

Abstract
This Review explores the dynamic behavior of water within nanopores and biological channels in lipid bilayer membranes. We focus on molecular simulation studies, alongside selected structural and other experimental investigations. Structures of biological nanopores and channels are reviewed, emphasizing those high-resolution crystal structures, which reveal water molecules within the transmembrane pores, which can be used to aid the interpretation of simulation studies. Different levels of molecular simulations of water within nanopores are described, with a focus on molecular dynamics (MD). In particular, models of water for MD simulations are discussed in detail to provide an evaluation of their use in simulations of water in nanopores. Simulation studies of the behavior of water in idealized models of nanopores have revealed aspects of the organization and dynamics of nanoconfined water, including wetting/dewetting in narrow hydrophobic nanopores. A survey of simulation studies in a range of nonbiological nanopores is presented, including carbon nanotubes, synthetic nanopores, model peptide nanopores, track-etched nanopores in polymer membranes, and hydroxylated and functionalized nanoporous silica. These reveal a complex relationship between pore size/geometry, the nature of the pore lining, and rates of water transport. Wider nanopores with hydrophobic linings favor water flow whereas narrower hydrophobic pores may show dewetting. Simulation studies over the past decade of the behavior of water in a range of biological nanopores are described, including porins and β-barrel protein nanopores, aquaporins and related polar solute pores, and a number of different classes of ion channels. Water is shown to play a key role in proton transport in biological channels and in hydrophobic gating of ion channels. An overall picture emerges, whereby the behavior of water in a nanopore may be predicted as a function of its hydrophobicity and radius. This informs our understanding of the functions of diverse channel structures and will aid the design of novel nanopores. Thus, our current level of understanding allows for the design of a nanopore which promotes wetting over dewetting or vice versa. However, to design a novel nanopore, which enables fast, selective, and gated flow of water de novo would remain challenging, suggesting a need for further detailed simulations alongside experimental evaluation of more complex nanopore systems.
1. Introduction
Biological channel proteins form nanoscale pores in cell membranes.1,2 They are of intrinsic physiological importance and also provide design templates for controllable nanopores in synthetic membranes. Biological channels and nanopores typically have an internal radius of ∼0.5 nm and a length of ∼5 nm. They are filled with water, thus providing permeation pathways across a lipid bilayer membrane for selected ions and/or uncharged low-molecular-weight solutes. Their functional properties (i.e., conductance, selectivity and gating) are therefore dependent on the behavior of water in a nanoconfined environment. To understand the relationship between structure and function of ion channels, and also to aid the design of novel bioinspired nanopores3,4 which may form components of “smart” membranes for e.g. water treatment and biosensing,5,6 it is therefore important to understand how such nanoconfined water behaves.
Molecular simulations play a key role in understanding the dynamic behavior of water in channels and nanopores. We will review simulation studies alongside key selected experimental investigations of the behavior of water in membrane-spanning nanopores and channels. Our bias will be toward biological or biomimetic pores, especially in lipid bilayer membranes (both synthetic and cellular). We will not cover nanopores in graphene sheets, which have recently been discussed elsewhere (e.g., see He et al.7). Our focus will be largely on the past decade during which advances in molecular simulation have been paralleled by advances in our understanding of the structures of many ion channels and biological pores.
The overall aim of this Review is to explore the relationship between the molecular structure of channels and nanopores, and the dynamic structural behavior of water within them. In particular, we wish to understand the fundamental behavior of water in membrane nanopores and channels, and how this differs from the behavior of bulk liquid water under ambient conditions. This will help us to annotate both existing and newly discovered ion channel and pore structures with respect to their biological function.8 Furthermore, it will aid and enable the design of novel nanopores such as artificial water channels.9 Our account is, therefore, complementary to recent accounts of nanopores in a range of advanced materials and biotechnological applications.10,11
2. Nanopores and Channels: Structures and Simulations
2.1. Basic Properties
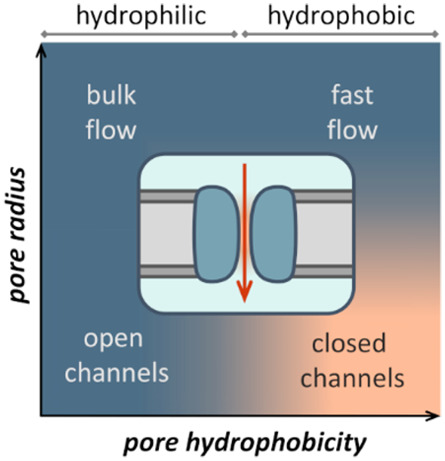
The interactions of water and ions with nanopores and ion channel proteins determine their key functional properties, namely, their permeability and selectivity (Figure 1). As we shall see, the relationship between nanopore dimensions, the chemical nature of the pore-lining surface and the pore occupancy with water is of particular importance. The dynamic behavior of water within nanopores is such that they can support high, near-diffusion-limited water permeation rates, for example, ∼10–13 cm3/s (i.e., ∼3 water molecules/ns) for a single aquaporin pore.12 Many biological nanopores exhibit selectivity for water and for other solutes or ions passing through a given pore. Water molecules may also play a key role in “gating” mechanisms of biological nanopores, that is, mechanisms for switching a pore between an open (permeable) and a closed (impermeable) state. This transition may correspond either to changes in the physical dimensions of a pore or to changes in the free energy landscape of water within a pore (e.g., in hydrophobic gating—see below).
Figure 1.
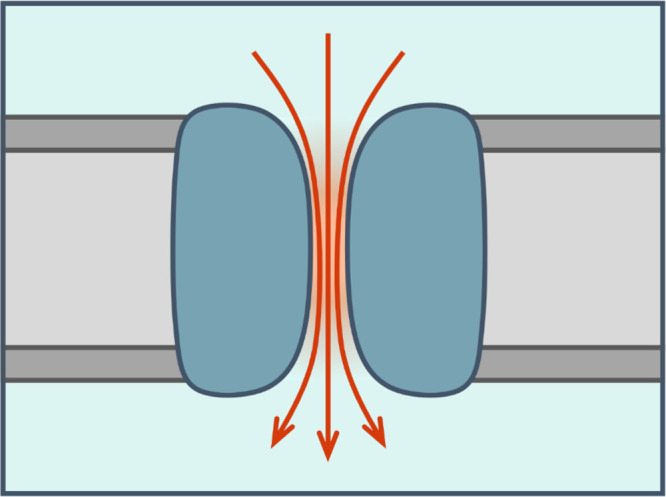
Schematic diagram of a nanopore or channel (dark blue) in a membrane (gray; lipid headgroups in dark gray). Water is shown in pale blue, with the orange region inside the nanopore indicating the region of confined water. Flow of ions and water molecules through the pore is indicated by the red arrows.
2.2. Systems
Nanopores and channels studied via molecular simulations may be classified as follows: (i) simplified models of nanoconfined water which omit most molecular details; (ii) nonbiological nanopores, including carbon nanotubes (CNTs), synthetic (biomimetic) nanopores, synthetic peptide nanopores, and track-etched nanopores in polymer membranes; (iii) porins and related nanopores formed by β-barrel membrane proteins; (iv) aquaporins and related protein pores permeable to water and small polar solutes; and (v) ion channels. This classification mirrors the structural complexity of nanopores and also broadly reflects the emphasis and development of simulation studies of behavior within them.
2.3. Structures and Experimental Approaches
The main experimental approach for determining high-resolution structures of biological nanopores and channels is X-ray crystallography. More recently, cryo-electron microscopy is having an enormous impact on the determination of the structures of channels13 and related proteins, albeit sometimes at medium resolution (∼3 Å). There are limitations of this resolution when using such structures as the basis for detailed simulation studies, including uncertainties of exact side chain conformations, and lack of experimentally defined initial positions for bound water molecules and ions. It is likely that these limitations will become less of an issue as cryo-electron microscopy of membrane proteins improves and more high-resolution structures (i.e., 2.5 Å and better) become available. For smaller membrane protein channels, NMR structures are also available.14
In a number of structures of ion channels and pores determined by high (better than 2 Å) resolution crystallography, the low energy positions (temperatures of ∼100 K) of most of the water molecules in pores are revealed. A selection of channel structures (some of which have formed the basis of simulation studies of water—see below) are shown in Figure 2 in order to illustrate the data which structural studies provide, and against which simulation studies may be compared. In a high-resolution (sub-Å) structure of the yeast aquaporin pore Aqy1 (PDB ID 3ZOJ(15)), there is a well-defined single file of water within the pore (which has a minimum radius RMIN < 0.1 nm) and details of the water–water and water–protein hydrogen bonds (albeit at ∼100 K) can be reliably defined. As discussed below, these interactions play a key role in the high water permeability and selectivity of aquaporins. The 1.5 Å resolution structure of a bacterial porin (see section 6.1 below) Omp32 (PDB ID 2FGR(16)) corresponds to a wider (RMIN = 0.22 nm) protein nanopore. This structure of an anion-selective porin shows that water molecules in the crystal structure nearly completely fill this wider transbilayer pore. Several structures of potassium channels have been determined at high resolutions and, thus, provide details of water molecules within a highly ion-selective pore. These structures include that of the canonical bacterial potassium channel KcsA at 2.0 Å resolution.17 The structure of the related bacterial potassium channel MthK at 1.45 Å resolution (PDB ID 3LDC(18)) is shown in Figure 2C. The positions of both potassium ions and of the water molecules solvating the ions within the pore are thus defined. However, one should recall that all three of the above structures were determined at low (∼100 K) temperatures, as is true for most crystal structures of membrane proteins. Recently, the use of an X-ray free-electron laser (XFEL) source has enabled the structure of the proton-selective influenza M2 channel to be determined both at an exceptionally high (1.45 Å) resolution (for a membrane protein) and at room temperature (PDB ID 5JOO(19)). This structure is notable in that a continuous hydrogen-bonded network of water molecules is seen to span the length of the channel (see below for further discussion of M2) even at room temperature. This helps to support the inference that details of water occupancy of membrane pore and channel proteins seen in low-temperature X-ray structures are relevant to the function of these proteins at room temperature. More generally, from these high-resolution structures of membrane channel proteins we can judge both the strengths and weaknesses of structural studies of water in biological nanopores and channels. High-resolution structures provide details of water–water and water–protein hydrogen bonds within pores. However, they provide at best limited information concerning the dynamics of these interactions and the implications of the dynamic behavior for function. For this, one must turn both to other experimental techniques and, particularly, to molecular simulations.
Figure 2.
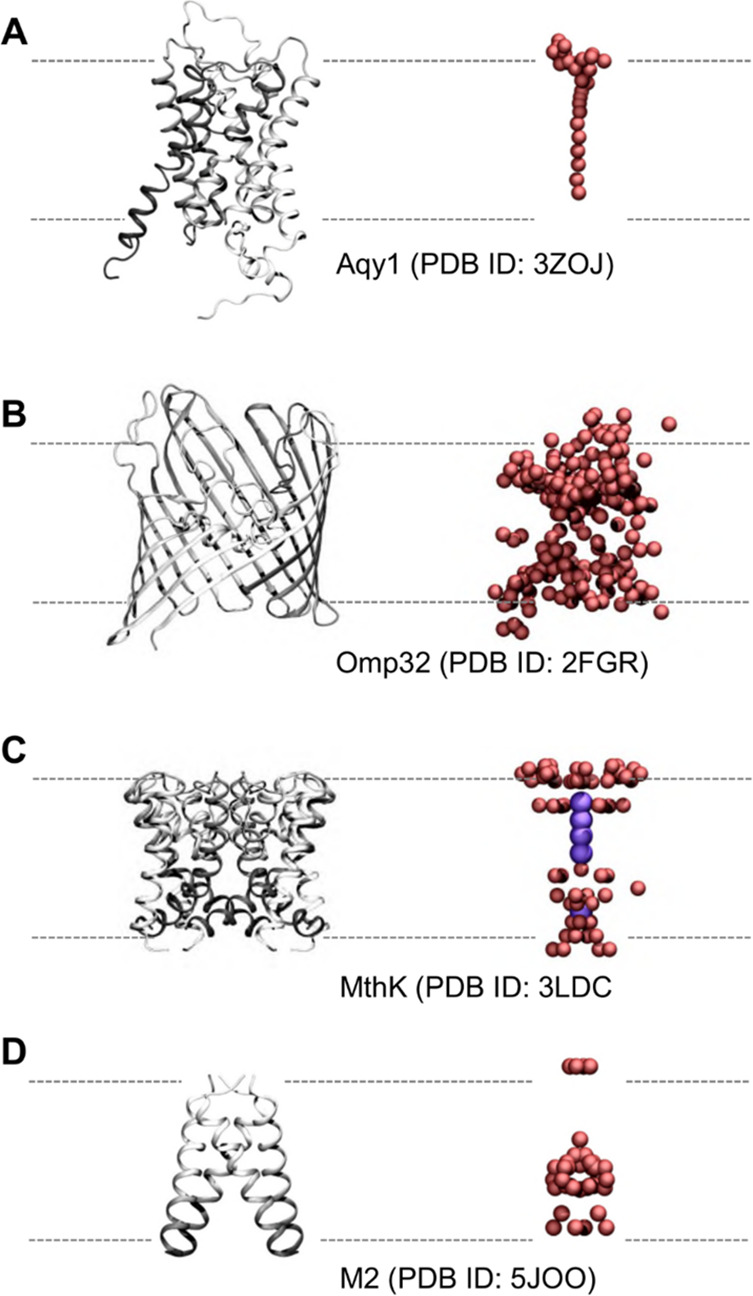
Selected pore and channel proteins showing water molecules present in crystal structures (see also Table 1). (A) Aquaporin Aqy1 (PDB ID 3ZOJ) at a resolution of 0.88 Å, (B) the porin Omp32 (PDB ID 2FGR) at resolution 1.5 Å, and (C, D) two ion channels MthK (PDB ID 3LDC) and influenza A M2 (PDB ID 5JOO), resolved at 1.45 and 1.41 Å, respectively. Each protein backbone is in ribbon representation, colored from light to dark gray in the direction of each polypeptide chain. Oxygen atoms of water molecules within the pore and around the mouth region are shown as red spheres. In the case of MthK, potassium ions in the selectivity filter and central cavity of the pore are shown in purple. All other water molecules, ions, and ligands present in the crystal structures are omitted for clarity.
A number of other experimental techniques have been used to identify bound waters within protein pores, including, for example, neutron diffraction for detecting functionally important wet spots in ion channels. This has been applied to KcsA and influenza M2 channels, yielding one-dimensional water profiles along the length of the pore while embedded in a lipid bilayer.20 Synchrotron-based radiolytic labeling combined with mass spectroscopy may be used to identify water molecules bound within cavities in pore and channel proteins21 and have been applied to, for example, a bacterial potassium channel to explore differences in water exposure of the pore of closed and open states of the channel.22 Insights into the local conformational dynamics of ion channels and their interactions with water and permeant ions can be obtained by two-dimensional infrared spectroscopy, as seen for, for example, gramicidin A and protons23 and for KcsA and potassium ions.24,25 When combined with structural studies, these investigations provide a range of experimental data sets against which to compare the outcomes of simulation studies of water within protein nanopores and channels.
2.4. Simulations
Levels of molecular simulation range from those employing classical mechanics to quantum mechanics, that is, from modeling the atoms as charges with accompanying short- and long-range interactions to modeling the nuclei as point charges with electronic interactions modeled explicitly through the inclusion of electronic wave functions. These can be used to access different length and time scales (Figure 3; after Cipcigan et al.26). Quantum mechanical simulations, while able to model explicitly a wider range of interactions, including electron transfer, tunnelling and photoexcitations, are currently computationally expensive for biological systems. Instead, MD simulations are favored as these provide a compromise between computational accuracy and efficiency.
Figure 3.

Schematic diagram of different simulation methods, along with typical time and length scales (modified from Cipcigan et al.26). Quantum mechanical methods are either time-independent or they can be coupled with molecular dynamics (for example via ab initio molecular dynamics) to access time scales of the order of fs or ps. In the context of modeling water: quantum mechanical methods are discussed in section 3.4, electronically coarse-grained methods in section 3.3.4, atomistic methods in section 3.3, atomically coarse-grained methods in section 3.2, and continuum methods in section 3.1. Reprinted with permission from ref (26). Copyright 2016 Elsevier.
Hybrid quantum mechanical/molecular mechanical (QM/MM) techniques have also been used to simulate biological systems. In this case, the region of interest is treated quantum mechanically and embedded in a surrounding classical region. For information on the mathematical formalisms, the associated practical pitfalls, and applications, the reader is directed toward the review of Brunk and Röthlisberger.27 Further reviews on quantum mechanical and hybrid quantum-classical mechanical approaches available for simulating biological molecules include those by Cui28 and Senn and Thiel.29 In terms of complete quantum mechanical treatments, a number of large-scale density functional theory (DFT) calculations have been conducted on biological systems, including enzymes and metalloproteins, as discussed in depth by Cole and Hine.30 However, there have not been many studies of channels and pores.
Water is a difficult substance to model because of the competing effects of hydrogen bonding (which tends to open up the molecular structure) and van der Waals interactions (which tend to close the structure). An accurate description of hydrogen bonding involves consideration of the electronic structure of water, including the effects of electrostatics, charge transfer and polarization.31 Nuclear quantum effects also play an important role and they are often not adequately included in traditional MD force fields.32 With the use of MD and corresponding water models, there is at best a cancellation of errors which mitigates for the lack of inclusion of quantum effects.33 A recent paper34 on a quantum water model (q-SPC/Fw) concluded that nuclear quantum effects are important, possibly as important as polarization, and should be included in subsequent polarizable water models. When comparing their quantum model to previous nonquantum models, the authors concluded that quantum reparameterization of rigid water models may be sufficient for modeling the quantum effects of water.
The multitude of interactions and methodologies for modeling these have important repercussions for accurately reproducing the water phase diagram both for the liquid phase and the multitude of ice phases (for experimental phase diagrams see, for example, those by Wagner, Feistel, and Soper,35−37 and for a comparison of simulations to experimental data see, for example, the study by Abascal and Vega38). By extension, this may also have a significant effect on the behavior of water in simulations of biological systems. For example, Anandakrishnan et al. have compared the influence of two explicit solvent water models and one implicit solvent model on protein folding of the CLN025 mini-protein.39 The free energy landscapes were found to differ substantially, even between the two explicit water models, with the largest difference in contribution to the folding free energy arising from the water–water interactions (in particular from long-range electrostatics). The choice of water model also has a notable effect on the behavior of water in various ion channels (see below).
The majority of recent studies still focus on MD simulations using well-established water models. While these have provided invaluable insights on the structure and function of ion channels and nanopores, their suitability for modeling nanoconfined water has yet to be systematically evaluated. The development of classical water models has been predominantly motivated by the accurate modeling of bulk water. However, it remains unclear how transferrable a well-performing bulk water model is to the modeling of water behavior in novel nanoscale environments. Furthermore, more advanced water models and methodologies, such as polarizable water models and quantum mechanical treatments, potentially offer more detailed and accurate information, in situations where this is required, although these methods remain limited by their significant computational cost. As we progress toward exascale computing,40,41 large-scale quantum mechanical calculations will become increasingly feasible, but as yet we fall short of being able to treat large protein/membrane systems in this way.
3. Water Models in Molecular Dynamics Simulations
Within the scope of MD simulations, the treatment of water can be broadly categorized into implicit solvent, coarse-grained, and explicit atomistic models (Figure 4). As of 2019, there are over 130 water models (a number of which are summarized in ref (42)).
Figure 4.
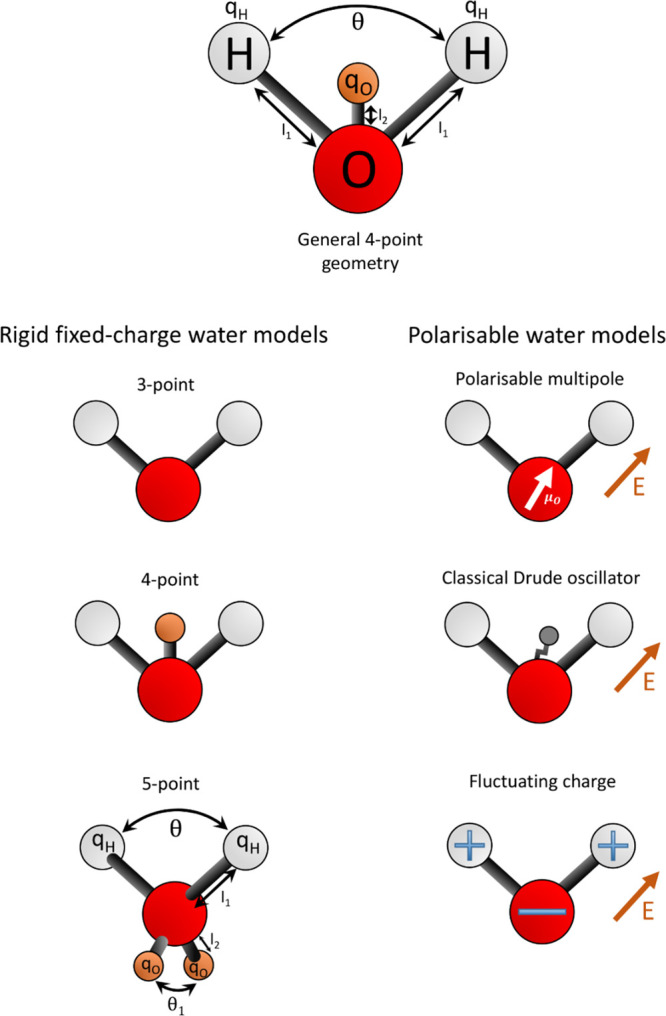
Schematic diagram of selected water model geometries. The left column shows the common geometries for rigid fixed charge models; the right is a schematic of the three key polarization schemes applied to a general 3-point geometry. It should be noted that there are various permutations to these, depending on the model. The labels for bond lengths, angles, and charges correspond to those reported in Table 2.
Some water models are developed to better describe a particular physical property, such as the dielectric constant, over a range of temperatures and pressures. Others are more transferable, aiming to provide a compromise between accuracy and applicability over a wide range of properties and conditions. In the case of biological simulations, the latter class of water model is of more use. This is because comparisons with experimental data are not straightforward and so a model designed to be accurately predictive in a wide range of environments and properties is needed. It is important to note that these water models are parametrized with respect to a given MD force field. Therefore, when conducting MD simulations with a given force field, more favorable agreement to experiment might be found with an older water model developed specifically with a given protein force field than with a more recent water model developed with a different force field. Explicit solvent tends to give a more accurate description of the system of interest but at significant computational cost compared with coarse-grained or implicit solvent models. An overview of different water models, discussing their uses and limitations, is provided by Onufriev and Izadi.43
3.1. Implicit Solvent
Implicit solvent models provide a continuum approach for treating the presence of water and any solutes as a mean field. The free energy of solvation is typically decomposed into polar and nonpolar components which model the electrostatic and hydrophobic behavior of the solvent, respectively.44 Formalisms are typically based on the Generalized Born equation45 or the Poisson–Boltzmann model.46 There are also extensions to these approaches that include missing elements of the physics, elevating implicit solvent approaches to being closer to explicit solvent methods, sometimes with relatively little increase in computational cost.43 In the case of calculating the behavior of water in ion channels, the use of implicit solvent models yields at best limited insights into the molecular structure of water inside the channel. There have been some multiscale models of ions in model nanopores focusing on comparison of explicit with implicit water models, for example in the study by Valiskó et al.47
3.2. Coarse-Grained Water Models
Within coarse-grained simulations, one or several water molecules are grouped together into a single “bead”. This increases the computational efficiency of the model but reduces its physical detail. Care must be taken when analyzing time scales, as the diffusivity of each particle is much increased compared with experimental or atomistic MD values. Furthermore, the structure and energetics of the system may not be able to be modeled to a sufficient degree of accuracy using the same coarse-grained method.48
There are several coarse-grained water models available49,50 including, for example, the model included within the widely used MARTINI coarse-grained force field,51 which contains four water molecules per bead, as well as others containing three waters per bead.52,53 Further adaptive coarse-grained water models have been developed which are able to model polarization effects by combining multiple water molecules into a network of multiple beads. These include the polarizable water model for MARTINI54 and the big multipole water (BMW) model,55 which both combine four water molecules into three beads, and the WAT FOUR water model which combines approximately 11 water molecules into a tetrahedral cluster of four beads.56 The use of polarizable water models comes at the cost of computational efficiency due to the increased number of bead–bead interactions and degrees of freedom. For example, the WAT FOUR model was shown to be ∼50% slower than a water model based on the MARTINI force field.57 Furthermore, the WAT FOUR water model cannot be used for channels with a small radius, as is the case for many biological systems.
The majority of simulations of channels using coarse-grained water models have focused on the protein or lipids. For example, the MARTINI representation of water was used to investigate the effect of varying the length and diameter of CNT porins on a surrounding POPC lipid bilayer and the tilt of the nanotube.58 Coarse-grained simulations have also been used to find a permeation pathway along which to determine the free energies of ion conductance through the NanC porin.59
3.3. Atomistic Water Models
These are by far the most widely used group of water models to investigate the effects of solvation in ion channels and nanopores. There are a wide range of types including rigid nonpolarizable models with 3–7 points, upon which charges or Lennard-Jones sites are placed to model the directional and nondirectional intermolecular interactions, respectively. In general, the Lennard-Jones site is placed on the oxygen, and the negative charge is either placed on the oxygen (for 3-point models) or is displaced away from the oxygen (for higher point models). Flexible and polarizable variants have also been developed: these are typically deemed to be more accurate but at increased computational cost. A summary of key nonpolarizable water models, often used in conjunction with ion channels and nanopores, is included in Table 2.
Table 2. Water Models.
| model | point charges | l1 (Å) | l2 (Å) | θ (deg) | qH (e) | σO (Å) | εO (kJ mol–1) | dipole moment μ (D) | dielectric constant ε | density ρ (g cm–3) | TMD (K) | surface tension σ (mN m–1) | self-diffusion coefficient D (10−9 m2 s–1) | refs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| exp (liquid) | 2.5–3 | 78.36 | 0.997 | 277 | 71.73 (at 300 K) | 2.3 | (36, 60−65) | |||||||
| exp (gas) | 0.9572 | 104.52 | 1.85 | (64, 66) | ||||||||||
| SPC | 3 | 1.0 | 109.47 | 0.41 | 3.166 | 0.65 | 2.274 | 65.6 | 0.97841 | 228 | 53.4 (at 300 K) | 4.3 | (67−69) | |
| TIP3P | 3 | 0.9572 | 104.52 | 0.417 | 3.15061 | 0.6364 | 2.348 | 94 | 0.980 | 182 | 52.3 | 5.51 | (63, 64, 68, 70, 71) | |
| TIP4P | 4 | 0.9572 | 0.150 | 104.52 | 0.520 | 3.15365 | 0.6485 | 2.18 | 50 | 0.988 | 253 | 59 | 3.22 | (38, 63, 64, 68, 70, 72) |
| TIP5P | 5 | 0.9572 | 0.70 | 104.52 | 0.241 | 3.12 | 0.6694 | 2.29 | 92 | 0.979 | 285 | 52.6 (at 300 K) | 2.77 | (63, 64, 68, 70, 71) |
| TIP4P/2005 | 4 | 0.9572 | 0.1546 | 104.52 | 0.5564 | 3.1589 | 0.7749 | 2.305 | 60 | 0.9979 | 278 | 69.3 (at 300 K) | 2.07 | (38, 63, 64) |
| TIP4P-Ew | 4 | 0.9572 | 0.1250 | 104.52 | 0.5242 | 3.16435 | 0.680946 | 2.32 | 63.9 | 0.9954 | 276 | 61.8 | 2.4 | (71, 73, 74) |
| H20-DC | 3 | 0.95800 | 109.47 | 0.45495 | 3.18400 | 0.593000 | 2.417 | 78.7 | 0.9975 | 255 | 2.17 | (66, 69) | ||
| SPC/E | 3 | 1.0 | 109.47 | 0.4238 | 3.166 | 0.65 | 2.35 | 68 | 0.994 | 247 | 63.6 (at 300 K) | 2.5 (at 300 K) | (64, 67, 69) | |
| OPC | 4 | 0.8724 | 0.1594 | 103.6 | 0.6791 | 3.16655 | 0.89036 | 2.48 | 78.4 | 0.997 | 272 | 2.3 | (71) | |
| OPC3 | 3 | 0.97888 | 109.47 | 0.447585 | 3.17427 | 0.68369 | 2.43 | 78.4 | 0.996 | 260 | (66, 71, 75) | |||
| TIP3P-FB | 3 | 1.0118 | 108.15 | 0.42422 | 3.1780 | 0.65214 | 2.419 | 81.3 | 0.995 | 261 | 64 | 2.28 | (66, 75) | |
| TIP4P-FB | 4 | 0.9572 | 0.10527 | 104.52 | 0.52587 | 3.1655 | 0.74928 | 2.429 | 77.3 | 0.996 | 277 | 70 | 2.21 | (75, 76) |
| SPC/DC | 3 | 1.00000 | 109.47 | 0.43681 | 3.15767 | 0.822882 | 2.42 | 78.3 | 0.99869 | 239 | 2.48 | (69, 76) |
This shows
parameters for common water models, along with selected
physical properties (at 298 K and 1 atm, unless otherwise stated)
of relevance to this review. The σO and εO values on the oxygen site
are defined according to the Lennard-Jones interaction energy, 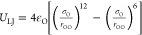 , where rOO is
the separation between two oxygen sites. qH denotes the charge on the hydrogen site. To maintain charge neutrality,
the charge on the oxygen site, qO, is
equal to −2qH except in the case
of 5-point models (TIP5P above) where qO = −qH. In the case of 4-point
models, qO is displaced away from the
oxygen site. θ denotes the angle between the hydrogen and oxygen
sites. In the case of 5-point models there is an additional angle
between the oxygen and displaced charge sites, denoted θ1 in Figure 4. For TIP5P, θ1 = 109.47°. l1 and l2 denote the oxygen–hydrogen
and oxygen−displaced-charge separations, respectively. A graphical
summary of these parameters is given in Figure 4.
, where rOO is
the separation between two oxygen sites. qH denotes the charge on the hydrogen site. To maintain charge neutrality,
the charge on the oxygen site, qO, is
equal to −2qH except in the case
of 5-point models (TIP5P above) where qO = −qH. In the case of 4-point
models, qO is displaced away from the
oxygen site. θ denotes the angle between the hydrogen and oxygen
sites. In the case of 5-point models there is an additional angle
between the oxygen and displaced charge sites, denoted θ1 in Figure 4. For TIP5P, θ1 = 109.47°. l1 and l2 denote the oxygen–hydrogen
and oxygen−displaced-charge separations, respectively. A graphical
summary of these parameters is given in Figure 4.
3.3.1. Rigid Nonpolarizable Water Models
The two most common families of water model are the “simple point charge” models (SPC)77 and “transferable intermolecular potential” models (TIP), which are based on the geometry of molecules within ice and water vapor, respectively. Within the TIP family, the 3-point model TIP3P and 4-point model TIP4P78,79 are widely used. SPC was used for the parametrization of the GROMOS force field,80 whereas TIP3P was used for AMBER81 and TIP4P for OPLS.82,83 A modified version of the TIP3P model, mTIP3P (sometimes referred to as TIPS3P), was used for the CHARMM force field and includes Lennard-Jones sites on the hydrogens.84 Because of the close relationship between water model and force field, it is often the case that the most suitable water model to use for a given force field is the one used to originally parametrize that force field.
There have been various modifications of the water models mentioned above. The SPC model was modified to include a correction to the self-energy interaction of the dipole moments. This “extended simple point charge” model, SPC/E, produced an improvement in the oxygen–oxygen radial distribution function, the density, and the diffusion constant at 300 K and 1 bar.67 The TIP4P model was reparameterized to include long-range Lennard-Jones and Coulomb interactions using Ewald summation. The resulting TIP4P-Ew model produced a significant improvement in the bulk density of liquid water and enthalpies of vaporisation across 235.5–400 K at 1 atm.73
The ability of a water model to accurately reproduce densities close to experimental values is taken as an indication of quality and transferability of the model because it is required for the accurate modeling of hydrophobic effects.85 Vega and Abascal compared the temperature of maximum density (TMD) for a range of rigid water models.68 The TMD for TIP4P-Ew was found to be 273 K.73 By comparison, the TMDs for SPC, SPC/E, TIP4P, and TIP3P were at least 24 K below the experimental value of 277.13 K.68,86 The 5-point water model TIP5P was specifically parametrized to accurately reproduce liquid densities across 236–373 K and 1–10000 atm, and was found to reproduce a TMD close to the experimental value.70 (Vega and Abascal similarly obtained a TMD value for TIP5P of 285 K.68) A further modification of TIP5P for use with Ewald summations, TIP5P-E, yields a TMD similar to TIP5P.68,87 Most recently, the TIP5P/201888 and 7-point TIP7P89 models have been developed which both favorably model the density as a function of temperature.
Several water models have been developed to better reproduce the dielectric constant under ambient conditions. These include the 3-point SPC/DC,69 SPC/ε,90 and H2O–DC69 models, as well as the 4-point TIP4P/ε.91 These were able to provide improvements on some properties but sometimes at the expense of others, providing evidence for the limitations of rigid nonpolarizable models. More recently, the dielectric constants for 19 nonpolarizable water models have been recalculated, showing that these values are particularly sensitive to simulation parameters:75 this should be taken into account in the reading of Table 2.
