

Abstract
Ketamine exerts rapid antidepressant action in depressed and treatment resistant depressed patients within hours. At the same time, ketamine elicits a unique form of functional synaptic plasticity that shares several attributes and molecular mechanisms with well-characterized forms of homeostatic synaptic scaling. Lithium is a widely used mood stabilizer also proposed to act via synaptic scaling for its antimanic effects. Several studies to date have identified specific forms of homeostatic synaptic plasticity that are elicited by these drugs used to treat neuropsychiatric disorders. In the last two decades extensive work on homeostatic synaptic plasticity mechanisms have shown they diverge from classical synaptic plasticity mechanisms that process and store information, and thus present a novel avenue for synaptic regulation with limited direct interference with cognitive processes. In this article, we discuss the intersection of the findings from neuropsychiatric treatments and homeostatic plasticity studies to highlight a potentially wider paradigm for treatment advance.
Introduction
The National Institute of Mental Health estimates 21.4% of U.S. adults experience a mood disorder, these include Major Depressive Disorder or Bipolar Disorder, at some point in their lives (https://www.nimh.nih.gov/health/statistics/any-mood-disorder.shtml). Identification of rapid acting treatments with predictable outcomes for these disorders is an urgent societal need. Since the middle of the 20th century, neuropharmacological treatments primarily targeting monoaminergic neurotransmission have reached the clinic with varying degrees of efficacy for the treatment of certain mood disorders. While existing treatments are effective in some patients in their ability to alleviate symptoms and improve the quality of life, they are ineffective in other patients and have raised the possibility that better understanding their mechanisms of action may lead to the identification of approaches with wider applicability and fewer side effects (Monteggia et al., 2014). While a large number of studies have focused on neuronal mechanisms that underlie neuropsychiatric drug action the findings have not had a major impact on treatment advance.
The unexpected discovery that ketamine exerts rapid antidepressant action provides an opportunity to identify specific synaptic substrates that are key to triggering antidepressant effects, which may ultimately serve as new targets for drug development. In the past decade, clinical studies have demonstrated that intravenous administration of a low dose of ketamine triggers a rapid antidepressant response in patients with major depression including treatment resistant depression and bipolar depression (Berman et al, 2000; Zarate et al., 2006; Price et al., 2009; Daly et al., 2018). As classical monoaminergic antidepressants typically take several weeks to exert antidepressant effects, their mechanism of action has been posited to involve long-term processes that rewire neuronal circuits and are dependent on transcriptional processes as well as chromatin remodeling (Nestler et al., 2000). In contrast, ketamine exerts its action within hours, shortly after its clearance, thus indicating a fundamentally different mechanism of action. Ketamine’s ability to exert rapid antidepressant effects indicates that major symptoms of depression can be alleviated without a requirement for substantial circuit rewiring. The fast-acting nature of ketamine action provides an opportunity to elucidate the types of acute synaptic plasticity changes that can be recruited to counter depression symptoms. Multiple studies from our group as well as others have demonstrated that ketamine administration elicits a unique form of synaptic plasticity that shares several attributes and molecular mechanisms that were previously identified in homeostatic synaptic scaling (Kavalali and Monteggia, 2012; Kavalali and Monteggia, 2015). Recent studies have started to examine synaptic plasticity processes elicited by lithium, a well-documented mood stabilizer used for the treatment of bipolar disorder. While lithium administration takes several weeks to exert its mood stabilizing effects, rather unexpectedly it has been shown to induce specific effects on homeostatic synaptic scaling (Gideons et al., 2017). In this article, we examine recent findings demonstrating that ketamine and lithium, two well documented treatments for mood disorders, trigger homeostatic plasticity mechanisms and discuss the intriguing possibility that these homeostatic forms of synaptic plasticity mediate the therapeutic effects of these drugs and may be viable targets for the treatment of mood disorders.
A Comparison of Adaptive versus Homeostatic Forms Plasticity
Acute and chronic alterations in the environment can induce various forms of plasticity in neuronal networks. Some forms of plasticity act in an adaptive fashion where changes in neuronal excitability or synaptic strength follow the direction of the manipulation and thus act to reinforce the demand imposed on the system via positive feedback regulation of neuronal or synaptic biophysical parameters (Figure 1). An example of such adaptive regulation occurs with short-term synaptic plasticity and presynaptic function and is observed in numerous biological systems. Studies in mammalian central synapses and invertebrate neuromuscular junction preparations have shown that synaptic release dynamics can be converted from tonic to phasic (or vice versa) in response to chronic changes in activity to meet or adjust to the demands in neurotransmitter release output (Lnenicka and Atwood, 1985, Lnenicka et al., 1986; Reid et al., 2003; Virmani et al., 2006). In these forms of plasticity, neuronal activity or synaptic efficacy diverge from their initial set point to adapt to the extrinsic influences. In many respects, classical Hebbian forms of plasticity can be considered within this context as synaptic efficacy increases — as in the case of Long Term Potentiation (LTP) — or weakens — as in the case of Long Term Depression (LTD) — based on the strength of the incoming neuronal activity (Bienenstock et al., 1982; Malenka and Bear, 2004).
Figure 1. Hebbian versus Homeostatic Synaptic Plasticity.
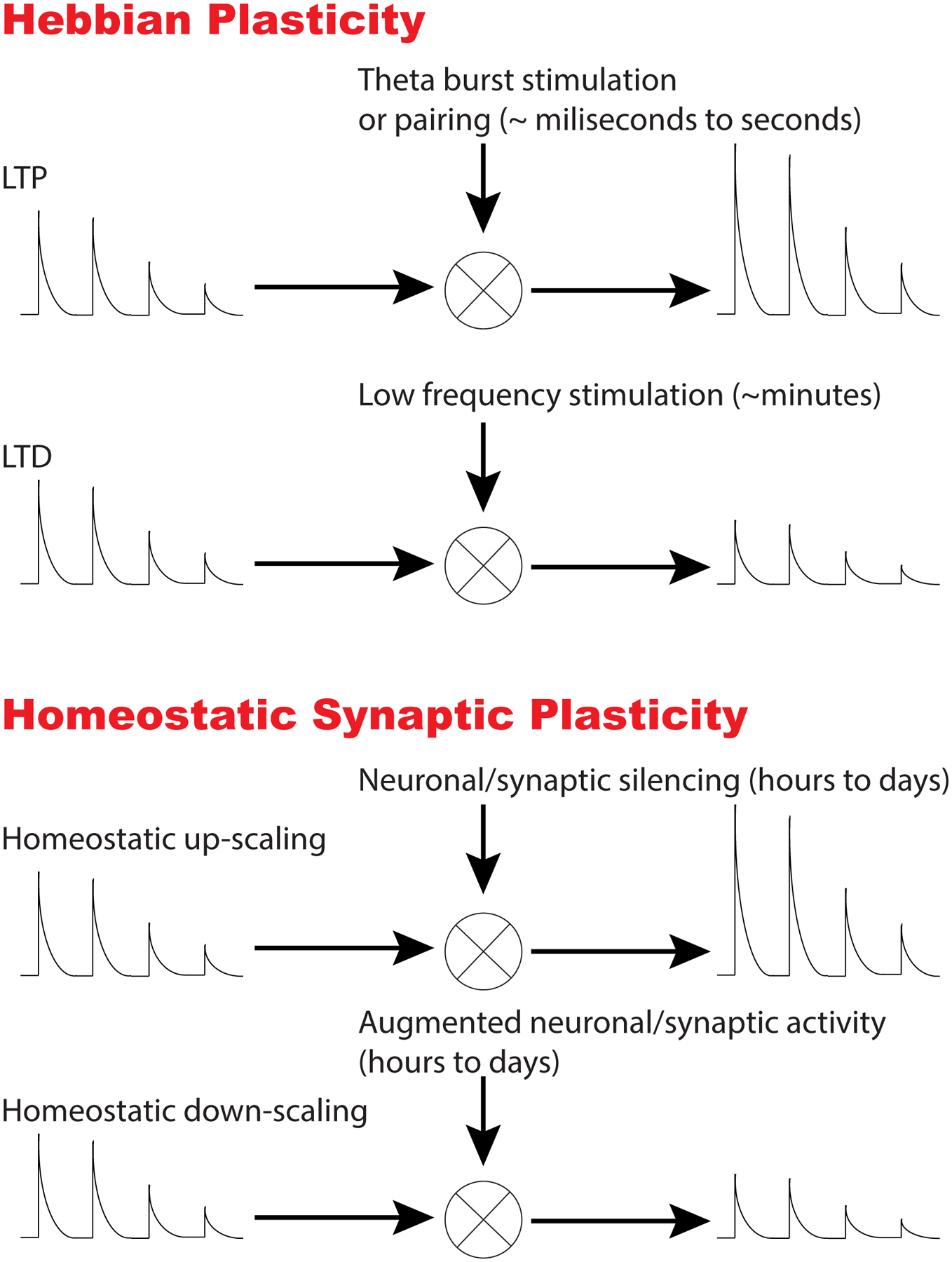
This figure depicts key distinguishing properties of Hebbian versus Homeostatic synaptic plasticity. During Hebbian plasticity, synaptic strength is modified in the same direction of the applied stimuli. For instance, strong stimulation (e.g. theta burst; 5 pulses at 100 Hz applied every second) or coincidence of presynaptic and postsynaptic activity (also called “pairing”) leads to an increase in synaptic strength resulting in Long Term Potentiation (LTP). In contrast, sustained low frequency stimulation (e.g. 1 Hz for 10–15 min) weakens synaptic strength leading to Long Term Depression (LTD). In contrast, homeostatic plasticity changes in neuronal network activity or alterations in synaptic inputs lead to an adjustment of synaptic strengths in the opposite direction (i.e. in a negative feedback manner) to bring neuronal activity patterns back to their initial set point. For instance, global silencing of activity leads to an increase in synaptic strength, whereas augmentation of activity results in downregulation of synaptic efficacy.
