

 The antileishmanial activity of a variety of 3-nitro and 3-amino-isoxazoles against promastigote and amastigote stages of L. donovani is described.
The antileishmanial activity of a variety of 3-nitro and 3-amino-isoxazoles against promastigote and amastigote stages of L. donovani is described.
Abstract
A chemical library comprising substituted 3-nitroisoxazoles and 3-aminoisoxazoles was prepared and screened for their antileishmanial activity against L. donovani. As compared to Miltefosine, the standard drug used in bioassays, several compounds displayed remarkably better inhibition of the promastigote and amastigote stages of parasites. The in vivo evaluation of a few compounds in a golden hamster model showed significant reduction of the parasite load post treatment via the intraperitoneal route by several compounds. The preliminary pharmacokinetic evaluation of a representative compound 4mfvia the oral route, however, indicated high systemic clearance from the body.
Introduction
Leishmaniasis is a neglected disease caused by protozoan parasites of the genus Leishmania. The major clinical forms of this disease are cutaneous leishmaniasis (CL) caused by L. major, mucocutaneous leishmaniasis (MCL) caused by L. braziliensis, and fatal visceral leishmaniasis (VL) caused by L. donovani.1 The most recent World Health Organization (WHO) report indicates that 310 million people are at risk of contracting leishmaniasis, while 0.7–1 million new infections and 26 000 to 65 000 deaths take place annually.2
Over the past few decades, the chemotherapy for treating VL has relied on the pentavalent antimonials meglumine antimonate and sodium stibogluconate.3,4 However, parenteral administration together with continual monitoring due to hepatic and cardiac toxicity and the problem of resistance make them far from ideal. Therefore, in the Indian subcontinent the antifungal drug amphotericin B as an injectable drug is currently recommended as a first-line treatment against VL.5,6 Unfortunately for this drug too, infusion reactions like chills and fever together with cases of serious nephrotoxicity are reported.7 The liposomal formulation of amphotericin B, AmBisome, which enhances drug delivery and reduces toxicity is better accepted but due to its high cost and low stability, pentavalent antimonials are still commonly used in several areas where a cold chain is absent. The phospholipid miltefosine, originally discovered as an anticancer drug, was repositioned as the first oral drug against VL.8,9 Initially with a high cure rate of 95% and only minor side effects it was extensively used in India, but subsequently its lower effectiveness or treatment failures and high toxicity were reported in the clinic.10 Another antileishmanial the aminoglycoside paromomycin shows cure rates of up to 85% which improves to 93% in combination with antimony-based drugs.11–13 However, long-term parenteral administration together with the resistance of the parasite against this class has also been reported. The last decade witnessed considerable efforts to discover new chemotypes as antileishmanial but most of them displayed some or other side effects. The drugs fexinidazole and DNDI-VL-2098 were discontinued from development due to their reduced efficacy and toxicity during the clinical trials.14–17 Therefore there is a pressing need to explore newer chemicals for antileishmanial activity. It is worthwhile to mention that several of the highly potent antileishmanial compounds are nitro-heterocycles, as evident from the structures of fexinidazole sulphone, DNDI-VL-2098, CGI-17341, (R)-PA-824, DNDI-0690 and nitrofurtimox (Fig. 1).18,19 It is suggested that bicyclic nitro-heterocyclic drugs act upon a NAD(P)H oxidase which activates nitroreductase (NTR2) whereas monocyclic nitro-heterocyclic compounds are activated by a mitochondrial oxygen-insensitive nitroreductase (NTR1).20 It is reported that the novel nitroreductase enzyme gives rise to reactive nitrogen species, including nitric oxide (NO) which can induce oxidative stress, either by producing radicals toxic to the parasite or by inhibiting enzymes which are involved in the cellular mechanism for detoxification of the free radicals. It was demonstrated that removing the nitro group from bioactive compounds results in inactive analogues underlining its contribution to antileishmanial activity.
Fig. 1. Nitro-heterocycles as antileishmanial agents.

Isoxazole, a privileged heterocycle, is present in many drugs, bioactive compounds and natural products.21–24 Isoxazole derivatives are cited to display antileishmanial properties too. Acivicin, an isoxazoline compound, was reported to act against L. donovani via intervention of the pyrimidine pathway.25 Suryavanshi et al. reported the in vivo efficacy of an isoxazole-containing heteroretinoid and its amides against L. donovani in a hamster model.26 Isoxazole-fused lupane derivatives were disclosed to elicit moderate inhibition of amastigotes of L. donovani.27 Recently, substituted 3,5-bisphenyl isoxazoles were reported to show moderate antileishmanial properties against L. amazonensis possibly via interfering with leishmanial trypanothione reductase (Fig. 2).28–30 In our project to discover new antileishmanial compounds, previously we have disclosed the promising in vitro efficacy of an isoxazole analogue against L. donovani.31 Earlier, we developed a facile route to the synthesis of substituted 3-nitroisoxazoles,32 and as nitro-heterocycles are known to display significant antileishmanial activity, we considered investigating the bioactivity of these compounds against L. donovani. During the study, we discovered that as compared to miltefosine, several compounds displayed better inhibition of the multiplication of amastigotes. In order to extend the scope, the antileishmanial activity of substituted 3-aminoisoxazoles has been also investigated. The study revealed that the bioactivity of isoxazole derivatives was independent of the nitro group. However, the in vivo investigation of a few analogues displayed them to be only moderately active. Preliminary pharmacokinetic evaluation of a representative compound suggested that high systemic clearance could be one of the reasons for the lower in vivo efficacy. The details of the results of this study are described herein.
Fig. 2. A few substituted isoxazoles cited for antileishmanial activity.26,28–30 .

Results and discussion
Chemistry
The substituted 3-nitroisoxazoles (1a–1q) were prepared by reacting appropriately substituted Morita–Baylis–Hillman acetates with NaNO2 in the presence of iodine as reported earlier (Scheme 1).32 The hydrolysis of esters 1b, 1e, 1k and 1m with LiOH·2H2O afforded the respective acids 2b, 2e, 2k and 2m.33 The compounds belonging to series 3 were prepared by treating acids 2b, 2e and 2k with thionyl chloride to obtain the respective acid chlorides, which without any purification were reacted with different amines (a–c, e, g–j, l–p) to afford 3-nitro-4-isoxazolecarboxamides 3ba–3bc, 3be, 3bg–3bi, 3l–3p, 3ea–3ec, 3ee, 3eg–3ej, 3el–3ep, 3ka–3kc, 3ke, and 3kg–3kj (Scheme 2). In order to assess the contribution of the nitro group towards bioactivity, compounds 1b, 1k and 1m were reacted with differently substituted amines (b–f, h, j–k, o–q), to furnish the corresponding secondary amines 4bb–4bf, 4bh, 4bj–4bk, 4bo–4bq, 4kb, 4kd–4kf, 4kj–4kk, 4ko–4kq, 4mb, 4md–4mf, 4mh, 4mj–4mk, and 4mo–4mq in good yields via SNAr reactions as reported earlier (Scheme 3).32
Scheme 1. Synthesis of 3-nitroisoxazoles (1).

Scheme 2. Synthesis of 3-nitro-4-isoxazolecarboxamides (3).

Scheme 3. Synthesis of 3-aminoisoxazoles (for key to amines refer to Scheme 2).