In addition to the development of water models which correctly reproduce a particular property (for example the dielectric constant or TMD as discussed above), Abascal and Vega have developed a 4-point rigid water model, which aims to be as predictive as possible across a wide range of properties and conditions.38 This new model, TIP4P/2005, was parametrized using the TMD, the enthalpy of vaporisation, the density of liquid water at ambient conditions, the densities of two ice polymorphs at particular temperatures and pressures, and the range of temperatures at which another ice polymorph (ice III) is thermodynamically stable at a pressure of 300 MPa.
There have been several studies testing the transferability and predictive quality of different water models. Five rigid nonpolarizable models (TIP3P, SPC/E, TIP4P, TIP4P/2005, and TIP5P) were analyzed using a scoring function which compared 17 simulated properties with experimental values.64 TIP4P/2005 scored highest with a value of 7.2 out of 10. SPC/E came second, scoring 5.1 out of 10. Additionally, the surface tension of seven rigid nonpolarizable water models has been evaluated.92 It was found that SPC/E, TIP4P/Ew, and TIP4P/2005 produced values which were in good agreement with experiments. The SPC, TIP3P, and TIP4P models underestimated the surface tension, whereas TIP4P/Ice93 overestimated it.
Izadi et al. employed a different method for developing new rigid nonpolarizable water models.71 Rather than deciding on an existing molecular geometry and fixing point charges to this framework, they instead positioned point charges to optimize the dipole, quadrupole, and octupole moments. The rationale for this approach arose from the observation that to optimally mimic the quantum mechanical charge distribution of liquid water, the three classical point charges would counterintuitively have to be placed far away from the oxygen and hydrogen nuclei. The resulting “optimal point charge” (OPC) model has a shorter OH bond length and HOH bond angle compared with the experimental values used for the TIP family of water models (see Table 2). The authors employed the scoring function of Vega et al.64 to evaluate the quality of OPC across a range of six physical properties. OPC was found to have a higher score (∼8.5) than previous water models. A 3-point version of OPC, OPC3, has also been developed using the same optimal point charge method.66 Using the same scoring system as for OPC, OPC3 obtained a score of 8.1 which was higher than other old and more recent rigid 3-point water models (TIP3P, SPC/E, H2O-DC, and TIP3P-FB). It is interesting to note that the OH bond lengths are longer than the experimental gas value; the bond angle was set to the tetrahedral value of 109.47° as a consequence of the fitting of the quadrupole moment.
Wang et al. have developed 3-point and 4-point water models based on the ForceBalance algorithm, which optimizes the model parameters using an adaptive combination of reference data and simulation results.76 The resulting models TIP3P-FB and TIP4P-FB produce improved densities, dielectric constants, self-diffusion coefficients and shear viscosities over a range of temperatures compared with TIP3P and TIP4P.76 Initial evaluations of their performance in simulations with amino acids94 and with lipid bilayers95 have been performed. The ForceBalance algorithm has been further used to develop water models based on data sets of surface tension.74 The subsequent 4-point model, TIP4P-ST, performed extremely well over a range of properties. The 3-point model, TIP3P-ST, while very accurately reproducing the TMD, leads to an overstructuring of water and a notably low self-diffusion coefficient.
Further reparameterization methods have been utilized. For example, O’Connor and English used the Design of Experiments (DoX) method96 to systematically investigate how site charge, OH bond length, bond angle, sigma, and epsilon values in SPC affected the self-diffusion coefficient and radial distribution functions (RDFs) at 298 K.97 It was found that the self-diffusion coefficient was primarily affected by the charge, epsilon, and OH bond length, and that the self-diffusion coefficient error with respect to experiment could be markedly reduced relative to the original SPC model. As pointed out by Vega and Abascal,64 any rigid nonpolarizable water model cannot properly describe the dielectric effects of liquid water and tends to underestimate dipole moments. This can be overcome by using polarizable water models.
3.3.2. Polarizable Water Models
An outline of the theory of polarization and how this can be applied to MD simulations is given by Yu and van Gunsteren.98 There are three key methods for introducing polarizability: induced dipole, classical Drude oscillator, and fluctuating charge.
-
1.
Induced dipole: Inducible dipoles are placed at specified points in the molecule. The dipole moments are often determined self-consistently within a field of isotropic atomic polarizabilities. Examples of such models include POL3 (developed to better reproduce liquid properties)99 and the BSV model.100 A set of water models have been developed for the AMOEBA force field. These use a modified Thole-type interaction101 in which the charge for one of the dipoles in each interacting pair is smeared over space instead of being treated as acting at a point. The earliest of these models is AMOEBA03.102 A more computationally efficient water model, iAMOEBA (inexpensive AMOEBA), was subsequently developed.103 This uses only one self-consistent cycle and has been parametrized with the ForceBalance algorithm. Although more computationally efficient than the original AMOEBA water model, there are possible limitations to the accuracy of iAMOEBA when modeling systems containing a high degree of heterogeneity and polarity (as discussed in the context of ion solvation104). The AMOEBA03 model has been reparameterized using ForceBalance, yielding the AMOEBA14 model, which gives excellent agreement with experiment, particularly in terms of the TMD and dielectric constant.105 A “coarse-grained” variant of the AMOEBA family, uAMOEBA, contains only polarizable sites on the oxygen centers, thereby producing a 3- to 5-fold computational acceleration with respect to the AMOEBA03 model.106 Most recently, the aniso-AMOEBA water model has been developed to incorporate the effect of anisotropic polarizability.107 A further variant on the induced dipole water model, GPCM, has been developed, which models the charges as Gaussian functions.108
-
2.
Classical Drude oscillator: A mobile point charge is attached to a fixed point charge by means of a harmonic spring. In this way, the changing geometry of the charge ensemble in response to an electric field (E-field) mimics an induced dipole. Models include the SWM4-DP109 and SWM4-NDP,110 which incorporate positively and negatively charged Drude particles respectively, and the six-site SWM6 model which has been developed from the SWM4-NDP model.111 For example, SWM4-DP has been used to explore the properties of the water–graphene interface,112 while the SWM4-NDP model has been used in development of a Drude polarizable force field for the simulation of lipid bilayer membranes113 and has been recently employed in simulations of model ion channels (see below, section 3.5).
-
3.
Fluctuating charge: The magnitudes of the charges are allowed to change in response to the E-field. Examples of this class of model include the SPC-FQ and TIP4P-FQ models which are based on the rigid geometries of SPC and TIP4P respectively.114 An extension of SPC-FQ and TIP4P-FQ models which incorporates an additional coupling between the Lennard-Jones parameters and the fluctuating charges on the oxygen sites has resulted in the SPC-pol and TIP4P-pol models.115
Some models contain a combination of the above approaches. For example, the POL5 model incorporates both induced dipoles and fluctuating charges, the effects of which are coupled together on a 5-point rigid geometry.116
Huang et al. have examined the behavior of a classical Drude oscillator model for proteins and water using the Drude-2013 polarizable force field and the SWM4-NDP water model.104 It was shown that water dipole moments were affected by the surrounding protein environment. The dipole moment was greater than its bulk value of 2.46 D when close to negatively charged amino acid side chains and was smaller when close to positively charged, polar, or hydrophobic side chains. Significantly, the dielectric constant of the hydrophobic interior of the protein was higher with the polarizable Drude model. Polarizable models (TIP4P-FQ and SWM4-NDP) have been compared with standard nonpolarizable models (TIP3P, TIP4P, and SPC/E) of hydrophobically confined water.117 The two polarizable models show a higher free energy barrier for water entering a dewetted region between two hydrophobic plates than for the nonpolarizable water models (also see discussion of hydrophobic gating below).
3.3.3. Flexible Water Models
In situations where the effects of intramolecular vibrations need to be taken into account, a set of potentials, typically harmonic functions, can be applied to the OH bond lengths and angles to produce a flexible water molecule. Such situations include the prediction of vibrational spectra.118,119 A consensus has yet to be reached regarding their effectiveness in nanopores and biological channels. For the flexible SPC/Fw model, the dielectric constant is improved if flexibility is taken into account.117 Comparison between the surface tension of seven different flexible water models suggests that the surface tension is determined by a combination of bond-stretching and Lennard-Jones interactions.120,121
Several flexible water models have been developed based on rigid model geometries. These include the SPC/Fw model based on SPC,122 SPCE-F based on SPCE,123 a flexible TIP3P,78 TIP4P/2005f based on TIP4P/2005,121 and a flexible SPC model (SPC-Fd), which includes an additional harmonic term for H–H interactions.124 Other flexible models include the F3C model125 which contains a rapid truncation of LJ and Coulomb interactions.
In the development of SPC/Fw, the influence of equilibrium bond length and angle on key properties was investigated.122 It was found that the self-diffusion constant and static dielectric constant were primarily affected by the equilibrium OH bond length and bond angle, respectively. The self-diffusion constant was found to decrease with increasing bond length. It was hypothesized that this was due to the hydrogen becoming less shielded and therefore more able to form hydrogen bonds as the OH bond length was increased. Similarly, the static dielectric constant decreased with increasing bond angle. While the latter could be hypothesized to be caused by the decrease in molecular dipole moment with increasing bond angle, an investigation into the correlation between dipole moment and dielectric constant showed that the dominant effect was directly from the changing bond angle, not the dipole moment.122 This is consistent with the findings of Höchtl et al.126
In the development of the TIP4P/2005f flexible model the fluctuations in bond angle are represented by a harmonic term, while bond length vibrations are represented by an anharmonic Morse potential.121 This model was shown to produce improved density–temperature characteristics and a melting point closer to experimental values than does the rigid TIP4P/2005 model. The dielectric constant is slightly worse compared with TIP4P/2005, in contrast to the SPC/Fw water model, which produced an improvement relative to the rigid SPC and SPC/E models.117,122 It should be noted that for flexible models the time step for MD simulations needs to be reduced by a factor of ∼5, which significantly increases the computational cost.121
3.3.4. Advanced Water Models
While flexibility can partially model the effects of molecular polarizability, it cannot model atomic polarizabilities and their response to the electrostatic environment. A class of models incorporating both flexibility and polarizability have, therefore, been developed. For example, TTM2.1-F,127 TTM3-F,128 and TTM4-F129 are Thole-type101 flexible and polarizable models. They have been modified from the earlier flexible TTM2-F130 and rigid TTM2-R131 models for use in liquid simulations. These TTM models are 4-point models with smeared charges on the hydrogen and M sites and smeared induced dipoles on the hydrogen and oxygen sites. Machine learning techniques have also recently been used to develop a flexible, polarizable, and multipolar electrostatic water model.132
To account for the quantum mechanical nature of bond flexibility and polarization, as well as for nuclear quantum effects, QM methods have been employed in the development of a further set of flexible water models. These include the q-SPC/Fw model, which was parametrized from SPC/Fw using normal-mode path integral MD and centroid MD simulations.34 A further flexible quantum model, q-TIP4P/F, has been developed using the rigid TIP4P/2005 as a basis.33 It was found that quantum effects on the dynamics of liquid water had been previously overestimated. The overall quantum effect is the sum of two mechanisms, which partially cancel each out: (i) the intermolecular zero point fluctuations and tunnelling, which weaken the hydrogen-bond network and result in an increased diffusion coefficient, and (ii) the intramolecular zero point fluctuations, which increases the molecular dipole moment, thereby strengthening intermolecular interactions and decreasing the diffusion coefficient.
More recently, advanced property analysis (for example energy decomposition analysis), automated parametrization methods, such as ForceBalance, and machine learning methods have been used to develop next-generation force fields. Many of these models, while yielding very accurate results, are computationally expensive. For example, MB-pol is regarded as one of the most accurate models, but it is ∼50 times more computationally expensive than AMOEBA, and has not yet been used for simulations beyond water133−136 and water-halide (using MB-nrg which is based on MB-pol137) systems. For further information about advanced water models, including parametrization and machine learning methods, the reader is directed to the recent review papers by Cisneros et al. and Demerdash et al.138,139
Finally, a particularly novel “Quantum Drude Oscillator” (QDO) model for water has been developed.26,140,141 The electrons are treated as a set of quantum Drude oscillators by including a harmonic oscillator operator into the Hamiltonian operator. In this way, the electrons are effectively treated as “coarse-grained”. While this model is able to model long-range effects, such as polarization, dispersion, and mixed interactions to orders higher than the dipole limit and is a potentially promising model for water simulations, it has yet to be developed for use with biological systems.
In conclusion, there are many water models available for MD simulations. The empirical pairwise-additive fixed-charge models, such as those in the SPC and TIP families, are the most commonly used due to being relatively computational inexpensive and transferable. These models have been reparametrized multiple times, yielding significant improvements in the reproducibility of experimental properties. Polarizable and flexible water models are also popular for liquid water simulations, although widespread use of these and other more advanced water models on simulations of nanopores and biological channels has yet to take place.
3.4. Quantum Mechanical Descriptions of Water
As discussed in section 2.4, it is possible to take a QM/MM approach to modeling water and biological regions of interest. For example, Shaw et al. conducted a systematic study of the interaction between a single water molecule, which was treated quantum mechanically, with a surrounding region of classical, that is, MM waters.142 Three QM theories were investigated: density functional theory (DFT) using the BLYP exchange-correlation functional, ab initio Hartree–Fock, and second-order Møller–Plesset perturbation theory. Four different MM water models were considered: TIP3P, TIPS3P, TIP4P, and TIP5P. By calculating the structural and energetic perturbations to the system, TIP4P was found to produce the smallest perturbation and was, therefore, the most compatible water model. By contrast, TIP5P was found to be the least suitable water model with the lone pairs becoming unphysically situated within in the electronic distribution of the quantum mechanical water molecule.
Rather than parametrizing classical water models to include quantum effects and/or using hybrid quantum/classical approaches, an ostensibly more accurate method is to use a full quantum mechanical approach. This does come at a significant computational cost. There are several studies on water using density functional theory (DFT) as reviewed by Gillan et al.143
Several studies report that ab initio MD (AIMD) simulations of water, in particular with the PBE144 and BLYP145,146 exchange correlation functionals, lead to an overstructuring of water as characterized by calculating the pair correlation functions between oxygen atoms.147−151 There has been speculation that this is due to incomplete convergence of the basis set, although Rempe et al. have demonstrated that this is not so when using the PBE functional.152
There has also been some discussion about flexible versus rigid water molecules in AIMD simulations. Allesch et al., using the PBE functional, found that fixing the OH bond length and HOH angle produced a structure that was not as overstructured as when using a flexible molecular representation, and that the resulting structural and dynamical properties were much closer to experiment.153 Leung et al. followed up on this by conducting a study varying the bond length and angles of the water molecule, again using the PBE functional.154 They found that increasing the bond length by as little as 2–3% had a significant effect on the structural and dynamical properties.
Several AIMD simulations have also been conducted of water in confined environments.155−157 Mann et al. demonstrated the stabilization of a water wire inside a (6, 6) single-walled CNT, and that this water wire had an affinity for accommodating an excess proton across the length of the nanotube.158 Meanwhile, Dellago et al. used both AIMD and an empirical bond valence (EVB) model to investigate how one-dimensional water wires inside CNTs enable proton conduction via a Grotthuss mechanism.159 It was found that proton mobility was enhanced by 40 times that of bulk water along perfect water wires but that it was reduced in the presence of defects.
In the context of ion channels and other biological nanopores, the channel geometry and electronic structure of gramicidin A has been investigated via large-scale DFT calculations of the entire channel in a vacuum.160 A large-scale DFT study of a complex ion channel in a lipid bilayer with surrounding water has yet to appear. The future of large-scale DFT on biological systems clearly points in this direction but current computational resources still delay their feasibility.
3.5. Evaluation of Water Models for Simulations of Channel and Nanopore Systems
There have been several investigations into the suitability of different water models for water in confined systems, including channels and nanopores. Prasad et al. compared the effect of water model on water transport through an OH-functionalized graphene nanopore.161 They found that the water flux between the models evaluated (SPC, SPC/E, SPC/Fw, TIP3P, TIP4P, and TIP4P/2005) varied by up to 84%. They hypothesized that this was due to the differences in partial charges influencing the hydrogen bond network and concluded that overall TIP4P/2005 was the most suitable water model for studying transport in such systems. Guerrero-Avilés et al. conducted DFT calculations and ab initio MD simulations to investigate the transport of water and sodium and chloride ions through “nanoslits” in graphene sheets.162 Their simulations showed that water molecules passed through the ∼0.8 nm diameter pores at a rate of ∼0.3 ps–1. This is in favorable agreement with experiments which produced a transport rate of 3 waters per picosecond through pores of ∼1 nm in diameter. Bauer et al. have compared TIP3P, TIP4P, and SPC/E against the polarizable TIP4P-FQ and SWM4-NDP models for a system of water confined between two hydrophobic plates.117 It was found that TIP4P-FQ behaved in a markedly different manner to the other water models, with nonbulk-like dynamics being observed at much larger plate separations than for the other models. In the context of hydrophobic gating, it is potentially important to accurately model the effects of induced polarization present at a vapor–liquid interface. The application of polarizable water models (using AMOEBA14) to simulations of ion channels containing a hydrophobic gate has been shown to have small but measurable effects on the wetting/dewetting behavior (ref (163) and also Lynch and Sansom ms. in preparation). Taken together, these studies argue for more widespread evaluation of different water models in nanopore and channel simulations.
4. Simplified Models of Nanopores
In the early days of simulations of nanopores, the absence of any high-resolution structures dictated the use of simplified, rather abstract models. These enabled a number of initial explorations (see, e.g., the studies by Breed et al. and Sansom et al.164−166), focusing on the diffusional and dielectric properties of water in nanoscale channel-like environments. Simulations of simplified models have continued; these have proved especially valuable for revealing general principles underlying the variation in water properties in nanoconfined environments compared with bulk water. More general studies of water at interfaces have been treated quite extensively (as reviewed by McCoustra167); simulation studies of water confined between two surfaces (a “nanoslit”; Figure 5A) or within a simple model nanopore (Figure 5B) will now be reviewed.
Figure 5.
Simple idealized models used in simulations of nanoconfined water: (A) water molecules (red) in a nanoslit (the confining surfaces are in blue) and (B) water molecules in a nanopore (shown in cross section). (C) Free energy density for water within a hydrophobic nanopore model (see below) as a function of n/nBULK, that is, the water density in the pore normalized to bulk density. The two minima correspond to the observed two-state (dry vs wet) behavior. The dry (i.e., vapor) state becomes metastable with increasing radius and for R > 0.55 nm the liquid (i.e., wet) state is globally stable. Curves are sketched from data reported in ref (168).
4.1. Physical Chemistry of Water in Nanopores
There have been a number of studies of the thermodynamics of water in nanopores and, more generally, in hydrophobic nanocavities within proteins and related environments, as reviewed in ref (169). A number of these studies have focused on the thermodynamics of hydration (i.e., of wetting vs. dewetting) of nanopores, which, in the context of gating of nanopores, is discussed more extensively below. Two key studies, of water in a hydrophobic nanopore168 and in a nonpolar nanocavity,170 used simulations of nanoconfined waters to estimate free energy landscapes of wetting/dewetting. As shown in Figure 5C, these landscapes display one or two minima: one for the empty pore (n/nBULK = 0) and one in the vicinity of the bulk density (n/nBULK = 1). As the radius of the nanopore is increased, the wetted state becomes globally stable relative to the dewetted state. Similar behavior has been seen in joint experimental and computational studies of the solvation of hydrophobic cavities in the enzyme T4 lysozyme L99A,171 for which wetting can be favored by increasing pressure, and in the tetrabrachion protein,172 for which wetting is disfavored at elevated temperatures. Comparable free energy curves are observed for water between two nanoscopic hydrophobic surfaces. For example, Remsing et al. showed that water density fluctuations played a key role in determining the pathways to dewetting for nanoconfined water.173
Experimentally, the existence of a dehydrated hydrophobic cavity within a protein (of a volume ∼0.3 nm3, that is, sufficient to accommodate up to ∼10 waters) has been demonstrated for bovine β-lactoglobulin using water 17O and 2H magnetic relaxation dispersion,174 alongside MD simulations and free energy calculations. More detailed thermodynamic analysis of the hydration of carbon nanotubes175 has suggested that wetting of hydrophobic CNTs is due mainly to increased entropy of nanoconfined water. This increase in entropy is rotational and translational for small CNTs but mainly translational for larger nanotubes.
In parallel with these studies of the thermodynamics of nanoconfined water, experimental studies based on ultrafast infrared pump–probe spectroscopy of water within reverse micelles176 have focused on hydrogen bond and orientational dynamics. In small reverse micelles (containing ∼50 water molecules), a single water ensemble is seen, which is dominated by waters interacting with the (hydrophilic) lining of the nanocavity within the reverse micelle. More generally, dynamic interfacial water density fluctuations also occur near convex-shaped nonpolar solutes.177
4.2. Nanoconfined Water
Simplified (i.e., idealized) models of nanotubes and nanopores have been investigated extensively in terms of the behavior of nanoconfined water within these systems. Recent studies have focused largely on two aspects: (i) water flow/transport as a function of the pore dimensions and the chemical properties of the pore-lining surface and (ii) the dielectric properties of water confined within model nanopores.
Simulations of TIP3P water in nanopores crossing a simple membrane model formed by a slab of immobile waters in a rigid ice-like structure178 focused on the difference between hydrophilic (ice-like water-lining) and hydrophobic (i.e., partial charges on the immobile waters removed) nanopores. The water flux under applied pressure was evaluated as a function of pore radius and geometry (cylindrical vs conical), and the results compared with the predictions of continuum fluid dynamic (CFD) theory via the Hagen–Poiseuille equation.179 For small (radius 0.6 nm) hydrophobic pores, water flow was greater than predicted by Hagen–Poiseuille, whereas lower water flux was seen for the hydrophilic pores, that is, less than predicted by the Hagen–Poiseuille equation. For the small hydrophilic pores, the structural and dynamical properties of water are strongly influenced by interactions with the pore-lining interfaces. Even in larger (radius 0.9 nm) pores the parabolic distribution of water velocities underlying the Hagen–Poiseuille equation was not observed. Comparable simulations180 of pressure-driven electrolyte flow through smooth, dipole-lined (i.e., hydrophilic) nanopores181 of radius 0.6 nm penetrating a model membrane yielded water fluxes which were comparable to those in CNT (see below) simulations. This suggests that reduced water flux in hydrophilic model pores may be related both to hydrogen bonding interactions and to the molecular-scale roughness of such pores. Other studies182 have employed a highly simplified kinetic generalization of a one-dimensional dipole model of water in a single-file nanopore to study the dielectric response of nanopore water to time-dependent E-fields in the direction of the pore axis (see below). This has helped relate studies of the microscopic properties of single-file water to, for example, data from dielectric spectroscopy experiments. Using a simple nanopore model (the particles of which were able to undergo thermal fluctuations) of radius ∼1 nm and containing a Lennard-Jones fluid183 in MD simulations under flow conditions, the velocity profile of the fluid particles was approximately parabolic as predicted by CFD theory. These simulations were combined with analytical approaches to provide a framework for exploring the relevance of fluctuation effects, including enhanced diffusion. Simulations of pressure-driven water flow through a silica nanochannel of radius ∼6 nm184 somewhat surprisingly suggested flow enhancement (relative to the Hagen–Poiseuille flow) similar to that of a CNT (see below). This suggests that the detailed chemical properties of a hydrophilic pore may be important in determining water behavior.
Effects of pore geometry on the pressure-induced flow of SPC/E water flow have been explored185 using hourglass-shaped (biconical) hydrophobic pores (minimum radius 0.15 nm) in MD simulations. The cone angle α and pore length were varied, revealing that a conical entrance reduces the hydrodynamic entrance hindrance and so leads to higher flow rates for water. Further MD simulations of water transport in these hourglass-shaped model pores186 explored the effect of the pore length on water energetics and transport, suggesting that water flux increased with pore length, reflecting the decreased importance of entrance effects as the pore length increases. Simulations of a simple hourglass shaped model (designed to mimic an AQP-like pore, see below)187 indicated that the water flow (albeit in rather short simulations) depended on the hydrophobicity of the pore walls, expressed as a contact angle, with a peak in water flux at θ ∼ 100°, that is, a hydrophobic pore. In addition to pressure-driven flow, MD simulations have been used to explore, for example, the effects of the surface roughness of the pore lining on electro-osmotic flow through nanochannels,188,189 indicating that surface topography (i.e., local pore shape) plays a significant role in electro-osmotic water flow. Simulations of electro-osmotic flow through idealized (i.e., smooth) nanopores show that both electro-osmotic and Poiseuille (i.e., pressure driven) flow agree with continuum transport descriptions in terms of, for example, comparable velocity profiles, apart from for the first layer of fluid close to the nanopore wall.190,191
In addition to water flow effects, the behavior of water in nanopores can also differ from the bulk in terms of water dielectric behavior.166 For example, studies combining MD simulations of SPC/E water with a linear response formalism to obtain a spatially resolved dielectric tensor considered water in generic hydrophobic cylindrical nanoconfinement, including cylindrical nanopores of radius ∼0.5–2.5 nm.192 Interestingly, the radial component of the dielectric constant showed oscillations near the center of the pore; this would be expected to influence the solvation of ions within such pores.
The studies discussed above have, by and large, used standard fixed point-charge water models (e.g., TIP3P or SPC/E; see section 3.3.1 above for a discussion of water models) to explore water behavior in nanopores. A number of other studies have either used simplified coarse-grained models or more complex representations of water in nanopores. Coarse-grained simulations193 using the ELBA water model194,195 have been used to explore capillary evaporation in simple nanopores (radius ∼2 nm) with either a moderately or a very hydrophilic pore-lining surface. This coarse-grained approach was extended to simple nanopores with different patterns of hydrophobic and hydrophilic walls, demonstrating that pore filling with water depends on the pattern of hydrophilicity/hydrophobicity.196 As we will see below (section 5.4), this is relevant to possible gating mechanisms for biomimetic pores.
An analytical model within a CFD framework197 has been used to explore water flow enhancement for nanopores with varying geometries and pore-lining hydrophobicity/hydrophilicity. Internal resistance to flow was reduced by increasing the contact angle (i.e., the hydrophobicity of the nanopore walls). Overall, water flow could be optimized by varying the contact angle and geometrical parameters including those for the conical pore entrance/exit. Earlier studies of idealized model nanopores198 used simplified finite-element calculations to demonstrate that, compared with a plain cylindrical nanotube, a biconical channel of optimal angle (mimicking AQPs—see section 7.1 below) provided increased hydrodynamic permeability. Therefore, the hourglass geometry, associated with small surface friction, optimizes the permeability. Comparison of these results with MD simulations of water flow in a biconical nanopore model based on “fused” CNTs (see section 5.1)199 yielded good agreement between MD simulations and continuum hydrodynamics predictions of entrance resistance. More recently, a hybrid continuum-molecular mechanics model has been used to correct the Hagen–Poiseuille model for water-surface interactions and surface wettability,200 deriving viscosity functions, which determine factors influencing flow enhancement/inhibition of confined water.
Switching to more complex water models, Zaragoza et al. have explored TIP4P/2005 water in nanoslits and nanopores (graphene and CNTs).201 As with simpler fixed point-charge models, enhanced diffusion of water is seen in hydrophobic nanotubes. These results and those discussed above agree with a CFD model202 in which the apparent viscosity of water decreases with an increasing contact angle (i.e., hydrophobicity), and tends to bulk viscosity with an increase of pore dimension. These effects have also been discussed in detail in the context of CNTs and related pores by Wu et al.203 (see section 5.1) and in a recent review.204
Moving beyond classical water models, ab initio MD simulations of water confined in model nanoslits between graphene sheets205 show that confining water in narrow slits (either ∼1 nm to give a water bilayer or ∼0.7 nm to give a water monolayer) changes both the hydrogen bonding and electronic structure of water.
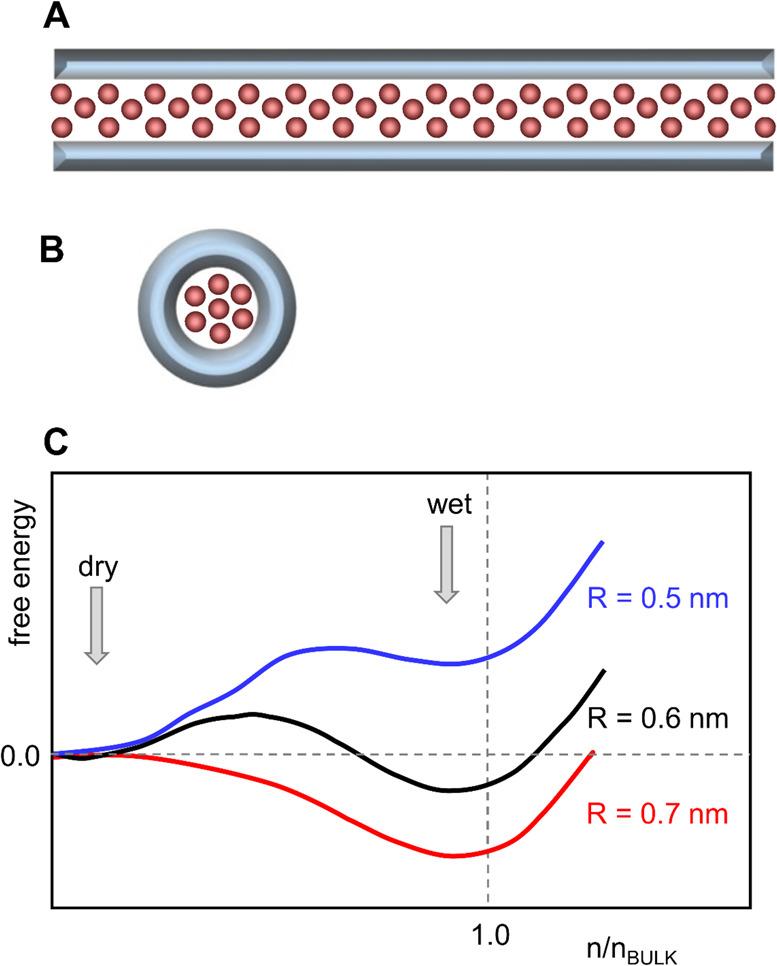
So, in summary, what have we learned from simplified models? A focus of many studies has been on water flow in nanopores. This is faster than Hagen–Poiseuille-predicted flow if the pore-lining is hydrophobic, while it is generally slower if the pore lining is hydrophilic (Figure 6). Thus, the strength with which water interacts with the pore-lining surface is crucial in determining its local behavior within nanopores. Pore-lining surface roughness and fluctuations as well as the overall pore geometry shape are also important. Therefore, to enhance nanopore design, characterization of the behavior of real “chemical” and “biological” nanopores may be required toward derivation of more general principles. We will now examine what simulations have taught us about the behavior of water in increasingly complex real pores.
Figure 6.
Water flow in a nanopore (diagrams redrawn from ref (203)). (A) Schematic diagram of water flow in nanopores. The left-hand panel shows a nanopore (in gray) with a hydrophilic (i.e., low contact angle) wall. This results in a negative effective slip length (lS < 0) corresponding to water–wall interactions, which are stronger than water–water interactions, and in turn results in a high viscosity region (orange), where water molecules are immobilized by the pore walls. The arrows show the water flow velocity profile within the nanopore. The right-hand panel shows a nanopore with a hydrophobic (i.e., high contact angle) wall. This results in a positive effective slip length (lS > 0) corresponding to water-wall interactions which are weaker than water–water interactions. Red arrows indicate regions with low viscosity. (B) Schematic graph of the water flow enhancement factor (ε, defined as the ratio of the water flow to that predicted by the Hagen–Poiseuille equation for bulk water viscosity and no-slip boundary conditions) versus pore wall water contact angle for an R = 4 nm pore. Thus, for hydrophilic pores, ε < 1 (the dotted line corresponds to ε = 1, that is, bulk flow), whereas for hydrophobic pores ε > 1. Reprinted with permission from ref (203). Copyright 2017 National Academy of Sciences.
4.3. Wetting and Dewetting: Hydrophobic Gates in Nanopores
Capillary evaporation or dewetting has been observed in a number of studies of simple model nanopores, and may be related to transient dewetting observed in CNTs (see the study by Hummer et al.206 and below). More generally, local dewetting has been discussed in terms of hydrophobicity and the folding and ligand-binding by proteins.207 Here, we focus on simple models of dewetting in nanopores, as this may be related to possible gating mechanisms for both biological and biomimetic nanopores.208−210 Crucially, simulations have been used to determine that such wetting/dewetting behavior may be modulated by the imposition of an external E-field, such as that which may be formed across a lipid bilayer membrane via an asymmetric distribution of ions.
