

Abstract
The immune system is armed with a broad range of receptors to detect and initiate the elimination of bacterial pathogens. Inflammasomes are molecular platforms that sense a diverse range of microbial insults to develop appropriate host response. In that context, non-canonical inflammasome arose as a sensor for Gram-negative bacteria derived LPS leading to the control of infections. This review describes the role of caspase-11/gasdermin-D-dependent immune response against Gram-negative bacteria and presents an overview of guanylate-binding proteins (GBPs) at the interface of non-canonical inflammasome activation. Indeed, caspase-11 acts as a receptor for LPS and this interaction elicits caspase-11 auto-proteolysis that is required for its optimal catalytic activity. Gasdermin-D is cleaved by activated caspase-11 generating an N-terminal domain that is inserted into the plasmatic membrane to form pores that induce pyroptosis, a cell death program involved in intracellular bacteria elimination. This mechanism also promotes IL-1β release and potassium efflux that connects caspase-11 to NLRP3 activation. Furthermore, GBPs display many features to allow LPS recognition by caspase-11, initiating the non-canonical inflammasome response prompting the immune system to control bacterial infections. In this review, we discuss the recent findings and nuances related to this mechanism and its biological functions.
Keywords: Caspase-11, Gasdermin-D, K+ efflux, NLRP3
Summary sentence:
Review on the role of GBPs as major players on non-canonical inflammasome activation
1. Introduction
The innate immune system is responsible for the first line of defense against bacterial infections [1]. At the molecular level, bacterial pathogens usually display an intricate molecular signature known as pathogen-associated molecular patterns (PAMPs). The most common PAMPs found in bacteria are lipopolysaccharide (LPS), peptidoglycan, lipoproteins, flagellin and nucleic acids [2]. Similarly to non-microbial (sterile) stressful insults, bacterial infection can also indirectly lead to physiological disturbance inducing host-derived damage-associated molecular patterns (DAMPs) [3]. The host can interact with this panoply of PAMPs and DAMPs using its own molecular sentinels called pattern-recognition receptors (PRRs) [4]. Host germ-line PRRs are located at extracellular membranes, intracellular vesicles/organelles or cytosolic space. Upon PAMPs recognition, PRRs activate multiple molecular cascades responsible to change gene expression profile leading to an efficient response against infection [5].
At the vanguard of PRRs are the Toll-like receptors (TLRs) discovered in the middle of the 1990s [6, 7]. Afterwards, several classes of cytosolic PRRs, including nucleic acid sensors RIG-I-like receptors (RLRs) [8], cGAS/STING pathway [9] and Nod-like receptors (NLRs) [10] were identified. In that context, inflammasomes have emerged as a group of PRRs acting as cytosolic multi-molecular platforms which trigger the activation of inflammatory caspases producing interleukin (IL)-1β and IL-18 [11]. The majority of the inflammasomes are NLRs, such as NLRP1 [Nod- leucine-rich repeat (LRR) and pyrin domain-containing 1], NLRP3, NLRP6, NLRP7, NLRP12 and NLRC4 (Nod- LRR and caspase recruitment domain (CARD)-containing 4) [12]. Additionally, a non-NLR termed AIM2 (absent in melanoma 2) which senses cytosolic DNA was described as a major inflammasome [13].
Inflammasome activation is triggered by a variety of factors that emerge during bacterial infection, such as cellular damage or homeostatic imbalance. For instance, while AIM2 inflammasome is activated by cytoplasmic DNA [14], NLRP3 can be activated by K+ efflux [15], mitochondrial reactive oxygen species (ROS) [16], lysosomal rupture [17] and oxidized mitochondrial DNA (mtDNA) [18]. Two signals are required for the production of IL-1β and IL-18. The signal 1 or priming signal regulates the expression of inflammasome components and both pro-IL-1β and pro-IL-18 synthesis. The signal usually initiates after PRRs recognition of PAMPs leading to nuclear factor (NF)-κB pathway activation. Signal 2 promotes inflammasome assembly and activation [19]. Once activated, inflammasomes recruit apoptosis-associated speck-like protein containing a CARD (PYCARD, also known as ASC). ASC acts as a molecular bridge between activated inflammasome sensors to the effector protein pro-caspase-1. Recruitment of pro-caspase-1 leads to its autocatalytic processing into caspase-1, which enzymatically converts the zymogenic forms of the pro-inflammatory cytokines pro-IL-1β and pro-IL-18 into their mature and biologically active forms [20]. In several occasions the activation of caspase-1 drives the secretion of pro-inflammatory cytokines and promotes a programmed cell death distinguished from the non-inflammatory apoptosis. Therefore, the term pyroptosis was suggested to describe this caspase-1-dependent cell death displaying pro-inflammatory features [21].
In the world of inflammatory caspases, caspase-11 used to live in the shadow of caspase-1. Initial mouse line genetically deficient in caspase-1 was also deficient in caspase-11, due to the close proximity of both caspases loci which prevented gene segregation during recombination. Thus, inflammasome data were to some extent misinterpreted [22]. After the genetic construction of caspase-11 deficient mice, the role of this protein in the triggering of a caspase-1-independent macrophage death was described [23]. In addition, under certain stimuli referred as non-canonical activators, caspase-11 promotes caspase-1-dependent IL-1β and IL-18 secretion. This novel form of inflammasome activation was entitled the non-canonical inflammasome to distinguish itself from the traditional canonical inflammasome activation [23]. The mechanism of cell death induced by the non-canonical inflammasome gained new insight when it was demonstrated that gasdermin D (GSDMD) is cleaved by caspase-11 resulting in a N-terminal fragment that induces pyroptosis [24]. In addition, the identification of LPS as a ligand for caspase-11 [25] and the description of the role of guanylate-binding proteins (GBPs) in non-canonical inflammasome activation shed light on this intricate pathway [26]. Here, we discuss how caspase-11 and GSDMD are activated and their role in bacterial infections. We also present an overview of how GBPs work at the interface of non-canonical inflammasome activation.
2. Caspase 11: the non-canonical inflammasome receptor
The classical mechanism of caspase‐1 activation mediated by NLRP3, NLRC4 and AIM2 was referred as the canonical inflammasome pathway. Caspase-11 arose as a member in the NLRP3 inflammasome pathway when macrophages derived from caspase-11−/− mice failed to secrete IL-1β when infected with live Gram‐negative bacteria Escherichia coli, Citrobacter rodentium or Vibrio cholera, leading to the identification of a non-canonical inflammasome pathway and its activators [23]. However, caspase-11 was dispensable for NLRP3-dependent IL-1β secretion in response to monosodium urate (MSU) or the ionophore nigericina, known as canonical inflammasome activators. In contrast, under non-canonical activators stimulation, caspase-11, rather than NLRP3, promoted the release of pyroptotic markers such as high mobility group box 1 (HMGB1), IL-1α and lactate dehydrogenase (LDH). Thus, pro-inflammatory caspase-11 activates caspase-1-independent cell death and caspase-1-dependent IL-1β secretion in response to non-canonical ligands. Moreover, caspase-11 deficiency protected mice from a lethal dose of LPS emphasizing a role for caspase-11 in response to clinically significant bacterial infections [23].
