

Abstract
Background:
Hepatocellular carcinoma (HCC) is a major cause of cancer death worldwide and establishment of new chemotherapies for HCC is urgently needed. GPR41 [free fatty acid receptor 3 (FFA3)] is a G protein-coupled receptor for short chain fatty acids, including acetate, propionate, and butyrate. In our previous study, we showed that propionate enhances the cytotoxic effect of cisplatin in HCC cells and that this mechanism is dependent on inhibition of histone deacetylases (HDACs) via GPR41/FFA3. However, the antitumor action of GPR41/FFA3 has not been elucidated.
Methods:
In this study, we examined AR420626 as a GPR41-selective agonist in HepG2 and HLE cells. Nude mice were used for HepG2 xenograft studies. The apoptotic effect of AR420626 was evaluated using flow cytometry analysis. Expression of apoptosis-related proteins and HDACs was evaluated by Western immunoblot. Gene silencing of HDAC 3/5/7 and GPR41 was performed using small interfering RNA. Expression of TNF-α mRNA was evaluated by TaqMan real-time polymerase chain reaction.
Results:
We found that AR420626, a selective GPR41/FFA3 agonist, suppressed growth of HepG2 xenografts and inhibited proliferation of HCC cells by inducing apoptosis. AR420626 induced proteasome activation through mTOR phosphorylation, which reduced HDAC proteins, and then increased expression of TNF-α.
Conclusion:
AR420626, a selective GPR41/FFA3 agonist, may be a candidate as a therapeutic agent for HCC.
Keywords: AR420626, FFA3, GPR41, histone deacetylase, mTORC1
Introduction
Hepatocellular carcinoma (HCC) is a common malignant cancer worldwide and the third leading cause of cancer death.1,2 Surgical resection followed by liver transplantation is a curative treatment for early stage HCC, but the tumor is prone to recurrence.3 Furthermore, the clinical symptoms of early stage HCC are frequently unclear, and diagnosis may be delayed until an advanced stage, resulting in a high mortality rate.4 Sorafenib, a multikinase inhibitor, is used as first-line treatment for patients with advanced-stage HCC, but may only provide a three-month survival benefit in comparison with a placebo.5 Thus, establishment of new chemotherapy is urgently needed, especially for patients with advanced HCC.
GPR41 (free fatty acid receptor 3; FFA3) is a G protein-coupled receptor for short chain fatty acids, including acetate, propionate, and butyrate. GPR41/FFA3 couples to the Gi/o signaling pathway6 and is expressed in the intestine, adipose tissue, pancreatic β cells, and peripheral nervous system.7 GPR41/FFA3 acts as a nutritional sensor to regulate the sympathetic nervous system8 and activation of GPR41/FFA3 regulates insulin secretion.9 A recent study also showed that the GPR41/FFA3 activation alters the breast cancer phenotype from invasive to noninvasive.10 However, the antitumor action of GPR41/FFA3 is unclear.
We have shown that GPR41/FFA3 is expressed at discernible levels in human HCC tissues of different pathological grades and in HCC cells, including HepG2, HuH-7, JHH-4, and HLE cells.11 We also found that sodium propionate enhances the cytotoxicity of cisplatin in HCC cells through a mechanism dependent on enhanced expression of TNF-α, which is induced by cisplatin through a histone deacetylase (HDAC) inhibitory pathway via GPR41/FFA3.11 Epigenetic modification, such as the acetylation and methylation of histones, plays critical roles in regulating gene expression. Acetylation of histones is maintained by a balance between histone acetyltransferase and HDAC. Currently, 18 HDACs have been identified and these are divided into four classes: class I (HDAC1, 2, 3, 8), class II (HDAC4, 5, 6, 7, 9, 10), class III (SIRT1–7), and class IV (HDAC11) based on homology with the primary structure of yeast HDAC. HDACs act as corepressors of transcription, and HDAC activity is associated with transcriptionally inactive chromatin, which favors gene expression patterns that promote tumor initiation and development.12 Aberrant expression of HDACs occurs in certain human cancers, including gastric cancer, lung cancer, and HCC. Thus, HDACs are promising anticancer therapeutic targets and HDAC inhibitors are considered to be potent anticancer agents through the promotion of acetylation of histones.
In this study, we show that AR420626, a GPR41/FFA3-selective agonist, induces apoptosis in HCC cells, including HepG2 and HLE cells. This mechanism was partially dependent on induction of the expression of TNF-α through an HDAC inhibitory pathway.
Methods and materials
Ethics statement
In all experiments in vitro, we did not conduct genetic recombination experiments, pathogen handling experiments, and did not use Human samples. Therefore, ethics approval was not sought for the present study. All animal experiments in vivo were carried out in accordance with the National Institutes of Health guidelines for the care and use of live animals and were approved by the University of Fukui animal care committees (29007).
Materials
N-(2,5-Dichlorophenyl)-4-(furan-2-yl)-2-methyl-5-oxo-1,4,5,6,7,8-hexahydro-quinoline-3-carboxamide (AR420626) and MG-132 (Sigma-Aldrich, St. Louis, MO, USA); TNF-α antagonist R-705013 (Calbiochem, Darmstadt, Germany); rapamycin (Selleckchem, Houston, TX, USA); polyclonal rabbit antibodies against human β-actin (Abcam, Cambridge, UK), acetyl-histone H3 (Lys9/Lys14), cleaved caspase-3 (Asp175), cleaved caspase-9, phospho-mTOR (Ser2448) (Cell Signaling Technology, Boston, MA, USA); monoclonal rabbit antibodies against HDAC1, HDAC2, HDAC4, HDAC5, HDAC6, HDAC7, mTOR (Cell Signaling Technology) and HDAC8 (Abcam); monoclonal mouse antibodies against HDAC3, caspase-8 (Cell Signaling Technology) and 20S Proteasome β5 (Santa Cruz Biotechnology, Dallas, TX, USA); and horseradish peroxidase (HRP)-conjugated antimouse and antirabbit immunoglobulins (Dako, Glostrup, Denmark) were used in the study.