Early studies of a simple model of a hydrophobic nanopore in a model membrane demonstrated hydrophobic gating, dependent on the radius of a pore of fixed transmembrane length. It was also shown that a closed hydrophobic nanopore could be opened by adding dipoles to its lining.211 A more extensive study of liquid–vapor oscillations of water in hydrophobic nanopores168 showed that the free-energy difference between the two states (liquid and vapor, i.e., wet vs dry) depended on the pore radius such that for hydrophobic nanopores with radii above ∼0.55 nm, a liquid-filled pore became the globally stable state (see Figure 7; also see Figure 5C and section 4.1 above). One-dimensional confinement was shown to affect the dynamic behavior of the water molecules, increasing self-diffusion compared with bulk water such that permeabilities for these simple model nanopores were of the same order of magnitude as for biological water pores. Subsequent studies focused on hydrophobic gating from an ionic perspective.212,213 Both for simple models of hydrophobic nanopores212 and for (6,6) vs (10,10) CNTs,214 there was a threshold radius of ∼0.5 nm below which ion permeation was not observed. At the same time comparable studies by Allen et al. showed that water (SPC/E) molecules penetrated a hydrophobic channel of fixed length only beyond a minimum radius and that near the threshold radius water permeation was intermittent and sensitive to, for example, the polarizability of the medium forming the channel.215 More recent studies have described how hydrophobic gating transitions from closed to open can be explained by a general thermodynamic analysis of nanoconfined water.216
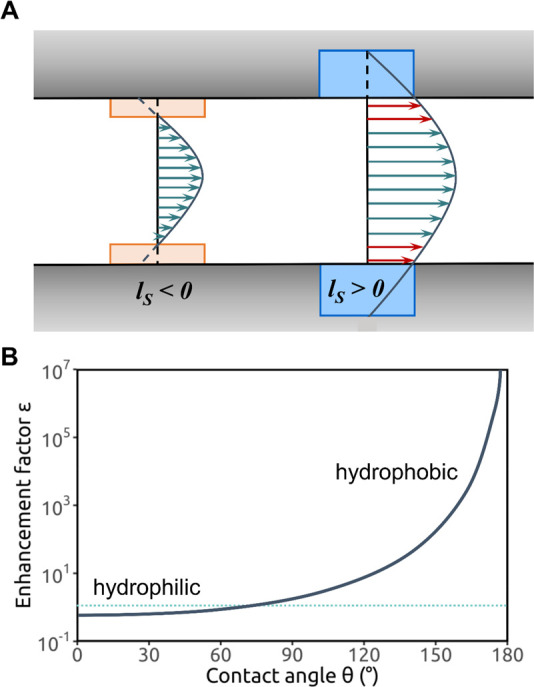
Figure 7.
Hydrophobic gating in nanopores. (A) Schematic of a hydrophobic gate within a biological ion channel. The hydrophobic gate region (dashed line) can dewet to form a closed state of the channel. Widening of the gate region of the pore leads to hydration and an open state of the channel. Water is represented by the pale blue background; ions, by red and blue spheres (from ref (217)). (B) A simple model of a nanopore (cyan), embedded in a membrane-mimetic slab (gold), with water molecules (red/white) on either side and within the pore (from ref (208)). (C) Water density (from black = 0 to red/orange = bulk) in hydrophobic nanopores with radii ranging from 1 to 0.4 nm. Modified from ref (168). Copyright 2003 National Academy of Sciences.
Building on initial observations of hydrophobic gating in a simplified model pore system,215 subsequent studies218,219 demonstrated how an E-field (generated by an ion concentration imbalance resulting in a field of ∼4 V/nm inside the pore) could wet a previously dry pore (radius ∼ 0.3 nm). The ionic charge imbalance across the membrane was shown to induce water permeation of the hydrophobic pore, thus making it permeable to ions. The movement of ions subsequently dissipated the charge imbalance and the pore once again dewetted. This is directly relevant to a number of studies of E-field-induced wetting of simple models of nanopores. Wetting of the narrow hydrophobic pore within a (6,6) CNT has been shown to be favored by an axial E-field of more than ∼0.3 V/nm. The free energy landscape of wetting/dewetting (see section 4.1 above and Figure 5C) was shown to be tilted in favor of wetting when the E-field was imposed.220
In a pioneering study, Wallqvist and Berne221 observed dewetting when two hydrophobic plates were brought closer than 0.95 nm together. Subsequently, in an impressive series of studies of dewetting/wetting of nanopores, Luzar and colleagues have used a simple model consisting of two parallel planar hydrophobic surfaces with water molecules in between. Early studies222,223 explored capillary evaporation, using a lattice gas model of liquid (water) confined between two planar surfaces. Changing the nature of the surfaces by combining hydrophilic and hydrophobic patches, it was shown that evaporation kinetics depends on this nanoscale structure, allowing estimation of the free energy barrier of vapor tube formation. A similar system was used in atomistic simulations (using both Monte Carlo and MD simulations)224 to explore capillary evaporation of (SPC) water confined in a smooth hydrocarbon-like slit. At a hydrophobic surface separation of 1.4 nm (i.e., ∼4 waters thick) the barrier to capillary evaporation was ∼19 kT. This corresponds to an evaporation rate of ∼105 nm2 s–1, indicating that water readily evaporated.
These investigations were extended to studies of “electro-wetting” as applied to a simple (nanoslit) model of a nanopore. This has proved to be directly relevant to subsequent experimental studies of (nonbiological) nanopores225 (see below for a more extended discussion) including hydrophobic nanopores in both silicon nitride226 and in polyethylene terephthalate.227 Initially, Daub et al. performed molecular simulations of nanosized water (SPC/E) droplets on a model hydrophobic (graphite) surface in the presence of an imposed E-field of 0.3 V/nm.228 The contact angle of the water nanodroplet on the hydrophobic surface (normally 96° in the absence of the E-field) was sensitive to the applied E-field direction relative to the liquid/solid interface. A parallel E-field had the largest effect, reducing the contact angle by ∼20° at the leading edge of the nanodrop. Similar studies showed a decrease in contact angle as the E-field was increased up to more than 1 V/nm.229 Subsequently, simulations were used to explore SPC/E water between parallel hydrophobic plates with a separation, d, of ∼0.9 to ∼4 nm and with applied E-fields up to 4 V/nm.230 Electrostriction was observed, i.e. an increase in water density, especially when the E-field was parallel to the planes, but no freezing of water was observed. Importantly, for smaller plate separations (d < 1.3 nm) capillary evaporation (which was observed in the absence of an E-field) was suppressed upon application of an E-field. Overall, these studies showed that an E-field induced a transition from strongly hydrophobic to strongly hydrophilic behavior in a system containing water confined in a hydrophobic nanoslit and in equilibrium with zero-field bulk water. Additional simulations231 showed that water molecules in nanoconfinement could maintain their interactions while polarized by the imposed field. Thus, if a field-exposed nanosized confined system is equilibrated with field-free bulk water, water molecules are attracted to the confined region. Simulations of two molecular hydrophobic plates formed by butyl-functionalized graphene232 with a separation between the terminal methyl groups of ∼1.1 or ∼1.9 nm, exposed to SPC/E water and ∼1 M NaCl, showed that in an E-field perpendicular to the plane (equivalent to a total voltage across the slab of the order of 0.1 V) the model nanopore wetted within <1 ns. Switching the field off caused a rapid drop in water density (i.e., the reversal of electrostriction) followed by dewetting within a few nanoseconds. Therefore, an applied E-field was shown to reversibly control the wetting and dewetting of a simple hydrophobic nanoslit. It was also shown that imposing an external E-field could switch the state between a narrow channel from salt depletion to salt excess.233 Electrofreezing of confined water has also been reported, for water confined between two hydrophilic surfaces (separation ∼0.9 nm) with an E-field of 5 V/nm applied parallel to the confinement plane.
In related studies, the effect of an E-field on TIP4P/2005 water in a 4 nm channel between two graphene sheets was examined.234 A field of up to 0.37 V/nm applied perpendicular to the sheets was imposed via placing opposing surface charges on the graphene sheets, and an external force was used to drive water flow. The water velocity profile became more parabolic as the E-field was increased and the apparent viscosity of water increased, reducing the flow rate. Therefore, the walls of the nanochannel became effectively more hydrophilic in the presence of the E-field.
The sensitivity of such effects of imposed E-fields on nanopores to the water model employed has not been widely investigated. The effect of E-fields up to 2 V/nm on “bulk” water (i.e., a box of 512 molecules) was investigated through comparison of a fixed point-charge (SPC/E) and two polarizable (SW4-NDP and BK3) water models.235 E-field effects were found to be stronger in the polarizable water models, with slower switching of hydrogen bond partners and reduced translational mobility compared to with the nonpolarizable water model. Extending these studies to alternating E-fields236 showed a nonmonotonic frequency dependence of changes in water hydrogen bonding with peak dynamics at ∼200 GHz, where the period of the E-field oscillation is comparable to the time it takes a water proton to switch between hydrogen bond acceptors. A polarizable model (BK3-AH) of water within either a smooth wall or graphene nanoslit (see above)237 was explored in response to a perpendicular external field of up to 3 V/nm. Regardless of the E-field, the mean dipoles of interfacial water molecules were lower (by ∼10%) than in the bulk phase with the polarizable model. Simulations of the nanopore-bulk phase equilibrium showed that the polarizable water model was more strongly absorbed to the nanopore in the absence of the applied E-field, while the nonpolarizable water showed greater uptake into the nanopore in response to an E-field.
The importance of such considerations of E-field effects is discussed in a recent review of wettability of nanopores.10 In particular, wetting/dewetting via changes in externally applied E-fields is an attractive prospect for direct control of gating in novel nanopores. An experimental study238 has demonstrated the influence of an applied electric potential on water affinity for the interior walls of a CNT channel 14 nm in diameter. Simulation studies of the effect of an external E-field on the wetting of a hydrophobic gate in a simplified model of a membrane protein239 (discussed in more detail below) demonstrate that these effects of E-field on wetting can also be seen in a protein nanopore environment. However, the studies discussed above on relatively simple systems show a perhaps unexpected complexity of behavior. This in turn suggests that quantitative, predictive design of E-field control of gating for more complex nanopores (whether based on naturally occurring proteins or on biomimetic materials) remains a major challenge and will need careful application to a number of simulation approaches.
5. Nonbiological Nanopores
Nonbiological nanopores, especially carbon nanotubes (CNTs), are an active area of research on the behavior of nanoconfined water in pores (Figure 8). A recent review,204 mainly focused on CNTs, compares MD and continuum approaches to water flow in such nanopores. In this section we will first discuss more recent simulation studies of water in CNTs, before examining a range of other nonbiological nanopores.
Figure 8.
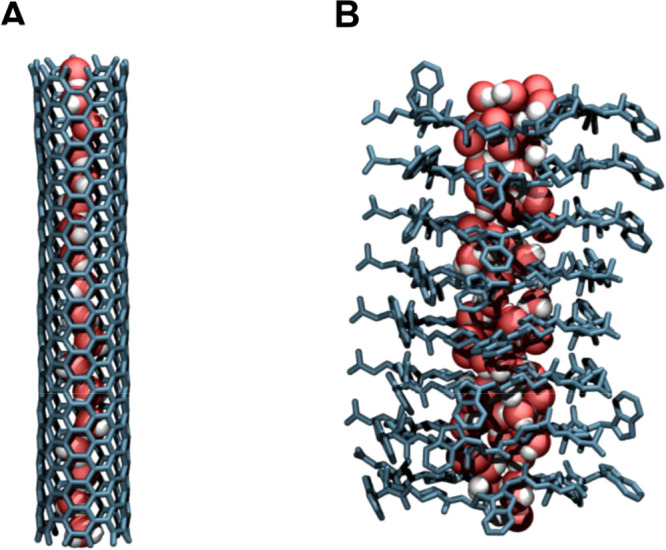
Water molecules (oxygens in red; hydrogen in white) as simulated in membrane embedded models of (A) a carbon nanotube and (B) an α,δ-cyclic peptide nanotube. Coordinates for this diagram were provided by Rebeca Garcia-Fandino and Martin Calvelo (Santiago de Compostela University; see ref (250) for simulation details).
5.1. CNTs
Following on from early suggestions of their wettability240 and from early MD simulations,206,241 there have been many computational studies of water in CNTs.242 Here we will focus on more recent studies, which have gained additional relevance following the experimental demonstration that short CNTs can insert into lipid bilayers to form “CNT porins”. These porins can transport water and ions across membranes,243 thereby providing a biomimetic platform for synthetic nanopores. Further experimental studies have built upon this landmark work:244 enhanced water permeability has been demonstrated in narrow (of radius 0.4 nm) CNTs which support single-file water,245 and more recently CNT porins have been incorporated into synthetic membranes formed by amphiphilic block copolymers. The demonstration that the electronic properties of single walled CNTs are sensitive to internal wetting, provides an additional motivation for understanding the behavior of water within CNT nanopores.246
The earliest simulation studies of water in CNTs206 showed a pulse-like motion of water through the nanotube, and indicated that small changes in channel hydrophobicity could lead to wet–dry transitions. An extensive review of this and related work169 concluded that the combination of strong water–water interactions with weak attraction of water to the confining wall resulted in rapid flow of water. Subsequent theoretical investigations247 focused on single-file water, both in CNTs248 and in nanopores in general.249 Simulations of single-file TIP3P water in (6,6) CNTs (of radius ∼0.4 nm) suggested that their filling with water was (just) favorable and was driven by a decrease in energy alongside a loss in water entropy. Recent first-principles MD studies of narrow CNTs have confirmed the one-dimensional single-file hydrogen bonding pattern seen in earlier studies.245
A number of studies have addressed enhanced water flow in CNTs, as is also seen in studies of idealized hydrophobic nanopores—see section 4.1 above. Experimentally, enhancement of water flow was estimated to be ∼100 times greater than those predicted by the Hagen–Poiseuille equation for CNTs with radius ∼0.4 to ∼0.8 nm.251 MD simulations of pressure-driven flow of TIP3P waters in CNT pores252 showed that water flow was enhanced (relative to the Hagen–Poiseuille equation) and exhibited a nonparabolic radial distribution of velocities. For pore radii from ∼0.4 to 0.6 nm (corresponding to (8,8) to (12,12) CNTs) enhancement factors of ∼10-fold were seen (i.e., somewhat lower than those suggested by experimental studies).251 A recent comparative study of pressure-induced flow through CNTs with radii between ∼0.4 and ∼0.9 nm predicted by different water models (SPC, SPC/E, TIP3P, TIP4P, and TIP4P/2005) suggested a nearly 3-fold difference between, for example, TIP3P and TIP4P/2005. Pressure-driven flow of water and ions through a (9,9) CNT has been compared for TIP3P and TIP4P/2005 water.253 TIP3P water flow rates agreed with CFD calculations. In contrast, for TIP4P/2005 water, flow was slowed by the water structure within the CNT. Furthermore, a dependence of the degree of enhancement on the thermostat used in the simulations has been suggested.254 Thus, while we should not overinterpret quantitative differences, the overall picture remains of significant water flow enhancement within CNTs, as seen by a combined analysis of published MD data and continuum theory to address water flow in CNTs.255 A decrease in the CNT radius was shown to be accompanied by a decrease in the water–CNT interfacial viscosity and an increase in the water core viscosity. Simulated water flow in CNTs with similar radii but different chirality (obtained by altering the chiral indices (n,m) of the nanotubes)256 showed that water molecules experienced the lowest resistance to flow in armchair (i.e., n = m) nanotubes. The force exerted by the CNT surface on the water molecules increased as the CNT type was changed from armchair to chiral and to zigzag. Therefore, the details of pore-lining surface structure can influence water flow even in “smooth” CNT nanopores.
Given the rapid flow of water through CNTs, it is of interest to explore how this might be regulated or “gated”. For example, it has been suggested that water flow might be switched off via lateral deformation of a (6,6) CNT.257 A deformation of ≥0.25 nm switched off water flux and resulted in local dewetting of the CNT pore. Water flow can also be slowed by mechanically “squashing” a CNT.258 Other studies have explored the effects of a point charge just outside a (6,6) CNT to increase the flow rate of water259 and how rotation of a chiral CNT260 can drive water flux through the pore. Clearly there is considerable potential for biomimetic modification of CNTs to control their ability to enable water flow.
In the context of both water and ionic flow, there have been a range of studies on the effects of an electrostatic field in water in CNTs. The overall picture is complex, with water behavior dependent on the magnitude and direction of the field and on the CNT dimensions. Broadly, lower-strength (≤1 V/nm) fields along the pore axis can enhance water permeation whereas greater (≥2 V/nm) fields can lead to ice-nanotube formation and loss of permeation. A study of a (6,6) CNT with single-file TIP3P water (but no membrane)220 showed that a field of 0.33 or 1.0 V/nm favored nanotube wetting. Simulations of various CNTs in a phospholipid bilayer (using CHARMM27 and TIP3P waters) with and without an E-field along the nanopore (z) direction of 65 mV/nm261 revealed that applying an E-field accelerated permeation approximately 2-fold in a (5,5) CNT, but slowed water self-diffusion in larger CNTs. For narrow CNTs with single-file TIP3P waters,262 water permeation was substantially reduced for high E-field intensities of 2–3 V/nm perpendicular to the pore axis. Pressure-driven flow of TIP3P water along a (6,6) CNT263 was shown to be modulated by a 1 V/nm field. By varying the angle of the field relative to the pore, it was shown that a field along z (the nanopore axis) gave a 4-fold increase in water flux, while a field along x gave approximately zero flux. The effect of E-fields on pressure-driven TIP4P/2005 water flux through a wider (15,14) CNT has also been explored.264 With an axial field of up to 1 V/nm, water flux only increases if the field is applied in the pore mouth region, and not if applied along the whole length of the pore or beyond. This suggests that an applied E-field may reduce the flow of molecules within the CNT but increase flow rates by aligning molecules close to the entry of the CNT. High fields (greater than 1.5 V/nm) were able to change the properties of water from liquid to solid. Fluctuating E-fields may also have an effect: positioning a vibrational charge outside a CNT pore can increase water flux due to changes in the hydrogen bonds.265 THz E-fields and enhanced water flow have also been explored.263
Looking beyond single CNTs, Kalra et al. employed MD simulations of a two-compartment system in which, driven by the osmotic imbalance, water flowed from a pure-water compartment into a salt solution through two membranes formed entirely by stacked CNTs.266 These enabled single-file water flow in an almost frictionless fashion.
A number of studies have suggested that imposed E-fields may induce ice formation within CNTs. For example, in a study267 of the phase transition of water inside a (4,13) (d = 1.2 nm) CNT, an ice–ice phase transition occurred discontinuously as the strength of the E-field changed. A further study of TIP4P water inside a wider (3,15) single-walled CNT (d = 1.31 nm)268 observed populations of three different polygonal ice nanotubes modulated by both temperature and E-field along the pore axis. An exploration of the effect of axial E-fields on the structure of SPC water in CNTs from (7,7) to (10,10)269 showed that E-fields of 1–2 V/nm along z induced a transition from liquid to ice nanotubes. Similarly, simulations of SPC and of TIP4P waters in a (8,8) CNT270 showed that an axial E-field of 1 V/nm caused water to form a solid-like structure at all simulation temperatures up to 350 K. This suggested that the E-field induced a phase transition from liquid to ice-nanotube. Applying a high piston pressure (∼500 MPa) can lead to water flow though the CNT even in the presence of an axial electrostatic field. When such a pressure is applied to a water/methanol mixture, along with an electrostatic field along the length of the CNT, only water is able to flow through the CNT due to the ordering of water molecules within the CNT.271 This results in an effective separation of the mixture.
Much of the motivation for studies of water in CNTs arises from a desire to design novel biomimetic nanopores based on CNT structures,272,273 especially with a view to potential applications in water desalination.9,274−276 In particular, such studies have focused on how smaller CNTs support rapid water transport while rejecting ions (as reviewed by Thomas et al.277). Simulation studies have also shown that functionalization of CNTs with charged groups (which is admittedly easier to demonstrate computationally than to achieve experimentally) can modify ion and water interactions with CNTs.272,275 This is of interest as, for example, recent studies of pressure-induced water flow through (unmodified) CNTs have suggested that increasing ionic strength (from 0 to 2 and to 4 M) decreases flow, most likely due to ions blocking water entry to the nanotube.278 Taken together, these studies suggest that modifications to CNTs may be used to optimize water flow and ion rejection, with MD simulations playing a key role in designing such modifications.
Other recent studies have focused on more disparate aspects of water in CNTs. Thus, simulations of SPC/Ew water have suggested superpermittivity of nanoconfined water279 with high (∼700) values of εZ (i.e., the dielectric constant parallel to the pore axis) near the pore surface arising from fluid structuring and preorientation of water molecules upon confinement. QM/MM simulations (with water treated via QM and the CNT by MM)280 of water in a (6,6) CNT suggested that the dissociation constant of water Kw drops by 3 orders of magnitude with respect to the bulk phase value due to under-coordination of OH– and H3O+ within the CNT. Of possible relevance to hydrophobic gating (see above), formation of nanobubbles has been studied281 in wide ((20,20) of radius ∼ 1.3 nm, and (26,26) of radius ∼ 1.8 nm) CNTs. Stabilization of the nanobubble was seen to be assisted by the adsorption of vapor-like molecules close to the hydrophobic surface and by strong hydrogen bonds at the vapor–liquid interface.
Taken together, this extensive body of research clearly demonstrates that water within CNTs behaves differently from bulk water, especially in terms of the rapid flow of water through CNTs. More recent studies have emphasized that water flow and behavior can be modulated both by local modifications to CNT structure and by external factors such as applied electrostatic fields. It is encouraging that such rich behavior of water can be observed in “simple” nanopores, both for future designs of novel nanopores and for an indication of how water behavior is likely to be “tuned to function” in biological nanopores.
5.2. Synthetic Nanopores
Water behavior has been studied in a number of synthetic biomimetic nanopores, including organic and metal–organic structures, as well as synthetic cyclic peptides and pores in polymer membranes. Experimental and computational studies into the effects of water structure and permeability through a range of synthetic nanopore-like structures have been reported, including imidazole I-quartet superstructures, metal–organic nanotubes, zeolites, and metal organic frameworks (MOFs). A useful survey of experimental studies is provided by the papers from a recent Faraday Discussion meeting.273
Peptide-appended pillar[5]arene synthetic nanopores have been synthesized and their selectivity investigated as a function of pore geometry.282,283 Most recently, peptide-appended hybrid[4]arene has been synthesized, exhibiting water transport rates of greater than 109 waters per second, similar to aquaporins (see below).284
Experimental studies on water channels formed by stacked imidazole I-quartet superstructures have indicated that water permeability increases with increasing organization in the water structure.285 In a later study,286 sum-frequency generation spectroscopy was used to characterize the hydrogen bond network within the channels, revealing the existence of chiral dipolar water wires. This was supported by MD simulations of the structures in lipid bilayers using the CHARMM36 force field for the lipids and the TIP3P water model. Further MD simulations via two different setup procedures suggest that in addition to ordered water wires through the channels, there may be an alternative disordered mechanism of water transport involving perturbations in the surrounding membrane.287
The diffusion of water into metal organic nanotubes containing UO2 (UMON) has been measured experimentally.288 It was found that the rate of diffusion was slower than most porous materials and that the water adopted a structure similar to amorphous ice. Interestingly, the UMON nanotubes demonstrated specificity to water over other common solvents; this is in contrast to many other porous materials. Single-crystal X-ray diffraction has also revealed structured waters in other novel metal organic nanotubes, for example Zn(ATIBDC)(bpy)·3H2O (MONT-1).289 More systematic simulation studies of water in metal organic nanotubes have yet to be undertaken.
There is also much interest in water permeation in (nano)porous materials such as zeolites and MOFs, which has been reviewed recently.290−293 A recent experimental and MD study on zeolites showed that surface defects (which increase the surface hydrophilicity) play a dominant role in decreasing the diffusivity of water through the crystal.294 This is in agreement with studies of simple nanopores (see above). Simulations of infrared spectra of the MOF MIL-53(Cr) using the TTMF-3 polarizable water model295 showed that the competing water-framework and water–water interactions considerably reduced water transport rates through the framework compared with in bulk water.
A series of simulation and experimental studies have focused on the behavior of water in biomimetic nanopores based on hydroxylated and functionalized nanoporous silica with pore radii down to <1 nm.296 A combined quantum (ab initio MD) and MD approach was used to characterize water in 0.6 nm radius pores created in amorphous silica: GCMC was used to determine SPC/E water content and ab initio MD used to evaluate the stability of the solvated model pore. Both hydroxylated (i.e., with SiOH groups lining the pore) and functionalized (with ∼30% of the pore-lining SiOH groups replaced by Si-CH2NH3+) pores were simulated, with water behavior and ion exclusion/permeation characterized. Subsequent studies focused on the potential of membranes formed by this class of nanoporous materials for desalination297 and on hybrid membranes combining nanoporous silica with a porous alumina support, with potential applications for desalination,297 and for CO2 separation exploiting nanocontained molecules of the enzyme carbonic anhydrase.298
5.3. Model Peptide Nanopores
A range of synthetic peptides have been shown to form model nanopores in lipid bilayers. Such peptide nanopores have been studied for a number of years299 as relatively simple models of more complex ion channel proteins. We will not discuss naturally occurring peptides, for example, gramicidin channels,300 which have been well reviewed in the past. There are a number of simulations of their stability and conformational dynamics, and of the behavior and conduction properties of the single-file water wire within gramicidin channels.301 There has also been interest in the behavior of water in channels formed by bundles of amphipathic α-helical peptides,302−304 especially in the context of proton conduction.305,306 It should be noted that these simulation studies have been limited by the difficulty of obtaining high-resolution structures. Recent structural and biophysical studies307−309 may enable such models to be revisited from a computational perspective.
There has been considerable interest in transmembrane nanotubes formed by stacks of cyclic peptides.310−316 Hsieh and Liaw have conducted a recent review of applications, including water and ion transport.317 Recent experimental and MD studies on cyclic peptides have focused on water and ion permeation to aid future biomimetic nanopore design.318,319 Ruiz et al. have investigated the effect of different pore linings of cyclic peptide nanotubes (CPNTs) on water transport.320 Using MD, it was determined that the functional group significantly affected the electrostatic potential and therefore the orientation of water molecules within the channel. The nanotube functionalized with glycine (mCPN-Gly) was found to produce the highest water flux out of the nanotubes studied. A further study employed forward osmosis to investigate water permeation through three types of CPNTs of different degrees of pore-lining hydrophobicity.321 These simulations showed that modifying the pore-lining with hydrophobic functional groups reduced the pore affinity for water due to the hydrophobic groups preventing entry of water, whereas once inside, water transport through the pore was rapid. This may be compared with simple models where increased hydrophobicity of the pore lining was correlated with higher water permeability.
5.4. Track-Etched Nanopores in Polymer Membranes
A number of studies have examined nanopores in polymer membranes and confirmed experimentally the existence of hydrophobic gating, for example, in track-etched pores in polyethylene terephthalate (PET) membranes using ionic-current recordings.227,322 It was found that pores chemically modified to have a more hydrophobic lining could be switched between ion nonconducting and conducting states by application of an E-field (Figure 9). Related studies have been conducted for PET pores in which hydrophobic gating can be achieved by modifying the surface charge density instead of through the application of hydrophobic molecules.323 Azobenzene-derivative-modified polymer nanochannels have been designed to exhibit hydrophobic gating in the presence of both E-fields and light.324 Ion selectivity has been investigated in pores etched into PET films.325 MD experiments using the TIP3P water model and CHARMM36 force field agreed well with experiment. These studies illustrate how design principles derived from computational studies (in this case of hydrophobic gating) may be experimentally tested in biomimetic nanopore systems.
Figure 9.

Hydrophobic gating observed via electro-wetting of track-etched nanopores in PET membranes. Modification of the (hydrophilic) pore surface using (trimethylsilyl)diazomethane resulted in local hydrophobic clusters inducing formation of local water vapor pockets. Applying an electric field electrowets the pore (green) with water (cyan) and thus enables ionic transport. (Modified from ref (227). Original figure courtesy of Zuzanna Siwy, University of California, Irvine). Reprinted with permission from ref (221). Copyright 2011 Springer Nature.
5.5. What Have We Learned so Far?
Two major outcomes can be drawn from reviewing the simulation and experimental studies of nonbiological pores, which parallel those from simulations of simplified systems. The first is that there is a complex relationship between pore size/geometry, the nature of the pore lining, and rates of water transport. Overall, a wider pore and a more hydrophobic lining favor water flow, provided the pore is wettable. The second is that narrow hydrophobic pores may show dewetting, as suggested by simple models of hydrophobic gating of pores and channels. Some of these concepts have been reviewed recently, drawing together simulation and experimental studies203 to provide an integrated view of wettability and nanoconfined water flow. A model is presented for confined water flow based on the concept of “effective slip”, which is expressed as a linear sum of true slip, depending on contact angle, and apparent slip, caused by a spatial variation of the confined water viscosity as a function of wettability and nanopore dimension. The flow rates for water through CNTs with diameters of 1.3 to 7.0 nm are two to five orders of magnitude greater than those calculated using the no-slip Hagen–Poiseuille equation. This indicates that the behavior of nanoconfined water is dominated by the interaction between water molecules and the lining of the nanopore. The “enhancement factor” (for flow through a nanopore relative to the ideal value from the Hagen–Poiseuille equation) thus depends on both hydrophobicity and the diameter of the nanopore. While this integrative model is formulated in the language of fluid mechanics, the underlying molecular details are embedded within the enhancement factor.
6. Porins and Related β-Barrel Protein Nanopores
6.1. Porins
Porins are a major class of membrane protein nanopores found in the outer membranes of Gram-negative bacteria and mitochondria. They form transmembrane pores based upon a β-barrel architecture (Figure 2B). They generally have high permeabilities to water, ions, and small solute molecules. Their β-barrel architecture confers structural stability, making them attractive templates for possible biotechnological manipulation and exploitation.
The structures of a large number of porins have been determined, mainly by X-ray crystallography. Several have been at high resolution (see Table 1), thus revealing details of bound water molecules within their pores. For example, the high-resolution (1.8 Å) structures of two monomeric porins, CymA (PDB ID 4V3H; see Figure 10) and NanC (PDB ID 2WJR), reveal a substantial number of water molecules bound to the walls of the respective pores. Indeed, the highest resolution porin structure, for the anion-selective porin Omp32 at 1.5 Å (PDB ID 3FGR), shows so many waters that the narrowest region of the pore is (almost) completely occluded by bound waters (albeit in the crystal structure at ∼100 K). This is evidence of favorable interactions between water molecules and the pore-lining residues of porins.
Table 1. Selected Biological Pore and Channel Proteins with Waters in Crystal Structures.
| protein | PDB ID | resolution (Å) | RMIN (Å) | comments on water |
|---|---|---|---|---|
| porins, etc. | ||||
| Omp32 anion-selective porin: Delftia acidovorans | 2FGR | 1.5 | 2.2 | waters largely resolved |
| OmpF porin: Escherichia coli | 2OMF | 2.4 | 3.5 | waters on pore walls |
| OmpC osmoporin: Escherichia coli | 2J1N | 2.0 | 2.8 | waters on pore walls |
| OmpU porin: Vibrio cholerae | 6EHB | 1.55 | 3.0 | waters largely resolved |
| NanC monomeric porin: Escherichia coli | 2WJR | 1.8 | 2.8 | waters largely resolved |
| CymA monomeric porin: Klebsiella oxytoca | 4V3H | 1.83 | 4.3 | waters largely resolved |
| α-hemolysin: Staphylococcus aureus | 7AHL | 1.9 | 5.1 | a few pore waters |
| aquaporins and other pore proteins | ||||
| yeast aquaporin: Pichia pastoris | 3ZOJ | 0.88 | <1 | single file water |
| FocA formate channel: Vibrio cholerae | 3KLY | 2.1 | <1 | water at pore mouths |
| urea transporter: Desulfovibrio vulgaris | 3K3F | 2.3 | 1.1 | some waters in pore |
| UT-B urea transporter: Bos taurus | 4EZC | 2.36 | 1.4 | some waters |
| AmtB ammonia channel (mutant): Escherichia coli | 1U7G | 1.4 | <1 | water at pore mouths |
| human Rh C ammonia channel: Homo sapiens | 3HD6 | 2.1 | <1 | some waters |
| ion channels | ||||
| GLIC pLGIC: Gloeobacter violaceus | 6HZW | 2.22 | 2.5 | several waters are present in the pore, 5 of which form a ring around a Na+ ion |
| KcsA potassium channel: Steptomyces lividans | 1R3J | 1.9 | <1 | several waters are present in the pore, 8 of which are bipyramidal around a K+ ion in the central cavity |
| MthK potassium channel: Methanothermobacter thermautotrophicus | 3LDC | 1.45 | <1 | several waters are present in the pore, 8 of which are bipyramidal around a K+ ion in the central cavity |
Figure 10.
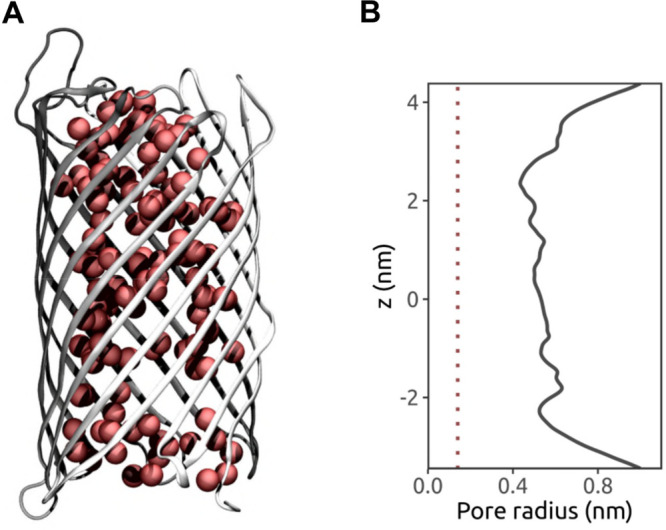
(A) Structure and (B) pore radius profile of the porin CymA (PDB ID 4V3H; see Table 1). Oxygen atoms of resolved water molecules within the pore are shown as red spheres. In the pore radius profile the broken line indicates the radius of a water molecule.
