

Abstract
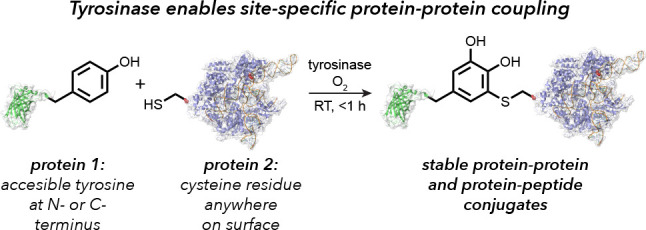
The synthesis of protein–protein and protein–peptide conjugates is an important capability for producing vaccines, immunotherapeutics, and targeted delivery agents. Herein we show that the enzyme tyrosinase is capable of oxidizing exposed tyrosine residues into o-quinones that react rapidly with cysteine residues on target proteins. This coupling reaction occurs under mild aerobic conditions and has the rare ability to join full-size proteins in under 2 h. The utility of the approach is demonstrated for the attachment of cationic peptides to enhance the cellular delivery of CRISPR-Cas9 20-fold and for the coupling of reporter proteins to a cancer-targeting antibody fragment without loss of its cell-specific binding ability. The broad applicability of this technique provides a new building block approach for the synthesis of protein chimeras.
Short abstract
Enzymatic oxidation of tyrosine residues followed by reaction with cysteine thiols allows the covalent coupling of proteins and peptides using only native amino acid side chains.
Introduction
The covalent modification of proteins while preserving native function has long been a goal of chemical biology research.1−3 Unfortunately, it can be difficult to target chemically distinct functional groups in protein sequences, leading to heterogeneous product mixtures with many modification strategies. This challenge is particularly notable for the synthesis of protein–protein and protein–peptide conjugates, which can serve as highly valuable constructs for vaccines, immunotherapy agents, signaling peptides with enhanced circulation properties, and targeted delivery vehicles. Currently, the coupling of two biomolecules is usually achieved using heterobifunctional cross-linking agents (which are rarely site-selective for both reactive species)1 or click-type reactions4 between bioorthogonal functional groups5 that must be introduced using artificial amino acid incorporation, metabolic engineering, or additional synthetic steps. The coupling of native functional groups on proteins and peptides can be achieved through native chemical ligations and sortase-based techniques,6,7 but these approaches can only be used at the N- and C-termini and may limit the quantities of bioconjugates that can be obtained. In some cases it is possible to fuse multiple protein sequences at the genetic level for recombinant expression.8,9 While this is a time-tested approach for producing chimeric proteins, such methods often require significant engineering to determine optimal linker lengths,10 can encounter folding problems during the expression of constructs of increasing size,11 and are again limited to attachment via the N- and C-termini.
This report describes a new method for the efficient generation of protein–peptide and protein–protein conjugates through the direct coupling of solvent-exposed tyrosine residues to cysteine sulfhydryl groups. This reaction requires only catalytic amounts of tyrosinase and adventitious oxygen and affords complex, multicomponent bioconjugates in under 1 h under mild reaction conditions with low stoichiometric excess. This strategy is demonstrated as a convenient method for the installation of cell-penetrating peptides on proteins, such as Cas9, and the coupling of entire protein domains. Most importantly, this reaction demonstrates the very rare ability to couple full-size proteins together through positionally defined linkages using native amino acids, providing a powerful new tool for the construction of multifunctional biomolecular constructs.
Results and Discussion
In previous work, our lab has shown that proteins bearing aniline and N-terminal proline nucleophiles can be modified with o-quinone and o-iminoquinone electrophiles in high yields.12,13 The oxidized intermediates in these reactions have typically been generated through the in situ oxidation of o-aminophenols or similar species using periodate12 or ferricyanide anions.13 While effective in many contexts, these stoichiometric reagents can exhibit collateral oxidation of methionine thioethers and surface-exposed cysteines to varying degrees. A recent breakthrough in this chemistry involves the use of the tyrosinase enzymes to generate the same reactive o-quinone electrophiles using simple phenols and oxygen.14 The small amount of molecular oxygen that is dissolved in the buffers is sufficient for the reaction to proceed, and water is the only byproduct. This reaction addresses two important limitations of the previous versions of this chemistry. First, it is no longer necessary to prepare and install the oxygen-sensitive o-aminophenol and o-catechol groups; instead, simple tyramine and tyrosine derivatives can be used. Second, the need for an additional oxidant that must later be removed is avoided. Importantly, the enzymatic method for triggering these reactions is selective for phenols that are exposed in solution and does not cause off-target oxidation of sensitive functional groups.
