

Abstract
Light-driven 3D printing to convert liquid resins into solid objects (i.e., photocuring) has traditionally been dominated by engineering disciplines, yielding the fastest build speeds and highest resolution of any additive manufacturing process. However, the reliance on high-energy UV/violet light limits the materials scope due to degradation and attenuation (e.g., absorption and/or scattering). Chemical innovation to shift the spectrum into more mild and tunable visible wavelengths promises to improve compatibility and expand the repertoire of accessible objects, including those containing biological compounds, nanocomposites, and multimaterial structures. Photochemistry at these longer wavelengths currently suffers from slow reaction times precluding its utility. Herein, novel panchromatic photopolymer resins were developed and applied for the first time to realize rapid high-resolution visible light 3D printing. The combination of electron-deficient and electron-rich coinitiators was critical to overcoming the speed-limited photocuring with visible light. Furthermore, azo-dyes were identified as vital resin components to confine curing to irradiation zones, improving spatial resolution. A unique screening method was used to streamline optimization (e.g., exposure time and azo-dye loading) and correlate resin composition to resolution, cure rate, and mechanical performance. Ultimately, a versatile and general visible-light-based printing method was shown to afford (1) stiff and soft objects with feature sizes <100 μm, (2) build speeds up to 45 mm/h, and (3) mechanical isotropy, rivaling modern UV-based 3D printing technology and providing a foundation from which bio- and composite-printing can emerge.
Short abstract
Liquid resins that rapidly harden upon exposure to low-energy visible light are developed and applied to high-resolution projection-based 3D printing across the color spectrum.
Introduction
3D printing has revolutionized the way the world creates, influencing nearly every aspect of modern society—from the consumer market to aerospace and medical technologies.1−4 With the help of computer-aided design, digital objects with customized form factors are realized through successive layering of material (i.e., additive manufacturing). The technology has gained traction in part by overcoming efficiency issues and intermediary processes, such as milling, carving, and machining, affiliated with conventional manufacturing. To date, engineering feats have dominated the arena of 3D printing, providing a number of elegant methods via extrusion, powder bed fusion, jetting, and light induced polymerization (e.g., stereolithography, SLA, and digital light processing, DLP).1−4 Among them, SLA and DLP utilize light to transform matter from liquid resins to solid objects (i.e., photocuring).5−12 DLP in particular has drawn wide attention owing to several attractive features, namely, some of the fastest build rates (>100 mm/h or <5 s/layer), highest feature resolution (<100 μm features), wide ink viscosity tolerance (up to ∼5000 cP), small footprint (fits on a standard benchtop), and low cost (starting at ∼$300).2,5,7 However, the demand for faster printing, milder operating conditions, better resolution, and a wider materials scope continues to drive research efforts and provides an opportunity for chemical innovation.
Contemporary photocuring processes in 3D printing are initiated by high-energy ultraviolet (UV) light, which provides rapid polymerization and correspondingly short build times (∼seconds). As an alternative, visible light offers numerous benefits, including reduced cost and energy of irradiation from readily available and modular light emitting diodes (LEDs), improved biocompatibility and functional group tolerance, greater depth of penetration, and reduced scattering (Figure 1).10,12−16 As such, visible light photocuring has the potential to enable next-generation designer material fabrication, including hydrogels containing live cells,17 opaque composites,16 and wavelength-selective multimaterial structures18−23 that promise to advance a range of applications, from structural plastics to tissue engineering and soft robotics.
Figure 1.
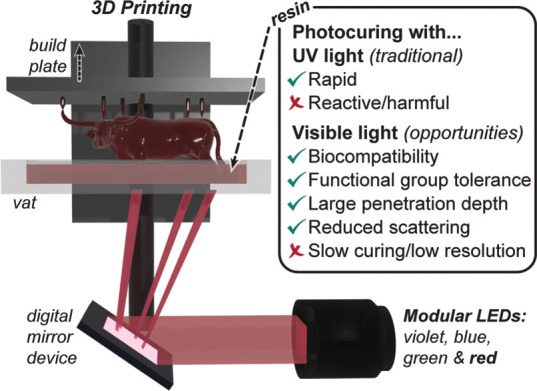
Presented method and opportunities offered by visible light photocuring for 3D printing. Illustration shows the general mechanism for digital light processing (DLP), with exchangeable light emitting diodes (LEDs). For UV and visible light photocuring, a green check mark indicates an attractive feature, while a red “X” represents a current challenge.
Nonetheless, the utility of low-energy visible LEDs (>420 nm), to the best of our knowledge, has never been demonstrated for rapid high-resolution DLP-based 3D printing. In part, this is due to a lack of commercially available 3D printers with monochromatic visible LEDs, but the grand challenge is to achieve efficient photocuring at these wavelengths to facilitate rapid builds (>10 mm/h or <50 s/layer) with high resolution (<100 μm features) and compete with contemporary UV/violet-light-based additive manufacturing. To improve visible light photocuring efficiency requires a chemical understanding of how reactive curing agents (e.g., radicals or ions) are generated.
Following absorption of light by a chromophore, photocuring can occur by one of two mechanisms: (i) direct photolysis of a photoinitiator (PI) or (ii) electron transfer from a photoredox catalyst (PRC) to a coinitiator followed by bond scission to generate radicals or ions.24−28 Although PI compounds often provide rapid photocuring, they rely on a “forbidden” n → π* transition, which generally corresponds to weak absorption that is restricted to short wavelengths of light (<420 nm, violet)—exceptions being titanocenes10 and acylgermanes29 that absorb up to ∼500 nm (blue/green). In contrast, the use of a PRC enables excitation via a π → π* transition, providing strong absorption that can extend to longer wavelengths (>500 nm, green to NIR). Leaders in materials photochemistry have demonstrated visible–NIR photocuring of a variety of resins, primarily comprising acrylic and epoxy monomers and cross-linkers.10,30−32 However, due to the multistep reaction mechanism (e.g., electron transfer), the cure times are typically slow (>60 s) and require relatively high irradiation intensities (>20 mW/cm2), making them impractical for standard DLP technology.
Herein, rapid visible-light-driven chemistry using a novel three-component system was developed, characterized, and systematically implemented in high-resolution 3D printing. Resin optimization was facilitated by real time Fourier transform infrared (RT-FTIR) spectroscopy and photorheology, and printing was enabled by a DLP system housing visible LEDs (Figure 1). Comprehensive studies on photocuring rate, feature resolution, and mechanical properties were conducted to inform future development and utility of the present chemistry in materials synthesis for 3D printing and other emerging areas.
Results and Discussion
To replicate traditional UV-based rapid high-resolution DLP 3D printing with low-energy visible LEDs necessitated the development of reactive panchromatic resins. These light sensitive polymer resins incorporated monomer, cross-linker, and a PI or PRC + coinitiators (i.e., donor, D, and/or acceptor, A). The monomer, cross-linker, and PI were held constant, while various PRC and coinitiator compounds were rationally combined to obtain a mixture that enables photocuring on the order of seconds. Specifically, three-component systems (PRC + two coinitiators) were examined to both promote PRC regeneration and potentially double the concentration of radicals produced per photon absorbed (Figure 2a).