Cell cultures
HepG2 and HLE (human hepatoma) cells from the Japanese Collection of Research Bioresources Cell Bank were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C in a humidified atmosphere of 5% CO2 and 18% O2. Upon reaching the logarithmic phase of growth, the cells were digested with trypsin and seeded in different culture plates as required experimentally.
Cell proliferation assay
Cell proliferation was evaluated by MTS assay (Promega, Madison, WI, USA). HepG2 and HLE cells (5 × 103 cells per well) were seeded in 96-well plates and treated with AR420626 (10, 25 µM) at 37°C under 5% CO2 for 24, 48, and 72 h. After each treatment, 20 µl of MTS reagent (from the CellTiter 96 Aqueous Assay kit) was added to each well, and the plates were incubated for a further 4 h at 37°C under 5% CO2. Absorbance of the product was measured with a microplate reader at 490 nm, with a reference wavelength of 650 nm.
TaqMan real-time PCR
TaqMan real-time reverse transcription polymerase chain reaction (PCR) was performed14 using specific primers and TaqMan MGB probes (Applied Biosystems) for human TNF-α (assay ID: Hs00174128_m1). Expression of TNF-α mRNA was normalized against β-actin mRNA, which was detected with a TaqMan human β-actin MGB control reagent kit (Applied Biosystems).
Knockdown of HDACs 3, 5, and 7 in HepG2 cells
Gene silencing of HDACs 3, 5, and 7 in HepG2 cells was performed14 using small interfering RNA (siRNA) against each HDAC and control siRNA [all ON-TARGET plus Human, GE Healthcare Dharmacon, Lafayette, CO, USA. HepG2 cells (70% confluence)] were transfected with control or HDAC 3, 5, or 7 siRNA at a final concentration of 50 nmol/l using DharmaFECT transfection reagent (Dharmacon). After incubation for 48 h, HepG2 cells were examined by immunoblotting and TaqMan real-time PCR.
Immunoblot analysis
HepG2 and HLE cells were lysed in radioimmunoprecipitation assay buffer with phosphatase inhibitors (Sigma-Aldrich). Lysates (5 µg protein) were analyzed by immunoblotting with antibodies for β-actin (1:1000), cleaved caspase-3 (1:1000), caspase-8 (1:1000), caspase-9 (1:1000), acetyl-H3 (1:1000), mTOR (1:1000), phospho-mTOR (1:1000), 20S proteasome β5 (1:1000), HDAC1 (1:1000), HDAC2 (1:1000), HDAC3 (1:1000), HDAC4 (1:1000), HDCA5 (1:1000), HDAC6 (1:1000), HDAC7 (1:1000), and HDAC8 (1:5000) for 24 h, and then with appropriate HRP-conjugated secondary antibodies (1:1000) at room temperature for 1 h. Immunoreactive bands were visualized as previously described.14
Quantification of apoptosis
An annexin V kit (MBL, Nagoya, Japan) was used to evaluate apoptosis in HepG2 and HLE cells resuspended in 85 µl of binding buffer. The suspension was incubated with 10 µl of annexin V-FITC and 5 µl of propidium iodide for 15 min at room temperature in the dark. Then, 400 µl of binding buffer was added to each tube and the percentage of apoptotic cells was determined by flow cytometry using a BD FACSCanto™ II Flow Cytometer.
In vivo xenograft study
Animals were housed in pathogen-free conditions. All experimental procedures were in conformity with the Regulations for Animal Research at the University of Fukui and were reviewed by the Animal Research Committee. Mice were housed under standard conditions (12 h light/12 h dark). Six-week-old male SHO nude mice (Charles River, Japan) were subcutaneously injected with 1.0 × 106 HepG2 cells into the right flank of each mouse. After the tumor reached 500–1000 mm3, mice were randomly allocated to two groups (n = 5 per group) treated with intraperitoneal injection of AR420626 dissolved in saline at 0.1 mg/kg on days 0–4 and 0.2 mg/kg on days 7–11 (AR group) and with saline alone (control group). The tumor size was measured with calipers and the weight was estimated as (length × width2)/2. To standardize tumor weights between the groups, relative tumor weights at different times were obtained from the formula TWi/TWo, in which TWi and TWo are the mean tumor weights of a group on days n and 0, respectively. Body weights were measured on the dates shown in the results. After treatments, blood samples were collected to measure laboratory data, and kidney, liver, and heart were obtained to analyze their histology. All animals were humanely killed by CO2 asphyxiation at 18 days.
Determination of laboratory data
Counts of red blood cells, white blood cells and platelets were measured by an automated cellular analyzer: XN-9000 (Sysmex). Concentrations of creatinine (Cr) in mouse blood samples were determined using standard enzymatic methods (Cygnus Auto CRE, Shino-Test Corp., Tokyo, Japan) with a Toshiba 2000FR analyzer (Toshiba Medical Systems Corp., Tokyo, Japan) in duplicate in a central laboratory. Serum activities of aspartate aminotransferase (AST), alanine aminotransferase (ALT), lactate dehydrogenase (LDH), and creatine kinase-MB isoform (CK-MB) were precisely and accurately determined by commercial assay kits using nicotinamide adenine dinucleotide methods (L-type AST.J2, L-type ALT.J2, and L-type LD.J, FUJIFILM Wako Pure Chemical Corp., Osaka, Japan and Cygnus Auto CK-MB MtO, SHINO-TEST Corp., Tokyo, Japan, respectively) with the Toshiba 2000FR analyzer, in duplicate in a central laboratory.
Statistical analysis
Data are expressed as the mean ± standard deviation. For all experiments, gaussian distribution was determined using a Kolmogorov–Smirnov test. The significance of differences between two groups was evaluated by Mann–Whitney U test or Student’s t test. Differences among more than two groups were assessed by analysis of variance with a Tukey post hoc test. p values of < 0.05 were considered to indicate statistical significance.