Later, the role of caspase-11 sensing cytoplasmic LPS and leading to septic shock was established. The lethal sepsis occurred bypassing the need for TLR4, the cell-surface LPS receptor [27, 28]. Afterwards, binding assays confirmed that caspase-11 and its human homologue caspase-4 bound to LPS with high specificity and affinity [25]. At the molecular level, the interaction between cytoplasmic LPS and the CARD domain of caspase-11 is crucial for non-canonical inflammasome activation. This interaction elicits caspase-11 auto-proteolysis which is required for its optimal catalytic activity, leading to pyroptosis and IL-1β release [29]. Recent data demonstrated that both caspase-11 and its human homologues caspase-4/5 bound hexa-acylated lipid A, the lipid portion of LPS. Remarkably, the under-acylated (tetra-acylated) lipid A of the Gram-negative bacterium Francisella novicida escaped caspase-11 recognition in mice but was sensed by caspase-4 in humans [30]. Thus, caspase-4 and caspase-11 display distinct specificity for LPS. In fact, CARD domains of caspase-11 and caspase-4 shares 51% identity, which may explain the broader sensitivity of caspase-4 in sensing LPS compared to caspase-11. In addition, it was demonstrated that GBP2 promotes efficient sensing of under-acylated LPS by caspase-4, although GBP2 was dispensable for hexa-acylated LPS recognition [30]. In this case, GBP2 might act as human-specific co-factor assisting under-acylated LPS detection by caspase-4 [30]. Taken together, the acylation state of lipid A may be used to predict the level of non-canonical inflammasome activation. For instance, E. coli and Shigella flexneri contain hexa-acylated lipid A that strongly activates caspase-11 [31]. In addition, hexa-acylated lipid A derived from Yersinia pestis grown at 25°C is detected by caspase-11, in contrast with tetra-acylated lipid A from pathogen grown at 37°C. Thus, this bacterium may exploit temperature-dependent lipid A acylation state in order to escape caspase-11 recognition during infection in the host [27].
The activation of non-canonical inflammasome prompts host immune system to control bacterial infections in different scenarios (Table 1). For instance, Klebsiella pneumoniae, a Gram-negative bacterium, induces a caspase-11-dependent pyroptosis in bone-marrow-derived macrophages (BMDMs), leading to IL-1α and IL-1β secretion. Indeed, caspase-11−/− mice showed impaired neutrophil recruitment and bacterial clearance during K. pneumoniae infection, accompanied by a reduction in IL-1α production, a critical cytokine for the recruitment of neutrophil in this model. These results suggest that pyroptosis leads to IL-1α secretion that recruits neutrophils to control K. pneumoniae infection [32]. Moreover, the pathogenic bacteria Salmonella enterica serovar Typhimurium colonize the intestinal epithelium of caspase-11-deficient mice more efficiently than wild-type animals. The increased pathogen burden was correlated with the lack of pyroptotic cell death, a host defense mechanism that assists infected cells extrusion in an attempt to restore gut homeostasis [33]. In addition, a role for caspase-11 controlling bacterial infection was determined for Acinetobacter baumannii, a pathogenic bacterium that can cause severe pulmonary infection. In that case, caspase-11 deficiency augmented bacterial burden in lungs, spleen and liver [34]. Similarly, caspase-11 controlled Brucella melitensis joint infection and exacerbated joint inflammatory response against this pathogen [35]. Moreover, recent data revealed that during B. abortus systemic infection, caspase-11−/− mice are more susceptible compared to wild-type animals and they recruited fewer immune cells such as neutrophils, macrophages and dendritic cells (DCs) in mouse spleens [36]. Additionally, Gram-negative bacteria such as Burkholderia pseudomallei and B. thailandensis, that naturally invade the cytosol, are targeted by caspase-11. Indeed, caspase-11 protected mice from lethal Burkholderia species challenge [37].
Table 1.
Caspase-11-dependent host protection during bacterial infections in mice.
| Bacteria | Bacterial number and via of infection | Caspase-11−/− mice phenotype compared to wild-type animals |
|---|---|---|
| Acinetobacter baumannii | 2 × 108 intranasal | + liver, lung, spleen and BALF [34] |
| Brucella abortus | 1 × 106 intraperitoneal | + spleen [36] |
| Brucella melitensis | 1×105 in each rear footpad | + joint [35] |
| Burkholderia thailandensis | 2×107 intraperitoneal | increased mortality [37] |
| Burkholderia pseudomallei | 100 intranasal | increased mortality [37] |
| Klebsiella pneumoniae | 2 × 107 intranasal | + lung and BALF [32] |
| Salmonella Typhimurium | 3 × 106 oral |
= spleen, liver and mesenteric lymph nodes; + cecal tissues and intestinal lumen [33] |
| Legionella pneumophila | 105 intranasal | = lung [108] |
| Legionella gratiana | 105 intranasal | = lung [108] |
increased bacterial burden;
no difference in bacterial burden.
The activation of caspase-11 is not restricted to the recognition of LPS. Recent data demonstrated that caspase-11 is crucial for controlling Leishmania infection [38]. The activation of non-canonical inflammasome was attributed to the membrane molecule termed lipophosphoglycan, formed by a lipid part attached to a polysaccharide moiety, which envelops Leishmania species. Moreover, parasites lacking lipophosphoglycan are unable to trigger caspase-11 activation. Intriguingly, cell-free binding assays failed to show lipophosphoglycan direct interaction with caspase-11, implying that additional cytoplasmic molecules might participate in this process [38]. On the other hand, under oxidative stress that might be generated during microbial infections, endogenous oxidized phospholipids formed, such as 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine (oxPAPC) [39], can bind to caspase-11 catalytic domain inhibiting its enzymatic activity [40]. Moreover, oxPAPC-caspase-11 interaction mediates IL-1β secretion but does not promote pyroptosis in DCs [40]. Thus, caspase-11 proteolytic activity is required to induce cell death, but not IL-1β release from DC. A model in which oxPAPC may induce the formation of a caspase-1/11 hetero-complex leading to IL-1β processing and secretion was proposed [40]. Furthermore, oxPAPC appeared as a caspase-11 regulator in macrophages, but not in DCs. Caspase-11 binds to oxPAPC competing with cytoplasmic LPS, thus abrogating LPS-induced pyroptosis and IL-1β release [41]. Hence, oxPAPC may modulate the non-canonical inflammasome offering a novel therapeutic approach against Gram-negative bacteria-induced septic shock.
3. GBPs: Revealing LPS to caspase-11
As previously described, several non-cytosolic Gram-negative bacteria trigger caspase-11 activation [23, 28, 42, 43], raising the question of how LPS from these bacteria gain access to the cytosol.
Interestingly, during infection, caspase-11 activation requires the production of type-I interferons (IFN) as well as the induction of IFN-stimulated genes [43, 44]. Among the most strongly upregulated IFN-stimulated genes, the GBPs, a class of guanosine 5′-triphosphatases (GTPases) spanning 65 to 73 kDa, are prominent [42, 45, 46]. GBPs are extremely conserved among vertebrates and seven human GBPs genes (GBP1-GBP7) located within a single cluster on chromosome 1 have been identified [47]. Genes encoding murine GBPs, mGBP1, mGBP2, mGBP3, mGBP5, and mGBP7 are clustered on chromosome 3 (GBPchr3), whereas genes encoding mGBP4, mGBP6, mGBP8, mGBP9, mGBP10, and mGBP11 are located on chromosome 5 [45, 48–50]. GBPs are primarily found in the cytoplasm, associated with intracellular membranes, within vesicle‐like structures or in the nucleus [50–53], and exhibit anti-microbial effects against intracellular bacteria, viruses and protozoa [45, 54, 55].
GBPs-dependent effects on non-canonical inflammasome activation have been shown in several vacuolar and cytosolic bacterial pathogens, including S. typhimurium, Legionella pneumophila, V. cholerae, S. flexneri, C. rodentium, Chlamydia trachomatis, Chlamydia muridarum and type three secretion system (T3SS)-negative Pseudomonas aeruginosa [26, 42, 44, 56, 57]. For instance, mice macrophages deficient in GBPchr3 showed reduced levels of non-canonical inflammasome activation and pyroptosis during infection with S. typhimurium and L. pneumophila [42, 56, 58].
