients had UCB.
TABLE 1.
Baseline Characteristics of Patients With UTUC and UCB
| Characteristic | UTUC N = 8 | UCB N = 59 | Total N = 67 |
|---|---|---|---|
| Age, no. (%) | |||
| <65 y | 4 (50.0) | 25 (42.4) | 29 (43.3) |
| ≥65 y | 4 (50.0) | 34 (57.6) | 38 (56.7) |
| Sex, no. (%) | |||
| Male | 7 (87.5) | 39 (66.1) | 46 (68.7) |
| Female | 1 (12.5) | 20 (33.9) | 21 (31.3) |
| WHO performance status, no. (%) | |||
| 0 | 2 (25.0) | 19 (32.2) | 21 (31.3) |
| 1 | 6 (75.0) | 30 (50.8) | 36 (53.7) |
| 2 | 0 | 10 (16.9) | 10 (14.9) |
| Bellmunt risk score, no. (%) | |||
| 0 | 2 (25.0) | 10 (16.9) | 12 (17.9) |
| 1 | 3 (37.5) | 24 (40.7) | 27 (40.3) |
| 2 | 3 (37.5) | 22 (37.3) | 25 (37.3) |
| 3 | 0 | 3 (5.1) | 3 (4.5) |
| Visceral disease, no. (%) | |||
| Lung | 5 (62.5) | 36 (61.0) | 41 (61.2) |
| Liver | 2 (25.0) | 23 (39.0) | 25 (37.3) |
| Lymph node metastases, no. (%) | |||
| Yes | 2 (25.0) | 26 (44.1) | 28 (41.8) |
| No | 6 (75.0) | 33 (55.9) | 39 (58.2) |
| Bony metastases, no. (%) | |||
| Yes | 3 (37.5) | 23 (39.0) | 26 (38.8) |
| No | 5 (62.5) | 36 (61.0) | 41 (61.2) |
Abbreviations: UCB, urothelial carcinoma of the bladder; UTUC, upper tract urothelial carcinoma; WHO, World Health Organization.
TABLE 2.
Previous Treatment for Patients With UTUC and UCB
| UTUC N = 8 | UCB N = 59 | Total N = 67 | |
|---|---|---|---|
| Total no. of lines of prior therapy (%) | |||
| 0 | 0 | 13 (22.0) | 13 (19.4) |
| 1 | 5 (62.5) | 19 (32.2) | 24 (35.8) |
| ≥2 | 3 (37.5) | 27 (45.8) | 30 (44.8) |
| Total no. of prior anticancer regimens (%) | |||
| 0 | 0 | 1 (1.7) | 1 (1.5) |
| 1 | 2 (25.0) | 17 (28.8) | 19 (28.4) |
| ≥2 | 6 (75.0) | 41 (69.5) | 47 (70.1) |
| Best response to prior anticancer regimen, no. (%) | |||
| CR (confirmed) | 0 | 1 (1.7) | 1 (1.5) |
| CR (unconfirmed) | 0 | 1 (1.7) | 1 (1.5) |
| PR | 2 (25.0) | 8 (13.6) | 10 (14.9) |
| SD | 2 (25.0) | 21 (35.6) | 23 (34.3) |
| PD | 2 (25.0) | 14 (23.7) | 16 (23.9) |
| Missing data | 2 (25.0) | 14 (23.7) | 16 (23.9) |
Abbreviations: CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease; UCB, urothelial carcinoma of the bladder; UTUC, upper tract urothelial carcinoma.
Four confirmed responses (CR or PR) were observed with infigratinib among the 8 patients with UTUC, for an ORR of 50% (Table 3). The other 4 patients with UTUC achieved a best response of SD for a DCR of 100%. The median progression-free survival was 8.54 months (95% CI, 3.68–12.91 months) and the median OS was 21.82 months (95% CI, 11.63 months to not estimable) in patients with UTUC (Figs. 1A and 1B). Among the 59 patients with UCB, 13 confirmed responses (all PRs) were observed for an ORR of 22% (Table 3). An additional 22 patients had a best response of SD for a DCR of 59%. In patients with UCB, the median progression-free survival was 3.65 months (95% CI, 2.76–4.91 months) and the median OS was 7.00 months (95% CI, 5.52–11.53 months) (Figs. 1 and 2). No differences with regard to toxicity were observed between the UTUC and UCB cohorts (see Supporting Tables 1 and 2).
TABLE 3.
Efficacy Results for Treatment With Infigratinib Among Patients With UTUC and UCB
| UTUC N = 8 | UCB N = 59 | Total N = 67 | |
|---|---|---|---|
| Response assessment, no. (%) | |||
| CR, confirmed | 1 (12.5) | 0 | 1 (1.5) |
| PR, confirmed | 3 (37.5) | 13 (22.0) | 16 (23.9) |
| CR or PR, unconfirmed | 1 (12.5) | 10 (16.9) | 11 (16.4) |
| SD | 4 (50.0) | 22 (37.3) | 26 (38.8) |
| PD | 0 | 18 (30.5) | 18 (26.9) |
| Unknown/not done | 0 | 6 (10.2) | 6 (9.0) |
| Confirmed objective response (CR or PR), no. (%) | 4 (50.0) | 13 (22.0) | 17 (25.4) |
| 95% CI | 15.7–84.3 | 12.3–34.7 | 15.5–37.5 |
| Best overall response (CR or PR, confirmed/unconfirmed), no. (%) | 5 (62.5) | 23 (39.0) | 28 (41.8) |
| 95% CI | 24.5–91.5 | 26.5–52.6 | 29.8–54.5 |
| Disease control rate (CR/PR or SD), no. (%) | 8 (100.0) | 35 (59.3) | 43 (64.2) |
| 95% CI | 63.1–100.0 | 45.7–71.9 | 51.5–75.5 |
| Median duration of response, mo | 6.77 | 5.04 | 5.62 |
| Range | 3.32–11.01 | 2.33–8.08 | 2.33–11.01 |
Abbreviations: 95% CI, 95% confidence interval; CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease; UCB, urothelial carcinoma of the bladder; UTUC, upper tract urothelial carcinoma.