Results
AR420626 inhibits growth of HepG2 xenografts
We previously showed that propionate, a physiological GPR41/FFA3 agonist, enhanced the cytotoxicity of cisplatin in HCC cells, and that cisplatin and propionate in combination significantly suppressed growth of HepG2 xenografts in comparison with cisplatin alone.11 Similarly, AR420626, a selective GPR41/FFA3 agonist, significantly suppressed growth of HepG2 xenografts (p < 0.01) (Figure 1). There was no significant difference in daily food intake or body weight changes between the control and AR groups (Supplementary Figure S1), and no deaths in either group. In addition, there were no significant differences in the serum levels of Cr, AST, ALT, CK-MB, LDH, and blood cell counts between the control and AR groups. (Supplementary Table S1). In addition, there were no histological changes of kidney, liver, and heart between the two groups (data not shown).
Figure 1.
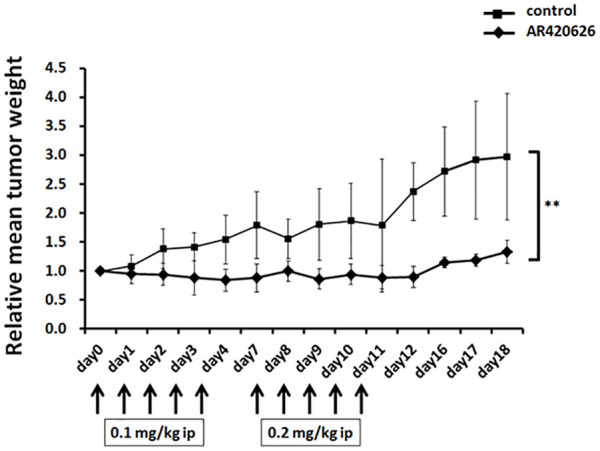
AR420626 inhibition of growth of HepG2 xenografts. Six-week-old male SHO nude mice were subcutaneously injected with 1.0 × 106 cells. When the tumor size reached 500–1000 mm3, mice were randomly allocated to two groups (n = 5 per group) and treated with intraperitoneal injection of AR420626 dissolved in saline at doses of 0.1 mg/kg on days 0–4 and 0.2 mg/kg on days 7–11. Xenografts were measured as described in the Materials and Methods. Results are shown as the mean ± SD (n = 5). **p < 0.01 by Student’s t test. ip, intraperitoneal.
AR420626 suppresses proliferation of HCC cells by inducing apoptosis
To determine the molecular mechanism of AR420626 inhibition of growth of HepG2 xenografts (Figure 1), in vitro analysis was performed in HepG2 and HLE cells. AR420626 significantly inhibited proliferation of HepG2 cells at 25 µM for 24 h and at 10 µM and 25 µM for 48 and 72 h (Figure 2a), and of HLE cells at 25 µM for 48 h and at 10 µM and 25 µM for 72 h (Figure 2b). As shown in Supplementary Figure S2, the IC50 of AR420626 in HepG2 was about 25 µM. In fluorescence activated cell sorting (FACS) analysis, the apoptosis rate at 48 h was significantly higher in each cell line and increased in a dose-dependent manner (Figure 2c, d).
Figure 2.

AR420626 suppression of proliferation of HCC cells by inducing apoptosis. (a) MTS assay of the effects of AR420626 (10, 25 µM) on proliferation of HepG2 cells for 24, 48, and 72 h. (b) MTS assay of the effects of AR420626 (10, 25 µM) on proliferation of HLE cells for 24, 48, and 72 h. Data are shown as the mean ± SD of 3 independent experiments. *p < 0.05, **p < 0.01, NS: not significant by one-way analysis of variance (ANOVA) with a Tukey–Kramer multiple comparison test. (c) HepG2 cells and (d) HLE cells were treated with AR420626 (10, 25 µM) for 48 h. Cells were then stained with annexin V and PI, and cytometry was performed. Data are shown as the mean ± SD of % apoptosis from 3 independent experiments. **p < 0.01 by one-way ANOVA with a Tukey–Kramer multiple comparison test.
AR420626 elicits an extrinsic apoptotic pathway and histone H3 acetylation
Apoptosis is regulated by highly coordinated activation of caspase cysteine proteases.15 The level of activated (cleaved) caspase-3, which causes DNA fragmentation and myonuclear apoptosis15,16 was determined in HCC cells by Western blotting (Figure 3a). AR420626 significantly increased expression of cleaved caspase-3 in a dose-dependent manner. Immunoblotting also showed that AR420626 increased cleaved caspase-8 in HCC cells in a dose-dependent manner, and slightly increased cleaved caspase-9 in HepG2 cells at 25 µM (Figure 3a). Cleaved caspase-8 is induced by activation of death receptors including CD95 (Fas) and tumor necrosis factor receptor 1 (TNFR1) in the extrinsic apoptosis pathway.17
Figure 3.

AR420626 elicits an extrinsic apoptotic pathway and histone deacetylase inhibition. (a, b) HepG2 and HLE cells were treated with vehicle or AR420626 (10, 25 µM) for 48 h. Western blotting was used to detect histone H3 acetylation and the apoptosis-related proteins, cleaved caspase-3, -8, and -9, in cell lysates. Levels of cleaved caspase-3, -8, and -9 and acetyl-H3 (all relative to β-actin) were set to 1.0 in cells treated with vehicle. Data are shown as the mean ± SD of 3 separate experiments. *p < 0.05, **p < 0.01, NS: not significant by one-way analysis of variance with a Tukey–Kramer multiple comparison test. (c) HepG2 and HLE cells were treated with AR420626 (25 µM) for 1, 3, 12, and 24 h. Western blotting was used to detect histone H3 acetylation and cleaved caspase-3 and -8 in cell lysates. (d) Immunoblotting of GPR41 in cells with small interfering RNA (siRNA)-mediated GPR41 knockdown for 48 h. (e) HepG2 cells treated with control or GPR41 siRNA were treated with vehicle or AR420626 (25 µM). Western blotting was used to detect histone H3 acetylation and cleaved caspase-3 in cell lysates.
Next, time course experiments were performed to investigate the detailed molecular mechanism of AR420626-induced apoptosis in HCC cells. Significant increases in protein levels of cleaved caspase-3 and -8 were observed at 24 h, while a significant increase in histone H3 acetylation was found at 3 h and persisted for 24 h in HepG2 cells (Figure 3b). Furthermore, cleaved caspase-3 and -8 levels were significantly increased at 12 h and persisted for 24 h, and histone H3 acetylation was significantly increased at 1 h and persisted for 24 h in HLE cells (Figure 3b).