Mechanistically, GBPs may induce inflammasome assembly directly or indirectly. Directly, GBPs may physically interact with inflammasome machinery, facilitating its assembly in both canonical and non-canonical pathways [46, 59]. Indirectly, GBPs might promote lysis of pathogen-containing vacuoles (PCVs), thereby destroying the microbial niche and exposing bacterial LPS for downstream detection by caspase-11 in the cytosol. Indeed, an experiment demonstrated that caspase-11 activation and cell death induced by S. typhimurium required GBPchr3 proteins expression confirming the early role of GBPs in the activation of non-canonical inflammasome [42]. The mechanisms by which GBPs detect and bind to PCVs remain poorly characterized; however, in murine cells infected with Yersinia or Legionella, GBP recruitment to the PCVs seems to be linked to the vacuolar damage sensor galectin-3. In this case, bacterial secretion systems insertion into PCVs destabilizes its membrane structure exposing luminal side to host cytosolic proteins. Indeed, the cytosolic exposure of intravacuolar host glycans derived from vacuolar damage drives the recruitment of galectin-3 which targets mGBP2 to PCVs in a coordinate immune response [60].
Murine macrophages infected with B. abortus present mGBP2 aggregates located close to the PCVs. Moreover, a decreased number of disrupted PCVs was observed in cells deficient in GBPchr3 when compared to wild-type cells [61], suggesting that GBPs orchestrate host surveillance during B. abortus infection. Besides, GBPchr3 machinery is essential for B. abortus induction of non-canonical inflammasome activation and pyroptosis in BMDMs [36]. Further experiments established that among the GBPs contained in mouse chromosome 3, mGBP5 is the most relevant for the recognition of Brucella LPS by caspase-11, triggering BMDM pore formation and IL-1β secretion [36]. Collectively, these results suggest that the GBPs are central to target and disrupt the PCV revealing B. abortus LPS to caspase-11 [36].
Furthermore, induction of caspase 11-dependent pyroptosis by cytoplasmic LPS derived from L. pneumophila likewise required GBPchr3 proteins. Similarly, pyroptosis in macrophages infected with a cytosol-invading L. pneumophilia mutant was also dependent on GBPchr3 proteins, supporting a role for GBPs in the detection of cytoplasmic LPS and/or in the subsequent activation of non-canonical inflammasome [56]. However, cytoplasmic LPS derived from E. coli and Salmonella triggered pyroptosis even in the absence of GBPchr3 proteins, although with a slight reduced efficiency. These observations may potentially be explained by structural differences in the lipid A moiety of the LPS variants derived from these distinct bacterial species [56].
Moreover, a following study suggested that GBPs were not only involved in vacuolar damage, but also in direct bacterial lysis (bacteriolysis) [62]. Indeed, emerging evidences have shown that cytoplasmic mGBP2 and mGBP5 interact with F. novicida membrane after its release from the PCVs and promote cytosolic lysis of this pathogen [63]. Accordingly, GBPs recruit the immunity-related GTPase family member b10 (IRGB10) that directly targets intracellular bacteria promoting bacteriolysis. Hence, IRGB10 contributes to the release of cytosolic bacterial PAMPs for recognition by inflammasome sensors: LPS sensing by caspase-11 and DNA sensing by AIM2 inflammasome. [64]. Interestingly, GBPs mediated-bacteriolysis of cytosolic F. novicida leads to the release of bacterial genomic DNA into the cytosol which activates both canonical and non-canonical inflammasome [63, 65]. It is unclear whether the caspase-11-dependent activation observed is due to sensing of F. novicida LPS or another ligand, since caspase-11 does not recognize the under-acylated Francisella LPS [27]. Notably, T3SS-negative P. aeruginosa proliferates in cytoplasm avoiding NLRP3 and NLRC4 detection, but is recognized by mGBP2 and IRGB10. Interestingly, only mGBP2 promotes bacterial killing, while both mGBP2 and IRGB10 enables caspase-11 activation leading to host cell death. Thus, during T3SS-negative P. aeruginosa infection, allegedly bacterial killing and exposure of bacterial ligands are two uncouple processes [26]. Taken together, GBPs are crucial not only during vacuolar pathogens infections but also in presenting concealed ligands by directly targeting cytosolic bacteria. Besides this lytic function displayed by murine GBPs over targeted pathogens, human GBPs mark and restrict bacteria spread by a different mechanism. The human GBP1 recognizes the O-antigen present at the S. flexneri LPS and promotes the corecruitment of four additional GBP paralogs (GBP2, GBP3, GBP4, and GBP6). GBP1 displays a specific polybasic protein motif which contains a triple-arginine essential for the recognition of S. flexneri. The GBP1-decorated S. flexneri lack the ability to form actin tails and hence are nonmotile, which avoids their cell-to-cell spread [66, 67]. Interestingly, the ubiquitin ligase IpaH9.8 secreted by the Shigella type 3 secretion system interferes with GBP1 targeting to cytosolic bacteria. Indeed, IpaH9.8 ubiquitylates human GBPs for proteasomal degradation and hence restores actin-mediated motility promoting bacterial spreading [66, 67].
Additional mechanisms of antimicrobial activity are also related to GBPs. Gram-negative bacteria produce and release outer membrane vesicles (OMVs) during infection that are internalized by macrophages and function as a vehicle that delivers LPS (and several other surface antigens) into the host citosol. LPS delivered by OMVs can trigger caspase-11 promoting non-canonical inflammasome activation, playing a center role in host defense and bacterial pathogenesis [68, 69]. In addition, the OMVs from Gram-negative bacteria E. coli, Pseudomonas, Salmonella and Shigella activated the non-canonical inflammasome in a GBPchr3-dependent manner. In that context, GBPs may disrupt OMV and therefore release LPS to engage caspase-11 activation [70]. Indeed, due to the hydrophobic nature of lipid A moiety of LPS, GBPs may facilitate caspase-11 binding to intracellular LPS itself and also LPS from experimental transfected liposomal membranes [70]. Accordingly, our recent data support the idea that mGBP5 contributes to the recognition of B. abortus LPS incorporated within liposomal membranes by caspase-11 [36].
4. Gasdermin: The executor of pyroptosis
As described above, caspase-11 triggers the non-canonical inflammasome pathway leading to pyroptotic cell death in response to intracellular LPS independently of NLRP3/ASC/caspase-1 [23, 27, 28]. However, the exact mechanism involved in pyroptosis remained unclear until the identification of gasdermin-D (GSDMD), a substrate of caspase-1 and caspase-11 [71, 72]. GSDMD belongs to a family of proteins first identified in the gastrointestinal tract and dermis, therefore it was named “gas-dermin” [73, 74]. These molecules are expressed in a variety of cell types and tissues and encompass a group of six proteins in humans and ten proteins in mice [75]. All GSDMs, except Pejvakin (PJVK), can be potentially cleaved by caspases, triggering oligomerization and membrane pore formation. This mechanism is well known for GSDMD and GSDME (DFNA5). GSDME is cleaved by the apoptotic caspase-3 which releases the N-terminal domain that targets the plasma membrane, leading to a secondary necrosis process [76]. GSDMD is cleaved by caspase-1 and caspase-11 generating a C-terminal (p20) and an N-terminal active domain (p30) [71, 72, 76]. In addition, the inhibition of transforming growth factor β-activated kinase 1 (TAK1) activity elicits caspase-8 cleavage of GSDMD similarly to caspase-1 and caspase-11 [77, 78].