Figure 1.
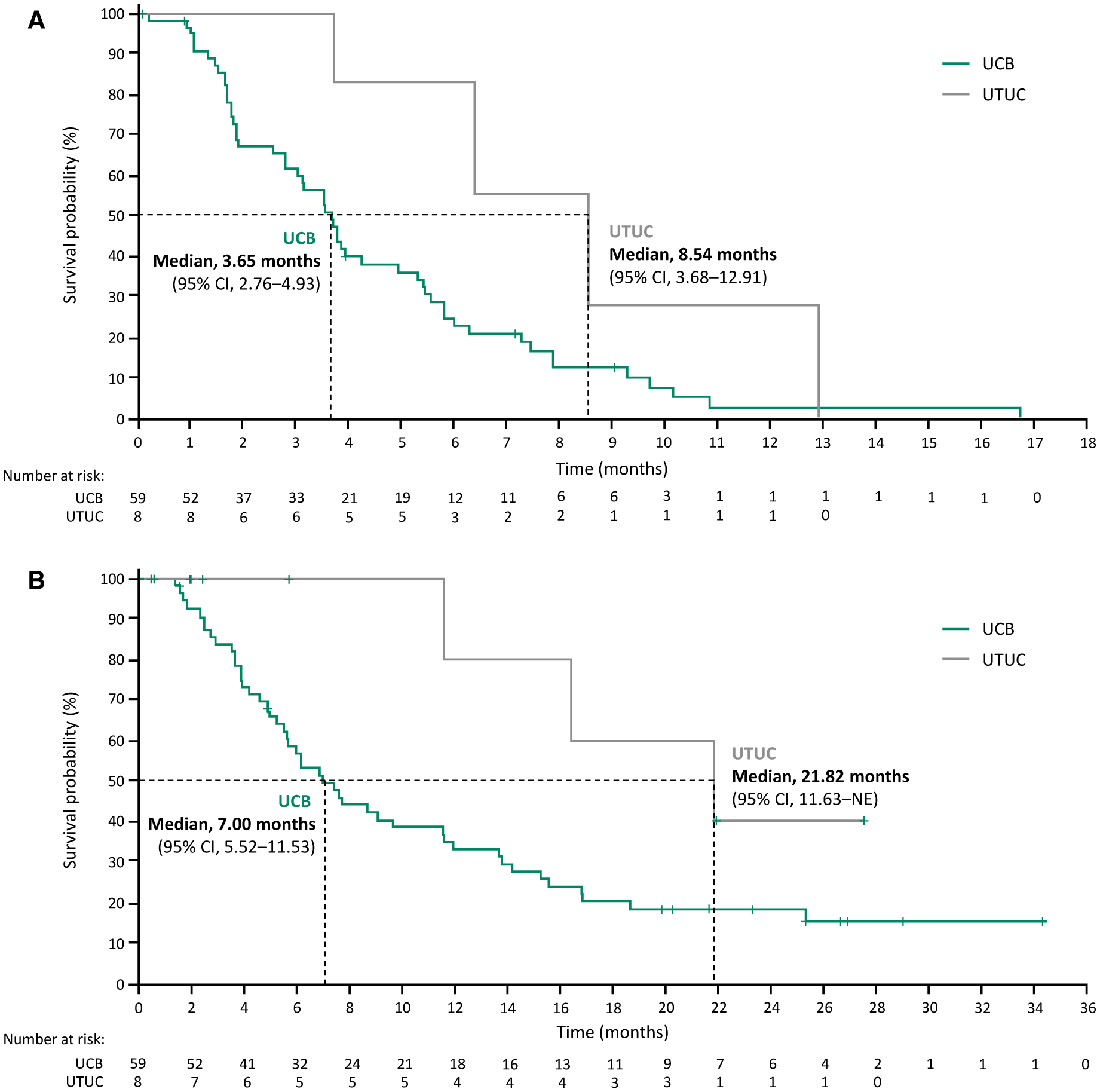
(A) Progression-free survival and (B) overall survival for patients with upper tract urothelial carcinoma (UTUC) and urothelial carcinoma of the bladder (UCB). 95% CI indicates 95% confidence interval.
Figure 2.