GPR41/FFA3 gene silencing in HepG2 cells using siRNA was performed to determine whether AR420626-induced apoptosis is mediated by a GPR41/FFA3 pathway. This silencing significantly reduced GPR41/FFA3 protein expression (Figure 3c) and significantly blocked 25 µM AR420626-induced cleavage of caspase-3 in HepG2 cells (Figure 3d). These results show that AR420626 induces apoptosis in a GPR41/FFA3-mediated manner. We previously showed that propionate enhances the effect of cisplatin through an HDAC inhibitory pathway via GPR41/FFA3.11 AR420626 significantly induced histone H3 acetylation in a dose-dependent manner in HCC cells (Figure 3a), and GPR41/FFA3 silencing in HepG2 cells significantly blocked 25 µM AR420626-induced acetylation of histone H3 (Figure 3d).
AR420626 induces apoptosis via a TNF-α pathway
AR420626 elicited an extrinsic apoptotic pathway and induced histone H3 acetylation prior to increases in cleaved caspase-3 and -8 in HCC cells. Therefore, we hypothesized that AR420626 induced apoptosis via upregulation of a TNF-α pathway, and we performed time course experiments to investigate expression of TNF-α mRNA, a pro-apoptotic cytokine, in HCC cells. At 25 µM, AR420626 significantly increased the TNF-α mRNA level in HepG2 cells from 1 to 24 h, with a peak around 24 h (Figure 4a), and in HLE cells from 1 to 24 h in a dose-dependent manner, with a peak at around 3 h (Figure 4b). AR420626 induction of cleaved caspase-3 was significantly blocked by a TNF-α antagonist in HepG2 cells (Figure 4c) and HLE cells (Figure 4d). Collectively, these results show that AR420626 induces apoptosis by upregulating TNF-α expression.
Figure 4.

AR420626 induces apoptosis via the TNF-α-induced extrinsic apoptotic pathway. (a) HepG2 cells were treated with AR420626 (25 µM) for 1, 3, 12, and 24 h. (b) HLE cells were treated with AR420626 (25 µM) for 1, 3, 12, and 24 h. TNF-α mRNA levels were quantified by TaqMan real-time polymerase chain reaction, with the level in cells treated with vehicle set to 1.0. Data are shown as the mean ± SD of 3 separate experiments. *p < 0.05, **p < 0.01, NS: not significant by one-way analysis of variance with a Tukey–Kramer multiple comparison test. (c) HepG2 cells and (d) HLE cells were treated with AR420626 (10, 25 µM) with or without a TNF-α antagonist (10 µM) for 24 h. Western blotting was used to detect cleaved caspase-3 in cell lysates.
AR420626 reduces expression of HDACs
We hypothesized that the increase in acetylation of histone H3 induced by AR420626 indicated a decrease in HDAC activity in HCC cells. Therefore, the protein levels of HDACs 1–8 were determined in HCC cells. At 25 µM, AR420626 distinctly reduced the levels of HDACs 3, 4, 5 and 7 in HepG2 cells and those of HDACs 3, 4, 6, 7, and 8 in HLE cells at 48 h (Figure 5a). In parallel, histone H3 acetylation was significantly enhanced in HCC cells at 48 h (Figure 5a). A time course experiment showed that the levels of HDACs 4–7 were reduced at 1 h and gradually decreased until 24 h in HepG2 cells (Figure 5b, d), those of HDACs 1–3 were significantly reduced at 24 h, and that of HDAC8 was significantly reduced at 1 h and gradually decreased until 24 h (Figure 5b, c). Histone H3 acetylation was significantly upregulated at 3 h and gradually further increased to 24 h, in parallel with the reduced HDAC levels (Figure 5b). Collectively, these results indicate that AR420626 increases histone H3 acetylation via reduction of levels of several HDACs, particularly class II HDACs.
Figure 5.

AR420626 reduces expression of histone deacetylases (HDACs). (a) HepG2 and HLE cells were treated with vehicle or AR420626 (10, 25 µM) for 48 h. Western blotting was used to detect HDACs 1–8 in cell lysates. (b) HepG2 cells were treated with vehicle or AR420626 (25 µM) for 1, 3, 12, and 24 h. Western blotting was used to detect HDACs 1–8 in cell lysates. (c, d) Levels of HDACs (all relative to β-actin) were set to 1.0 in cells treated with vehicle. Data are shown as the mean ± SD of 3 separate experiments. *p < 0.05, **p < 0.01, NS not significant by one-way analysis of variance with a Tukey–Kramer multiple comparison test.
AR420626 induces TNF-α-induced apoptosis through HDAC inhibition
The effect of reduction of HDAC levels on expression of TNF-α in HepG2 cells was investigated using gene silencing of HDAC3, as a representative class I HDAC, and of HDACs 5 and 7, as representative class II HDACs, in HepG2 cells. Silencing of all three HDACs significantly reduced their expression (Figure 6a) and significantly increased TNF-α mRNA levels in comparison with control siRNA treatment at 24 h (Figure 6b). These results show that reduction of HDAC levels is sufficient to induce expression of TNF-α.
Figure 6.

The histone deacetylase (HDAC) inhibitory pathway induces apoptosis through a TNF-α-induced pathway. (a) HepG2 cells were treated with control, HDAC3, HDAC5, and HDAC7 siRNA. Western blotting was used to detect HDACs 3, 5, and 7 in cell lysates from cells with control and HDAC 3, 5, and 7 siRNA knockdown. (b) TNF-α mRNA levels in the knockdown cells were quantified by TaqMan real-time polymerase chain reaction, with the level in cells treated with control small interfering RNA set to 1.0. Data are shown as the mean ± SD of 3 separate experiments. **p < 0.01 by Mann–Whitney U test.
