

Abstract
Dystrophin plays a crucial role in maintaining sarcolemma stability during muscle contractions, and mutations that prevent the expression of a functional protein cause Duchenne muscular dystrophy (DMD). Antisense oligonucleotide-mediated manipulation of pre-messenger RNA splicing to bypass Duchenne-causing mutations and restore functional dystrophin expression has entered the clinic for the most common DMD mutations. The rationale of “exon skipping” is based upon genotype-phenotype correlations observed in Becker muscular dystrophy, a milder allelic disorder generally characterized by in-frame deletions and internally truncated but semi-functional dystrophin isoforms. However, there is a lack of genotype-phenotype correlations downstream of DMD exon 55, as deletions in this region are rare and most single exon deletions would disrupt the reading frame. Consequently, the amenability of mutations in this region of the DMD gene to exon skipping strategies remains unknown. Here, we induced “Becker muscular dystrophy-like” in-frame dystrophin isoforms in vivo by intraperitoneal injection of peptide-conjugated phosphorodiamidate morpholino oligomers targeting selected exons. The dystrophin isoform encoded by the transcript lacking exons 56+57 appears to be more functional than that encoded by the 58+59-deleted transcript, as determined by higher dystrophin expression, stabilized β-dystroglycan, and less severe dystrophic pathology, indicating some potential for the strategy to address Duchenne-causing mutations affecting these exons.
Keywords: Duchenne muscular dystrophy, dystrophin isoforms, antisense oligonucleotides, splice-switching, exon skipping, phosphorodiamidate morpholino oligomer
Graphical Abstract
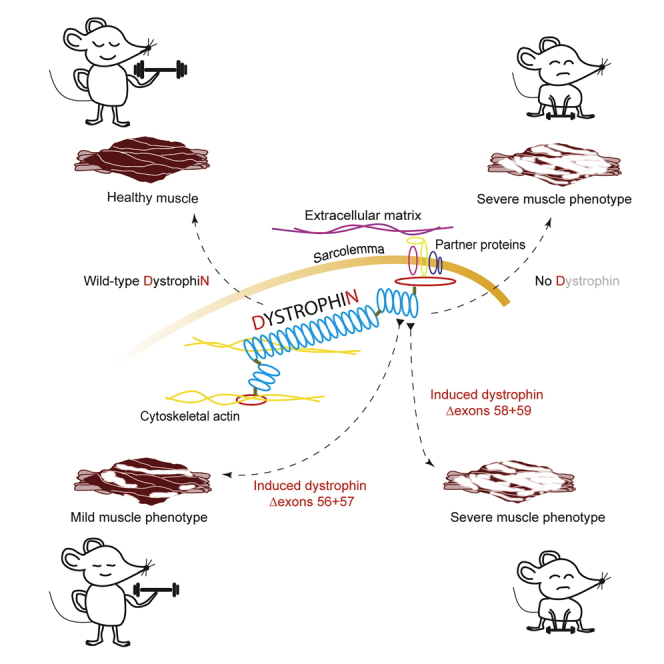
Antisense oligonucleotide-mediated exon skipping is used to induce different dystrophin isoforms, one isoform missing amino acids encoded by DMD exons 56+57 and the other missing exons 58+59. Initial studies indicate that the latter is less functional, suggesting that mutations involving these exons may not respond to an exon skipping therapy.
Introduction
Duchenne muscular dystrophy (DMD) is a devastating inherited neuromuscular disorder affecting 15.9–19.5 cases per 100,000 live male births in the USA and the UK1 and leads to death in the late teens or early 20s.2 This disorder is most commonly caused by protein truncating mutations and subsequent dystrophin deficiency. Although multiple dystrophin isoforms are expressed, it appears that the full-length 427 kDa muscle-specific protein is most significant. Dystrophin has multiple roles including functioning as a molecular shock absorber that stabilizes the sarcolemma during muscle contractions and acting as scaffolding for various signaling pathways such as the neuronal nitric oxide synthase pathway.3,4 Muscle fibers with no functional dystrophin are more prone to mechanical damage that initiates secondary changes in pathophysiological signaling, ultimately causing severe muscle wasting.5 The 79 exons of the DMD gene are spread across nearly 2.3 Mbp, and it is apparent that some exons are not essential for the production of a near-normal, functional dystrophin. Becker muscular dystrophy (BMD) generally arises from genomic deletions in the DMD gene that do not disrupt the dystrophin mRNA reading frame, whereby the internally truncated dystrophin protein retains partial function and can stabilize the sarcolemma.6 The profound phenotype-genotype correlations seen in BMD and DMD and the concept of dispensable exons provide the basis for the development of molecular therapies to modify the dystrophin transcript as a treatment for selected DMD patients with amenable mutations.
Antisense oligonucleotide (AO)-mediated splice-switching has been used to manipulate the splicing of pre-messenger RNAs through targeting splicing motifs to reinforce exon selection, or excise exons that contain nonsense mutations or those that flank frameshifting rearrangements, to produce in-frame transcripts as a potential treatment for several inherited disorders.7 Around 83% of all DMD patients are predicted to respond to exon skipping interventions,8 either through single exon skipping or multiple exon skipping strategies. Eteplirsen, a phosphorodiamidate morpholino oligomer (PMO) designed to induce skipping of DMD exon 51, received accelerated FDA approval as a treatment for the 13%–14% of DMD patients carrying amenable mutations that flank exon 51.9 With the FDA accelerated approvals of Golodirsen in December 201910 and Vitolarsen in August 202011 and the ongoing clinical trial of Casimersen, these antisense drugs could address around 30% of all DMD-causing mutations.