Figure 2.
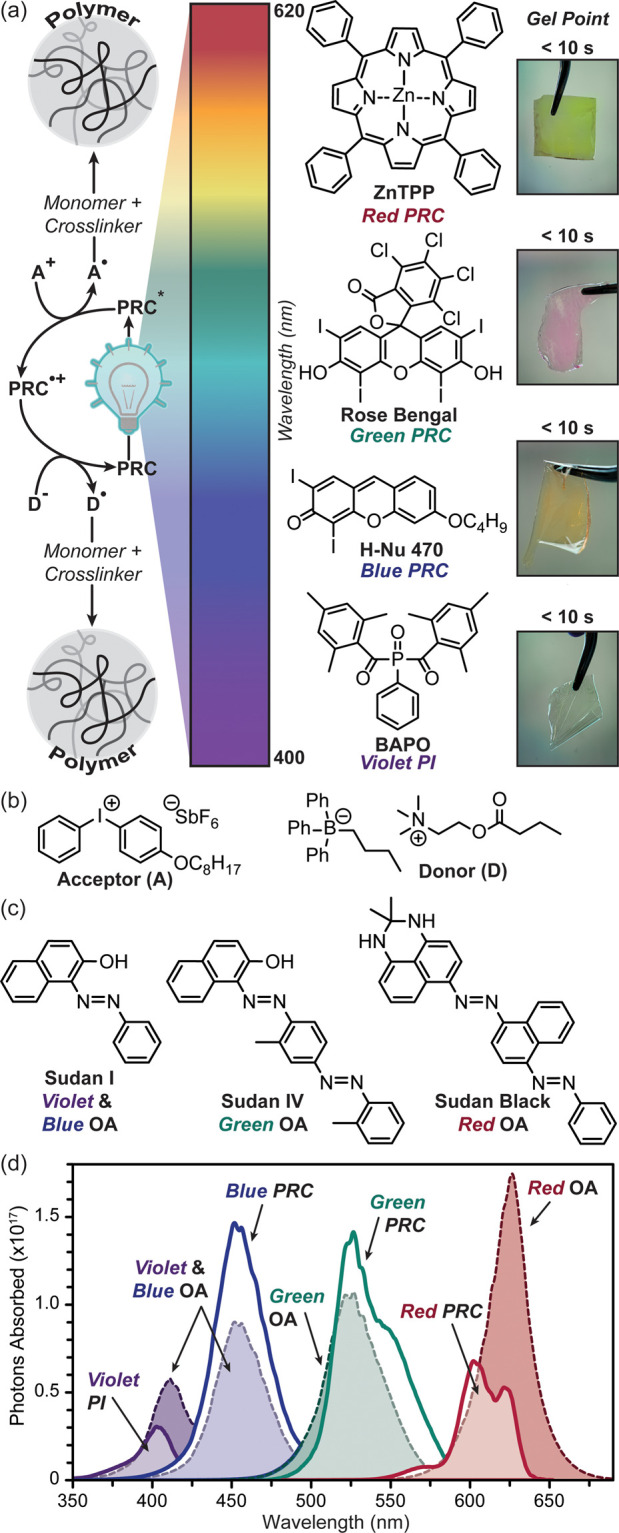
Visible light curing. (a) General mechanism (oxidative quenching) for a three-component system (left). Chemical structures of photoinitiator (PI) and photoredox catalysts (PRCs), and corresponding pictures of photocured films with qualitative gel times (right). (b) Chemical structures of iodonium acceptor (A) and borate donor (D) coinitiators. (c) Chemical structures of opaquing agents (OAs). (d) Photons absorbed vs wavelength for PI and PRC compounds at optimal photocuring concentration, and OA at 0.5 mM (red) and 1 mM (green, blue, and violet). Light exposure was from calibrated violet (405 nm), blue (460 nm), green (525 nm), and red (615 nm) LEDs at the DLP 3D printer image plane.
Resins comprised dimethyl acrylamide and trimethylolpropane triacrylate as monomer and cross-linker in a 4:1 ratio, respectively. This resin mixture was selected for its good solubilizing characteristics for PRC and coinitiator compounds in addition to fast photocuring rate, as demonstrated with a potent PI, bis-acylphosphine oxide (BAPO), used here as a violet light (405 nm) control. For reference, BAPO comprised 0.12 mol % (=0.5 wt % relative to monomer + cross-linker) of the resin (contents provided as mol % for quantitative analysis and wt % for direct comparison with the prior formulation literature).11
Rapid photocuring with blue (∼460 nm), green (∼525 nm), or red (∼615 nm) light was accomplished by replacing the violet absorbing PI with a visible absorbing PRC and a donor (D) and acceptor (A) coinitiator. In the present study, all resins contain the same electron-deficient radical coinitiator (A), [4-(octyloxy)phenyl](phenyl)iodonium hexafluoroantimonate diphenyliodonium (0.3 mol % = 2 wt %). To find the optimal partner for A, different electron-rich coinitiators (D) (e.g., amine, silane, and organoborate derivatives) were screened together with various PRCs (e.g., camphorquinone, xanthene, cyanine, and porphyrin derivatives). Initial qualitative assessments of visible light curing (e.g., blue, green, or red) were accomplished by irradiating 100 μm thick, argon degassed resins between glass microscope slides using a low-intensity light (∼5 mW/cm2) until film formation was noted; for comparison, this light source was ∼2–20× less intense than an unfocused commercial laser pointer (≈10–100 mW/cm2). Subsequently, RT-FTIR spectroscopy was used to more closely analyze a subset of samples that showed solidification in ≲60 s (Table S5 in the Supporting Information). The organoborate derivative, 2-(butyryloxy)-N,N,N-trimethylethan-1-aminium butyltriphenylborate,33,34 consistently resulted in the fastest curing rates and was thus used in all subsequent resin formulations (0.04 mol % = 0.2 wt %) (Figure 2b). In combination with PRC compounds (optimized concentration), H-Nu470 (0.02 mol % = 0.1 wt %), Rose Bengal (0.01 mol % = 0.1 wt %), and zinc tetraphenylporphyrin (ZnTPP, 0.05 mol % = 0.3 wt %), solidification in <10 s was demonstrated upon exposure to blue, green, and red light irradiation, respectively. For comparison, curing in <10 s was observed for the violet resin, but with 2–10× the concentration of PI (BAPO, 0.12 mol % = 0.5 wt %) relative to each PRC (Figure 2a, inset images). These components serve as the “active” ingredients in all subsequent resins for photocuring.