In addition to the forms of plasticity guided by positive feedback regulation, studies in the last two decades uncovered an array of mechanisms that counter global changes in neuronal network activity or alterations in synaptic inputs by adjusting synaptic strengths in a negative feedback manner to bring neuronal activity patterns back to their initial set point (Turrigiano et al., 1998; Styr et al 2019; Turrigiano and Nelson, 2004). These homeostatic plasticities respond to deviations from specific network activity patterns by triggering a negative feedback response. In this way, they either directly adjust neuronal excitability, to counter environmental or cell autonomous challenges (such as genetic mutations), or up- or down-regulate synaptic weights on a given neuron by a multiplicative factor in a process commonly referred to as “synaptic scaling”. During these forms of plasticity, synaptic weights are scaled up or down to counter the chronic activity levels detected in the cell. As a rule, “homeostatic synaptic scaling” is expected to preserve relative strengths of synapses and not alter the “information” content of synaptic inputs (Figures 1 and 2). This feature of homeostatic synaptic scaling makes it a desirable target for the treatment of nervous system disorders as the cognitive functions — that are thought to rely on relative values of synaptic weights — are expected to be minimally impaired.
Figure 2. Examples of Multiplicative Synaptic Scaling.
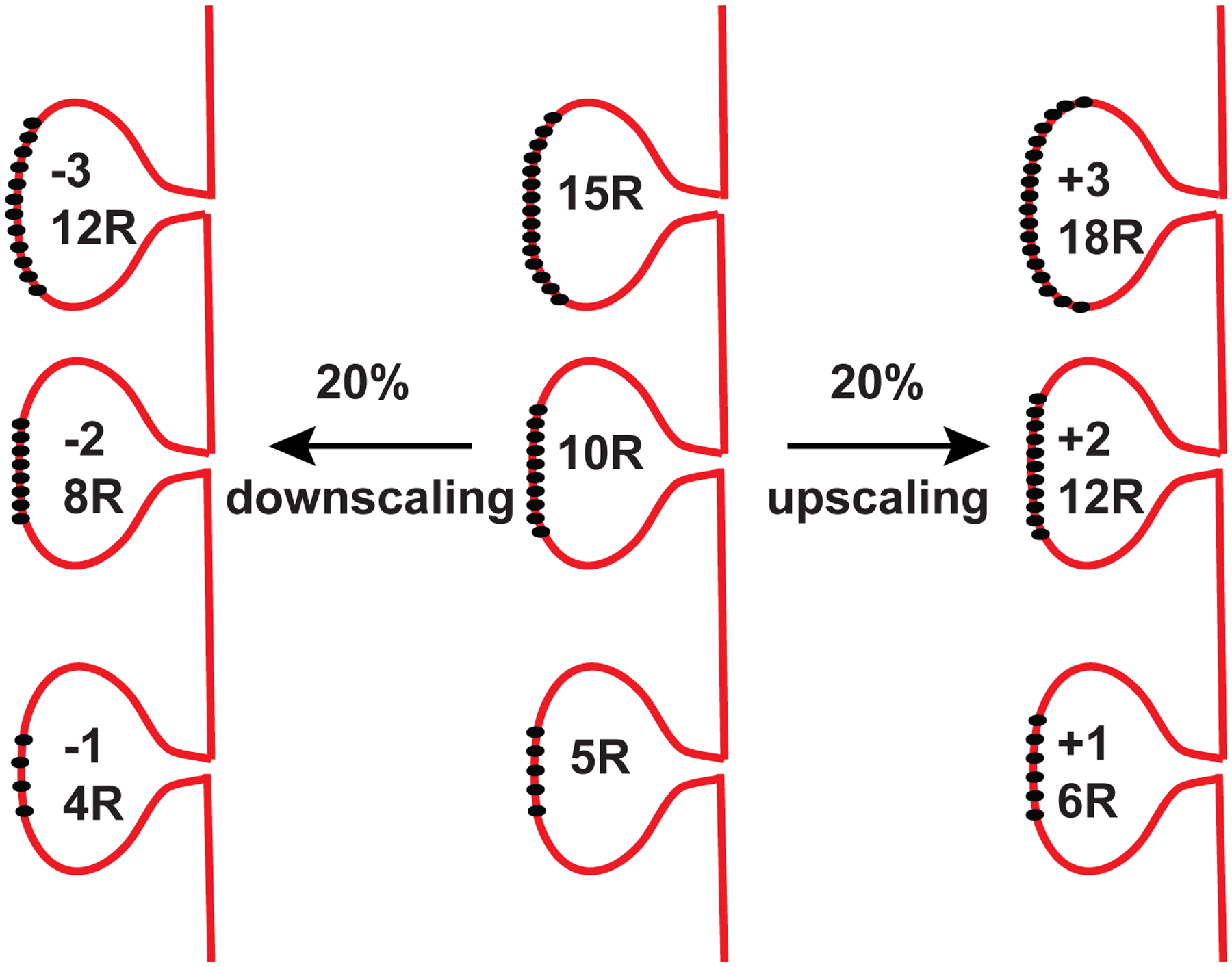
Multiplicative synaptic scaling involves up- or down-regulation of synaptic weights on a given neuron by a constant factor. During these forms of plasticity, synaptic weights are scaled up or down in a negative feedback manner to counter the chronic activity levels detected in the cell. As a rule, “homeostatic synaptic scaling” is expected to preserve relative strengths of synapses. Unlike Hebbian forms of plasticity, synaptic scaling does not alter the relative strength of a synapse with respect to its neighbors on a neuron. The figure depicts three representative postsynaptic spines on a neuron which possess 15, 10 and 5 receptors on their surface. If these synapses go through 20% synaptic downscaling or upscaling, the receptor numbers decrease (left) or increase (right) by 20% (±3, ±2, ±1) respectively.
Homeostatic Synaptic Plasticity Exists in Multiple Forms
Homeostatic synaptic scaling comprises a specific form of homeostatic synaptic plasticity where in response to long-lasting changes in neuronal activity quantal amplitude of all synapses onto a postsynaptic neuron are scaled up or down in a multiplicative manner. In mammalian central synapses scaling can be assessed using rank order plots of postsynaptic quantal amplitudes where unitary synaptic amplitudes (typically carried out by α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors) line up in a linear fashion and the slope increases or decreases without a significant change in the shape of the linear plot (Figure 2 and 3) (Turrigiano et al., 1998, Kim et al., 2012). This linear transformation is critical as it preserves the relative strengths of synaptic inputs. In this manner, neurons are expected to alter the strength of incoming synaptic inputs to arrive at a specific set point of action potential firing or metabolic activity (Styr et al 2019).
Figure 3. Ketamine and Lithium Elicit Multiplicative Synaptic Scaling.
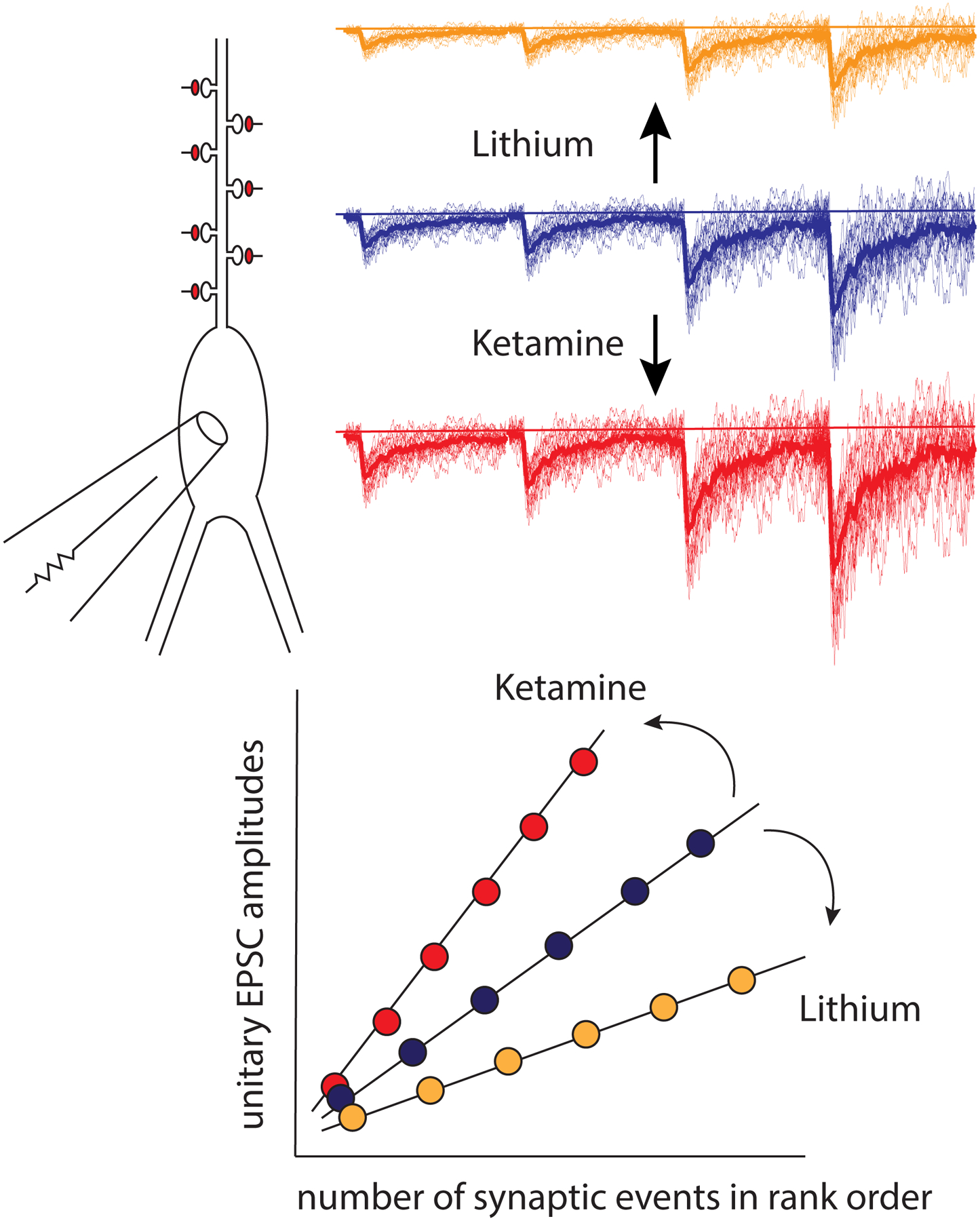
This figure depicts the synaptic action of ketamine and lithium on a neuron detected via whole cell voltage clamp recordings. Brief (1–3 hour) application of ketamine or other NMDA receptor blockers results in homeostatic upscaling of unitary synaptic responses (red traces). In contrast, chronic (~10–11 days) treatment of neurons with 1 mM lithium chloride results in marked synaptic downscaling (yellow traces). Despite their widely divergent targets and mechanisms of action, in preclinical models both treatments utilize the same synaptic process (i.e. synaptic scaling), albeit in opposite directions. The graph shows the tabulation of results from experiments where unitary excitatory postsynaptic current amplitudes are ranked from lowest to highest. These plots can be fitted with a line and ketamine or lithium treatment selectively increases or decreases the slope of the respective linear plot. The difference between the slopes of linear plots depicting unitary EPSC amplitudes after drug treatment and controls correspond to the “multiplicative factor” underlying the change in synaptic strength.