Genomic deletions of one or more dystrophin exons that disrupt the reading frame are the most common type of DMD-causing mutation and two deletion hot-spots have been identified; the minor mutation region encompassing exons 2–20 (18.7% of total deletions) and the major deletion hotspot spanning exons 45–55 (73.2% of all deletions).12,13 However, genomic deletions downstream of DMD exon 55 are rare, and the exonic arrangement is such that most deletions would be expected to disrupt the reading frame and cause premature termination of translation, consistent with a severe phenotype. Consequently, very few BMD mutations have been reported in this region of the gene and there is a lack of phenotype-genotype correlations. Therefore, the applicability of exon skipping strategies to by-pass DMD mutations in this region remains to be determined. Here, we report selected exon skipping strategies to induce in vivo murine models that allow for functional assessment of specific dystrophin isoforms to indicate any therapeutic potential.
Results
Dystrophin Exon Map Downstream of Exon 55 and the Dystrophin-Glycoprotein Complex
Genomic deletions downstream of DMD exon 5514 are not common, and the loss of most single exons in this region would disrupt the reading frame and therefore cause premature termination of translation, as shown in Figure 1A. Potential exon skipping strategies to generate in-frame dystrophin isoforms in this region are shown in Table S1. Figure 1B shows that most of the dystrophin structures encoded by the distal third of the DMD gene are essential for assembly of the dystrophin-glycoprotein complex and contain important domains such as the coiled-coil domains that interact with dystrobrevin, the syntrophin binding domain, the WW domain, the ZZ domain, and the EF-hands for the direct stabilization of β-dystroglycan within the sarcolemma.15
Figure 1.

Dystrophin Exon Map, Downstream Exon 55, and the Dystrophin Glycoprotein Complex
(A) The majority of exons downstream of dystrophin exon 55 are out-of-frame. These exons encode important domains for the assembly of the dystrophin glycoprotein complex. SLR, spectrin-like repeats. (B) The distal third of dystrophin interacts, either directly or indirectly with proteins in the dystrophin glycoprotein complex, including α/β dystroglycans, α/β/γ/δ/ε sarcroglycans, α/β syntrophins, and α/β dystrobrevins to stabilize the sarcolemma. ABD, actin binding domain; H, hinge; DYG, dystroglycan; SYB, syntrophin binding site; CC1 and CC2, coiled coil 1 and 2 (image not to scale).
Induction of Dystrophin Transcripts In Vitro
H2K mdx myogenic cells16 were transfected with peptide-conjugated PMOs (PPMOs) at an equal oligomer ratio for preparations to skip exons 23+58+59 or exons 23+70, and the ratio of compounds to skip exons 23+56+57 was 5:4:6. The PMO sequences are shown in Table S2 and were chosen after in vitro screening outlined in Table S3 and Figure S1. It was considered important to use a dystrophin null model to ensure subsequent analyses were not compromised by traces of wild-type dystrophin. Robust skipping of exon 23, exons 56+57, exons 58+59, and exon 70 was detected by single round RT-PCR (exon 23) or nested RT-PCR, as shown in Figure 2.
Figure 2.

Analysis of Dystrophin Isoforms Induced by PPMO-Mediated Exon Skipping In Vitro
Single-round and nested RT-PCR analysis of DMD transcripts confirming robust skipping of exon 23, exons 56+57, 58+59, and exon 70 in vitro. An RT-PCR no-template negative control was loaded in the final lane. Transcript product size in base pairs (bp) are indicated by 100 bp DNA ladder. (A) The level of exon 23 skipping after individual exon 23 PPMO treatment. (B) Analysis of exon 23 skipping after treatment with PPMO cocktails targeting exons 23+56+57 and 23+58+59. (C) Analysis of exon 23 skipping after exon 23+70 exon skipping PPMO cocktail treatment. (D) Exons 56+57 and 58+59 skipping after PPMO cocktail treatment. (E) Exon 70 skipping after treatment with the AO cocktail targeting exons 23+70. Cocktail 1, a combination of PPMOs targeting exon 23+58+59 skipping; cocktail 2, a combination of PPMOs to skip exons 23+56+57; cocktail 3, a preparation of PPMOs to skip exons 23+70. FL, full-length amplicon; Δ23, exon 23 skipped amplicon; Δ56+57, exons 56+57 excised amplicon; Δ58+59, exons 58+59 removed amplicon; Δ70, exon 70 skipped amplicon; GTC, gene-tools control PPMO; UT, untreated; -Ve, no template negative control.