Once GSDMD cleavage occurs, the N-terminal domain is inserted into the plasmatic membrane and oligomerizes forming 10–16 nm-diameter pores [79, 80]. The efflux of potassium can occur through these pores triggering NLRP3/ASC inflammasome activation [15, 81–85]. In addition, smaller diameter substrates like mature IL-1β [86, 87], IL-18 and inflammatory mediators such as eicosanoids can be released through the pores, leading to neutrophil recruitment to the site of infection [88]. For instance, Gsdmd−/− mice are more susceptible to B. abortus infection and show impaired neutrophil recruitment to spleen. Interestingly, the levels of CD62L on neutrophils were higher in Gsdmd−/− infected animals compared to wild-type, what is related to less activated neutrophils. In addition, depletion of neutrophils in wild-type mice increases susceptibility to bacterial infection, indicating a prominent role of GSDMD in B. abortus infection restriction [36].
Moreover, GSDMD-dependent pore formation renders water influx that causes an osmotic imbalance which might culminate in cell rupture and death [89]. However, novel data demonstrated that, at some extension, cells can repair membrane pores to regulate pyroptosis and cytokine release [87, 90]. This process is coordinated by the endosomal sorting complexes required for transport (ESCRT) machinery that removes pores shedding bud vesicles, known as ectosomes, from the plasma membrane to the extracellular space [91–93]. ESCRT acts downstream of GSDMD dependently on Ca2+ influx through GSMDM-membrane pores, which triggers the recruitment of ESCRT proteins to damaged membrane sites [90].
Since the cleavage of GSDMD is executed by caspase-1 or caspase-11, both canonical and non-canonical inflammasomes can trigger GSDMD-dependent pyroptosis. Gram-negative bacteria such as E. coli, S. typhimurium, S. flexneri, and B. thailandensis activate caspase-11 promoting GSDMD cleavage and pore formation [71]. Similarly, GSDMD-dependent pyroptosis can also be induced by canonical inflammasomes such as AIM2 and NLRC4. For instance, AIM2 inflammasome is required for GSDMD cleavage in response to F. novicida. Indeed, Gsdmd−/− mice are more susceptible to F. novicida infection, suggesting that GSDMD is required for infection restriction of this pathogen [94]. Concerning NLRC4 inflammasome activation, flagellin failed to induce pore formation in Gsdmd−/− BMDMs at early time points, although pyroptosis progressed at later time points. Hence, this result suggests that caspase-1 can cleave other pro-pyroptotic substrates apart from GSDMD [71].
In fact, recent data revealed alternative forms of GSDMD-independent cell death and IL-1β secretion. Canonical inflammasomes activators induce caspase-1 promoting not only cleavage of IL-1β but also its early secretion by GSDMD pores [95]. In that scenario, caspase-8 activation is inhibited [96]. However, GSDMD-deficient cells exhibit a delayed form of lytic death in which caspase-8 is activated and might drive the secretion of similar levels of IL-1β compared to wild-type cells. Interestingly, cells deficient in caspase-1 secrete reduced levels of mature IL-1β even at late time points. The GSDMD-independent mechanisms of IL-1β release may promote host protection against pathogens, at least to some extent, since GSDMD-deficient mice show a less severe F. novicida infection than caspase-1-deficient mice [96]. Intriguingly, the mechanism of IL-1β maturation itself empowers its relocation from the cytosol to the phosphatidylinositol 4,5-bisphosphate microdomains in the plasma membrane. This effect enables IL-1β fast exit via GSDMD pore or its slow secretion in a GSDMD-independent manner [97].
Several mechanisms by which GSDMD-dependent pyroptosis can contribute to bacterial infection restriction were proposed. One of them suggests that when pyroptosis leads to cell death, intracellular bacteria remain trapped into the cell forming structures named pore-induced intracellular traps (PITs) [98]. In addition, the secretion of cytokines and mediators through GSDMD pores recruits neutrophils which remove by efferocytosis these PITs, assisting the control of bacterial infection [88, 98]. Furthermore, recent studies linked GSDMD to the release of neutrophil extracellular traps (NETs), a cell death process termed NETosis [99, 100]. These structures are composed of neutrophil DNA and granular proteins such as neutrophil elastase (NE), myeloperoxidase (MPO) and cathepsin G. These NETs act serving as a barrier that avoids bacterial spreading and also kill pathogens by degradation their virulent factors [101]. In view of that, GSDMD can be cleaved by serine proteases such as NE [99]. Once it is cleaved, it leads to pore formation in the membrane of neutrophil azurophilic granules, thus contributing to enhance NE release in the cytosol [99]. NE released in the cytosol may enter nucleus where it processes histones to promote chromatin decondensation, triggering nuclear expansion which is an important stage during NETosis process [102]. Furthermore, NE released in the cytosol can also cleave more GSDMD promoting a positive feedback as GSDMD triggers more pore formation in the neutrophil azurophilic granules membranes. Finally, cleaved GSDMD also inserts into plasmatic cell membrane culminating in cell lysis and release of NETs [99]. Notably, this is a process in which GSDMD is involved in pore formation independent of inflammasome activation. On the other hand, PAMPs that trigger non-canonical inflammasome such as intracellular LPS can trigger GSDMD-dependent NET formation, but independently of classical proteins such as NE and MPO, describing a new pathway of NET release [100]. Moreover, N-terminal fragments of GSDMD were found in these traps after release, suggesting a possible role for GSDMD in pathogens restriction by directly killing microbes outside the cells [99]. Remarkably, this direct killing may constitute a novel manner that GSDMD reduces the spread of viable pathogens from pyroptotic cells, although it lacks confirmation by in vivo studies. In addition, GSDMD attacking bacterial membranes may result in release of PAMPs into the cytosol. This mechanism was recently hypothesized to explain F. novicida response during AIM2 inflammasome activation [94]. In response to F. novicida, GSDMD is required for optimal caspase-1 activation. Since Gsdmd−/− BMDMs display residual caspase-1 activation, it was suggested that during F. novicida initial phase of infection, it activates caspase-1 independent of GSDMD. However, once caspase-1 is active, it cleaves GSDMD, which can form pores in the intracellular bacteria causing its lysis and release of DNA which, in a positive feedback, activates AIM2 leading to more caspase-1 activation and pyroptosis [94]. In view of that, GSDMD N-terminal fragment can interact with bacterial cardiolipin-rich membrane forming pores causing bacteriolysis, as shown when N-terminal fragment was incubated in vitro with E. coli and Staphylococcus aureus [103].
5. Potassium efflux: connecting caspase-11 to NLRP3 activation
As mentioned before, caspase-11 induces NLRP3 inflammasome assembly leading to caspase-1 activation and IL-1β secretion [23]. However, the mechanism persisted undefined until new data allowed a better comprehension of this pathway. The NLRP3 inflammasome is activated by a variety of structurally and chemically distinct stimuli, including changes in cell volume, Ca2+ and Na+ influx, K+ efflux, pore-forming channels toxins, ROS, mtDNA, bacterial mRNA, translocation of cardiolipin from the inner mitochondrial membrane to the outer mitochondrial membrane, phagosomal destabilization and cathepsin release [15, 104]. Although it remains unclear if and how these distress signals are causally linked, an increasing body of evidence indicates that K+ efflux alone, acting on or upstream of NLRP3, is the minimal common cellular event that is necessary and sufficient to activate the canonical NLRP3 inflammasome [15, 105]. Accordingly, it was demonstrated that caspase‐11 activates a canonical NLRP3 inflammasome by promoting K+ efflux, suggesting that canonical and non-canonical inflammasomes work in concert to protect the host against intracellular pathogens [84]. In fact, GSDMD depletes intracellular K + via membrane pores formation [36, 106] and, hence, it is tempt to hypothesize that GSDMD activated by caspase-11 induces K+ efflux leading to NLRP3 activation [31].