AR420626 induces apoptosis via proteasome activation in HepG2 cells
Time course experiments showed reduced levels of HDACs, and especially class II HDACs, which are mainly localized in the cytoplasm,18 in the early phase. We hypothesized that GPR41/FFA3 might affect HDACs through regulation of HDAC expression via the proteasome system, and we examined whether MG-132, a specific proteasome inhibitor, could attenuate the AR420626-induced reduction of HDACs. MG-132 significantly reduced AR420626-induced cleavage of caspase-3 and histone H3 acetylation in HepG2 cells at 24 h (Figure 7a). At 24 h after treatment with MG-132, this reduction of HDACs (especially HDACs 1, 3, 4, 5, 6, and 8) was significantly attenuated and that of HDAC7 was partially attenuated (Figure 7b). In addition, at 6 h after AR420626 treatment, the mRNA levels for HDACs 1–8 were significantly upregulated in HepG2 cells (data not shown). Collectively, these results show that AR420626 enhances proteasome-mediated breakdown of HDACs, leading to histone acetylation and apoptosis in HepG2 cells.
Figure 7.

AR420626 induces apoptosis via proteasome activation in HepG2 cells. (a, b) HepG2 cells were treated with AR420626 (25 µM) with or without MG-132 (20 µM) for 24 h. Western blotting was performed to detect (a) cleaved caspase-3 and histone H3 acetylation and (b) HDACs 1–8 in cell lysates.
AR420626 induces apoptosis via activation of the mTORC1 pathway in HepG2 cells
A recent study showed that the mammalian target of rapamycin complex 1 (mTORC1) pathway enhances protein degradation through proteasome activation.19 Therefore, we investigated the time course of mTOR phosphorylation. At 25 µM, AR420626 significantly induced mTOR phosphorylation at 1 h and this persisted for 24 h (Figure 8a). Rapamycin, an mTORC1-selective inhibitor, inhibited the AR420626-induced cleavage of caspase-3 and acetylation of histone H3 in a dose-dependent manner at 24 h (Figure 8b). Furthermore, the cotreatment of Rapamycin at 1 µM and MG-132 at 1 µM was completely inhibited the AR420626-induced cleavage of caspase-3 (Supplementary Figure S3). In addition, consistent with the previous report,19 AR420626 significantly induced expression of proteasome β5 at 12 h, and this expression gradually increased until 24 h (Figure 8a). Taken together, these results show that AR420626 enhances mTOR phosphorylation and induces proteasome activation, resulting in reduction of levels of HDAC proteins in HepG2 cells.
Figure 8.

AR420626 induces apoptosis via activation of the mammalian target of rapamycin complex 1 (mTORC1) pathway in HepG2 cells. (a) HepG2 cells were treated with vehicle or AR420626 (25 µM) for 1, 3, 12, and 24 h. Western blotting was used to detect phospho-mTOR, mTOR, and proteasome β5 in cell lysates. (b) Levels of proteasome β5 treated with AR420626 (25 µM; all relative to β-actin) were set to 1.0 in cells treated with vehicle. Data are shown as the mean ± SD of 3 separate experiments. *p < 0.05 by one-way analysis of variance (ANOVA) with a Tukey–Kramer multiple comparison test. (c) HepG2 cells were treated with AR420626 (25 µM) with or without rapamycin (1, 10 µM) for 24 h. Western blotting was used to detect histone H3 acetylation, cleaved caspase-3, and proteasome β5 in cell lysates. (d) Levels of proteasome β5 treated with AR420626 (25 µM) with or without rapamycin (1, 10 µM) (all relative to β-actin) were set to 1.0 in cells treated with vehicle. Data are shown as the mean ± SD of 3 separate experiments. *p < 0.05, NS not significant by one-way ANOVA with a Tukey–Kramer multiple comparison test.
Discussions
In this study, we showed that AR420626, a selective agonist of GPR41/FFAR3, induces apoptosis in HCC cells by upregulating expression of TNF-α via HDAC inhibition.
A key finding was that AR420626 induced apoptosis by reducing expression of HDACs, since aberrant expression of HDACs is associated with cancer aggressiveness and a poor prognosis.20 In a previous study, an expression of a subset of HDACs in liver cancer was shown to be higher than that in normal liver tissues, and to be associated with the presence of cirrhotic and dysplastic nodules.21 In particular, HDAC1, 2, 3, 4, 5, and 8 are upregulated in many HCC cells.22 Inactivation of HDAC1 reduces tumor cell growth and induces autophagic cell death in Hep3B cells,23 aberrant expression of HDAC2 induces proliferation by dysregulation of expression of G1/S cell cycle proteins in Hep3B cells24 and HDAC2 expression plays an important role in the prognosis of HCC.25 HDAC3 plays a significant role in regulating proliferation and invasion of liver cancer cells.26 Furthermore, knockdown of HDAC4 suppresses proliferation of HCC cells,27 knockdown of HDAC5 inhibits cancer cell proliferation by induction of G1/S cell cycle arrest and leads to apoptosis in HCC cells,28 knockdown of HDAC6 reduces migration and invasion of HCC cells,29 and knockdown of HDAC8 represses tumor cell growth and induces apoptosis in BEL-7402 and HepG2 cells.30 Thus, suppression of HDACs is likely to be effective for treatment of HCC.
We also found that AR420626 reduced expression of HDACs (especially HDACs 2–7), in addition to increasing histone H3 acetylation, which suggests that GPR41/FFA3 may have an HDAC-inhibitory effect, regulating expression of HDACs via the ubiquitin-proteasome system in HepG2 cells. In support of this hypothesis, treatment with MG-132, a specific proteasome inhibitor, reversed AR420626-elicited downregulation of HDACs 1, 3, 4, 5, 6, and 8 in HepG2 cells and significantly reduced cleavage of caspase-3 and acetylation of histone H3. mTORC1 signaling enhances protein degradation through an increase in proteasome levels,19 and in the current study, AR420626 induced mTOR phosphorylation within 1 h and this persisted until at least 24 h. Rapamycin, an mTORC1 selective inhibitor, inhibited the AR420626-induced cleavage of caspase 3 in a dose-dependent manner. These results indicate that GPR41/FFA3 stimulation evoked mTORC1 phosphorylation to activate proteasomes to induce HDAC degradation, leading to apoptosis in HepG2 cells.