In contrast to synaptic scaling, homeostatic or negative feedback responses to chronic alterations in network activity may also involve presynaptic forms of plasticity (Murthy et al., 2001, Jakawich et al., 2010) possibly via retrograde regulation following postsynaptic signaling (Jakawich et al., 2010; Lindskog et al., 2010). However, presynaptic alterations in synaptic strength — typically based on changes in presynaptic release probability — violate the “scaling” rule as they do not necessarily preserve the relative strengths of postsynaptic efficacy. This is a critical distinction between pre- versus postsynaptic forms of homeostatic plasticity as alterations in presynaptic release probability not only up- or downregulate synaptic strength but also alter its frequency response thus modifying information processing dynamics in a synapse (Tsodyks and Markram, 1997). Thus, presynaptic forms of plasticity do not necessarily preserve relative strengths of synaptic input and may interfere with the transfer of information (Turrigiano and Nelson, 2004).
Early studies identified neuron wide activity as a key regulator of incoming synaptic inputs. However, subsequent work in a variety of systems —including hippocampal neurons, embryonic spinal cord, and the drosophila neuromuscular junction —also revealed a critical role of neurotransmitter release in regulating synaptic inputs. These findings have led to the hypothesis that a critical role for spontaneous release is in homeostatic synaptic plasticity, wherein synaptic strength is adjusted in response to sustained changes in the extent of neurotransmitter input independent of neuronal action potential firing (Gonzalez-Islas et al., 2017). Studies have isolated the contribution of neurotransmitter release events to this form of plasticity either by targeting spontaneous release-specific regulatory mechanisms to elicit direct and selective molecular interference with spontaneous release mediated signaling (Crawford et al., 2017; Ramirez et al., 2017), or by employing optogenetic stimulation to clamp cell wide action potential firing levels while manipulating neurotransmitter input pharmacologically. Collectively, these studies demonstrate a specific role for neurotransmitter input, in particular spontaneous release, in the regulation of synaptic strength (Fong et al., 2015; Garcia-Bereguiain et al., 2016).
These forms of homeostatic plasticity, independent of whether they sense and respond to cell wide activity or the level of neurotransmitter release input, are not directly involved in information storage or processing. The impairment of homeostatic plasticity may alter the dynamic range to elicit conventional forms of Hebbian plasticity (Yee et al., 2017; Turrigiano et al., 2017), however they typically play a modulatory role via global or local adjustment of synaptic strength. There is long standing evidence that these processes play a key role in recovery or plasticity of peripheral or central nervous system function in response to denervation, neuronal degeneration (Cannon and Rosenblueth, 1949), or during visual system plasticity (Turrigiano and Nelson, 2004), yet their precise function in normal physiological processes remains largely unclear. Recent studies have suggested that distinct phases of sleep may activate similar mechanisms to maintain neuronal network stability (de Vivo et al., 2017; Diering et al., 2017, but see Hengen et al., 2016) but there is more work needed in this area of study.
To date, research efforts have largely emphasized the malfunction in homeostatic plasticity processes identified in models of neurological, neuropsychiatric or neurodevelopmental disorders. However, treatment strategies targeting homeostatic processes have received relatively little attention. The increasing number of studies that have suggested impairments in homeostatic plasticity as underlying particular disorders (see below) may conversely provide therapeutic targets for treatment advance.
Chemical and Genetic Manipulations that Elicit Homeostatic Synaptic Plasticity
As indicated above, homeostatic forms of plasticity occur in response to negative feedback signals from neuronal circuits to re-establish a set point of activity or of synaptic strengths. These forms of plasticity can also operate at a local level in neurons targeting a set of synapses on a particular dendritic branch or can be synapse specific thus targeting the strength of a particular synapse rather than the firing of a neuron. Although, physiological circumstances that trigger homeostatic plasticity are expected to show neuronal circuit and/or synapse specificity, this is not necessarily the case for systemically administered drugs as they act on their targets and can modify neuronal circuits globally. Small molecules targeting key ion channels or synaptic signaling cascades can also trigger the same type of homeostatic mechanisms on a more global scale. Indeed, most experimental approaches to examine homeostatic plasticity forms have employed blockers of voltage-gated Na+ channels and postsynaptic ionotropic receptors (e.g. AMPA, NMDA, GABA receptors) in a variety of systems (see Table 1). These same receptors have been pharmacologically targeted for the treatment of numerous neurological and neuropsychiatric conditions. Despite the potential global effects of these pharmacological agents, a subset of these drugs act on their targets in a state-dependent manner selectively affecting open channels, inactivated channels or distinct allosteric states of receptors. The state dependence of these targets can provide circuit specificity versus simply targeting every receptor in a given circuit. An example of such state selective action is exhibited by NMDA or AMPA receptor antagonists that specifically block open channels via accessing the ion conduction pore (Huettner and Bean, 1988, Sara et al., 2011; Peled et al., 2014). NMDA receptor blockers such as MK-801, ketamine and memantine or the AMPA receptor blocker philanthotoxin possess this property and thus are expected to preferentially impact synapses with relatively high neurotransmitter release probability and postsynaptic channel open probability (Huettner and Bean, 1988; Gideons et al., 2014; Johnson et al., 2015, Sara et al., 2011; Peled et al., 2014).
Table 1.
An outline of experimental approaches used to examine forms of homeostatic synaptic plasticity.
| Manipulation | Agent | Synaptic Change | Translation/Transcription Dependence | Citation |
|---|---|---|---|---|
| Sodium channel block | TTX | Upscaling | Transcription | Turrigiano et al., 1998; Schaukowitch et al., 2017 |
| GABA receptor block | PtcrotoxirVbicuculline | Downscaling | Transcription? | Tyssowski et al., 2019 |
| NMDA receptor block | MK801/AP5/Ketamine | Upscaling | Translation | Sutton et al., 2006, 2007; Autry et al., 2011; Nosyreva et al., 2013 |
| AMPA receptor block | CNOX/NBQX | Increase in presynaptic function | Translation | Murthy et al., 2001; Jakawich et al., 2010; Lindskog et al., 2010 |
| Calcium chelation | EGTA-AM/BAPTA-AM | Upscaling | Translation | Wang et al., 2011 |
| L-type VGCC block | Nimodipine | Upscaling | Translation | Wang et al., 2011 |
| Ryanodine receptor block | Ryanodine/Dantrolene | Upscaling | Translation | Reese and Kavalali, 2015 |
| K+ channel overexpression | Kir1.2 | Reduction in synapses/increase in Pr | ND | Burrone et al., 2002 |
| Optogenetic stimulation | Channelrhodopsin | Postsynaptic elimination | Transcription | Goold and Nicoll, 2010 |
| Lithium | LiCI | Downscaling/restoration of homeostatic plasticity | Transcription | Gideons et al., 2017; Tatavarty et al., 2020 |
Most approaches have employed blockers of voltage-gated Na+ or Ca2+ channels and postsynaptic ionotropic receptors (e.g. AMPA, NMDA, GABA receptors) in a variety of systems. In addition, molecular expression of inward rectifier K+ channels or light-gated ion channels such as channelrhodopsin have been used to trigger multiple forms of homeostatic synaptic plasticity. It is interesting to note that Li+ administration elicits a similar effect as these more commonly used direct manipulations of neuronal activity or synaptic signaling.
Extrinsic pharmacological manipulation of neurotransmitter receptors or voltage-gated ion channels can lead to either chronic suppression or elevation of neuronal activity. Previous experiments suppressed neuronal activity using the voltage-gated Na+ channel blocker tetrodotoxin (TTX) or by genetic overexpression of an inwardly rectifying K+ channel that impedes membrane depolarization (Burrone et al., 2002). In contrast, neuronal activity can be enhanced by blocking GABAA receptors with bicuculline or picrotoxin (Turrigiano et al., 1998; O’Brien et al., 1998) as well as by optogenetic activation of neurons (Goold and Nicoll, 2010). The application of these compounds to neuronal cultures elicits multiplicative up- or down-scaling of miniature synaptic currents within 24 to 48 hours. Extensive evidence has shown synaptic upscaling triggered after sustained block of Na+ block depends on transcriptional regulation and thus operates on a slower time scale (Ibata et al., 2008, Schaukowitch et al., 2017). However, re-establishment of neuronal firing rates during persistent pharmacological stimulation (e.g. via chronic suppression of GABAergic inputs) may not strictly require activity-dependent gene transcription (Tyssowski et al., 2019).