An additional component often present in lithographic resins is an opaquing agent (OA), which serves as a “passive” absorber (i.e., does not elicit a chemical reaction) to control the optical path length of incident light and, in-turn, improve resolution and homogeneity of curing (particularly in the z-dimension).35 Ideally, OAs (e.g., dyes and pigments) operate by absorbing light in the same wavelength range as the PI or PRC within the emission profile of the incident light source. Rapid excited state relaxation is desirable for OAs to preclude electron transfer. Effective OAs in 3D printing reduce the penetration depth of light to mitigate cure through (i.e., curing unwanted regions within previous layers of an object). A series of azo-dyes were selected as OAs given that cis/trans isomerization and intramolecular proton transfer (when phenol functionality is present ortho to the azo group) provide the desired rapid relaxation.36 Specifically, Sudan I, IV, and Black (Figure 2c) were chosen for their good absorption overlap with both PI/PRC absorption and LED emission profiles (Figure 2d).
To correlate the effect of optical attenuation on photocuring parameters, the number of photons absorbed by PI and PRC relative to the corresponding OA was determined. This was accomplished by measuring the extinction coefficients for each component using UV–vis absorption spectroscopy (Figures S5–S11 in the Supporting Information) and integrating that with respect to the emission profile for the different LEDs at a particular intensity (see the Calculating Photons Absorbed section in the SI for details). Quantitative LED absorption by PI and PRC compounds at their optimized concentrations (vide supra) was calculated and compared to that for OAs at 0.5 mM (Sudan Black) and 1 mM (rest) (for reference, 1 mM = 0.01 mol % in the prescribed resin). This analysis revealed the following percent absorption of each LED: (violet) 8% PI and 19% OA, (blue) 54% PRC and 33% OA, (green) 57% PRC and 43% OA, and (red) 17% PRC and 42% OA (see Table S3 in the Supporting Information). Overall, the three OAs and the blue and green PRC compounds absorbed significantly more than the violet PI and red PRC. The red light absorbing OA was particularly potent, as can be seen in Figure 2d as the tallest peak at half the concentration relative to the other OAs. Notably, the green light absorbing OA had reduced overlap with the corresponding PRC at longer LED wavelengths (∼570+ nm) (Figure 2d), which may result in unwanted cure through. Based on these values, we hypothesized that the resin activated by blue light would need the least amount of OA to prevent cure through and that the red light sensitive resin would be greatly influenced by small fluctuations in [OA]. Notably, while the red PRC (ZnTPP) absorbed a fraction of the photons (∼3× fewer) relative to the other PRC compounds at different wavelengths, it maintained rapid photocuring (<10 s) at low light intensity (∼5 mW/cm2), making it an excellent candidate for 3D printing.
To demonstrate the utility of these novel resins in 3D printing, a custom DLP (Figure S3 in the Supporting Information) with modular visible LEDs (peak emission ≈ 405, 460, 525, 615 nm) was constructed. The minimum volume element (voxel) on the printer has lateral dimensions of 20 × 20 μm2 and a vertical dimension of 25 μm (i.e., layer thickness). To examine resolution and mechanical properties of prints in conjunction with curing rates necessitated the use of a 100 μm layer thickness, since RT-FTIR and photorheology monitoring required sample thickness ≥100 μm for adequate signal. Beneficially, the thicker layers facilitate faster builds (vide infra). A novel printing method, referred to here as “resolution print”, was developed to efficiently optimize resolution and build speed. Critically, the method provides control over exposure time within different locations of a single layer, which facilitated rapid screening (Figure 3a). This printing method was used to correlate exposure time/layer and OA concentration, [OA], to lateral (x, y) and vertical (z) resolution. In this assessment, each resolution print contained a set of squares that were simultaneously printed, varying exposure time/layer. In the bottom half of each square was an array of smaller patterns that were 1–16 pixels wide, used to characterize resolution. As shown in Figure 3b, a 12 square array was printed (4 × 4 mm2 each square) with 4 × 100 μm layers, and exposure times from 1 to 12 s/layer (time in seconds engraved in each square). For ease of handling, the square array was printed on a rectangular base (12 layers at 12 s/layer). Thus, a single print contained resolution information about 12 exposure times at a particular [OA], streamlining optimization.
Figure 3.

3D print optimization protocol using the “resolution print” method. (a) Digital projection layer at one second when all squares are illuminated simultaneously (white regions correspond to exposure) (top left). Expanded square for 11 s exposure/layer showing the line and pixel array for the projection (bottom left) and red light resolution print taken with optical profilometry (bottom right). Chemical composition for photocured stiff resin comprising dimethyl acrylamide and trimethylolpropane triacrylate (top right). (b) Optical images of prints from the four resins by exposure color (100 μm layers, 4 layers for each numbered square on top of 12 base layers). (c) z-resolution analysis: 3D image of a corner on the 11 s square outlined as box i in the profilometry image above in part a. Height and sidewall angle (SWA) were determined using an average of 10 line traces per corner (with one shown for reference). (d) Graph of thickness and SWA vs exposure time for the red resin. The light and dark red shaded regions represent exposure times (or processing windows) to provide optimal z-resolution without OA and with optimized [OA], respectively (theoretical height = 400 μm). (e) x-, y-resolution analysis: Plot of surface areas measured for 16, 8, 4, and 2 pixel wide squares using optical profilometry. Surface areas are averages from 3, 5, 8, and 10 squares for the 2, 4, 8, and 16 pixel wide features, respectively (error represents ±1 standard deviation). Dashed lines represent theoretical surface areas, and error bars represent ±1 standard deviation, showing how OA enhances print fidelity and reproducibility of features below 100 μm in length.
Resolution prints were accomplished using violet, blue, green, and red resins with varying amounts of the corresponding OA to identify optimal resin composition and exposure time/layer (Figure 2c). As the [OA] was increased, the first square pattern to appear occurred at longer exposure times, indicating an inverse relationship between build speed and [OA]. This suggests that the OA was effectively competing for photons with that of the PI or PRC present (i.e., passive absorption). Even with OA present, patterns were observed within 2 s for all resin formulations, demonstrating the ability to perform rapid visible light curing (Figure 3b). Of note, for green and red light sensitive samples a blanket of inert gas (e.g., nitrogen or argon) during the print was necessary to achieve these speeds, while violet and blue light printing was less affected. Under ambient conditions, polymerizations are delayed for varying lengths of time (i.e., inhibition period). The delay was attributed to quenching of triplet excited states by oxygen, supported by longer inhibition periods observed for the red light PRC (ZnTPP37) known to provide high triplet yields.