Another major finding in the study was that AR420626 induced apoptosis by producing TNF-α via HDAC inhibition in HepG2 cells. Death receptors, including CD95 (Fas) and TNFR1, mediate the extrinsic pathway for apoptosis by activating caspase-8.17 In the current study, we found that 25 µM AR420626 significantly enhanced cleavage of caspase-3 and -8. Time course experiments showed that 25 µM AR420626 significantly induced expression of TNF-α mRNA in HepG2 cells from 1 to 24 h, with a peak at around 24 h, and in HLE cells from 1 to 24 h, with a peak at around 3 h. In addition, induction of caspase-3 cleavage by AR420626 was completely blocked by a TNF-α antagonist in HCC cells. Finally, knockdown of HDAC3, a representative class I HDAC, and HDACs 5 and 7, representative class II HDACs, significantly induced expression of TNF-α mRNA in HepG2 cells. These findings suggest that AR420626 induces apoptosis by increasing expression of autocrine TNF-α via inactivation of HDACs. Suppression of HDACs and the resulting histone acetylation leads to the upregulation of gene expression. Therefore, AR420626-induced HDACs downregulation might directly upregulate TNF-α gene expression through modifying the chromatin states of its gene regulatory region. However, we can’t rule out the possibility that the reduction of HDACs indirectly affects to TNF-α gene expression by modulation of other relative transcriptional factors. Chromatin immunoprecipitation (ChIP) assays might need to demonstrate the regulatory mechanism of TNF-α gene expression. In addition, genome-wide ChIP-on-chip analysis will help to evaluate the global influence of AR420626-induced HDACs reduction.
Currently, sorafenib, an oral multikinase inhibitor, has been approved as first-line treatment for patients with advanced-stage HCC, but may only provide a three-month survival benefit in comparison with a placebo.5 Furthermore, HCC becomes resistant to sorafenib within 6 months.31,32 Therefore, the signaling pathway of AR420626 targeting may be a candidate therapeutic agent for HCC. However, the current study is associated with some limitations. First, there is the possibility that AR420626 acts directly on proteins other than GPR41/FFA3. Second, the toxicities of AR420626 is not clearly shown. The previous study reported that AR420626 (0.1 mg/kg, ip) significantly inhibited exogenous serotonin-induced defecation.33 Another study showed that AR420626 (0.1 mg/kg, ig) significantly inhibited the indomethacin-induced small intestinal ulcers.34 In addition, AR420626 at 10 µM significantly elevated peptide YY ex vivo.35 In this present study, we could not specify the toxicities of AR420626 in bone marrow, heart, kidney, and liver in laboratory tests and histological changes in treated AR420626 (0.1~0.2 mg/kg, ip) in vivo. Furthermore, IC50 of AR420626 in HepG2 cells was 25 µM in vitro. Therefore, in this study, we used 2–2.5 fold of higher concentrations compared with the previous reports. Further toxicity tests are needed for clinical application of AR420626.
Conclusion
Our current findings show that AR420626 significantly inhibited proliferation of HCC cells by inducing apoptosis by expression of TNF-α via reduction of HDACs in a GPR41/FFA3-dependent manner (summarized in Figure 9). Our results suggest that AR420626 may exert therapeutic effects on HCC and that a selective agonist of GPR41/FFAR3 might serve as a novel therapeutic agent for HCC.
Figure 9.
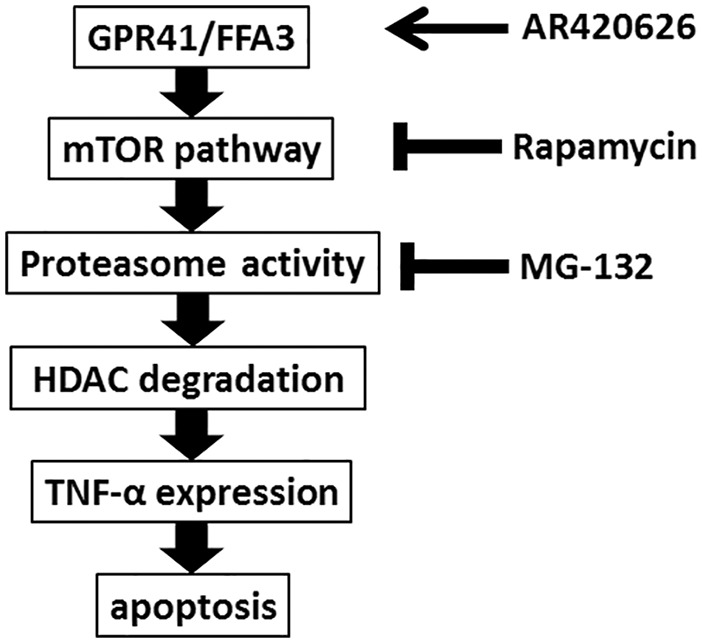
Model of AR420626-induced apoptosis in HepG2 cells. AR420626, a selective GPR41/FFA3 agonist, induces mammalian target of rapamycin (mTOR) phosphorylation and proteasome activity and reduces pan-histone deacetylase proteins, which results in extrinsic apoptosis via increased expression of autocrine TNF-α.