Induction of Different Dystrophin Isoforms In Vivo
Upon confirmation of PPMO combinations capable of inducing efficient targeted exon skipping in vitro, neonatal C57BL/10ScSn (C57) mice (n = 2 for each treatment group) and C57BL/10ScSnmdx (mdx) mice (n = 5 for each treatment group), 1–2 days old, were treated by intraperitoneal injection with PPMO preparations (20 mg/kg) targeting Dmd exon 23, exons 56+57, exons 58+59, or exon 70 in C57 mice. The mdx model carries a nonsense mutation in exon 23 and excising this exon would allow production of a near-normal dystrophin. PPMO preparations targeting Dmd exon 23, exons 23+56+57, exons 23+58+59, or exons 23+70 were administered to mdx mice, biweekly for 6 weeks. Mice were sacrificed and dissected at 8 weeks of age. Frozen diaphragm cryosections were homogenized and lysed for RNA extraction and protein isolation to analyze dystrophin and β-dystroglycan expression. Four dystrophin isoforms were induced in both mouse strains after the PPMO treatment and characteristics of the induced isoforms are summarized in Table 1. Skipping of the target exons at the RNA level can be seen in Figures 3A–3C. Simultaneous skipping of exons 22+23 was observed after treatment with exon 23-targeting PPMO (Figure 3A), consistent with observations reported previously.17 Excision of Dmd exon 23 in C57 mouse muscle did not significantly alter dystrophin expression (Figure 3D), while the removal of Dmd exon 23 in mdx mice that carry a nonsense mutation restored around 80% of healthy dystrophin levels, relative to sham-treated C57 mice (Figure 3E). The skipping of exons 56+57 in C57 mice and the exclusion of exons 23+56+57 in mdx mice was not as efficient as in the in vitro experiments (Figure 2D). However, an increase in dystrophin expression, analyzed by western blot and immunofluorescence staining, was evident in mdx mice treated with PPMOs targeting exons 23+56+57, compared to normal saline sham-treated mdx mice. Dystrophin expression was similar to that in mdx mice treated with the exon 23-targeting PPMO (Figures 3D–3F). The removal of exons 58+59 or exons 23+58+59 in C57 and mdx mice, respectively was pronounced, as demonstrated by RT-PCR showing the absence of the full-length transcript product (Figure 3B). Skipping exons 58+59 substantially reduced the level of dystrophin protein, as shown by both western blot (Figures 3D and 3E) and sarcolemmal dystrophin immunofluorescence intensity (Figure 3F), and was indistinguishable from mice expressing the out-of-frame dystrophin transcript induced by Dmd exon 70 skipping (Figures 3D–3F).
Table 1.
Characteristics of Induced Dystrophin Isoforms
| Dystrophin Isoforms | Skipped Exons | Dystrophin Protein Level | Dystrophin Immunofluorescent Intensity | β-Dystroglycan Immunofluorescent Intensity | Centrally Nucleated Fibers (Total Fibers Counted) | Fibrosis | Muscle Pathology |
|---|---|---|---|---|---|---|---|
| C57 | |||||||
| Sham-treated | N.A | 100% | 100% | 100% | 0 (538) | 36.10% | – |
| Del23 | 23 | 96.7% | 76.7% | 88.4% | 1.79% (502) | 54.98% | + |
| Del56/57 | 56+57 | 82.3% | 77.4% | 81.0% | 1.84% (489) | 51.33% | + |
| Del58/59 | 58+59 | 27.5% | 48.5% | 71.3% | 25.75% (532) | 80.99% | + + + + |
| Del70 | 70 | 31.8% | 44.1% | 57.4% | 26.85% (517) | 85.41% | + + + + |
| mdx | |||||||
| Sham-treated | N.A | 0 | 30.2% | 56.1% | 28.14% (501) | 100% | + + + + + |
| Del23 | 23 | 81.4% | 79.2% | 78.0% | 6.12% (555) | 63.59% | + + |
| Del23/56/57 | 23+56+57 | 67.5% | 77.3% | 74.5% | 4.60% (717) | 59.9% | + + |
| Del23/58/59 | 23+58+59 | 22.3% | 44.4% | 59.9% | 19.20% (552) | 88.97% | + + + + |
| Del23/70 | 23+70 | 10.1% | 35.8% | 58.2% | 23% (500) | 81.21% | + + + + |
Dystrophin protein level was measured by densitometry and compared to sham-treated C57 (set as 100%) and sham-treated mdx mice (set as 0). Mean immunofluorescent intensity was measured by ImageJ to show the expression level of the dystrophin and β-dystroglycan; immunofluorescence intensity was normalized to sham-treated C57. Total muscle fibers and central nucleated fibers were counted and the percentage of fibers with central nucleation was shown. The degree of fibrosis was determined by ImageJ measuring the intensity of collagen-rich fibrotic regions in blue and was normalized to sham-treated mdx; “+” indicates the severity of muscle pathology; “+ + + + +” indicates the most severe pathology; “–” indicates negative; Del: deletion; N.A., not applicable.
Figure 3.

Analysis of the Expression of Induced Dystrophin Isoforms in Mice Diaphragm after Biweekly, Systemic PPMO Treatment
(A) Single round RT-PCR analysis of the level of exon 23 skipping across different PPMO treatments. (B) Nested RT-PCR analysis of the level of exons 56+57 and 58+59 skipping after PPMO treatment. (C) Analysis of exon 70 skipping by nested RT-PCR in mice treated with PPMOs. (D) Western blot analysis of the expression of dystrophin isoforms and corresponding β-dystroglycan in PPMO treated C57 mouse diaphragm. (E) Analysis of the expression of induced dystrophin isoforms and β-dystroglycan in PPMO treated mdx mouse diaphragm by western blot. An RT-PCR no template negative control was loaded in the final lane. Transcript product size in base pairs (bp) are indicated by a 100 bp DNA ladder. FL, full-length amplicon; Δ22+23, exon 22+23 removed amplicon; Δ23, exon 23 skipped amplicon; Δ56+57, exons 56+57 excised amplicon; Δ58+59, exons 58+59 removed amplicon; Δ70, exon 70 skipped amplicon; -Ve, no template negative control. C57, C57BL/10ScSn; mdx, C57BL/10ScSnmdx, 50% and 10% C57 sham; the total protein concentration of sham-treated C57 are diluted into 50% or 10% and used as controls. The protein expression level was standardized according to alpha-actinin expression. Images were captured by Vilber Lourmat Fusion FX system using Fusion software. (F) Immunofluorescent analysis of the expression of dystrophin isoforms on diaphragm cryosections (scale bar, 50 μm). Sham-treated C57 and mdx mouse diaphragm samples were included for comparison. Images were captured under the Nikon microscope with the NIS-Elements software (Nikon Instruments).