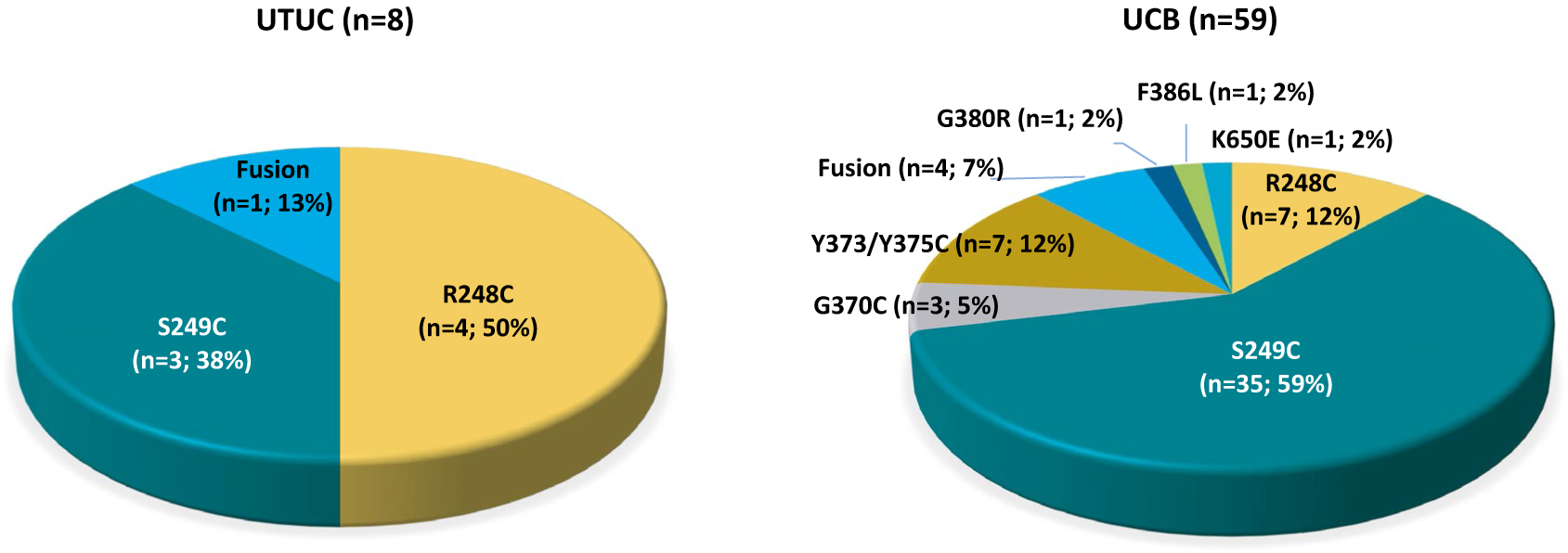
Genomic differences in tumor tissue samples between patients with upper tract urothelial carcinoma (UTUC) and urothelial carcinoma of the bladder (UCB).
When examining the entire population (both the UTUC and UBC cohorts), patients without visceral disease (9 patients) had a higher ORR (33.3%) compared with those with visceral disease of the lung and/or the liver (58 patients; 24.1%).
Comparison of FGFR3 in Patients With UTUC Versus UCB
Tumor tissue analysis demonstrated notable differences in FGFR3 alterations between patients with UTUC and UCB. These included a smaller collection of FGFR3 alterations among patients with UTUC and significant differences in the frequency of FGFR3 R248C mutations (50% vs 12%) and FGFR3 S249C mutations (38% vs 59%) in patients in the UTUC cohort compared with the UCB cohort, respectively (Fig. 2). Oncoplots demonstrated a less complex genomic profile with an increased number of known or likely deleterious mutations and copy number alterations in tissue from patients with UCB versus those with UTUC (Fig. 3A). Amplification of genes involved in FGFR signaling, including the FGFR3 ligands FGF3, FGF4, and FGF19 on chromosome 11 and the FGFR adaptor protein FRS2, was more common in patients with UCB (10 of 39 patients; 26%) compared with those with UTUC (1 of 7 patients; 14%). In addition, mutations in the telomerase reverse transcriptase (TERT) promoter that are known to activate gene transcription were more common in patients with UCB (29 of 39 patients; 74%) compared with those with UTUC (3 of 7 patients; 43%). Only one patient with UTUC was found to have a high mutation load; however, this patient likely was deficient in mismatch repair due to a frameshift mutation in mutS homolog 2 (MSH2).
Figure 3.
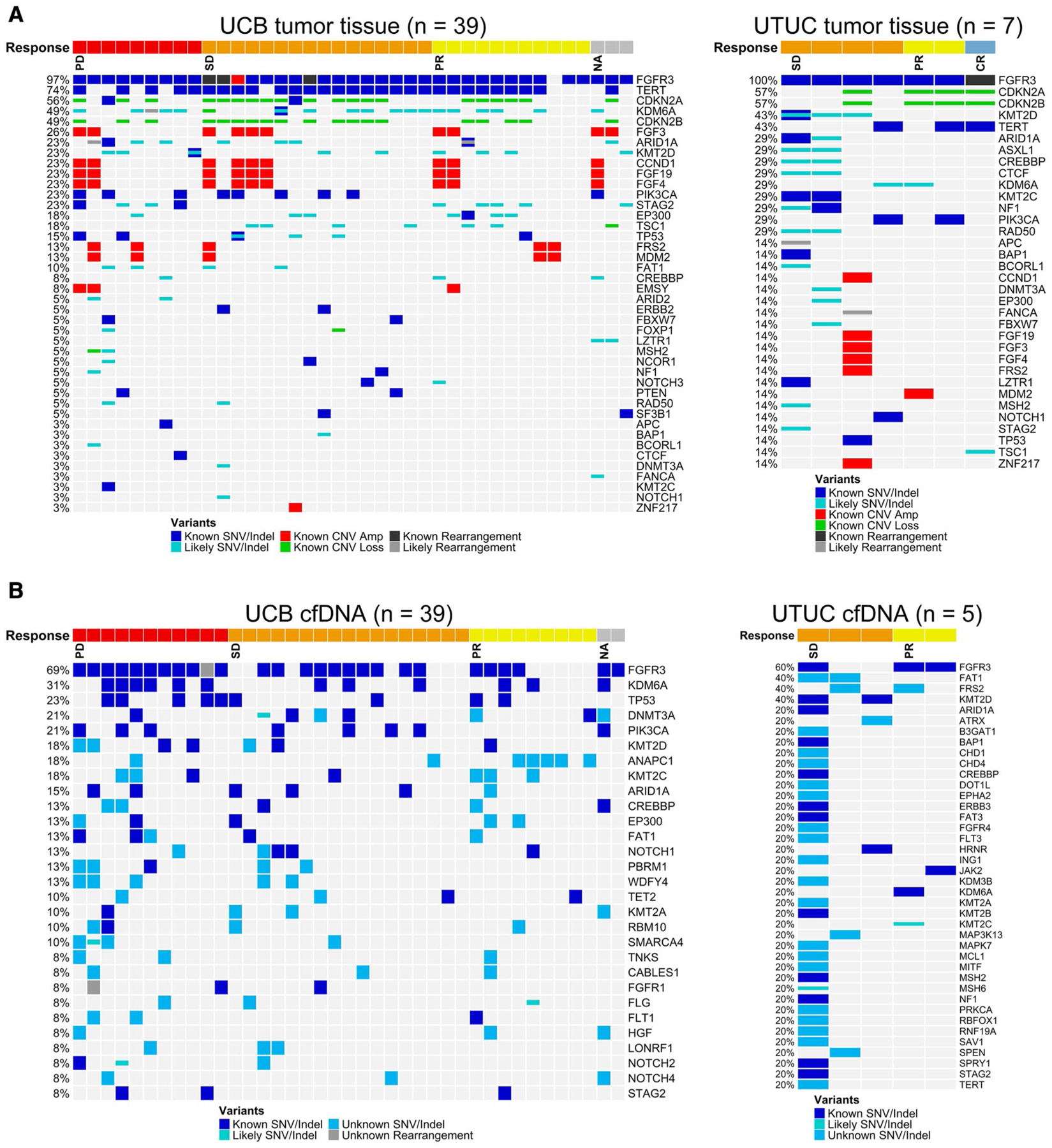