Supplemental Material
Supplemental material, Fig.S1 for AR420626, a selective agonist of GPR41/FFA3, suppresses growth of hepatocellular carcinoma cells by inducing apoptosis via HDAC inhibition by Daisuke Mikami, Mamiko Kobayashi, Junsuke Uwada, Takashi Yazawa, Kazuko Kamiyama, Kazuhisa Nishimori, Yudai Nishikawa, Sho Nishikawa, Seiji Yokoi, Takanobu Taniguchi and Masayuki Iwano in Therapeutic Advances in Medical Oncology
Supplemental material, Fig.S2 for AR420626, a selective agonist of GPR41/FFA3, suppresses growth of hepatocellular carcinoma cells by inducing apoptosis via HDAC inhibition by Daisuke Mikami, Mamiko Kobayashi, Junsuke Uwada, Takashi Yazawa, Kazuko Kamiyama, Kazuhisa Nishimori, Yudai Nishikawa, Sho Nishikawa, Seiji Yokoi, Takanobu Taniguchi and Masayuki Iwano in Therapeutic Advances in Medical Oncology
Supplemental material, Fig.S3 for AR420626, a selective agonist of GPR41/FFA3, suppresses growth of hepatocellular carcinoma cells by inducing apoptosis via HDAC inhibition by Daisuke Mikami, Mamiko Kobayashi, Junsuke Uwada, Takashi Yazawa, Kazuko Kamiyama, Kazuhisa Nishimori, Yudai Nishikawa, Sho Nishikawa, Seiji Yokoi, Takanobu Taniguchi and Masayuki Iwano in Therapeutic Advances in Medical Oncology
Supplemental material, Table.S1 for AR420626, a selective agonist of GPR41/FFA3, suppresses growth of hepatocellular carcinoma cells by inducing apoptosis via HDAC inhibition by Daisuke Mikami, Mamiko Kobayashi, Junsuke Uwada, Takashi Yazawa, Kazuko Kamiyama, Kazuhisa Nishimori, Yudai Nishikawa, Sho Nishikawa, Seiji Yokoi, Takanobu Taniguchi and Masayuki Iwano in Therapeutic Advances in Medical Oncology
Acknowledgments
Daisuke Mikami and Mamiko Kobayashi contributed equally to this work.
Footnotes
Author contributions: MK, SY, YN, KN, and SN performed the cell culture experiments, Western blotting and flow cytometry. DM designed the study, analyzed the data, and wrote the paper. JU, TY, and DM performed the siRNA and TaqMan PCR experiments. KK performed the xenograft experiments. TT and MI designed the study, analyzed the data, and wrote the paper. All authors reviewed and approved the final manuscript.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by JSPS KAKENHI (grant numbers 18K06946 [Grant-in-Aid for Scientific Research (C)], 18K15971, 19K17735, 19K17702, and 19K17395 [Grant-in-Aid for Young Scientists]) provided by the Japan Society for the Promotion of Science and the National Center for Child Health and Development (29-1).
Conflict of interest statement: The authors declare that there is no conflict of interest.
Supplemental material: Supplemental material for this article is available online.
Contributor Information
Daisuke Mikami, Department of Nephrology, Faculty of Medical Sciences, University of Fukui, 23-3 Matsuoka-shimoaizuki, Eiheiji, Yoshida, Fukui 910-1193 Japan.
Mamiko Kobayashi, Department of Nephrology, Faculty of Medical Sciences, University of Fukui, Fukui, Japan.
Junsuke Uwada, Department of Biochemistry, Division of Cellular Signal Transduction, Asahikawa Medical University, Asahikawa, Japan.
Takashi Yazawa, Department of Biochemistry, Division of Cellular Signal Transduction, Asahikawa Medical University, Asahikawa, Japan.
Kazuko Kamiyama, Department of Nephrology, Faculty of Medical Sciences, University of Fukui, Fukui, Japan.
Kazuhisa Nishimori, Department of Nephrology, Faculty of Medical Sciences, University of Fukui, Fukui, Japan.
Yudai Nishikawa, Department of Nephrology, Faculty of Medical Sciences, University of Fukui, Fukui, Japan.
Sho Nishikawa, Department of Nephrology, Faculty of Medical Sciences, University of Fukui, Fukui, Japan.
Seiji Yokoi, Department of Nephrology, Faculty of Medical Sciences, University of Fukui, Fukui, Japan.
Takanobu Taniguchi, Department of Biochemistry, Division of Cellular Signal Transduction, Asahikawa Medical University, Asahikawa, Japan.
Masayuki Iwano, Department of Nephrology, Faculty of Medical Sciences, University of Fukui, Fukui, Japan.
References
- 1. Ayoub WS, Steggerda J, Yang JD, et al. Current status of hepatocellular carcinoma detection: screening strategies and novel biomarkers. Ther Adv Med Oncol 2019; 11: 1758835919869120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. He B, Dai L, Zhang X, et al. The HDAC inhibitor Quisinostat (JNJ-26481585) supresses hepatocellular carcinoma alone and synergistically in combination with Sorafenib by G0/G1 phase arrest and Apoptosis induction. Int J Biol Sci 2018; 14: 1845–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet 2003; 362: 1907–1917. [DOI] [PubMed] [Google Scholar]
- 4. Yang JD, Roberts LR. Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol 2010; 7: 448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008; 359: 378–390. [DOI] [PubMed] [Google Scholar]
- 6. Brown AJ, Goldsworthy SM, Barnes AA, et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem 2003; 278: 11312–11319. [DOI] [PubMed] [Google Scholar]
- 7. Miyamoto J, Hasegawa S, Kasubuchi M, et al. Nutritional signaling via free fatty acid receptors. Int J Mol Sci 2016; 17: 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kimura I, Inoue D, Maeda T, et al. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc Natl Acad Sci U S A 2011; 108: 8030–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tang C, Ahmed K, Gille A, et al. Loss of FFA2 and FFA3 increases insulin secretion and improves glucose tolerance in type 2 diabetes. Nat Med 2015; 21: 173–177. [DOI] [PubMed] [Google Scholar]
- 10. Thirunavukkarasan M, Wang C, Rao A, et al. Short-chain fatty acid receptors inhibit invasive phenotypes in breast cancer cells. PLoS One 2017; 12: e0186334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kobayashi M, Mikami D, Uwada J, et al. A short-chain fatty acid, propionate, enhances the cytotoxic effect of cisplatin by modulating GPR41 signaling pathways in HepG2 cells. Oncotarget 2018; 9: 31342–31354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med 2011; 17: 330–339. [DOI] [PubMed] [Google Scholar]
- 13. Gururaja TL, Yung S, Ding R, et al. A class of small molecules that inhibit TNFα-induced survival and death pathways via prevention of interactions between TNFαRI, TRADD, and RIP1. Chem Biol 2007; 14: 1105–1118. [DOI] [PubMed] [Google Scholar]