The Interaction between Induced Dystrophin Isoforms and β-Dystroglycan
The presence of β-dystroglycan at the sarcolemma was detected in sham-treated mdx mouse diaphragm cross-sections (Figure 4), as reported elsewhere in mdx mouse skeletal muscles.18,19 Compared to sham-treated C57 mice, the dystrophin isoform lacking exon 23 in C57 mice did not significantly change the abundance of β-dystroglycan, as assessed by immunohistochemical staining, while the removal of exon 23 in mdx mice restored the staining of β-dystroglycan, which is compromised in mdx mice. β-dystroglycan staining at the sarcolemma was reduced in mice expressing the dystrophin variant missing the domain encoded by exons 58+59, which resembled that seen in mice expressing the out-of-frame exon 70-skipped transcript and sham-treated mdx mice. Somewhat improved β-dystroglycan and dystrophin staining was achieved after excising Dmd exons 56+57, as shown in Figures 3F and 4. This observation was confirmed by β-dystroglycan western blotting and comparison to β-dystroglycan levels in diaphragm from sham-treated mdx mice (Figures 3D and 3E).
Figure 4.

Immunofluorescent Analysis of the Expression of β-Dystroglycan
Immunofluorescent analysis of the expression of sarcolemmal β-dystroglycan on diaphragm cryosections (scale bar, 50 μm). β-Dystroglycan was visualized by anti-β-dystroglycan (Santa Cruz) labeled with Zenon Alexa Fluor 488 dye (Thermo Fisher Scientific). Images were captured under the Nikon microscope with the NIS-Elements software (Nikon Instruments). C57, C57BL/10ScSn; mdx, C57BL/10ScSnmdx.
Muscle Pathology after Induction of Dystrophin Isoforms
The exclusion of Dmd exon 23 from mdx mice reversed the dystrophic pathology in diaphragm, including central nucleation, muscle fiber necrosis, mononuclear cell infiltrate, and fibrosis, as shown by hematoxylin and eosin staining in sham-treated mdx mice (Figure 5). However, severe dystrophic pathology was evident in mouse diaphragm cryosections after exclusion of dystrophin exons 58+59 or exon 70 in both C57 and mdx mice (Figure 5). A much lower percentage of central nucleation and degree of muscle fiber necrosis was seen in the diaphragm cryosections from mice expressing the dystrophin variant encoded by the transcript missing exons 56+57, compared to sham-treated mice (Figure 5), suggesting a milder dystrophic pathology. Picro Mallory trichrome staining was also applied to show connective tissue, where intense fibrosis was demonstrated in mouse muscles in which exons 58+59 and exon 70 were skipped. Moderate connective tissue deposition was evident in mdx mouse diaphragm after skipping of exons 23+56+57 (Figure 6). Muscle pathology related to the induced dystrophin isoforms is summarized in Table 1.
Figure 5.

Evaluation of Muscle Pathology by Hematoxylin and Eosin Staining
Diaphragm sections were stained by hematoxylin and eosin to reveal pathogenic changes in muscle architecture (scale bar, 100 μm). Images were captured using a Nikon microscope with the NIS-Elements software (Nikon Instruments). C57, C57BL/10ScSn; mdx, C57BL/10ScSnmdx.
Figure 6.

Analysis of Pathogenic Changes in Muscle by Picro Mallory Trichrome Staining
Mouse diaphragm cryosections were stained by Picro Mallory trichrome to indicate fibrosis (scale bar, 50 μm). Images were captured using a Nikon microscope with the NIS-Elements software (Nikon Instruments). C57, C57BL/10ScSn; mdx, C57BL/10ScSnmdx.
Discussion
Genomic DMD deletions downstream of exon 55 only account for around 7% of all DMD mutations (http://www.treat-nmd.org) and when intra-exonic (nonsense and indel mutations) are included, this number increases to 16%. Personalized medicines to address these genetic variations could benefit a relatively large cohort of DMD patients. However, as this DMD region encodes important protein motifs for the assembly of the dystrophin-glycoprotein complex, developing antisense compounds to skip exons downstream of exon 55 needs to be justified, since removing exons that encode important protein motifs may seriously compromise dystrophin functionality and the induction of a non-functional isoform would be counterproductive. If it can be shown that certain dystrophin isoforms are functional, some exon skipping strategies could be viable therapeutic options for selected DMD patients who have no functional dystrophin.