While global activity manipulation on synaptic scaling is rather slow and transcription dependent, direct interference of synaptic inputs generates rapid synaptic scaling within hours via a mechanism that requires dendritic protein translation. Previous studies have shown that block of NMDA receptor-mediated transmission (Sutton et al., 2006, Autry et al., 2011, Nosyreva et al., 2013; Reese and Kavalali, 2015) or AMPA receptor-mediated transmission (Thiagarajan et al., 2005; Jakawich et al., 2010, Lindskog et al., 2010) results in local, synapse-specific, forms of homeostatic plasticity that occur within hours rather than days. NMDA receptor blockade by MK-801 or (2R)-amino-5-phosphonovaleric acid (AP5), independent of neuronal activity (i.e. in the presence of TTX), augments rapid protein synthesis through dephosphorylation of eukaryotic elongation factor-2 (eEF2), a critical catalytic factor for ribosomal translocation during protein synthesis (Sutton et al., 2007). Notably, application of TTX alone was not sufficient to trigger eEF2 dephosphorylation or the homeostatic plasticity (Nosyreva et al., 2013) demonstrating these events are activity independent. Taken together, these data show NMDA receptor activity at rest causes chronic activation of eEF2 kinase (eEF2K; also called CaMKIII), which phosphorylates eEF2, effectively suppressing translation. In contrast, acute NMDA receptor blockade at rest prevents eEF2 phosphorylation, thereby increasing rapid protein translation of target transcripts, including AMPA receptor subunits. These findings are consistent with genetic manipulations in which cell autonomous deletion of postsynaptic NMDA receptors leads to a similar augmentation of AMPA receptor mediated responses (Adesnik et al., 2008). Taken together, these studies provide a causal link between neurotransmission occurring at rest, in the absence of action potentials, and synaptic homeostasis. A key aspect of these studies is that spontaneous neurotransmitter release, rather than evoked neurotransmission, is a specific regulator of postsynaptic sensitivity to neurotransmitters by suppressing dendritic protein translation machinery locally and maintaining receptor composition of synapses (Sutton et al., 2004; Kavalali, 2015). This link between postsynaptic receptor block and its specific effects on homeostatic synaptic responses appears to be evolutionarily conserved. At the Drosophila neuromuscular junction, postsynaptic suppression of glutamate receptor signaling leads to retrograde signaling and increases in presynaptic input strength thus counteracting the diminished postsynaptic sensitivity (Davis and Muller, 2015). A similar form of presynaptic plasticity can also be triggered in mammalian synapses by acute block of postsynaptic AMPA receptors or genetic deletion of GluA4 AMPA receptors (Jakawich et al., 2010, Lindskog et al., 2010, Devendahl et al., 2019). In addition to pharmacological block of postsynaptic excitatory neurotransmitter receptors, denervation or application of clostridial neurotoxins, which proteolytically cleave synaptic vesicle fusion machinery components, in particular at the neuromuscular junction leads to suppression of neurotransmitter release and elicits similar homeostatic plasticity responses (Axelsson and Thesleff, 1959; Kim et al, 1984). Furthermore, genetic deletion of presynaptic voltage gated P/Q type Ca2+ channels also elicits compensatory changes in presynaptic output (Piedras-Renteria et al., 2004). Taken together, these studies demonstrate that although manipulations altering neuronal activity provided the initial insight into homeostatic plasticity, even subtle manipulations that directly interfere with neurotransmission can lead to robust homeostatic synaptic scaling (Nosyreva et al., 2013; Crawford et al., 2017). Thus, synapse targeted manipulations may provide a more viable path for development of novel therapeutics with expedient property profiles.
Homeostatic Synaptic Plasticity Deficits in Nervous System Disorders
Homeostatic synaptic plasticity deficits have been reported in a variety of animal models of neuropsychiatric, neurological and neurodevelopmental disorders. Impaired homeostatic responses to alterations of neuronal activity has been proposed as a key factor underlying the pathophysiology of Alzheimer’s Disease (Frere and Slutsky, 2018; Styr and Slutsky, 2018). Specifically, loss of presenilin — a key component of the gamma secretase complex associated with familial versions of Alzheimer’s Disease — in a mouse model of leads to specific deficits in homeostatic synaptic plasticity (Pratt et al., 2011). Studies in the field of drug addiction have shown incubation of drug craving can be driven by a maladaptive form of homeostatic plasticity (Conrad et al., 2008). Homeostatic synaptic deficiencies have been observed in a variety of preparations, including the invertebrate neuromuscular junction and the mammalian visual cortex, highlighting the evolutionary conserved nature of the deficits and may suggest they play a role in the disease pathophysiology (Davis and Bezprozvanny, 2001). Despite the reports of homeostatic plasticity deficits in nervous system disorders, the identification of specific disease related molecular targets that underlie many of these disorders have remained elusive.
Mouse models to study diseases of monogenic origin have started to yield insight into specific genes that may impact homeostatic plasticity. Mutations in a number of synaptic proteins linked to autism have shown particular proteins play critical roles in distinct forms of homeostatic plasticity (Zoghbi, 2003; Südhof, 2008). These proteins include synaptic cell adhesion molecules, such as neuroligins and neurexins, or synaptic scaffold proteins, such as Shank3 where mutations identified in patients lead to neurodevelopmental disorder profiles. While the majority of these mutations have been identified in only a few patients, they represent a starting point to examine whether there are common impairments, and potential points of convergence, in synaptic and circuit function that may ultimately underlie these disorders. In contrast to the limited number of patients affected by synaptic protein mutations, Rett Syndrome and Fragile X syndrome provide a wider clinical framework as they are two prevalent neurodevelopmental disorders of monogenic origin in which mouse models show specific deficits in homeostatic synaptic plasticity.
Rett syndrome is a neurological disorder caused by loss of function mutations in the transcriptional regulator methyl-CpG binding protein 2 (MECP2) gene. Rett syndrome is a rare disorder with estimates of 1:10,000 live female births. However, MeCP2 mutations have also been identified in autism patients as well as individuals with other neurodevelopmental disorders (Chahrour and Zoghbi, 2007) suggesting alterations in MeCP2 function contribute to neuronal dysfunction. Indeed, MeCP2 has been shown to act in a cell-autonomous manner to regulate synaptic scaling (Blackman et al., 2012; Zhong et al., 2012). Mouse models that express mutant Mecp2, or delete MeCP2, recapitulate similar key phenotypes of Rett syndrome making them powerful tools to examine how loss of Mecp2 function impacts transcriptional and synaptic processes (Nelson et al., 2006). Studies have shown Mecp2 knockout mice are unable to undergo homeostatic scaling in neocortex following visual deprivation suggesting that alterations in homeostatic plasticity may underlie aspects of Rett syndrome (Blackman et al., 2012). Recently generated non-human primate models with impaired Mecp2 function recapitulate several key phenotypes and will hopefully contribute to a more detailed understanding of the pathophysiology of this disorder (Chen et al., 2017).
Fragile X mental retardation protein (FMRP) is a dendritic RNA-binding protein encoded by the Fmr1 gene. Loss-of-function of FMRP in human patients causes Fragile X syndrome, the most common inherited form of mental retardation (Bassell and Warren, 2008). FMRP is associated with both translationally repressed messenger ribonucleoprotein particles and actively translating polyribosomes (Corbin et al., 1997; Zalfa et al., 2003) and is thought to specifically bind to mRNAs and regulate their translation (Laggerbauer et al., 2001; Li et al., 2001; Bassell and Warren, 2008). FMRP knock-out mice exhibit normal baseline synaptic transmission but show altered dendritic spine morphology (Comery et al., 1997; Irwin et al., 2000) and impairments in certain forms of long-term potentiation (LTP) (Li et al., 2002; Larson et al., 2005) and long-term depression (LTD) (Huber et al., 2002). Recent work has demonstrated that FMRP is required postsynaptically for a form of homeostatic synaptic scaling that requires Retinoic Acid signaling. In the absence of FMRP, Retinoic Acid-induced local translation of specific mRNAs is impaired resulting in suppression of the increase in synaptic strength seen after activity blockade or Retinoic Acid treatment (Soden and Chen, 2010). These findings further bolster the link between postsynaptic protein synthesis and acute regulation of homeostatic synaptic scaling in central neurons. Importantly, a recent study provides compelling evidence that homeostatic synaptic plasticity is actually important in the pathophysiology of Fragile X syndrome by demonstrating that in neurons from human patients, the FMR1 mutation suppresses this form of plasticity (Zhang et al. 2018).
While Rett syndrome or Fragile X syndrome are linked to individual genes, the specific impairments in homeostatic plasticity linked to transcriptional (in the case of MeCP2) or translational (FMRP) function of the underlying protein targets strongly supports the instructive role played by the transcriptional or translational mechanisms mediating homeostatic plasticity. However, most neuropsychiatric disorders are not linked to one gene but rather likely involve many genes. Therefore, it may be difficult to fully genetically model many of these disorders to examine potential alterations in baseline homeostatic plasticity processes, however these processes could still be targeted for treatment advance. Indeed, the diversity of homeostatic plasticity mechanisms that may coexist in neurons suggests that certain forms of homeostatic plasticity may be impaired, while others may be intact, allowing for the possibility of precise targeting to correct or bypass homeostatic plasticity deficiencies to reinstate specific homeostatic responses.
Homeostatic Synaptic Plasticity Can be Elicited by Neuropsychiatric Treatments
Recent studies have started to report the rather unexpected findings that neuropsychiatric treatments trigger homeostatic synaptic scaling similar to global regulation of activity or alterations in synaptic inputs. Ketamine and lithium are pharmacological treatments which trigger homeostatic synaptic upscaling and downscaling respectively (Figure 3). Importantly, loss-of-function genetic models that impair their mechanism of action and suppress their behavioral outcomes also hinder their impact on synaptic scaling (Autry et al., 2011, Nosyreva et al., 2013, Gideons et al., 2017). Taken together these findings implicate synaptic scaling as the primary synaptic target for the action of these drugs. Overall, despite their widely divergent targets, both treatments appear to focus on the same synaptic process (i.e. synaptic scaling), albeit in opposite directions in preclinical models.
Ketamine elicits homeostatic synaptic upscaling
Ketamine, is an NMDA receptor antagonist, that exerts rapid antidepressant effects in patients with treatment resistant depression and depression (Monteggia and Zarate, 2015). Clinical studies have shown that ketamine exerts rapid antidepressant effects within a few hours with effects persisting for days or in some patients for more than a week. Preclinical studies have started to examine the mechanism for ketamine’s rapid effects. While there have been various suggested targets for ketamine’s antidepressant action, its role as an antagonist of the NMDA receptor is the most widely accepted and the only target which has been linked to specific intracellular signaling pathways.