There have been a number of simulations of porins, largely focused on their ion and antibiotic permeation properties (see the recent review by Pothula et al.326). Although there has not been a systematic survey of the behavior of water within porins, a number of individual studies of water behavior within these pores provide an emerging overall picture. Early simulations of the canonical porin OmpF327 showed that water molecules within the pore diffused more slowly than in bulk, and structuring of waters in the narrow charged region of the pore was observed. Subsequent simulations of water diffusion through OmpF328 (using a modified SPC water model) used a first-passage-time approach and compared water behavior in OmpF with that in a (12,12) CNT. The overall diffusive behavior of water in OmpF was similar to that within a homogeneous CNT channel but a small fraction of the water molecules appeared to be trapped by the protein wall for considerable lengths of time. This is consistent with the structures of porins discussed above. The water diffusion coefficient (D) within the hydrophilic pore was 1.35 nm2/ns (compared with D = 4.25 nm2/ns for bulk SPC water). This is also lower than in for example the hydrophobic pores of CNTs (D = 2.7 nm2/ns). This correlates with structural observations of waters bound to the pore walls in a number of porin X-ray structures (see above and Table 1). Analysis of the effect of the transverse E-field (generated by ionizable residues in the central constriction of the hourglass shaped OmpF pore) on dielectric saturation of water within the pore329 suggested a space-dependent reduction of the dielectric constant of water within the pore down to values of ∼20. MD simulations (using the TIP3P water model) have been used to compare water behavior in the pores OmpF and OmpC,330 from which residence times suggest that there are more slow-moving waters in the central constricted region of the respective pores. Simulations have been used to explore a relatively simple cylindrical pore formed by CymA (see Figure 10) from which the N-terminus was removed.331 Electro-osmotic flow of water associated with ions moving in response to an external transmembrane voltage was observed. MD simulations have also been used to explore water behavior in Wzi,332 the pore of which is largely occluded by long loops that block the pore.
From these various simulations of porins it is evident that even though the pores are relatively wide (see Table 1), water within the pores behaves differently from bulk water, both in terms of its diffusion rate and dielectric behavior. Bound layers of water are seen both in simulations and structures, and correlate with the reduced diffusion of water within porins. These effects are due to charged side chains lining the pores and hence strong local electrostatic fields experienced within the pore by water molecules.
6.2. β-Barrel Nanopores
In addition to studies of porins, there is considerable interest in structurally related nanopores formed by β-barrels.4,333 These include the heptameric transmembrane pore formed by α-hemolysin (αHL),334 which has been widely investigated in terms of DNA detection335 and other stochastic sensing336 technologies based on measurement of ionic currents through single nanopores. There has also been parallel interest in the redesign of nanopores based on simplified β-barrel architectures,337 both in terms of experimental redesign of bacterial outer membrane proteins for stability338 and computational redesign for selectivity and other aspects of function339 as biomimetic protein nanopores.
These studies have been paralleled by computational studies of water within both αHL and in designed β-barrel nanopores. For example, Markosyan et al. used a number of different simulation granularities340 to investigate the effect of confinement within αHL nanopores on the behavior of water and single-stranded DNA. MD simulations of flexible TIP3P water were used to estimate the one-dimensional profile of the dielectric constant along the length of the αHL nanopore. The water dielectric constant dropped from ∼80 (in bulk water and in the wide extracellular mouth of the nanopore) to ∼55 in the narrower (radius ∼ 0.8 nm) transmembrane pore region. A decrease in water density (∼3-fold) was also shown within the pore in the more constricted regions. This aspect was analyzed in more detail in MD simulations of the behavior of SPC water in pores formed by α-HL molecules embedded in a phospholipid bilayer.341 These studies revealed fast water transport (66 waters/ns corresponding to D = 2.7 nm2/ns) comparable to diffusion of water in a CNT 1.2 nm in diameter (D = 2.7 nm2/ns). Analysis of water density along the αHL nanopore axis revealed two drops in density corresponding to two rings of hydrophobic amino acid side chains lining the pore. The free energy profile of water along the pore length revealed barriers of ∼3 to 4 kJ/mol corresponding to these hydrophobic rings. More recently electro-osmotic flow of TIP3P water through an αHL pore has been studied by MD simulations.342 Perhaps surprisingly, simple continuum models were shown to be able to capture the order of magnitude of the simulated electro-osmotic flow. Simulations of electro-osmotic flow have also been performed for the porin CymA.331
There have been a number of attempts to redesign simplified β-barrel nanopores (recently reviewed by Slusky343). For example, FhuA has been re-engineered344 to provide a physically robust transmembrane protein for the sampling of biomolecular events within a nanopore. Likewise, a “quiet” OmpG nanopore has been engineered which exhibits negligible spontaneous gating and which can be functionalized using cysteine chemistry.345 PoreDesigner346 is a workflow to redesign the β-barrel OmpF as a scaffold for specific pore designs. More purely computational studies of nanopore designs based on porins have also been undertaken.339,347
The behavior of water has been explored in simple models of β-barrel nanopores, varying the shape and radius of the pores, and the nature of the pore-lining side chains, and focusing on the transport of water through the resultant pores.347 It was shown that a hydrophobic barrier could be introduced into a β-barrel protein nanopore, and that the height of the energetic barrier to water could be adjusted by modifying the number of consecutive rings of hydrophobic side chains. As with simplified models (see section 4.2 above) a hydrophobic barrier prevented both water and ion permeation even though the pore was sterically unoccluded. It was subsequently shown that a hydrophobic gate in a model β-barrel nanopore could be functionally opened by electro-wetting239 (see Figure 11). Voltage-induced alignment of water dipoles was produced within the hydrophobic gate region of the nanopore, resulting in water entry followed by the permeation of ions through the opened nanopore. When the ionic imbalance that generated a transbilayer potential was dissipated, water was expelled from the hydrophobic gate (i.e., dewetting took place) and the nanopore functionally reclosed. These studies indicate that the properties observed in simple models of nanopores (see above) can be computationally “transplanted” to biomimetic designs of protein nanopore structures. Similar models of β-barrel nanopores containing hydrophobic barriers may provide template structures for DNA nanosensing.348
Figure 11.

Electrowetting of a hydrophobic gate in a model β-barrel nanopore. (A) Water entry into the nanopore at ΔV = 1.2 V. Water molecules are shown as cyan (oxygen) and white (hydrogen) spheres; the protein is shown as a molecular surface (sliced to show the interior of the pore). Ions and lipid molecules are omitted for clarity. The pore is shown dewetted at the start of the simulation, and after 0.32 ns when the pore has become fully wetted. (B) Trajectories for water molecules (pale blue) and ions (red = Na+; blue = Cl–) entering the β-barrel nanopore at ΔV = 1.2 V. The positions along the pore (z) axis of water molecules (pale blue) and of ions (red = Na+; blue = Cl–) within the nanopore are shown as a function of simulation time. The dashed horizontal lines indicate the average z position of the phosphate atoms of the lipid headgroups. The white region which persists until ∼0.3 ns corresponds to the initial dewetted region within the pore formed by three rings of leucine side chains. Modified from ref (239). Copyright 2017 American Chemical Society.
7. Aquaporins and Related Water and Polar Solute Pores
7.1. Aquaporins
Aquaporins (AQPs) are α-helical membrane proteins which form water-permeable pores in cell membranes in a wide range of organisms, including animals, plants, and bacteria. They provide a high permeability to water (water permeation through a single aquaporin pore is at rates of ∼109 water molecules/s). They are highly selective in that many aquaporins (e.g., AqpZ) are permeable only to water. Others, such as the glycerol-aquaporin GlpF are permeable both to water and to small polar (but uncharged) solutes, such as glycerol. Importantly, despite a high permeability to water, aquaporins are not permeable to protons.
Structures of many (∼50) aquaporins have been determined, both by X-ray diffraction and by cryo-electron microscopy. Several of these structures are at high resolution and reveal details of the water molecules within the central aquaporin pore. Currently, the highest resolution structure is for a yeast aquaporin Aqy1, 0.88 Å in resolution (PDB ID 3ZOJ).15 This structure reveals well-defined positions for water molecules along the pore of the Aqy1 monomer (Figure 2A), albeit at the low temperature (∼100 K) of structure determination by macromolecular X-ray crystallography. The overall structure of all aquaporins is tetrameric, with four identical pore subunits arranged around a central axis. The functional unit is an AQP monomer which is formed by a bundle of six transmembrane α-helices (plus two shorter α-helices each of which spans half of the bilayer) surrounding a central pore. The pore lining is formed by a mixture of peptide backbone atoms and side chains, and contains both polar and hydrophobic regions. Of particular importance is the presence of two asparagine (N) side chains (in a pair of NPA (asparagine-proline-alanine) sequence motifs characteristic of AQPs), which lie close to the middle of the pore and which can hydrogen bond to water molecules.
There have been numerous simulation studies of AQPs and water over the past couple of decades since the first structures were determined. We will not discuss all of these but shall rather focus on key earlier studies alongside more recent work in this area. Reviews of some of the older simulation studies are provided by Roux and Schulten349 and by de Groot and Grubmüller350 with a particular focus on possible mechanisms of proton exclusion alongside rapid water permeation in the latter review. More recent reviews have focused on, for example, comparison of AQPs with nanopores351 and in a recent Chemical Reviews article on Aqp0.352
Early simulations (based on relatively low-resolution AQP structures) focused on the organization of water within the AQP pore in relation to permeation mechanisms353 and explored pressure-induced water flow.354 As seen in these and other simulations, the single-file column of water molecules within the central pore is hydrogen-bonded such that there is a bipolar arrangement of the water dipoles, with a switch in their direction at the position of the NPA motifs (see above). The bipolar arrangement of waters appears to be maintained by hydrogen bonds of waters to the protein in the vicinity of the NPA motifs along with the overall protein electrostatic field. At the NPA motifs midway along the pore, water–protein electrostatic interactions facilitate this inversion. Simulations have been used to estimate both the osmotic and diffusion permeability of AQPs,355 and it has been shown that differences in permeability between different AQP species are not explained simply by (small) differences in pore size.356 Recent simulations to estimate osmotic permeabilities of aquaporins AQP4, AQP5, and GlpF yielded values in good agreement with experimental estimates.357 Simulations comparing AqpZ (a highly water-selective AQP) and GlpF (which also allows glycerol to pass through the pore) suggest there is a trade-off between water selectivity and permeability.358 In analyzing simulations, an osmotic permeability matrix approach has been used to relate permeability to the local structure of the pore lining in different AQP species.359 Simulations of permeability of H2O versus D2O and comparison with experiments suggested that, in both cases, water permeability of Aqp1 was reduced by 15–20% in D2O, most likely due to the lower self-diffusion coefficient of D2O.360
Simulations have been used to explore the selectivity of AQPs for water. For example, estimation of free energy profiles has been used to explore the selectivity for water over other small polar solutes of AQP1,361 revealing a negative correlation between permeability and solute hydrophobicity. Of particular importance and interest in terms of the behavior of water within the AQP pore is the mechanism of proton exclusion. Their behavior differs from other single-file water pores (e.g., that within gramicidin A, see above) which conduct protons efficiently362 via the Grotthuss mechanism.363 There have been a number of simulation studies of the mechanism of blocking proton permeation by AQPs, resulting in some debate over the relative contribution of different factors (including the water dipolar reversal mentioned above and other electrostatic effects).364−369 Most of this analysis has focused on MD simulations with “standard” water models (generally TIP3P). Ab initio Car–Parrinello MD simulations370 suggested that polarization of water molecules varied along the pore axis, and the water wire was broken by the NPA motifs. A more general study of long-range proton transfer in biomolecular pumps by QM/MM371 revealed that GlpF produced water structure in its interior in agreement with previous MD studies.
Initially it was thought that AQPs were permanently open to water permeation. However, several simulation studies have emphasized the role of the conformational dynamics of certain pore-lining side chains in controlling rates of water permeation.372,373 Furthermore, higher-resolution experimental structures have revealed alternative local conformations of the NPA motifs374 in addition to suggesting possible gating at the N-terminal mouth of, for example, the yeast Aqy1.375 For AqpZ, the possible role of an arginine (R189) side chain conformation in relation to water permeability and gating has been discussed.376 This may in turn be linked to the protonation state of a histidine side chain.377,378 Side chain motions and gating have also been explored by simulations of Aqp5379 and Aqp4.380 What emerges from these studies is a picture of a pore far from static, with local conformational transitions able to modify rates of water permeation.
AQPs have been the focus of a number of simulation studies of how imposed E-fields may modulate the behavior of water within their pores. For example, relatively large voltages (from a physiological perspective, for example, ± 1.5 V) have been shown to affect water permeability via a switch in the position of an arginine side chain.381 This indirect effect of water behavior is different from that on water in CNTs where imposing an E-field can align all the water molecules and increase water flow.382 Thus, effects of fields on both water dipole alignments (as in CNTs—also see above261) and on side chain flipping may have effects on AQPs.383 Pulsed E-fields have been shown to influence the histidine side chain (see above) conformation.384 Interestingly, THz fields have been shown to influence water permeability in CNTs via the breaking of hydrogen bonds,263 and E-fields have been shown to have effects on water dipole alignment in cyclic peptide nanotube pores (see above).385 Brief E-field pulses both perpendicular to386 and axial to387 the AQP pore have been shown to have effects on water diffusivity and on pore-lining side chain conformations.388 It has been suggested that circularly polarized E-fields could yield enhanced water permeation, thus resulting in the “electro-pumping” of water through AQP pores.389 Overall, these studies indicate how the behavior of water confined within a conformationally dynamic nanopore may be modulated both directly and indirectly by externally applied E-fields.
Simulation studies of AQPs have provided considerable insights into the behavior of water confined within a complex and dynamic protein pore. A narrow (radius ∼0.1 nm) pore with a varied lining (containing both hydrophobic and hydrophilic regions) accommodates a single file of water molecules (see Figure 12), which are aligned with a midway dipole switch. This complex pore structure helps to enable proton exclusion. Side chain dynamics may modulate water permeation through the pore, and may in turn be altered by externally applied E-fields. In terms of possible applications, there has been some discussion of the possible uses of AQP-containing membranes for water purification and desalination.390,391 In this context, a recent in silico feasibility study392 has suggested truncation of the AQP structure as a possible route to a “streamlined functional analogue” of the native protein.
Figure 12.
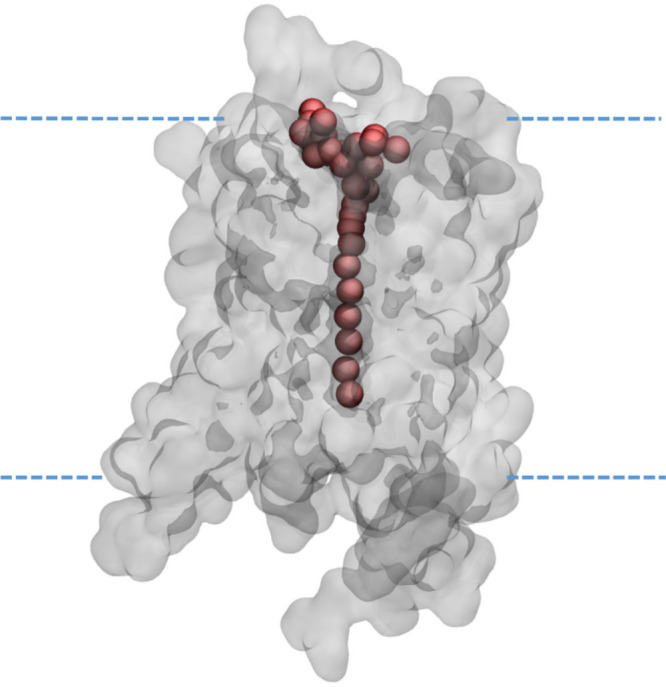
Water molecules (red) resolved within the crystal structure of yeast aquaporin Aqy1 (PDB ID 3ZOJ; see Table 1), with the protein in a semitransparent surface representation, and the broken horizontal lines indicating the approximate location of a bilayer membrane relative to the protein.
7.2. Other Protein Pores for Polar Solutes
There are a number of pores which enable the transport of small solutes (e.g., formate, urea, and ammonia) across cell membranes (Table 1). These are generally oligomeric, with the monomeric pore as the functional unit. They include the pentameric formate-nitrite transporter (FNT) family, the trimeric urea transporter (UT) family, hexameric pores for urea and acetate (UreI and SatP), and the Amt/Mep/Rh family of trimeric ammonia transporters. The FNTs have an AQP-like fold.393 The others have more complex folds, but all have pore-like structures surrounded by α-helices (e.g., for the UT-B fold394). A number of the higher-resolution crystal structures reveal water molecules within the pores (Table 1).
Although simulation studies of these pores have focused on their solute transport mechanisms, a number of these studies have also examined the properties of pore water in passing. Early simulations of FocA focused on the protonation state of a key histidine residue in the center of the pore.395 Subsequent, more detailed, studies of FNT pores combined MD, QM, and pKa calculations to explore the energetics of the complete permeation cycle, showing that anion binding into the pore is coupled to protonation of the central histidine.396 Significantly, water was efficiently translocated only when the central histidine side chain was in a protonated state, indicating that water behavior can be modulated by the charge state of residues within a nanopore.
There have been a number of simulation studies of urea-permeable pores of the UT-B family, mainly focusing on the energetics of urea permeation (e.g., the studies by Levin et al., Azouzi et al. and Ariz-Extreme et al.394,397,398). A combined experimental and computational study of UT-B from red blood cells demonstrated that the per-monomer water permeability of UT-B is similar to that of AQP1.397 Simulations of water permeability were in good agreement with experiments, and free energy calculations showed a central barrier to water permeation of ∼8 kJ/mol. A study of a bacterial UT showed that the urea selectivity filter in the center of the pore is largely dehydrated due to a hydrophobic constriction.399 A comparison of UT-B and Aqp1 revealed that despite their different folds, the pore constriction radius in both pores was <0.2 nm, presenting a central energetic barrier to water in UT-B of ∼10 kJ/mol.398
A detailed simulation study of the hexameric Ure1 pore revealed a low barrier to water (∼4 kJ/mol) and a water transport rate (∼4 × 109 s–1) comparable to that of AQPs.400 Simulations of the structurally related SatP acetate channel showed that water hydrates all parts of the pore, including the central constriction site which forms an energetic barrier for acetate.401
A number of studies have explored water behavior in the ammonia channels of the Amt and Rh proteins. Early MD studies showed that the pore of AmtB can accommodate water molecules.402 Subsequent studies revealed different hydration patterns in Amt and Rh proteins in relation to their transport mechanisms.403 Waters were seen to be stable in the AmtB pore but significantly less so in the Rh pore. This was suggested to be due to differences in phenylalanine side chain conformations. Interestingly, free energy studies of water and solutes in Rh50 channels showed a barrier for water of ∼25 kJ/mol, suggesting that Rh supports NH3 but not water transport.404
These simulation studies extend our understanding of water in narrow protein pores beyond just AQPs. In many cases, water permeability in simulations of these “other pores” is comparable to that seen for AQPs. Similarly, the local conformation (for example of phenylalanine in Amt/RH) or charge state (of histidine in UTs) of amino acid side chains within a narrow pore can control water permeability and hydration of the pore. As discussed in a recent simulation study of the SecY pore,405 water within a narrow protein nanopore can exhibit a slowing down in structural relaxation dynamics, which may be compared to the effects of lowering the temperature of bulk water.
8. Ion Channels
Ion channel proteins function as water-filled pores that control the passage of selected ions across biological membranes, commonly involving conformational transitions in response to intra- and/or intercellular signals. Ion channel pores are typically subnanometer-sized and lined by rings of different amino acid residues whose side chains may be oriented toward or away from the central pore, thus conferring nonuniformity both in pore dimensions and hydrophobicity. Such molecular features of the permeation pathway differentially modulate water behavior in distinct regions of the channel, which in some instances may, in turn, constitute part of its physiological function.
8.1. Water in Relation to Potassium Channel Permeation
In a large family of cation-selective channels, the tetrameric pore-loop channels (which include potassium channels as well as voltage-gated sodium and calcium channels), ion selectivity is controlled by a filter region lined by a short polypeptide segment from each of the four subunits on one side of the membrane. There have been extensive simulation studies of ion permeation and selectivity mechanisms of these channels.24,349,406−409 The exact mechanisms of selectivity and permeation remain unresolved and are areas of active research by a number of groups (see, e.g., ref (410) for an important collection of reviews and ref (411) for a more recent survey of advances and challenges). Here, we will focus more specifically on the behavior of water which remains central to our understanding of permeation mechanisms.
MD simulations and electrostatic calculations of TIP3P water inside the canonical potassium channel KcsA showed that the presence of water in the selectivity filter enhanced the depth of potential energy wells occupied by a potassium ion, suggesting a structural role for water in the region.412 Water molecules (TIP3P and SPC) have also been observed to travel through alternative pathways “behind” the KcsA selectivity filter in the absence of ions, giving rise to a high osmotic permeability of the channel when in a potassium-depleted state.413 This is consistent with experimental studies of water transport through the KcsA channel.414,415 Coupling of ion and water flux has also been observed experimentally in other potassium channels.416,417
More recently, there has been some debate about the interplay of water and ions in conduction through potassium channels. The “canonical” model envisages the narrow (single-file) selectivity filter being occupied by alternating potassium ions and water molecules.406 However, recent simulations using computational electrophysiology418 (i.e., in the presence of an E-field due to an ionic charge imbalance across the membrane) suggest a direct knock-on mechanism in which K+ ions move through the selectivity filter in the absence of intervening water molecules.407 Data from two-dimensional infrared spectroscopy experiments have been interpreted as implicating water in the filter,24,25 although this has been contested,419 and solid-state NMR experiments suggest the conduction pathway in the filter may be water-free.420 Although details remain to be resolved (see ref (411) for a measured review), it is clear that there is a complex role of conformational flexibility within the selectivity filter and elsewhere in the KcsA channel,25 which determines the relative occupancies of the filter by ions or water in its different conformational states. This is consistent with the more general emerging view of biological nanopores (see above) whereby local changes in conformation and/or electrostatics of narrow protein pores can modulate their water permeability.
8.2. Water and Proton Transport in Ion Channels
Water behavior is of particular interest in those ion channels and related membrane proteins which form proton-permeable pores. This has been studied in the past421,422 in simple model peptide nanopores such as gramicidin A (see section 5.2 above), which contains a single file of water and allows rapid proton flux via a Grotthuss mechanism. This remains a valuable test system for the direct simulation of proton conduction, for example, via multistate EVB simulations423 or by combining classical MD simulations with a stochastic model for proton hopping.424 EVB simulations have also been used to explore proton transport through water-filled CNTs,159 and yield similar results to ab initio MD simulations. Proton-conducting water wires are rather more complex in a number of proton-permeable ion channels for which structural data have recently become available, including the voltage-gated proton channel Hv1 and the low-pH-activated proton channel M2 from influenza A.
Early studies of Hv1 function, combined with homology model-based simulations, suggested that a water wire existed within the channel and played a role in proton permeation.425,426 The determination of the crystal structure427 and subsequent MD simulations revealed fluctuations in the water density within the pore.428 These results suggested that formation of a continuous water wire along the entire length of the (dynamic) channel was unlikely to enable a simple Grotthuss-like proton conduction mechanism. Subsequent studies have focused on the interplay of water molecules and charged side chains in the proton conduction mechanism.429,430
The low-pH-activated M2 proton channel from influenza A virus has been the subject of intensive structural and computational studies of the relationship between channel structure, the nature of water within the channel, and the likely mechanism(s) of proton permeation. Initial simulations of water within M2 were based on plausible models of the tetrameric bundle of M2 helices spanning a membrane431,432 and included simulations of proton permeation using multistate EVB simulations.433 Subsequently, structures of the M2 channel were determined using X-ray diffraction,434 solution NMR of the protein in a detergent micelle,435 and by solid-state NMR in a lipid bilayer.436 These and subsequent studies have emphasized the conformational flexibility of this “simple” ion channel.437 Subsequent simulations based on these structures438,439 explored the interplay of the protonation state of the key pH-sensing His37 side chain, the channel conformation, and the behavior of water within the transmembrane pore, suggesting that a simple water wire Grotthuss mechanism might not apply. More recent high-resolution structures (see Figure 2D; PDB ID 5JOO resolved at 1.4 Å) obtained using XFELs19 reveal that at low pH there is a continuous hydrogen-bonded network of water molecules through the length of the channel, consistent with a Grotthuss mechanism for proton transport. Overall these studies reveal the complexities of a dynamic channel structure and its effects on water within a fluctuating charged pore and emphasize the need for further simulations based on recent higher-resolution structures440 to better understand water behavior in such a complex dynamic pore. An example is provided by recent simulations of a Grotthuss-like mechanism from the Voth group.441
Proton-permeable channels are also seen in a number of mitochondrial and related proteins, including the proton-translocating transhydrogenase for which structures442,443 and simulations442,443 reveal a dry, hydrophobic region in the channel. Wetting is promoted by protonation and the change in conformation of a key histidine residue. In complex I, a redox-driven proton pump, channel hydration has been suggested by simulation to be sensitive to the protonation states of buried lysine residues.444
In summary, these studies of proton-permeable pores suggest that the influence of pore size, hydrophobicity, and local conformation on water behavior seen in simpler nanopores is exploited by nature/evolution to control water hydrogen bonding and proton permeation through more complex channels and related structures.
8.3. Water and Hydrophobic Gating of Ion Channels
Most ion channels form a continuous pathway for conduction of their permeating species only when in an active (i.e., open) state, with water and ion flow interrupted at one or more positions (i.e., gates) when at rest or inactivated. In the case of a hydrophobic gate (see section 4.2 above), which favors a shift in equilibrium from the liquid to the vapor phase, closure to conduction is achieved energetically without requiring steric occlusion of the pore. Initially characterized using model nanopores, observations of dewetting in hydrophobic regions of channel pores have been increasingly reported in structural studies of ion channels and implicated as an important feature to consider in the mechanistic elucidation of these proteins.209
The first direct lines of evidence for hydrophobic gating in a biological ion channel were provided by studies on the small-conductance mechanosensitive channel, MscS, one member of a family of bacterial proteins that protect against hypo-osmotic shock.445 During atomistic MD simulations of TIP3P water in a crystal structure of the protein,446 liquid–vapor transitions were observed within the channel in a hydrophobic region where the central pore radius was ≥0.25 nm.447,448 The dewetting region was surrounded by side chains of leucine residues and considered to form a closed hydrophobic gate in the structure, preventing ion conduction in such a conformation.447 By contrast, a hydrophilic mutation at the constriction, substituting polar serine amino acids for one of the two leucine rings, yielded stable hydration through the channel under the same simulation conditions.447 An open conformation was subsequently resolved for the protein, showing a widened channel pore with radius >0.6 nm.449 Further simulation studies examined pore dewetting and hydration using TIP3P and SPC water in models of both MscS and the related large-conductance mechanosensitive channel, MscL, supporting the role of channel pore hydrophobicity (and consequent wettability) in the gating of both proteins.450
8.3.1. Pentameric Ligand-Gated Ion Channels (pLGICs)
Members of the pLGIC family mediate fast synaptic transmission and are the subject of considerable biomedical interest. The transmembrane portion of their pores is each lined by five M2 helices that contain a highly conserved hydrophobic residue in their 9′ position.451 This region was first demonstrated to constitute a hydrophobic gate by atomistic simulations of SPC water in the M2 helix bundle of a nicotinic acetylcholine receptor structure,452 in which narrowing of the permeation pathway to ∼0.3 nm in radius by two consecutive pore-lining rings formed by the 9′ leucine and 13′ valine residues was calculated to present an energetic barrier to water and ion passage.453 Consistent with such simulation results, effects of mutagenesis of the hydrophobic rings on channel electrophysiology were reported by a number of experimental studies.454−457 Later studies on two prokaryotic homologues of the channel, ELIC and GLIC, respectively, having phenylalanine and isoleucine in the 9′ position, showed dewetting of both proteins around this region in their simulated structures, conferring closure to the putative hydrophobic gate.458,459 For GLIC, liquid-to-vapor transitions were reported during string-method MD simulations of the isolated transmembrane domain from its putative open conformation to a nonconductive state.460 Comparable studies have also been performed for the entire GLIC channel.461,462 For the isolated transmembrane domain, the free-energy cost of hydrating the hydrophobic gate (with TIP3P water) was estimated.463 The hydration state at such a region was suggested to be a principal determinant of the functional (ion conduction) state of the channel based on calculations from the latter study, whereby pore hydration was associated with an increase in free energy of ∼46 kJ mol–1, compared with the smaller energetic cost (∼5 kJ mol–1) of transferring a sodium ion into a hydrated gate.463 Functioning of pore-lining hydrophobic side chains located midway through the transmembrane pathway as a gate in putative closed conformations of pLGICs, with conduction prevented even in the absence of steric obstruction, has since also been substantiated by MD simulations of the serotonin 5-HT3 receptor (Figure 13).8,464 Using a structure of a glycine receptor,465 another member of the pLGIC family, the sensitivity of the analysis to various nonpolarizable water models (SPCE, TIP3P, and TIP4P) was also examined, yielding similar water density and free energy profiles.8 The comparison of water models was further extended in MD simulations of the 5-HT3 receptor in multiple (both closed and open) states.163 The open (i.e., wetted) state conformation of the pore exhibited degrees of hydration ranging from partial wetting using the TIP4P/2005 water model to complete wetting for the polarizable AMOEBA14 model. These results indicate that induced polarization can influence the behavior of water and ions within hydrophobic gates of nanoscale pores. The same 5-HT3 pore has been used to explore electrowetting (see section 4.3 above), revealing that a (supra-physiological) potential difference of ∼0.85 V across the membrane was required to hydrate the hydrophobic gate of the closed (i.e., dewetted) state of the pore.466
Figure 13.

Structure and simulation of water in the transmembrane pore domain of the serotonin 5-HT3 receptor (PDB ID 4PIR). The overall protein architecture is in ribbon representation, with its transmembrane pore surface determined using CHAP and colored by hydrophobicity,467 and with pore-lining amino acid side chains shown as sticks. Only two out of the five protein subunits are displayed in the zoomed-in view for clarity. Two snapshots from an equilibrium simulation of water (red-and-white spheres) within the membrane-embedded protein (gray surface) are shown. Initially (at t = 0) the pore is hydrated (waters in red/white) but shortly afterward (t = 1 ns) it has dewetted.
8.3.2. Hydrophobic Gating in Other Channel Families
Dewetting of a hydrophobic region in the inner pore has been implicated to play a role in gating among members of several potassium channel families (as reviewed by Aryal et al.209). This is supported by atomistic MD simulations of SPC and TIP4P water in the voltage-gated potassium channel,468,469 of the SPC and TIP4P models in the TWIK-1 channel of the two-pore-domain (K2P) family,470 and of SPC and TIP3P in a homology model of the large-conductance calcium-activated potassium (BK) channel structure.471 In the case of TWIK-1, significance of the hydrophobic barrier was further validated by electrophysiological measurements, where a hydrophilic substitution led to enhanced channel activity in whole-cell current recordings.470 Evidence is also emerging for the presence of hydrophobic residues at the gates of TRP (transient receptor potential) channels, a class of nonselective cation channels whose members fulfill diverse functions in sensory physiology.472,473 The temperature dependence of the hydration equilibrium within TRPV1 has been suggested to contribute to the biological function of the protein as a temperature sensor.474
The presence of hydrophobic gates and their functional significance continue to be reported for other cation and anion channels as well. Investigated using TIP3P water, pore wetting events in the CorA magnesium channel, concurrent with small increases in pore diameter, have been suggested to mediate control of ion conduction by regulatory ions.475 A combination of spectroscopy and electrophysiology have demonstrated coupling of ion conduction to hydration of transmembrane helices in a channelrhodopsin.476,477 In bestrophin-1, a calcium-activated chloride channel, TIP4P water molecules were expelled from the closed state of the hydrophobic gate478 but hydrated the pore in simulations of in silico mutants of the protein with polar amino acid substitutions in the gate region.479
8.3.3. Integrative View of Water and Hydrophobic Gating of Ion Channels
An exhaustive survey of water behavior in biological channels has provided an integrative view of hydrophobic gating. MD simulations using the TIP4P water model were run for nearly 200 ion channel structures.217 The hydration/dewetting behavior of the channels was analyzed as a function of local radius and hydrophobicity of the pores (Figure 14). This revealed a clear relationship between the radius and local hydrophobicity of the pore and the free energy of water within the pore. Narrow (R < 0.4 nm) hydrophobic pores show a clear barrier to water entry. Fitting the simulation data from this analysis allowed a simple (simulation-free) heuristic model to be developed that enables accurate prediction of the presence of a closed hydrophobic gate in an ion channel structure based solely on measurement of pore radius and hydrophobicity profiles.467 This in turn enables rapid functional annotation of new channel structures as soon as they are determined.
Figure 14.