Most of our exon skipping PPMOs induce robust skipping of targeted exons, as seen in our previous studies, as well as the skipping of exons 56+57 in vitro. The induction of dystrophin isoform missing exons 56+57 in vivo was not as efficient and this was not unexpected and might be influenced by the high GC content (69.5%) of the sequence designed to skip exon 5720,21 or not achieving high enough PPMO levels into the target muscle. Mdx mice were used in the current study to eliminate any confounding effects of residual wild-type dystrophin that may remain after the AO-mediated isoform induction in C57 mice. Notwithstanding the reduced efficiency of the exon 56+57 PPMO preparation, the moderate skipping resulted in higher dystrophin levels, localization of sarcolemmal β-dystroglycan, and milder dystrophic pathology, evident from muscle fiber de/regeneration, central nucleation, and fibrosis analysis. Compared to the highly conserved C-terminal of dystrophin across species, especially the region encoded by exons 68–73 as shown in Figure S2A, viability in the sequences encoded by DMD exons 56+57 (Figure S2B) may indicate a less importance of this region regarding to the biological functions of the protein. Dystrophin phosphorylation is one of the key mechanisms regulating the interaction of dystrophin with the dystrophin-glycoprotein complex.22 Due to the absence of predicted phosphorylation sites in the exons 56+57 coding region (Figure S3; http://www.phosphosite.org), it might be speculated that this part of dystrophin protein is less important in the assembly of the dystrophin-glycoprotein complex. Our data suggest that the in-frame exon block, exons 56+57, are somewhat expendable and have some therapeutic potential of exon skipping to address DMD-causing mutations in these two exons.
Removing in-frame exons or exon blocks does not guarantee the generation of a functional protein. DMD exon 5 is an in-frame exon and encodes the second actin binding site. The skipping of exon 5 results in a phenotype that is more severe than excising exons 3–9, which removes the second and third actin binding sites.23 It was suggested that skipping exon 5 causes spatial disruption of the remaining actin binding sites or even the secondary actin binding region encoded by exons 31–40.23, 24, 25 The skipping of both exons 58 and 59 lead to omission of 130 amino acids immediately before dystrophin hinge 4. Although no known partner protein binding motifs are missing from this particular induced isoform, the dystrophic phenotype appears severe. The dystrophic pathology evident in mouse diaphragm expressing this dystrophin isoform resembled that in sham-treated mdx mice and mice missing out-of-frame exon 70 that eliminates all the dystrophin isoforms. Although skipping exons 58+59 preserves the reading frame, the marked dystrophic pathology may be caused by altered dystrophin tertiary structure. The latter would then interfere with the direct interaction between the induced dystrophin isoform and β-dystroglycan, or other components in the dystrophin-glycoprotein complex that ultimately makes muscle fibers prone to degeneration during contraction. In addition, as exon 56 and a small part of exon 57 encode part of spectrin-like repeat (SLR) 22, most of exon 57, all of exon 58, and some of exon 59 encode SLR 23, and most of exon 59, all of exon 60, and a small part of exon 61 encode SLR 24, we could speculate that fusing part of SLR 23 to part of SLR 24 through skipping exons 58+59 may be less tolerated than fusing part of SLR 22 to part of SLR 23 through skipping exons 56+57.
On the other hand, although missense mutations causing DMD are rare, around 1.5% of all DMD mutations, they are clustered in either the actin binding domain or β-dystroglycan binding domain,26 indicating that small changes in these regions would substantially disrupt dystrophin function. The region encoded by exons 58+59 is adjacent to β-dystroglycan binding domain, thus removing exons 58+59 is speculated to alter the protein tertiary structure to a greater extent than missense mutations and destabilize the binding between dystrophin and β-dystroglycan. As the protein structure change caused by DMD-causing missense mutations in the N-terminal region was reported to decrease its thermodynamic stability and increase dystrophin misfolding,27 one could expect that the stability of this internally truncated dystrophin generated by removing exons 58+59 may be compromised. Subsequently, the misfolded dystrophin isoform undergoes rapid proteasomal degradation,28, 29, 30 leading to decreased dystrophin expression as detected by both the western blot and immunofluorescence.
In summary, we have shown that the dystrophin reading frame rule may not be entirely applicable to exons in the latter third of the dystrophin gene. Large differences in dystrophin expression and localization, and muscle pathology are evident in muscle after in-frame exon 23 skipping and exon 58+59 skipping. Loss of exon 56+57 appears less detrimental, but still results in more severe pathology than only loss of exon 23, hence exon skipping may be hard to justify for these patients. Currently, exon skipping is the only FDA-approved treatment that addresses the primary cause of DMD, with 3 compounds given accelerated approval and an investigational new drug assessment initiated for a fourth (Casimersen to skip exon 45) developed from our group.31 While this is of great significance to potentially 20%–25% of all DMD patients, it is not applicable to those individuals with other mutations. Many compounds addressing other frameshifting exons and in-frame exons within the rod domain have been optimized. These are expected to induce highly functional dystrophin isoforms and will take time and effort to get to the clinic. Those dystrophin isoforms of questionable function will be of lower priority and individuals carrying such mutations are likely to have to rely on mutation-independent therapies such as micro-dystrophin gene replacement therapy, some of which are showing exceptional promise.32
This pilot study mapped functional domains encoded by the first four exons in the distal third of dystrophin gene. Additional studies are needed to confirm the functionality of these induced dystrophin isoforms, for example ex vivo muscle strength testing. The mdx mouse was used in the current study to minimize confounding effects of residual wild-type dystrophin that might remain in C57 mouse muscle; however, the phenotype is less severe in mdx mouse compared to human. Although it is less of an issue in mouse diaphragm,33 the compensatory effects of utrophin to dystrophin may suggest that the mdx mouse is not a perfect model to recapitulate all aspects of DMD. Dystrophin-utrophin double knock-out mice might be an alternative option to characterize functions of the induced isoforms, since these mice exhibit a spectrum of degenerative changes that are similar to DMD patients.34 Making transgenic DMD animal models missing those exon combinations is another strategy to better reflect the dystrophinopathy that characterizes the human disease and could provide stronger evidence to develop exon-skipping therapies for patients with mutations in this region. However, making dozens of specific transgenic animals may not be cost-effective. Additional studies will determine which other DMD exons downstream of exon 55 could be excluded to generate functional dystrophin variations; however, severe phenotypes are expected since this region is the most highly conserved.