Preclinical studies have shown the antidepressant effects of ketamine are dependent on rapid protein synthesis of brain-derived neurotrophic factor (BDNF). Ketamine produces a rapid and robust increase in BDNF protein expression that returns to baseline levels by 24 hours. This rapid increase in protein expression is required for ketamine’s action as protein translation inhibitors block ketamine’s rapid effects (Autry et al., 2011) (Figure 4). This rapid increase in BDNF protein translation, which appears to occur at dendrites and involves the increased expression of other synaptic proteins, can produce rapid effects on synaptic transmission and provide an explanation for how ketamine can exert such rapid behavioral effects. The rapid increase in BDNF protein expression is not maintained suggesting its key effects may be in triggering downstream effects. The rapid synthesis of BDNF protein expression is downstream of eEF2 kinase, specifically requiring deactivation of eEF2 kinase and decreased eEF2 phosphorylation. In agreement with the studies outlined above, we have shown ketamine-mediated blockade of NMDA receptors at rest deactivates eEF2 kinase, resulting in a reduction of eEF2 phosphorylation and desuppression of BDNF translation (Autry et al., 2011; Suzuki et al., 2017). It is worth noting that ketamine has a relatively short half-live, so it is not persistent blockade of NMDA receptors that mediates the behavioral effects, but rather a more transient blockade that then resets the system by engaging eEF2 kinase and the downstream intracellular signaling pathway. These findings provide a framework for how ketamine, by blocking synaptic NMDA receptors, leads to a rapid increase in BDNF protein expression. A key finding of these studies is that these effects were not mimicked by alterations in neuronal activity levels in vivo, suggesting that spontaneous glutamate release and the subsequent NMDA receptor activation that occurs independent of action potentials, comprise the primary substrate for ketamine action. These data support the hypothesis that suppression of spontaneous neurotransmission-mediated NMDA receptor activation is necessary and sufficient to trigger antidepressant-like responses. These results also indicate that rapid up-regulation of BDNF, as well as other dendritic proteins, trigger multiplicative synaptic upscaling mechanisms that encompass the augmentation of evoked transmission to mediate long-term antidepressant effects (Nosyreva et al, 2013; Nosyreva et al, 2014). In a more recent study, although selective presynaptic inhibition of spontaneous neurotransmitter occluded this ketamine-induced plasticity, the same manipulation did not alter LTP indicating that ketamine induced potentiation and Hebbian LTP do not directly influence each other (Crawford et al., 2017). Nevertheless, there is evidence that ketamine administration may elicit metaplastic effects on modulating LTP and potentially other processes in the long term (Izumi and Zorumski, 2014). The exact nature of the intersection between ketamine-induced potentiation and other forms of plasticity that include LTP and LTD requires further investigation.
Figure 4. Model for Ketamine Action and Regulation of Protein Synthesis.
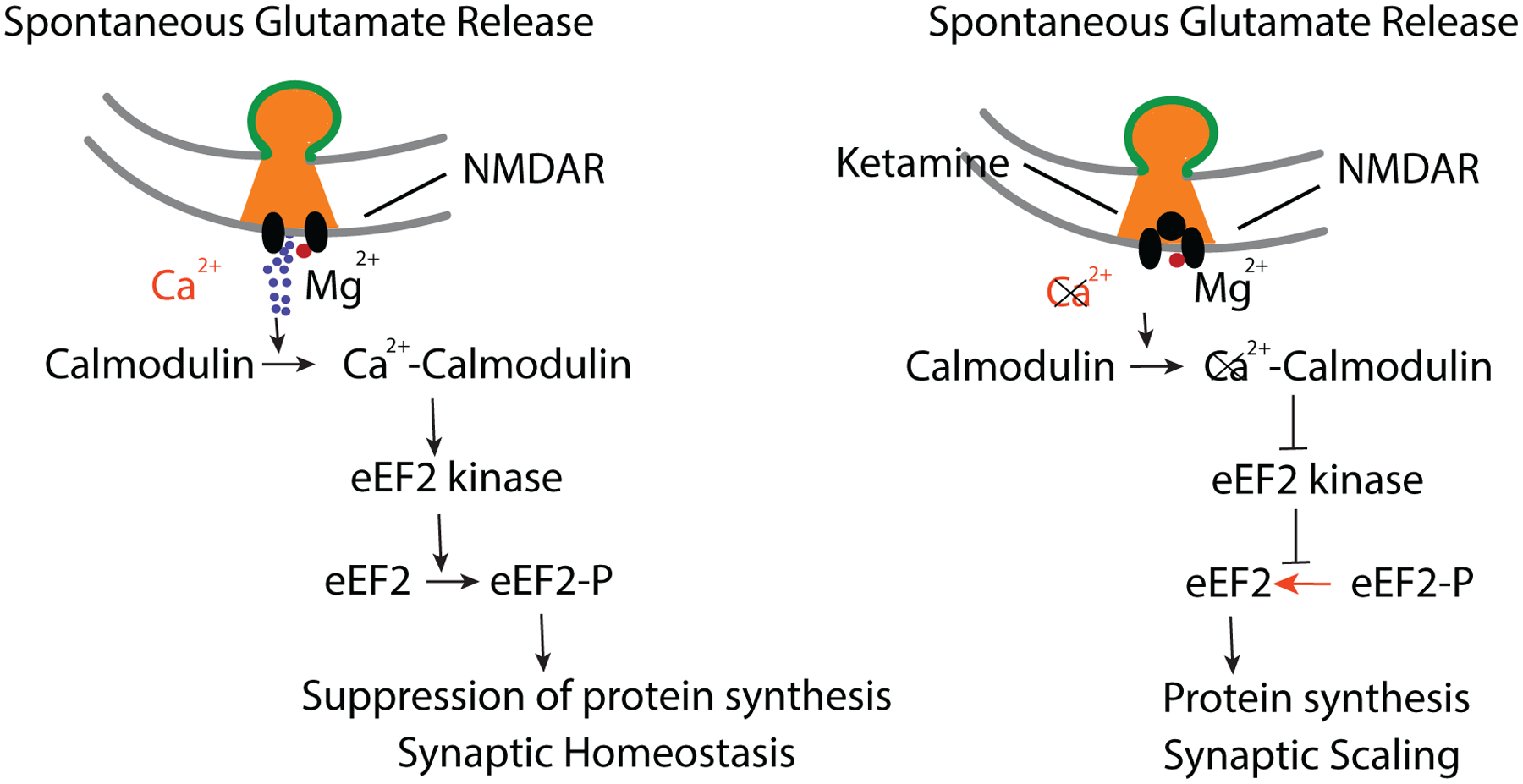
Left, Spontaneous glutamate release and NMDA receptor activation leads Ca2+ influx (even when Mg2+ is present see Reese and Kavalali, 2015). In turn, Ca2+-Calmodulin complexes activate eEF2 kinase leading to phosphorylation of eEF2 and suppression of protein translation. Right, Ketamine blocks this resting NMDA receptor activity leading to a decrease in Ca2+-Calmodulin complexes and deactivation of eEF2 kinase. Subsequent decrease in the amount of phosphorylated eEF2 elicits desuppression of dendritic protein translation, ultimately triggering synaptic upscaling (Sutton et al., 2006; Autry et al., 2011; Nosyreva et al., 2013, Reese and Kavalali, 2015).
Lithium elicits homeostatic synaptic downscaling
Lithium was initially reported as a mood stabilizer over 60 years ago (Cade, 1949) and is still widely used for the treatment of Bipolar Disorder (Poolsup et al., 2000; Vieta and Valentí, 2013). Lithium has effects on a variety of targets and its mechanism of action is not well understood. Some of the most widely replicated findings have implicated inhibition of GSK3, increased expression of neurotrophins, and decreased expression of AMPA receptors (Hashimoto et al., 2002; Pisanu et al., 2016).
Recent work examining the synaptic mechanisms elicited by lithium action have shown a robust and significant decrease on mEPSC amplitudes (Wei et al., 2010; Gideons et al., 2017). This decrease in glutamatergic neurotransmission may counteract increases in glutamatergic signaling seen in bipolar patients during mania (Lan et al., 2009; Ongür et al., 2008). This premise is consistent with other evidence from post-mortem brain tissue of individuals with bipolar disorder (Eastwood and Harrison, 2010; Hashimoto et al., 2007) or electrophysiological recordings from human induced pluripotent stem cell derived neurons from patients with bipolar disorder reporting molecular and functional alterations consistent with increase in glutamatergic neurotransmission (Mertens et al., 2015). In addition, there is evidence that lithium administration can restore homeostatic synaptic plasticity deficits seen in Shank3 loss-of-function mutants associated with autism spectrum disorders (Tatavarty et al. 2019).
Subsequent experiments from our group demonstrated that chronic lithium treatment decreases AMPA receptor surface expression in the hippocampus of treated mice, as well as primary hippocampal cultures, by targeting AMPAR trafficking (Figure 5). These findings are consistent with previous in vivo and in vitro studies that have shown lithium decreases GluA1 and GluA2 surface expression (Du et al., 2003; Du et al., 2008; Wei et al., 2010). Rather intriguingly this decrease in AMPA receptor surface expression requires BDNF and signaling via its high affinity receptor TrkB, similar to the anti-manic effects of lithium which also require BDNF and TrkB (Gideons et al., 2017). Lithium exerts effects on BDNF expression, but in contrast to ketamine, it produces gradual effects on increasing BDNF mRNA. The increase in BDNF mRNA is stable, long-lasting and results in a prolonged increase in BDNF protein expression.
Figure 5. A synaptic model illustrating a putative mechanism by which Li+ treatment leads to enhanced AMPAR endocytosis and downscaling of synaptic responses.