It is reported that N′-(7-chloroquinolin-4-yl)-4-methyl benzenesulfonohydrazide displays a significant antileishmanial effect.34 This report prompted us to expand the SAR to quinolone-tethered isoxazoles 6 which were prepared by reacting 3-nitroisoxazoles 1b, 1g, 1h, 1k and 1m with N1-(quinolin-4-yl)alkyl-diamines 5a–d. These reactions were performed in the presence of Et3N (1.5 equiv.) under heating at 80 °C to afford the quinoline derivatives 6ba–6bd, 6ga–6gc, 6ha–6hd, 6ka–6kd and 6ma–6mc in 74–92% yields (Scheme 4).
Scheme 4. Synthesis of quinoline-tethered 3-aminoisoxazoles.

In vitro antileishmanial screening
The in vitro assay was referenced with miltefosine which displayed significant antipromastigote (IC50 = 2.52 ± 0.14 μM) and moderate antiamastigote (IC50 = 9.25 ± 0.17 μM) activities and cytotoxicity toward the J-774A.1 macrophage cell line (CC50 values of 53.33 ± 1.10 μM). Initially, substituted 3-nitroisoxazoles 1a–1q were subjected to in vitro screening against the promastigote stage of L. donovani and the results are summarized in Table 1. Strikingly, all compounds except for 1g, 1i and 1p displayed IC50 values in the range between 0.34 ± 0.23 μM and 2.72 ± 0.04 μM, which were either equipotent or better than the standard drug. Encouraged by the initial activity, the compounds were further screened against intracellular amastigotes residing within murine macrophages. It was observed that compounds 1k, 1o and 1n displayed IC50 values in the range of 4.38 ± 0.09 μM to 7.10 ± 0.21 μM, which were better than miltefosine, whereas compounds 1h, 1i and 1m, displayed activity with IC50 values in the range of 16.05 ± 1.6 to 17.35 ± 0.07 μM. All other compounds did not show any significant inhibition. The cytotoxicity evaluation of these compounds revealed that except compound 1m with a selectivity index (SI) of 6.51, all analogues displayed SI values below 5.0, suggesting them to be potentially toxic for the in vivo assay. Examining the antileishmanial activity of acids 2b and 2m, we discovered reduction or loss of activity indicating that the presence of the ester group supports bioactivity.
Table 1. In vitro antileishmanial activity of 3-nitroisoxazoles (1 and 2) against L. donovani and cytotoxicity against the J-774A.1 cell line.
| Compd. no | IC50 (μM)
a
|
CC50 (μM) | SI b | |
| Promastigote | Amastigote | |||
| 1a | 2.17 ± 0.22 | NI c | ND d | ND |
| 1b | 0.67 ± 0.20 | NI | ND | ND |
| 1c | 0.66 ± 0.19 | NI | ND | ND |
| 1d | 2.10 ± 0.26 | NI | ND | ND |
| 1e | 1.79 ± 0.04 | NI | ND | ND |
| 1f | 2.49 ± 0.14 | NI | ND | ND |
| 1g | 4.01 ± 0.19 | NI | ND | ND |
| 1h | 2.87 ± 0.09 | 16.05 ± 1.06 | ± 0.07 | 2.19 |
| 1i | 2.72 ± 0.04 | 16.6 ± 0.14 | 80.05 ± 1.34 | 4.82 |
| 1j | 0.34 ± 0.23 | 32.25 ± 0.07 | 34.8 ± 0.042 | 1.08 |
| 1k | 1.82 ± 0.31 | 4.38 ± 0.09 | 19.7 ± 0.56 | 4.49 |
| 1l | 1.11 ± 0.14 | NI | ND | ND |
| 1m | 0.67 ± 0.21 | 17.35 ± 0.07 | 113 ± 0 | 6.51 |
| 1n | 0.86 ± 0.071 | 7.10 ± 0.21 | 18.45 ± 0.77 | 2.5 |
| 1o | 0.77 ± 0.063 | 4.80 ± 0.007 | 19.7 ± 0.56 | 3.6 |
| 1p | 38.4 ± 2.12 | NI | ND | ND |
| 1q | 0.76 ± 0.15 | 21.25 ± 2.73 | 79.9 ± 1.13 | 3.76 |
| 2b | 19 ± 0.91 | NI | ND | ND |
| 2m | NI | NI | ND | ND |
| Miltefosine e | 2.52 ± 0.14 | 9.25 ± 0.17 | 53.33 ± 1.10 | 5.76 |
aIC50 (μM): concentration corresponding to 50% growth inhibition of the parasite. IC50 and CC50 values are the average of three independent assays expressed as average ± standard error.
bSelectivity index (SI): IC50 values of cytotoxic activity/IC50 values of the anti-amastigote antileishmanial activity.
cNI = no inhibition.
dND = not determined.
eMiltefosine used as the standard.
These results implied that analogues bearing an ortho-substituted phenyl ring at C-5 of the isoxazole displayed better antiamastigote activity as compared to compounds with a para-substituted ring. Moreover, replacing the ester group with an acid group was found to be detrimental to the activity. Subsequently, the ester group at the 4-position in isoxazoles 1b, 1e and 1k was transformed into carboxamide to prepare compounds 3ba–3bc, 3be, 3bg–3bj, 3bl–3bp, 3ea–3ec, 3ee, 3eg–3ej, 3el–3ep, 3ka–3kc, 3ke, and 3kg–3kj which were investigated for their antileishmanial effect and the results are summarized in Table 2. From the first set of carboxamides bearing a 4-nitrophenyl group at C-5, compounds 3be and 3bj having a phenyl or benzyl subunit, displayed IC50 values of 3.52 ± 0.04 and 1.3 ± 0.04 μM, respectively, whereas compounds 3ba–3bc and 3bg–3bi did not elicit significant inhibition against the promastigotes. On the other hand compounds 3bl–3bn and 3bp bearing an aliphatic amino subunit in the carboxamide group were equipotent to miltefosine. Against the amastigotes, 3be displayed significant inhibition with IC50 of 2.06 ± 0.01 μM, but 3bj was found to be toxic. Conversely compounds 3bl–3bn and 3bp showed moderate inhibition of the amastigote stage but with low SI values. Compound 3bo in this set was not evaluated. Next the group having a 4-chlorophenyl unit at the C-5 of isoxazole was investigated. Unlike the above, from compounds 3ea–3ec, 3ee, and 3eg–3ej bearing a phenyl or benzyl subunit, except 3eb and 3eg, all analogues displayed antipromastigote activity with IC50 values from 1.16 ± 0.49 to 3.05 ± 0.06 μM. The evaluation of the antiamastigote activity of these compounds revealed that compounds 3ea, 3eh and 3ej had IC50 values of 3.56 ± 0.02, 3.43 ± 0.02 and 3.22 ± 0.21 μM, respectively, which were at least two fold better than that of miltefosine. However, carboxamides 3el–3en and 3ep prepared from the aliphatic amines did not show any significant inhibition against any stage. Among this set 3eo was not evaluated for its bioactivity. The last group included compounds originating from 1k bearing 2,4-dichlorophenyl at C-5 of the isoxazole core. In this set, carboxamides 3ka–3kc, 3ke, and 3kg–3kj containing phenyl or benzyl subunits were only prepared. We discovered that all compounds displayed significant activity against the promastigote stage with IC50 values in the range between 1.32 ± 0.03 and 5.24 ± 0.10 μM. More importantly compounds 3ke and 3ki, 3kj showed antiamastigote activity in the range between 1.41 ± 0.01 and 2.03 ± 0.02 μM with improved SI values (39.65–58.01).