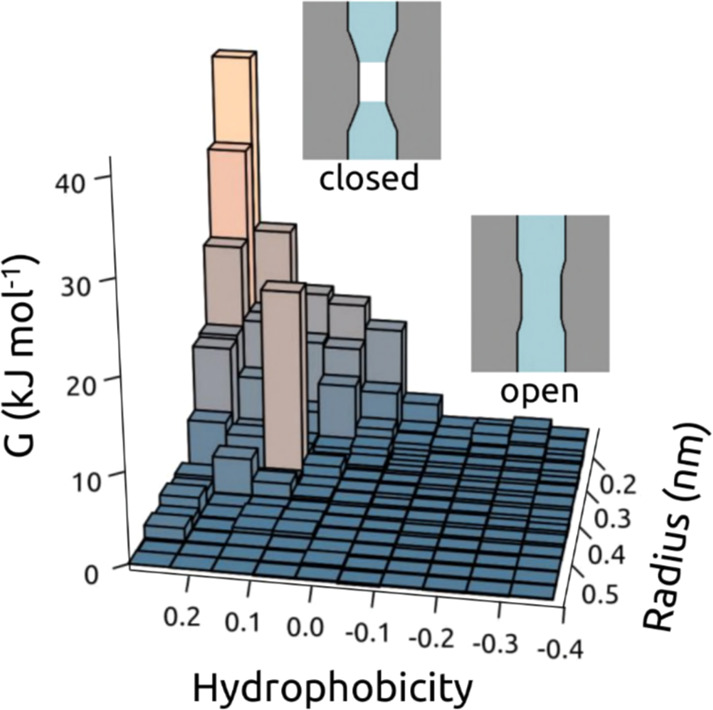
Hydrophobic gating of biological ion channels depicted schematically as a function of (hydrophobicity, radius) values of the transmembrane pore. The surface shows the free energy of water within a channel as a function of the local (hydrophobicity, radius) derived from molecular dynamics simulations (see ref (217) for details). Schematic depictions of dewetted (closed; low radius and high hydrophobicity) and hydrated (open; higher radius and hydrophilic) channels are shown. Modified from ref (211). Copyright 2010 National Academy of Sciences.
9. Conclusions and Critical Reflections
The studies reviewed above range from simulations of water in idealized models of nanopores through to complex biological ion channels. For the idealized models, a focus of many studies has been on water flow in nanopores. This has been shown to be potentially faster than Hagen–Poiseuille-predicted flow if the pore-lining is sufficiently hydrophobic. Simulations of nonbiological nanopores reinforce this view, revealing a complex relationship between pore size and geometry, the nature of the pore lining, and rates of water transport. Overall, a wider pore and a more hydrophobic lining favor water flow, provided the pore is wettable. Thus, the strength with which water interacts with the pore-lining surface is crucial in determining its local behavior within nanopores. Pore-lining surface roughness and dynamic fluctuations, as well as the overall pore geometry, are also important. Biological pores and channels exhibit a range of behaviors. β-Barrel bacterial outer membrane proteins confine water within hydrophilic pores with multiple water binding/interaction sites. Aquaporins are narrow and less hydrophilic, and have high water permeation rates. Ion channels confine water and may exhibit hydrophobic gating, which was first predicted in simple models of nanopores, and has been demonstrated experimentally in nonbiological nanopores.
The overall picture emerging from these studies is summarized in Figure 15 where the behavior of water within a nanopore is considered as a function of the hydrophobicity and radius of the pore. There are four regions, as exemplified by (i) wide (radius > 1 nm) and hydrophilic nanopores, which will exhibit behavior comparable to bulk water; (ii) wide (radius > 1 nm) and hydrophobic nanopores, which should enable water to flow faster than is predicted by the Hagen–Poiseuille theory, with enhancement factors substantially >1 possible for nanopores; (iii) many biological pores and (open) channels are narrow (radius < 1 nm) and hydrophilic, and thus contain water which interacts with the pore-lining residues; and (iv) some ion channels exhibit hydrophobic gating, with dewetting in their narrow (radius < 1 nm) and hydrophobic constrictions, which in turn closes the channel to the flow of ions.
Figure 15.
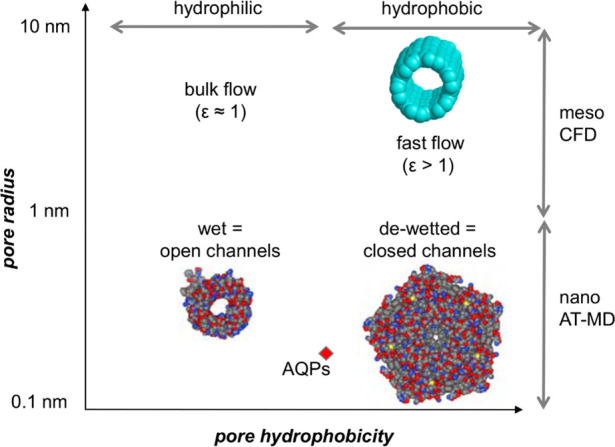
Schematic summary of the behavior of water within nanopores as a function of the hydrophobicity and radius of the pores. There are four regions, as exemplified in (i) wide (R > 1 nm) hydrophilic nanopores, exhibiting behavior comparable to bulk water; (ii) wide (R > 1 nm) hydrophobic nanopores, which enable water to flow faster than predicted by the Hagen–Poiseuille theory; (iii) narrow (R < 1 nm) hydrophilic nanopores, corresponding to many biological pores; and (iv) biological channels which exhibit hydrophobic gating, with dewetting in their narrow (R < 1 nm) and hydrophobic constrictions. In this figure, ε is the ratio of the water flow compared to that predicted by the Hagen–Poiseuille equation for bulk water viscosity and no-slip boundary conditions.
The biological relevance of these wide-ranging simulation studies is in helping us to understand the emerging diversity of channel structures. Being able to position a new channel structure into its corresponding region in Figure 15, and to characterize how conformational changes in the pore may move a channel from one region to another, will inform our mechanistic understanding of the regulation of biological channels, for example by hydrophobic gating. Such a schematic view may be used as a “template” to aid the design of novel nanopores such as those functioning as artificial water channels.480 There is also further work to be done regarding the elucidation of the effects of electric fields on water behavior, when designing more complex pores, as a possible mechanism for the switching on/off of novel nanopores.
It is informative to reflect critically on the progress summarized in this review. Two main questions arise, one concerning optimal systems for simulation studies of water in nanopores, and the other concerning simulation methodologies and models.
Nanopore systems for simulations span a spectrum from highly simplified idealized models, offering more general insights into the behavior of nanoconfined water, through to complex biological pores which are gated, that is, switchable between open and closed states. Simplified models provide general insights into e.g. the relationship between pore radius, hydrophobicity, and flow rate of water. However, it remains uncertain to what extent such generalizations can be used in a quantitative and predictive fashion. The latter is an important consideration if we wish to exploit our general understanding in the design of novel nanopores with desired functional properties, and in annotation of the functional properties of novel biological channels and pores (as an increasing number of structures of these are determined).
A more methodological consideration is whether the simulation methodologies and models used to study water in nanopores are of sufficient granularity and/or accuracy for the task. Many studies to date have used conventional MD with well-established water models (e.g., TIP3P or SPC/E). Perhaps surprisingly, continuum models (modified by insights from atomistic MD simulations) have also provided valuable insights into the behavior of water in simple nanopores. It is likely that for the design of novel nanopores and in order to understand complex biological nanopores, accurate atomistic simulations of water properties are needed. It remains uncertain whether fixed point-charge MD models of water are sufficient, or whether polarizable (or even QM) models are needed in some situations (e.g., hydrophobic gating). This is of particular importance in the context of confined water systems as classical (i.e., MM) water models are typically developed and validated for how accurately they reproduce bulk water properties. It, therefore, may be timely to use more advanced water models and to evaluate the robustness to changes in water model when running simulation studies of water behavior in complex nanopores, particularly as preliminary studies indicate that the nanoscale behavior of water is sensitive to water model. Quantum mechanical approaches offer tantalizingly even higher levels of accuracy. This comes at a significant increase in computational cost, and there are still many levels of theory and parametrizations to evaluate. Substantial progress is being made in rendering quantum mechanical methods more computationally feasible for biological systems. There is still some way to go before it will be possible to perform quantum mechanical calculations on system sizes and time scales comparable to those accessed via classical methodologies.
Returning to the schematic summary (Figure 15) in light of these reflections, we are currently able to design a nanopore which promotes wetting over dewetting or vice versa. But can we design a novel nanopore which enables fast, selective flow of water, that is, can we match aquaporins by de novo design? And can we design such a nanopore that it is also switchable in a controlled fashion? Fully answering these two key questions will require more detailed simulations alongside experimental evaluation, as exemplified by, for example, a recent study of gating of hydrophobic nanopores with large anions.481
Acknowledgments
We thank the following agencies for funding research in M.S.P.S.’s group: BBSRC (BB/R00126X/1; BB/N000145/1), EPSRC (EP/R004722/1; EP/R029407/1), and Wellcome (208361/Z/17/Z). Our thanks to our colleagues for many discussions of channels and water, especially Dr. Gianni Klesse and Prof. Stephen Tucker.
Biographies
Charlotte I. Lynch read for an MEng in Materials Science at Trinity College, University of Oxford. She then completed her DPhil in (Computational) Materials Science, also at Trinity College, Oxford, before moving to a postdoctoral position in the group of Mark Sansom, where she researches the behavior of water in ion channels and nanopores.
Shanlin Rao read for an MBiochem degree at Hertford College, University of Oxford. She is now reading for a DPhil in (Computational) Biochemistry, studying hydrophobic gating of ion channels.
Mark S. P. Sansom read for a B.A. in Biochemistry at University College, University of Oxford, followed by a DPhil in Molecular Biophysics. Following a postdoctoral fellowship and a junior faculty position at the University of Nottingham, he returned to the Department of Biochemistry, University of Oxford, where he is the David Phillips Professor of Molecular Biophysics. His research interests are in the computational biophysics of membrane proteins and their interactions.
The authors declare no competing financial interest.
References
- Hille B.Ionic Channels of Excitable Membranes, 3rd ed.; Sinauer Associates, Inc.: Sunderland, MA, 2001. [Google Scholar]
- Gouaux E.; MacKinnon R. Principles of selective ion transport in channels and pumps. Science 2005, 310, 1461–1465. 10.1126/science.1113666. [DOI] [PubMed] [Google Scholar]
- Hou X.; Guo W.; Jiang L. Biomimetic smart nanopores and nanochannels. Chem. Soc. Rev. 2011, 40, 2385–2401. 10.1039/c0cs00053a. [DOI] [PubMed] [Google Scholar]
- Ayub M.; Bayley H. Engineered transmembrane pores. Curr. Opin. Chem. Biol. 2016, 34, 117–126. 10.1016/j.cbpa.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adiga S. P.; Brenner D. W. Toward designing smart nanovalves: modeling of flow control through nanopores via the helix-coil transition of grafted polypeptide chains. Macromolecules 2007, 40, 1342–1348. 10.1021/ma0617522. [DOI] [Google Scholar]
- Zhang Z.; Li P.; Kong X. Y.; Xie G. H.; Qian Y. C.; Wang Z. Q.; Tian Y.; Wen L. P.; Jiang L. Bioinspired heterogeneous ion pump membranes: Unidirectional selective pumping and controllable gating properties stemming from asymmetric ionic group distribution. J. Am. Chem. Soc. 2018, 140, 1083–1090. 10.1021/jacs.7b11472. [DOI] [PubMed] [Google Scholar]
- He Z. J.; Zhou J.; Lu X. H.; Corry B. Bioinspired graphene nanopores with voltage-tunable ion selectivity for Na+ and K+. ACS Nano 2013, 7, 10148–10157. 10.1021/nn4043628. [DOI] [PubMed] [Google Scholar]
- Trick J. L.; Chelvaniththilan S.; Klesse G.; Aryal P.; Wallace E. J.; Tucker S. J.; Sansom M. S. P. Functional annotation of ion channel structures by molecular simulation. Structure 2016, 24, 2207–2216. 10.1016/j.str.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W.; Kumar M. Artificial water channels: toward and beyond desalination. Curr. Opin. Chem. Eng. 2019, 25, 9–17. 10.1016/j.coche.2019.06.007. [DOI] [Google Scholar]
- Zhang X. Q.; Liu H. L.; Jiang L. Wettability and applications of nanochannels. Adv. Mater. 2019, 31, 1804508. 10.1002/adma.201804508. [DOI] [PubMed] [Google Scholar]
- Danda G.; Drndic M. Two-dimensional nanopores and nanoporous membranes for ion and molecule transport. Curr. Opin. Biotechnol. 2019, 55, 124–133. 10.1016/j.copbio.2018.09.002. [DOI] [PubMed] [Google Scholar]
- Horner A.; Siligan C.; Cornean A.; Pohl P. Positively charged residues at the channel mouth boost single-file water flow. Faraday Discuss. 2018, 209, 55–65. 10.1039/C8FD00050F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M. F.; Cao E. H.; Julius D.; Cheng Y. F. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandala V. S.; Williams J. K.; Hong M. Structure and dynamics of membrane proteins from solid-state NMR. Annu. Rev. Biophys. 2018, 47, 201–222. 10.1146/annurev-biophys-070816-033712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosinska Eriksson U.; Fischer G.; Friemann R.; Enkavi G.; Tajkhorshid E.; Neutze R. Subangstrom resolution X-ray structure details aquaporin-water interactions. Science 2013, 340, 1346–1349. 10.1126/science.1234306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariae U.; Kluhspies T.; De S.; Engelhardt H.; Zeth K. High resolution crystal structures and molecular dynamics studies reveal substrate binding in the porin Omp32. J. Biol. Chem. 2006, 281, 7413–7420. 10.1074/jbc.M510939200. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Morais-Cabral J. H.; Kaufman A.; MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature 2001, 414, 43–48. 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- Ye S.; Li Y.; Jiang Y. X. Novel insights into K+ selectivity from high-resolution structures of an open K+ channel pore. Nat. Struct. Mol. Biol. 2010, 17, 1019–1865. 10.1038/nsmb.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomaston J. L.; Woldeyes R. A.; Nakane T.; Yamashita A.; Tanaka T.; Koiwai K.; Brewster A. S.; Barad B. A.; Chen Y. J.; Lemmin T.; Uervirojnangkoorn M.; Arima T.; Kobayashi J.; Masuda T.; Suzuki M.; Sugahara M.; Sauter N. K.; Tanaka R.; Nureki O.; Tono K.; Joti Y.; Nango E.; Iwata S.; Yumoto F.; Fraser J. S.; DeGrado W. F. XFEL structures of the influenza M2 proton channel: room temperature water networks and insights into proton conduction. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 13357–13362. 10.1073/pnas.1705624114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasic J. R.; Worcester D. L.; Gawrisch K.; Gurnev P.; Mihailescu M. Pore hydration states of KcsA potassium channels in membranes. J. Biol. Chem. 2015, 290, 26765–U26472. 10.1074/jbc.M115.661819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S.; Feng J.; Chan L. J. G.; Petzold C. J.; Ralston C. Y. Synchrotron X-ray footprinting as a method to visualize water in proteins. J. Synchrotron Radiat. 2016, 23, 1056–1069. 10.1107/S1600577516009024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S.; Bavro V. N.; D’Mello R.; Tucker S. J.; Venien-Bryan C.; Chance M. R. Conformational Changes During the Gating of a Potassium Channel Revealed by Structural Mass Spectrometry. Structure 2010, 18, 839–846. 10.1016/j.str.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C. W.; Jansen T. L. C.; Knoester J. Proton transport in biological systems can be probed by two-dimensional infrared spectroscopy. J. Chem. Phys. 2011, 134, 044502. 10.1063/1.3522770. [DOI] [PubMed] [Google Scholar]
- Kratochvil H. T.; Carr J. K.; Matulef K.; Annen A. W.; Li H.; Maj M.; Ostmeyer J.; Serrano A. L.; Raghuraman H.; Moran S. D.; Skinner J. L.; Perozo E.; Roux B.; Valiyaveetil F. I.; Zanni M. T. Instantaneous ion configurations in the K+ ion channel selectivity filter revealed by 2D IR spectroscopy. Science 2016, 353, 1040–1044. 10.1126/science.aag1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratochvil H. T.; Maj M.; Matulef K.; Annen A. W.; Ostmeyer J.; Perozo E.; Roux B.; Valiyaveetil F. I.; Zanni M. T. Probing the effects of gating on the ion occupancy of the K+ channel selectivity filter using two-dimensional infrared spectroscopy. J. Am. Chem. Soc. 2017, 139, 8837–8845. 10.1021/jacs.7b01594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipcigan F. S.; Sokhan V. P.; Crain J.; Martyna G. J. Electronic coarse graining enhances the predictive power of molecular simulation allowing challenges in water physics to be addressed. J. Comput. Phys. 2016, 326, 222–233. 10.1016/j.jcp.2016.08.030. [DOI] [Google Scholar]
- Brunk E.; Röthlisberger U. Mixed quantum mechanical/molecular mechanical molecular dynamics simulations of biological systems in ground and electronically excited states. Chem. Rev. 2015, 115, 6217–6263. 10.1021/cr500628b. [DOI] [PubMed] [Google Scholar]
- Cui Q. Perspective: Quantum mechanical methods in biochemistry and biophysics. J. Chem. Phys. 2016, 145, 140901. 10.1063/1.4964410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senn H. M.; Thiel W. QM/MM methods for biomolecular systems. Angew. Chem., Int. Ed. 2009, 48, 1198–1229. 10.1002/anie.200802019. [DOI] [PubMed] [Google Scholar]
- Cole D. J.; Hine N. D. M. Applications of large-scale density functional theory in biology. J. Phys.: Condens. Matter 2016, 28, 393001. 10.1088/0953-8984/28/39/393001. [DOI] [PubMed] [Google Scholar]
- Hassanali A. A.; Cuny J.; Verdolino V.; Parrinello M. Aqueous solutions: state of the art in ab initio molecular dynamics. Philos. Trans. R. Soc., A 2014, 372, 20120482. 10.1098/rsta.2012.0482. [DOI] [PubMed] [Google Scholar]
- Pereyaslavets L.; Kurnikov I.; Kamath G.; Butin O.; Illarionov A.; Leontyev I.; Olevanov M.; Levitt M.; Kornberg R. D.; Fain B. On the importance of accounting for nuclear quantum effects in ab initio calibrated force fields in biological simulations. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 8878–8882. 10.1073/pnas.1806064115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habershon S.; Markland T. E.; Manolopoulos D. E. Competing quantum effects in the dynamics of a flexible water model. J. Chem. Phys. 2009, 131, 024501 10.1063/1.3167790. [DOI] [PubMed] [Google Scholar]
- Paesani F.; Zhang W.; Case D. A.; Cheatham T. E.; Voth G. A. An accurate and simple quantum model for liquid water. J. Chem. Phys. 2006, 125, 184507. 10.1063/1.2386157. [DOI] [PubMed] [Google Scholar]
- Wagner W.; et al. International equations for the pressure along the melting and along the sublimation curve of ordinary water substance. J. Phys. Chem. Ref. Data 1994, 23, 515–525. 10.1063/1.555947. [DOI] [Google Scholar]
- Feistel R.; Wagner W. A new equation of state for H2O ice Ih. J. Phys. Chem. Ref. Data 2006, 35, 1021–1047. 10.1063/1.2183324. [DOI] [Google Scholar]
- Soper A. K. Structural transformations in amorphous ice and supercooled water and their relevance to the phase diagram of water. Mol. Phys. 2008, 106, 2053–2076. 10.1080/00268970802116146. [DOI] [Google Scholar]
- Abascal J. L. F.; Vega C. A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem. Phys. 2005, 123, 234505. 10.1063/1.2121687. [DOI] [PubMed] [Google Scholar]
- Anandakrishnan R.; Izadi S.; Onufriev A. V. Why computed protein folding landscapes are sensitive to the water model. J. Chem. Theory Comput. 2019, 15, 625–636. 10.1021/acs.jctc.8b00485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pall S.; Abraham M. J.; Kutzner C.; Hess B.; Lindahl E.. Tackling exascale software challenges in molecular dynamics simulations with GROMACS. In Solving Software Challenges for Exascale; Markidis S., Laure E., Eds.; Lecture Notes in Computer Science; Springer, Cham, 2015; Vol. 8759; pp 3–27. [Google Scholar]
- Hoekstra A. G.; Portegies Zwart S.; Coveney P. V. Multiscale modelling, simulation and computing: from the desktop to the exascale. Philos. Trans. R. Soc., A 2019, 377, 20180355. 10.1098/rsta.2018.0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://www.sklogwiki.org/SklogWiki/index.php/Water_models.
- Onufriev A. V.; Izadi S. Water models for biomolecular simulations. WIREs Comput. Mol. Sci. 2018, 8, e1347 10.1002/wcms.1347. [DOI] [Google Scholar]
- Onufriev A. Chapter 7 - Implicit solvent models in molecular dynamics simulations: a brief overview. Annu. Rep. Comput. Chem. 2008, 4, 125–137. 10.1016/S1574-1400(08)00007-8. [DOI] [Google Scholar]
- Still W. C.; Tempczyk A.; Hawley R. C.; Hendrickson T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. 10.1021/ja00172a038. [DOI] [Google Scholar]
- Warwicker J.; Watson H. C. Calculation of the electric-potential in the active-site cleft due to a-helix dipoles. J. Mol. Biol. 1982, 157, 671–679. 10.1016/0022-2836(82)90505-8. [DOI] [PubMed] [Google Scholar]
- Valisko M.; Matejczyk B.; Hato Z.; Kristof T.; Madai E.; Fertig D.; Gillespie D.; Boda D. Multiscale analysis of the effect of surface charge pattern on a nanopore’s rectification and selectivity properties: From all-atom model to Poisson-Nernst-Planck. J. Chem. Phys. 2019, 150, 179902. 10.1063/1.5104347. [DOI] [PubMed] [Google Scholar]
- Toth G. Effective potentials from complex simulations: a potential-matching algorithm and remarks on coarse-grained potentials. J. Phys.: Condens. Matter 2007, 19, 335222. 10.1088/0953-8984/19/33/335222. [DOI] [PubMed] [Google Scholar]
- Hadley K. R.; McCabe C. Coarse-grained molecular models of water: a review. Mol. Simul. 2012, 38, 671–681. 10.1080/08927022.2012.671942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darre L.; Machado M. R.; Pantano S. Coarse-grained models of water. WIREs-Comput. Mol. Sci. 2012, 2, 921–930. 10.1002/wcms.1097. [DOI] [Google Scholar]
- Marrink S. J.; Risselada J.; Yefimov S.; Tieleman D. P.; de Vries A. H. The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- Shelley J. C.; Shelley M. Y.; Reeder R. C.; Bandyopadhyay S.; Klein M. L. A coarse grain model for phospholipid simulations. J. Phys. Chem. B 2001, 105, 4464–4470. 10.1021/jp010238p. [DOI] [Google Scholar]
- Groot R. D.; Rabone K. L. Mesoscopic simulation of cell membrane damage, morphology change and rupture by nonionic surfactants. Biophys. J. 2001, 81, 725–736. 10.1016/S0006-3495(01)75737-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yesylevskyy S. O.; Schäfer L. V.; Sengupta D.; Marrink S. J. Polarizable water model for the coarse-grained MARTINI force field. PLoS Comput. Biol. 2010, 6, e1000810 10.1371/journal.pcbi.1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.; Cui Q. A.; Yethiraj A. A new coarse-grained model for water: the importance of electrostatic interactions. J. Phys. Chem. B 2010, 114, 10524–10529. 10.1021/jp1019763. [DOI] [PubMed] [Google Scholar]
- Darre L.; Machado M. R.; Dans P. D.; Herrera F. E.; Pantano S. Another coarse grain model for aqueous solvation: WAT FOUR?. J. Chem. Theory Comput. 2010, 6, 3793–3807. 10.1021/ct100379f. [DOI] [Google Scholar]
- Stachiewicz A.; Molski A. A coarse-grained MARTINI-like force field for DNA unzipping in nanopores. J. Comput. Chem. 2015, 36, 947–956. 10.1002/jcc.23874. [DOI] [PubMed] [Google Scholar]
- Vogele M.; Kofinger J.; Hummer G. Molecular dynamics simulations of carbon nanotube porins in lipid bilayers. Faraday Discuss. 2018, 209, 341–358. 10.1039/C8FD00011E. [DOI] [PubMed] [Google Scholar]
- Dreyer J.; Strodel P.; Ippoliti E.; Finnerty J.; Eisenberg B.; Carloni P. Ion permeation in the NanC porin from Escherichia coli: free energy calculations along pathways identified by coarse-grain simulations. J. Phys. Chem. B 2013, 117, 13534–13542. 10.1021/jp4081838. [DOI] [PubMed] [Google Scholar]
- Wagner W.; Saul A.; Pruss A. International equations for the pressure along the melting and along the sublimation curve of ordinary water substance. J. Phys. Chem. Ref. Data 1994, 23, 515–527. 10.1063/1.555947. [DOI] [Google Scholar]
- Gregory J. K.; Clary D. C.; Liu K.; Brown M. G.; Saykally R. J. The water dipole moment in water clusters. Science 1997, 275, 814–817. 10.1126/science.275.5301.814. [DOI] [PubMed] [Google Scholar]
- CRC Handbook of Chemistry and Physics, 84th ed.; Lide D. R., Ed.; CRC Press, Inc.: Boca Raton, FL, 2004. [Google Scholar]
- Vega C.; Abascal J. L. F.; Conde M. M.; Aragones J. L. What ice can teach us about water interactions: a critical comparison of the performance of different water models. Faraday Discuss. 2009, 141, 251–276. 10.1039/B805531A. [DOI] [PubMed] [Google Scholar]
- Vega C.; Abascal J. L. F. Simulating water with rigid non-polarizable models: a general perspective. Phys. Chem. Chem. Phys. 2011, 13, 19663–19688. 10.1039/c1cp22168j. [DOI] [PubMed] [Google Scholar]
- Skinner L. B.; Huang C.; Schlesinger D.; Pettersson L. G. M.; Nilsson A.; Benmore C. J. Benchmark oxygen-oxygen pair-distribution function of ambient water from x-ray diffraction measurements with a wide Q-range. J. Chem. Phys. 2013, 138, 074506 10.1063/1.4790861. [DOI] [PubMed] [Google Scholar]
- Izadi S.; Onufriev A. V. Accuracy limit of rigid 3-point water models. J. Chem. Phys. 2016, 145, 074501 10.1063/1.4960175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Grigera J. R.; Straatsma T. P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. 10.1021/j100308a038. [DOI] [Google Scholar]
- Vega C.; Abascal J. L. F. Relation between the melting temperature and the temperature of maximum density for the most common models of water. J. Chem. Phys. 2005, 123, 144504. 10.1063/1.2056539. [DOI] [PubMed] [Google Scholar]
- Fennell C. J.; Li L. B.; Dill K. A. Simple liquid models with corrected dielectric constants. J. Phys. Chem. B 2012, 116, 6936–6944. 10.1021/jp3002383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney M. W.; Jorgensen W. L. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J. Chem. Phys. 2000, 112, 8910–8922. 10.1063/1.481505. [DOI] [Google Scholar]
- Izadi S.; Anandakrishnan R.; Onufriev A. V. Building water models: a different approach. J. Phys. Chem. Lett. 2014, 5, 3863–3871. 10.1021/jz501780a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lísal M.; Smith W. R.; Nezbeda I. Accurate vapour–liquid equilibrium calculations for complex systems using the reaction Gibbs ensemble Monte Carlo simulation method. Fluid Phase Equilib. 2001, 181, 127–146. 10.1016/S0378-3812(01)00489-7. [DOI] [Google Scholar]
- Horn H. W.; Swope W. C.; Pitera J. W.; Madura J. D.; Dick T. J.; Hura G. L.; Head-Gordon T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. 10.1063/1.1683075. [DOI] [PubMed] [Google Scholar]
- Qiu Y. D.; Nerenberg P. S.; Head-Gordon T.; Wang L. P. Systematic optimization of water models using liquid/vapor surface tension data. J. Phys. Chem. B 2019, 123, 7061–7073. 10.1021/acs.jpcb.9b05455. [DOI] [PubMed] [Google Scholar]
- Zarzycki P.; Gilbert B. Temperature-dependence of the dielectric relaxation of water using non-polarizable water models. Phys. Chem. Chem. Phys. 2020, 22, 1011–1018. 10.1039/C9CP04578C. [DOI] [PubMed] [Google Scholar]
- Wang L. P.; Martinez T. J.; Pande V. S. Building force fields: an automatic, systematic, and reproducible approach. J. Phys. Chem. Lett. 2014, 5, 1885–1891. 10.1021/jz500737m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; Hermans J.: Interaction models for water in relation to protein hydration. In Intermolecular Forces; Pullman B., Ed.; Reidel: Dordrecht, 1981; pp 331–342. [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Jorgensen W. L.; Madura J. D. temperature and size dependence for Monte-Carlo simulations of TIP4P water. Mol. Phys. 1985, 56, 1381–1392. 10.1080/00268978500103111. [DOI] [Google Scholar]
- Scott W. R. P.; Hunenberger P. H.; Tironi I. G.; Mark A. E.; Billeter S. R.; Fennen J.; Torda A. E.; Huber T.; Kruger P.; van Gunsteren W. F. The GROMOS biomolecular simulation program package. J. Phys. Chem. A 1999, 103, 3596–3607. 10.1021/jp984217f. [DOI] [Google Scholar]
- Weiner P. K.; Kollman P. A. AMBER - Assisted model-building with energy refinement - a general program for modeling molecules and their interactions. J. Comput. Chem. 1981, 2, 287–303. 10.1002/jcc.540020311. [DOI] [Google Scholar]
- Jorgensen W. L.; Tirado-Rives J. The OPLS potential functions for proteins - energy minimizations for crystals of cyclic-peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- Kaminski G. A.; Friesner R. A.; Tirado-Rives J.; Jorgensen W. L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6474–6487. 10.1021/jp003919d. [DOI] [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiorkiewicz-Kuczera J.; Yin D.; Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Paschek D. Temperature dependence of the hydrophobic hydration and interaction of simple solutes: An examination of five popular water models. J. Chem. Phys. 2004, 120, 6674–6690. 10.1063/1.1652015. [DOI] [PubMed] [Google Scholar]
- Poole P. H.; Sciortino F.; Essmann U.; Stanley H. E. Phase-behavior of metastable water. Nature 1992, 360, 324–328. 10.1038/360324a0. [DOI] [Google Scholar]
- Rick S. W. A reoptimization of the five-site water potential (TIP5P) for use with Ewald sums. J. Chem. Phys. 2004, 120, 6085–6093. 10.1063/1.1652434. [DOI] [PubMed] [Google Scholar]
- Khalak Y.; Baumeier B.; Karttunen M. Improved general-purpose five-point model for water: TIP5P/2018. J. Chem. Phys. 2018, 149, 224507. 10.1063/1.5070137. [DOI] [PubMed] [Google Scholar]
- Zhao C. L.; Zhao D. X.; Bei C. C.; Meng X. N.; Li S. M.; Yang Z. Z. Seven-site effective pair potential for simulating liquid water. J. Phys. Chem. B 2019, 123, 4594–4603. 10.1021/acs.jpcb.9b03149. [DOI] [PubMed] [Google Scholar]
- Fuentes-Azcatl R.; Mendoza N.; Alejandre J. Improved SPC force field of water based on the dielectric constant: SPC/e. Phys. A 2015, 420, 116–123. 10.1016/j.physa.2014.10.072. [DOI] [PubMed] [Google Scholar]
- Fuentes-Azcatl R.; Alejandre J. Non-polarizable force field of water based on the dielectric constant: TIP4P/e. J. Phys. Chem. B 2014, 118, 1263–1272. 10.1021/jp410865y. [DOI] [PubMed] [Google Scholar]
- Vega C.; de Miguel E. Surface tension of the most popular models of water by using the test-area simulation method. J. Chem. Phys. 2007, 126, 154707. 10.1063/1.2715577. [DOI] [PubMed] [Google Scholar]
- Abascal J. L. F.; Sanz E.; García Fernández R. G.; Vega C. A potential model for the study of ices and amorphous water: TIP4P/Ice. J. Chem. Phys. 2005, 122, 234511. 10.1063/1.1931662. [DOI] [PubMed] [Google Scholar]
- Zhang H. Y.; Yin C. H.; Jiang Y.; van der Spoel D. Force field benchmark of amino acids: I. hydration and diffusion in different water models. J. Chem. Inf. Model. 2018, 58, 1037–1052. 10.1021/acs.jcim.8b00026. [DOI] [PubMed] [Google Scholar]
- Sajadi F.; Rowley C. N. Simulations of lipid bilayers using the CHARMM36 force field with the TIP3P-FB and TIP4P-FB water models. PeerJ 2018, 6, e5472 10.7717/peerj.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery D. C.Design and Analysis of Experiments, 7th ed.; Wiley: New York, 2008. [Google Scholar]