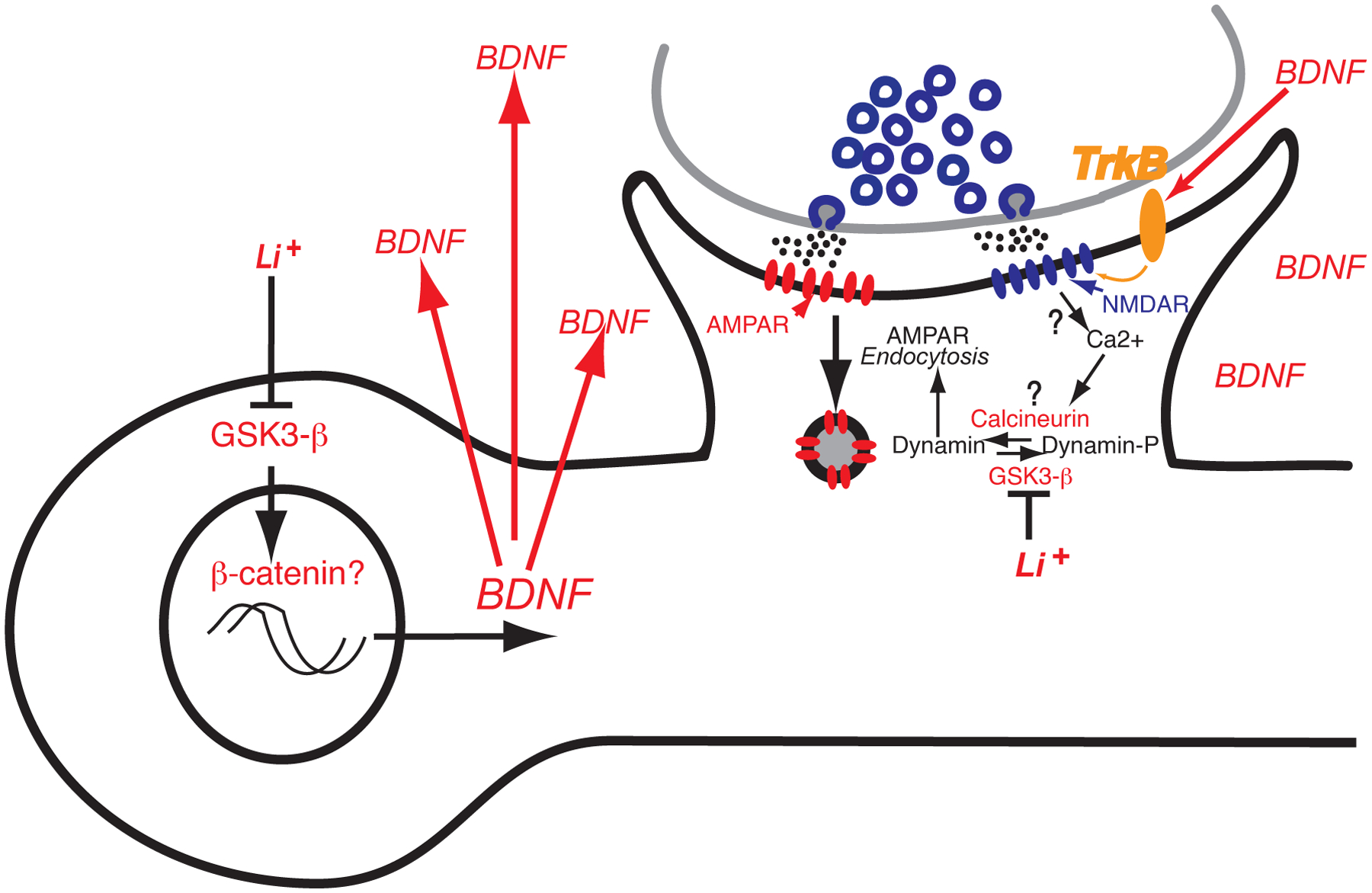
Several studies suggest that lithium can impact gene transcription via GSK3-β and β-catenin signaling. There is evidence that chronic lithium exposure leads to sustained elevation in BDNF levels, which in turn impacts surface levels of AMPA receptors via activation of its high affinity receptor TrkB. Importantly, recent work has identified dynamin-dependent endocytosis acting in conjunction with BDNF-TrkB signaling as a key target of lithium’s effects on synaptic function. According to this model, chronic lithium administration — that triggers anti-manic behavior — elicits synaptic downscaling via dynamin-dependent endocytosis of AMPA receptors. Here, in addition to alterations in gene transcription, lithium may directly impact AMPA receptor endocytosis by regulation of dynamin phosphorylation due to its inhibition of GSK3-β (see Gideons et al., 2017).
Further analysis revealed that lithium produces a significant downward multiplicative synaptic scaling of all AMPAR-mEPSC amplitudes consistent with an impact on post-synaptic homeostatic plasticity (Figure 3). In agreement with this result, we found that in vivo lithium treatment caused a significant decrease in input-output curves in hippocampal slices consistent with synaptic downscaling due to decreased GluA subunit surface expression. A key distinction between the actions of acute ketamine administration and chronic lithium treatment is the duration of elevations in BDNF they trigger. The findings so far indicate that acute transient elevations in BDNF levels elicit synaptic upscaling, while sustained elevations in BDNF may exert the opposite effect by downscaling postsynaptic AMPA responses. Here, it is important to note that the difference in BDNF action on synaptic efficacy may not only be a consequence of the duration of elevation but also due to subcellular location of BDNF action during a specific treatment (Reimers et al., 2014; Lin et al., 2018; Song et al., 2017). Despite the current advances in our understanding of the relationship of these neuropsychiatric treatments and BDNF signaling as well as their synaptic targets, future studies are needed to address how a single ligand-receptor couple such as BDNF and TrkB can elicit such versatile effects at the synaptic and behavioral levels.
Summary and Conclusion
Given the well-established efficacy of ketamine and lithium in the clinic, it is possible that previously unidentified homeostatic mechanisms — multiplicative synaptic upscaling in the case of ketamine and synaptic downscaling in the case of lithium — may also be involved in the therapeutic response to these drugs. However, the role of synaptic scaling in these compounds’ mechanism of action does not necessarily mean that depression or bipolar disorder is due to a defect in homeostatic plasticity, but rather that eliciting or augmenting homeostatic plasticity may have therapeutic benefits. While these two drugs provide a strong rationale for the hypothesis that synaptic scaling may play a key role in the treatment response, these findings likely represent a potentially wider paradigm applicable to other pharmacological treatments. It is possible that other neuropsychiatric treatments lead to global suppression or augmentation of synaptic activity and elicit similar forms of homeostatic synaptic plasticity. These include electroconvulsive shock therapy (Zhao et al., 2012), anesthetics such as isoflurane (Antila et al., 2017) and nitrous oxide (Nagele et al., 2015), that globally alter neuronal activity and have all been shown to have antidepressant effects in the clinic or in preclinical models. Although there had been much discussion over the years for the role of ‘plasticity’ in the brain in mediating treatment responses of numerous mental disorders, it has been difficult to identify common types of plasticity mechanisms that may be involved. The identification that ketamine and lithium can exert effects on homeostatic mechanisms in both rapid and sustained effects provides novel perspective on the types of plasticity that may actually be involved with the clinical efficacy.
This article provides an overview of our current knowledge on homeostatic synaptic plasticity as a substrate for the treatment of neuropsychiatric disorders. Ketamine as a rapid antidepressant and lithium as a mood stabilizer elicit homeostatic plasticity albeit via different mechanisms. Nevertheless, we posit that the convergence of these drugs on homeostatic mechanisms may suggest a common synaptic substrate for their therapeutic action. Directly eliciting homeostatic synaptic signaling can strengthen or weaken synaptic inputs, which in turn results in functional rewiring of circuits without structural reorganization. However, these functional changes may also be eventually coupled to alterations in synaptic structure that ensure maintenance of the functional modification for extended periods (Moda-Sava et al., 2019). Importantly, this form of functional rewiring via synaptic scaling can modify circuit dynamics without substantially altering information storage. Future approaches directly targeting the signaling pathways underlying homeostatic synaptic plasticity may widely expand the repertoire of available treatments. For instance, signal transduction cascades elicited by BDNF-TrkB signaling, translational regulation by eEF2-kinase (Autry et al., 2011; Nosyreva et al., 2013), retinoic acid signaling (Aoto et al., 2008; Wang et al., 2011) or other signaling pathways associated with homeostatic synaptic plasticity (e.g. Stellwagen and Malenka, 2006) may constitute viable options for therapy. Moreover, there is evidence that divergent vesicle trafficking pathways regulate homeostatic synaptic plasticity both presynaptically by targeting distinct forms of release and postsynaptically by mediating non-overlapping pathways for glutamate receptor trafficking (Arendt et al., 2015; Crawford et al., 2017). In future studies, specific signaling or vesicle trafficking pathways and their potential for treatment advance can be evaluated within the context of their impact on homeostatic plasticity.
The pathophysiology of neuropsychiatric disorders may be caused by a variety of genetic and environmental processes and therefore treatments may not necessarily be targeting the underlying abnormality but instead may be triggering processes that compensate the existing pathophysiology. Therefore, without direct evidence it is premature to expect that a neuropsychiatric drug actually “fixes” the underlying deficiency associated with a particular disease. This premise is consistent with the common observation that in most circumstances cessation of drug treatment leads to re-emergence of symptoms. Current neuropsychiatric drugs may instead be activating compensatory mechanisms that at least temporarily counteract or “mask” disease processes rather than cure the fundamental pathology. In this regard, multiplicative up- or downscaling of individual synaptic strengths may be a unifying feature of treatment strategies. As homeostatic plasticities rely on multiple parallel forms of signaling that converge at a synaptic end point, these treatments can bypass the inherent defects associated with a particular disorder and restore or compensate circuit function. Alternatively, they can activate dormant mechanisms that are not evoked under normal physiological circumstances that lead to treatment. The evaluation of existing and future therapies within the context of the rapidly developing literature on homeostatic synaptic plasticity may provide valuable novel insight for the treatment of neuropsychiatric disorders.
At this time, homeostatic synaptic plasticity is primarily studied as an in vitro phenomenon, as studying these forms of plasticities with the same level of mechanistic insight in vivo is subject to multiple obstacles. These include relatively longer time scales involved in these processes and the technical limits associated with monitoring a large number of synapses all at once for long periods. Nevertheless, testing the behavioral impact of manipulations targeting molecular players identified in vitro have provided some key links between cell culture and in vivo studies (e.g. Autry et al., 2011; Nosyreva et al., 2013). Moreover, while it is difficult to monitor synapse specific processes in vivo, it is easier to assess alterations in neuronal firing patterns (e.g. Hengen et al., 2016). Despite the current setbacks, the access to novel probes and approaches that can report synaptic function and molecular composition with exquisite spatial and temporal specificity will significantly improve our ability to examine homeostatic synaptic plasticities under behaviorally relevant settings.
Finally, we hope the concepts we covered in this article will facilitate the dialogue between synaptic transmission and translational neuropsychiatry fields. We believe results from clinical and translational neuropsychiatry studies may provide a much-needed validation for emerging synaptic transmission concepts, whereas basic synaptic transmission research, especially those focused on homeostatic synaptic plasticities, may provide new perspectives and targets for treatment advance.