Table 2. In vitro antileishmanial activity of 3-nitro-isoxazole-4-carboxamides (3) against L. donovani.
| Compd. no | IC50 (μM) |
CC50 (μM) | SI index | |
| Promastigote | Amastigote | |||
| 3ba | NI | NI | ND | ND |
| 3bb | NI | NI | ND | ND |
| 3bc | NI | NI | ND | ND |
| 3be | 3.52 ± 0.04 | 2.06 ± 0.01 | 69.6 ± 1.55 | 33.78 |
| 3bg | NI | NI | ND | ND |
| 3bh | NI | NI | ND | ND |
| 3bi | NI | NI | ND | ND |
| 3bj | 1.3 ± 0.04 | Toxic | ND | ND |
| 3bl | 3.07 ± 0.08 | 21.3 ± 0.28 | 76.2 ± 0.14 | 3.57 |
| 3bm | 3.01 ± 0.06 | 17.35 ± 0.07 | 66.95 ± 0.49 | 3.85 |
| 3bn | 2.83 ± 0.03 | 21.9 ± 0.28 | 76.45 ± 0.21 | 3.49 |
| 3bo | ND | ND | ND | ND |
| 3bp | 2.71 ± 0.26 | 15.6 ± 0.14 | 65.95 ± 0.63 | 4.22 |
| 3ea | 2.81 ± 0.19 | 3.56 ± 0.02 | 89.3 ± 0.14 | 25.08 |
| 3eb | NI | NI | ND | ND |
| 3ec | 1.16 ± 0.49 | 19.45 ± 0.35 | 91.9 ± 0.98 | 4.72 |
| 3ee | 2.67 ± 0.05 | 9.41 ± 0.07 | 89.49 ± 0.07 | 9.50 |
| 3eg | NI | NI | ND | ND |
| 3eh | 2.79 ± 0.08 | 3.43 ± 0.02 | 68.55 ± 0.49 | 19.98 |
| 3ei | 2.90 ± 0.03 | 19.6 ± 0.42 | 73.7 ± 0.70 | 3.76 |
| 3ej | 3.05 ± 0.06 | 3.22 ± 0.21 | 106.35 ± 0.49 | 33.02 |
| 3el | NI | NI | ND | ND |
| 3em | NI | NI | ND | ND |
| 3en | NI | NI | ND | ND |
| 3eo | ND | ND | ND | ND |
| 3ep | NI | NI | ND | ND |
| 3ka | 2.68 ± 0.05 | 5.97 ± 0.23 | 67.3 ± 0.84 | 11.27 |
| 3kb | 1.32 ± 0.03 | Toxic | ND | ND |
| 3kc | 2.46 ± 0.02 | 50.6 ± 0.98 | 86.35 ± 0.91 | 1.70 |
| 3ke | 3.42 ± 0.04 | 2.03 ± 0.02 | 80.5 ± 1.27 | 39.65 |
| 3kg | 3.05 ± 0.03 | 7.74 ± 0.04 | 71.2 ± 0.28 | 9.19 |
| 3kh | 3.02 ± 0.02 | 11.3 ± 0.28 | 61.15 ± 0.77 | 5.41 |
| 3ki | 3.49 ± 0.05 | 1.41 ± 0.01 | 81.8 ± 1.37 | 58.01 |
| 3kj | 5.24 ± 0.10 | 1.95 ± 0.02 | 81.75 ± 0.35 | 41.92 |
These results suggested that having 2,4-dichlorophenyl at C-5 in the isoxazole is favoured for bioactivity as compared to 4-nitrophenyl or 4-chlorophenyl groups and a substituent at the ortho position is preferred. Amongst various substitutions in the amino group of the carboxamide, the presence of 3,4-dimethoxy or 4-methoxy groups on the phenyl ring of the benzyl group elicited better bioresponse.
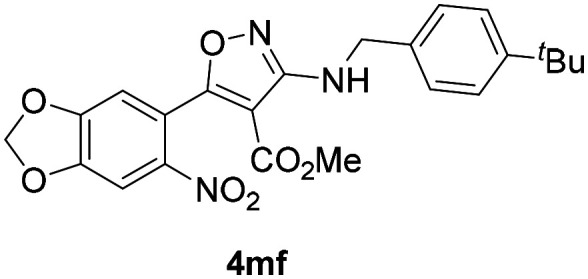
Aiming to evaluate the influence of the nitro group present at C-3 of the isoxazole if any, substituted 3-aminoisoxazoles having an ester group at C-4 were screened and the results are presented in Table 3. As described above, for the synthesis of this series of isoxazoles, different benzyl amines and aliphatic amines (b–d–f, h, j–n, o) were used. Evaluation of compounds 4bb–4bd, 4bf, 4bh, 4bj–4bo, and 4bq bearing a 4-nitrophenyl group at C-5 of the isoxazole delineated that except 4bb (IC50 = 3.9 ± 0.09 μM) and 4bd (IC50 = 3.79 ± 0.05 μM), no compound displayed promising inhibition of the promastigote multiplication. In contrast four analogues 4bd, 4bf, 4bn and 4bo (IC50 = 4.53 ± 0.16 to 8.16 ± 0.02 μM) displayed antiamastigote activity better than or similar to miltefosine (9.25 ± 0.17 μM). Amongst the analogues 4kb, 4kd–4kf, 4kj–4kk, 4ko, and 4kq bearing a 2,4-dichlorophenyl group at C-5 of the isoxazole, compounds bearing methoxy group(s) on the phenyl ring of the benzyl unit at C-3 (4ke and 4kj) or the morpholino propyl group at C-3 (4ko) were almost equipotent to the standard in inhibiting the promastigotes with IC50 values of 3.09 ± 0.23 μM, 4.49 ± 0.042 μM and 2.58 ± 0.27 μM, respectively. Unlike for compounds bearing 6-nitro-3,4-methylenedioxyphenyl at C-5, compounds 4mb, 4mh, 4mj, and 4mk bearing methoxy group(s) on the phenyl ring of the benzyl moiety did not display promising activity. However, compound 4mf having a 4-tert-butyl group on the phenyl ring of the benzylamine subunit displayed significant activity against promastigote growth with an IC50 value of 2.14 ± 0.15 μM. Another compound 4md bearing 4-chlorophenyl in the benzylamine subunit displayed an IC50 value of 15.45 ± 1.48 μM. Evaluating 4mf and 4md against the amastigotes revealed that 4mf was highly potent with IC50 of 0.54 ± 0.01 μM (SI = 14) which was better than miltefosine by 18 fold. On the other hand 4md displayed IC50 of 4.59 ± 0.11 (SI = 7) which was at least 2 fold better than that of the standard. These results implied that the presence of a nitro group at the C-3 position of the isoxazole is not essential for antileishmanial activity. Also the pronounced activity in 4mf endorsed the usefulness of having an ortho-substituent on the phenyl ring.