However, in addition to NLRP3, other inflammasomes could be involved in the caspase-11-dependent non-canonic inflammasome response. In a Legionella infection model, it was demonstrated that the AIM2 inflammasome also cooperates with caspase-11 to induce host resistance to bacterial infection. AIM2 engages active caspase-1 to induce pore formation and K+ efflux, thus amplifying the infection signals to trigger activation of non-canonical NLRP3 inflammasome [107].
6. Concluding remarks
Host cells are often challenge by pathogenic bacteria that are capable to survive and replicate intracellularly. The past years witnessed great advances in our understanding of molecular mechanisms underlying innate immune system activation by cytosolic insults, mainly through inflammasome biology comprehension. The recognition of several PAMPs and DAMPs by inflammasome sensors was established and crucial downstream signaling pathways were identified. Moreover, inflammasome activation is known as an essential part of the immune response to control bacterial infections and thus acknowledged as a potential target to therapeutic drug interventions. Additionally, the non-canonical inflammasome arose as a key player that promotes host surveillance during cytosolic Gram-negative bacteria infection. Indeed, recent data shed light into the intricate network of signaling pathways during non-inflammasome activation (Fig. 1). Caspase-11, with the guidance of GBPs, is activated by cytosolic LPS derived from PCVs or OMVs, initiating the non-canonical inflammasome pathway. Subsequently, the cleavage of GSDMD by activated caspase-11 yields an N-terminal fragment that induces pore formation in cell membranes. This process ultimately leads to pyroptosis, a cell death program important for the elimination of intracellular bacteria. In addition, K+ efflux may potentially induce NLRP3 inflammasome assembly, which results in caspase-1 activation and IL-1β secretion. It is worthy of notice that bacteria can interact with several PRRs during their infection, and activation or inhibition of one signal transduction pathway may affect other pathways. For instance, K+ efflux can be a result of different insults apart of GSDMD-dependent pore formation. Moreover, canonical inflammasomes such as AIM2 or NLRC4 trigger pyroptosis in a GSDMD-dependent manner, suggesting that a complex signaling pathway cross talk occurs regularly. In that context, GBPs attacking PCVs may release different factors apart from LPS, such as DNA, enabling multiple pathways activation concomitantly. In addition, secretion of mediators during pyroptosis may recruit cells such as neutrophils, which are prominent participants in bacterial clearance. Thus, non-canonical inflammasome activation claims to be known as a coordinating process designed to restore tissue integrity and function. Additionally, recent data indicate a variety of endogenous regulatory mechanisms on the activation of non-canonical inflammasome pathway. These findings argue in favor of a pyroptotic process not necessarily committed to cell death that could be exploited by pathogens for their own benefit. Finally, since non-canonical inflammasomes are in the interface of pyroptotic cell death and inflammation mechanisms, and considering the role of this pathway in sensing cytoplasmic LPS, these novel findings may support innovative treatments for inflammasome-related diseases and sepsis.
Figure 1: Molecular basis of non-canonical inflammasome activation leading to pyroptosis and bacterial infections restriction.
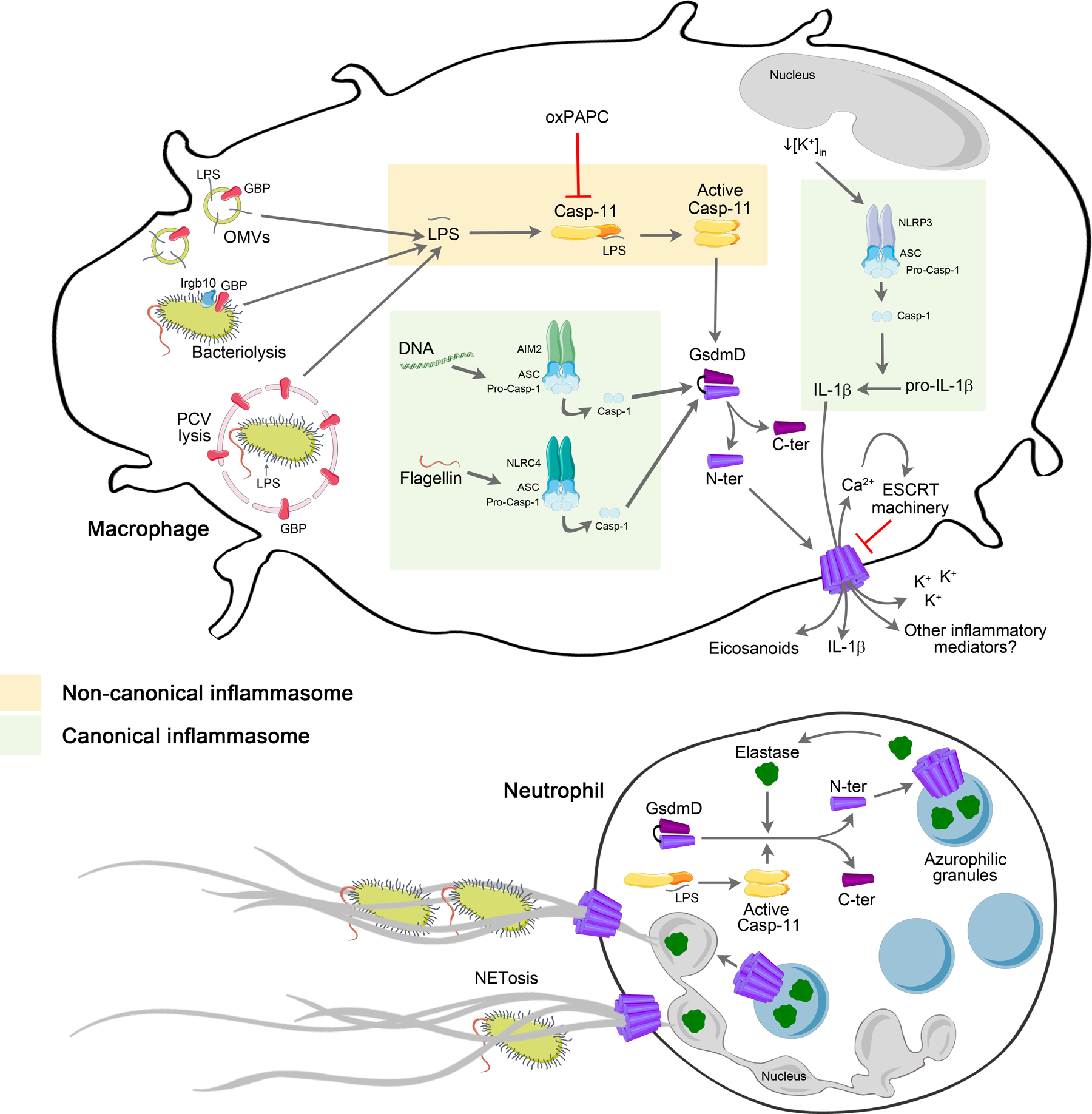
GBPs contribute to the cytosolic delivery of LPS from OMVs and may recruit IRGB10 to promote bacteriolysis. GBPs also promote PCVs damage destroying the microbial niche and exposing bacterial LPS for detection by caspase-11 in the cytosol. Cytoplasmic LPS activates caspase-11 that cleaves GSDMD releasing its N-terminal which forms membrane pores. OxPAPC can regulate this process as it binds to caspase-11 inhibiting its activation. The canonical inflammasomes AIM2 and NLRC4 lead to caspase-1 activation which also culminates in GSDMD cleavage, via recognition of bacterial DNA and flagellin, respectively. GSDMD pore acts as a conduit for potassium efflux that potentially triggers NLRP3 inflammasome activation. NLRP3-ASC complex activates caspase-1 leading to cleavage and release of IL-1β. ESCRT machinery dependent on Ca2+ influx acts controlling GSDMD pores. Inflammatory mediators (eicosanoids and possible other factors) released through membrane pore contribute to recruitment of immune cells such as neutrophils to the local of infection. Concerning neutrophils, GSDMD is cleaved by elastase and forms pores in the membrane of azurophilic granules releasing more elastase which can cleave more GSDMD. Elastase may enter nucleus triggering NETosis while GSDMD forms pores in the plasmatic membrane releasing NETs. Furthermore, LPS activates caspase-11 which also cleaves GSDMD leading to NET release.