It has been known for some time that tyrosinases can act upon tyrosine residues in proteins and peptides.15 Typically these reactions have been used to achieve protein cross-linking at high substrate concentrations involving indiscriminate conversion of tyrosine residues within the peptide backbone to o-quinines followed by reactions with a variety of nucleophiles.16−19 Previous reports have followed the reaction progress using UV or other spectroscopic analyses and have explored the impact of tyrosine oxidation on enzyme activity; however, the sequence specificities and sites of oxidation have not been characterized well. In terms of site-selective coupling chemistry, other laboratories have recently shown that tyrosine-tagged immunoglobulins can be oxidized and subjected to subsequent hetero-Diels–Alder reactions with cyclooctynes,20 and abTYR has been used to activate an ovalbumin peptide tag for coupling to substituted boronic acids.21 Work from our lab has shown that abTYR and a new tyrosinase derived from Bacillus megatarium can activate phenols in both small-molecule and protein substrates for coupling to N-terminal proline residues and anilines (Figure 1a, top and bottom reaction pathways).14,22 Taken together, these studies highlight the utility of tyrosinase enzymes to generate transient o-quinone electrophiles in dilute aqueous solutions, allowing highly chemoselective couplings that are not realized at the higher substrate concentrations evaluated previously.
Figure 1.

Tyrosinase-mediated oxidative coupling reactions. (a) Phenols are oxidized by tyrosinase to yield o-quinone intermediates that couple with a variety of nucleophiles on biomolecules. This work explored the addition of cysteine thiolates (shown in red). (b) Structure of the MS2 viral capsid (PDB ID 2MS2), with 180 interior cysteine residues (N87C) indicated in red. Pores in the capsid shell allow the entry of peptides and small molecules. (c) Structure of α-endorphin, with the targeted tyrosine residue shown in blue. (d–f) Coupling reactions were screened using ESI-TOF MS, showing full modification of the MS2 N87C capsid without off-target oxidation. Reaction conditions: pH 6.5 phosphate buffer, RT, 30 min. Expected mass values: MS2 N87C [M + H]+ = 13719; MS2 N87C–N-ethylmaleimide (NEM) adduct [M + H]+ = 13844; MS2 N87C–endorphin o-hydroquinone product [M + H]+ = 15521.
Key to the present work is the observation that cysteine thiols on full-size biomolecules participate readily in this chemistry at low equivalencies, allowing the first general coupling reaction of two native and readily encodable functional groups on protein surfaces (Figure 1a, middle pathway). Although we have known that these residues can participate in oxidative coupling reactions to varying degrees, the reliance on stoichiometric oxidants in our previous studies led to numerous reaction byproducts that are completely avoided using the new tyrosinase oxidation system. Other reports have noted reactivity between small-molecule thiols and o-quinones, but with minimal product characterization.23,24
In the context of site-selective bioconjugation, this reactivity pathway was first tested on genome-free MS2 bacteriophage capsids bearing the N87C mutation in each of the subunit proteins.25,26 Each of these assemblies provides 180 cysteine residues facing the inside of a 27 nm sphere.27 The capsid structures possess a series of 2 nm pores that are large enough for peptides and small molecules to access the inner surface (Figure 1b).26,28,29 Modification was achieved by exposing 10 μM (based on monomer) MS2 N87C to 200 μM α-endorphin peptide (Figure 1c) in the presence of 167 nM abTYR for 30 min at room temperature in 20 mM phosphate buffer (pH 6.5). Clean conversion of each capsid monomer to a single peptide conjugate was observed (Figure 1d). This represents the installation of >170 peptide groups inside each capsid despite the high degree of steric crowding. Subsequent exposure to the common cysteine capping reagent N-ethylmaleimide (NEM) showed no further modification (Figures 1e and S1), supporting the cysteine selectivity of the reaction. Similarly, no α-endorphin addition was observed for MS2 N87C that had been previously coupled to NEM, and there was no modification of wild-type MS2 capsids lacking exposed cysteine residues (Figure S1). Equally importantly, no background oxidation was observed on any of the MS2 proteins, each of which contains four tyrosine residues (Figures 1c and S1). This is presumably because tyrosine residues embedded in secondary and tertiary structural elements cannot access the deep binding pocket of the enzyme active site.30 These results demonstrate that covalent bonds can be formed between these two native amino acid residues using an efficient and operationally simple procedure.
In order to characterize the chemical structure of the bioconjugation product, we conducted NMR experiments for a small-molecule analogue. Somewhat surprisingly, the product results from thiol addition at the 5-position of the o-quinone ring (numbering from the amino acid connection point as shown in Figure 1a), and it remains predominantly in the catechol form (Figures 2a, S2, and S3). Additional MS data further support the catechol structure. This product contrasts with the previously observed structures resulting from proline and aniline additions to o-quinone species, where the addition occurs at the 6-position and the product tends to remain in the oxidized quinone form (Figure 1a).12−14 MS data confirmed that the thiol addition products on larger biomolecules also existed predominantly in the reduced catechol form.