Table 3. In vitro antileishmanial activity of 3-amino-isoxazole-4-carboxylates (4) against L. donovani.
| Compd. no | IC50 (μM) |
CC50 (μM) | SI index | |
| Promastigote | Amastigote | |||
| 4bb | 3.9 ± 0.09 | 13.85 ± 0.77 | 85.35 ± 0.77 | 6.16 |
| 4bc | NI | NI | ND | ND |
| 4bd | 3.79 ± 0.05 | 7.88 ± 0.11 | 78.3 ± 0.28 | 9.93 |
| 4be | NI | NI | ND | ND |
| 4bf | 49.7 ± 1.55 | 8.16 ± 0.02 | 81.35 ± 0.21 | 9.69 |
| 4bh | NI | NI | ND | ND |
| 4bj | NI | NI | ND | ND |
| 4bk | NI | NI | ND | ND |
| 4bo | 33.75 ± 1.20 | 4.53 ± 0.16 | 71.8 ± 1.27 | 15.84 |
| 4bq | 26 ± 0.84 | 8.15 ± 0.13 | 69.25 ± 0.35 | 8.49 |
| 4kb | 35.35 ± 0.63 | 1.65 ± 0.07 | 54.15 ± 0.37 | 32.81 |
| 4kd | 14.05 ± 0.35 | 3.10 ± 0.03 | 175.5 ± 3.53 | 56.61 |
| 4ke | 3.09 ± 0.23 | 23.85 ± 0.07 | 34.65 ± 0.21 | 1.5 |
| 4kf | >50 | >50 | ND | ND |
| 4kj | 4.49 ± 0.042 | 2.37 ± 0.07 | 7.58 ± 0.07 | 3 |
| 4kk | 8.34 ± 0.16 | 5.48 ± 0.16 | 32.15 ± 0.07 | 6 |
| 4ko | 2.58 ± 0.27 | 21.2 ± 0.28 | 72.95 ± 0.21 | 3.44 |
| 4kq | NI | NI | ND | ND |
| 4mb | >50 | NI | ND | ND |
| 4md | 15.45 ± 1.48 | 4.59 ± 0.11 | 32.95 ± 0.07 | 7 |
| 4me | NI | NI | ND | ND |
| 4mf | 2.14 ± 0.15 | 0.54 ± 0.01 | 7.49 ± 0.01 | 14 |
| 4mh | NI | NI | ND | ND |
| 4mj | NI | NI | ND | ND |
| 4mk | NI | NI | ND | ND |
| 4mo | 27.75 ± 1.34 | 17 ± 0.84 | 65.05 ± 0.63 | 3.82 |
| 4mq | NI | NI | ND | ND |
Continuing with this series, for the next group the 4-position of the isoxazole was substituted with aminoalkyl-(4-aminoquinoline) and interestingly almost all compounds displayed potent to moderate inhibitory activity against both the promastigote and amastigote forms and several of them displayed better profile as compared to miltefosine (Table 4). From the first set of compounds 6ba–6bd bearing a 4-nitrophenyl unit at C-5, 6ba–6bc had an N1-(7-chloroquinolin-4-yl)alkane-1,2-diamine subunit with ethyl, propyl and butyl groups at C-3 of the isoxazole. These compounds displayed antipromastigote activity with IC50 values in the range of 1.87 ± 0.20 and 3.37 ± 0.04 μM. Notably, compounds 6ba–6bc displayed inhibition against the amastigotes too with IC50 values of 0.91 ± 0.028, 3.14 ± 0.04 and 1.84 ± 0.09 μM, respectively. Whereas 6ba displayed an SI of only 4.16, 6bb and 6bc had corresponding SI values of 33.91 and 73.77. However, compound 6bd having N1-(2,7-bis(trifluoromethyl)quinolin-4-yl)ethane-1,2-diamine at C-3 displayed lower activity. The promising antiamastigote activity for this set prompted the synthesis and evaluation of compounds 6ga–6gc having 2-nitrophenyl at C-5 of the isoxazole. 6gb was inactive in the amastigote model, and 6ga was two-fold less active than miltefosine. Compound 6gc with IC50 of 8.63 ± 0.39 μM was equipotent to the standard but had a better SI. Installing a 2-fluorophenyl substituent at C-5 of the isoxazole resulted in compounds 6ha–6hd out of which only 6ha displayed an IC50 value better than the standard but the SI was too low. Here 6hd, too, having N1-(2,7-bis(trifluoromethyl)quinolin-4-yl)ethane-1,2-diamine at C-3 was several fold less active against the promastigote stage confirming that the chloro-group on the quinoline ring is more suited for the antileishmanial effect. From 6ka–6kd bearing a 2,4-dichlorophenyl unit at C-5 of the isoxazole, 6ka and 6kb with IC50 values of 0.85 ± 0.077 and 3.21 ± 0.063, respectively, were 11 and 3 fold better than the standard drug against the amastigotes though the SI of 6kb (9.67) was relatively better than that of 6ka (4.05). Amongst 6ma–6mc having a 6-nitro-3,4-methylenedioxyphenyl group at C-5, 6ma displayed significant inhibition of the amastigote stage with IC50 of 1.49 ± 0.084 μM, whereas 6mc was equipotent to the standard. But the SI of 6mc (7.97) was relatively better than that of 6ma (2.48). The remarkable activity of 3-aminoisoxazoles against the amastigote stage of L. donovani further reflected that the antileishmanial effect of the compounds described herein is independent of the nitro group at the C-3 position.
Table 4. In vitro antileishmanial activity of isoxazole–quinoline hybrids (6) against L. donovani and cytotoxicity against the J-774A.1 cell line.
| Compd. no | IC50 (μM) |
CC50 (μM) | SI index | |
| Promastigote | Amastigote | |||
| 6ba | 1.87 ± 0.20 | 0.91 ± 0.028 | 3.79 ± 0.007 | 4.16 |
| 6bb | 3.37 ± 0.04 | 3.14 ± 0.04 | 106.5 ± 2.12 | 33.91 |
| 6bc | 2.85 ± 0.03 | 1.84 ± 0.09 | 135.75 ± 1.7 | 73.77 |
| 6bd | 10.05 ± 0.48 | 18.1 ± 0.56 | 73 ± 0.56 | 4.03 |
| 6ga | 3.45 ± 0.11 | 18.65 ± 0.35 | 73.6 ± 1.97 | 3.94 |
| 6gb | 1.98 ± 0.22 | NI | ND | ND |
| 6gc | 1.95 ± 0.45 | 8.63 ± 0.39 | 77.65 ± 1.06 | 8.99 |
| 6ha | 1.73 ± 0.01 | 1.47 ± 0.0 | 3.42 ± 0.028 | 2.32 |
| 6hb | 16.75 ± 0.21 | NI | ND | ND |
| 6hc | 10.6 ± 0.14 | 19.9 ± 0.14 | 70.85 ± 1.20 | 3.56 |
| 6hd | 43.25 ± 0.35 | 17.75 ± 0.21 | 80.35 ± 0.77 | 4.52 |
| 6ka | 1.86 ± 0.12 | 0.85 ± 0.077 | 3.45 ± 0.02 | 4.05 |
| 6kb | 3.82 ± 0.14 | 3.21 ± 0.063 | 31.05 ± 0.63 | 9.67 |
| 6kc | 5.93 ± 0.021 | 27.55 ± 0.49 | 93.55 ± 0.63 | 3.39 |
| 6kd | 48.25 ± 1.34 | 20.3 ± 0.21 | 99 ± 0.14 | 4.86 |
| 6ma | 2.04 ± 0.07 | 1.49 ± 0.084 | 3.70 ± 0.021 | 2.48 |
| 6mb | 7.75 ± 0.44 | 16.15 ± 0.35 | 73.6 ± 1.97 | 4.55 |
| 6mc | 4.5 ± 0.07 | 8.49 ± 0.69 | 67.7 ± 0.98 | 7.97 |
A summary of the SAR based on the in vitro activity in both the promastigote and amastigote stages of the three different series of compounds is provided in Fig. 3.
Fig. 3. Summary of the SAR based on the in vitro activity in both the promastigote and amastigote stages of L. donovani.

In vivo antileishmanial screening
Only a few compounds35 were evaluated for in vivo efficacy against L. donovani in a golden hamster model at a dose of 50 mg kg–1 × 5 days through the intraperitoneal (IP) route or 100 mg kg–1 × 5 days via the oral route and the results are summarized in Table 5. Miltefosine (40 mg kg–1) was used as the standard drug for the in vivo study too.