- 14. Mikami D, Kimura H, Kamiyama K, et al. Telmisartan activates endogenous peroxisome proliferator-activated receptor-δ and may have anti-fibrotic effects in human mesangial cells. Hypertens Res 2014; 37: 422–431. [DOI] [PubMed] [Google Scholar]
- 15. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 2007; 35: 495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Powers SK, Hudson MB, Nelson WB, et al. Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit Care Med 2011; 39: 1749–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Horssen R, Ten Hagen TL, Eggermont AM. TNF-α in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist 2006; 11: 397–408. [DOI] [PubMed] [Google Scholar]
- 18. Pandian GN, Sugiyama H. Strategies to modulate heritable epigenetic defects in cellular machinery: lessons from nature. Pharmaceuticals (Basel) 2012; 6: 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Y, Nicholatos J, Dreier JR, et al. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 2014; 513: 440–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 2006; 5: 769–784. [DOI] [PubMed] [Google Scholar]
- 21. Lachenmayer A, Toffanin S, Cabellos L, et al. Combination therapy for hepatocellular carcinoma: additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J Hepatol 2012; 56: 1343–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim HS, Shen Q, Nam SW. Histone deacetylases and their regulatory MicroRNAs in hepatocarcinogenesis. J Korean Med Sci 2015; 30: 1375–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xie HJ, Noh JH, Kim JK, et al. HDAC1 inactivation induces mitotic defect and caspase-independent autophagic cell death in liver cancer. PLoS One 2012; 7: e34265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Noh JH, Jung KH, Kim JK, et al. Aberrant regulation of HDAC2 mediates proliferation of hepatocellular carcinoma cells by deregulating expression of G1/S cell cycle proteins. PLoS One 2011; 6: e28103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Quint K, Agaimy A, Di Fazio P, et al. Clinical significance of histone deacetylases 1, 2, 3, and 7: HDAC2 is an independent predictor of survival in HCC. Virchows Arch 2011; 459: 129–139. [DOI] [PubMed] [Google Scholar]
- 26. Wu LM, Yang Z, Zhou L, et al. Identification of histone deacetylase 3 as a biomarker for tumor recurrence following liver transplantation in HBV-associated hepatocellular carcinoma. PLoS One 2010; 5: e14460. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27. Zhang J, Yang Y, Yang T, et al. microRNA-22, downregulated in hepatocellular carcinoma and correlated with prognosis, suppresses cell proliferation and tumourigenicity. Br J Cancer 2010; 103: 1215–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fan J, Lou B, Chen W, et al. Down-regulation of HDAC5 inhibits growth of human hepatocellular carcinoma by induction of apoptosis and cell cycle arrest. Tumour Biol 2014; 35: 11523–11532. [DOI] [PubMed] [Google Scholar]
- 29. Kanno K, Kanno S, Nitta H, et al. Overexpression of histone deacetylase 6 contributes to accelerated migration and invasion activity of hepatocellular carcinoma cells. Oncol Rep 2012; 28: 867–873. [DOI] [PubMed] [Google Scholar]
- 30. Wu J, Du C, Lv Z, et al. The up-regulation of histone deacetylase 8 promotes proliferation and inhibits apoptosis in hepatocellular carcinoma. Dig Dis Sci 2013; 58: 3545–3553. [DOI] [PubMed] [Google Scholar]
- 31. Chen J, Jin R, Zhao J, et al. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett 2015; 367: 1–11. [DOI] [PubMed] [Google Scholar]
- 32. Ray EM, Sanoff HK. Optimal therapy for patients with hepatocellular carcinoma and resistance or intolerance to sorafenib: challenges and solutions. J Hepatocell Carcinoma 2017; 4: 131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kaji I, Akiba Y, Furuyama T, et al. Free fatty acid receptor 3 activation suppresses neurogenic motility in rat proximal colon. Neurogastroenterol Motil 2018; 30: e13157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Said H, Akiba Y, Narimatsu K, et al. FFA3 activation stimulates duodenal bicarbonate secretion and prevents NSAID-induced enteropathy via the GLP-2 pathway in rats. Dig Dis Sci 2017; 62: 1944–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Christiansen CB, Gabe MBN, Svendsen B, et al. The impact of short-chain fatty acids on GLP-1 and PYY secretion from the isolated perfused rat colon. Am J Physiol Gastrointest Liver Physiol 2018; 315: G53–G65. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Fig.S1 for AR420626, a selective agonist of GPR41/FFA3, suppresses growth of hepatocellular carcinoma cells by inducing apoptosis via HDAC inhibition by Daisuke Mikami, Mamiko Kobayashi, Junsuke Uwada, Takashi Yazawa, Kazuko Kamiyama, Kazuhisa Nishimori, Yudai Nishikawa, Sho Nishikawa, Seiji Yokoi, Takanobu Taniguchi and Masayuki Iwano in Therapeutic Advances in Medical Oncology
Supplemental material, Fig.S2 for AR420626, a selective agonist of GPR41/FFA3, suppresses growth of hepatocellular carcinoma cells by inducing apoptosis via HDAC inhibition by Daisuke Mikami, Mamiko Kobayashi, Junsuke Uwada, Takashi Yazawa, Kazuko Kamiyama, Kazuhisa Nishimori, Yudai Nishikawa, Sho Nishikawa, Seiji Yokoi, Takanobu Taniguchi and Masayuki Iwano in Therapeutic Advances in Medical Oncology
Supplemental material, Fig.S3 for AR420626, a selective agonist of GPR41/FFA3, suppresses growth of hepatocellular carcinoma cells by inducing apoptosis via HDAC inhibition by Daisuke Mikami, Mamiko Kobayashi, Junsuke Uwada, Takashi Yazawa, Kazuko Kamiyama, Kazuhisa Nishimori, Yudai Nishikawa, Sho Nishikawa, Seiji Yokoi, Takanobu Taniguchi and Masayuki Iwano in Therapeutic Advances in Medical Oncology
Supplemental material, Table.S1 for AR420626, a selective agonist of GPR41/FFA3, suppresses growth of hepatocellular carcinoma cells by inducing apoptosis via HDAC inhibition by Daisuke Mikami, Mamiko Kobayashi, Junsuke Uwada, Takashi Yazawa, Kazuko Kamiyama, Kazuhisa Nishimori, Yudai Nishikawa, Sho Nishikawa, Seiji Yokoi, Takanobu Taniguchi and Masayuki Iwano in Therapeutic Advances in Medical Oncology