Acknowledgments
This study was funded by grants from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, PNPD Post Doctoral Fellowship), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, #406883/2018-1, #402527/2013-5 and #302660/2015-1), Fundação de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG, APQ #01945/17, APQ # 00837/15 and Rede Mineira de Imunobiologicos #00140-16) and National Institute of Health R01 AI116453.
Abbreviations Page:
- AIM2
absent in melanoma 2
- ASC
apoptosis-associated speck-like protein containing a caspase recruitment domain
- BALF
bronchoalveolar lavage fluid
- BMDM
bone-marrow-derived macrophage
- CARD
caspase recruitment domain
- DAMP
host-derived damage-associated molecular pattern
- DC
dendritic cell
- ESCRT
endosomal sorting complexes required for transport
- GBP
guanylate-binding protein
- GSDMD
gasdermin D
- GTPases
guanosine 5′-triphosphatases
- HMGB1
high mobility group box 1
- IFN
interferons
- IL
interleukin
- IRGB10
immunity-related GTPase family member b10
- LDH
lactate dehydrogenase
- LPS
lipopolysaccharide
- LRR
leucine-rich repeat
- MPO
myeloperoxidase
- MSU
monosodium urate
- mtDNA
mitochondrial DNA
- NE
neutrophil elastase
- NET
neutrophil extracellular trap
- NF
nuclear factor
- NLR
Nod-like receptors
- NLRC4
Nod- leucine-rich repeat and caspase recruitment domain-containing 4
- NLRP
Nod- leucine-rich repeat and pyrin domain-containing
- OMV
outer membrane vesicle
- oxPAPC
oxidized 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine
- PAMP
pathogens-associated molecular pattern
- PCV
pathogen-containing vacuole
- PIT
intracellular trap
- PJVK
Pejvakin
- PRR
pattern-recognition receptor
- RLR
RIG-I-like receptors
- ROS
reactive oxygen species
- T3SS
type three secretion system
- TAK1
Transforming growth factor β-activated kinase 1
- TLR
Toll-like receptor
Footnotes
Disclosures
The authors declare no conflicts of interest.
References
- 1.Rivera A, Siracusa MC, Yap GS, Gause WC (2016) Innate cell communication kick-starts pathogen-specific immunity. Nature immunology 17, 356–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kieser KJ and Kagan JC (2017) Multi-receptor detection of individual bacterial products by the innate immune system. Nature reviews. Immunology 17, 376–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Land WG (2015) The Role of Damage-Associated Molecular Patterns in Human Diseases: Part I - Promoting inflammation and immunity. Sultan Qaboos University medical journal 15, e9–e21. [PMC free article] [PubMed] [Google Scholar]
- 4.Medzhitov R (2001) Toll-like receptors and innate immunity. Nature reviews. Immunology 1, 135–45. [DOI] [PubMed] [Google Scholar]
- 5.Newton K and Dixit VM (2012) Signaling in innate immunity and inflammation. Cold Spring Harbor perspectives in biology 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. (1997) A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388, 394–7. [DOI] [PubMed] [Google Scholar]
- 7.Kawai T and Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature immunology 11, 373–84. [DOI] [PubMed] [Google Scholar]
- 8.Yoneyama M and Fujita T (2009) RNA recognition and signal transduction by RIG-I-like receptors. Immunological reviews 227, 54–65. [DOI] [PubMed] [Google Scholar]
- 9.Marinho FV, Benmerzoug S, Oliveira SC, Ryffel B, Quesniaux VFJ (2017) The Emerging Roles of STING in Bacterial Infections. Trends in microbiology 25, 906–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinon F, Mayor A, Tschopp J (2009) The inflammasomes: guardians of the body. Annual review of immunology 27, 229–65. [DOI] [PubMed] [Google Scholar]
- 11.Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular cell 10, 417–26. [DOI] [PubMed] [Google Scholar]
- 12.Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nature reviews. Immunology 13, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES (2009) AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G (2013) K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–5. [DOI] [PubMed] [Google Scholar]
- 17.He Y, Hara H, Nunez G (2016) Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends in biochemical sciences 41, 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, Wong J, Ding S, Seki E, Schnabl B, Hevener AL, Greenberg HB, Kisseleva T, Karin M (2018) New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jo EK, Kim JK, Shin DM, Sasakawa C (2016) Molecular mechanisms regulating NLRP3 inflammasome activation. Cellular & molecular immunology 13, 148–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoss F, Rodriguez-Alcazar JF, Latz E (2017) Assembly and regulation of ASC specks. Cellular and molecular life sciences : CMLS 74, 1211–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cookson BT and Brennan MA (2001) Pro-inflammatory programmed cell death. Trends in microbiology 9, 113–4. [DOI] [PubMed] [Google Scholar]
- 22.Man SM, Karki R, Kanneganti TD (2017) Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunological reviews 277, 61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–21. [DOI] [PubMed] [Google Scholar]
- 24.Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, Dueber EC (2016) GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proceedings of the National Academy of Sciences of the United States of America 113, 7858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–92. [DOI] [PubMed] [Google Scholar]
- 26.Balakrishnan A, Karki R, Berwin B, Yamamoto M, Kanneganti TD (2018) Guanylate binding proteins facilitate caspase-11-dependent pyroptosis in response to type 3 secretion system-negative Pseudomonas aeruginosa. Cell death discovery 4, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA (2013) Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, Forsberg LS, Carlson RW, Dixit VM (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–9. [DOI] [PubMed] [Google Scholar]
- 29.Lee BL, Stowe IB, Gupta A, Kornfeld OS, Roose-Girma M, Anderson K, Warming S, Zhang J, Lee WP, Kayagaki N (2018) Caspase-11 auto-proteolysis is crucial for noncanonical inflammasome activation. The Journal of experimental medicine 215, 2279–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lagrange B, Benaoudia S, Wallet P, Magnotti F, Provost A, Michal F, Martin A, Di Lorenzo F, Py BF, Molinaro A, Henry T (2018) Human caspase-4 detects tetra-acylated LPS and cytosolic Francisella and functions differently from murine caspase-11. Nature communications 9, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Russo AJ, Behl B, Banerjee I, Rathinam VAK (2018) Emerging Insights into Noncanonical Inflammasome Recognition of Microbes. Journal of molecular biology 430, 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J, Shao Y, Wang W, Li S, Xin N, Xie F, Zhao C (2017) Caspase-11 deficiency impairs neutrophil recruitment and bacterial clearance in the early stage of pulmonary Klebsiella pneumoniae infection. International journal of medical microbiology : IJMM 307, 490–496. [DOI] [PubMed] [Google Scholar]
- 33.Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, Ernst RK, Steele-Mortimer O, Celli J, Vallance BA (2014) Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell host & microbe 16, 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang W, Shao Y, Li S, Xin N, Ma T, Zhao C, Song M (2017) Caspase-11 Plays a Protective Role in Pulmonary Acinetobacter baumannii Infection. Infection and immunity 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lacey CA, Mitchell WJ, Dadelahi AS, Skyberg JA (2018) Caspases-1 and caspase-11 mediate pyroptosis, inflammation, and control of Brucella joint infection. Infection and immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerqueira DM, Gomes MTR, Silva ALN, Rungue M, Assis NRG, Guimaraes ES, Morais SB, Broz P, Zamboni DS, Oliveira SC (2018) Guanylate-binding protein 5 licenses caspase-11 for Gasdermin-D mediated host resistance to Brucella abortus infection. PLoS pathogens 14, e1007519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A, Miao EA (2013) Caspase-11 protects against bacteria that escape the vacuole. Science 339, 975–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Carvalho RVH, Andrade WA, Lima-Junior DS, Dilucca M, de Oliveira CV, Wang K, Nogueira PM, Rugani JN, Soares RP, Beverley SM, Shao F, Zamboni DS (2019) Leishmania Lipophosphoglycan Triggers Caspase-11 and the Non-canonical Activation of the NLRP3 Inflammasome. Cell reports 26, 429–437.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freigang S (2016) The regulation of inflammation by oxidized phospholipids. European journal of immunology 46, 1818–25. [DOI] [PubMed] [Google Scholar]