Figure 2.
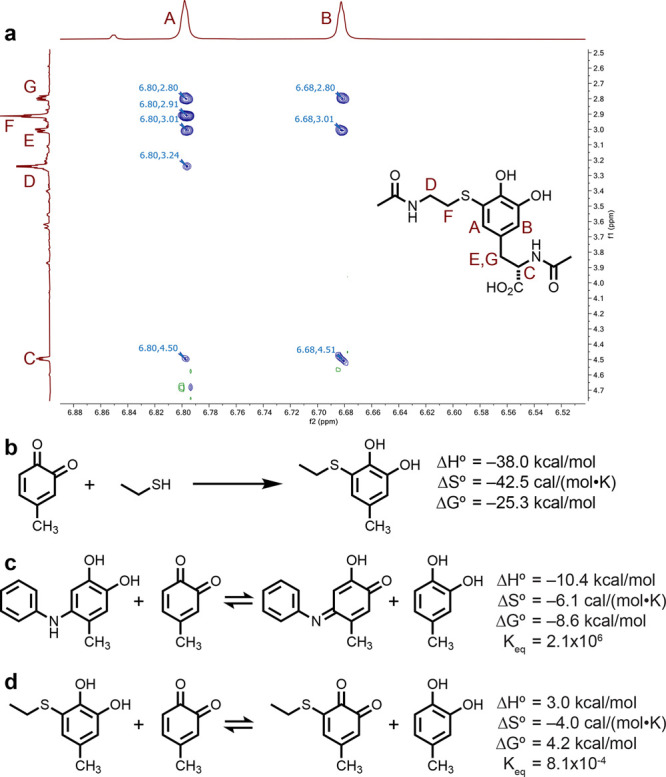
Structural analysis of thiol–o-quinone coupling products. (a) ROESY 1H NMR data (900 MHz) show a clear correlation between proton A and the corresponding signals from both the alkyl chain in N-acetylcysteamine (F) and the N-acetyltyrosine alkyl chain (E, G). (b) Energetics of the coupling reaction as estimated using DFT calculations. Geometry optimization and vibrational spectral calculations were conducted at the B3LYP-D3/6-31G** level, and final electronic energies were calculated at the ωB97M-V/6-311G++(3df,3pd) level. A CPCM solvation model was used in these calculations (see the Supporting Information for details and molecular coordinates). These data were combined to obtain the reported thermodynamic values. For comparison purposes, the reaction energies and equilibrium constants were estimated using this approach for the oxidation of the initial (c) aniline and (d) thiol coupling products with a methyl-substituted o-quinone.
To establish the degree of quinoid versus catechol nature of the product further, we exposed both thiol and N-terminal proline coupling adducts of Y200C green fluorescent protein (GFP) to methoxyamine (CH3ONH2) overnight to effect oxime formation with any ketone groups that were present, as has been recently shown.31 Notably, only the proline adducts exhibited any detectable reactivity to the methoxyamine (Figure S4).
The energetics of the o-quinone–thiol coupling reaction were investigated using DFT calculations. Briefly, geometry-optimized structures were evaluated at the B3LYP-D3/6-31G** level to obtain vibrational spectra, and ωB97M-V/6-311G++(3df,3pd) calculations were used to obtain accurate ground-state electronic energies.32 These results were combined to yield estimates of the thermodynamic parameters for the reaction. A CPCM solvent model33−35 was used for all of the calculated results appearing in Figure 2b; gas-phase versions of the same calculations are included in Figure S5. This study indicated that the reaction is highly favorable (ΔH° = −38.0 kcal/mol and ΔG° = −25.3 kcal/mol), which is a common feature associated with click-type reactions and serves to validate why the reactions proceed despite the high degree of building steric bulk.4 For comparison, the addition of a thiol nucleophile to a maleimide group affords more modest values (ΔH° = −24.4 kcal/mol and ΔG° = −10.1 kcal/mol) using the same computational approach (Figure S5). Calculations were also used to compare the relative oxidation propensities for the initial thiol and aniline addition products relative to a methyl-substituted o-quinone. While such an equilibrium was strongly favorable for the formation of the oxidized aniline product (Figure 2c), it was disfavored for the thiol addition product (Figure 2d). This relative comparison is in line with the observation that the thiol addition product remains predominantly in the reduced catechol form.
To test the stability of the protein conjugation product, we subjected α-endorphin-modified MS2 to buffers ranging from pH 5 to pH 8, potassium ferricyanide, 5 mM tris(carboxyethyl)phosphine (TCEP), and 5 mM n-mercaptobutanol for up to 6 days at room temperature. None of the conditions led to greater than 5% loss of the attached peptides (Table S1 and Figure S6). Additionally, GFP modified with an RRRRY peptide showed no reversion when kept at 37 °C for up to 7 days, even when exposed to up to 4 mM glutathione (GSH). It should be noted that small amounts of GSH and other small-molecule thiols were shown to add to the o-hydroquinone ring over the course of several days (Figure S7), presumably after aerobic oxidation to form small equilibrium amounts of the o-quinone; however, these adducts did not appear to affect the integrity of the linkage formed in the initial coupling reaction.