- O’Connor D.; English N. J. Systematic design-of-experiments, factorial-design approaches for tuning simple empirical water models. Mol. Simul. 2019, 1. 10.1080/08927022.2019.1626987. [DOI] [Google Scholar]
- Yu H. B.; van Gunsteren W. F. Accounting for polarization in molecular simulation. Comput. Phys. Commun. 2005, 172, 69–85. 10.1016/j.cpc.2005.01.022. [DOI] [Google Scholar]
- Caldwell J. W.; Kollman P. A. Structure and properties of neat liquids using nonadditive molecular-dynamics - water, methanol, and N-methylacetamide. J. Phys. Chem. 1995, 99, 6208–6219. 10.1021/j100016a067. [DOI] [Google Scholar]
- Brodholt J.; Sampoli M.; Vallauri R. Parameterizing a polarizable intermolecular potential for water. Mol. Phys. 1995, 86, 149–158. 10.1080/00268979500101901. [DOI] [Google Scholar]
- Thole B. T. Molecular polarizabilities calculated with a modified dipole interaction. Chem. Phys. 1981, 59, 341–350. 10.1016/0301-0104(81)85176-2. [DOI] [Google Scholar]
- Ren P. Y.; Ponder J. W. Polarizable atomic multipole water model for molecular mechanics simulation. J. Phys. Chem. B 2003, 107, 5933–5947. 10.1021/jp027815+. [DOI] [Google Scholar]
- Wang L. P.; Head-Gordon T.; Ponder J. W.; Ren P.; Chodera J. D.; Eastman P. K.; Martinez T. J.; Pande V. S. Systematic improvement of a classical molecular model of water. J. Phys. Chem. B 2013, 117, 9956–9972. 10.1021/jp403802c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Lopes P. E. M.; Roux B.; MacKerell A. D. Recent advances in polarizable force fields for macromolecules: microsecond simulations of proteins using the classical drude oscillator model. J. Phys. Chem. Lett. 2014, 5, 3144–3150. 10.1021/jz501315h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laury M. L.; Wang L. P.; Pande V. S.; Head-Gordon T.; Ponder J. W. Revised parameters for the AMOEBA polarizable atomic multipole water model. J. Phys. Chem. B 2015, 119, 9423–9437. 10.1021/jp510896n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi R.; Wang L. P.; Wang Q. T.; Pande V. S.; Ren P. Y. United polarizable multipole water model for molecular mechanics simulation. J. Chem. Phys. 2015, 143, 014504 10.1063/1.4923338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A. K.; Demerdash O. N.; Head-Gordon T. Improvements to the AMOEBA force field by introducing anisotropic atomic polarizability of the water molecule. J. Chem. Theory Comput. 2018, 14, 6722–6733. 10.1021/acs.jctc.8b00978. [DOI] [PubMed] [Google Scholar]
- Paricaud P.; Predota M.; Chialvo A. A.; Cummings P. T. From dimer to condensed phases at extreme conditions: accurate predictions of the properties of water by a Gaussian charge polarizable model. J. Chem. Phys. 2005, 122, 244511. 10.1063/1.1940033. [DOI] [PubMed] [Google Scholar]
- Lamoureux G.; MacKerell A. D.; Roux B. A simple polarizable model of water based on classical Drude oscillators. J. Chem. Phys. 2003, 119, 5185–5197. 10.1063/1.1598191. [DOI] [Google Scholar]
- Lamoureux G.; Harder E.; Vorobyov I. V.; Roux B.; MacKerell A. D. A polarizable model of water for molecular dynamics simulations of biomolecules. Chem. Phys. Lett. 2006, 418, 245–249. 10.1016/j.cplett.2005.10.135. [DOI] [Google Scholar]
- Yu W. B.; Lopes P. E. M.; Roux B.; MacKerell A. D. Six-site polarizable model of water based on the classical Drude oscillator. J. Chem. Phys. 2013, 138, 034508. 10.1063/1.4774577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho T. A.; Striolo A. Polarizability effects in molecular dynamics simulations of the graphene-water interface. J. Chem. Phys. 2013, 138, 054117 10.1063/1.4789583. [DOI] [PubMed] [Google Scholar]
- Li H.; Chowdhary J.; Huang L.; He X. B.; MacKerell A. D.; Roux B. Drude polarizable force field for molecular dynamics simulations of saturated and unsaturated zwitterionic lipids. J. Chem. Theory Comput. 2017, 13, 4535–4552. 10.1021/acs.jctc.7b00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rick S. W.; Stuart S. J.; Berne B. J. Dynamical fluctuating charge force-fields - application to liquid water. J. Chem. Phys. 1994, 101, 6141–6156. 10.1063/1.468398. [DOI] [Google Scholar]
- Chen B.; Xing J. H.; Siepmann J. I. Development of polarizable water force fields for phase equilibrium calculations. J. Phys. Chem. B 2000, 104, 2391–2401. 10.1021/jp993687m. [DOI] [Google Scholar]
- Stern H. A.; Rittner F.; Berne B. J.; Friesner R. A. Combined fluctuating charge and polarizable dipole models: Application to a five-site water potential function. J. Chem. Phys. 2001, 115, 2237–2251. 10.1063/1.1376165. [DOI] [Google Scholar]
- Raabe G.; Sadus R. J. Molecular dynamics simulation of the dielectric constant of water: The effect of bond flexibility. J. Chem. Phys. 2011, 134, 234501. 10.1063/1.3600337. [DOI] [PubMed] [Google Scholar]
- Bakker H. J.; Skinner J. L. Vibrational spectroscopy as a probe of structure and dynamics in liquid water. Chem. Rev. 2010, 110, 1498–1517. 10.1021/cr9001879. [DOI] [PubMed] [Google Scholar]
- Li F.; Skinner J. L. Infrared and Raman line shapes for ice Ih. II. H2O and D2O. J. Chem. Phys. 2010, 133, 244504. 10.1063/1.3516460. [DOI] [PubMed] [Google Scholar]
- Yuet P. K.; Blankschtein D. Molecular dynamics simulation study of water surfaces: comparison of flexible water models. J. Phys. Chem. B 2010, 114, 13786–13795. 10.1021/jp1067022. [DOI] [PubMed] [Google Scholar]
- Gonzalez M. A.; Abascal J. L. F. A flexible model for water based on TIP4P/2005. J. Chem. Phys. 2011, 135, 224516. 10.1063/1.3663219. [DOI] [PubMed] [Google Scholar]
- Wu Y. J.; Tepper H. L.; Voth G. A. Flexible simple point-charge water model with improved liquid-state properties. J. Chem. Phys. 2006, 124, 024503 10.1063/1.2136877. [DOI] [PubMed] [Google Scholar]
- Lopez-Lemus J.; Chapela G. A.; Alejandre J. Effect of flexibility on surface tension and coexisting densities of water. J. Chem. Phys. 2008, 128, 174703. 10.1063/1.2907845. [DOI] [PubMed] [Google Scholar]
- Dang L. X.; Pettitt B. M. Simple intramolecular model potentials for water. J. Phys. Chem. 1987, 91, 3349–3354. 10.1021/j100296a048. [DOI] [Google Scholar]
- Levitt M.; Hirshberg M.; Sharon R.; Laidig K. E.; Daggett V. Calibration and testing of a water model for simulation of the molecular dynamics of proteins and nucleic acids in solution. J. Phys. Chem. B 1997, 101, 5051–5061. 10.1021/jp964020s. [DOI] [Google Scholar]
- Hochtl P.; Boresch S.; Bitomsky W.; Steinhauser O. Rationalization of the dielectric properties of common three-site water models in terms of their force field parameters. J. Chem. Phys. 1998, 109, 4927–4937. 10.1063/1.477104. [DOI] [Google Scholar]
- Fanourgakis G. S.; Xantheas S. S. The flexible, polarizable, thole-type interaction potential for water (TTM2-F) revisited. J. Phys. Chem. A 2006, 110, 4100–4106. 10.1021/jp056477k. [DOI] [PubMed] [Google Scholar]
- Fanourgakis G. S.; Xantheas S. S. Development of transferable interaction potentials for water. V. Extension of the flexible, polarizable, Thole-type model potential (TTM3-F, v. 3.0) to describe the vibrational spectra of water clusters and liquid water. J. Chem. Phys. 2008, 128, 074506. 10.1063/1.2837299. [DOI] [PubMed] [Google Scholar]
- Burnham C. J.; Anick D. J.; Mankoo P. K.; Reiter G. F. The vibrational proton potential in bulk liquid water and ice. J. Chem. Phys. 2008, 128, 154519. 10.1063/1.2895750. [DOI] [PubMed] [Google Scholar]
- Burnham C. J.; Xantheas S. S. Development of transferable interaction models for water. IV. A flexible, all-atom polarizable potential (TTM2-F) based on geometry dependent charges derived from an ab initio monomer dipole moment surface. J. Chem. Phys. 2002, 116, 5115–5124. 10.1063/1.1447904. [DOI] [Google Scholar]
- Burnham C. J.; Xantheas S. S. Development of transferable interaction models for water. III. Reparametrization of an all-atom polarizable rigid model (TTM2-R) from first principles. J. Chem. Phys. 2002, 116, 1500–1510. 10.1063/1.1423942. [DOI] [Google Scholar]
- Hughes Z. E.; Ren E.; Thacker J. C. R.; Symons B. C. B.; Silva A. F.; Popelier P. L. A. A FFLUX water model: flexible, polarizable and with a multipolar description of electrostatics. J. Comput. Chem. 2020, 41, 619–628. 10.1002/jcc.26111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babin V.; Leforestier C.; Paesani F. Development of a ″first principles″ water potential with flexible monomers: dimer potential energy surface, VRT spectrum, and second virial coefficient. J. Chem. Theory Comput. 2013, 9, 5395–5403. 10.1021/ct400863t. [DOI] [PubMed] [Google Scholar]
- Babin V.; Medders G. R.; Paesani F. Development of a ″first principles″ water potential with flexible monomers. II: trimer potential energy surface, third virial coefficient, and small clusters. J. Chem. Theory Comput. 2014, 10, 1599–1607. 10.1021/ct500079y. [DOI] [PubMed] [Google Scholar]
- Medders G. R.; Babin V.; Paesani F. Development of a ″first-principles″ water potential with flexible monomers. III. liquid phase properties. J. Chem. Theory Comput. 2014, 10, 2906–2910. 10.1021/ct5004115. [DOI] [PubMed] [Google Scholar]
- Reddy S. K.; Straight S. C.; Bajaj P.; Huy Pham C.; Riera M.; Moberg D. R.; Morales M. A.; Knight C.; Gotz A. W.; Paesani F. On the accuracy of the MB-pol many-body potential for water: Interaction energies, vibrational frequencies, and classical thermodynamic and dynamical properties from clusters to liquid water and ice. J. Chem. Phys. 2016, 145, 194504. 10.1063/1.4967719. [DOI] [PubMed] [Google Scholar]
- Paesani F.; Bajaj P.; Riera M. Chemical accuracy in modeling halide ion hydration from many-body representations. Adv. Physics-X 2019, 4, 1631212. 10.1080/23746149.2019.1631212. [DOI] [Google Scholar]
- Cisneros G. A.; Wikfeldt K. T.; Ojamäe L.; Lu J. B.; Xu Y.; Torabifard H.; Bartók A. P.; Csányi G.; Molinero V.; Paesani F. Modeling molecular interactions in water: from pairwise to many body potential energy functions. Chem. Rev. 2016, 116, 7501–7528. 10.1021/acs.chemrev.5b00644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demerdash O.; Wang L. P.; Head-Gordon T. Advanced models for water simulations. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018, 8, e1355 10.1002/wcms.1355. [DOI] [Google Scholar]
- Cipcigan F. S.; Sokhan V. P.; Jones A. P.; Crain J.; Martyna G. J. Hydrogen bonding and molecular orientation at the liquid-vapour interface of water. Phys. Chem. Chem. Phys. 2015, 17, 8660–8669. 10.1039/C4CP05506C. [DOI] [PubMed] [Google Scholar]
- Cipcigan F.; Sokhan V.; Martyna G.; Crain J. Structure and hydrogen bonding at the limits of liquid water stability. Sci. Rep. 2018, 8, 1718. 10.1038/s41598-017-18975-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw K. E.; Woods C. J.; Mulholland A. J. Compatibility of quantum chemical methods and empirical (MM) water models in quantum mechanics/molecular mechanics liquid water simulations. J. Phys. Chem. Lett. 2010, 1, 219–223. 10.1021/jz900096p. [DOI] [Google Scholar]
- Gillan M. J.; Alfe D.; Michaelides A. Perspective: How good is DFT for water?. J. Chem. Phys. 2016, 144, 130901. 10.1063/1.4944633. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Lee C. T.; Yang W. T.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Asthagiri D.; Pratt L. R.; Kress J. D. Free energy of liquid water on the basis of quasichemical theory and ab initio molecular dynamics. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top. 2003, 68, 041505 10.1103/PhysRevE.68.041505. [DOI] [PubMed] [Google Scholar]
- Grossman J. C.; Schwegler E.; Draeger E. W.; Gygi F.; Galli G. Towards an assessment of the accuracy of density functional theory for first principles simulations of water. J. Chem. Phys. 2004, 120, 300–311. 10.1063/1.1630560. [DOI] [PubMed] [Google Scholar]
- Kuo I. F. W.; Mundy C. J.; McGrath M. J.; Siepmann J. I.; VandeVondele J.; Sprik M.; Hutter J.; Chen B.; Klein M. L.; Mohamed F.; Krack M.; Parrinello M. Liquid water from first principles: Investigation of different sampling approaches. J. Phys. Chem. B 2004, 108, 12990–12998. 10.1021/jp047788i. [DOI] [Google Scholar]
- Izvekov S.; Voth G. A. Ab initio molecular-dynamics simulation of aqueous proton solvation and transport revisited. J. Chem. Phys. 2005, 123, 044505 10.1063/1.1961443. [DOI] [PubMed] [Google Scholar]
- Mantz Y. A.; Chen B.; Martyna G. J. Structural correlations and motifs in liquid water at selected temperatures: Ab initio and empirical model predictions. J. Phys. Chem. B 2006, 110, 3540–3554. 10.1021/jp054789h. [DOI] [PubMed] [Google Scholar]
- Rempe S. B.; Mattsson T. R.; Leung K. On ″the complete basis set limit″ and plane-wave methods in first-principles simulations water. Phys. Chem. Chem. Phys. 2008, 10, 4685–4687. 10.1039/b810017a. [DOI] [PubMed] [Google Scholar]
- Allesch M.; Schwegler E.; Gygi F.; Galli G. A first principles simulation of rigid water. J. Chem. Phys. 2004, 120, 5192–5198. 10.1063/1.1647529. [DOI] [PubMed] [Google Scholar]
- Leung K.; Rempe S. B. Ab initio rigid water: Effect on water structure, ion hydration, and thermodynamics. Phys. Chem. Chem. Phys. 2006, 8, 2153–2162. 10.1039/b515126k. [DOI] [PubMed] [Google Scholar]
- Coudert F. X.; Vuilleumier R.; Boutin A. Dipole moment, hydrogen bonding and IR spectrum of confined water. ChemPhysChem 2006, 7, 2464–2467. 10.1002/cphc.200600561. [DOI] [PubMed] [Google Scholar]
- Cicero G.; Grossman J. C.; Schwegler E.; Gygi F.; Galli G. Water confined in nanotubes and between graphene sheets: A first principle study. J. Am. Chem. Soc. 2008, 130, 1871–1878. 10.1021/ja074418+. [DOI] [PubMed] [Google Scholar]
- Munoz-Santiburcio D.; Wittekindt C.; Marx D. Nanoconfinement effects on hydrated excess protons in layered materials. Nat. Commun. 2013, 4, 2349. 10.1038/ncomms3349. [DOI] [PubMed] [Google Scholar]
- Mann D. J.; Halls M. D. Water alignment and proton conduction inside carbon nanotubes. Phys. Rev. Lett. 2003, 90, 195503. 10.1103/PhysRevLett.90.195503. [DOI] [PubMed] [Google Scholar]
- Dellago C.; Naor M. M.; Hummer G. Proton transport through water-filled carbon nanotubes. Phys. Rev. Lett. 2003, 90, 105902. 10.1103/PhysRevLett.90.105902. [DOI] [PubMed] [Google Scholar]
- Todorovic M.; Bowler D. R.; Gillan M. J.; Miyazaki T. Density-functional theory study of gramicidin A ion channel geometry and electronic properties. J. R. Soc., Interface 2013, 10, 20130547. 10.1098/rsif.2013.0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad K V.; Kannam S. K.; Hartkamp R.; Sathian S. P. Water desalination using graphene nanopores: influence of the water models used in simulations. Phys. Chem. Chem. Phys. 2018, 20, 16005–16011. 10.1039/C8CP00919H. [DOI] [PubMed] [Google Scholar]
- Guerrero-Aviles R.; Orellana W. Energetics and diffusion of liquid water and hydrated ions through nanopores in graphene: ab initio molecular dynamics simulation. Phys. Chem. Chem. Phys. 2017, 19, 20551–20558. 10.1039/C7CP03449K. [DOI] [PubMed] [Google Scholar]
- Klesse G.; Rao S.; Tucker S. J.; Sansom M. S. P. Induced polarization in molecular dynamics simulations of the 5-HT3 receptor channel. J. Am. Chem. Soc. 2020, 142, 9415–9427. 10.1021/jacs.0c02394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breed J.; Sankararamakrishnan R.; Kerr I. D.; Sansom M. S. P. Molecular dynamics simulations of water within models of transbilayer pores. Biophys. J. 1996, 70, 1643–1661. 10.1016/S0006-3495(96)79727-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom M. S. P.; Kerr I. D.; Breed J.; Sankararamakrishnan R. Water in channel-like cavities: structure and dynamics. Biophys. J. 1996, 70, 693–702. 10.1016/S0006-3495(96)79609-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom M. S. P.; Smith G. R.; Adcock C.; Biggin P. C. The dielectric properties of water within model transbilayer pores. Biophys. J. 1997, 73, 2404–2415. 10.1016/S0006-3495(97)78269-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoustra M. Water at interfaces (editorial). Phys. Chem. Chem. Phys. 2008, 10, 4676–4677. 10.1039/b812223g. [DOI] [PubMed] [Google Scholar]
- Beckstein O.; Sansom M. S. P. Liquid–vapor oscillations of water in hydrophobic nanopores. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 7063–7068. 10.1073/pnas.1136844100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasaiah J. C.; Garde S.; Hummer G. Water in nonpolar confinement: from nanotubes to proteins and beyond. Annu. Rev. Phys. Chem. 2008, 59, 713–740. 10.1146/annurev.physchem.59.032607.093815. [DOI] [PubMed] [Google Scholar]
- Vaitheeswaran S.; Yin H.; Rasaiah J. C.; Hummer G. Water clusters in nonpolar cavities. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 17002–17005. 10.1073/pnas.0407968101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins M. D.; Hummer G.; Quillin M. L.; Matthews B. W.; Gruner S. M. Cooperative water filling of a nonpolar protein cavity observed by high-pressure crystallography and simulation. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 16668–16671. 10.1073/pnas.0508224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H.; Hummer G.; Rasaiah J. C. Metastable water clusters in the nonpolar cavities of the thermostable protein tetrabrachion. J. Am. Chem. Soc. 2007, 129, 7369–7377. 10.1021/ja070456h. [DOI] [PubMed] [Google Scholar]
- Remsing R. C.; Xi E.; Vembanur S.; Sharma S.; Debenedetti P. G.; Garde S.; Patel A. J. Pathways to dewetting in hydrophobic confinement. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 8181–8186. 10.1073/pnas.1503302112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qvist J.; Davidovic M.; Hamelberg D.; Halle B. A dry ligand-binding cavity in a solvated protein. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 6296–6301. 10.1073/pnas.0709844105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal T. A.; Goddard W. A.; Jung Y. Entropy and the driving force for the filling of carbon nanotubes with water. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 11794–11798. 10.1073/pnas.1108073108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayer M. D.; Levinger N. E. Analysis of water in confined geometries and at interfaces. Annu. Rev. Anal. Chem. 2010, 3, 89–107. 10.1146/annurev-anchem-070109-103410. [DOI] [PubMed] [Google Scholar]
- Mittal J.; Hummer G. Static and dynamic correlations in water at hydrophobic interfaces. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 20130–20135. 10.1073/pnas.0809029105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith J.; Martens C. C. Pressure-induced water flow through model nanopores. Phys. Chem. Chem. Phys. 2009, 11, 528–533. 10.1039/B807823H. [DOI] [PubMed] [Google Scholar]
- Schaschke C.A Dictionary of Chemical Engineering; Oxford University Press: Oxford, 2014. [Google Scholar]
- Leung K.; Rempe S. B. Ion rejection by nanoporous membranes in pressure-driven molecular dynamics simulations. J. Comput. Theor. Nanosci. 2009, 6, 1948–1955. 10.1166/jctn.2009.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung K. Ion-dipole interactions are asymptotically unscreened by water in dipolar nanopores, yielding patterned ion distributions. J. Am. Chem. Soc. 2008, 130, 1808–1809. 10.1021/ja076229x. [DOI] [PubMed] [Google Scholar]
- Kofinger J.; Dellago C. Microscopic properties of nanopore water from its time-dependent dielectric response. Phys. Rev. B: Condens. Matter Mater. Phys. 2010, 82, 205416. 10.1103/PhysRevB.82.205416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detcheverry F.; Bocquet L. Thermal fluctuations of hydrodynamic flows in nanochannels. Phys. Rev. E 2013, 88, 012106 10.1103/PhysRevE.88.012106. [DOI] [PubMed] [Google Scholar]
- Richard R.; Anthony S.; Aziz G. Pressure-driven molecular dynamics simulations of water transport through a hydrophilic nanochannel. Mol. Phys. 2016, 114, 2655–2663. 10.1080/00268976.2016.1170219. [DOI] [Google Scholar]
- Tang D.; Yoo Y. E.; Kim D. Molecular dynamics simulations on water permeation through hourglass-shaped nanopores with varying pore geometry. Chem. Phys. 2015, 453, 13–19. 10.1016/j.chemphys.2015.04.002. [DOI] [Google Scholar]
- Shahbabaei M.; Kim D. Effect of hourglass-shaped nanopore length on osmotic water transport. Chem. Phys. 2016, 477, 24–31. 10.1016/j.chemphys.2016.08.005. [DOI] [Google Scholar]
- Han C.; Tang D.; Kim D. Molecular dynamics simulation on the effect of pore hydrophobicity on water transport through aquaporin-mimic nanopores. Colloids Surf., A 2015, 481, 38–42. 10.1016/j.colsurfa.2015.04.012. [DOI] [Google Scholar]
- Liu J.; Wang M.; Chen S. Y.; Robbins M. O. Molecular simulations of electroosmotic flows in rough nanochannels. J. Comput. Phys. 2010, 229, 7834–7847. 10.1016/j.jcp.2010.06.042. [DOI] [Google Scholar]
- Lu P. F.; Liu X. D.; Zhang C. B. Electroosmotic flow in a rough nanochannel with surface roughness characterized by fractal cantor. Micromachines 2017, 8, 190. 10.3390/mi8060190. [DOI] [Google Scholar]
- Thompson A. P. Nonequilibrium molecular dynamics simulation of electro-osmotic flow in a charged nanopore. J. Chem. Phys. 2003, 119, 7503–7511. 10.1063/1.1609194. [DOI] [Google Scholar]
- Yoshida H.; Mizuno H.; Kinjo T.; Washizu H.; Barrat J. L. Molecular dynamics simulation of electrokinetic flow of an aqueous electrolyte solution in nanochannels. J. Chem. Phys. 2014, 140, 214701. 10.1063/1.4879547. [DOI] [PubMed] [Google Scholar]
- Schaaf C.; Gekle S. Spatially resolved dielectric constant of confined water and its connection to the non-local nature of bulk water. J. Chem. Phys. 2016, 145, 084901 10.1063/1.4960775. [DOI] [PubMed] [Google Scholar]
- Yamashita K.; Daiguji H. Coarse-grained molecular dynamics simulations of capillary evaporation of water confined in hydrophilic mesopores. Mol. Phys. 2016, 114, 884–894. 10.1080/00268976.2015.1133858. [DOI] [Google Scholar]
- Orsi M. Comparative assessment of the ELBA coarse-grained model for water. Mol. Phys. 2014, 112, 1566–1576. 10.1080/00268976.2013.844373. [DOI] [Google Scholar]
- Ding W.; Palaiokostas M.; Orsi M. Stress testing the ELBA water model. Mol. Simul. 2016, 42, 337–346. 10.1080/08927022.2015.1047367. [DOI] [Google Scholar]
- Kashiwagi K.; Suh D.; Hwang J.; Hsu W. L.; Daiguji H. Molecular simulations of water adsorption and transport in mesopores with varying hydrophilicity arrangements. Nanoscale 2018, 10, 11657–11669. 10.1039/C8NR02016G. [DOI] [PubMed] [Google Scholar]
- Wu K. L.; Chen Z. X.; Li J.; Xu J. Z.; Wang K.; Li R.; Wang S. H.; Dong X. H. Ultrahigh water flow enhancement by optimizing nanopore chemistry and geometry. Langmuir 2019, 35, 8867–8873. 10.1021/acs.langmuir.9b01179. [DOI] [PubMed] [Google Scholar]
- Gravelle S.; Joly L.; Detcheverry F.; Ybert C.; Cottin-Bizonne C.; Bocquet L. Optimizing water permeability through the hourglass shape of aquaporins. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 16367–16372. 10.1073/pnas.1306447110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravelle S.; Joly L.; Ybert C.; Bocquet L. Large permeabilities of hourglass nanopores: From hydrodynamics to single file transport. J. Chem. Phys. 2014, 141, 18C526. 10.1063/1.4897253. [DOI] [PubMed] [Google Scholar]
- Shaat M.; Zheng Y. M. Fluidity and phase transitions of water in hydrophobic and hydrophilic nanotubes. Sci. Rep. 2019, 9, 5689. 10.1038/s41598-019-42101-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaragoza A.; Gonzalez M. A.; Joly L.; Lopez-Montero I.; Canales M. A.; Benavides A. L.; Valeriani C. Molecular dynamics study of nanoconfined TIP4P/2005 water: how confinement and temperature affect diffusion and viscosity. Phys. Chem. Chem. Phys. 2019, 21, 13653–13667. 10.1039/C9CP02485A. [DOI] [PubMed] [Google Scholar]
- Wang H.; Su Y. L.; Wang W. D.; Sheng G. L.; Li H.; Zafar A. Enhanced water flow and apparent viscosity model considering wettability and shape effects. Fuel 2019, 253, 1351–1360. 10.1016/j.fuel.2019.05.098. [DOI] [Google Scholar]
- Wu K. L.; Chen Z. X.; Li J.; Li X. F.; Xu J. Z.; Dong X. H. Wettability effect on nanoconfined water flow. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 3358–3363. 10.1073/pnas.1612608114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H. J.; Wang Y. Y.; Fan Y. Q.; Xu J. B.; Yang C. Structure and transport properties of water and hydrated ions in nano-confined channels. Adv. Theor. Simul. 2019, 2, 1900016. 10.1002/adts.201900016. [DOI] [Google Scholar]
- Ruiz-Barragan S.; Munoz-Santiburcio D.; Marx D. Nanoconfined water within graphene slit pores adopts distinct confinement-dependent regimes. J. Phys. Chem. Lett. 2019, 10, 329–334. 10.1021/acs.jpclett.8b03530. [DOI] [PubMed] [Google Scholar]
- Hummer G.; Rasaiah J. C.; Noworyta J. P. Water conduction through the hydrophobic channel of a carbon nanotube. Nature 2001, 414, 188–190. 10.1038/35102535. [DOI] [PubMed] [Google Scholar]
- Berne B. J.; Weeks J. D.; Zhou R. H. Dewetting and hydrophobic interaction in physical and biological systems. Annu. Rev. Phys. Chem. 2009, 60, 85–103. 10.1146/annurev.physchem.58.032806.104445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckstein O.; Biggin P. C.; Bond P. J.; Bright J. N.; Domene C.; Grottesi A.; Holyoake J.; Sansom M. S. P. Ion channel gating: insights via molecular simulations. FEBS Lett. 2003, 555, 85–90. 10.1016/S0014-5793(03)01151-7. [DOI] [PubMed] [Google Scholar]
- Aryal P.; Sansom M. S. P.; Tucker S. J. Hydrophobic gating in ion channels. J. Mol. Biol. 2015, 427, 121–130. 10.1016/j.jmb.2014.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trick J. L.; Aryal P.; Tucker S. J.; Sansom M. S. P. Molecular simulation studies of hydrophobic gating in nanopores and ion channels. Biochem. Soc. Trans. 2015, 43, 146–150. 10.1042/BST20140256. [DOI] [PubMed] [Google Scholar]
- Beckstein O.; Biggin P. C.; Sansom M. S. P. A hydrophobic gating mechanism for nanopores. J. Phys. Chem. B 2001, 105, 12902–12905. 10.1021/jp012233y. [DOI] [Google Scholar]
- Beckstein O.; Tai K.; Sansom M. S. P. Not ions alone: barriers to ion permeation in nanopores and channels. J. Am. Chem. Soc. 2004, 126, 14694–14695. 10.1021/ja045271e. [DOI] [PubMed] [Google Scholar]
- Beckstein O.; Sansom M. S. P. The influence of geometry, surface character and flexibility on the permeation of ions and water through biological pores. Phys. Biol. 2004, 1, 42–52. 10.1088/1478-3967/1/1/005. [DOI] [PubMed] [Google Scholar]
- Peter C.; Hummer G. Ion transport through membrane-spanning nanopores studied by molecular dynamics simulations and continuum electrostatics calculations. Biophys. J. 2005, 89, 2222–2234. 10.1529/biophysj.105.065946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen R.; Hansen J. P.; Melchionna S. Molecular dynamics investigation of water permeation through nanopores. J. Chem. Phys. 2003, 119, 3905–3919. 10.1063/1.1590956. [DOI] [Google Scholar]
- Roth R.; Gillespie D.; Nonner W.; Eisenberg R. Bubbles, gating, and anaesthetics in ion channels. Biophys. J. 2008, 94, 4282–4298. 10.1529/biophysj.107.120493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S.; Klesse G.; Stansfeld P. J.; Tucker S. J.; Sansom M. S. P. A heuristic derived from analysis of the ion channel structural proteome permits the rapid identification of hydrophobic gates. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 13989–13995. 10.1073/pnas.1902702116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzubiella J.; Allen R. J.; Hansen J. P. Electric field-controlled water permeation coupled to ion transport through a nanopore. J. Chem. Phys. 2004, 120, 5001–5004. 10.1063/1.1665656. [DOI] [PubMed] [Google Scholar]
- Dzubiella J.; Hansen J. P. Electric-field-controlled water and ion permeation of a hydrophobic nanopore. J. Chem. Phys. 2005, 122, 234706. 10.1063/1.1927514. [DOI] [PubMed] [Google Scholar]
- Vaitheeswaran S.; Rasaiah J. C.; Hummer G. Electric field and temperature effects on water in the narrow nonpolar pores of carbon nanotubes. J. Chem. Phys. 2004, 121, 7955–7965. 10.1063/1.1796271. [DOI] [PubMed] [Google Scholar]
- Wallqvist A.; Berne B. J. Computer-simulation of hydrophobic hydration forces on stacked plates at short-range. J. Phys. Chem. 1995, 99, 2893–2899. 10.1021/j100009a053. [DOI] [Google Scholar]
- Luzar A.; Leung K. Dynamics of capillary evaporation. I. Effect of morphology of hydrophobic surfaces. J. Chem. Phys. 2000, 113, 5836–5844. 10.1063/1.1290478. [DOI] [Google Scholar]
- Leung K.; Luzar A. Dynamics of capillary evaporation. II. Free energy barriers. J. Chem. Phys. 2000, 113, 5845–5852. 10.1063/1.1290479. [DOI] [Google Scholar]
- Leung K.; Luzar A.; Bratko D. Dynamics of capillary drying in water. Phys. Rev. Lett. 2003, 90, 065502 10.1103/PhysRevLett.90.065502. [DOI] [PubMed] [Google Scholar]
- Rant U. Water flow at the flip of a switch. Nat. Nanotechnol. 2011, 6, 759–760. 10.1038/nnano.2011.215. [DOI] [PubMed] [Google Scholar]
- Smirnov S. N.; Vlassiouk I. V.; Lavrik N. V. Voltage-gated hydrophobic nanopores. ACS Nano 2011, 5, 7453–7461. 10.1021/nn202392d. [DOI] [PubMed] [Google Scholar]
- Powell M. R.; Cleary L.; Davenport M.; Shea K. J.; Siwy Z. S. Electric-field-induced wetting and dewetting in single hydrophobic nanopores. Nat. Nanotechnol. 2011, 6, 798–802. 10.1038/nnano.2011.189. [DOI] [PubMed] [Google Scholar]
- Daub C. D.; Bratko D.; Leung K.; Luzar A. Electrowetting at the nanoscale. J. Phys. Chem. C 2007, 111, 505–509. 10.1021/jp067395e. [DOI] [Google Scholar]
- Song F. H.; Li B. Q.; Liu C. Molecular dynamics simulation of nanosized water droplet spreading in an electric field. Langmuir 2013, 29, 4266–4274. 10.1021/la304763a. [DOI] [PubMed] [Google Scholar]
- Bratko D.; Daub C. D.; Leung K.; Luzar A. Effect of field direction on electrowetting in a nanopore. J. Am. Chem. Soc. 2007, 129, 2504–2510. 10.1021/ja0659370. [DOI] [PubMed] [Google Scholar]
- Bratko D.; Daub C. D.; Luzar A. Field-exposed water in a nanopore: liquid or vapour?. Phys. Chem. Chem. Phys. 2008, 10, 6807–6813. 10.1039/b809072f. [DOI] [PubMed] [Google Scholar]
- Vanzo D.; Bratko D.; Luzar A. Dynamic control of nanopore wetting in water and saline solutions under an electric field. J. Phys. Chem. B 2015, 119, 8890–8899. 10.1021/jp506389p. [DOI] [PubMed] [Google Scholar]