Oncoplots of (A) tumor tissue genomic profiles and (B) cell-free DNA (cfDNA) genomic profiles in upper tract urothelial carcinoma (UTUC) and urothelial carcinoma of the bladder (UCB). Amp indicates amplification; CNV, copy number variation; CR, complete response; Indel, insertion and deletion; NA, not available; PD, progressive disease; PR, partial response; SD, stable disease; SNV, single-nucleotide variant.
Genomic analysis of patients with available tumor tissue demonstrated notable differences in FGFR3 alterations between patients with UTUC and those with UCB.
Assessment of cfDNA
FGFR3 alterations were concordant in 30 of 38 patients (79%) for whom both tumor tissue and cfDNA samples were available at screening. As identified in tissue, oncoplots of cfDNA data alluded to a less complex genomic profile and increased mutation in UTUC (Fig. 3B). The one patient with UTUC with a high number of point mutations in the cfDNA was likely to be deficient in mismatch repair caused by a frameshift mutation in MSH2. A correlative analysis of FGFR3 allele frequency in cfDNA and clinical characteristics, including the sum of the longest dimensions, Eastern Cooperative Oncology Group performance status, and sites of tumor metastasis, is shown in Figure 4, including 53 patients for whom cfDNA data were available.
Figure 4.
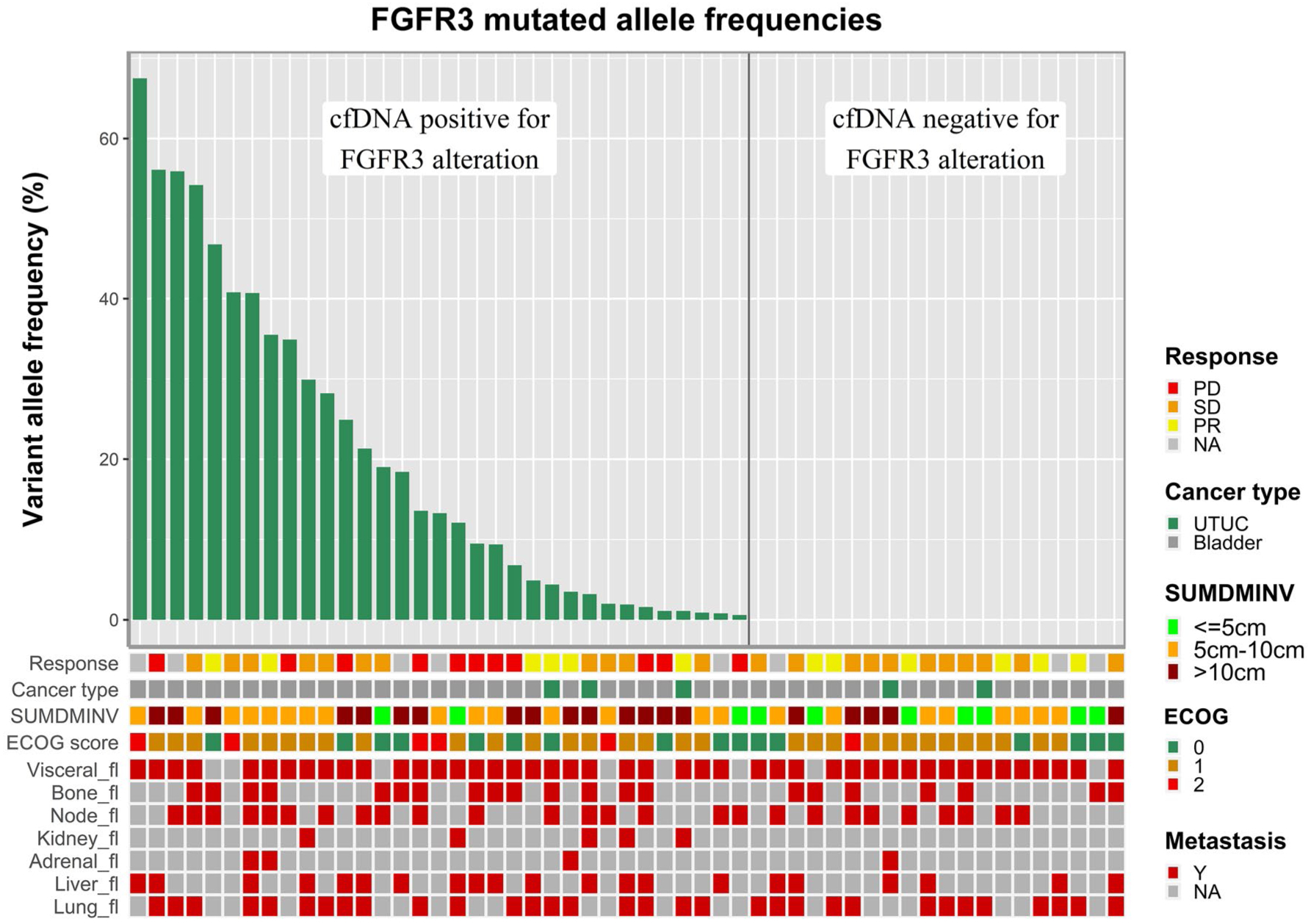
Correlative analysis of fibroblast growth factor receptor 3 (FGFR3) allele frequency in cell-free DNA (cfDNA) and clinical characteristics, including sum of the longest dimensions, Eastern Cooperative Oncology Group (ECOG) performance status, and sites of tumor metastasis in 53 patients for whom cfDNA was available. NA indicates not available; PD, progressive disease; PR, partial response; SD, stable disease; SUMDMINV, sum of diameters per investigator; UTUC, upper tract urothelial carcinoma; Y, yes.