Materials and Methods
Antisense Oligomers
Sequences (PMO) targeting exons 23,17,35 56, 57, 58, 59, and 70 are shown in Table S2. The peptide conjugated to the 5′ end of oligomers is HO-(CH2CH2O)3-CO-(pip-PDA), and the peptide conjugate linked to the 3′ end is Ac-RXR RXR RXR RXR XB-OH.36 Peptide synthesis and peptide conjugation to PMOs were kindly supported by Sarepta Therapeutics (Cambridge, MA, USA).
Myogenic Cells and In Vitro Transfection
H-2Kb-tsA58 (H2K) mdx mouse myoblasts16 were seeded at a cell density of 28,600 cells per well in 24-well plates, pre-coated with 50 μg/mL poly-D-lysine (Sigma) and 100 μg/mL matrigel (Becton Dickinson) for 1 h each at 37°C. Cells were allowed to differentiate in 5% horse serum, DMEM for 24 h before PPMO transfection and were collected for RNA extraction 72 h after transfection.
Animals and In Vivo Oligomer Injections
Mice were supplied by the Animal Resources Centre (Murdoch, Australia) and housed according to the National Health and Medical Research Council (Australia) guidelines. The use of animals and all the experiments conducted on the animals were approved by the Animal Ethics Committee of Murdoch University (approval numbers R2625/13 and R2829/16). Neonatal mice were treated by twice-weekly, intraperitoneal injection of PPMOs in normal saline (oligomer dosage: 20 mg/kg) targeting selected exons for 6 weeks. All the mice were sacrificed 2 weeks after the final injection (8 weeks of age).
RNA Extraction and RT-PCR
Total RNA was extracted from tissue cryosections using Magmax-96 Total RNA isolation kit (Thermo Fisher Scientific), according to the manufacturer’s protocol. Single round RT-PCR was performed across exons 20–26 for 35 cycles to detect exon 23 skipping; nested RT-PCR was performed across exons 53–60 or exons 66–77 to analyze the skipping of exons 56, 57, 58, 59, or 70. Primer sequences used to amplify gene segments for exon skipping analysis are shown Table S2.
Western Blotting
Frozen muscle cryosections were homogenized on the day of cryo-sectioning and prepared for western blot as described previously.37 Proteins extracts (7.5 μg total protein for each sample) was loaded onto Criterion 4%–12% precast Bis Tris gradient gels (Bio-Rad) for electrophoresis, staining, and densitometry. The abundance of the myosin heavy chain was used to standardize loading of a second gel that was blotted as previously described.35 Dystrophin was detected using NCL-DYS1 monoclonal anti-dystrophin (Novacastra, Newcastle-upon-Tyne, UK) at a dilution of 1: 1,500 and incubation for 2 h at room temperature. Sarcomeric alpha-actinin, used as a housekeeping protein, was detected with a monoclonal antibody (Sigma A7811) at a dilution of 1:1,500,000. Both monoclonal antisera were visualized using the Western Breeze Chemiluminescent anti-mouse Kit (Invitrogen) according to the manufacturer's recommendations. β-dystroglycan was detected by polyclonal rabbit anti-β-dystroglycan antibody (Santa Cruz, sc-28535) at a dilution of 1:500 and incubation for 2 h at room temperature, followed by horseradish peroxidase (HRP) labeled anti-rabbit immunoglobulins (Dako P0448) secondary antibody for 1 h. Images were captured by Vilber Lourmat Fusion FX system using Fusion software (Vilber Lourmat, Marne-la-Vallee, France), and ImageJ software (NIH, MD, USA) was used for densitometry analysis.
Immunofluorescence and Histology
Dystrophin and β-dystroglycan expression on unfixed cryosections (6 μm) from diaphragm of C57 and mdx mice were detected by polyclonal anti-dystrophin (Abcam 15277) at a dilution of 1:200 and polyclonal anti-β-dystroglycan (Santa Cruz sc-28535) at dilution of 1:300, respectively. Both primary antibodies were detected using Alexa Fluor 488 dye (Thermo Fisher Scientific) at a dilution of 1:400. Hematoxylin and eosin and Picro Mallory trichrome staining protocols were slightly modified from the method described elsewhere.38 Sections were viewed under the Nikon microscope with the NIS-Elements software (Nikon Instruments, South Australia, Australia).
Author Contributions
D.L. designed the antisense oligomers, performed most of the in vitro and in vivo experiments, and drafted the manuscript. A.M.A. coordinated mouse in vivo studies (intraperitoneal injections, dissections, and analyses), and contributed to editing of the manuscript. R.D.J. provided expertise and feedback on muscle sample preparations, western blot and immunofluorescent studies, and revised the manuscript. S.F. and S.D.W. conceived of the study, secured funding, and revised the manuscript. S.F. designed the in vivo studies and performed all injections and dissections. All the authors read and approved the final manuscript.