Stability tests were also conducted in human blood serum after coupling of Y200C sfGFP to a biotin moiety through either maleimide- or tyrosinase-based conjugation. The resulting samples were kept in the serum at room temperature or at 37 °C for up to 7 days. At several time points during incubation, aliquots were removed and subjected to streptavidin bead capture followed by a final elution using 80% acetonitrile (ACN), 5% formic acid (FA), and 2 mM biotin (Figure S8).36 We found that the maleimide-coupled product decreased over time and at 7 days was no longer detectable in either sample (Figure S9). This result is consistent with literature observations for these types of adducts on antibodies and other proteins.37 In contrast, the tyrosinase-coupled product was recovered throughout the course of the experiment, indicating that this product has substantially greater stability in serum.
In addition to α-endorphin (Figure 3a), the tyrosine–cysteine coupling chemistry has proven remarkably successful for the site-selective attachment of additional peptides. Both superfolder GFP (sfGFP) bearing the Y200C mutation (GFP Y200C) (Figure 3a,b) and the N87C MS2 coat protein (Figure S10) were coupled to tyrosines at the ends of peptides derived from interleukin-13 (IL-13), simian vacuolating virus 40 (SV40) large T-antigen,38,39 HIV Tat,40 and polyarginine40 with excellent conversion (Figure 3b). It is important to note that N-terminal acylation or an additional glycine was needed in these cases to prevent dopachrome-like cyclization of the oxidized N-terminal tyrosine residues. Substrate compatibility tests using the tyrosinase activation/thiol addition protocol also showed that small-molecule phenols, including biotin phenol, rhodamine phenol, and 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) phenol, all resulted in high conversion of the starting protein to the expected products within 30 min (Figure S11). Some secondary adducts were observed with these small-molecule substrates for sfGFP, which are believed to be the products of competing N-terminal modification (Figure S11).14 The diversity of small molecules and peptides compatible with this coupling strategy demonstrates the utility of the reaction as a general tool for protein modification. Our previous studies of aniline-based oxidation coupling reactions have indicated that o-quinone formation is rate-limiting.41 This is likely the case for the thiol additions as well, complicating the calculation of the second-order rate constant for the key coupling step. Even considering the enzymatic activation requirement, these reactions still reach completion in 1 h with coupling partner concentrations well below 100 μM.
Figure 3.

Synthesis of protein–peptide and protein–protein conjugates using thiol-directed enzymatic oxidative coupling reactions. (a, b) A superfolder GFP (sfGFP) thiol mutant (Y200C) was used as a protein component to evaluate peptide coupling reactions. All of the reactions were run for 30 min at RT and characterized using ESI-TOF MS. (c–e) A CRISPR-Cas9 variant with two C-terminal and four N-terminal nuclear localization sequences (Cas9-4NLS) was modified with peptide and protein coupling partners. Cas9 has two surface thiols (Cys 80 and Cys 574, red in the attached structure) in the native sequence. (c) ESI-TOF MS data indicated complete coupling at both sites with the α-endorphin peptide. The coupling reaction was successful even when the coupling partner was sfGFP bearing an exposed tyrosine residue near the N-terminus (MYGGS-GFP). All Cas9 couplings were run on ice for 1 h. (d) The site-specific DNA cleaving ability of the Cas9–peptide conjugate was unchanged relative to an untreated control. This was true when the preassembled ribonucleoprotein (RNP) was modified directly or when the RNA-free protein (apo-Cas9) was used for the modification reaction. In the latter case, the single guide RNA strand was added before the DNA cleavage experiment. (e) The Cas9–GFP conjugate retained site-specific DNA cleaving ability despite the high degree of added steric bulk.
An application of this chemistry for the modification of large proteins and enzymes was demonstrated for CRISPR-Cas9 ribonucleoprotein (RNP) complexes. Cas9, an RNA-guided DNA endonuclease used widely for genome editing, contains two surface cysteines at positions 80 and 574, thus providing two native bioconjugation handles for peptide attachment. To generate a modified form of Cas9 bearing two peptides, coupling reactions were conducted using Cas9 bearing two copies of the SV40 large T-antigen nuclear localization sequence (NLS) to facilitate nuclear entry. Quantitative conversion was observed to the doubly modified protein in both the apo and RNA-bound forms (Figure 3c). Importantly, tyrosinase-modified Cas9 RNPs fully retained their target cleavage activity compared to unmodified Cas9 RNPs (Figure 3d). Building off the success of peptide conjugates, we applied the tyrosinase conjugation method to the coupling of whole protein domains. We found that tyrosinase could be used to ligate Cas9 to green fluorescent protein with no detriment to the DNA cleavage ability (MYGGS-GFP; Figures 2c,e and S12). It is important to note that the mass spectral data revealed no evidence of Cas9 oxidation, nor did they indicate significant amounts of off-target modification. These experiments underscore the exquisite chemoselectivity of this reaction, as the Cas9 constructs contain 158 lysine and 58 tyrosine residues in addition to representatives of the 18 other native amino acids, providing ample opportunities for off-target reactions. Despite having a highly reactive system, this method has exceptionally predictable conjugation outcomes.