Concomitant with reduced cure rates upon increasing [OA] was an improved z-resolution as a result of diminished cure through. The z-resolution was characterized by measuring the thickness and angle between the top surface and connecting vertical wall (i.e., sidewall angle, SWA) for each time point on a print using 3D imaging (optical profilometry) (Figure 3c). A representative topographical profile for an 11 s/layer square on an optimized red light resolution print (with OA) is shown in Figure 3a. Heights and SWA values were determined from an average of 10 positions, using a line measurement tool as depicted in Figure3c (i). Ideal values for the resolution prints reflect the imported digital images, which equated to a height of 400 μm (4 layers at 100 μm/layer) and an SWA of 90°. Without OA, the maximum thickness and SWA did not always occur at the same exposure time/layer (Figures S14–S18 in the Supporting Information). For example, the red resin without OA had a maximum thickness of 384 μm at 5 s/layer and an SWA of 78° at 4 s/layer (Figure 3c, bottom). Additionally, at shorter and longer exposure times, both thickness and SWA rapidly decrease, providing a narrow processing window, hindering reproducibility. This was exemplified by setting a tolerance of ±5% for thickness (400 ± 20 μm) and ±17% for SWA (90 ± 15°), which results in a processing window of ∼1 s without OA present (exposure time/layer ∼5 s). This short processing window (and corresponding drop in thickness) was attributed to incomplete curing and cure through prior to and after 5 s, respectively. Increasing [OA] resulted in a convergence in exposure time for both optimal thickness and SWA, along with a much wider and more well-defined processing window (≥6 s). Consistent with our hypothesis, blue resins did not require any OA to mitigate cure through, which was attributed to the strong absorption by the blue PRC. Also in line with our prior observations was the small amount of OA necessary to mitigate cure through for prints with the green resin (0.003 mol % OA) and, to a greater effect, the red resin (0.0005 mol % OA) (Table S1 in the Supporting Information). At the optimal [OA] for the red resin, an average thickness of 405 μm and SWA of 85° were observed across a wide exposure window from 7 to 12 s/layer (Figure 3c). Complete thickness, SWA, and processing window data for all resins are provided in the Supporting Information (Figures S14–S18). Optimal exposure times/layer fell between 8 and 11 s for all resins, which corresponds to a build rate of 45–33 mm/h (not including the recoating process). Visible light printing was shown to offer wide processing windows to facilitate reproducibility, while providing objects with z-resolution and build speeds that rival contemporary UV-based DLP 3D printers.2
To assess x-, y-resolution, surface areas for the pixel arrays were characterized from the topographical images, as shown in Figure 3a (ii). Specifically, 16, 8, 4, and 2 pixel wide pixel arrays were analyzed by imaging the surfaces for both red (Figure 3e) and violet (Figure S20 in the Supporting Information) resolution prints. Samples with and without OA were characterized and compared at optimal exposure times (8 and 4 s/layer for violet with and without OA, and 11 and 5 s/layer for red with and without OA, respectively). Surface area analysis revealed that the presence of OA results in a better match to theoretical, corresponding with larger SWAs (dashed lines in Figure 3d). The improved x-, y-resolution was hypothesized to result from attenuating light outside of predefined irradiation zones (e.g., scattered light). Additionally, it was noted that deviation of surface area from theoretical values increased as feature size decreased. For red prints with optimized [OA] at an 11 s exposure time/layer, the surface area for decreasing features (320 × 320 to 40 × 40 μm2) deviated from theoretical values by 6%, 20%, 46%, and 78%, respectively. The diminished fidelity for smaller features was attributed to incomplete curing. As these polymerizations are exothermic, small features do not generate as much heat as larger features, which can autoaccelerate curing (as seen with frontal polymerizations).38,39 Irrespective of the feature size, reproducibility was excellent, as made evident by the small standard deviations in surface area values between same size squares. The present study clearly illustrates that high-resolution features (<100 μm) can be accomplished reproducibly using visible light 3D printing.
The photopolymerization rates and times to gelation were determined to inform future resin development for low-energy visible light 3D printing, among other photocuring applications in imaging, lithography, coatings, and adhesives.9 To this end, RT-FTIR spectroscopy and photorheology were accomplished, carefully matching 3D printing conditions (e.g., thickness, atmosphere, and light intensity) (Figure 4a). Specifically, the samples were 100 μm thick, under an inert environment (e.g., degassed with N2 or argon), and LED exposure intensities matched those of the 3D printer at the resin vat: 3.3, 3.4, 1.8, and 2.1 mW/cm2 for violet, blue, green, and red, respectively. As a control, data were collected for 10 s in the dark, showing no polymerization prior to light exposure, which demonstrates the efficient temporal nature of these photosystems. The near IR (NIR) absorption region (8000–4000 cm–1) was monitored using RT-FTIR to determine monomer/cross-linker conversion by measuring the disappearance of C=C vinylic stretches found at ∼6160 cm–1.40 The inherently weak absorption signals in the NIR (e.g., overtones of mid-IR signals) required that samples were ∼100 μm thick but enabled the utility of disposable glass slides for facile preparation and measurement (note: glass is NIR transparent, but mid-IR opaque).
Figure 4.
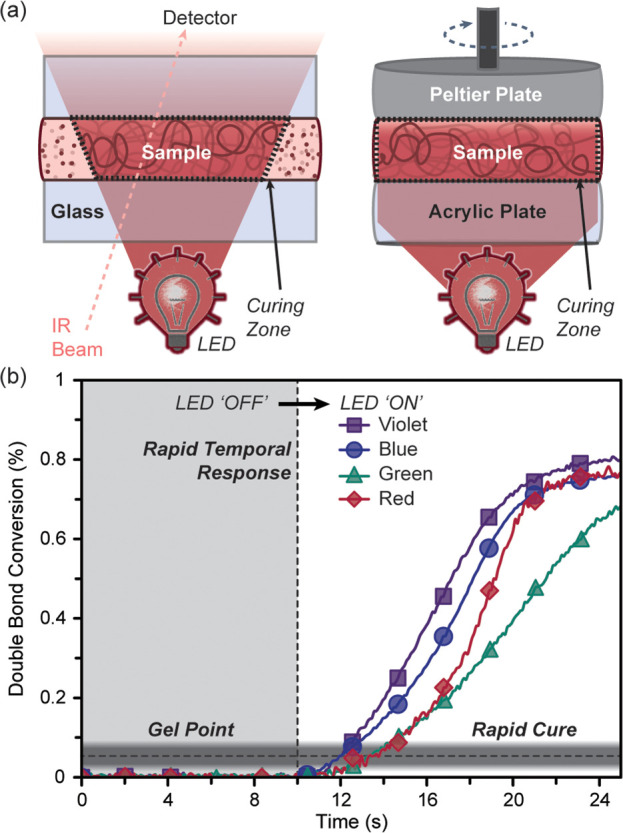
Photopolymerization rate and gel point characterization. (a) Schematic representations of the RT-FTIR and photorheology setups. (b) Plot of double bond conversion vs time for violet, blue, green, and red photopolymerizations. Experiments were performed under an inert atmosphere at an exposure intensity matching the 3D printer. Light exposure began at 10 s, highlighting the rapid temporal response. The gradient bar represents the gel point range found for all four samples (∼5–8% conversion), which was determined using a combination of photorheology and RT-FTIR.
Under the optimized printing conditions, each resin rapidly polymerized, reaching a maximum monomer conversion within 20 s of turning the light on (Figure 4b). Notably, the C=C conversion peaks at ∼80%, which was likely due to a reduction in molecular motion postgelation. Moreover, under ambient conditions a distinct induction period was observed after turning the light on, consistent with the slower printing speed noted previously. Specifically, inhibition times of 2, 5, 8, and 83 s were observed for violet, blue, green, and red, respectively (Figure S23 in the Supporting Information), confirming the distinct sensitivity of the red PRC (ZnTPP) to oxygen. However, a similar maximum polymerization rate was reached upon oxygen consumption. The similar rate was attributed to either efficient regeneration of the PRC in the tricomponent photosystem (Figure 2a) and/or a rate limiting step other than electron transfer, such that removing a fraction of PRC upon reaction with oxygen does not alter the apparent rate. These findings reinforce that oxygen removal is an effective strategy to increase photocuring rate and accordingly print speeds.