Conflicts of Interest
S.D.W. and S.F. are consultants to Sarepta Therapeutics. They are named inventors on patents licensed through the University of Western Australia to Sarepta Therapeutics and as such are entitled to milestone and royalty payments.
Acknowledgments
This work was supported by funding to S.D.W. and S.F. from the National Health and Medical Research Council (grant number 1144791). D.L. receives a postgraduate scholarship from Muscular Dystrophy Western Australia. The authors thank Sarepta Therapeutics for generously providing the PPMOs. This work was conducted in Perth, Australia.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.08.019.
Supplemental Information
References
- 1.Birnkrant D.J., Bushby K., Bann C.M., Apkon S.D., Blackwell A., Brumbaugh D., Case L.E., Clemens P.R., Hadjiyannakis S., Pandya S., DMD Care Considerations Working Group Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17:251–267. doi: 10.1016/S1474-4422(18)30024-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Ruiten H., Bushby K., Guglieri M. State of the art advances in Duchenne muscular dystrophy. EMJ. 2017;2:90–99. [Google Scholar]
- 3.Le S., Yu M., Hovan L., Zhao Z., Ervasti J., Yan J. Dystrophin As a Molecular Shock Absorber. ACS Nano. 2018;12:12140–12148. doi: 10.1021/acsnano.8b05721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nichols B., Takeda S., Yokota T. Nonmechanical Roles of Dystrophin and Associated Proteins in Exercise, Neuromuscular Junctions, and Brains. Brain Sci. 2015;5:275–298. doi: 10.3390/brainsci5030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deconinck N., Dan B. Pathophysiology of duchenne muscular dystrophy: current hypotheses. Pediatr. Neurol. 2007;36:1–7. doi: 10.1016/j.pediatrneurol.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 6.Muntoni F., Torelli S., Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2:731–740. doi: 10.1016/s1474-4422(03)00585-4. [DOI] [PubMed] [Google Scholar]
- 7.Li D., Mastaglia F.L., Fletcher S., Wilton S.D. Precision medicine through antisense oligonucleotide-mediated exon skipping. Trends Pharmacol. Sci. 2018;39:982–994. doi: 10.1016/j.tips.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 8.Aartsma-Rus A., Fokkema I., Verschuuren J., Ginjaar I., van Deutekom J., van Ommen G.J., den Dunnen J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009;30:293–299. doi: 10.1002/humu.20918. [DOI] [PubMed] [Google Scholar]
- 9.Lim K.R.Q., Maruyama R., Yokota T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Devel. Ther. 2017;11:533–545. doi: 10.2147/DDDT.S97635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drug, USFa . 2019. FDA grants accelerated approval to first targeted treatment for rare Duchenne muscular dystrophy mutation.https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation [Google Scholar]
- 11.DRUG USF . 2020. FDA Approves Targeted Treatment for Rare Duchenne Muscular Dystrophy Mutation.https://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation [Google Scholar]
- 12.Bladen C.L., Salgado D., Monges S., Foncuberta M.E., Kekou K., Kosma K., Dawkins H., Lamont L., Roy A.J., Chamova T. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015;36:395–402. doi: 10.1002/humu.22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Juan-Mateu J., Gonzalez-Quereda L., Rodriguez M.J., Baena M., Verdura E., Nascimento A., Ortez C., Baiget M., Gallano P. DMD mutations in 576 dystrophinopathy families: a step forward in genotype-phenotype correlations. PLoS ONE. 2015;10:e0135189. doi: 10.1371/journal.pone.0135189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prior T.W., Bartolo C., Pearl D.K., Papp A.C., Snyder P.J., Sedra M.S., Burghes A.H., Mendell J.R. Spectrum of small mutations in the dystrophin coding region. Am. J. Hum. Genet. 1995;57:22–33. [PMC free article] [PubMed] [Google Scholar]
- 15.Michele D.E., Campbell K.P. Dystrophin-glycoprotein complex: post-translational processing and dystroglycan function. J. Biol. Chem. 2003;278:15457–15460. doi: 10.1074/jbc.R200031200. [DOI] [PubMed] [Google Scholar]
- 16.Morgan J.E., Beauchamp J.R., Pagel C.N., Peckham M., Ataliotis P., Jat P.S., Noble M.D., Farmer K., Partridge T.A. Myogenic cell lines derived from transgenic mice carrying a thermolabile T antigen: a model system for the derivation of tissue-specific and mutation-specific cell lines. Dev. Biol. 1994;162:486–498. doi: 10.1006/dbio.1994.1103. [DOI] [PubMed] [Google Scholar]
- 17.Gebski B.L., Mann C.J., Fletcher S., Wilton S.D. Morpholino antisense oligonucleotide induced dystrophin exon 23 skipping in mdx mouse muscle. Hum. Mol. Genet. 2003;12:1801–1811. doi: 10.1093/hmg/ddg196. [DOI] [PubMed] [Google Scholar]