Table 5. In vivo antileishmanial activity against L. donovani in a golden hamster model.
| Compd. no | Percentage inhibition |
Route of administration | Dose mg kg–1 | |
| 7th day | 28th day | |||
| 1h | 36.0 ± 8.48 | NI | IP | 50 |
| 1k | 39.5 ± 0.70 | 29.0 ± 0.0 | IP | 50 |
| 1m | 36.0 ± 20.20 | 71.25 ± 10.56 | IP | 50 |
| 1m | 48.41 ± 11.44 | 66.03 ± 8.74 | Oral | 100 |
| 4bb | 44.02 ± 12.63 | 75.20 ± 6.64 | IP | 50 |
| 4kd | 29.22 ± 41.32 | NI | IP | 50 |
| 4kk | 50.92 ± 8.48 | 38.04 ± 2.6 | IP | 50 |
| 4md | 13.51 ± 0.6 | 2.08 ± 3.6 | IP | 50 |
| 4mf | 71.08 ± 11.11 | 28.63 ± 11.04 | IP | 50 |
| 4mf | 69.47 ± 8.34 | 12.70 ± 8.91 | Oral | 100 |
| 6ba | NI | NI | IP | 50 |
| 6gc | NI | NI | IP | 50 |
| 6ka | 19.32 ± 3.06 | NI | IP | 50 |
| 6kb | 61.99 ± 2.46 | 9.23 ± 6.12 | IP | 50 |
| 6mc | 20.01 ± 8.22 | 4.38 ± 5.28 | IP | 50 |
| Miltefosine | 97.32 ± 0.55 | 98.41 ± 1.41 | Oral | 30 |
Among the 3-nitroisoxazoles from series 1, compounds 1h, 1k and 1m were investigated and it was found that compound 1m bearing 6-nitro-3,4-methylenedioxylphenyl at C-5 of the isoxazole was the most potent. When administered via the IP route, 1m displayed 36.0 ± 20.20% inhibition of parasite burden on the 7th day post treatment which improves to 71.25 ± 10.56% on the 28th day. This prompted oral evaluation of 1m and it was discovered that at 100 mg kg–1 dose the compound displayed 48.41 ± 11.44% inhibition of parasite burden on the 7th day post treatment which improved to 66.03 ±8.74% on the 28th day. Next from the series of 3-aminoisoxazoles (4), compounds 4bb, 4kd, 4kk, 4md, and 4mf were evaluated. Compound 4bb showed moderate inhibition (44.02 ± 12.63%) of parasite burden on the 7th day, however, it improved to 75.20 ± 6.64% by the 28th day post treatment. Likewise, compounds 4kd and 4kk displayed moderate inhibition on the 7th day which was not sustained by the 28th day post treatment. Compound 4md displayed insignificant inhibition, but 4mf produced 71.08 ± 11.11% inhibition on the 7th day which was decreased significantly (28.63 ± 11.04%) by the 28th day. As the objective was to discover an orally active antileishmanial agent, 4mf was assessed orally too at a dose of 100 mg kg–1 × 5 d. In this experiment, the inhibition of parasite load by the 7th day was 69.47 ± 8.34%, but it reduced to 12.70 ± 8.91% by the 28th day post treatment. From compounds 6ba, 6gc, 6ka, 6kb and 6mc of series 6 only compound 6kb displayed significant inhibition (61.99 ± 2.46%) on day 7 but decreased by the 28th day. Unfortunately out of the investigated compounds, none was found to have more than 90% antileishmanial efficacy via either the IP or oral route.
Pharmacokinetic study
A representative compound 4mf was subjected to in vivo pharmacokinetic assessment in a Syrian hamster at 100 mg kg–1 oral dose to probe its moderate in vivo efficacy as compared to the in vitro efficacy. The plasma concentration versus time profile in orally administered suspension of 4mf is presented in Fig. 4, and the pharmacokinetic parameters are shown in Table 6. The compound reached systemic circulation within five minute post oral administration and was observed for 24 h within the detection limit. After the oral administration of 4mf suspension the 100 mg kg–1 dose reached the maximum plasma concentration (Cmax) of 15.40 ± 0.28 ng mL–1 and the area under the curve (AUC0-∞) was 148.94 ± 17.88 h ng mL–1. The half-life of the compound was found to be 3.78 ± 1.22 h, while the clearance of the drug was 676.30 ± 81.19 (L h–1 kg–1). The blood level of 4mf however was very low and erratic and the compound was metabolized in the biological system which explains the moderate activity.
Fig. 4. Mean plasma concentration vs. time profile of 4mf upon oral administration at a dose of 100 mg kg–1 to a Syrian hamster (n = 3).

Table 6. Pharmacokinetic parameters of 4mf after oral administration to the Syrian hamster (n = 3).
| |
| Pharmacokinetic parameters a | 4mf (dose 100 mg kg–1) |
| C max (ng mL–1) | 15.40 ± 0.28 |
| T max (h) | 3.00 ± 1.41 |
| AUC0-∞ (h ng mL–1) | 148.94 ± 17.88 |
| t 1/2 (h) | 3.78 ± 1.22 |
| MRT (h) | 9.53 ± 1.89 |
| Cl/F (L h–1 K–1) | 676.30 ± 81.19 |
aEach value represented in mean ± SD.
Conclusions
In summary, we have presented the synthesis and assessment of a series of 3-nitro and 3-amino-isoxazole derivatives as antileishmanial agents to treat VL caused by L. donovani. Several analogues displayed remarkable bioactivity against the promastigote and amastigote stages of the parasite. It was encouraging to discover that at least 75% inhibition of the parasite load of L. donovani on the 28th day post treatment was achieved in a golden hamster model via the IP route. But as a result of the preliminary pharmacokinetic study with a representative compound reflected high systemic clearance of the compound in the body, we did not pursue the in vivo investigations of all the compounds that showed potent in vitro efficacy. We are now in the process of preparing new compounds which exhibit significant in vivo efficacy and are metabolically more stable in the biosystem. Studies towards understanding the possible mode of action of these compounds are also being initiated and will be reported separately.
Experimental
Unless otherwise stated all reactions were performed in non-dry glassware under an air atmosphere and were monitored by analytical thin layer chromatography (TLC). TLC was performed on pre-coated silica gel plates. After elution, the plate was visualized under UV illumination at 254 nm for UV active materials. Further visualization was achieved via an iodine chamber. The melting points were recorded on a hot stage apparatus and are uncorrected. IR spectra were recorded using a FTIR spectrophotometer. 1H NMR and 13C NMR spectra were recorded on a 400 or 500 MHz NMR spectrometer with CDCl3 or d6-DMSO as a solvent, using TMS as an internal standard (chemical shifts in δ). Peak multiplicities of the 1H-NMR signals were designated as s (singlet), brs (broad singlet), d (doublet), dd (doublet of doublet), t (triplet), q (quartet), m (multiplet), etc. Coupling constants (J) are in Hz. The LC-ESI-MS were recorded on a triple quadrupole mass spectrometer. Column chromatography was performed using silica gel (100–200 mesh). Analytical grade solvents for the column chromatography were used as received.
All the alkyl 3-nitro-5-(aryl/alkyl)isoxazole-4-carboxylates (1) were synthesized via iodine-mediated reaction of Morita–Baylis–Hillman acetate with sodium nitrite following our previously reported procedures with no deviation.32 As a consequence the experimental procedure and spectroscopic data for substituted 3-nitroisoxazoles 1 are not provided.