In addition to increasing photopolymerization rate by oxygen removal, the effect of light intensity was examined as a complementary route to further improve curing rate and associated build speeds. At the optimized printing conditions, the maximum apparent polymerization rates were 1.33 ± 0.05, 1.16 ± 0.05, 1.09 ± 0.09, and 1.9 ± 0.3 M/s, for violet, blue, green, and red, respectively. Increasing the light intensity to 10 mW/cm2 resulted in a ∼1.6–2× polymerization rate enhancement relative to those obtained at the printer intensities (Table S7 and Figures S27–S34 in the Supporting Information).
Photorheology was used to determine gel times and corresponding double bond conversion at the gel point to inform future resin development. Gelation was identified as the crossover between storage and loss modulus, which occurred within 2–4 s of light exposure. This short gel time was consistent with the first appearance of features on the resolution prints (Figure 3b). Specifically, gel points were 1.8 ± 0.2, 1.9 ± 0.1, 3.4 ± 0.2, and 4.2 ± 0.2 s for violet, blue, green, and red respectively (Figures S35–S38 and Table S8 in the Supporting Information). The measured double conversions at the gel points fell within a similar range for all resins (∼5–8%), which was expected given the constant monomer and cross-linker composition in each resin (Figure 4b, semitransparent gray bar). The C=C conversion for each resin at the optimized 3D printing exposure time/layer (∼8–11 s) occurred at ∼50–70%, which falls below the maximum conversion (∼80%). Therefore, polymerizable vinylic residues should be present at the interface of each layer during the 3D printing process, which can facilitate covalent bonding and increase mechanical strength.
To push the boundaries of additive manufacturing from traditional hobbyist printing and models to applications that require both precision and structural integrity necessitated a thorough understanding of mechanical performance. To this end, dogbones (ASTM D638) from each resin formulation were 3D printed under optimized conditions and subjected to tensile testing (Figure 5). Mechanical properties of interest included stiffness (E, Young’s modulus), yield strength (σy), and strain at fracture (εf). Following standard protocols, all objects were postcured with UV light prior to tensile testing (20 min, centered at ∼370 nm). As a control, postcuring red light printed dogbones with red light in place of UV light provided nearly identical mechanical performance (Figure S39 in the Supporting Information). Excitingly, E, σy, and εf were consistent for all resins, suggesting that the presence of a tricomponent photoactive system does not alter mechanical performance: E = 997 ± 62 MPa (violet), 1022 ± 63 MPa (blue), 986 ± 102 MPa (green), and 1043 ± 94 MPa (red) (Figure 5a and Table S9).
Figure 5.
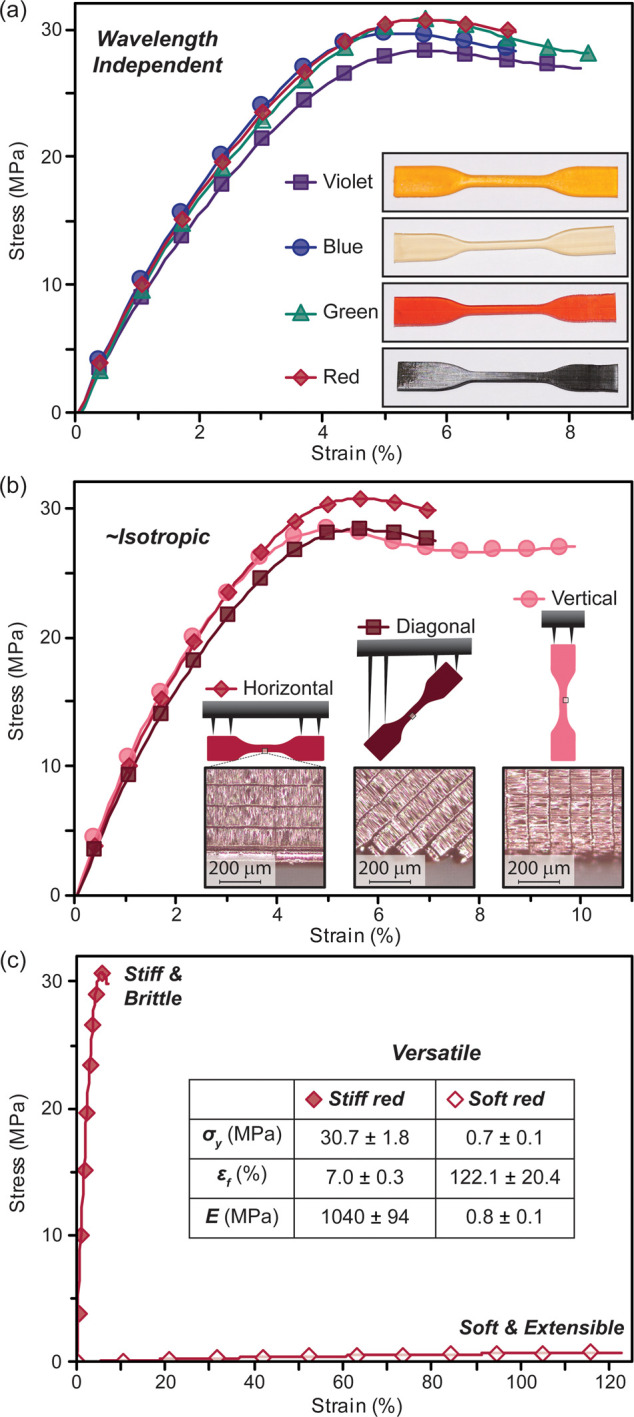
Mechanical testing of 3D printed dogbones. (a) Stress–strain curves of violet, blue, green, and red dogbones showing wavelength independent properties. (b) Red dogbones printed at three different edge-on angles (horizontal, diagonal, and vertical), demonstrating near isotropic mechanical properties. Insets are drawings of each dogbone orientation, with supports to the build platform shown, and close up images to visualize each layer. (c) Stress–strain curves of stiff and elastic red resin. The inset shows a table highlighting the disparate mechanical properties, and associated versatility of the platform.