With these results in hand, we examined whether we could attach peptides capable of mediating Cas9 delivery. A genetic fusion of four consecutive SV40 NLSs to the N-terminus of Cas9 bearing two additional C-terminal NLSs has been shown to enable delivery of Cas9 RNPs to human neural progenitor cells (NPCs) in culture and in vivo.38 However, the production of such complexes is lengthy, requires dedicated cloning and protein purification, and limits the locations of the delivery peptides to the native N- and C-termini. Moreover, most commercially produced Cas9 RNPs demonstrate little to no cell-penetrating capability.38 Thus, we examined whether standard Cas9 RNP complexes could be chemically modified with peptides or proteins for editing in human NPCs.38 Three different cell-penetrating peptides were coupled to the native cysteine residues of a Cas9 RNP bearing two C-terminal NLS sequences, which does not appreciably edit cells in the NPC assay on its own (Figure 4a). After the reaction, ESI-TOF MS analysis confirmed that two copies of each tested peptide coupled quantitatively to Cas9 (Figure S13). These constructs, plus Cas9RNP with sfGFP, were then assayed for their editing efficiencies.42−45 Cas9 conjugates bearing +36 GFP proteins precipitated upon guide RNA addition and thus could not be evaluated in this experiment. The Cas9RNP construct bearing two 2SV40 peptides (where each peptide is composed of two consecutive SV40 NLSs) showed a 20-fold increase in editing efficiency (Figures 4a and S14) relative to the unmodified 2NLS-Cas9RNP. This indicates that the delivery of Cas9 RNP can be mediated by direct peptide coupling to the protein surface of a readily available standard Cas9 protein. The addition of other peptides and sfGFP did not improve the editing efficiency. In addition, experiments involving the original 4NLS Cas9 construct showed successful modification, but the added protein domains did not increase the editing activity further (Figure S15).
Figure 4.

Evaluation of tyrosine–cysteine bioconjugates in cell culture assays. (a) Evaluation of modified CRISPR-Cas9 ribonucleoproteins (RNPs) for gene editing in neural progenitor cells. The assay measured the increase in fluorescent protein expression following successful editing at a tdTomato locus. Cas9 modified with two copies of the SV40 nuclear localization sequence (Cas9-2SV40) showed a 20-fold increase in editing efficiency compared with unmodified Cas9. Data were collected 72 h after treatment. Error bars show standard deviations of triplicates. (b) The coupling reaction was successful for the coupling of GFP Y200C to a HER2-binding scFv-GGY construct, as measured by ESI-TOF. The observed product mass was consistent with the linkage depicted in (c). (d) Flow cytometry analysis of SK-BR-3 (HER2+) and MDA-MB-468 (HER2−) breast cancer cells treated with the trastuzumab scFv–sfGFP construct showed HER2-specific cell binding. Gating and statistics are shown in Figure S18.
Though Cas9–GFP conjugates showed no change in cell uptake, the capability to couple two protein domains so efficiently presents an attractive prospect that has been pursued for some time by the field. To test the scope of the reaction, we generated versions of nanoluciferase (nanoLuc) and a HER2-binding short-chain variable fragment (scFv) with tyrosine tags and combined them with Y200C sfGFP. The nanoLuc was produced with either an N-terminal or C-terminal tag, and coupling was performed using 10 μM nanoLuc and 20 μM Y200C GFP in pH 6.5 phosphate buffer for 30 min at RT. Both constructs yielded over 80% conversion (as determined using ESI-TOF MS) to the singly modified nanoLuc–GFP conjugates (Figure S16). The reaction was similarly successful for the coupling of Y200C sfGFP (∼7 μM by absorbance at 485 nm using an extinction coefficient of 83 300 M–1 cm–1) to a HER2-binding scFv–GGSY construct (5 μM) in pH 6.5 phosphate buffer at room temperature for 30 min. These conditions quantitatively converted the scFv into a tagged GFP (Figure 4b,c). Tagged GFP constructs were then incubated with resuspended HER2+ and HER2– cells (SK-BR-3 and MDA-MB-468, respectively), at 2 μg per 100 μL of cell suspension. Binding was tracked via flow cytometry, where the scFv–GFP construct bound specifically to HER2-expressing cells but not HER2-negative cells (Figure 4d). No cellular fluorescence was observed with either GFP alone or in controls where GFP was not coupled to the scFv (Figures S17 and S18). These and corresponding control experiments showed that only highly exposed tyrosines near the N- or C-terminus are capable of participating in the reaction because of the lack of further substrate oxidation.22 This was quite important, as the scFv is known to have 15 native tyrosine residues, with eight located in the antigen binding site.46 Furthermore, these results demonstrate the utility of tyrosinase-mediated conjugation for site-specific protein–protein coupling while preserving targeted immunoglobulin binding affinity.