General procedure for the synthesis of carboxamides (3) as exemplified for the synthesis of 5-(4-chlorophenyl)-3-nitro-N-phenylisoxazole-4-carboxamide (3ea)
To a solution of 5-(4-chlorophenyl)-3-nitroisoxazole-4-carboxylic acid 2e (0.2 g, 0.74 mmol) in toluene (8 mL) was added thionyl chloride (0.165 mL, 2.22 mmol) at room temperature and the mixture was heated at 110 °C for 2 h. On completion, the solvent was removed under vacuum to obtain the crude acid chloride as a yellow oil, which was used immediately without purification and characterization.
To a solution of aniline (0.067 mL, 0.74 mmol) in DCM (6 mL) was added Et3N (0.104 mL, 0.74 mmol) at 0 °C and stirred for 10 min. To this mixture, a solution of acid chloride in DCM (2 mL) was added dropwise at 0 °C and stirring was continued for another 3 h. After completion of the reaction (as monitored by TLC), the solvent was removed under reduced pressure. The crude residue was purified by column chromatography over silica gel using hexanes/EtOAc (85 : 15, v/v) as eluent to afford the desired product 3ea (0.202 g from 0.2 g, 79%) as a white solid. 5-(4-Chlorophenyl)-3-nitro-N-phenylisoxazole-4-carboxamide (3ea). Yield: 79% (0.202 g from 0.2 g); a white solid, mp 157–159 °C; Rf = 0.42 (hexanes : EtOAc, 7 : 3, v/v); IR (KBr) νmax: 1644 (C O) cm–1. 1H NMR (400 MHz, d6-DMSO): δ (ppm) = 7.19 (t, J = 7.4 Hz, 1H), 7.41 (t, J = 8.3 Hz, 2H), 7.62 (d, J = 7.5 Hz, 2H), 7.74 (d, J = 8.7 Hz, 2H), 7.95 (d, J = 8.7 Hz, 2H), 10.87 (s, 1H); 13C NMR (100 MHz, d6-DMSO): δ (ppm) = 108.9, 120.2, 124.2, 125.2, 129.1, 129.6, 130.4, 137.9, 138.4, 156.5, 165.1, 170.5. MS (ESI+): m/z = 344.0. ESI-HR-MS calculated for C16H10ClN3O4 (M+ + H): 344.0438, found: 344.0435.
General procedure for the synthesis of substituted 3-aminoisoxazoles (4) as exemplified for the synthesis of methyl 3-(benzylamino)-5-(4-nitrophenyl)isoxazole-4-carboxylate (4bb)
To a stirred solution of methyl 3-nitro-5-(4-nitrophenyl)isoxazole-4-carboxylate 1b (0.2 g, 0.68 mmol) in MeCN (5 mL) was added benzylamine (0.11 mL, 1.02 mmol) and the reaction was continued at room temperature for 12 h. After completion (as monitored by TLC), the solvent was evaporated and the residue was extracted with EtOAc (3 × 30 mL) and water (20 mL). The combined organic fractions were dried over Na2SO4, and the solvent was removed under vacuum. The crude product thus obtained was purified via silica gel column chromatography (hexanes/EtOAc, 9 : 1, v/v) to obtain 4bb (0.216 g from 0.2 g, 90%) as a yellow solid. Methyl 3-(benzylamino)-5-(4-nitrophenyl)isoxazole-4-carboxylate (4bb). Mp = 112–114 °C; Rf = 0.65 (hexanes : EtOAc, 6 : 4, v/v); IR (KBr) νmax: 1730 (CO2Me), 3310 (NH) cm–1; 1H NMR (400 MHz, CDCl3): δ (ppm) = 3.76 (s, 3H), 4.46 (d, J = 5.9 Hz, 2H), 5.91 (t, J = 5.6 Hz, 1H), 7.22–7.25 (m, 1H), 7.28–7.35 (m, 4H), 7.96 (d, J = 9.0 Hz, 2H), 8.26 (d, J = 9.0 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 47.3, 52.1, 100.2, 123.5, 127.7, 127.8, 128.8, 130.4, 133.2, 138.4, 149.2, 162.8, 163.3, 170.1. MS (ESI+): m/z = 354.1. ESI-HR-MS calculated for C18H15N3O5 (M+ + H): 354.1090, found: 354.1093.
General procedure for the synthesis of isoxazole-tethered quinolones (6) as exemplified for the synthesis of methyl 3-((2-((7-chloroquinolin-4-yl)amino)ethyl)amino)-5-(4-nitrophenyl)isoxazole-4-carboxylate (6ba)
To a stirred solution of methyl 3-nitro-5-(4-nitrophenyl)isoxazole-4-carboxylate 1b (0.2 g, 0.68 mmol) in MeCN (8 mL) was added Et3N (0.141 mL, 1.02 mmol) and N1-(7-chloroquinolin-4-yl)ethane-1,2-diamine 5a (0.151 g, 0.68 mmol) and the mixture was heated at 80 °C for 12 h. After completion, the solvent was removed under reduced pressure and the residue was extracted with EtOAc (3 × 30 mL) and water (30 mL). The combined organic fractions were dried over Na2SO4, and the solvent was removed under vacuum. The crude product thus obtained was purified via silica gel column chromatography (hexanes/EtOAc, 3 : 7, v/v) to obtain 6ba (0.265 g from 0.2 g, 83%) as a brown solid, which was further crystallized from ethanol. Methyl 3-((2-((7-chloroquinolin-4-yl)amino)ethyl)amino)-5-(4-nitrophenyl)isoxazole-4-carboxylate (6ba). Mp = 181–184 °C; Rf= 0.51 (hexanes : EtOAc, 1 : 9, v/v); IR (KBr) νmax: 1743 (CO2Me), 3392 (NH) cm–1; 1H NMR (400 MHz, d6-DMSO): δ (ppm) = 3.57 (s, 4H), 3.75 (s, 3H), 6.57 (t, J = 5.1 Hz, 1H), 6.71 (d, J = 5.4 Hz, 1H), 7.51 (d, J = 8.6 Hz, 1H), 7.59 (t, J = 4.2 Hz, 1H), 7.80 (s, 1H), 8.10 (d, J = 8.4 Hz, 2H), 8.28 (d, J = 8.9 Hz, 1H), 8.36 (d, J = 8.5 Hz, 2H), 8.44 (d, J = 5.1 Hz, 1H); 13C NMR (100 MHz, d6-DMSO): δ (ppm) = 41.4, 41.5, 52.4, 99.2, 100.9, 117.8, 123.9, 124.6, 124.8, 127.4, 130.9, 133.0, 134.2, 148.7, 149.2, 151.1, 151.7, 162.2, 163.0, 170.1. MS (ESI+): m/z = 468.1. ESI-HR-MS calculated for C22H18ClN5O5 (M+ + H): 468.1075, found 468.1078.
In vitro antileishmanial screening
Parasite
Leishmania donovani promastigotes (WHO designation MHOM/IN/80/Dd8; originally obtained from Imperial College, London as a gift by the kind courtesy of (the late) Prof. P. C. C. Garnham) transfected with a luciferase reporter gene were maintained at 25 ± 1 °C in medium 199 (Sigma Chemical Co., USA) supplemented with 10% heat-inactivated foetal calf serum (Gibco, Gaithersburg, MD) and G418 (20 μg ml–1) and were used for the in vitro evaluation of antileishmanial activity.