Mechanical uniformity of visible light printed objects was examined, as layered production is often challenged with an undesirable anisotropic mechanical response with weakness at the boundaries between adjacent layers.41−43 To this end, dogbones from the optimized red resin were printed at three different edge-on angles—horizontal (0°), vertical (90°), and diagonal (45°) (Figure 5b). Optical profilometry images of the dogbones printed at different angles clearly show the layers and demonstrates excellent thickness accuracy (102 ± 1, 100 ± 2, and 99 ± 2 μm/layer for horizontal, vertical, and diagonal angles, respectively) (Figure 5b, inset). Compellingly, E values were unperturbed by changing print angle (average E = 1083 ± 47 MPa), and only small fluctuations in σy and εf were observed: σy = 31 ± 2, 31 ± 2, and 29 ± 2 MPa for horizontal, diagonal, and vertical angles, respectively (Figure 5b and Table S10 in the Supporting Information). These minor differences are in accord with other objects printed via UV-based vat photopolymerization.41−43
To demonstrate the versatility of visible light 3D printing, mechanically and chemically disparate objects were prepared (e.g., stiff vs soft and hydrophobic vs hydrophilic). The previous resin provided a stiff and brittle object, which was attributed to the high glass transition temperature for poly(dimethyl acrylamide) (Tg ≈ 90 °C) in conjugation with a large amount of cross-linker (i.e., 20 wt % triacrylate relative to monomer). 3D printing soft and extensible objects remain an active area of research due to the challenges that arise from their natural tendency to deform during prints.44−47 A resin comprising 2-hydroxyethyl acrylate monomer with a correspondingly low polymer Tg (≈−15 °C) and a small amount of cross-linker, tetra(ethylene glycol) diacrylate (0.1 wt % relative to monomer), was formulated to provide a softer and more extensible material. With the optimized red light photosystem, dogbones were 3D printed with the new soft resin formulation. Tensile testing revealed an E = 0.8 ± 0.1 MPa, which is 3 orders of magnitude lower than those from the stiff resin (Figure 5c). Moreover, swelling tests in water for six cubes printed with either stiff or soft resin qualitatively highlight the difference in chemical makeup (Figure S41). The soft cubes had a much larger water uptake (177 ± 1% increase in weight) compared to the stiff resin (54 ± 2% increase in weight), which is attributed to a combination of lower cross-link density and increased hydrophilicity of the alcohol-laden soft network (Table S13 in the Supporting Information). The contrasting mechanical and chemical properties of objects printed with the same visible light photosystem demonstrate the versatility of the present method, which will provide access to a myriad of materials without the need for harmful UV light.
As a final demonstration, an octet truss was printed to show how visible light additive manufacturing can be used to rapidly provide high-resolution complex (e.g., hierarchical) 3D objects that are challenging or impossible to produce using traditional manufacturing processes. The octet truss was printed with the original stiff red resin using both 100 μm (Figure S42) and 25 μm (Figure 6) layer thickness. Decreasing the layer thickness facilitated reduced exposure times/layer, from 11 s at 100 μm layers to 8 s at 25 μm layers, correlating to a build rate of ∼7 mm/h. Scanning electron microscopy (SEM) images show the high printing fidelity along with individual layers at a pitch of 25.1 ± 0.6 μm. Interestingly, the final object was also red emissive under a hand-held 365 nm UV light (Figure S43 in the Supporting Information), which demonstrates that a certain amount of embedded PRC (ZnTPP) remains intact, providing an avenue toward functional 3D prints (i.e., “smart” plastics). Additional 3D prints can be found in the Supporting Information (Figures S44–S47). These sophisticated structures with custom form factors emphasize the utility of visible light 3D printing to seamlessly reproduce complex digital inputs with excellent feature fidelity.
Figure 6.

Hierarchical octet truss as a complex 3D print demonstration. (a) Digital rendering. (b) Photograph of the printed object using the stiff red resin composition and red light exposure (∼2.1 mW/cm2) for 8 s/25 μm layer. (c, d) Scanning electron microscope images at different magnifications showing the structural hierarchy.
Concluding Remarks
Novel photopolymer resins that rapidly react upon irradiation with visible light across the spectrum were described and systematically examined for use in high-resolution DLP 3D printing. Key discoveries and developments include rapid photocuring from combined iodonium (donor) and borate (acceptor) coinitiators, resolution enhancements from visible light absorbing azo-dyes (OA), and efficient optimization using a custom “resolution print” method. A DLP 3D printer with exchangeable LEDs provided rapid build speeds from 33 to 45 mm/h for low-intensity violet, blue, green, and red light exposure (∼2–3 mW/cm2). RT-FTIR and photorheology were used to judiciously correlate chemical composition of resins to photocuring rate, x-, y- ,z-resolution, build speed, and mechanical properties. Inclusion of OA compounds into the resin formulation was shown to enhance resolution, with features <100 μm in the lateral dimension and 25 μm layer thickness, while simultaneously enhancing reproducibility by widening the processing window prior to cure through from ∼1 to ≥6 s. Mechanical properties were shown to be nearly independent of curing wavelength and the print orientation, demonstrating the production of relatively isotropic parts. Versatility was highlighted by rapidly producing both stiff and soft objects with stiffness ranging from ∼1480 to 0.8 MPa. Finally, objects with intricate form factors were printed as a testament to the 3D capabilities offered by the present technique. As a future endeavor, increasing light intensity (up to ∼10 mW/cm2) provides an avenue toward even faster visible-light-based 3D printing (∼1.5–2×), and different additives (e.g., oxygen scavengers)48 may be incorporated in the resin formulation to mitigate air sensitivity and enable ambient printing. The foundational studies provided herein will serve as a road map for further development of visible and NIR photocuring with excellent spatial control, material compatibility, and utility that extends beyond 3D printing (e.g., coatings, adhesives, imaging, and lithography). As a result, we envision numerous exciting and transformative interdisciplinary research opportunities in academic, industrial, and medical sectors, including hierarchical multimaterial fabrication, fiber reinforced composites, and cell-laden hydrogels.
Acknowledgments
We thank the ARO STIR program of the Department of Defense (W911NF1910310) and Robert A. Welch Foundation (F-2007) for financial support. The authors acknowledge the use of shared research facilities supported in part by the Texas Materials Institute, the Center for Dynamics and Control of Materials, an NSF MRSEC (DMR-1720595), and the NSF National Nanotechnology Coordinated Infrastructure (ECCS-1542159). We thank Dr. Yun-Ho Jang at MonoPrinter for unwavering help with the custom DLP 3D printer and software design.
Glossary
Abbreviations
- SLA
stereolithography
- DLP
digital light processing
- UV
ultraviolet
- LED
light emitting diode
- PI
photoinitiator
- PRC
photoredox catalyst
- RT-FTIR
real time Fourier transform infrared
- OA
opaquing agent
- ASTM
American Society for Testing and Materials
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c00929.