DISCUSSION
In a cohort already selected for FGFR3 alterations, the data from the current study have suggested that infigratinib may have greater activity in patients with UTUC compared with those with UCB, with DCRs of 100% and 59%, respectively. The current analysis included 8 patients with UTUC and 59 patients with UCB who were enrolled in an expansion cohort of the phase 1 study of infigratinib. It is worth noting that in the dose escalation phase of this study, 2 additional patients with FGFR3-altered UTUC were enrolled.16 One of these patients had a durable CR, and remained on treatment 6 years after the initiation of infigratinib. Including these 2 additional patients (total UTUC cohort of 10 patients) gave an ORR of 50% among patients with UTUC, with a CR rate of 20%.
To our knowledge, these are the highest response rates achieved in a UTUC cohort to date and this is the only targeted therapy found to have differentiated efficacy in patients with UTUC compared with those with UCB. There are several other small molecule FGFR3 inhibitors currently in clinical development. Erdafitinib recently received US Food and Drug Administration accelerated approval for the treatment of patients with locally advanced or mUC with susceptible FGFR3 or FGFR2 genetic alterations who have developed disease progression during or after at least 1 line of prior platinum-based chemotherapy. The approval was based on a study of 99 patients with FGFR3-mutant advanced UC; an ORR of 34% was observed, with 3% of patients achieving a CR.18 Response rates in patients with UTUC and lower tract urothelial carcinoma appear comparable at 43% and 39%, respectively, but to the best of our knowledge the CR rates and DCR rates in these subsets of patients have not been reported as yet. It is interesting to note that infigratinib has higher specificity for FGFR3 compared with erdafitinib for patients with the more “FGFR3-driven” upper tract tumors, and this may be why a higher response rate was encountered.19 Another small molecule, rogaratinib, was tested in a cohort of patients with mUC with either increased FGFR1 to FGFR3 messenger RNA expression or FGFR3 mutation.20 A response rate of 24% was observed among the 51 patients who were evaluable for response. Distinct from these small molecule inhibitors, vofatamab is a monoclonal antibody directed at FGFR3. In a recently reported phase 2 study including 21 patients with mUC who received monotherapy with vofatamab, the DCR was 21% at 6 months.21 Across these studies of distinct FGFR inhibitors, there are limited data comparing activity in UTUC versus UCB and it would be an important proof of concept to confirm the consistency of the current study findings across other trials.
In addition to suggesting a higher response rate to infigratinib in patients with UTUC versus those with UCB, genomic profiling of available tumor tissue revealed several biologic differences between these patient groups. A different distribution of FGFR3 alterations was observed, with FGFR3 mutations in patients with UTUC restricted to the extracellular immunoglobulin-like domain whereas activating mutations in patients with UCB also were identified in the transmembrane region and kinase domain. Similar to the tissue-based genomic profiling in the current study, pretreatment cfDNA analysis demonstrated an increase in mutations and gain and/or loss of gene copy that are known to alter protein function among patients with UCB versus those with UTUC. Together, these data suggest a less heterogenous mutation profile in patients with UTUC compared with those with UCB, which may lead to a greater likelihood that FGFR3 is the primary alteration driving the cancer in UTUC22 and therefore a greater likelihood of response to infigratinib. The finding of less heterogeneity in patients with UTUC versus those with UCB is consistent with prior reports.23
In what to our knowledge is the largest study to date to assess cfDNA in patients with UC, Agarwal et al used a commercially available, 73-gene platform to assess 369 patients with advanced disease.24 In contrast to tissue-based studies, no significant difference was found in the frequency of genomic alterations between 294 patients with UCB versus 75 patients with UTUC. Although the study reported that approximately 91% of patients had detectable alterations, it is interesting that the vast majority were mutations in TP53, giving rise to the possibility that these results could be affected by clonal hematopoiesis.25 Irrespective of these results, blood-based genomic screening tests will be increasingly attractive as a noninvasive means of characterizing eligibility for targeted therapy. In the current study, FGFR3 alterations were found to be concordant in nearly 80% of patients with both tumor tissue and cfDNA samples available at screening, suggesting that blood-based cfDNA may play an important role in the detection of FGFR3 alterations among patients with UC in the future. However, to the best of our knowledge, blood-based genomic tests and other platforms (eg, urine cfDNA) are not yet technologically advanced enough to fully characterize FGFR3 status and other series have pointed toward significant discordance between tissue and blood26; therefore, tissue-based genomic sequencing continues to be the preferred diagnostic method.27 Beyond blood and urine DNA assessment, it remains to be seen whether RNA assessment (as in the aforementioned rogaratinib experience) could broaden the spectrum of eligible patients for therapy. Currently, the reverse transcriptase–polymerase chain reaction–based companion diagnostic for erdafitinib only detects mutations and select fusions. The test uses RNA as a substrate to improve sensitivity through allele-specific complementary DNA amplification and allele-specific polymerase chain reaction to improve mutation detection.