Antipromastigote activity
The effect of the compounds on the growth of promastigotes was assessed by monitoring the luciferase tagged activity of viable cells after compound treatment.36 The transgenic promastigotes of late log phase (5 × 105 cells per well) were seeded in 96-well flat bottomed microtitre (MT) plates (Cellstar, Greiner Bio-One GmbH, Monroe, NC) and were allowed to grow for 72 h in medium alone or in the presence of different concentrations of the antileishmanial compound dissolved in DMSO. Parallel dilutions of DMSO were used as controls. After incubation, an aliquot (50 μl) of promastigote suspension was aspirated from each well and mixed with an equal volume of Steady-Glo reagent (Promega, Madison, WI), and the luminescence was measured using a luminometer. The values were expressed as relative luminescence unit (RLU). The inhibition of parasitic growth was determined by comparing the luciferase activity of drug treated parasites with that of untreated controls using the formula:Percentage Inhibition = [N – n/N] × 100where N is the average relative luminescence unit (RLU) of control wells and n is the average RLU of treated wells.
Antiamastigote activity
For assessing the activity of the compounds against the amastigote stage of the parasite, the J-774A.1 mouse macrophage cell line (obtained from NCCS, Pune) was infected with luciferase expressing transgenic promastigotes.36 The macrophage cells were seeded in a 96-well plate (2 × 105 cells per 200 μl per well) in RPMI-1640 containing 10% foetal calf serum. The plates were incubated at 37 °C in a CO2 incubator. After 24 h, the medium was replaced with a fresh medium containing stationary phase transgenic promastigotes in a 1 : 10 ratio (2 × 106 cells per 200 μl per well). Promastigotes invade the macrophage and are transformed into amastigotes. The test compounds in appropriate concentrations in complete medium were added after replacing the previous medium and the plates were incubated at 37 °C in a CO2 incubator for 72 hours. After incubation, the drug containing medium was replaced with 50 μL PBS, mixed with an equal volume of Steady-Glo reagent, and gently shaken for 1–2 min, and then reading was taken on a luminometer. The inhibition of amastigote growth was determined as described above.
Cytotoxicity against the J774A cell line (MTT method)
The J774.1A cells were seeded in a 96-well plate at a concentration of 0.2 × 106 cells per well and incubated overnight in a CO2 incubator, with a supply of 5% CO2 at 37 °C. Different concentrations of the test compounds and reference drugs dissolved in DMSO were added. Parallel dilutions of DMSO alone were used as controls. The plates were incubated further for 48 h and the number of viable cells per well was determined by formation of a blue colour formazan product of MTT, generated as a result of the mitochondrial dehydrogenase activity of viable cells.37 The relative O. D. was measured at 600 nm against the blank using an automated microplate reader (Spectra max, Molecular Devices). The cytotoxic effect was expressed as 50% lethal dose (CC50), i.e. the concentration of a compound which results in 50% reduction in cell viability compared to that of a cell in culture medium alone.
Data analysis
The IC50 and CC50 values were calculated by regression obtained through Probit analysis38 of the log dose/response of the drug. The data were presented as mean ± standard deviation of three independent experiments. A P-value of <0.05 was considered as significant.
In vivo antileishmanial screening
Male golden hamsters (inbred strain) weighing 40 to 45 g were used as experimental hosts for the in vivo study and acclimatized for one week before the experiment under standard environment conditions. The hamsters were housed at 23 ± 2 °C, with humidity of 60 to 63%, fed with standard rodent pellets and fresh drinking water and subjected to a particular light/dark cycle of 12 hours. The animals were infected with 1 × 107 amastigotes per animal intra-cardially and infection was allowed to establish within visceral organs for 20 days. Pre-treatment spleen biopsy in all the animals was carried out to assess the degree of infection. The animals with +1 infection (5–15 amastigotes/100 spleen cell nuclei) were randomized into several groups (five animals/group). Drug treatment both by the IP and oral routes was initiated after 2 days of pre-treatment biopsy and continued for 5 consecutive days. Untreated infected controls received only the vehicle. Post-treatment biopsies were done on the 7th day and 28th day of the last drug administration and amastigote counts were assessed by Giemsa staining. Intensification of infection in both treated and untreated animals, as well as the initial count, was compared and the efficacy was expressed in terms of percentage inhibition (PI) using the following formula:PI = 100 – [ANAT × 100/(INAT × TIUC)]where PI is the percent inhibition of amastigote multiplication, ANAT is the actual number of amastigotes in treated animals, INAT is the initial number of amastigotes in treated animals and TIUC is the times increase of parasites in untreated control animals.
For the in vivo evaluation, aqueous solutions of the test compounds were prepared by suspending the accurately weighed sample in a standard suspension vehicle of 10% Tween-80/ethanol (70 : 30) in ddH2O. The final volume contains 10% of the vehicle for inoculation by the IP route.
For oral administration, aqueous solutions of the test compounds were prepared by suspending the accurately weighed sample in a standard suspension vehicle of 5% carboxymethyl cellulose in double distilled water. The final volume contains 5% of the vehicle for inoculation.
Pharmacokinetic assessment
Hamsters were segregated in four groups, with each group containing three hamsters per cage (n = 3). The suspension of 4mf was prepared in 0.5% sodium carboxy methylcellulose (NaCMC) as a suspending agent and was orally administered to the animals. The plasma concentration was analysed with an LC–MS/MS system using electrospray ionization. The data were processed using Analyst 1.6.2 software (AB Sciex, Canada). The pharmacokinetic parameters were calculated with Phoenix WinNonlin software version 6.3 (Pharsight Corporation, Mountain View, CA, USA) using a non-compartmental model.
Ethical statement
All procedures involving experimental animals were approved by the Institutional Animal Ethics Committee (IAEC) of CSIR-Central Drug Research Institute, Lucknow. The animal protocol was reviewed and approved (IAEC/2018/F37/dated 09/03/2018) and renewed (IAEC/2018/F37/renew1/dated 05/04/2019). They also adhered to the National Guidelines CPCSEA (Committee For the Purpose of Control and Supervision of Experiments on Animals) of Government of India. The animals were housed in plastic cages at 23 ± 2 °C and 60–63% humidity, and fed with standard rodent pellets and water ad libitum and fed with standard rodent food pellets (Lipton India, Bombay).
Conflicts of interest
There is no conflict of interest to declare.
Supplementary Material
Acknowledgments
Two of the authors (SM and DSB) gratefully acknowledge the financial support from the Council of Scientific and Industrial Research, New Delhi in the form of fellowship. The authors thank the SAIF Division of this institute for providing the spectroscopic data. The present work was supported by the SERB grant EMR-2016/002162. This is CDRI communication number 10088.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/d0md00083c
References
- Burza S., Croft S. L., Boelaert M. Lancet. 2018;392:951–970. doi: 10.1016/S0140-6736(18)31204-2. [DOI] [PubMed] [Google Scholar]
- WHO, WHO fact sheet on Leishmaniasis, https://www.who.int/en/news-room/fact-sheets/detail/leishmaniasis, 4 March 2019. (accessed on 17 Feb 2020).
- WHO, World Health Organisation Technical Report Series, 2012, p. 949.