Additional experimental (materials and instrumentation) and characterization details (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Narupai B.; Nelson A. 100th Anniversary of Macromolecular Science Viewpoint: Macromolecular Materials for Additive Manufacturing. ACS Macro Lett. 2020, 9, 627–638. 10.1021/acsmacrolett.0c00200. [DOI] [PubMed] [Google Scholar]
- Wallin T. J.; Pikul J.; Shepherd R. F. 3D Printing of Soft Robotic Systems. Nat. Rev. Mater. 2018, 3 (6), 84–100. 10.1038/s41578-018-0002-2. [DOI] [Google Scholar]
- Ligon S. C.; Liska R.; Stampfl J.; Gurr M.; Mülhaupt R. Polymers for 3D Printing and Customized Additive Manufacturing. Chem. Rev. 2017, 117 (15), 10212–10290. 10.1021/acs.chemrev.7b00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truby R. L.; Lewis J. A. Printing Soft Matter in Three Dimensions. Nature 2016, 540 (7633), 371–378. 10.1038/nature21003. [DOI] [PubMed] [Google Scholar]
- Tumbleston J. R.; Shirvanyants D.; Ermoshkin N.; Janusziewicz R.; Johnson A. R.; Kelly D.; Chen K.; Pinschmidt R.; Rolland J. P.; Ermoshkin A.; et al. Continuous Liquid Interface Production of 3D Objects. Science 2015, 347 (6228), 1349–1352. 10.1126/science.aaa2397. [DOI] [PubMed] [Google Scholar]
- Kelly B. E.; Bhattacharya I.; Heidari H.; Shusteff M.; Spadaccini C. M.; Taylor H. K. Volumetric Additive Manufacturing via Tomographic Reconstruction. Science 2019, 363 (6431), 1075–1079. 10.1126/science.aau7114. [DOI] [PubMed] [Google Scholar]
- Walker D. A.; Hedrick J. L.; Mirkin C. A. Rapid, Large-Volume, Thermally Controlled 3D Printing Using a Mobile Liquid Interface. Science 2019, 366 (6463), 360–364. 10.1126/science.aax1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung K.; Corrigan N.; Ciftci M.; Xu J.; Seo S. E.; Hawker C. J.; Boyer C. Designing with Light: Advanced 2D, 3D, and 4D Materials. Adv. Mater. 2020, 32, 1903850. 10.1002/adma.201903850. [DOI] [PubMed] [Google Scholar]
- del Barrio J.; Sánchez-Somolinos C. Light to Shape the Future: From Photolithography to 4D Printing. Adv. Opt. Mater. 2019, 7 (16), 1900598. 10.1002/adom.201900598. [DOI] [Google Scholar]
- Bagheri A.; Jin J. Photopolymerization in 3D Printing. ACS Appl. Polym. Mater. 2019, 1 (4), 593–611. 10.1021/acsapm.8b00165. [DOI] [Google Scholar]
- Zhang J.; Xiao P. 3D Printing of Photopolymers. Polym. Chem. 2018, 9 (13), 1530–1540. 10.1039/C8PY00157J. [DOI] [Google Scholar]
- Chatani S.; Kloxin C. J.; Bowman C. N. The Power of Light in Polymer Science: Photochemical Processes to Manipulate Polymer Formation, Structure, and Properties. Polym. Chem. 2014, 5 (7), 2187–2201. 10.1039/C3PY01334K. [DOI] [Google Scholar]
- Xiao P.; Zhang J.; Dumur F.; Tehfe M. A.; Morlet-Savary F.; Graff B.; Gigmes D.; Fouassier J. P.; Lalevée J. Visible Light Sensitive Photoinitiating Systems: Recent Progress in Cationic and Radical Photopolymerization Reactions under Soft Conditions. Prog. Polym. Sci. 2015, 41 (C), 32–66. 10.1016/j.progpolymsci.2014.09.001. [DOI] [Google Scholar]
- Bagheri A.; Bainbridge C.; Jin J. Visible Light-Induced Transformation of Polymer Networks. ACS Appl. Polym. Mater. 2019, 1 (7), 1896–1904. 10.1021/acsapm.9b00458. [DOI] [Google Scholar]
- Dumur F. Recent Advances on Visible Light Metal-Based Photocatalysts for Polymerization under Low Light Intensity. Catalysts 2019, 9 (9), 736. 10.3390/catal9090736. [DOI] [Google Scholar]
- Strehmel B.; Schmitz C.; Cremanns K.; Göttert J. Photochemistry with Cyanines in the Near Infrared: A Step to Chemistry 4.0 Technologies. Chem. - Eur. J. 2019, 25 (56), 12855–12864. 10.1002/chem.201901746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K. S.; Galarraga J. H.; Cui X.; Lindberg G. C. J.; Burdick J. A.; Woodfield T. B. F.. Fundamentals and Applications of Photo-Cross-Linking in Bioprinting. Chem. Rev. 2020, in press. 10.1021/acs.chemrev.9b00812 [DOI] [PubMed] [Google Scholar]
- Schwartz J. J.; Boydston A. J. Multimaterial Actinic Spatial Control 3D and 4D Printing. Nat. Commun. 2019, 10 (1), 791. 10.1038/s41467-019-08639-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialas S.; Michalek L.; Marschner D. E.; Krappitz T.; Wegener M.; Blinco J.; Blasco E.; Frisch H.; Barner-Kowollik C. Access to Disparate Soft Matter Materials by Curing with Two Colors of Light. Adv. Mater. 2019, 31 (8), 1807288. 10.1002/adma.201807288. [DOI] [PubMed] [Google Scholar]
- Dolinski N. D.; Page Z. A.; Callaway E. B.; Eisenreich F.; Garcia R. V.; Chavez R.; Bothman D. P.; Hecht S.; Zok F. W.; Hawker C. J. Solution Mask Liquid Lithography (SMaLL) for One-Step, Multimaterial 3D Printing. Adv. Mater. 2018, 30 (31), 1800364. 10.1002/adma.201800364. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Xi W.; Huang S.; Long K.; Bowman C. N. Wavelength-Selective Sequential Polymer Network Formation Controlled with a Two-Color Responsive Initiation System. Macromolecules 2017, 50 (15), 5652–5660. 10.1021/acs.macromol.7b01117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottisch V.; Michaudel Q.; Fors B. P. Photocontrolled Interconversion of Cationic and Radical Polymerizations. J. Am. Chem. Soc. 2017, 139 (31), 10665–10668. 10.1021/jacs.7b06661. [DOI] [PubMed] [Google Scholar]
- Peterson B. M.; Kottisch V.; Supej M. J.; Fors B. P. On Demand Switching of Polymerization Mechanism and Monomer Selectivity with Orthogonal Stimuli. ACS Cent. Sci. 2018, 4 (9), 1228–1234. 10.1021/acscentsci.8b00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garra P.; Dietlin C.; Morlet-Savary F.; Dumur F.; Gigmes D.; Fouassier J. P.; Lalevée J. Redox Two-Component Initiated Free Radical and Cationic Polymerizations: Concepts, Reactions and Applications. Prog. Polym. Sci. 2019, 94, 33–56. 10.1016/j.progpolymsci.2019.04.003. [DOI] [Google Scholar]