Limitations of the current study included the small sample size of this single-arm, nonrandomized trial. We acknowledge that it was challenging to draw robust conclusions from the patients with UTUC who were included in the current study and therefore we consider these findings to be hypothesis generating. The conclusions surrounding the genomic findings in cfDNA also were tempered by the lack of available blood specimens in several patients. However, it should be noted that the findings in the cfDNA and tissue specimens were somewhat consistent (ie, a lesser degree of genomic diversity in UTUC vs UCB). Furthermore, we were limited in our ability (given the small sample size) to correct for potentially confounding clinical and demographic variables that could affect outcome.
Conclusions
The updated results presented in the current study have demonstrated that infigratinib retains significant activity in patients with mUC. Furthermore, to the best of our knowledge, it provides one of the most robust signals of activity in UTUC presented to date, with an ORR of 50% and a DCR of 100%. The nature of the mutations noted in UTUC appears to differ from that of mutations in UCB, with the caveat of the small sample size, a higher rate of FGFR3 S249C mutations, and a smaller repertoire of mutations observed in the current study. The results of the current study regarding infigratinib in patients with UTUC offer support to a planned phase 3, adjuvant study performed predominantly in this population.
Supplementary Material
FUNDING SUPPORT
Sponsored and funded by Novartis and QED Therapeutics Inc.
We thank the patients participating in the trial and their families, the investigators, and the staff at participating centers.
Footnotes
CONFLICT OF INTEREST DISCLOSURES
Sumanta K. Pal has acted as a paid consultant or in an advisory role for Genentech/Roche, Aveo Pharmaceuticals, Eisai, Pfizer, Novartis, Exelixis, Ipsen, Bristol-Myers Squibb, Astellas Pharma, and Myriad Pharmaceuticals; has received honoraria from Astellas Pharma, Medivation, and Novartis; and has received research funding from Medivation for work performed outside of the current study. Dean Bajorin has acted as a paid consultant or in an advisory role for Bristol-Myers Squibb, Eisai, EMD Serono, Fidia Farmaceutica S.p.A., Eli Lilly, Merck, Novartis, Pfizer, Genentech/Roche, and UroGen Pharma; has received travel and accommodations expenses from Bristol-Myers Squibb, Eli Lilly, Merck, Genentech/Roche, and UroGen Pharma; has received honoraria from Merck, Sharp & Dohme; and has received research funding to his institution from Amgen, Astellas Pharma, AstraZeneca, Bristol-Myers Squibb, Dendreon, Merck, Novartis, Genentech/Roche, and Seattle Genetics for work performed outside of the current study. Dr. Bajorin also is supported by Memorial Sloan Kettering Cancer Center core grant P30 CA008748. Jean Hoffman-Censits has acted as a paid consultant or in an advisory role for AstraZeneca, Oncology, Foundation Medicine, and Genentech/Roche and has received research funding from Foundation Medicine and Sanofi for work performed outside of the current study. David I. Quinn has acted as a paid consultant or in an advisory role for Amgen, Astellas Pharma, AstraZeneca, Bayer, Bristol-Myers Squibb, Clovis Oncology, Dendreon, Eisai, EMD Serono, Exelixis, Janssen Oncology, Merck Sharp & Dohme, Novartis, Pfizer, Genentech/Roche, Sanofi, Seattle Genetics, and US Biotest; has received honoraria from Astellas Pharma, AstraZeneca, Bayer, Bristol-Myers Squibb, Clovis Oncology, Dendreon, Exelixis, Janssen Oncology, Merck Sharp & Dohme, Mundipharma, Novartis, Pharmacyclics, Pfizer, Genentech/Roche, and Sanofi; and has received research funding to his institution from GlaxoSmithKline, Millennium, Genentech/Roche, and Sanofi for work performed outside of the current study. Daniel P. Petrylak has received grant support to his institution from and acted as a paid consultant or in an advisory role for Advanced Accelerator Applications, Astellas Pharma, AstraZeneca, Bayer, Bristol-Myers Squibb, Clovis Oncology, Eli Lilly, Pfizer, Genentech/Roche, and Seattle Genetics; has acted as a paid consultant for Amgen, Boehringer Ingelheim, Exelixis, Incyte, Janssen, Pharmacyclics, UroGen, Bellicum Pharmaceuticals, Dendreon, Ferring, Johnson & Johnson, Medivation, Millennium, Sanofi, and Tyme; has provided expert testimony for Celgene and Sanofi; has stock and other ownership interests in Bellicum Pharmaceuticals and Tyme; and has received research funding to his institution from Agensys, Astellas Medivation, Dendreon, Endocyte, Genentech/Roche, Innocrin Pharma, Johnson & Johnson, MedImmune, Merck, Millennium, Novartis, Progenics Pharmaceuticals, Sanofi Aventis, and Sotio for work performed outside of the current study. Matthew D. Galsky has acted as a paid consultant or in an advisory role for and received research funding from Merck, Genentech/Roche, AstraZeneca, and Bristol-Myers Squibb; has acted as a paid consultant or in an advisory role for Aileron Therapeutics, Astellas Pharma, BioMotiv, Dendreon, Dracen, Dragonfly, EMD Serono, GlaxoSmithKline, Incyte, Inovio Pharmaceuticals, Janssen, Lilly, Novartis, Numab Therapeutics, Pfizer, and Seattle Genetics; has patents, royalties, or other intellectual property (Methods and compositions