Conclusion
This study establishes that tyrosinase activation can achieve site-specific protein–protein coupling through the formation of cysteine–tyrosine linkages. This simple approach allows for the targeted modification of proteins with a diverse array of molecules in potentially any desired location using an inexpensive, commercially available enzyme as the catalyst. It does not require additional modification steps or exposure to aggressive reaction conditions, conserving protein folding and function. It also offers the advantage that the attachment site can be moved by changing the location of the introduced thiol component, which is an inherent advantage of cysteine-based modification chemistry. In particular, the success of Cas9 delivery via peptide conjugation highlights a potential application for tyrosinase-mediated coupling to facilitate the screening of candidate peptides for the cellular entry of large biomolecules without the need to clone, express, and purify individual genetic fusions for each combination. Finally, its ability to couple full-sized proteins together with high yields and minimal stoichiometric excesses suggests broad potential for the generation of protein chimeras for biotherapeutic, vaccine, and bioanalytical applications.
Acknowledgments
The authors thank Jamie Gleason for the kind gift of the plasmid containing GFP Y200C, Dr. Jeffrey Pelton for help with 900 MHz NMR data collection and analysis, and the UCB Cell Culture Facility, which is supported by The University of California, Berkeley, for providing cell cultures.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c00940.
Author Contributions
M.J.L. developed the project idea, conducted coupling reactions, ran NMR experiments, and prepared the manuscript and figures. C.F. ran cell culture assays with the help of R.J.L., assisted with writing, assisted with flow cytometry data analysis, and aided in experimental design. A.M.M. synthesized and purified the small-molecule product, assisted in the structural characterization, provided trastuzumab scFv samples, and ran scFv–GFP cell coupling studies. E.N.K. ran MS2 couplings, stability studies, and NEM capping experiments. J.C.M. contributed to experimental design, provided tyrosinase stocks, and assisted in data analysis. C.S.M. assisted in reagent preparation and conducted stability studies. S.A.R. assisted in Cas9 activity assays and the initial Cas9 coupling reactions. B.T.S. provided initial samples of 4NLS Cas9 protein for NPC assays and established the NPC line and assay protocols. C.U. ran NPC assays. J.A.D. provided funding support, assisted in experimental design, and contributed to the text and figures. M.B.F. provided funding support, assisted in data analysis, assisted with experimental design, and contributed to the text and figures.
M.B.F. was supported by the NSF (CHE-1808189). Funds for the 900 MHz NMR spectrometer were provided by the NIH (Grant GM68933). C.F. was supported by an NIH Pathway to Independence Award (K99GM118909, R00GM118909) from the National Institute of General Medical Sciences (NIGMS). J.A.D. received funding from the William M. Keck Foundation, a Collaborative MS Research Center Award from the National Multiple Sclerosis Society, the Centers for Excellence in Genomic Science of the NIH (Award RM1HG009490), the Somatic Cell Genome Editing Program of the Common Fund of the NIH (Award U01AI142817-02), and the NSF (Award 1817593). J.A.D. is an Investigator of the Howard Hughes Medical Institute and a Paul Allen Distinguished Investigator.
The authors declare the following competing financial interest(s): J.A.D. is an Investigator of the Howard Hughes Medical Institute; executive director of the Innovative Genomics Institute at the University of California, Berkeley and the University of California, San Francisco; a co-founder of Editas Medicine, Intellia Therapeutics, and Caribou Biosciences; and a scientific adviser to Caribou, Intellia, eFFECTOR Therapeutics, and Driver. The Regents of the University of California have patents pending for CRISPR technologies and the use of tyrosinase in the manner described herein on which the authors are inventors.