- 40.Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, Donado CA, Shao F, Wu H, Springstead JR, Kagan JC (2016) An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352, 1232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chu LH, Indramohan M, Ratsimandresy RA, Gangopadhyay A, Morris EP, Monack DM, Dorfleutner A, Stehlik C (2018) The oxidized phospholipid oxPAPC protects from septic shock by targeting the non-canonical inflammasome in macrophages. Nature communications 9, 996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S, Roose-Girma M, Bumann D, Kayagaki N, Takeda K, Yamamoto M, Broz P (2014) Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 509, 366–70. [DOI] [PubMed] [Google Scholar]
- 43.Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA (2012) TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150, 606–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, Monack DM (2012) Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490, 288–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim BH, Shenoy AR, Kumar P, Das R, Tiwari S, MacMicking JD (2011) A family of IFN-gamma-inducible 65-kD GTPases protects against bacterial infection. Science 332, 717–21.21551061 [Google Scholar]
- 46.Kim BH, Chee JD, Bradfield CJ, Park ES, Kumar P, MacMicking JD (2016) Interferon-induced guanylate-binding proteins in inflammasome activation and host defense. Nature immunology 17, 481–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Man SM, Place DE, Kuriakose T, Kanneganti TD (2017) Interferon-inducible guanylate-binding proteins at the interface of cell-autonomous immunity and inflammasome activation. Journal of leukocyte biology 101, 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olszewski MA, Gray J, Vestal DJ (2006) In silico genomic analysis of the human and murine guanylate-binding protein (GBP) gene clusters. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research 26, 328–52. [DOI] [PubMed] [Google Scholar]
- 49.Degrandi D, Konermann C, Beuter-Gunia C, Kresse A, Wurthner J, Kurig S, Beer S, Pfeffer K (2007) Extensive characterization of IFN-induced GTPases mGBP1 to mGBP10 involved in host defense. J Immunol 179, 7729–40. [DOI] [PubMed] [Google Scholar]
- 50.Shenoy AR, Kim BH, Choi HP, Matsuzawa T, Tiwari S, MacMicking JD (2007) Emerging themes in IFN-gamma-induced macrophage immunity by the p47 and p65 GTPase families. Immunobiology 212, 771–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kravets E, Degrandi D, Ma Q, Peulen TO, Klumpers V, Felekyan S, Kuhnemuth R, Weidtkamp-Peters S, Seidel CA, Pfeffer K (2016) Guanylate binding proteins directly attack Toxoplasma gondii via supramolecular complexes. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tripal P, Bauer M, Naschberger E, Mortinger T, Hohenadl C, Cornali E, Thurau M, Sturzl M (2007) Unique features of different members of the human guanylate-binding protein family. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research 27, 44–52. [DOI] [PubMed] [Google Scholar]
- 53.Vestal DJ, Gorbacheva VY, Sen GC (2000) Different subcellular localizations for the related interferon-induced GTPases, MuGBP-1 and MuGBP-2: implications for different functions? Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research 20, 991–1000. [DOI] [PubMed] [Google Scholar]
- 54.Ngo CC and Man SM (2017) Mechanisms and functions of guanylate-binding proteins and related interferon-inducible GTPases: Roles in intracellular lysis of pathogens. Cellular microbiology 19. [DOI] [PubMed] [Google Scholar]
- 55.Yamamoto M, Okuyama M, Ma JS, Kimura T, Kamiyama N, Saiga H, Ohshima J, Sasai M, Kayama H, Okamoto T, Huang DC, Soldati-Favre D, Horie K, Takeda J, Takeda K (2012) A cluster of interferon-gamma-inducible p65 GTPases plays a critical role in host defense against Toxoplasma gondii. Immunity 37, 302–13. [DOI] [PubMed] [Google Scholar]
- 56.Pilla DM, Hagar JA, Haldar AK, Mason AK, Degrandi D, Pfeffer K, Ernst RK, Yamamoto M, Miao EA, Coers J (2014) Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proceedings of the National Academy of Sciences of the United States of America 111, 6046–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finethy R, Jorgensen I, Haldar AK, de Zoete MR, Strowig T, Flavell RA, Yamamoto M, Nagarajan UM, Miao EA, Coers J (2015) Guanylate binding proteins enable rapid activation of canonical and noncanonical inflammasomes in Chlamydia-infected macrophages. Infection and immunity 83, 4740–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thurston TL, Matthews SA, Jennings E, Alix E, Shao F, Shenoy AR, Birrell MA, Holden DW (2016) Growth inhibition of cytosolic Salmonella by caspase-1 and caspase-11 precedes host cell death. Nature communications 7, 13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shenoy AR, Wellington DA, Kumar P, Kassa H, Booth CJ, Cresswell P, MacMicking JD (2012) GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science 336, 481–5. [DOI] [PubMed] [Google Scholar]
- 60.Feeley EM, Pilla-Moffett DM, Zwack EE, Piro AS, Finethy R, Kolb JP, Martinez J, Brodsky IE, Coers J (2017) Galectin-3 directs antimicrobial guanylate binding proteins to vacuoles furnished with bacterial secretion systems. Proceedings of the National Academy of Sciences of the United States of America 114, E1698–E1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Costa Franco MM, Marim F, Guimaraes ES, Assis NRG, Cerqueira DM, Alves-Silva J, Harms J, Splitter G, Smith J, Kanneganti TD, de Queiroz N, Gutman D, Barber GN, Oliveira SC (2018) Brucella abortus Triggers a cGAS-Independent STING Pathway To Induce Host Protection That Involves Guanylate-Binding Proteins and Inflammasome Activation. J Immunol 200, 607–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Man SM, Karki R, Malireddi RK, Neale G, Vogel P, Yamamoto M, Lamkanfi M, Kanneganti TD (2015) The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nature immunology 16, 467–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meunier E, Wallet P, Dreier RF, Costanzo S, Anton L, Ruhl S, Dussurgey S, Dick MS, Kistner A, Rigard M, Degrandi D, Pfeffer K, Yamamoto M, Henry T, Broz P (2015) Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nature immunology 16, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, Temirov J, Frase S, Zhu Q, Malireddi RKS, Kuriakose T, Peters JL, Neale G, Brown SA, Yamamoto M, Kanneganti TD (2016) IRGB10 Liberates Bacterial Ligands for Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell 167, 382–396 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wallet P, Benaoudia S, Mosnier A, Lagrange B, Martin A, Lindgren H, Golovliov I, Michal F, Basso P, Djebali S, Provost A, Allatif O, Meunier E, Broz P, Yamamoto M, Py BF, Faudry E, Sjostedt A, Henry T (2017) IFN-gamma extends the immune functions of Guanylate Binding Proteins to inflammasome-independent antibacterial activities during Francisella novicida infection. PLoS pathogens 13, e1006630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wandel MP, Pathe C, Werner EI, Ellison CJ, Boyle KB, von der Malsburg A, Rohde J, Randow F (2017) GBPs Inhibit Motility of Shigella flexneri but Are Targeted for Degradation by the Bacterial Ubiquitin Ligase IpaH9.8. Cell host & microbe 22, 507–518 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Piro AS, Hernandez D, Luoma S, Feeley EM, Finethy R, Yirga A, Frickel EM, Lesser CF, Coers J (2017) Detection of Cytosolic Shigella flexneri via a C-Terminal Triple-Arginine Motif of GBP1 Inhibits Actin-Based Motility. mBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kaparakis-Liaskos M and Ferrero RL (2015) Immune modulation by bacterial outer membrane vesicles. Nature reviews. Immunology 15, 375–87. [DOI] [PubMed] [Google Scholar]