- Sundar S., Chakravarty J. Int. J. Environ. Res. Public Health. 2010;7:4267–4277. doi: 10.3390/ijerph7124267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappuis F., Sundar S., Hailu A., Ghalib H., Rijal S., Peeling R. W., Alvar J., Boelaert M. Nat. Rev. Microbiol. 2007;5:873–882. doi: 10.1038/nrmicro1748. [DOI] [PubMed] [Google Scholar]
- Mishra M., Singh M. P., Choudhury D., Singhand V. P., Khan A. B. Lancet. 1991;337:926–927. doi: 10.1016/0140-6736(91)90268-t. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S. K., Sinha P. K., Sundar S., Thakur C. P., Jha T. K., Pandey K., Das V. R., Kumar N., Lal C., Verma N., Singh V. P., Ranjan A., Verma R. B., Anders G., Sindermann H., Ganguly N. K. J. Infect. Dis. 2007;196:591–598. doi: 10.1086/519690. [DOI] [PubMed] [Google Scholar]
- Balasegaram M., Ritmeijer K., Lima M. A., Burza S., Ortiz G. G., Milani B., Gaspani S., Potet J., Chappuis F. Expert Opin. Emerging Drugs. 2012;17:493–510. doi: 10.1517/14728214.2012.748036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar S., Chakravarty J. J. Global Infect. Dis. 2010;2:159–166. doi: 10.4103/0974-777X.62886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorlo T. P. C., Huitema A. D. R., Beijnen J. H., de Vries P. J. Antimicrob. Agents Chemother. 2012;56:3864–3872. doi: 10.1128/AAC.00292-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari V., Kulshrestha A., Deep D. K., Stark O., Prajapati V. K., Ramesh V., Sundar S., Schonian G., Dujardin J. C., Salotra P. PLoS Neglected Trop. Dis. 2012;6:e1657. doi: 10.1371/journal.pntd.0001657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey K., Pun S., Pandey B. Indian J. Med. Microbiol. 2012;30:227–229. doi: 10.4103/0255-0857.96703. [DOI] [PubMed] [Google Scholar]
- Obonaga R., Fernández O. L., Valderrama L., Rubiano L. C., Castro M. D. M., Barrera M. C., Gomez M. A., Gore Saravia N. Antimicrob. Agents Chemother. 2014;58:144–152. doi: 10.1128/AAC.01023-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie S., Patterson S., Stojanovski L., Simeons F. R. C., Norval S., Kime R., Rea K. D., Fairlamb A. H. Sci. Transl. Med. 2012;4:119re1. doi: 10.1126/scitranslmed.3003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://clinicaltrials.gov/ct2/show/NCT01980199 (accessed on 18 Feb, 2020).
- Gupta S., Yardley V., Vishwakarma P., Shivahare R., Sharma B., Launay D., Martin D., Puri S. K. J. Antimicrob. Chemother. 2015;70:518–527. doi: 10.1093/jac/dku422. [DOI] [PubMed] [Google Scholar]
- Thompson A. M., O'Connor P. D., Marshall A. J., Yardley V., Maes L., Gupta S., Launay D., Braillard S., Chatelain E., Franzblau S. G., Wan B., Wang Y., Ma Z., Cooper C. B., Denny W. A. J. Med. Chem. 2017;60:4212–4233. doi: 10.1021/acs.jmedchem.7b00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson S., Fairlamb A. H. Curr. Med. Chem. 2019;26:4454–4475. doi: 10.2174/0929867325666180426164352. [DOI] [PubMed] [Google Scholar]
- Petri e Silva S. C., Palace-Berl F., Tavares L. C., Soares S. R., Lindoso J. A., Exp. Parasitol., 2016, 163 , 68 –75 , , and references cited therein . [DOI] [PubMed] [Google Scholar]
- Wyllie S., Roberts A. J., Norval S., Patterson S., Foth B. J., Berriman M., Read K. D., Fairlamb A. H. PLoS Pathog. 2016;12:e1005971. doi: 10.1371/journal.ppat.1005971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giomi D., Cordero F. M. and Machetti F., Comprehensive Heterocyclic Chemistry III, ed. A. R. Katrizky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, UK, 2008, pp. 365–486. [Google Scholar]
- Barmade M. A., Murumkar P. R., Sharma M. K., Yadav M. R. Curr. Top. Med. Chem. 2016;16:2863–2883. doi: 10.2174/1568026616666160506145700. [DOI] [PubMed] [Google Scholar]
- Sysak A., Obmińska-Mrukowicz B. Eur. J. Med. Chem. 2017;137:292–309. doi: 10.1016/j.ejmech.2017.06.002. [DOI] [PubMed] [Google Scholar]
- Zhu J., Mo J., Lin H. Z., Chen Y., Sun H. P. Bioorg. Med. Chem. 2018;23:3065–3075. doi: 10.1016/j.bmc.2018.05.013. [DOI] [PubMed] [Google Scholar]
- Mukherjee T., Roy K., Bhaduri A. Biochem. Biophys. Res. Commun. 1990;170:426–432. doi: 10.1016/0006-291x(90)92109-d. [DOI] [PubMed] [Google Scholar]
- Suryavanshi S. N., Tiwari A., Chandra N., Ramesh, Gupta S. Bioorg. Med. Chem. Lett. 2012;22:6559–6562. doi: 10.1016/j.bmcl.2012.09.024. [DOI] [PubMed] [Google Scholar]
- Haavikko R., Nasereddin A., Sacerdoti-Sierra N., Kopelyanskiy D., Alakurtti S., Tikka M., Jaffe C. L., Yli-Kauhaluoma J. Med. Chem. Commun. 2014;5:445–451. [Google Scholar]
- Trefzger O. S., Barbosa N. V., Scapolatempo R. L., das Neves A. R., Ortale M. L. F. S., Carvalho D. B., Honorato A. M., Fragoso M. R., Shuiguemoto C. Y. K., Perdomo R. T., Matos M. F. C., Chang M. R., Arruda C. C. P. and Baroni A. C. M., Arch. Pharm. Chem. Life Sci., 2019, p. e1900241. [DOI] [PubMed] [Google Scholar]
- Trefzger O. S., das Neves A. R., Barbosa N. V., Carvalho D. B., Pereira I. C., Perdomo R. T., Matos Maria F. C., Yoshida N. C., Kato M. J., de Albuquerque S., Arruda C. C. P., Baroni A. C. M. Chem. Biol. Drug Des. 2019;93:313–324. doi: 10.1111/cbdd.13417. [DOI] [PubMed] [Google Scholar]
- da Rosa R., de Moraes M. H., Almida Zimmermann L., Schenkel E. P., Steindel M., Bernardes L. S. Campos. Eur. J. Med. Chem. 2017;128:25–35. doi: 10.1016/j.ejmech.2017.01.029. [DOI] [PubMed] [Google Scholar]
- Gangwar S., Baig M. S., Shah P., Biswas S., Batra S., Siddiqi M. I., Goyal N. Chem. Biol. Drug Des. 2012;79:149–156. doi: 10.1111/j.1747-0285.2011.01262.x. [DOI] [PubMed] [Google Scholar]
- Dighe S. U., Mukhopadhyay S., Kolle S., Kanojiya S., Batra S. Angew. Chem., Int. Ed. 2015;54:10926–10930. doi: 10.1002/anie.201504529. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S., Barak D. S., Avasthi I., Batra S. Adv. Synth. Catal. 2017;359:4050–4056. [Google Scholar]
- Antinarelli L. M. R., Dias R. M. P., Souza I. O., Lima W. P., Gameiro J., da Silva A. D., Coimbra E. S. Chem. Biol. Drug Des. 2015;86:704–714. doi: 10.1111/cbdd.12540. [DOI] [PubMed] [Google Scholar]
- .
- Ashutosh S., Gupta R., Sundar S., Goyal N. Antimicrob. Agents Chemother. 2005;49:3776–3783. doi: 10.1128/AAC.49.9.3776-3783.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann T. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Finney D. J., Probit Analysis, Cambridge University Press, 3rd edn, 1971. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.