- Moucka F.; Bratko D.; Luzar A. Salt and water uptake in nanoconfinement under applied electric field: an open ensemble Monte Carlo study. J. Phys. Chem. C 2015, 119, 20416–20425. 10.1021/acs.jpcc.5b04725. [DOI] [Google Scholar]
- Celebi A. T.; Barisik M.; Beskok A. Electric field controlled transport of water in graphene nano-channels. J. Chem. Phys. 2017, 147, 164311. 10.1063/1.4996210. [DOI] [PubMed] [Google Scholar]
- Shafiei M.; von Domaros M.; Bratko D.; Luzar A. Anisotropic structure and dynamics of water under static electric fields. J. Chem. Phys. 2019, 150, 074505 10.1063/1.5079393. [DOI] [PubMed] [Google Scholar]
- Shafiei M.; Ojaghlou N.; Zamfir S. G.; Bratko D.; Luzar A. Modulation of structure and dynamics of water under alternating electric field and the role of hydrogen bonding. Mol. Phys. 2019, 117, 3282–3296. 10.1080/00268976.2019.1651919. [DOI] [Google Scholar]
- Moucka F.; Zamfir S.; Bratko D.; Luzar A. Molecular polarizability in open ensemble simulations of aqueous nanoconfinements under electric field. J. Chem. Phys. 2019, 150, 164702. 10.1063/1.5094170. [DOI] [PubMed] [Google Scholar]
- Sheng J. D.; Zhu Q.; Zeng X.; Yang Z. H.; Zhang X. H. Promotion of water channels for enhanced ion transport in 14 nm diameter carbon nanotubes. ACS Appl. Mater. Interfaces 2017, 9, 11009–11015. 10.1021/acsami.7b00174. [DOI] [PubMed] [Google Scholar]
- Trick J. L.; Song C.; Wallace E. J.; Sansom M. S. P. Voltage gating of a biomimetic nanopore: electrowetting of a hydrophobic barrier. ACS Nano 2017, 11, 1840–1847. 10.1021/acsnano.6b07865. [DOI] [PubMed] [Google Scholar]
- Dujardin E.; Ebbesen T. W.; Hiura H.; Tanigaki K. Capillarity and wetting of carbon nanotubes. Science 1994, 265, 1850–1852. 10.1126/science.265.5180.1850. [DOI] [PubMed] [Google Scholar]
- Gordillo M. C.; Marti J. Hydrogen bond structure of liquid water confined in nanotubes. Chem. Phys. Lett. 2000, 329, 341–345. 10.1016/S0009-2614(00)01032-0. [DOI] [Google Scholar]
- Alexiadis A.; Kassinos S. Molecular simulation of water in carbon nanotubes. Chem. Rev. 2008, 108, 5014–5034. 10.1021/cr078140f. [DOI] [PubMed] [Google Scholar]
- Geng J.; Kim K.; Zhang J. F.; Escalada A.; Tunuguntla R.; Comolli L. R.; Allen F. I.; Shnyrova A. V.; Cho K. R.; Munoz D.; Wang Y. M.; Grigoropoulos C. P.; Ajo-Franklin C. M.; Frolov V. A.; Noy A. Stochastic transport through carbon nanotubes in lipid bilayers and live cell membranes. Nature 2014, 514, 612–615. 10.1038/nature13817. [DOI] [PubMed] [Google Scholar]
- Tunuguntla R. H.; Escalada A.; Frolov V. A.; Noy A. Synthesis, lipid membrane incorporation, and ion permeability testing of carbon nanotube porins. Nat. Protoc. 2016, 11, 2029–2047. 10.1038/nprot.2016.119. [DOI] [PubMed] [Google Scholar]
- Tunuguntla R. H.; Henley R. Y.; Yao Y. C.; Pham T. A.; Wanunu M.; Noy A. Enhanced water permeability and tunable ion selectivity in subnanometer carbon nanotube porins. Science 2017, 357, 792–796. 10.1126/science.aan2438. [DOI] [PubMed] [Google Scholar]
- Cao D.; Pang P.; He J.; Luo T.; Park J. H.; Krstic P.; Nuckolls C.; Tang J. Y.; Lindsay S. Electronic sensitivity of carbon nanotubes to internal water wetting. ACS Nano 2011, 5, 3113–3119. 10.1021/nn200251z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofinger J.; Hummer G.; Dellago C. Macroscopically ordered water in nanopores. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 13218–13222. 10.1073/pnas.0801448105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waghe A.; Rasaiah J. C.; Hummer G. Entropy of single-file water in (6,6) carbon nanotubes. J. Chem. Phys. 2012, 137, 044709. 10.1063/1.4737842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofinger J.; Hummer G.; Dellago C. Single-file water in nanopores. Phys. Chem. Chem. Phys. 2011, 13, 15403–15417. 10.1039/c1cp21086f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Fandino R.; Pineiro A.; Trick J. L.; Sansom M. S. P. Lipid bilayer membrane perturbation by embedded nanopores: A simulation study. ACS Nano 2016, 10, 3693–3701. 10.1021/acsnano.6b00202. [DOI] [PubMed] [Google Scholar]
- Qin X. C.; Yuan Q. Z.; Zhao Y. P.; Xie S. B.; Liu Z. F. Measurement of the rate of water translocation through carbon nanotubes. Nano Lett. 2011, 11, 2173–2177. 10.1021/nl200843g. [DOI] [PubMed] [Google Scholar]
- Wang L. Y.; Dumont R. S.; Dickson J. M. Nonequilibrium molecular dynamics simulation of water transport through carbon nanotube membranes at low pressure. J. Chem. Phys. 2012, 137, 044102 10.1063/1.4734484. [DOI] [PubMed] [Google Scholar]
- Liu L.; Patey G. N. A molecular dynamics investigation of the influence of water structure on ion conduction through a carbon nanotube. J. Chem. Phys. 2017, 146, 074502. 10.1063/1.4975690. [DOI] [PubMed] [Google Scholar]
- Thomas M.; Corry B. Thermostat choice significantly influences water flow rates in molecular dynamics studies of carbon nanotubes. Microfluid. Nanofluid. 2015, 18, 41–47. 10.1007/s10404-014-1406-y. [DOI] [Google Scholar]
- Shaat M. Viscosity of water interfaces with hydrophobic nanopores: application to water flow in carbon nanotubes. Langmuir 2017, 33, 12814–12819. 10.1021/acs.langmuir.7b02752. [DOI] [PubMed] [Google Scholar]
- Sam A.; Prasad K V.; Sathian S. P. Water flow in carbon nanotubes: the role of tube chirality. Phys. Chem. Chem. Phys. 2019, 21, 6566–6573. 10.1039/C9CP00429G. [DOI] [PubMed] [Google Scholar]
- Wan R. Z.; Li J. Y.; Lu H. J.; Fang H. P. Controllable water channel gating of nanometer dimensions. J. Am. Chem. Soc. 2005, 127, 7166–7170. 10.1021/ja050044d. [DOI] [PubMed] [Google Scholar]
- Mendonca B. H. S.; de Freitas D. N.; Kohler M. H.; Batista R. J. C.; Barbosa M. C.; de Oliveira A. B. Diffusion behaviour of water confined in deformed carbon nanotubes. Phys. A 2019, 517, 491–498. 10.1016/j.physa.2018.11.042. [DOI] [Google Scholar]
- Li J. Y.; Gong X. J.; Lu H. J.; Li D.; Fang H. P.; Zhou R. H. Electrostatic gating of a nanometer water channel. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 3687–3692. 10.1073/pnas.0604541104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J. W.; Ding H. M.; Ren C. L.; Ma Y. Q. Pumping of water by rotating chiral carbon nanotube. Nanoscale 2014, 6, 13606–13612. 10.1039/C4NR03407D. [DOI] [PubMed] [Google Scholar]
- Garate J. A.; English N. J.; MacElroy J. M. D. Carbon nanotube assisted water self-diffusion across lipid membranes in the absence and presence of electric fields. Mol. Simul. 2009, 35, 3–12. 10.1080/08927020802353491. [DOI] [PubMed] [Google Scholar]
- Figueras L.; Faraudo J. Competition between hydrogen bonding and electric field in single-file transport of water in carbon nanotubes. Mol. Simul. 2012, 38, 23–25. 10.1080/08927022.2011.599032. [DOI] [Google Scholar]
- Zhang Q. L.; Yang R. Y. Fast transport of water molecules across carbon nanotubes induced by static electric fields. Chem. Phys. Lett. 2016, 644, 201–204. 10.1016/j.cplett.2015.11.061. [DOI] [Google Scholar]
- Ritos K.; Borg M. K.; Mottram N. J.; Reese J. M. Electric fields can control the transport of water in carbon nanotubes. Philos. Trans. R. Soc., A 2016, 374, 20150025. 10.1098/rsta.2015.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kou J. L.; Yao J.; Lu H. J.; Zhang B.; Li A. F.; Sun Z. X.; Zhang J. G.; Fang Y. Z.; Wu F. M.; Fan J. T. Electromanipulating water flow in nanochannels. Angew. Chem., Int. Ed. 2015, 54, 2351–2355. 10.1002/anie.201408633. [DOI] [PubMed] [Google Scholar]
- Kalra A.; Garde S.; Hummer G. Osmotic water transport through carbon nanotube membranes. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 10175–10180. 10.1073/pnas.1633354100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z. M.; Luo Y.; Ma J. P.; Wei G. H. Phase transition of nanotube-confined water driven by electric field. J. Chem. Phys. 2011, 134, 154507. 10.1063/1.3579482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z. Y.; Fu Z. M.; Wei G. H. Influence of electric fields on the structure and structure transition of water confined in a carbon nanotube. J. Chem. Phys. 2014, 140, 154508. 10.1063/1.4871625. [DOI] [Google Scholar]
- Winarto; Takaiwa D.; Yamamoto E.; Yasuoka K. Structures of water molecules in carbon nanotubes under electric fields. J. Chem. Phys. 2015, 142, 124701. 10.1063/1.4914462. [DOI] [PubMed] [Google Scholar]
- Winarto; Yamamoto E.; Yasuoka K. Water molecules in a carbon nanotube under an applied electric field at various temperatures and pressures. Water 2017, 9, 473. 10.3390/w9070473. [DOI] [Google Scholar]
- Winarto; Yamamoto E.; Yasuoka K. Separation of water-alcohol mixtures using carbon nanotubes under an electric field. Phys. Chem. Chem. Phys. 2019, 21, 15431–15438. 10.1039/C9CP01799B. [DOI] [PubMed] [Google Scholar]
- Garcia-Fandiño R.; Sansom M. S. P. Designing biomimetic pores based on carbon nanotubes. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 6939–6944. 10.1073/pnas.1119326109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artificial Water Channels, Faraday Discussions 209; Barboiu M., Eds.; Royal Society of Chemistry, 2018; pp 450. [Google Scholar]
- Corry B. Designing carbon nanotube membranes for efficient water desalination. J. Phys. Chem. B 2008, 112, 1427–1434. 10.1021/jp709845u. [DOI] [PubMed] [Google Scholar]
- Corry B. Water and ion transport through functionalised carbon nanotubes: implications for desalination technology. Energy Environ. Sci. 2011, 4, 751–759. 10.1039/c0ee00481b. [DOI] [Google Scholar]
- Thomas M.; Corry B. A computational assessment of the permeability and salt rejection of carbon nanotube membranes and their application to water desalination. Philos. Trans. R. Soc., A 2016, 374, 20150020. 10.1098/rsta.2015.0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M.; Corry B.; Hilder T. A. What have we learnt about the mechanisms of rapid water transport, ion rejection and selectivity in nanopores from molecular simulation?. Small 2014, 10, 1453–1465. 10.1002/smll.201302968. [DOI] [PubMed] [Google Scholar]
- Su Z. L.; Chen J. Y.; Zhao Y. Z.; Su J. Y. How ions block the single-file water transport through a carbon nanotube. Phys. Chem. Chem. Phys. 2019, 21, 11298–11305. 10.1039/C9CP01714C. [DOI] [PubMed] [Google Scholar]
- Renou R.; Szymczyk A.; Maurin G.; Malfreyt P.; Ghoufi A. Superpermittivity of nanoconfined water. J. Chem. Phys. 2015, 142, 184706. 10.1063/1.4921043. [DOI] [PubMed] [Google Scholar]
- Sirkin Y. A. P.; Hassanali A.; Scherlis D. A. One-dimensional confinement inhibits water dissociation in carbon nanotubes. J. Phys. Chem. Lett. 2018, 9, 5029–5033. 10.1021/acs.jpclett.8b02183. [DOI] [PubMed] [Google Scholar]
- Desgranges C.; Delhommelle J. Stabilization of nanobubbles under hydrophobic confinement. J. Phys. Chem. C 2019, 123, 11707–11713. 10.1021/acs.jpcc.9b01440. [DOI] [Google Scholar]
- Shen Y.-x.; Si W.; Erbakan M.; Decker K.; De Zorzi R.; Saboe P. O.; Kang Y. J.; Majd S.; Butler P. J.; Walz T.; Aksimentiev A.; Hou J.-l.; Kumar M. Highly permeable artificial water channels that can self-assemble into two-dimensional arrays. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 9810–9815. 10.1073/pnas.1508575112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W.; Shen Y. X.; Lang C.; Saha P.; Zenyuk I.; Hickey R. J.; Kumar M. Unique selectivity trends of highly permeable PAP 5 water channel membranes. Faraday Discuss. 2018, 209, 193–204. 10.1039/C8FD00043C. [DOI] [PubMed] [Google Scholar]
- Song W.; Joshi H.; Chowdhury R.; Najem J. S.; Shen Y.-x.; Lang C.; Henderson C. B.; Tu Y.-M.; Farell M.; Pitz M. E.; Maranas C. D.; Cremer P. S.; Hickey R. J.; Sarles S. A.; Hou J.-l.; Aksimentiev A.; Kumar M. Artificial water channels enable fast and selective water permeation through water-wire networks. Nat. Nanotechnol. 2020, 15, 73. 10.1038/s41565-019-0586-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licsandru E.; Kocsis I.; Shen Y. X.; Murail S.; Legrand Y. M.; van der Lee A.; Tsai D.; Baaden M.; Kumar M.; Barboiu M. Salt-excluding artificial water channels exhibiting enhanced dipolar water and proton translocation. J. Am. Chem. Soc. 2016, 138, 5403–5409. 10.1021/jacs.6b01811. [DOI] [PubMed] [Google Scholar]
- Kocsis I.; Sorci M.; Vanselous H.; Murail S.; Sanders S. E.; Licsandru E.; Legrand Y. M.; van der Lee A.; Baaden M.; Petersen P. B.; Belfort G.; Barboiu M. Oriented chiral water wires in artificial transmembrane channels. Sci. Adv. 2018, 4, eaao5603. 10.1126/sciadv.aao5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murail S.; Vasiliu T.; Neamtu A.; Barboiu M.; Sterpone F.; Baaden M. Water permeation across artificial I-quartet membrane channels: from structure to disorder. Faraday Discuss. 2018, 209, 125–148. 10.1039/C8FD00046H. [DOI] [PubMed] [Google Scholar]
- Jayasinghe A. S.; Payne M. K.; Unruh D. K.; Johns A.; Leddy J.; Forbes T. Z. Diffusion and selectivity of water confined within metal-organic nanotubes. J. Mater. Chem. A 2018, 6, 1531–1539. 10.1039/C7TA06741K. [DOI] [Google Scholar]
- Dai F. N.; He H. Y.; Sun D. F. A metal-organic nanotube exhibiting reversible adsorption of (H2O)12 Cluster. J. Am. Chem. Soc. 2008, 130, 14064–14605. 10.1021/ja805920t. [DOI] [PubMed] [Google Scholar]
- Jiang J. W.; Babarao R.; Hu Z. Q. Molecular simulations for energy, environmental and pharmaceutical applications of nanoporous materials: from zeolites, metal-organic frameworks to protein crystals. Chem. Soc. Rev. 2011, 40, 3599–3612. 10.1039/c0cs00128g. [DOI] [PubMed] [Google Scholar]
- Canivet J.; Fateeva A.; Guo Y. M.; Coasne B.; Farrusseng D. Water adsorption in MOFs: fundamentals and applications. Chem. Soc. Rev. 2014, 43, 5594–5617. 10.1039/C4CS00078A. [DOI] [PubMed] [Google Scholar]
- Rangnekar N.; Mittal N.; Elyassi B.; Caro J.; Tsapatsis M. Zeolite membranes - a review and comparison with MOFs. Chem. Soc. Rev. 2015, 44, 7128–7154. 10.1039/C5CS00292C. [DOI] [PubMed] [Google Scholar]
- Hasan Z.; Jhung S. H. Removal of hazardous organics from water using metal-organic frameworks (MOFs): Plausible mechanisms for selective adsorptions. J. Hazard. Mater. 2015, 283, 329–339. 10.1016/j.jhazmat.2014.09.046. [DOI] [PubMed] [Google Scholar]
- Fasano M.; Humplik T.; Bevilacqua A.; Tsapatsis M.; Chiavazzo E.; Wang E. N.; Asinari P. Interplay between hydrophilicity and surface barriers on water transport in zeolite membranes. Nat. Commun. 2016, 7, 12762. 10.1038/ncomms12762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medders G. R.; Paesani F. Water dynamics in metal-organic frameworks: effects of heterogeneous confinement predicted by computational spectroscopy. J. Phys. Chem. Lett. 2014, 5, 2897–2902. 10.1021/jz5013998. [DOI] [PubMed] [Google Scholar]
- Leung K.; Rempe S. B.; Lorenz C. D. Salt permeation and exclusion in hydroxylated and functionalized silica pores. Phys. Rev. Lett. 2006, 96, 095504 10.1103/PhysRevLett.96.095504. [DOI] [PubMed] [Google Scholar]
- Rempe S. B.; Rogers D. M.; Jiang Y. B.; Yang S.; Leung K.; Brinker C. J.; Lorenz C.; Varma S.; Sabo D.; Chen Z.; Singh S.; Rempe C. S.; Mayer T.; Alam T. M.; Feibelman P. J.; Merson J. Computational and experimental platform for understanding and optimizing water flux and salt rejection in nanoporous membranes. SANDIA Report 2010, SAND2010-6735 10.2172/1008105. [DOI] [Google Scholar]
- Fu Y. Q.; Jiang Y. B.; Dunphy D.; Xiong H. F.; Coker E.; Chou S.; Zhang H. X.; Vanegas J. M.; Croissant J. G.; Cecchi J. L.; Rempe S. B.; Brinker C. J. Ultra-thin enzymatic liquid membrane for CO2 separation and capture. Nat. Commun. 2018, 10.1038/s41467-018-04642-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom M. S. P. The biophysics of peptide models of ion channels. Prog. Biophys. Mol. Biol. 1991, 55, 139–236. 10.1016/0079-6107(91)90004-C. [DOI] [PubMed] [Google Scholar]
- Andersen O. S. Gramicidin channels. Annu. Rev. Physiol. 1984, 46, 531–548. 10.1146/annurev.ph.46.030184.002531. [DOI] [PubMed] [Google Scholar]
- de Groot B. L.; Tieleman D. P.; Pohl P.; Grubmuller H. Water permeation through gramicidin A: Desformylation and the double helix: A molecular dynamics study. Biophys. J. 2002, 82, 2934–2942. 10.1016/S0006-3495(02)75634-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Q. F.; Jiang Q.; Moore P. B.; Newns D. M.; Klein M. L. Molecular dynamics simulation of a synthetic ion channel. Biophys. J. 1998, 74, 3–10. 10.1016/S0006-3495(98)77761-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randa H. S.; Forrest L. R.; Voth G. A.; Sansom M. S. P. Molecular dynamics of synthetic leucine-serine ion channels in a phospholipid membrane. Biophys. J. 1999, 77, 2400–2410. 10.1016/S0006-3495(99)77077-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G. R.; Sansom M. S. P. Free energy of a potassium ion in a model of the channel formed by an amphipathic leucine-serine peptide. Eur. Biophys. J. 2002, 31, 198–206. 10.1007/s00249-002-0206-6. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Voth G. A. A computer simulation study of the hydrated proton in a synthetic proton channel. Biophys. J. 2003, 85, 864–875. 10.1016/S0006-3495(03)74526-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. J.; Ilan B.; Voth G. A. Charge delocalization in proton channels, II: The synthetic LS2 channel and proton selectivity. Biophys. J. 2007, 92, 61–69. 10.1529/biophysj.106.091942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccai N. R.; Chi B.; Thomson A. R.; Boyle A. L.; Bartlett G. J.; Bruning M.; Linden N.; Sessions R. B.; Booth P. J.; Brady R. L.; Woolfson D. N. A de novo peptide hexamer with a mutable channel. Nat. Chem. Biol. 2011, 7, 935–941. 10.1038/nchembio.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess N. C.; Sharp T. H.; Thomas F.; Wood C. W.; Thomson A. R.; Zaccai N. R.; Brady R. L.; Serpell L. C.; Woolfson D. N. Modular design of self-assembling peptide-based nanotubes. J. Am. Chem. Soc. 2015, 137, 10554–10562. 10.1021/jacs.5b03973. [DOI] [PubMed] [Google Scholar]
- Mahendran K. R.; Niitsu A.; Kong L. B.; Thomson A. R.; Sessions R. B.; Woolfson D. N.; Bayley H. A monodisperse transmembrane α-helical peptide barrel. Nat. Chem. 2017, 9, 411–419. 10.1038/nchem.2647. [DOI] [PubMed] [Google Scholar]
- Engels M.; Bashford D.; Ghadiri M. R. Structure and dynamics of self-assembling peptide nanotubes and the channel-mediated water organization and self-diffusion—A molecular-dynamics study. J. Am. Chem. Soc. 1995, 117, 9151–9158. 10.1021/ja00141a005. [DOI] [Google Scholar]
- Liu J.; Fan J. F.; Tang M.; Zhou W. Q. Molecular dynamics simulation for the structure of the water chain in a transmembrane peptide nanotube. J. Phys. Chem. A 2010, 114, 2376–2383. 10.1021/jp910624z. [DOI] [PubMed] [Google Scholar]
- Liu J. A.; Fan J. F.; Tang M.; Cen M.; Yan J. F.; Liu Z.; Zhou W. G. Water diffusion behaviors and transportation properties in transmembrane cyclic hexa-, octa- and decapeptide nanotubes. J. Phys. Chem. B 2010, 114, 12183–12192. 10.1021/jp1039207. [DOI] [PubMed] [Google Scholar]
- Liu J.; Fan J. F.; Cen M.; Song X. Z.; Liu D. Y.; Zhou W. Q.; Liu Z.; Yan J. F. Dependences of water permeation through cyclic octa-peptide nanotubes on channel length and membrane thickness. J. Chem. Inf. Model. 2012, 52, 2132–2138. 10.1021/ci300185c. [DOI] [PubMed] [Google Scholar]
- Liu D. Y.; Fan J. F.; Song X. Z.; Li R.; Li H. MD simulations on the influences of an external force on the water transportation behavior through a cyclic peptide nanotube. Comput. Mater. Sci. 2013, 78, 47–54. 10.1016/j.commatsci.2013.05.013. [DOI] [Google Scholar]
- Comer J.; Dehez F.; Cai W. S.; Chipot C. Water conduction through a peptide nanotube. J. Phys. Chem. C 2013, 117, 26797–26803. 10.1021/jp4088223. [DOI] [Google Scholar]
- Maroli N.; Kolandaivel P. Structure, stability and water permeation of (D-Leu-L-Lys-(D-Gln-L-Ala)(3)) cyclic peptide nanotube: a molecular dynamics study. Mol. Simul. 2018, 44, 225–235. 10.1080/08927022.2017.1366653. [DOI] [Google Scholar]
- Hsieh W. H.; Liaw J. Applications of cyclic peptide nanotubes (cPNTs). J. Food Drug Analysis 2019, 27, 32–47. 10.1016/j.jfda.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvelo M.; Vazquez S.; Garcia-Fandino R. Molecular dynamics simulations for designing biomimetic pores based on internally functionalized self-assembling α,γ-peptide nanotubes. Phys. Chem. Chem. Phys. 2015, 17, 28586–28601. 10.1039/C5CP04200C. [DOI] [PubMed] [Google Scholar]
- Calvelo M.; Granja J. R.; Garcia-Fandino R. Competitive double-switched self-assembled cyclic peptide nanotubes: a dual internal and external control. Phys. Chem. Chem. Phys. 2019, 21, 20750–20756. 10.1039/C9CP02327E. [DOI] [PubMed] [Google Scholar]
- Ruiz L.; Wu Y. Q.; Keten S. Tailoring the water structure and transport in nanotubes with tunable interiors. Nanoscale 2015, 7, 121–132. 10.1039/C4NR05407E. [DOI] [PubMed] [Google Scholar]
- Wu H. C.; Yoshioka T.; Nakagawa K.; Shintani T.; Tsuru T.; Saeki D.; Chen Y. R.; Tung K. L.; Matsuyama H. Water transport and ion rejection investigation for application of cyclic peptide nanotubes to forward osmosis process: A simulation study. Desalination 2017, 424, 85–94. 10.1016/j.desal.2017.09.008. [DOI] [Google Scholar]
- Pevarnik M.; Healy K.; Davenport M.; Yen J.; Siwy Z. S. A hydrophobic entrance enhances ion current rectification and induces dewetting in asymmetric nanopores. Analyst 2012, 137, 2944–2950. 10.1039/c2an16139g. [DOI] [PubMed] [Google Scholar]
- Xiao K.; Zhou Y. H.; Kong X. Y.; Xie G. H.; Li P.; Zhang Z.; Wen L. P.; Jiang L. Electrostatic-charge- and electric-field-induced smart gating for water transportation. ACS Nano 2016, 10, 9703–9709. 10.1021/acsnano.6b05682. [DOI] [PubMed] [Google Scholar]
- Xie G. H.; Li P.; Zhao Z. J.; Zhu Z. P.; Kong X. Y.; Zhang Z.; Xiao K.; Wen L. P.; Jiang L. Light- and electric-field-controlled wetting behavior in nanochannels for regulating nanoconfined mass transport. J. Am. Chem. Soc. 2018, 140, 4552–4559. 10.1021/jacs.7b13136. [DOI] [PubMed] [Google Scholar]
- Wang P. F.; Wang M.; Liu F.; Ding S. Y.; Wang X.; Du G. H.; Liu J.; Apel P.; Kluth P.; Trautmann C.; Wang Y. G. Ultrafast ion sieving using nanoporous polymeric membranes. Nat. Commun. 2018, 9, 569. 10.1038/s41467-018-02941-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothula K. R.; Solano C. J. F.; Kleinekathofer U. Simulations of outer membrane channels and their permeability. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1760–1771. 10.1016/j.bbamem.2015.12.020. [DOI] [PubMed] [Google Scholar]
- Tieleman D. P.; Berendsen H. J. C. A molecular dynamics study of the pores formed by Escherichia coli OmpF porin in a fully hydrated palmitoyloleoylphosphatidylcholine bilayer. Biophys. J. 1998, 74, 2786–2801. 10.1016/S0006-3495(98)77986-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hijkoop V. J.; Dammers A. J.; Malek K.; Coppens M. O. Water diffusion through a membrane protein channel: A first passage time approach. J. Chem. Phys. 2007, 127, 085101 10.1063/1.2761897. [DOI] [PubMed] [Google Scholar]
- Aguilella-Arzo M.; Andrio A.; Aguilella V. M.; Alcaraz A. Dielectric saturation of water in a membrane protein channel. Phys. Chem. Chem. Phys. 2009, 11, 358–365. 10.1039/B812775A. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Hajjar E.; Ruggerone P.; Ceccarelli M. Structural and dynamical properties of the porins OmpF and OmpC: insights from molecular simulations. J. Phys.: Condens. Matter 2010, 22, 454125. 10.1088/0953-8984/22/45/454125. [DOI] [PubMed] [Google Scholar]
- Bhamidimarri S. P.; Prajapati J. D.; van den Berg B.; Winterhalter M.; Kleinekathofer U. Role of electroosmosis in the permeation of neutral molecules: CymA and cyclodextrin as an example. Biophys. J. 2016, 110, 600–611. 10.1016/j.bpj.2015.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdeva S.; Kolimi N.; Nair S. A.; Rathinavelan T. Key diffusion mechanisms involved in regulating bidirectional water permeation across E. coli outer membrane lectin. Sci. Rep. 2016, 6, 28157. 10.1038/srep28157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayley H. Designed membrane channels and pores. Curr. Opin. Biotechnol. 1999, 10, 94–103. 10.1016/S0958-1669(99)80017-2. [DOI] [PubMed] [Google Scholar]
- Song L.; Hobaugh M. R.; Shustak C.; Cheley S.; Bayley H.; Gouaux J. E. Structure of staphylococcal a-hemolysin, a heptameric transmembrane pore. Science 1996, 274, 1859–1866. 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- Howorka S.; Cheley S.; Bayley H. Sequence-specific detection of individual DNA strands using engineered nanopores. Nat. Biotechnol. 2001, 19, 636–639. 10.1038/90236. [DOI] [PubMed] [Google Scholar]
- Bayley H.; Cremer P. S. Stochastic sensors inspired by biology. Nature 2001, 413, 226–230. 10.1038/35093038. [DOI] [PubMed] [Google Scholar]
- Chen M.; Khalid S.; Sansom M. S. P.; Bayley H. Outer membrane protein G: engineering a quiet pore for biosensing. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 6272–6277. 10.1073/pnas.0711561105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton J. A.; Whitehead T. A.; Nanda V. Computational redesign of the lipid-facing surface of the outer membrane protein OmpA. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 9632–9637. 10.1073/pnas.1501836112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongprayoon P.; Beckstein O.; Sansom M. S. P. Biomimetic design of a brush-like nanopore: simulation studies. J. Phys. Chem. B 2012, 116, 462–468. 10.1021/jp206754w. [DOI] [PubMed] [Google Scholar]
- Markosyan S.; De Biase P. M.; Czapla L.; Samoylova O.; Singh G.; Cuervo J.; Tieleman D. P.; Noskov S. Y. Effect of confinement on DNA, solvent and counterion dynamics in a model biological nanopore. Nanoscale 2014, 6, 9006–9016. 10.1039/C3NR06559F. [DOI] [PubMed] [Google Scholar]
- Wong-ekkabut J.; Karttunen M. Molecular dynamics simulation of water permeation through the α-hemolysin channel. J. Biol. Phys. 2016, 42, 133–146. 10.1007/s10867-015-9396-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonome E. L.; Cecconi F.; Chinappi M. Electroosmotic flow through an a-hemolysin nanopore. Microfluid. Nanofluid. 2017, 21, 96. 10.1007/s10404-017-1928-1. [DOI] [Google Scholar]
- Slusky J. S. G. Outer membrane protein design. Curr. Opin. Struct. Biol. 2017, 45, 45–52. 10.1016/j.sbi.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad M. M.; Iyer R.; Howard K. R.; McPike M. P.; Borer P. N.; Movileanu L. Engineering a rigid protein tunnel for biomolecular detection. J. Am. Chem. Soc. 2012, 134, 9521–9531. 10.1021/ja3043646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanganna Gari R. R.; Seelheim P.; Liang B.; Tamm L. K. Quiet outer membrane protein G (OmpG) nanopore for biosensing. ACS Sensors 2019, 4, 1230–1235. 10.1021/acssensors.8b01645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury R.; Ren T. W.; Shankla M.; Decker K.; Grisewood M.; Prabhakar J.; Baker C.; Golbeck J. H.; Aksimentiev A.; Kumar M.; Maranas C. D. PoreDesigner for tuning solute selectivity in a robust and highly permeable outer membrane pore. Nat. Commun. 2018, 9, 3661. 10.1038/s41467-018-06097-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trick J. L.; Wallace E. J.; Bayley H.; Sansom M. S. P. Designing a hydrophobic barrier within biomimetic nanopores. ACS Nano 2014, 8, 11268–11279. 10.1021/nn503930p. [DOI] [PubMed] [Google Scholar]
- Haynes T.; Smith L. P. S.; Wallace E. J.; Trick J. L.; Sansom M. S. P.; Khalid S. Electric-field-driven translocation of ssDNA through hydrophobic nanopores. ACS Nano 2018, 12, 8208–8213. 10.1021/acsnano.8b03365. [DOI] [PubMed] [Google Scholar]
- Roux B.; Schulten K. Computational studies of membrane channels. Structure 2004, 12, 1343–1351. 10.1016/j.str.2004.06.013. [DOI] [PubMed] [Google Scholar]
- de Groot B. L.; Grubmuller H. The dynamics and energetics of water permeation and proton exclusion in aquaporins. Curr. Opin. Struct. Biol. 2005, 15, 176–183. 10.1016/j.sbi.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Wan R. Z.; Fang H. P. Water transportation across narrow channel of nanometer dimension. Solid State Commun. 2010, 150, 968–975. 10.1016/j.ssc.2010.01.016. [DOI] [Google Scholar]
- Hall J. E.; Freites J. A.; Tobias D. J. Experimental and simulation studies of Aquaporin 0 water permeability and regulation. Chem. Rev. 2019, 119, 6015–6039. 10.1021/acs.chemrev.9b00106. [DOI] [PubMed] [Google Scholar]
- de Groot B. L.; Grubmuller H. Water permeation across biological membranes: Mechanism and dynamics of aquaporin-1 and GlpF. Science 2001, 294, 2353–2357. 10.1126/science.1062459. [DOI] [PubMed] [Google Scholar]
- Zhu F. Q.; Tajkhorshid E.; Schulten K. Pressure-induced water transport in membrane channels studied by molecular dynamics. Biophys. J. 2002, 83, 154–160. 10.1016/S0006-3495(02)75157-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F. Q.; Tajkhorshid E.; Schulten K. Theory and simulation of water permeation in aquaporin-1. Biophys. J. 2004, 86, 50–57. 10.1016/S0006-3495(04)74082-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashido M.; Ikeguchi M.; Kidera A. Comparative simulations of aquaporin family: AQP1, AQPZ, AQP0 and GlpF. FEBS Lett. 2005, 579, 5549–5552. 10.1016/j.febslet.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Wambo T. O.; Rodriguez R. A.; Chen L. Y. Computing osmotic permeabilities of aquaporins AQP4, AQP5, and GlpF from near-equilibrium simulations. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 1310–1316. 10.1016/j.bbamem.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen M. O.; Mouritsen O. G. Single-channel water permeabilities of Escherichia coli aquaporins AqpZ and GlpF. Biophys. J. 2006, 90, 2270–2284. 10.1529/biophysj.105.073965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashido M.; Kidera A.; Ikeguchi M. Water transport in aquaporins: osmotic permeability matrix analysis of molecular dynamics simulations. Biophys. J. 2007, 93, 373–385. 10.1529/biophysj.106.101170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamonov A. B.; Coalson R. D.; Zeidel M. L.; Mathai J. C. Water and deuterium oxide permeability through aquaporin 1: MD predictions and experimental verification. J. Gen. Physiol. 2007, 130, 111–116. 10.1085/jgp.200709810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hub J. S.; de Groot B. L. Mechanism of selectvity in aquaporins and aquaglyceroporins. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 1198–1203. 10.1073/pnas.0707662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomes R.; Roux B. Molecular mechanism of H+ conduction in the single-file water chain of the gramicidin channel. Biophys. J. 2002, 82, 2304–2316. 10.1016/S0006-3495(02)75576-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx D. Proton transfer 200 years after von Grotthuss: Insights from ab initio simulations. ChemPhysChem 2006, 7, 1848–1870. 10.1002/cphc.200600128. [DOI] [PubMed] [Google Scholar]