- 69.Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, Rathinam VAK (2016) Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 165, 1106–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Santos JC, Dick MS, Lagrange B, Degrandi D, Pfeffer K, Yamamoto M, Meunier E, Pelczar P, Henry T, Broz P (2018) LPS targets host guanylate-binding proteins to the bacterial outer membrane for non-canonical inflammasome activation. The EMBO journal 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM (2015) Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–71. [DOI] [PubMed] [Google Scholar]
- 72.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–5. [DOI] [PubMed] [Google Scholar]
- 73.Saeki N, Kuwahara Y, Sasaki H, Satoh H, Shiroishi T (2000) Gasdermin (Gsdm) localizing to mouse Chromosome 11 is predominantly expressed in upper gastrointestinal tract but significantly suppressed in human gastric cancer cells. Mamm Genome 11, 718–24. [DOI] [PubMed] [Google Scholar]
- 74.Tamura M, Tanaka S, Fujii T, Aoki A, Komiyama H, Ezawa K, Sumiyama K, Sagai T, Shiroishi T (2007) Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics 89, 618–29. [DOI] [PubMed] [Google Scholar]
- 75.Feng S, Fox D, Man SM (2018) Mechanisms of Gasdermin Family Members in Inflammasome Signaling and Cell Death. J Mol Biol 430, 3068–3080. [DOI] [PubMed] [Google Scholar]
- 76.Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES (2017) Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun 8, 14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, Berger SB, Gough PJ, Bertin J, Proulx MM, Goguen JD, Kayagaki N, Fitzgerald KA, Lien E (2018) Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR, Poltorak A (2018) Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci U S A 115, E10888–E10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Broz P (2015) Immunology: Caspase target drives pyroptosis. Nature 526, 642–3. [DOI] [PubMed] [Google Scholar]
- 80.Ruan J, Xia S, Liu X, Lieberman J, Wu H (2018) Cryo-EM structure of the gasdermin A3 membrane pore. Nature 557, 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D’Silva DB, Tanzer MC, Monteleone M, Robertson AA, Cooper MA, Alvarez-Diaz S, Herold MJ, Bedoui S, Schroder K, Masters SL (2015) NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. European journal of immunology 45, 2918–26. [DOI] [PubMed] [Google Scholar]
- 82.Perregaux D and Gabel CA (1994) Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem 269, 15195–203. [PubMed] [Google Scholar]
- 83.Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J (2007) Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 14, 1583–9. [DOI] [PubMed] [Google Scholar]
- 84.Ruhl S and Broz P (2015) Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. European journal of immunology 45, 2927–36. [DOI] [PubMed] [Google Scholar]
- 85.Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V (2015) Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur J Immunol 45, 2911–7. [DOI] [PubMed] [Google Scholar]
- 86.He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J (2015) Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res 25, 1285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC (2018) The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48, 35–44 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jorgensen I, Lopez JP, Laufer SA, Miao EA (2016) IL-1beta, IL-18, and eicosanoids promote neutrophil recruitment to pore-induced intracellular traps following pyroptosis. Eur J Immunol 46, 2761–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gaidt MM and Hornung V (2016) Pore formation by GSDMD is the effector mechanism of pyroptosis. EMBO J 35, 2167–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ruhl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P (2018) ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960. [DOI] [PubMed] [Google Scholar]
- 91.Andrews NW, Almeida PE, Corrotte M (2014) Damage control: cellular mechanisms of plasma membrane repair. Trends Cell Biol 24, 734–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jimenez AJ, Maiuri P, Lafaurie-Janvore J, Divoux S, Piel M, Perez F (2014) ESCRT machinery is required for plasma membrane repair. Science 343, 1247136. [DOI] [PubMed] [Google Scholar]
- 93.Scheffer LL, Sreetama SC, Sharma N, Medikayala S, Brown KJ, Defour A, Jaiswal JK (2014) Mechanism of Ca(2)(+)-triggered ESCRT assembly and regulation of cell membrane repair. Nat Commun 5, 5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhu Q, Zheng M, Balakrishnan A, Karki R, Kanneganti TD (2018) Gasdermin D Promotes AIM2 Inflammasome Activation and Is Required for Host Protection against Francisella novicida. J Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, Broz P (2018) The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. European journal of immunology 48, 584–592. [DOI] [PubMed] [Google Scholar]
- 96.Schneider KS, Gross CJ, Dreier RF, Saller BS, Mishra R, Gorka O, Heilig R, Meunier E, Dick MS, Cikovic T, Sodenkamp J, Medard G, Naumann R, Ruland J, Kuster B, Broz P, Gross O (2017) The Inflammasome Drives GSDMD-Independent Secondary Pyroptosis and IL-1 Release in the Absence of Caspase-1 Protease Activity. Cell reports 21, 3846–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Monteleone M, Stanley AC, Chen KW, Brown DL, Bezbradica JS, von Pein JB, Holley CL, Boucher D, Shakespear MR, Kapetanovic R, Rolfes V, Sweet MJ, Stow JL, Schroder K (2018) Interleukin-1beta Maturation Triggers Its Relocation to the Plasma Membrane for Gasdermin-D-Dependent and -Independent Secretion. Cell reports 24, 1425–1433. [DOI] [PubMed] [Google Scholar]
- 98.Jorgensen I, Zhang Y, Krantz BA, Miao EA (2016) Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med 213, 2113–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, Menninger S, Eickhoff J, Nussbaumer P, Klebl B, Kruger R, Herzig A, Zychlinsky A (2018) Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol 3. [DOI] [PubMed] [Google Scholar]
- 100.Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, von Pein JB, Broz P, Sweet MJ, Schroder K (2018) Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Science immunology 3. [DOI] [PubMed] [Google Scholar]
- 101.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303, 1532–5. [DOI] [PubMed] [Google Scholar]
- 102.Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A (2010) Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. The Journal of cell biology 191, 677–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J (2016) Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Man SM and Kanneganti TD (2015) Regulation of inflammasome activation. Immunological reviews 265, 6–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kanneganti TD and Lamkanfi M (2013) K(+) drops tilt the NLRP3 inflammasome. Immunity 38, 1085–8. [DOI] [PubMed] [Google Scholar]
- 106.Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Menoret A, Ghosh A, Vella AT, Vanaja SK, Sarkar SN, Fitzgerald KA, Rathinam VAK (2018) Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity 49, 413–426 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cunha LD, Silva ALN, Ribeiro JM, Mascarenhas DPA, Quirino GFS, Santos LL, Flavell RA, Zamboni DS (2017) AIM2 Engages Active but Unprocessed Caspase-1 to Induce Noncanonical Activation of the NLRP3 Inflammasome. Cell reports 20, 794–805. [DOI] [PubMed] [Google Scholar]
- 108.Cerqueira DM, Pereira MS, Silva AL, Cunha LD, Zamboni DS (2015) Caspase-1 but Not Caspase-11 Is Required for NLRC4-Mediated Pyroptosis and Restriction of Infection by Flagellated Legionella Species in Mouse Macrophages and In Vivo. J Immunol 195, 2303–11. [DOI] [PubMed] [Google Scholar]