- 18.Johnson E.K., Li B., Yoon J.H., Flanigan K.M., Martin P.T., Ervasti J., Montanaro F. Identification of new dystroglycan complexes in skeletal muscle. PLoS ONE. 2013;8:e73224. doi: 10.1371/journal.pone.0073224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han R., Rader E.P., Levy J.R., Bansal D., Campbell K.P. Dystrophin deficiency exacerbates skeletal muscle pathology in dysferlin-null mice. Skelet. Muscle. 2011;1:35. doi: 10.1186/2044-5040-1-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan J.H., Lim S., Wong W.S. Antisense oligonucleotides: from design to therapeutic application. Clin. Exp. Pharmacol. Physiol. 2006;33:533–540. doi: 10.1111/j.1440-1681.2006.04403.x. [DOI] [PubMed] [Google Scholar]
- 21.Shao Y., Wu Y., Chan C.Y., McDonough K., Ding Y. Rational design and rapid screening of antisense oligonucleotides for prokaryotic gene modulation. Nucleic Acids Res. 2006;34:5660–5669. doi: 10.1093/nar/gkl715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swiderski K., Shaffer S.A., Gallis B., Odom G.L., Arnett A.L., Scott Edgar J., Baum D.M., Chee A., Naim T., Gregorevic P. Phosphorylation within the cysteine-rich region of dystrophin enhances its association with β-dystroglycan and identifies a potential novel therapeutic target for skeletal muscle wasting. Hum. Mol. Genet. 2014;23:6697–6711. doi: 10.1093/hmg/ddu388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toh Z.Y., Thandar Aung-Htut M., Pinniger G., Adams A.M., Krishnaswarmy S., Wong B.L., Fletcher S., Wilton S.D. Deletion of dystrophin in-frame exon 5 leads to a severe phenotype: guidance for exon skipping strategies. PLoS ONE. 2016;11:e0145620. doi: 10.1371/journal.pone.0145620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rybakova I.N., Amann K.J., Ervasti J.M. A new model for the interaction of dystrophin with F-actin. J. Cell Biol. 1996;135:661–672. doi: 10.1083/jcb.135.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amann K.J., Renley B.A., Ervasti J.M. A cluster of basic repeats in the dystrophin rod domain binds F-actin through an electrostatic interaction. J. Biol. Chem. 1998;273:28419–28423. doi: 10.1074/jbc.273.43.28419. [DOI] [PubMed] [Google Scholar]
- 26.Tuffery-Giraud S., Béroud C., Leturcq F., Yaou R.B., Hamroun D., Michel-Calemard L., Moizard M.P., Bernard R., Cossée M., Boisseau P. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum. Mutat. 2009;30:934–945. doi: 10.1002/humu.20976. [DOI] [PubMed] [Google Scholar]
- 27.Singh S.M., Kongari N., Cabello-Villegas J., Mallela K.M. Missense mutations in dystrophin that trigger muscular dystrophy decrease protein stability and lead to cross-β aggregates. Proc. Natl. Acad. Sci. USA. 2010;107:15069–15074. doi: 10.1073/pnas.1008818107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Talsness D.M., Belanto J.J., Ervasti J.M. Disease-proportional proteasomal degradation of missense dystrophins. Proc. Natl. Acad. Sci. USA. 2015;112:12414–12419. doi: 10.1073/pnas.1508755112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ciechanover A., Kwon Y.T. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp. Mol. Med. 2015;47:e147. doi: 10.1038/emm.2014.117. e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amm I., Sommer T., Wolf D.H. Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim. Biophys. Acta. 2014;1843:182–196. doi: 10.1016/j.bbamcr.2013.06.031. [DOI] [PubMed] [Google Scholar]
- 31.Drugs.com . 2020. Sarepta Therapeutics Completes Submission of New Drug Application Seeking Approval of Casimersen (SRP-4045) for Patients with Duchenne Muscular Dystrophy Amenable to Skipping Exon 45.https://www.drugs.com/nda/casimersen_200626.html [Google Scholar]
- 32.Duan D. Micro-Dystrophin Gene Therapy Goes Systemic in Duchenne Muscular Dystrophy Patients. Hum. Gene Ther. 2018;29:733–736. doi: 10.1089/hum.2018.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stedman H.H., Sweeney H.L., Shrager J.B., Maguire H.C., Panettieri R.A., Petrof B., Narusawa M., Leferovich J.M., Sladky J.T., Kelly A.M. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352:536–539. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- 34.Isaac C., Wright A., Usas A., Li H., Tang Y., Mu X., Greco N., Dong Q., Vo N., Kang J. Dystrophin and utrophin “double knockout” dystrophic mice exhibit a spectrum of degenerative musculoskeletal abnormalities. J. Orthop. Res. 2013;31:343–349. doi: 10.1002/jor.22236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fletcher S., Honeyman K., Fall A.M., Harding P.L., Johnsen R.D., Steinhaus J.P., Moulton H.M., Iversen P.L., Wilton S.D. Morpholino oligomer-mediated exon skipping averts the onset of dystrophic pathology in the mdx mouse. Mol. Ther. 2007;15:1587–1592. doi: 10.1038/sj.mt.6300245. [DOI] [PubMed] [Google Scholar]
- 36.Iversen P.L., Weller D.D., Hassinger J.N. Google Patents; 2014. Peptide conjugated, inosine-substituted antisense oligomer compound and method. [Google Scholar]
- 37.Fletcher S., Honeyman K., Fall A.M., Harding P.L., Johnsen R.D., Wilton S.D. Dystrophin expression in the mdx mouse after localised and systemic administration of a morpholino antisense oligonucleotide. J. Gene Med. 2006;8:207–216. doi: 10.1002/jgm.838. [DOI] [PubMed] [Google Scholar]
- 38.Carleton H.M. Carleton’s Histological Technique. In: Drury R.A.B., Wallington E.A., editors. 1967. New York. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.