Acknowledgements
We would like to thank current and former members of Kavalali and Monteggia laboratories for numerous invaluable discussions. This work was supported by National Institute of Mental Health grants, MH081060 and MH070727 (LMM), and MH66198 (ETK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adesnik H, Li G, During MJ, Pleasure SJ, and Nicoll RA (2008) NMDA receptors inhibit synapse unsilencing during brain development. Proc Natl Acad Sci U S A. 105, 5597–5602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antila H, Ryazantseva M, Popova D, Sipilä P, Guirado R, Kohtala S, Yalcin I, Lindholm J, Vesa L, Sato V, et al. (2017) Isoflurane produces antidepressant effects and induces TrkB signaling in rodents. Sci Rep. 7, 7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto J, Nam CI, Poon MM, Ting P and Chen L (2008) Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron 60, 308–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt KL, Zhang Y, Jurado S, Malenka RC, Südhof TC, and Chen L (2015) Retinoic Acid and LTP Recruit Postsynaptic AMPA Receptors Using Distinct SNARE-Dependent Mechanisms. Neuron 86, 442–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, and Monteggia LM (2011). NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson J and Thesleff S (1959) A study of supersensitivity in denervated mammalian skeletal muscle. J. Physiol. (Lond.) 147, 178–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassell GJ, and Warren ST (2008) Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60, 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, and Krystal JH (2000) Antidepressant effects of ketamine in depressed patients. Biological Psychiatry 47, 351–354. [DOI] [PubMed] [Google Scholar]
- Bienenstock EL, Cooper LN, Munro PW (1982) Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci. 2, 32–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman MP, Djukic B, Nelson SB, and Turrigiano GG (2012) A critical and cell-autonomous role for MeCP2 in synaptic scaling up. J Neurosci. 32, 13529–13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J, O’Byrne M, and Murthy VN (2002). Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature 420, 414–418. [DOI] [PubMed] [Google Scholar]
- Cade JFJ (1949). Lithium salts in the treatment of psychotic excitement. Med. J. Aust 2, 349–352. [DOI] [PubMed] [Google Scholar]
- Cannon WB and Rosenblueth A (1949) The Supersensitivity of Denervated Structures: A Law of Denervation (Macmillan, New York: ). [Google Scholar]
- Chahrour M, and Zoghbi HY (2007) The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437. [DOI] [PubMed] [Google Scholar]
- Chen Y, Yu J, Niu Y, Qin D, Liu H, Li G, Hu Y, Wang J, Lu Y, Kang Y, et al. (2017) Modeling Rett Syndrome Using TALEN-Edited MECP2 Mutant Cynomolgus Monkeys. Cell 169, 945–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, and Greenough WT (1997) Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A 94, 5401–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, and Wolf ME (2008) Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454, 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin F, Bouillon M, Fortin A, Morin S, Rousseau F, and Khandjian EW (1997) The fragile X mental retardation protein is associated with poly(A)+ mRNA in actively translating polyribosomes. Hum Mol Genet 6, 1465–1472. [DOI] [PubMed] [Google Scholar]
- Crawford DC, Ramirez DMO, Trauterman B, Monteggia LM, and Kavalali ET (2017). Selective molecular impairment of spontaneous neurotransmission modulates synaptic efficacy. Nature Communications 8, 14436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly EJ, Singh JB, Fedgchin M, Cooper K, Lim P, Shelton RC, Thase ME, Winokur A, Van Nueten L, Manji H, and Drevets WC (2018) Efficacy and Safety of Intranasal Esketamine Adjunctive to Oral Antidepressant Therapy in Treatment-Resistant Depression: A Randomized Clinical Trial. JAMA psychiatry 75, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, and Bezprozvanny I (2001) Maintaining the stability of neural function: a homeostatic hypothesis. Annu Rev Physiol 63, 847–869. [DOI] [PubMed] [Google Scholar]
- Davis GW, and Müller M (2015) Homeostatic control of presynaptic neurotransmitter release. Annu Rev Physiol. 77, 251–270. [DOI] [PubMed] [Google Scholar]
- de Vivo L, Bellesi M, Marshall W, Bushong EA, Ellisman MH, Tononi G, and Cirelli C (2017) Ultrastructural evidence for synaptic scaling across the wake/sleep cycle. Science 355, 507–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvendahl I, Kita K, and Müller M (2019) Rapid and sustained homeostatic control of presynaptic exocytosis at a central synapse. Proc Natl Acad Sci USA. 116, 23783–23789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diering GH, Nirujogi RS, Roth RH, Worley PF, Pandey A, and Huganir RL (2017) Homer1a drives homeostatic scaling-down of excitatory synapses during sleep. Science 355, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Creson TK, Wu L-J, Ren M, Gray NA, Falke C, Wei Y, Wang Y, Blumenthal R, Machado-Vieira R, et al. (2008). The role of hippocampal GluR1 and GluR2 receptors in manic-like behavior. J. Neurosci 28, 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Gray NA, Falke C, Yuan P, Szabo S, and Manji HK (2003). Structurally dissimilar antimanic agents modulate synaptic plasticity by regulating AMPA glutamate receptor subunit GluR1 synaptic expression. Ann. N. Y. Acad. Sci 1003, 378–380. [DOI] [PubMed] [Google Scholar]
- Eastwood SL, and Harrison PJ (2010) Markers of glutamate synaptic transmission and plasticity are increased in the anterior cingulate cortex in bipolar disorder Biological Psychiatry 67, 1010–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong MF, Newman JP, Potter SM, and Wenner P (2015). Upward synaptic scaling is dependent on neurotransmission rather than spiking. Nat Commun 6, 6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frere S, and Slutsky I (2018) Alzheimer’s Disease: From Firing Instability to Homeostasis Network Collapse Neuron 97, 32–58. [DOI] [PubMed] [Google Scholar]
- Garcia-Bereguiain MA, Gonzalez-Islas C, Lindsly C, and Wenner P (2016). Spontaneous Release Regulates Synaptic Scaling in the Embryonic Spinal Network In Vivo. J Neurosci. 36, 7268–7682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gideons ES, Kavalali ET, and Monteggia LM (2014). Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc. Natl. Acad. Sci. U. S. A 111, 8649–8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gideons ES, Lin PY, Mahgoub M, Kavalali ET, and Monteggia LM (2017). Chronic lithium treatment elicits its antimanic effects via BDNF-TrkB dependent synaptic downscaling. eLife 6, e25480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Islas C, Bülow P, Wenner P (2018). Regulation of synaptic scaling by action potential-independent miniature neurotransmission. J Neurosci Res. 96, 348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold CP, and Nicoll RA (2010). Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron 68, 512–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Sawa A, and Iyo M (2007). Increased levels of glutamate in brains from patients with mood disorders. Biological Psychiatry 62, 1310–1316. [DOI] [PubMed] [Google Scholar]
- Hashimoto R, Takei N, Shimazu K, Christ KL, Lu B and Chuang DM (2002) Lithium induces brain-derived neurotrophic factor and activates TrkB in rodent cortical neurons: an essential step for neuroprotection against glutamate excitotoxicity. Neuropharmacology 43, 1173–1179. [DOI] [PubMed] [Google Scholar]
- Hengen KB, Torrado Pacheco A, McGregor JN, Van Hooser SD, and Turrigiano GG (2016). Neuronal Firing Rate Homeostasis Is Inhibited by Sleep and Promoted by Wake. Cell 165, 180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, and Bear MF (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A 99, 7746–7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettner JE, and Bean BP (1988) Block of N-methyl-D-aspartate-activated current by the anticonvulsant MK-801: selective binding to open channels. Proc Natl Acad Sci U S A. 85, 1307–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, and Turrigiano GG (2008). Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron 57, 819–826. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, and Greenough WT (2000) Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex 10, 1038–1044. [DOI] [PubMed] [Google Scholar]
- Izumi Y, and Zorumski CF (2014). Metaplastic effects of subanesthetic ketamine on CA1 hippocampal function. Neuropharmacology 86, 273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakawich SK, Nasser HB, Strong MJ, McCartney AJ, Perez AS, Rakesh N, Carruthers CJL, and Sutton MA (2010). Local presynaptic activity gates homeostatic changes in presynaptic function driven by dendritic BDNF synthesis. Neuron 68, 1143–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JW, Glasgow NG, and Povysheva NV (2015). Recent insights into the mode of action of memantine and ketamine. Curr Opin Pharmacol. 20, 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET (2015). The mechanisms and functions of spontaneous neurotransmitter release. Nat Rev Neurosci 16, 5–16. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, and Monteggia LM (2012). Synaptic mechanisms underlying rapid antidepressant action of ketamine. The American Journal of Psychiatry 169, 1150–1156. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, and Monteggia LM (2015) How does ketamine elicit a rapid antidepressant response. Current Opinion in Pharmacology 20, 35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Tsien RW, and Alger BE (2012). An improved test for detecting multiplicative homeostatic synaptic scaling. PLoS One. 7, e37364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YI, Lømo T, Lupa MT, and Thesleff S (1984) Miniature end-plate potentials in rat skeletal muscle poisoned with botulinum toxin. J Physiol. 356, 587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, and Fischer U (2001) Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet 10, 329–338. [DOI] [PubMed] [Google Scholar]
- Lan MJ, McLoughlin GA, Griffin JL, Tsang TM, Huang JT, Yuan P, Manji H, Holmes E, and Bahn S, (2009). Metabonomic analysis identifies molecular changes associated with the pathophysiology and drug treatment of bipolar disorder Molecular Psychiatry 14, 269–279. [DOI] [PubMed] [Google Scholar]