Supplementary Material
References
- Hermanson G. T.Bioconjugate Techniques, 2nd ed.; Elsevier, 2008. 10.1016/B978-0-12-370501-3.X0001-X. [DOI] [Google Scholar]
- Witus L. S.; Francis M. B. Using synthetically modified proteins to make new materials. Acc. Chem. Res. 2011, 44, 774–783. 10.1021/ar2001292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephanopoulos N.; Francis M. B. Choosing an effective protein bioconjugation strategy. Nat. Chem. Biol. 2011, 7, 876–884. 10.1038/nchembio.720. [DOI] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. . [DOI] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem., Int. Ed. 2009, 48, 6974–6998. 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson P.; Muir T.; Clark-Lewis I.; Kent S. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- Theile C. S.; et al. Site-specific N-terminal labeling of proteins using sortase-mediated reactions. Nat. Protoc. 2013, 8, 1800–1807. 10.1038/nprot.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. G.; Chandrasegaran S. Chimeric restriction endonuclease. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 883–887. 10.1073/pnas.91.3.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolukbasi M. F.; et al. Orthogonal Cas9–Cas9 chimeras provide a versatile platform for genome editing. Nat. Commun. 2018, 9, 4856. 10.1038/s41467-018-07310-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes B. L.; et al. CRISPR-Cas9 Circular Permutants as Programmable Scaffolds for Genome Modification. Cell 2019, 176, 254–267. 10.1016/j.cell.2018.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusion Protein Technologies for Biopharmaceuticals: Applications and Challenges; Schmidt S. R., Ed.; Wiley, 2013. 10.1002/9781118354599. [DOI] [Google Scholar]
- Behrens C. R.; et al. Rapid chemoselective bioconjugation through oxidative coupling of anilines and aminophenols. J. Am. Chem. Soc. 2011, 133, 16398–16401. 10.1021/ja2033298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermeyer A. C.; Jarman J. B.; Francis M. B. N-Terminal Modification of Proteins with o -Aminophenols. J. Am. Chem. Soc. 2014, 136, 9572–9579. 10.1021/ja500728c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maza J. C.; et al. Enzymatic Modification of N-Terminal Proline Residues Using Phenol Derivatives. J. Am. Chem. Soc. 2019, 141, 3885–3892. 10.1021/jacs.8b10845. [DOI] [PubMed] [Google Scholar]
- Cory J. G.; Frieden E. Differential Reactivities of Tyrosine Residues of Proteins to Tyrosinase. Biochemistry 1967, 6, 121–126. 10.1021/bi00853a021. [DOI] [PubMed] [Google Scholar]
- Ito S.; Kato T.; Shinpo K.; Fujita K. Oxidation of tyrosine residues in proteins by tyrosinase. Formation of protein-bonded 3,4-dihydroxyphenylalanine and 5-S-cysteinyl-3,4-dihydroxyphenylalanine. Biochem. J. 1984, 222, 407–411. 10.1042/bj2220407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selinheimo E.; Autio K.; Kruus K.; Buchert J. Elucidating the Mechanism of Laccase and Tyrosinase in Wheat Bread Making. J. Agric. Food Chem. 2007, 55, 6357–6365. 10.1021/jf0703349. [DOI] [PubMed] [Google Scholar]
- Lantto R.; Puolanne E.; Kruus K.; Buchert J.; Autio K. Tyrosinase-Aided Protein Cross-Linking: Effects on Gel Formation of Chicken Breast Myofibrils and Texture and Water-Holding of Chicken Breast Meat Homogenate Gels. J. Agric. Food Chem. 2007, 55, 1248–1255. 10.1021/jf0623485. [DOI] [PubMed] [Google Scholar]
- Thalmann C. R.; Lötzbeyer T. Enzymatic cross-linking of proteins with tyrosinase. Eur. Food Res. Technol. 2002, 214, 276–281. 10.1007/s00217-001-0455-0. [DOI] [Google Scholar]
- Bruins J. J.; et al. Inducible, Site-Specific Protein Labeling by Tyrosine Oxidation–Strain-Promoted (4 + 2) Cycloaddition. Bioconjugate Chem. 2017, 28, 1189–1193. 10.1021/acs.bioconjchem.7b00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montanari E.; et al. Tyrosinase-Mediated Bioconjugation. A Versatile Approach to Chimeric Macromolecules. Bioconjugate Chem. 2018, 29, 2550–2560. 10.1021/acs.bioconjchem.8b00227. [DOI] [PubMed] [Google Scholar]
- Marmelstein A. M.; et al. Tyrosinase-Mediated Oxidative Coupling of Tyrosine Tags on Peptides and Proteins. J. Am. Chem. Soc. 2020, 142, 5078–5086. 10.1021/jacs.9b12002. [DOI] [PubMed] [Google Scholar]
- Ito S.; Prota G. A facile one-step synthesis of cysteinyldopas using mushroom tyrosinase. Experientia 1977, 33, 1118–1119. 10.1007/BF01946005. [DOI] [PubMed] [Google Scholar]
- Agrup G.; Hansson C.; Rorsman H.; Rosengren E. The effect of cysteine on oxidation of tyrosine dopa, and cysteinyldopas. Arch. Dermatol. Res. 1981, 272, 103–115. 10.1007/BF00510400. [DOI] [PubMed] [Google Scholar]