- de Groot B. L.; Frigato T.; Helms V.; Grubmuller H. The mechanism of proton exclusion in the aquaporin-1 water channel. J. Mol. Biol. 2003, 333, 279–293. 10.1016/j.jmb.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Jensen M. O.; Tajkhorshid E.; Schulten K. Electrostatic tuning of permeation and selectivity in aquaporin water channels. Biophys. J. 2003, 85, 2884–2899. 10.1016/S0006-3495(03)74711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burykin A.; Warshel A. What really prevents proton transport through aquaporin? Charge self-energy versus proton wire proposals. Biophys. J. 2003, 85, 3696–3706. 10.1016/S0006-3495(03)74786-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilan B.; Tajkhorshid E.; Schulten K.; Voth G. A. The mechanism of proton exclusion in aquaporin channels. Proteins: Struct., Funct., Genet. 2004, 55, 223–228. 10.1002/prot.20038. [DOI] [PubMed] [Google Scholar]
- Chen H. N.; Wu Y. J.; Voth G. A. Origins of proton transport behavior from selectivity domain mutations of the aquaporin-1 channel. Biophys. J. 2006, 90, L73–L75. 10.1529/biophysj.106.084061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti N.; Roux B.; Pomes R. Structural determinants of proton blockage in aquaporins. J. Mol. Biol. 2004, 343, 493–510. 10.1016/j.jmb.2004.08.036. [DOI] [PubMed] [Google Scholar]
- Jensen M. O.; Rothlisberger U.; Rovira C. Hydroxide and proton migration in aquaporins. Biophys. J. 2005, 89, 1744–1759. 10.1529/biophysj.104.058206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig P. H.; Ghosh N.; Hoffmann M.; Elstner M.; Tajkhorshid E.; Frauenheim T.; Cui Q. Toward theoretical analyis of long-range proton transfer kinetics in biomolecular pumps. J. Phys. Chem. A 2006, 110, 548–563. 10.1021/jp052328q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolin N.; Li B.; Beck D. A. C.; Daggett V. Side-chain dynamics are critical for water permeation through aquaporin-1. Biophys. J. 2008, 95, 1089–1098. 10.1529/biophysj.107.125187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto E.; Akimoto T.; Hirano Y.; Yasui M.; Yasuoka K. 1/f fluctuations of amino acids regulate water transportation in aquaporin 1. Phys. Rev. E 2014, 89, 022718. 10.1103/PhysRevE.89.022718. [DOI] [PubMed] [Google Scholar]
- Ho J. D.; Yeh R.; Sandstrom A.; Chorny I.; Harries W. E. C.; Robbins R. A.; Miercke L. J. W.; Stroud R. M. Crystal structure of human aquaporin 4 at 1.8 angstrom and its mechanism of conductance. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 7437–7442. 10.1073/pnas.0902725106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G.; Kosinska-Eriksson U.; Aponte-Santamaria C.; Palmgren M.; Geijer C.; Hedfalk K.; Hohmann S.; de Groot B. L.; Neutze R.; Lindkvist-Petersson K. Crystal structure of a yeast aquaporin at 1.15 angstrom reveals a novel gating mechanism. PLoS Biol. 2009, 7, e1000130. 10.1371/journal.pbio.1000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin L.; Su H. B.; Nielsen C. H.; Tang C. Y.; Torres J.; Mu Y. G. Water permeation dynamics of AqpZ: A tale of two states. Biochim. Biophys. Acta, Biomembr. 2011, 1808, 1581–1586. 10.1016/j.bbamem.2011.02.001. [DOI] [PubMed] [Google Scholar]
- Hu G. D.; Chen L. Y.; Wang J. H. Insights into the mechanisms of the selectivity filter of Escherichia coli aquaporin Z. J. Mol. Model. 2012, 18, 3731–3741. 10.1007/s00894-012-1379-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G. D.; Qi L. S.; Dou X. H.; Wang J. H. The influences of protonation state of histidine on aromatic/arginine region of aquaporin-1 protein. Mol. Simul. 2013, 39, 261–269. 10.1080/08927022.2012.718438. [DOI] [Google Scholar]
- Janosi L.; Ceccarelli M. The gating mechanism of the human aquaporin 5 revealed by molecular dynamics simulations. PLoS One 2013, 8, e59897. 10.1371/journal.pone.0059897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberga D.; Nicolotti O.; Lattanzi G.; Nicchia G. P.; Frigeri A.; Pisani F.; Benfenati V.; Mangiatordi G. F. A new gating site in human aquaporin-4: Insights from molecular dynamics simulations. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 3052–3060. 10.1016/j.bbamem.2014.08.015. [DOI] [PubMed] [Google Scholar]
- Hub J. S.; Aponte-Santamaria C.; Grubmuller H.; de Groot B. L. Voltage-regulated water flux through aquaporin channels in silico. Biophys. J. 2010, 99, L97–L99. 10.1016/j.bpj.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J. Y.; Guo H. X. Control of unidirectional transport of single-file water molecules through carbon nanotubes in an electric field. ACS Nano 2011, 5, 351–359. 10.1021/nn1014616. [DOI] [PubMed] [Google Scholar]
- Garate J. A.; English N. J.; MacElroy J. M. D. Human aquaporin 4 gating dynamics in dc and ac electric fields: A molecular dynamics study. J. Chem. Phys. 2011, 134, 055110. 10.1063/1.3529428. [DOI] [PubMed] [Google Scholar]
- Reale R.; English N. J.; Garate J. A.; Marracino P.; Liberti M.; Apollonio F. Human aquaporin 4 gating dynamics under and after nanosecond-scale static and alternating electric-field impulses: A molecular dynamics study of field effects and relaxation. J. Chem. Phys. 2013, 139, 205101. 10.1063/1.4832383. [DOI] [PubMed] [Google Scholar]
- Li H.; Fan J. F. F.; Li R.; Yu Y.; Yan X. L. L. Molecular dynamics studies on the influences of a gradient electric field on the water chain in a peptide nanotube. J. Mol. Model. 2014, 20, 2370. 10.1007/s00894-014-2370-x. [DOI] [PubMed] [Google Scholar]
- Marracino P.; Liberti M.; Trapani E.; Burnham C. J.; Avena M.; Garate J. A.; Apollonio F.; English N. J. Human aquaporin 4 gating dynamics under perpendicularly-oriented electric-field impulses: a molecular dynamics study. Int. J. Mol. Sci. 2016, 17, 1133. 10.3390/ijms17071133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi M.; Marracino P.; Ghaani M. R.; Liberti M.; Del Signore F.; Burnham C. J.; Garate J. A.; Apollonio F.; English N. J. Human aquaporin 4 gating dynamics under axially oriented electric-field impulses: A non-equilibrium molecular-dynamics study. J. Chem. Phys. 2018, 149, 245102. 10.1063/1.5044665. [DOI] [PubMed] [Google Scholar]
- English N. J.; Garate J. A. Near-microsecond human aquaporin 4 gating dynamics in static and alternating external electric fields: non-equilibrium molecular dynamics. J. Chem. Phys. 2016, 145, 085102 10.1063/1.4961072. [DOI] [PubMed] [Google Scholar]
- Burnham C. J.; English N. J. Electropumping of water through human aquaporin 4 by circularly polarized electric fields: dramatic enhancement and control revealed by non-equilibrium molecular dynamics. J. Phys. Chem. Lett. 2017, 8, 4646–4651. 10.1021/acs.jpclett.7b02323. [DOI] [PubMed] [Google Scholar]
- Tang C. Y.; Wang Z. N.; Petrinic I.; Fane A. G.; Helix-Nielsen C. Biomimetic aquaporin membranes coming of age. Desalination 2015, 368, 89–105. 10.1016/j.desal.2015.04.026. [DOI] [Google Scholar]
- Pedersen P. A.; Bjorkskov F. B.; Alvisse S.; Helix-Nielsen C. From channel proteins to industrial biomimetic membrane technology. Faraday Discuss. 2018, 209, 287–301. 10.1039/C8FD00061A. [DOI] [PubMed] [Google Scholar]
- Decker K.; Page M.; Boyd A.; MacAllister I.; Ginsberg M.; Aksimentiev A. Selective permeability of truncated aquaporin 1 in silico. ACS Biomater. Sci. Eng. 2017, 3, 342–348. 10.1021/acsbiomaterials.6b00583. [DOI] [PubMed] [Google Scholar]
- Waight A. B.; Czyzewski B. K.; Wang D. N. Ion selectivity and gating mechanisms of FNT channels. Curr. Opin. Struct. Biol. 2013, 23, 499–506. 10.1016/j.sbi.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin E. J.; Cao Y.; Enkavi G.; Quick M.; Pan Y. P.; Tajkhorshid E.; Zhou M. Structure and permeation mechanism of a mammalian urea transporter. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 11194–11199. 10.1073/pnas.1207362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z. W.; Hou T. J.; Li Y. Y. Concerted movement in pH-dependent gating of FocA from molecular dynamics simulations. J. Chem. Inf. Model. 2012, 52, 2119–2131. 10.1021/ci300250q. [DOI] [PubMed] [Google Scholar]
- Atkovska K.; Hub J. S. Energetics and mechanism of anion permeation across formate-nitrite transporters. Sci. Rep. 2017, 7, 12027. 10.1038/s41598-017-11437-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azouzi S.; Gueroult M.; Ripoche P.; Genetet S.; Colin Aronovicz Y.; Le Van Kim C.; Etchebest C.; Mouro-Chanteloup I. Energetic and molecular water permeation mechanisms of the human red blood cell urea transporter B. PLoS One 2013, 8, e82338 10.1371/journal.pone.0082338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariz-Extreme I.; Hub J. S. Potential of mean force calculations of solute permeation across UT-B and AQP1: a comparison between molecular dynamics and 3D-RISM. J. Phys. Chem. B 2017, 121, 1506–1519. 10.1021/acs.jpcb.6b11279. [DOI] [PubMed] [Google Scholar]
- Padhi S.; Priyakumar U. D. Urea-aromatic stacking and concerted urea transport: conserved mechanisms in urea transporters revealed by molecular dynamics. J. Chem. Theory Comput. 2016, 12, 5190–5200. 10.1021/acs.jctc.6b00602. [DOI] [PubMed] [Google Scholar]
- McNulty R.; Ulmschneider J. P.; Luecke H.; Ulmschneider M. B. Mechanisms of molecular transport through the urea channel of Helicobacter pylori. Nat. Commun. 2013, 4, 2900. 10.1038/ncomms3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M.; Sun L. P.; Zhou Q. T.; Peng Y.; Liu Z. J.; Zhao S. W. Molecular mechanism of acetate transport through the acetate channel SatP. J. Chem. Inf. Model. 2019, 59, 2374–2382. 10.1021/acs.jcim.8b00975. [DOI] [PubMed] [Google Scholar]
- Lamoureux G.; Klein M. L.; Berneche S. A stable water chain in the hydrophobic pore of the AmtB ammonium transporter. Biophys. J. 2007, 92, L82–L84. 10.1529/biophysj.106.102756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baday S.; Wang S. H.; Lamoureux G.; Berneche S. Different hydration patterns in the pores of AmtB and RhCG could determine their transport mechanisms. Biochemistry 2013, 52, 7091–7098. 10.1021/bi400015f. [DOI] [PubMed] [Google Scholar]
- Hub J. S.; Winkler F. K.; Merrick M.; de Groot B. L. Potentials of mean force and permeabilities for carbon dioxide, ammonia, and water flux across a Rhesus protein channel and lipid membranes. J. Am. Chem. Soc. 2010, 132, 13251–13263. 10.1021/ja102133x. [DOI] [PubMed] [Google Scholar]
- Capponi S.; White S. H.; Tobias D. J.; Heyden M. Structural relaxation processes and collective dynamics of water in biomolecular environments. J. Phys. Chem. B 2019, 123, 480–486. 10.1021/acs.jpcb.8b12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux B. Ion conduction and selectivity in K+ channels. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 153–171. 10.1146/annurev.biophys.34.040204.144655. [DOI] [PubMed] [Google Scholar]
- Koepfer D. A.; Song C.; Gruene T.; Sheldrick G. M.; Zachariae U.; de Groot B. L. Ion permeation in K+ channels occurs by direct Coulomb knock-on. Science 2014, 346, 352–355. 10.1126/science.1254840. [DOI] [PubMed] [Google Scholar]
- Medovoy D.; Perozo E.; Roux B. Multi-ion free energy landscapes underscore the microscopic mechanism of ion selectivity in the KcsA channel. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1722–1732. 10.1016/j.bbamem.2016.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan K. M.; Roux B. Molecular dynamics of ion conduction through the selectivity filter of the Na(V)Ab sodium Channela. J. Phys. Chem. B 2018, 122, 10126–10142. 10.1021/acs.jpcb.8b09678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen O. S. Perspectives on: Ion selectivity. J. Gen. Physiol. 2011, 137, 393–395. 10.1085/jgp.201110651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMarco K. R.; Bekker S.; Vorobyov I. Challenges and advances in atomistic simulations of potassium and sodium ion channel gating and permeation. J. Physiol. 2019, 597, 679–698. 10.1113/JP277088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compoint M.; Boiteux C.; Huetz P.; Ramseyer C.; Girardet C. Role of water molecules in the KcsA protein channel by molecular dynamics calculations. Phys. Chem. Chem. Phys. 2005, 7, 4138–4145. 10.1039/b508281a. [DOI] [PubMed] [Google Scholar]
- Furini S.; Beckstein O.; Domene C. Permeation of water through the KcsA K+ channel. Proteins: Struct., Funct., Genet. 2009, 74, 437–448. 10.1002/prot.22163. [DOI] [PubMed] [Google Scholar]
- Hoomann T.; Jahnke N.; Horner A.; Keller S.; Pohl P. Filter gate closure inhibits ion but not water transport through potassium channels. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 10842–10847. 10.1073/pnas.1304714110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner A.; Pohl P. Single-file transport of water through membrane channels. Faraday Discuss. 2018, 209, 9–33. 10.1039/C8FD00122G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcayaga C.; Cecchi X.; Alvarez O.; Latorre R. Streaming potential measurements in Ca2+-activated K+ channels from skeletal and smooth-muscle - coupling of ion and water fluxes. Biophys. J. 1989, 55, 367–371. 10.1016/S0006-3495(89)82814-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando H.; Kuno M.; Shimizu H.; Muramatsu I.; Oiki S. Coupled K+-water flux through the HERG potassium channel measured by an osmotic pulse method. J. Gen. Physiol. 2005, 126, 529–538. 10.1085/jgp.200509377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutzner C.; Kopfer D. A.; Machtens J. P.; de Groot B. L.; Song C.; Zachariae U. Insights into the function of ion channels by computational electrophysiology simulations. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1741–1752. 10.1016/j.bbamem.2016.02.006. [DOI] [PubMed] [Google Scholar]
- Kopec W.; Kopfer D. A.; Vickery O. N.; Bondarenko A. S.; Jansen T. L. C.; de Groot B. L.; Zachariae U. Direct knock-on of desolvated ions governs strict ion selectivity in K+ channels. Nat. Chem. 2018, 10, 813–820. 10.1038/s41557-018-0105-9. [DOI] [PubMed] [Google Scholar]
- Oster C.; Hendriks K.; Kopec W.; Chevelkov V.; Shi C. W.; Michl D.; Lange S.; Sun H.; de Groot B. L.; Lange A. The conduction pathway of potassium channels is water free under physiological conditions. Science Adv. 2019, 5, eaaw6756. 10.1126/sciadv.aaw6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagnella D. E.; Voth G. A. Structure and dynamics of hydronium in the ion channel gramicidin A. Biophys. J. 1996, 70, 2043–2051. 10.1016/S0006-3495(96)79773-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer M. L.; Schmitt U. W.; Voth G. A. The formation and dynamics of proton wires in channel environments. Biophys. J. 2001, 80, 1691–1702. 10.1016/S0006-3495(01)76140-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z.; Tepper H. L.; Voth G. A. Effect of membrane environment on proton permeation through gramicidin a channels. J. Phys. Chem. B 2007, 111, 9931–9939. 10.1021/jp0708998. [DOI] [PubMed] [Google Scholar]
- Lazaridis T.; Hummer G. Classical molecular dynamics with mobile protons. J. Chem. Inf. Model. 2017, 57, 2833–2845. 10.1021/acs.jcim.7b00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey I. S.; Mokrab Y.; Carvacho I.; Sands Z. A.; Sansom M. S. P.; Clapham D. E. An aqueous H+ permeation pathway in the voltage-gated proton channel Hv1. Nat. Struct. Mol. Biol. 2010, 17, 869–U121. 10.1038/nsmb.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood M. L.; Schow E. V.; Freites J. A.; White S. H.; Tombola F.; Tobias D. J. Water wires in atomistic models of the Hvl proton channel. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 286–293. 10.1016/j.bbamem.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita K.; Sakata S.; Yamashita E.; Fujiwara Y.; Kawanabe A.; Kurokawa T.; Okochi Y.; Matsuda M.; Narita H.; Okamura Y.; Nakagawa A. X-ray crystal structure of voltage-gated proton channel. Nat. Struct. Mol. Biol. 2014, 21, 352–U170. 10.1038/nsmb.2783. [DOI] [PubMed] [Google Scholar]
- Gianti E.; Delemotte L.; Klein M. L.; Carnevale V. On the role of water density fluctuations in the inhibition of a proton channel. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E8359–E8368. 10.1073/pnas.1609964114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Keulen S. C.; Gianti E.; Carnevale V.; Klein M. L.; Rothlisberger U.; Delemotte L. Does proton conduction in the voltage-gated H+ channel hHv1 involve Grotthuss-like hopping via acidic residues?. J. Phys. Chem. B 2017, 121, 3340–3351. 10.1021/acs.jpcb.6b08339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.; Bai C.; Feliks M.; Alhadeff R.; Warshel A. On the control of the proton current in the voltage-gated proton channel Hv1. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 10321–10326. 10.1073/pnas.1809766115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest L. R.; Kukol A.; Arkin I. T.; Tieleman D. P.; Sansom M. S. P. Exploring models of the influenza A M2 channel: MD simulations in a lipid bilayer. Biophys. J. 2000, 78, 55–69. 10.1016/S0006-3495(00)76572-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. J.; Voth G. A. Computational studies of proton transport through the M2 channel. FEBS Lett. 2003, 552, 23–27. 10.1016/S0014-5793(03)00779-8. [DOI] [PubMed] [Google Scholar]
- Chen H. N.; Wu Y. J.; Voth G. A. Proton transport behavior through the influenza a M2 channel: Insights from molecular simulation. Biophys. J. 2007, 93, 3470–3479. 10.1529/biophysj.107.105742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stouffer A. L.; Acharya R.; Salom D.; Levine A. S.; Di Costanzo L.; Soto C. S.; Tereshko V.; Nanda V.; Stayrook S.; DeGrado W. F. Structural basis for the function and inhibition of an influenza virus proton channel. Nature 2008, 451, 596–599. 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell J. R.; Chou J. J. Structure and mechanism of the M2 proton channel of influenza A virus. Nature 2008, 451, 591–595. 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M.; Yi M. G.; Dong H.; Qin H. J.; Peterson E.; Busath D. D.; Zhou H. X.; Cross T. A. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science 2010, 330, 509–512. 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross T. A.; Dong H.; Sharma M.; Busath D. D.; Zhou H.-X. M2 protein from Influenza A: from multiple structures to biophysical and functional insights. Curr. Opin. Virol. 2012, 2, 128–133. 10.1016/j.coviro.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana E.; Dal Peraro M.; DeVane R.; Vemparala S.; DeGrado W. F.; Klein M. L. Molecular dynamics calculations suggest a conduction mechanism for the M2 proton channel from influenza A virus. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 1069–1074. 10.1073/pnas.0811720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C. Y.; Pohorille A. Activation and proton transport mechanism in influenza A M2 channel. Biophys. J. 2013, 105, 2036–2045. 10.1016/j.bpj.2013.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomaston J. L.; Alfonso-Prieto M.; Woldeyes R. A.; Fraser J. S.; Klein M. L.; Fiorin G.; DeGrado W. F. High-resolution structures of the M2 channel from influenza A virus reveal dynamic pathways for proton stabilization and transduction. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 14260–14265. 10.1073/pnas.1518493112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins L. C.; Liang R. B.; Swanson J. M. J.; DeGrado W. F.; Voth G. A. Proton-induced conformational and hydration dynamics in the influenza A M2 channel. J. Am. Chem. Soc. 2019, 141, 11667–11676. 10.1021/jacs.9b05136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padayatti P. S.; Leung J. H.; Mahinthichaichan P.; Tajkhorshid E.; Ishchenko A.; Cherezov V.; Soltis S. M.; Jackson J. B.; Stout C. D.; Gennis R. B.; Zhang Q. H. Critical role of water molecules in proton translocation by the membrane-bound transhydrogenase. Structure 2017, 25, 1111–1119. 10.1016/j.str.2017.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampjut D.; Sazanov L. A. Structure and mechanism of mitochondrial proton-translocating transhydrogenase. Nature 2019, 573, 291–295. 10.1038/s41586-019-1519-2. [DOI] [PubMed] [Google Scholar]
- Di Luca A.; Gamiz-Hernandez A. P.; Kaila V. R. I. Symmetry-related proton transfer pathways in respiratory complex I. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E6314–E6321. 10.1073/pnas.1706278114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naismith J. H.; Booth I. R. Bacterial mechanosensitive channels-MscS: evolution’s solution to creating sensitivity in function. Annu. Rev. Biophys. 2012, 41, 157–177. 10.1146/annurev-biophys-101211-113227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass R. B.; Strop P.; Barclay M.; Rees D. C. Crystal structure of Escherichia coli MscS, a voltage-modulated and mechanosensitive channel. Science 2002, 298, 1582–1587. 10.1126/science.1077945. [DOI] [PubMed] [Google Scholar]
- Anishkin A.; Sukharev S. Water dynamics and dewetting transitions in the small mechanosensitive channel MscS. Biophys. J. 2004, 86, 2883–2895. 10.1016/S0006-3495(04)74340-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotomayor M.; Schulten K. Molecular dynamics study of gating in the mechanosensitive channel of small conductance MscS. Biophys. J. 2004, 87, 3050–3065. 10.1529/biophysj.104.046045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. J.; Black S. S.; Edwards M. D.; Miller S.; Morrison E. L.; Bartlett W.; Dong C. J.; Naismith J. H.; Booth I. R. The structure of an open form of an E. coli mechanosensitive channel at 3.45 angstrom resolution. Science 2008, 321, 1179–1183. 10.1126/science.1159262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anishkin A.; Akitake B.; Kamaraju K.; Chiang C. S.; Sukharev S. Hydration properties of mechanosensitive channel pores define the energetics of gating. J. Phys.: Condens. Matter 2010, 22, 454120. 10.1088/0953-8984/22/45/454120. [DOI] [PubMed] [Google Scholar]
- Jaiteh M.; Taly A.; Henin J. Evolution of pentameric ligand-gated ion channels: pro-loop receptors. PLoS One 2016, 11, e0151934 10.1371/journal.pone.0151934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa A.; Fujiyoshi Y.; Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature 2003, 423, 949–955. 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- Beckstein O.; Sansom M. S. P. A hydrophobic gate in an ion channel: the closed state of the nicotinic acetylcholine receptor. Phys. Biol. 2006, 3, 147–159. 10.1088/1478-3975/3/2/007. [DOI] [PubMed] [Google Scholar]
- Revah F.; Bertrand D.; Galzi J. L.; Devillers-Thiery A.; Mulle C.; Hussy N.; Bertrand S.; Ballivet M.; Changeux J. P. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature 1991, 353, 846–849. 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- Labarca C.; Nowak M. W.; Tang L.; Deshpande P.; Davidson N.; Lester H. Leucine residues at the 9 position of the M2 domain in AChR govern EC50 independently and symmetrically. Biophys. J. 1995, 68, A233. [Google Scholar]
- Filatov G. N.; White M. M. The role of conserved leucines in the M2 domain of the acetylcholine receptor in channel gating. Mol. Pharmacol. 1995, 48, 379–384. [PubMed] [Google Scholar]
- Plazas P. V.; De Rosa M. J.; Gomez-Casati M. E.; Verbitsky M.; Weisstaub N.; Katz E.; Bouzat C.; Elgoyhen A. B. Key roles of hydrophobic rings of TM2 in gating of the a9a10 nicotinic cholinergic receptor. Br. J. Pharmacol. 2005, 145, 963–974. 10.1038/sj.bjp.0706224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X. L.; Ivanov I.; Wang H. L.; Sine S. M.; McCammon J. A. Molecular-dynamics simulations of ELIC-a prokaryotic homologue of the nicotinic acetylcholine receptor. Biophys. J. 2009, 96, 4502–4513. 10.1016/j.bpj.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nury H.; Poitevin F.; Van Renterghem C.; Changeux J. P.; Corringer P. J.; Delarue M.; Baaden M. One-microsecond molecular dynamics simulation of channel gating in a nicotinic receptor homologue. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 6275–6280. 10.1073/pnas.1001832107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F. Q.; Hummer G. Pore opening and closing of a pentameric ligand-gated ion channel. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 19814–19819. 10.1073/pnas.1009313107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev B.; Murail S.; Poitevin F.; Cromer B. A.; Baaden M.; Delarue M.; Allen T. W. String method solution of the gating pathways for a pentameric ligand-gated ion channel. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E4158–E4167. 10.1073/pnas.1617567114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev B.; Allen T. W. Simulating ion channel activation mechanisms using swarms of trajectories. J. Comput. Chem. 2020, 41, 387–401. 10.1002/jcc.26102. [DOI] [PubMed] [Google Scholar]
- Zhu F. Q.; Hummer G. Drying transition in the hydrophobic gate of the GLIC channel blocks ion conduction. Biophys. J. 2012, 103, 219–227. 10.1016/j.bpj.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S.; Filipek S.; Vogel H. A gating mechanism of the serotonin 5-HT3 receptor. Structure 2016, 24, 816–825. 10.1016/j.str.2016.03.019. [DOI] [PubMed] [Google Scholar]
- Du J.; Lu W.; Wu S. P.; Cheng Y. F.; Gouaux E. Glycine receptor mechanism elucidated by electron cryo-microscopy. Nature 2015, 526, 224–229. 10.1038/nature14853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klesse G.; Tucker S. J.; Sansom M. S. P. Electric field induced wetting of a hydrophobic gate in a model nanopore based on the 5-HT3 receptor channel. ACS Nano 2020, 10.1021/acsnano.0c04387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klesse G.; Rao S.; Sansom M. S. P.; Tucker S. J. CHAP: a versatile tool for the structural and functional annotation of ion channel pores. J. Mol. Biol. 2019, 431, 3353–3365. 10.1016/j.jmb.2019.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen M. O.; Borhani D. W.; Lindorff-Larsen K.; Maragakis P.; Jogini V.; Eastwood M. P.; Dror R. O.; Shaw D. E. Principles of conduction and hydrophobic gating in K+ channels. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 5833–5838. 10.1073/pnas.0911691107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen M. O.; Jogini V.; Borhani D. W.; Leffler A. E.; Dror R. O.; Shaw D. E. Mechanism of Voltage Gating in Potassium Channels. Science 2012, 336, 229–233. 10.1126/science.1216533. [DOI] [PubMed] [Google Scholar]
- Aryal P.; Abd-Wahab F.; Sansom M. S. P.; Tucker S. J.; et al. A hydrophobic barrier deep within the inner pore of the TWIK-1 K2P potassium channel. Nat. Commun. 2014, 5, 4377. 10.1038/ncomms5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z.; Yazdani M.; Zhang G.; Cui J.; Chen J. Hydrophobic gating in BK channels. Nat. Commun. 2018, 9, 3408. 10.1038/s41467-018-05970-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W.; Hu R. K.; Cai R. Q.; Hofmann L.; Hu Q. L.; Fatehi M.; Long W. T.; Kong T.; Tang J. F.; Light P.; Flockerzi V.; Cao Y.; Chen X. Z. Identification and characterization of hydrophobic gate residues in TRP channels. FASEB J. 2018, 32, 639–653. 10.1096/fj.201700599RR. [DOI] [PubMed] [Google Scholar]
- Zheng W.; Yang X.; Hu R.; Cai R.; Hofmann L.; Wang Z.; Hu Q.; Liu X.; Bulkley D.; Yu Y.; Tang J.; Flockerzi V.; Cao Y.; Cao E.; Chen X.-Z. Hydrophobic pore gates regulate ion permeation in polycystic kidney disease 2 and 2L1 channels. Nat. Commun. 2018, 9, 2302. 10.1038/s41467-018-04586-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasimova M. A.; Yazici A.; Yudin Y.; Granata D.; Klein M. L.; Rohacs T.; Carnevale V. Ion channel sensing: are fluctuations the crux of the matter?. J. Phys. Chem. Lett. 2018, 9, 1260–1264. 10.1021/acs.jpclett.7b03396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale C.; Chakrabarti N.; Pomorski P.; Pai E. F.; Pomes R. Hydrophobic gating of ion permeation in magnesium channel CorA. PLoS Comput. Biol. 2015, 11, e1004303 10.1371/journal.pcbi.1004303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz-Fonfria V. A.; Bamann C.; Resler T.; Schlesinger R.; Bamberg E.; Heberle J. Temporal evolution of helix hydration in a light-gated ion channel correlates with ion conductance. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E5796–E5804. 10.1073/pnas.1511462112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H. E.; Kim Y. S.; Paggi J. M.; Evans K. E.; Allen W. E.; Richardson C.; Inoue K.; Ito S.; Ramakrishnan C.; Fenno L. E.; Yamashita K.; Hilger D.; Lee S. Y.; Berndt A.; Shen K.; Kandori H.; Dror R. O.; Kobilka B. K.; Deisseroth K. Structural mechanisms of selectivity and gating in anion channelrhodopsins. Nature 2018, 561, 349–354. 10.1038/s41586-018-0504-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S.; Klesse G.; Stansfeld P. J.; Tucker S. J.; Sansom M. S. P. A BEST example of channel structure annotation by molecular simulation. Channels 2017, 11, 347–353. 10.1080/19336950.2017.1306163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S. L.; Lynch C. I.; Klesse G.; Oakley G. E.; Stansfeld P. J.; Tucker S. J.; Sansom M. S. P. Water and hydrophobic gates in ion channels and nanopores. Faraday Discuss. 2018, 209, 231–247. 10.1039/C8FD00013A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noy A.; Wanunu M. A new type of artificial water channels. Nat. Nanotechnol. 2020, 15, 9–10. 10.1038/s41565-019-0617-5. [DOI] [PubMed] [Google Scholar]
- Polster J. W.; Acar E. T.; Aydin F.; Zhan C.; Pham T. A.; Siwy Z. S. Gating of hydrophobic nanopores with large anions. ACS Nano 2020, 14, 4306–4315. 10.1021/acsnano.9b09777. [DOI] [PubMed] [Google Scholar]