for treating cancer and related methods; Mount Sinai School of Medicine. July 2012; application number: 20120322792); has stock and other ownership interests in Rappta Therapeutics; and has received research funding to his institution from Dendreon, Janssen, and Novartis for work performed outside of the current study. Ulka Vaishampayan has acted as a paid consultant or in an advisory role for and received research funding to his institution from Bristol-Myers Squibb, Exelixis, Pfizer, and Bayer; has acted as a paid consultant or in an advisory role for Astellas Pharma and Genentech/Roche; has received honoraria from Bayer, Bristol-Myers Squibb, Exelixis, Eisai, Pfizer, and Sanofi; and has received research funding to his institution from Astellas Pharma and Merck for work performed outside of the current study. Ugo De Giorgi has acted as a paid consultant or in an advisory role for Astellas Pharma, Bayer, Bristol-Myers Squibb, Ipsen, Janssen, Merck, Pfizer, and Sanofi; has received research funding to his institution from AstraZeneca, Roche, and Sanofi; and has received travel and accommodations expenses from Bristol-Myers Squibb, Ipsen, Janssen, and Pfizer for work performed outside of the current study. Sumati Gupta has an immediate family member that has acted as a paid consultant or in an advisory role for and received travel and accommodations expenses from Keryx Biopharmaceuticals and owns stock and other ownership interests in Salarius Pharmaceuticals. Dr. Gupta has received research funding to her institution from AstraZeneca, Bristol-Myers Squibb, Clovis Oncology, Five Prime Therapeutics, Hoosier Cancer Research Network, Immunocore, Incyte, LSK, MedImmune, Merck, Mirati Therapeutics, QED Therapeutics Inc, Novartis, Pfizer, Rexahn Pharmaceuticals, Viralytics, and Debiopharm for work performed outside of the current study. Howard A. Burris has received a grant to his institution from Novartis for work performed as part of the current study and is employed by HCA Healthcare/Sarah Cannon for which he also has stock ownership; has acted as a paid consultant or in an advisory role for which fees were paid to his institution from AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Eisai, FORMA Therapeutics, Incyte, Janssen, MedImmune, Mersana Therapeutics, Novartis, Genentech/Roche, TG Therapeutics, and Tolero Pharmaceuticals; has provided leadership at HCA Healthcare/Sarah Cannon; has been paid for expert testimony by Novartis; and has received research funding to his institution from AbbVie, Agios, Arch, Arvinas, AstraZeneca, Bayer, BioAtla, BioMed Valley Discoveries, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Celldex Therapeutics, CicloMed, CytomX Therapeutics, Daiichi Sankyo, eFFECTOR Therapeutics, Forma Therapeutics, Gilead Sciences, GlaxoSmithKline, H3 Biomedicine, Harpoon Therapeutics, Immunocore, Incyte, Janssen, Jiangsu Hengrui Medicine, Jounce Therapeutics, Kyocera, Lilly, Loxo, MacroGenics, MedImmune, Merck, Mersana Therapeutics, Millennium, Moderna Therapeutics, Neon Therapeutics, Novartis, OncoMed, Pfizer, PTC Therapeutics, Regeneron, Revolution Medicines, Genentech/Roche, Sanofi, Seattle Genetics, Tarveda Therapeutics, Takeda, Tesaro, TG Therapeutics, Valent Technologies, Verastem, and Vertex. Harris S. Soifer is employed by QED Therapeutics Inc and holds stock and other ownership interests in Biotheranostics and QED Therapeutics Inc. Gary Li, Hao Wang, Carl L. Dambkowski, and Susan Moran are employed by QED Therapeutics Inc. Siamak Daneshmand has acted as a paid consultant or in an advisory role for Bristol-Myers Squibb, Photocure, Taris, Ferring, QED Therapeutics Inc, Olympus, and Pacific Edge; has received travel and accommodations expenses from Bristol-Myers Squibb, Photocure, Taris, Ferring, Olympus, and Pacific Edge; has received honoraria from Bristol-Myers Squibb, Photocure, Taris, Ferring, QED Therapeutics Inc, Olympus, and Pacific Edge; and has received research funding to his institution from Photocure for work performed outside of the current study. Jonathan E. Rosenberg has received clinical trial funding from QED Therapeutics Inc and Novartis for work performed as part of the current study and has received clinical trial funding from Seattle Genetics, Astellas, and Genentech/Roche; acted as a paid consultant or in an advisory role for Adicet Bio, Agensys, AstraZeneca/MedImmune, Bayer, BioClin/Rainier Therapeutics, Bristol-Myers Squibb, EMD Serono, Fortress Biotech, GlaxoSmithKline, Inovio Pharmaceuticals, Janssen, Lilly, Merck, Pharmacyclics, QED Therapeutics Inc, Genentech/Roche, Sanofi, Seattle Genetics, Sensei Biotherapeutics, and Western Oncolytics; has received travel and accommodations expenses from Bristol-Myers Squibb and Genentech/Roche; owns patents, royalties, and other intellectual property (predictor of platinum sensitivity); has stock and other ownership interests in Illumina and Merck; and has received honoraria from Agensys, AstraZeneca, Bayer, Bristol-Myers Squibb, Chugai Pharma, Genentech/Roche, Incyte, Medscape, Mirati Therapeutics, Novartis, OncoGenex, PeerView, Seattle Genetics, UpToDate, Vindico, and Viralytics for work performed outside of the current study. Nazli Dizman made no disclosures.
Additional supporting information may be found in the online version of this article.