- Wu W.; Hsiao S. C.; Carrico Z. M.; Francis M. B. Genome-free viral capsids as multivalent carriers for taxol delivery. Angew. Chem., Int. Ed. 2009, 48, 9493–9497. 10.1002/anie.200902426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capehart S. L.; Coyle M. P.; Glasgow J. E.; Francis M. B. Controlled integration of gold nanoparticles and organic fluorophores using synthetically modified MS2 viral capsids. J. Am. Chem. Soc. 2013, 135, 3011–3016. 10.1021/ja3078472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valegård K.; Liljas L.; Fridborg K.; Unge T. The three-dimensional structure of the bacterial virus MS2. Nature 1990, 345, 36–41. 10.1038/345036a0. [DOI] [PubMed] [Google Scholar]
- Farkas M. E.; et al. PET Imaging and Biodistribution of Chemically Modified Bacteriophage MS2. Mol. Pharmaceutics 2013, 10, 69–76. 10.1021/mp3003754. [DOI] [PubMed] [Google Scholar]
- Carrico Z. M.; Romanini D. W.; Mehl R. A.; Francis M. B. Oxidative coupling of peptides to a virus capsid containing unnatural amino acids. Chem. Commun. 2008, 1205. 10.1039/b717826c. [DOI] [PubMed] [Google Scholar]
- Muñoz-Muñoz J. L.; et al. Catalytic oxidation of o-aminophenols and aromatic amines by mushroom tyrosinase. Biochim. Biophys. Acta, Proteins Proteomics 2011, 1814, 1974–1983. 10.1016/j.bbapap.2011.07.015. [DOI] [PubMed] [Google Scholar]
- Maza J. C.; et al. Secondary Modification of Oxidatively-Modified Proline N-termini for the Construction of Complex Bioconjugates. Org. Biomol. Chem. 2020, 18, 1881–1885. 10.1039/D0OB00211A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardirossian N.; Head-Gordon M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. 10.1080/00268976.2017.1333644. [DOI] [Google Scholar]
- Cossi M.; Rega N.; Scalmani G.; Barone V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- Barone V.; Cossi M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. 10.1021/jp9716997. [DOI] [Google Scholar]
- Takano Y.; Houk K. N. Benchmarking the conductor-like polarizable continuum model (CPCM) for aqueous solvation free energies of neutral and ionic organic molecules. J. Chem. Theory Comput. 2005, 1, 70–77. 10.1021/ct049977a. [DOI] [PubMed] [Google Scholar]
- Schiapparelli L. M.; et al. Direct detection of biotinylated proteins by mass spectrometry. J. Proteome Res. 2014, 13, 3966–3978. 10.1021/pr5002862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine S. D.; Reid R.; Robinson L.; Ashley G. W.; Santi D. V. Long-Term Stabilization of Maleimide–Thiol Conjugates. Bioconjugate Chem. 2015, 26, 145–152. 10.1021/bc5005262. [DOI] [PubMed] [Google Scholar]
- Staahl B. T.; et al. Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat. Biotechnol. 2017, 35, 431–434. 10.1038/nbt.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya H.; Gibo D. M.; Garg S.; Kridel S.; Debinski W. An interleukin 13 receptor α 2-specific peptide homes to human Glioblastoma multiforme xenografts. Neuro. Oncol. 2012, 14, 6–18. 10.1093/neuonc/nor141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender P. A.; et al. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 13003–13008. 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElSohly A. M.; et al. ortho-Methoxyphenols as Convenient Oxidative Bioconjugation Reagents with Application to Site-Selective Heterobifunctional Cross-Linkers. J. Am. Chem. Soc. 2017, 139, 3767–3773. 10.1021/jacs.6b12966. [DOI] [PubMed] [Google Scholar]
- Allen J.; Brock D.; Kondow-McConaghy H.; Pellois J.-P. Efficient Delivery of Macromolecules into Human Cells by Improving the Endosomal Escape Activity of Cell-Penetrating Peptides: Lessons Learned from dfTAT and its Analogs. Biomolecules 2018, 8, 50. 10.3390/biom8030050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronican J. J.; et al. Potent Delivery of Functional Proteins into Mammalian Cells in Vitro and in Vivo Using a Supercharged Protein. ACS Chem. Biol. 2010, 5, 747–752. 10.1021/cb1001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lönn P.; et al. Enhancing Endosomal Escape for Intracellular Delivery of Macromolecular Biologic Therapeutics. Sci. Rep. 2016, 6, 32301. 10.1038/srep32301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futaki S.; et al. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–40. 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- Eigenbrot C.; Randal M.; Presta L.; Carter P.; Kossiakoff A. A. X-ray structures of the antigen-binding domains from three variants of humanized anti-p185HER2 antibody 4D5 and comparison with molecular modeling. J. Mol. Biol. 1993, 229, 969–995. 10.1006/jmbi.1993.1099. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.