- Blasco E.; Wegener M.; Barner-Kowollik C. Photochemically Driven Polymeric Network Formation: Synthesis and Applications. Adv. Mater. 2017, 29 (15), 1604005. 10.1002/adma.201604005. [DOI] [PubMed] [Google Scholar]
- Matsui J. K.; Lang S. B.; Heitz D. R.; Molander G. A. Photoredox-Mediated Routes to Radicals: The Value of Catalytic Radical Generation in Synthetic Methods Development. ACS Catal. 2017, 7 (4), 2563–2575. 10.1021/acscatal.7b00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadashi-Silab S.; Doran S.; Yagci Y. Photoinduced Electron Transfer Reactions for Macromolecular Syntheses. Chem. Rev. 2016, 116 (17), 10212–10275. 10.1021/acs.chemrev.5b00586. [DOI] [PubMed] [Google Scholar]
- Chen M.; Zhong M.; Johnson J. A. Light-Controlled Radical Polymerization: Mechanisms, Methods, and Applications. Chem. Rev. 2016, 116 (17), 10167–10211. 10.1021/acs.chemrev.5b00671. [DOI] [PubMed] [Google Scholar]
- Haas M.; Radebner J.; Eibel A.; Gescheidt G.; Stueger H. Recent Advances in Germanium-Based Photoinitiator Chemistry. Chem. - Eur. J. 2018, 24 (33), 8258–8267. 10.1002/chem.201705567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri A.; Bainbridge C. W. A.; Engel K. E.; Qiao G. G.; Xu J.; Boyer C.; Jin J. Oxygen Tolerant PET-RAFT Facilitated 3D Printing of Polymeric Materials under Visible LEDs. ACS Appl. Polym. Mater. 2020, 2 (2), 782–790. 10.1021/acsapm.9b01076. [DOI] [Google Scholar]
- Zhang Z.; Corrigan N.; Bagheri A.; Jin J.; Boyer C. A Versatile 3D and 4D Printing System through Photocontrolled RAFT Polymerization. Angew. Chem., Int. Ed. 2019, 58 (50), 17954–17963. 10.1002/anie.201912608. [DOI] [PubMed] [Google Scholar]
- Bagheri A.; Engel K. E.; Bainbridge C. W. A.; Xu J.; Boyer C.; Jin J. 3D Printing of Polymeric Materials Based on Photo-RAFT Polymerization. Polym. Chem. 2020, 11 (3), 641–647. 10.1039/C9PY01419E. [DOI] [Google Scholar]
- Polykarpov A. Y.; Neckers D. C. Tetramethylammonium Phenyltrialkylborates in the Photoinduced Electron Transfer Reaction with Benzophenone. Generation of Alkyl Radicals and Their Addition to Activated Alkenes. Tetrahedron Lett. 1995, 36 (31), 5483–5486. 10.1016/0040-4039(95)01055-M. [DOI] [Google Scholar]
- Kabatc J.; Jȩdrzejewska B.; Pa̧czkowski J. Hemicyanine N-Butyltriphenylborate Salts as Effective Initiators of Free-Radical Polymerization Photoinitiated via Photoinduced Electron-Transfer Process. J. Polym. Sci., Part A: Polym. Chem. 2003, 41 (19), 3017–3026. 10.1002/pola.10886. [DOI] [Google Scholar]
- Pritchard Z. D.; Beer M. P.; Whelan R. J.; Scott T. F.; Burns M. A. Modeling and Correcting Cure-Through in Continuous Stereolithographic 3D Printing. Adv. Mater. Technol. 2019, 4, 1900700. 10.1002/admt.201900700. [DOI] [Google Scholar]
- Cui G.; Guan P. J.; Fang W. H. Photoinduced Proton Transfer and Isomerization in a Hydrogen-Bonded Aromatic Azo Compound: A CASPT2//CASSCF Study. J. Phys. Chem. A 2014, 118 (26), 4732–4739. 10.1021/jp503865y. [DOI] [PubMed] [Google Scholar]
- Shanmugam S.; Xu J.; Boyer C. Exploiting Metalloporphyrins for Selective Living Radical Polymerization Tunable over Visible Wavelengths. J. Am. Chem. Soc. 2015, 137 (28), 9174–9185. 10.1021/jacs.5b05274. [DOI] [PubMed] [Google Scholar]
- Hennessy M. G.; Vitale A.; Cabral J. T.; Matar O. K. Role of Heat Generation and Thermal Diffusion during Frontal Photopolymerization. Phys. Rev. E - Stat. Nonlinear, Soft Matter Phys. 2015, 92 (2), 022403. 10.1103/PhysRevE.92.022403. [DOI] [PubMed] [Google Scholar]
- O’Brien A. K.; Bowman C. N. Modeling Thermal and Optical Effects on Photopolymerization Systems. Macromolecules 2003, 36 (20), 7777–7782. 10.1021/ma034070c. [DOI] [Google Scholar]
- Park H. Y.; Kloxin C. J.; Fordney M. F.; Bowman C. N. Stress Relaxation of Trithiocarbonate-Dimethacrylate-Based Dental Composites. Dent. Mater. 2012, 28 (8), 888–893. 10.1016/j.dental.2012.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monzón M.; Ortega Z.; Hernández A.; Paz R.; Ortega F. Anisotropy of Photopolymer Parts Made by Digital Light Processing. Materials 2017, 10 (1), 64. 10.3390/ma10010064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dizon J. R. C.; Espera A. H.; Chen Q.; Advincula R. C. Mechanical Characterization of 3D-Printed Polymers. Addit. Manuf. 2018, 20, 44–67. 10.1016/j.addma.2017.12.002. [DOI] [Google Scholar]
- Anastasio R.; Peerbooms W.; Cardinaels R.; Van Breemen L. C. A. Characterization of Ultraviolet-Cured Methacrylate Networks: From Photopolymerization to Ultimate Mechanical Properties. Macromolecules 2019, 52 (23), 9220–9231. 10.1021/acs.macromol.9b01439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D. K.; Sakhaei A. H.; Layani M.; Zhang B.; Ge Q.; Magdassi S. Highly Stretchable and UV Curable Elastomers for Digital Light Processing Based 3D Printing. Adv. Mater. 2017, 29 (15), 1606000. 10.1002/adma.201606000. [DOI] [PubMed] [Google Scholar]
- Herzberger J.; Sirrine J. M.; Williams C. B.; Long T. E. Polymer Design for 3D Printing Elastomers: Recent Advances in Structure, Properties, and Printing. Prog. Polym. Sci. 2019, 97, 101144. 10.1016/j.progpolymsci.2019.101144. [DOI] [Google Scholar]
- Thrasher C. J.; Schwartz J. J.; Boydston A. J. Modular Elastomer Photoresins for Digital Light Processing Additive Manufacturing. ACS Appl. Mater. Interfaces 2017, 9 (45), 39708–39716. 10.1021/acsami.7b13909. [DOI] [PubMed] [Google Scholar]
- Sirrine J. M.; Zlatanic A.; Meenakshisundaram V.; Messman J. M.; Williams C. B.; Dvornic P. R.; Long T. E. 3D Printing Amorphous Polysiloxane Terpolymers via Vat Photopolymerization. Macromol. Chem. Phys. 2019, 220 (4), 1800425. 10.1002/macp.201800425. [DOI] [Google Scholar]
- Ligon S. C.; Husár B.; Wutzel H.; Holman R.; Liska R. Strategies to Reduce Oxygen Inhibition in Photoinduced Polymerization. Chem. Rev. 2014, 114 (1), 557–589. 10.1021/cr3005197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.