REFERENCES
- 1.von der Maase H, Sengelov L, Roberts JT, et al. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J Clin Oncol. 2005;23:4602–4608. [DOI] [PubMed] [Google Scholar]
- 2.Lattanzi M, Balar AV. Current status and future direction of immunotherapy in urothelial carcinoma. Curr Oncol Rep. 2019;21:24. [DOI] [PubMed] [Google Scholar]
- 3.Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017; 376:1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Powles T, Duran I, van der Heijden MS, et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2018;391:748–757. [DOI] [PubMed] [Google Scholar]
- 5.Sharma P, Retz M, Siefker-Radtke A, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017;18:312–322. [DOI] [PubMed] [Google Scholar]
- 6.Patel MR, Ellerton J, Infante JR, et al. Avelumab in metastatic urothelial carcinoma after platinum failure (JAVELIN Solid Tumor): pooled results from two expansion cohorts of an open-label, phase 1 trial. Lancet Oncol. 2018;19:51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Massard C, Gordon MS, Sharma S, et al. Safety and efficacy of durvalumab (MEDI4736), an anti–programmed cell death ligand–1 immune checkpoint inhibitor, in patients with advanced urothelial bladder cancer. J Clin Oncol. 2016;34:3119–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corrales L, Matson V, Flood B, Spranger S, Gajewski TF. Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 2017;27:96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santiago-Walker A, Chen F, Loriot Y, et al. Predictive value of fibroblast growth factor receptor mutations and gene fusions on anti-PD-(L)1 treatment outcomes in patients with advanced urothelial cancer [abstract]. J Clin Oncol 2019;37(suppl 7):419.30589599 [Google Scholar]
- 10.Sweis RF, Galsky MD. Emerging role of immunotherapy in urothelial carcinoma–immunobiology/biomarkers. Urol Oncol. 2016;34: 556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ross JS, Wang K, Khaira D, et al. Comprehensive genomic profiling of 295 cases of clinically advanced urothelial carcinoma of the urinary bladder reveals a high frequency of clinically relevant genomic alterations. Cancer. 2016;122:702–711. [DOI] [PubMed] [Google Scholar]
- 12.Wang L, Gong Y, Saci A, et al. Fibroblast growth factor receptor 3 alterations and response to PD-1/PD-L1 blockade in patients with metastatic urothelial cancer. Eur Urol. 2019;76:599–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moss TJ, Qi Y, Xi L, et al. Comprehensive genomic characterization of upper tract urothelial carcinoma. Eur Urol. 2017;72:641–649. [DOI] [PubMed] [Google Scholar]
- 14.Audenet F, Isharwal S, Cha EK, et al. Clonal relatedness and mutational differences between upper tract and bladder urothelial carcinoma. Clin Cancer Res. 2019;25:967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pal SK, Rosenberg JE, Hoffman-Censits JH, et al. Efficacy of BGJ398, a fibroblast growth factor receptor 1–3 inhibitor, in patients with previously treated advanced urothelial carcinoma with FGFR3 alterations. Cancer Discov. 2018;8:812–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nogova L, Sequist LV, Perez Garcia JM, et al. Evaluation of BGJ398, a fibroblast growth factor receptor 1–3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: results of a global phase I, dose-escalation and dose-expansion study [errata In: J Clin Oncol. 2017;35:926; J Clin Oncol. 2019;37:358.]. J Clin Oncol. 2017;35:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tate JG, Bamford S, Jubb HC, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019;47:D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loriot Y, Necchi A, Park SH, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med. 2019;381:338–348. [DOI] [PubMed] [Google Scholar]
- 19.Perera TPS, Jovcheva E, Mevellec L, et al. Discovery and pharmacological characterization of JNJ-42756493 (erdafitinib), a functionally selective small-molecule FGFR family inhibitor. Mol Cancer Ther. 2017; 16:1010–1020. [DOI] [PubMed] [Google Scholar]
- 20.Joerger M, Cassier PA, Penel N, et al. Rogaratinib in patients with advanced urothelial carcinomas prescreened for tumor FGFR mRNA expression and effects of mutations in the FGFR signaling pathway [abstract]. J Clin Oncol. 2018;36(suppl 15):4513. [Google Scholar]
- 21.Necchi A, Castellano DE, Mellado B, et al. Fierce-21: phase II study of vofatmab (B-701), a selective inhibitor of FGFR3, as salvage therapy in metastatic urothelial carcinoma (mUC) [abstract]. J Clin Oncol. 2019;37(suppl 7):409. [Google Scholar]
- 22.Robinson BD, Vlachostergios PJ, Bhinder B, et al. Upper tract urothelial carcinoma has a luminal-papillary T-cell depleted contexture and activated FGFR3 signaling. Nat Commun. 2019;10:2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winters BR, De Sarkar N, Arora S, et al. Genomic distinctions between metastatic lower and upper tract urothelial carcinoma revealed through rapid autopsy. JCI Insight. 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agarwal N, Pal SK, Hahn AW, et al. Characterization of metastatic urothelial carcinoma via comprehensive genomic profiling of circulating tumor DNA. Cancer. 2018;124:2115–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu Y, Ulrich BC, Supplee J, et al. False-positive plasma genotyping due to clonal hematopoiesis. Clin Cancer Res. 2018;24:4437–4443. [DOI] [PubMed] [Google Scholar]
- 26.Barata PC, Koshkin VS, Funchain P, et al. Next-generation sequencing (NGS) of cell-free circulating tumor DNA and tumor tissue in patients with advanced urothelial cancer: a pilot assessment of concordance. Ann Oncol. 2017;28:2458–2463. [DOI] [PubMed] [Google Scholar]
- 27.Hayashi Y, Fujita K, Matsuzaki K, et al. Diagnostic potential of TERT promoter and FGFR3 mutations in urinary cell-free DNA in upper tract urothelial carcinoma. Cancer Sci. 2019;110:1771–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.