- Larson J, Jessen RE, Kim D, Fine AK, and du Hoffmann J (2005) Age-dependent and selective impairment of long-term potentiation in the anterior piriform cortex of mice lacking the fragile X mental retardation protein. J Neurosci 25, 9460–9469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Pelletier MR, Perez Velazquez JL, and Carlen PL (2002) Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol Cell Neurosci 19, 138–151. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhang Y, Ku L, Wilkinson KD, Warren ST, and Feng Y (2001) The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res 29, 2276–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PY, Kavalali ET, and Monteggia LM (2018). Genetic Dissection of Presynaptic and Postsynaptic BDNF-TrkB signaling in synaptic efficacy of CA3-CA1 synapses. Cell Reports 24, 1550–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindskog M, Li L, Groth RD, Poburko D, Thiagarajan TC, Han X, and Tsien RW (2010). Postsynaptic GluA1 enables acute retrograde enhancement of presynaptic function to coordinate adaptation to synaptic inactivity. Proc Natl Acad Sci U S A. 107, 21806–21811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lnenicka GA, and Atwood HL (1985). Age-dependent long-term adaptation of crayfish phasic motor axon synapses to altered activity. J Neurosci 5, 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lnenicka GA, Atwood HL, and Marin L (1986). Morphological transformation of synaptic terminals of a phasic motoneuron by long-term tonic stimulation. J Neurosci 6, 2252–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, and Bear MF (2004). LTP and LTD: an embarrassment of riches. Neuron 44, 5–21. [DOI] [PubMed] [Google Scholar]
- Mertens J, Wang QW, Kim Y, Yu DX, Pham S, Yang B, Zheng Y, Diffenderfer KE, Zhang J, Soltani S, et al. , (2015) Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 527, 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moda-Sava RN, Murdock MH, Parekh PK, Fetcho RN, Huang BS, Huynh TN, Witztum J, Shaver DC, Rosenthal DL, et al. (2019). Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 364, (6436). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteggia LM, and Zarate C Jr. (2015) Antidepressant actions of ketamine: from molecular mechanisms to clinical practice. Current Opinion in Neurobiology 30, 139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteggia LM, Malenka RC, and Deisseroth K (2014) Depression: The best way forward. Nature 515, 200–201. [DOI] [PubMed] [Google Scholar]
- Murthy VN, Schikorski T, Stevens CF, and Zhu Y (2001). Inactivity produces increases in neurotransmitter release and synapse size. Neuron 32, 673–682. [DOI] [PubMed] [Google Scholar]
- Nagele P, Duma A, Kopec M, Gebara MA, Parsoei A, Walker M, Janski A, Panagopoulos VN, Cristancho P, Miller JP, Zorumski CF, and Conway CR (2015). Nitrous Oxide for Treatment-Resistant Major Depression: A Proof-of-Concept Trial. Biol Psychiatry. 78, 10–18. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, and Monteggia LM (2006). MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr. Biol 16, 710–716. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, and Monteggia LM (2002). Neurobiology of Depression. Neuron 34, 13–25. [DOI] [PubMed] [Google Scholar]
- Nosyreva E, Autry AE, Kavalali ET, and Monteggia LM (2014) Age dependence of the rapid antidepressant and synaptic effects of acute NMDA receptor blockade. Frontiers in molecular neuroscience 7, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva E, Szabla K, Autry AE, Ryazanov AG, Monteggia LM, and Kavalali ET (2013). Acute suppression of spontaneous neurotransmission drives synaptic potentiation. J Neurosci 33, 6990–7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, and Huganir RL (1998). Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron 21, 1067–1078. [DOI] [PubMed] [Google Scholar]
- Ongür D, Jensen JE, Prescot AP, Stork C, Lundy M, Cohen BM, and Renshaw PF (2008) Abnormal glutamatergic neurotransmission and neuronal-glial interactions in acute mania. Biol Psychiatry. 64, 718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peled ES, Newman ZL, and Isacoff EY (2014). Evoked and spontaneous transmission favored by distinct sets of synapses. Current Biology 24, 484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piedras-Rentería ES, Pyle JL, Diehn M, Glickfeld LL, Harata CN, Cao Y, Kavalali ET, Brown PO, and Tsien RW (2004). Presynaptic homeostasis at CNS nerve terminals compensates for lack of a key Ca2+ entry pathway. Proceedings of the National Academy of Sciences of the USA 101, 3609–3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisanu C, Melis C, and Squassina A (2016). Lithium Pharmacogenetics: Where Do We Stand? Drug Dev. Res 77, 368–373. [DOI] [PubMed] [Google Scholar]
- Poolsup N, Li Wan Po A, and de Oliveira IR (2000). Systematic overview of lithium treatment in acute mania. J. Clin. Pharm. Ther 25, 139–156. [DOI] [PubMed] [Google Scholar]
- Pratt KG, Zimmerman EC, Cook DG, and Sullivan JM (2011). Presenilin 1 regulates homeostatic synaptic scaling through Akt signaling. Nat Neurosci. 14, 1112–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price RB, Nock MK, Charney DS & Mathew SJ (2009). Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biological psychiatry 66, 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DMO, Crawford DC, Chanaday NL, Trauterman B, Monteggia LM, and Kavalali ET (2017). Loss of Doc2-dependent spontaneous neurotransmission augments glutamatergic synaptic strength. The Journal of Neuroscience 37, 6224–6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese AL, and Kavalali ET (2015). Spontaneous neurotransmission signals through store-driven Ca2+ transients to maintain synaptic homeostasis. eLife 4, 09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid B, Martinov VN, Njå A, Lømo T, and Bewick GS (2003). Activity-dependent plasticity of transmitter release from nerve terminals in rat fast and slow muscles. J Neurosci. 23, 9340–9348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimers JM, Loweth JA, and Wolf ME (2014). BDNF contributes to both rapid and homeostatic alterations in AMPA receptor surface expression in nucleus accumbens medium spiny neurons. Eur. J. Neurosci 39, 1159–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara Y, Bal M, Adachi M, Monteggia LM, and Kavalali ET (2011). Use-dependent AMPA receptor block reveals segregation of spontaneous and evoked glutamatergic neurotransmission. The Journal of Neuroscience 31, 5378–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaukowitch K, Reese AL, Kim SK, Kilaru G, Joo JY, Kavalali ET, and Kim TK (2017) An intrinsic transcriptional program underlying synaptic scaling during activity suppression. Cell Reports 18, 1512–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soden ME, and Chen L (2010) Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid. J Neurosci. 30, 16910–16921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Martinowich K, and Lee FS (2017). BDNF at the synapse: why location matters. Mol Psychiatry 22, 1370–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, and Malenka RC (2006). Synaptic scaling mediated by glial TNF-alpha. Nature 440, 1054–1059. [DOI] [PubMed] [Google Scholar]
- Styr B and Slutsky I (2018). Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer’s disease. Nat Neurosci. 21, 463–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styr B, Gonen N, Zarhin D, Ruggiero A, Atsmon R, Gazit N, Braun G, Frere S, Vertkin I, Shapira I, et al. (2019). Mitochondrial Regulation of the Hippocampal Firing Rate Set Point and Seizure Susceptibility Neuron 102, 1009–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC (2008). Neuroligins and Neurexins Link Synaptic Function to Cognitive Disease Nature 455, 903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC and Schuman EM (2006). Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell, 125, 785–99. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Taylor AM, Ito HT, Pham A, and Schuman EM (2007). Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron 55, 648–661. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Wall NR, Aakalu GN and Schuman EM (2004). Regulation of dendritic protein synthesis by miniature synaptic events. Science 304, 1979–1983. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Nosyreva E, Hunt KW, Kavalali ET, and Monteggia LM (2017). Effects of a ketamine metabolite on synaptic NMDAR function. Nature 546, E1–E3. [DOI] [PubMed] [Google Scholar]
- Tatavarty V, Torrado Pacheco A, Groves Kuhnle C, Lin H, Koundinya P, Miska NJ, Hengen KB, Wagner FF, Van Hooser SD, and Turrigiano GG (2020). Autism-Associated Shank3 Is Essential for Homeostatic Compensation in Rodent V1. Neuron 2020 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, and Tsien RW (2005). Adaptation to synaptic inactivity in hippocampal neurons. Neuron 47, 725–737. [DOI] [PubMed] [Google Scholar]
- Tsodyks MV, and Markram H (1997). The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc Natl Acad Sci USA. 94, 719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG (2017). The dialectic of Hebb and homeostasis. Philos Trans R Soc Lond B Biol Sci. 372(1715). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, and Nelson SB (2004). Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci 5, 97–107. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, and Nelson SB (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391, 892–896. [DOI] [PubMed] [Google Scholar]
- Tyssowski KM, Letai KC, Rendall SD, Tan C, Nizhnik A, Kaeser PS, and Gray JM (2019). Firing Rate Homeostasis Can Occur in the Absence of Neuronal Activity-Regulated Transcription. J Neurosci. 39, 9885–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieta E, and Valentí M (2013). Pharmacological management of bipolar depression: acute treatment, maintenance, and prophylaxis. CNS Drugs 27, 515–529. [DOI] [PubMed] [Google Scholar]
- Virmani T, Atasoy D, and Kavalali ET (2006). Synaptic vesicle recycling adapts to chronic changes in activity. The Journal of Neuroscience 26, 2197–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HL, Zhang Z, Hintze M & Chen L (2011) Decrease in calcium concentration triggers neuronal retinoic acid synthesis during homeostatic synaptic plasticity. The Journal of Neuroscience 31, 17764–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Liu W, and Yan Z (2010). Regulation of AMPA receptor trafficking and function by glycogen synthase kinase 3. J. Biol. Chem 285, 26369–26376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee AX, Hsu YT, and Chen L (2017). A metaplasticity view of the interaction between homeostatic and Hebbian plasticity. Philos Trans R Soc Lond B Biol Sci. 372(1715). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, Reis S, Oostra B, and Bagni C (2003) The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 112, 317–327. [DOI] [PubMed] [Google Scholar]
- Zarate CA Jr., Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, and Manji HK (2006). A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Archives of general psychiatry 63, 856–864. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Marro SG, Zhang Y, Arendt KL, Patzke C, Zhou B, Fair T, Yang N, Südhof TC, Wernig M, and Chen L (2018). The fragile X mutation impairs homeostatic plasticity in human neurons by blocking synaptic retinoic acid signaling. Sci Transl Med. 10(452). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Warner-Schmidt J, Duman RS, and Gage FH (2012). Electroconvulsive seizure promotes spine maturation in newborn dentate granule cells in adult rat. Dev Neurobiol. 72, 937–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X, Li H, and Chang Q (2012). MeCP2 phosphorylation is required for modulating synaptic scaling through mGluR5. J Neurosci. 32, 12841–12847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY (2003). Postnatal Neurodevelopmental Disorders: Meeting at the Synapse? Science 302, 826–830. [DOI] [PubMed] [Google Scholar]